Cilia-Localized Counterregulatory Signals as Drivers of Renal Cystogenesis
 Rebecca V. Walker1
Rebecca V. Walker1  Anthony Maranto1 Vivek Reddy Palicharla2 Sun-Hee Hwang2
Anthony Maranto1 Vivek Reddy Palicharla2 Sun-Hee Hwang2  Saikat Mukhopadhyay2
Saikat Mukhopadhyay2  Feng Qian1*
Feng Qian1*- 1Division of Nephrology, Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, United States
- 2Department of Cell Biology, UT Southwestern Medical Center, Dallas, TX, United States
Primary cilia play counterregulatory roles in cystogenesis—they inhibit cyst formation in the normal renal tubule but promote cyst growth when the function of polycystins is impaired. Key upstream cilia-specific signals and components involved in driving cystogenesis have remained elusive. Recent studies of the tubby family protein, Tubby-like protein 3 (TULP3), have provided new insights into the cilia-localized mechanisms that determine cyst growth. TULP3 is a key adapter of the intraflagellar transport complex A (IFT-A) in the trafficking of multiple proteins specifically into the ciliary membrane. Loss of TULP3 results in the selective exclusion of its cargoes from cilia without affecting their extraciliary pools and without disrupting cilia or IFT-A complex integrity. Epistasis analyses have indicated that TULP3 inhibits cystogenesis independently of the polycystins during kidney development but promotes cystogenesis in adults when polycystins are lacking. In this review, we discuss the current model of the cilia-dependent cyst activation (CDCA) mechanism in autosomal dominant polycystic kidney disease (ADPKD) and consider the possible roles of ciliary and extraciliary polycystins in regulating CDCA. We then describe the limitations of this model in not fully accounting for how cilia single knockouts cause significant cystic changes either in the presence or absence of polycystins. Based on available data from TULP3/IFT-A-mediated differential regulation of cystogenesis in kidneys with deletion of polycystins either during development or in adulthood, we hypothesize the existence of cilia-localized components of CDCA (cCDCA) and cilia-localized cyst inhibition (CLCI) signals. We develop the criteria for cCDCA/CLCI signals and discuss potential TULP3 cargoes as possible cilia-localized components that determine cystogenesis in kidneys during development and in adult mice.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is characterized by the formation of numerous fluid-filled cysts (Igarashi and Somlo, 2002; Harris and Torres, 2009) from all tubule segment origins. The cysts destroy normal parenchyma and cause kidney failure in more than half of the patients by the age of 60 (Gabow, 1993). ADPKD is caused primarily by mutations in PKD1 or PKD2, which encodes polycystin-1 (PC1) or polycystin-2 (PC2), respectively (polycystins or PCs collectively) (The European Polycystic Kidney Disease Consortium, 1994; Mochizuki et al., 1996). PC1 is a 4302 aa 11-transmembrane (TM) protein (Hughes et al., 1995) with a GPCR autoproteolysis-inducing (GAIN) domain located upstream of the first TM domain (Ponting et al., 1999; Arac et al., 2012). PC1 undergoes autoproteolytic cleavage at a G protein-coupled receptor (GPCR) proteolysis site (GPS) within the GAIN domain—a key post-translational modification of the protein (Qian et al., 2002; Yu et al., 2007; Chapin et al., 2010; Cai et al., 2014; Kim et al., 2014; Kurbegovic et al., 2014; Gainullin et al., 2015; Padovano et al., 2020). PC2 belongs to the transient receptor potential (TRP) channel superfamily of proteins (Koulen et al., 2002) and can form a homotetramer that functions as a non-selective cation channel (Shen et al., 2016; Grieben et al., 2017). PC1 and PC2 form a stable complex with a 1:3 stoichiometry (Yu et al., 2009; Zhu et al., 2011; Su et al., 2018). Recent studies have shown that PC1/PC2 complex has an ion channel function with properties that are distinct from that of the homomeric PC2 channel (Wang et al., 2019; Ha et al., 2020). Moreover, the PC1 subunit directly contributes to the channel pore and thereby affects its ion channel function (Wang et al., 2019). Polycystins transduce intracellular calcium signals in response to extracellular stimuli (Nauli et al., 2003; Delmas, 2004; Nauli and Zhou, 2004) by unknown mechanisms. Multiple downstream cellular pathways, such as extracellular regulated kinase (Shibazaki et al., 2008; Ma et al., 2013), mTOR (Shillingford et al., 2006; Boletta, 2009; Distefano et al., 2009), cAMP (Torres et al., 2004; Wang et al., 2018), WNT (Kim et al., 2016), Ca2+ (Koulen et al., 2002; Nauli et al., 2003; Cantiello, 2004; Anyatonwu et al., 2007; Kim et al., 2016) and G-protein signaling (Parnell et al., 1998; Hama and Park, 2016; Parnell et al., 2018; Zhang et al., 2018) are dysregulated in polycystic kidneys with mutated polycystins. How these downstream pathways are mechanistically linked to cystogenesis caused by a lack of polycystins remains largely unknown. Although current therapies target many of these pathways, they have achieved limited effects and showed significant side effects that restrict their usage (Torres et al., 2010; Torres and Harris, 2014; Torres et al., 2017; Torres et al., 2020). Many therapies are targeted toward preventing the progression of the disease rather than halting disease initiation. Indeed, distinguishing the changes that arise from initiation of, and are proximate to, cystogenesis from those that are secondary to the progression of cysts has been challenging. Key upstream signals that are regulated by polycystins and determine kidney cystogenesis in ADPKD remain unknown, preventing the development of more effective therapies.
Complex Role of the Primary Cilium in Cystogenesis
An important clue into the role of the polycystins in tubular homeostasis came from the findings that both proteins localize to the primary cilium (Pazour et al., 2002; Yoder et al., 2002; Nauli et al., 2003; Nauli and Zhou, 2004). The primary cilium is a small hair-like projection from the apical surface of almost all cell types (Rosenbaum and Witman, 2002; Wheatley, 2005). Primary cilia function as sensory antennae in most vertebrate cells playing fundamental roles in cell cycle control, cellular differentiation, and polarity (Goetz and Anderson, 2010; Anvarian et al., 2019). In the kidney, primary cilia jut out into the tubular lumen and are thought to sense urine flow-induced shear stress or ion composition (Nauli et al., 2003; Praetorius et al., 2003).
The ciliary localization of polycystins has led ADPKD to be categorized as a ciliopathy, a genetic disease caused by defective cilium-resident proteins and ciliary dysfunction. In fact, kidney cyst formation during development appears to be common pathogenesis for many other ciliopathies. These results suggest that cilia regulate tubular homeostasis and that ciliary dysfunction is a key upstream pathogenic step leading to tubular dilatation and cystogenesis (Pazour, 2004; Nauli et al., 2006). It has been suggested that ciliary polycystins may sense mechanical stimuli by mediating calcium entry into cilia, and loss of the mechanically induced cilia-initiated calcium signaling may underlie cystogenesis (Nauli et al., 2003). This view has recently been challenged by the Clapham laboratory who demonstrated that primary cilia are not calcium-responsive mechanosensors (Delling et al., 2016).
Genetic studies have shown a complex role of primary cilia in cystogenesis. Loss of cilia, by deletion of the intraflagellar transport complex-B proteins (Davenport et al., 2007; Jonassen et al., 2008) or kinesin-II (Lin et al., 2003), causes mild fibrocystic disease with slow cyst growth in the kidney. This finding suggested that cilia inhibit cyst formation in normal renal tubules. By comparison, loss of the polycystins caused significantly more severe cystogenesis marked by early and rapid cyst growth while keeping intact ciliary structure (Lu et al., 1997; Boulter et al., 2001; Piontek et al., 2004), for example see Figure 1A. In a pioneering study, the Somlo laboratory has shown that concomitant loss of polycystins and cilia results in an intermediate phenotype (Figure 1B), closer to that of loss of cilia alone (Figure 1C) (Ma et al., 2013). Therefore, polycystins play an important role in cilia signaling to suppress tubular dilation and cystogenesis. Furthermore, the extent of the suppression was directly related to the length of time between the initial loss of the polycystins and the subsequent involution of cilia—the disease worsened when this time interval was lengthened (Ma et al., 2013). The suppression of cyst growth by the loss of cilia was found to occur in all segments of the renal tubules following both early and late Pkd1 or Pkd2 gene deletion. The genetic epistasis data indicate that primary cilia play dual and opposing roles in cyst development dependent on the presence of polycystins: they inhibit cyst formation in the normal renal tubule but promote cyst growth when polycystins are lacking.
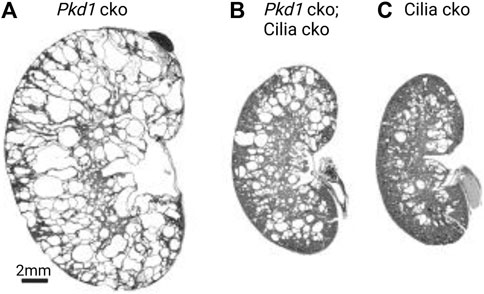
FIGURE 1. Complex roles of cilia in cystogenesis. Cysts from PC1 loss are severe and only partially suppressed from cilia loss. Images adapted from (Ma et al., 2013) with permission. Postnatal day 24 (P24) kidneys from (A) Pkd1 cko (Pkhd1-cre;Pkd1fl/fl), (B) Pkd1 cko; cilia cko (Pkhd1-cre;Kif3afl/-;pkd1fl/fl), and (C) cilia cko (Pkhd1-cre;Kif3afl/-) mice. Abbreviations: cko, conditional knockout.
PC1/PC2 Repressed Cilia-Dependent Cyst Activation Signal
Based on the above-mentioned genetic epistasis data from Pkd1/Pkd2 and cilia conditional knockout (cko) mice, the Somlo laboratory has proposed an unidentified cilia-dependent cyst activation (CDCA) signal(s) (Ma et al., 2013; Ma et al., 2017). The CDCA signal is dependent on intact cilia for activity and is normally inhibited by the functioning of polycystins (Figure 2A). The function of polycystins in suppressing CDCA might be part of a normal physiological or homeostatic cilia-dependent signaling pathway promoting functional tubule adaptation to either chemical or mechanical signals (Ma et al., 2013). Loss of polycystins in the presence of intact cilia—the condition for cyst initiation in ADPKD—leads to upregulation of CDCA, perhaps via ciliary translocation, and constitutive activation (Figure 2B). This leads to uncontrolled lumen diameter expansion and cyst growth (Ma et al., 2013; Ma et al., 2017; Ma, 2021). Ciliary localization of CDCA is required for full activation in Pkd1 cko; the CDCA signal is impeded when cilia are co-ablated, thus resulting in a reduction of the cystic burden in the Pkd1-cilia double mutants (Figure 2C).
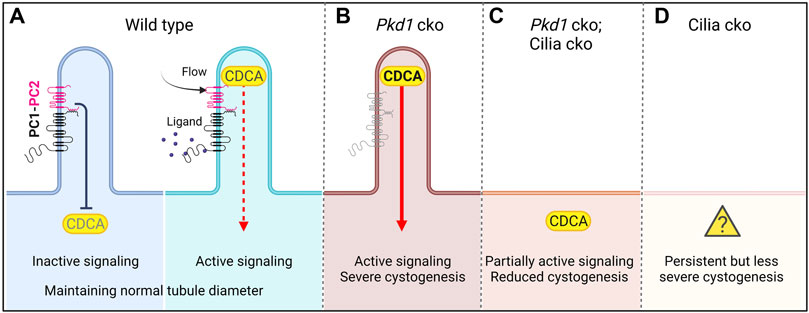
FIGURE 2. Cilia-dependent cyst activation (CDCA) model for cystogenesis in ADPKD (adapted from Ma, 2021). (A) The CDCA in the wild-type cell is suppressed by polycystins and is activated through an unknown mechanism, possibly ligand binding to polycystins or flow bending the cilium. (B) In Pkd1 cko, CDCA is derepressed and constitutively activated leading to severe cystogenesis. (C) If Pkd1 and cilia are co-ablated, the CDCA cannot be upregulated. The cystogenic signal is thus diminished compared with single Pkd1 cko, resulting in reduced cystogenesis. In this model, the CDCA signal must retain a partial “leaky” function in the cytoplasm to explain how Pkd1-cilia double mutants display significant cystic changes. (D) In cilia single mutants, there is persistent mild cystogenesis. Here the CDCA must have a leaky function despite the presence of intact polycystins and lack of cilia (“?”). Created in BioRender.
The molecular nature of CDCA components and effectors are unknown. The CDCA mechanism of cyst growth is active in all tubular segments following both early and late ADPKD gene inactivation (Ma et al., 2013). The CDCA signal(s) is likely distinct from MAPK/ERK (Shibazaki et al., 2008; Distefano et al., 2009), mTOR (Pema et al., 2016), cAMP (Torres and Harris, 2014; Bergmann et al., 2018), and phosphorylated cAMP Response Element-Binding Protein (pCREB) signaling (Kakade et al., 2016) at a cellular level (Ma et al., 2013), which are altered in cystic kidneys. These pathways are unlikely to be central to the CDCA mechanism because they show restricted nephron segment specificities and later activation (Ma et al., 2013). These pathways may be activated only in certain cell types or conditions to actively promote cyst growth, making them unlikely to be universally applicable targets for reducing cyst growth. A recent study has shown that signaling through the Hedgehog pathway is not required for cystic phenotype caused by loss of function of Pkd1 (Ma et al., 2019). In fact, neither activation nor repression of the Hedgehog signaling components (Smo, Gli2, and Gli3) influenced the progression of polycystic kidney disease in mouse Pkd1 models of developmental or adult-onset of ADPKD. These results suggested that the Hedgehog signaling pathway does not contribute to the CDCA or other ciliary signals that drive renal cystogenesis.
Roles of Ciliary and Extraciliary Polycystins in Regulating Cilia-Dependent Signaling
PC1 and PC2 localize to multiple subcellular compartments in the cell body in addition to cilia where they can exert their various functions (Boletta and Germino, 2003; Kottgen and Walz, 2005; Chapin and Caplan, 2010). This raises the question as to which of the polycystins pools may be involved in regulating the cilia-dependent signaling. Ciliary polycystins may inhibit the CDCA within the cilia, but direct evidence is lacking. Recent data suggest a critical role of the ciliary polycystins in this role. Walker et al. (2019) have analyzed Pkd2lrm4 mutant model, a missense (E442G) mutant variant that encodes a channel-functional (Yoshiba et al., 2012) but non-cilia localizing (Grimes et al., 2016; Walker et al., 2019) form of PC2. The mutant mice developed a Pkd2 null-like phenotype characterized by embryonic kidney cysts (Walker et al., 2019). This finding suggests that ciliary exclusion of PC2 is sufficient to cause kidney cystogenesis in a mouse model of ADPKD. Similarly, PC2W414G, a human pathogenic variant, retains its channel activity but fails to traffic to the cilia (Cai et al., 2014). Cai et al. (2014) examined the ciliary localization of a cohort of human missense pathogenic variants of PC1 and PC2 and found that ∼70% of them exhibit defects in ciliary trafficking. These data suggest that ciliary polycystins may be necessary to prevent kidney cyst formation and functional polycystins remaining in the cell body is not sufficient to counter the cystogenic signal from the cilium. The molecular mechanism by which the ciliary pool of polycystins inhibits the CDCA is unknown.
A critical role of the ciliary PC1 is further suggested by the finding of a GPS cleavage resistant mouse PC1 mutant with an amino acid substitution L3040H within the GAIN/GPS domain (Cai et al., 2014). This mutant did not reach cilia and its expression by a BAC transgene could not rescue embryonic cystogenesis and lethality in mouse Pkd1 mutant background. However, the PC1 mutant (L3040H) expressed in the cell body remains completely Endoglycosidase H (Endo H) sensitive implying that this mutant is retained in the ER and defective in the trafficking (Cai et al., 2014). It is thus unclear whether loss-of-function observed for the PC1-L3040H mutant may be a secondary consequence of the unfolding of the GAIN domain or global structural disruption, making it difficult to conclude a role of the extraciliary pool of PC1. Interestingly, studies of the hypomorphic Pkd1 knock-in model, Pkd1V/V, provided evidence for a role of the extraciliary pools of polycystins in regulating the cilia-dependent signals (Yu et al., 2007). The Pkd1V/V mouse contains a single amino acid substitution T3041V at the GAIN/GPS domain, which blocks autoproteolytic cleavage of PC1 (Qian et al., 2002; Yu et al., 2007; Trudel et al., 2016). The resulting non-cleavable PC1V mutant is excluded from cilia without disrupting the cilia (Kim et al., 2014). The Pkd1V/V mice escaped renal cystogenesis and lethality during embryonic stages that are seen in Pkd1 null models but started cystic dilation in distal nephron segments and collecting ducts at birth, culminating in death at ∼3 weeks of age (Yu et al., 2007). The lack of ciliary access of this mutant PC1 form supports a mechanism that a CDCA signal is generated in intact cilia that are devoid of polycystins. Molecular analyses of the uncleavable PC1V mutant protein showed that this mutant acquires Endo H resistance and thus can exit the ER and is functional for intracellular trafficking (Kim et al., 2014; Kurbegovic et al., 2014; Trudel et al., 2016). We recently showed that this extraciliary PC1 can form a functional ion channel complex with PC2 in Xenopus oocytes (Wang et al., 2019). The delayed and restricted cystogenesis in the Pkd1V/V model thus suggested that the extra-ciliary PC1 likely suppresses CDCA or its effector pathway(s), albeit less effectively than the wild-type protein. It remains to be seen whether the anti-cystogenic role of PC1V in the cell body is mediated through its function in the mitochondria (Padovano et al., 2017) or via interaction with the cell matrix (Lee et al., 2015) or by the ion channel function of the polycystin complex at extraciliary membranes (Wang et al., 2019; Ha et al., 2020; Vien et al., 2020).
Cyst Formation in Cilia Mutants: Cilia-Regulated Signals That Suppress Cystogenesis Independent of Polycystins
The concomitant ablation of cilia significantly suppressed rapid cyst growth in Pkd1 cko compared with Pkd1 cko single mutants. However, instead of completely resolving renal cyst development, this suppression was only partial, resulting in an intermediate phenotype of cyst growth, closer to that of loss of cilia alone (Ma et al., 2013). This was described by Ma et al. (2013), which shows that Pkd1-cilia double cko produced phenotypes more severe than cilia cko but much less severe than Pkd1 cko when measured by the kidney to body weight ratio, cystic index, and serum urea nitrogen. The intermediate phenotype was consistent for both Pkd1 and Pkd2 mutants when in combination with a cilia cko, suggesting that this is a consistent phenomenon for the interaction between polycystins and cilia. This observation highlights several gaps in the CDCA model as it stands. First, cilia single knockouts cause significant cystic changes in the presence of polycystins, which should otherwise inhibit the CDCA to prevent cystogenesis in normal tubules (Fig. 1C). Without cilia, the cilia-dependent signal should be abolished and the cyst inhibiting role of polycystins should prevail. Second, cyst growth continues to persist in Pkd1-cilia double mutants (Fig. 1B); however, loss of cilia should prevent CDCA from initiating cystogenesis if the cyst activation signal solely originates in the cilium. Moreover, neither concomitant reduction in dose (by half) of PC1 nor transgenic overexpression of PC1 were found to have an impact on the cystic burden in cilia mutants (Ma et al., 2013). These data indicate that cysts from cilia single knockouts arise independently of PC1.
The above observations have additional implications. First, and according to the model (Ma et al., 2017; Ma, 2021) (Figure 2), the CDCA signal has to retain a partial “leaky” function or quiescent state in the cytoplasm to explain how Pkd1-cilia double mutants display significant cystic changes. Second, in cilia single mutants the CDCA has to be activated in absence of cilia and in the presence of intact polycystins to develop cysts (Figure 2D, “?”). An alternative parsimonious interpretation would be that persistent cyst growth in Pkd1-cilia double mutants arises from additional ciliary roles independent of polycystins. Cilia-defective mutants are likely to cause fibrocystic kidney disease phenotype by mechanisms (Jonassen et al., 2008; Choi et al., 2011) that are likely divergent from loss of polycystins (Shibazaki et al., 2008). The cilia-regulated signal(s) could normally suppress cystogenesis, parallel to the CDCA. Such signal(s) would require localization to cilia for full activity and cannot be generated if cilia are ablated as in cilia-single and Pkd1-cilia double mutants to suppress cystogenesis. We annotate this polycystin-independent ciliary component as cilia-localized cyst inhibition (CLCI) signal(s).
Overall, these considerations suggest a more complex cilia-regulated mechanism in cyst growth involving a combination of positive and negative regulatory signals in ADPKD. These counterregulatory signals generated within the cilium are in a finely tuned balance to facilitate functional tubule adaptation to physiological inputs in normal kidneys. Dysregulation of these ciliary signals may underlie cystic kidney diseases.
Cilia-Dependent Signaling Within Cilia: Challenges of Assessment
Several difficulties have limited our ability to dissect the crucial ciliary signals. First, ciliary signals cannot be identified in experimental models that lack cilia. Ablation of cilia, an important cellular compartment, disrupts a great number of cellular pathways, including cell cycle regulation and cell polarity (Anvarian et al., 2019). The crude disruption of cilia, therefore, obscures our ability to determine the nature of the ciliary signals that are pertinent to cystogenesis. Identification of these ciliary signals will necessitate approaches that retain intact cilia. Second, uncoupling ciliary signals causing tissue phenotypes, such as cystogenesis, from the downstream pathways affected is difficult (Mukhopadhyay et al., 2017). Third, the small size of the cilium with respect to the cell (Delling et al., 2013) makes ciliary perturbations difficult to detect. Fourth, ciliary proteins, including PC1 and PC2 (Pisitkun et al., 2004; Hogan et al., 2009; Wang et al., 2015; Hardy and Tsiokas, 2020; Hu and Harris, 2020; Lea et al., 2020) have additional extraciliary roles necessitating approaches that selectively target ciliary pools of signaling proteins.
The components of the CDCA signal may be present in cilia and cytoplasm (Ma et al., 2017; Ma, 2021), but their relative contributions remain unclear. A recent transcriptomic study using Pkd1 single and Pkd1-cilia double mutant kidneys has identified non-ciliary cyclin-dependent kinase 1 as a driver of cyst cell proliferation from Pkd1 inactivation but did not find changes in ciliary drivers in cystogenesis (Zhang et al., 2021). The lack of detection of the ciliary drivers in the study may reflect heterogenous cystic mechanisms between polycystin and cilia loss. Alternatively, changes in ciliary signaling or trafficking of ciliary components may be too small to be detected at the global transcriptional level and are unlikely to be detected by the whole kidney analyses.
Tubby-Like Protein 3 is a key Adapter of the IFT-A Complex in Trafficking Multiple Proteins to the Ciliary Membrane
The tubby family member, Tubby-like protein 3 (TULP3) is a key adapter of the intraflagellar transport complex A (IFT-A) in the trafficking of multiple proteins specifically into the ciliary membrane. The IFT-A holo-complex is generally considered to be regulating the retrograde trafficking of cargoes including IFT-B complex in the cilia (Piperno et al., 1998; Iomini et al., 2001; Iomini et al., 2009). TULP3 interacts with the IFT-A core (consisting of IFT140/122/144 subunits) (Mukhopadhyay et al., 2010; Behal et al., 2012) through its N-terminus to enable trafficking of itself and TULP3 bound cargoes into cilia (Mukhopadhyay et al., 2010; Badgandi et al., 2017). Thus, the IFT-A core complex has an additional function in the pre-ciliary trafficking of TULP3 and cargoes, in addition to its established role in retrograde trafficking in cilia. TULP3 mediates ciliary trafficking by a 3-step mechanism: 1) capture of membrane cargo by the tubby domain in a PI(4,5)P2-dependent manner, 2) ciliary delivery by IFT-A core-binding to TULP3 N-terminus, and 3) release into PIP2-deficient ciliary membrane (Figure 3) (Badgandi et al., 2017). Loss of TULP3 results in the selective exclusion of its cargoes from cilia without affecting their extraciliary pools and without disrupting cilia or IFT-A complex integrity. Therefore, studying TULP3 provides a unique opportunity to investigate potential ciliary components that regulate cystogenesis from intact cilia.
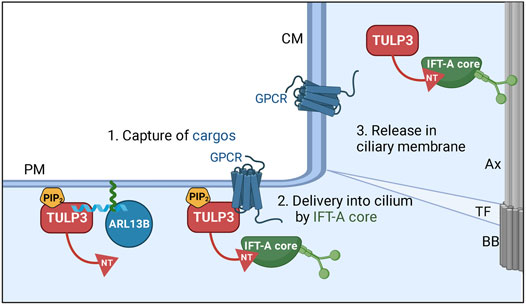
FIGURE 3. Model for TULP3 and IFT-A—mediated trafficking of cargoes into cilia. TULP3 tubby domain is anchored to the plasma membrane by PI(4,5)P2. The tubby domain captures short CLS (cilia localization signal) peptide regions in diverse cargoes. The N-terminus (NT) of TULP3 binds to the IFT-A core subunits (IFT140, IFT122, IFT144) and recruits the tubby domain-bound cargoes to cilia. After reaching cilia, the lack of PI(4,5)P2 in the ciliary membrane could dislodge the cargoes from the tubby domain. The cargoes are very diverse and include transmembrane proteins and membrane-associated proteins such as ARL13B. The N-terminus amphipathic helix in ARL13B that binds the tubby domain in TULP3 is shown in blue. ARL13B is anchored to the membrane by palmitoylation (green) inside the helix. Abbreviations: Ax, Axoneme; TF, transition fiber; BB, Basal body; CM, ciliary membrane; PM, plasma membrane. Created in BioRender.
The IFT-A core and peripheral subunit mutants also affect the ciliary localization of multiple TULP3 cargoes (Fu et al., 2016; Takahara et al., 2018; Picariello et al., 2019; Kobayashi et al., 2021; Quidwai et al., 2021) indicating that IFT-A is required for their ciliary trafficking. Substitution of Ift140 subunit in Chlamydomonas with a WD-40 repeat deleted Ift140 fragment restores flagella partially, as opposed to lack in Ift140 knockout. Instead, trafficking of multiple lipidated proteins, including ARL13 and other farnesylated and myristoylated proteins, are affected. Thus IFT-A could have an evolutionary function in the pre-ciliary trafficking of cargoes to cilia.
Recent genetic studies of Tulp3 provided compelling evidence for cilia-localized signals in determining cyst growth. Tulp3 depletion has been shown to be deleterious but also protective in developmental (Hwang et al., 2019) and adult-onset ADPKD models in mice (Legue and Liem, 2019). These results are described in detail below and they provide a unique approach to test potential TULP3 cargoes as possible components of the cilia-localized signals that determine cystogenesis.
TULP3/IFT-A Ciliary Cargoes as Cilia-Localized Cyst Inhibition signal(s) During Development
Conditional knockout (cko) of Tulp3 in renal tubules in mice using Ksp1-Cre or HoxB7-Cre caused cyst formation during development approximating ciliary loss (Hwang et al., 2019) (Figure 4A). This result indicated that a subset of TULP3-trafficked ciliary cargoes suppresses cystogenesis in normal tubules, and that lack of these molecules in the cilia underlies the disease in the Tulp3 cko. Furthermore, Tulp3 cko mutants showed cystic changes indistinguishable from Tulp3-cilia double cko mutants (Hwang et al., 2019). This result implies that TULP3/IFT-A is necessary to traffic most, if not all, crucial components of cilia-regulated cyst inhibitory signals to cilia.
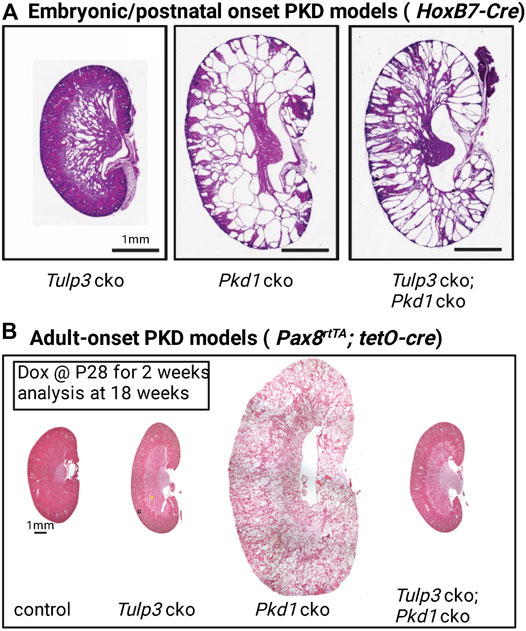
FIGURE 4. Tulp3 and Pkd1 double knockout models of PKD. (A) Kidneys from postnatal day 5 (P5). Tulp3 cko (HoxB7-cre; Tulp3fl/fl), Pkd1 cko (HoxB7-cre; Pkd1fl/fl), Tulp3 cko;Pkd1 cko (HoxB7-cre; Tulp3fl/fl;Pkd1fl/fl). cko: conditional knockout. Scale bar, 1 mm. Images adapted from (Hwang et al., 2019). (B) Adult-onset models. Pax8rtTA; tetO-cre doxycycline inducible mice were given doxycycline (Dox) starting at P28 for 2 weeks and analyzed at 18 weeks. Control (mice without tetO-cre), Tulp3 cko (Pax8rtTA; tetO-cre; Tulp3fl/fl), Pkd1 cko (Pax8rtTA; tetO-cre; Pkd1fl/fl), Tulp3 cko;Pkd1 cko (Pax8rtTA; tetO-cre; Tulp3fl/fl;Pkd1fl/fl). Images adapted from (Legue and Liem, 2019) with permission.
Cystic kidney disease in Tulp3 cko was slower to develop and less severe than that caused by loss of Pkd1. However, concomitant Tulp3 cko had a distinctive effect on cystogenesis of Pkd1 cko than from concomitant ciliary loss. Unlike loss of cilia, concomitant Tulp3 cko did not inhibit cystogenesis upon PC1 loss but rather caused earlier lethality than Pkd1 cko alone (Hwang et al., 2019), suggesting that Tulp3 inactivation accelerated loss of renal function in the Pkd1 cko. This genetic epistasis between Tulp3 and Pkd1 implied that some TULP3 ciliary cargoes suppress cystogenesis independently of polycystins during kidney development and that dysregulation of these signals may significantly contribute to the cyst growth in ADPKD.
A recent multi-group study led by the Bergmann laboratory found biallelic TULP3 mutations in patients with progressive fibrocystic kidney disease, degenerative liver fibrosis, and hypertrophic cardiomyopathy with atypical fibrotic patterns in histopathology (Devane et al., 2022). Another multi-group study led by the Harris laboratory found monoallelic loss-of-function IFT140 mutations in patients with mild polycystic kidney disease with limited kidney insufficiency. The authors analyzed the United Kingdom Biobank cystic kidney disease group and found probands with IFT140 loss-of-function variants as the third most common group after PKD1 and PKD2 (Senum et al., 2022). Conditional knockout of another IFT-A core subunit, Ift144, does not suppress cystogenesis from Pkd1 loss during embryogenesis, despite causing shortened or no cilia (Yu et al., 2022). Rather cyst number is increased arguing for a role of IFT144 cargoes in CLCI signaling (Yu et al., 2022). Although IFT140 and IFT144 are core IFT-A complex subunits (Mukhopadhyay et al., 2010; Behal et al., 2012) and regulate retrograde trafficking in cilia (Piperno et al., 1998; Iomini et al., 2001; Iomini et al., 2009), the mild polycystic kidney disease phenotype in patients with IFT140 mutations and the lack of cyst suppression in Pkd1; Ift144 double cko with respect to Pkd1 cko could partly arise from pre-ciliary function of the IFT-A core in trafficking TULP3 and its cargoes.
Based on these results, we propose that a subset of TULP3/IFT-A ciliary cargoes generate CLCI signal(s) and can be defined experimentally by the following criteria:
(i) It is trafficked to cilia.
(ii) It could be a ciliary cargo of TULP3.
(iii) Lack causes cystic changes but milder than Pkd1/2 cko.
(iv) Concomitant cko with Pkd1/2 cko enhances cystic kidney phenotype during development.
TULP3/IFT-A Ciliary Cargoes as Cilia-Localized CDCA signal(s) in Adult Kidneys
Recent epistasis data of Tulp3 and Pkd1 in adult mouse kidneys have provided compelling evidence for a critical role of Tulp3 in the trafficking of the ciliary component(s) of the CDCA signal in Pkd1 cko mice. The Liem laboratory showed that in adult mice, concomitant loss of Tulp3 completely suppressed cystogenesis in Pkd1 cko in adult mice at 18 weeks (Figure 4B) (Legue and Liem, 2019). Tulp3 cko by itself at this age caused no cystogenesis (Figure 4B). However, later at 42 weeks, Tulp3 cko did cause limited cystogenesis (Legue and Liem, 2019).
These results suggest that TULP3 traffics cilia localized cyst promoting signal(s) into cilia, which are suppressed by polycystins in normal tubules but are derepressed in adult-onset Pkd1 cko. These signals are equivalent to the ciliary components of the CDCA. Overall, these findings provide strong evidence for a cilia-localized component(s) of the CDCA signal and delineate those as a subset of Tulp3/IFT-A ciliary cargoes in adult kidneys. To highlight their cilia-specificity and distinctiveness in action within the cilia, we term the cilia-localized component ciliary CDCA or cCDCA.
The Pkd1 cko late-onset model offers a cyst suppressor system to test the cCDCA candidates from among the TULP3/IFT-A ciliary cargoes. The TULP3-dependent cCDCA signal(s) may be identified experimentally by the following criteria:
(i) It is trafficked to cilia.
(ii) It is a ciliary cargo of TULP3.
(iii) Lack causes no cystic changes in early adulthood.
(iv) Concomitant cko rescues adult-onset PKD in Pkd1/2 cko at early adulthood.
The Tran laboratory found a result similar to Tulp3 for the IFT-A peripheral subunit Ift139/Thm1. Concomitant loss of this IFT-A subunit suppresses cystogenesis from PC1 or PC2 loss in adult kidneys while retaining the cilia (Wang et al., 2022). TULP3 entry into cilia is not affected in Thm1 mutants, as only the IFT-A core is required for TULP3 trafficking (Mukhopadhyay et al., 2010; Hirano et al., 2017). However, TULP3 is accumulated in cilia or in ciliary tips upon loss of IFT139 and other IFT-A core subunits, from defects in the retrograde ciliary trafficking (Mukhopadhyay et al., 2010; Hirano et al., 2017). Whether mechanisms similar to that from loss of TULP3 underlie the suppression of cystogenesis from IFT139 loss in adult-onset ADPKD is unclear. It is possible that TULP3 cargoes functioning as cCDCA signals could show abnormal accumulation in end-state ciliary levels from defects in retrograde trafficking out of cilia in the Thm1 mutants, thereby affecting or disrupting the function of the cCDCA signals.
Regulation of Cilia-Localized Signals in Early-vs. Late-Onset Cystogenesis by TULP3
Recent studies of Tulp3 mouse models have indicated complex counterregulatory cilia localized signals that positively (cCDCA) and negatively (CLCI) impact cyst growth (Figure 5A). Given the differences in dependency of these signals on polycystins for activity, the CLCI and cCDCA are likely distinct molecular entities. If TULP3/IFT-A can traffic the components for both CLCI and cCDCA signals to cilia in the absence of polycystins, why does concomitant Tulp3 cko result in such different effects on cyst growth following early and late Pkd1 gene inactivation?
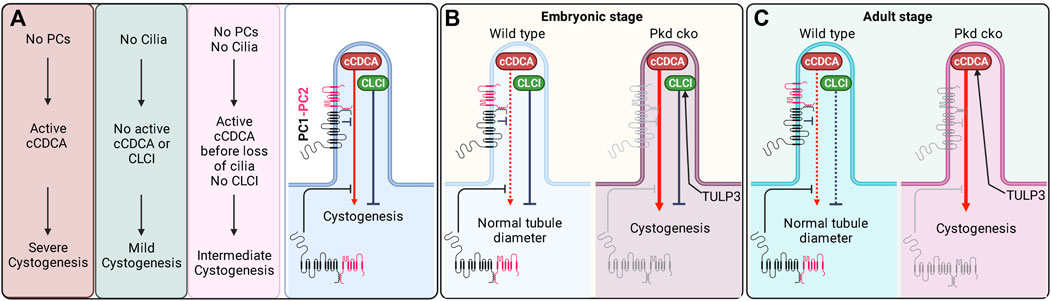
FIGURE 5. Dual roles of cilia in cystogenesis. (A) The genetic data (Figures 1, 4) implies (i) cilia-localized cilia-dependent cyst activation (cCDCA) signal(s) inhibited by PC1/2 in both the cell body and cilium and (ii) independent cilia-localized cyst inhibition (CLCI) signal(s). (B) In the embryonic stages, the cCDCA is normally weak and suppressed by polycystins. However, in the absence of polycystins, cCDCA is enhanced. TULP3 likely traffics components of the CLCI. (C) In the adult stage, the inhibitory arm is weaker and a lack of polycystins is sufficient to lead to strongly activated cCDCA. TULP3 likely traffics components of the cCDCA. Created in BioRender.
One possibility is that TULP3/IFT-A traffics different cargoes in developing vs. adult kidneys (Figures 5B,C), perhaps to meet specific needs and tasks of the kidneys that likely differ at each stage. TULP3 inactivation would result in the ciliary exclusion of a different set of cargoes in the two stages to account for the different effects. During kidney development, TULP3/IFT-A may predominantly traffic CLCI components to cilia but few cCDCA components, which are activated following the loss of polycystins (Figure 5B). There could be redundancy between TULP3 and Tubby (or other members of the family) in trafficking cCDCA components to cilia in the developing kidney. This may allow the signal(s) to be regulated by a broader number of inputs perhaps required during development when renal tubules are forming, elongating, and branching. However, unlike in adult kidney epithelia, Tubby is expressed at very low levels in the developing embryonic and perinatal kidney compared to Tulp3 (https://www.ebi.ac.uk/gxa/experiments/E-MTAB-6798/Results). In the adult kidney, TULP3/IFT-A might predominantly traffic the cCDCA component(s) unique to the adult state, but few CLCI components (until the later adult stages) (Figure 5C). The CLCI signal(s) might no longer be present or may be less effective to inhibit cyst growth at this stage when terminal tubule differentiation and maturation are complete.
Alternatively, there may be no fundamental differences in the TULP3/IFT-A regulated trafficking of cilia-signaling components between the two stages. The cCDCA may instead be trafficked by TULP3/IFT-A during development as in adult kidneys, but this regulation may be obscured by the rapid cyst growth following early inactivation of Pkd1. Conditional inactivation of Tulp3 causes a stop to the ongoing ciliary trafficking of the cCDCA components. However, the already elevated cCDCA may have delayed turnover from the cilia to halt rapid cyst growth following early Pkd1 inactivation. Residual activity of cCDCA is likely sufficient to drive considerable cyst growth that is rapidly ongoing, thus preventing a rescue of cyst growth in the Tulp3-Pkd1 double cko during development. This scenario is consistent with the previous finding that severity of cyst growth is highly sensitive to the length of time between the initial loss of the polycystins and the subsequent involution of cilia (Ma et al., 2013). Therefore, TULP3 may regulate cCDCA in addition to the CLCI during development, but fast cyst progression in absence of Pkd1 could make such regulation difficult to detect. Genetic epistasis between TULP3 cargo and Pkd1 mutants of varying severities will be required to unmask either cCDCA or CLCI signal during development (see next Section). In comparison, cysts grow at a much slower rate following Pkd1 cko in adult age. The elevated level of cCDCA may drop below a threshold that is required to promote cyst growth within a time interval that is not sufficient to drive cyst growth to a significant extent.
Such counterregulatory ciliary roles are the foundational basis of the Hedgehog (Hh) pathway (Anvarian et al., 2019; Kopinke et al., 2021). We and others (Zhang et al., 2001; Huangfu et al., 2003; Norman et al., 2009; Qin et al., 2011; Mukhopadhyay et al., 2013; Somatilaka et al., 2020) have shown that cilia play an essential role by localizing both positive (e.g., Smoothened) (Corbit et al., 2005; Rohatgi et al., 2007) and negative regulators of Hh pathway, e.g., GPCR Gpr161 (Mukhopadhyay et al., 2013) and adenylyl cyclases (Somatilaka et al., 2020). They modify Gli-transcriptional factors into repressors or activators by activating or repressing cAMP-regulated protein kinase A, strictly in a cilia-dependent manner. Even the Gli transcriptional factors localize (Haycraft et al., 2005) and transit through cilia in this process (Ocbina and Anderson, 2008; Kim et al., 2009).
Dissecting TULP3 Cargoes as Potential CLCI and cCDCA SIGNALS BY GENETIC EPISTASIS
The genetic epistasis approaches with TULP3 cargoes and Pkd1 mutants of varying severity during kidney development or in adult kidneys would inform whether TULP3 cargoes function as likely CLCI or cCDCA signals. A decrease in the severity of Pkd1 mutant cystic phenotype upon concomitant loss of a TULP3 cargo would argue for this cargo to function as a cCDCA signal. An increase in severity of Pkd1 mutant cystic phenotype upon concomitant loss of a TULP3 cargo would argue for this cargo to function as a CLCI signal.
Tulp3 deletion alone causes milder cystogenesis than Pkd1 loss. Concomitant Tulp3 cko in Pkd1 cko animals did not inhibit cystogenesis but caused earlier lethality than Pkd1 cko alone, suggesting that Tulp3 inactivation accelerated loss of renal function in the Pkd1 cko. Therefore, it is highly likely that TULP3 traffics cilia-localized cyst inhibition (CLCI) signal(s) during kidney development. Alternatively, Tulp3 could additionally regulate a cCDCA signal during development, but fast cyst progression in absence of Pkd1 could make such regulation difficult to detect. Genetic epistasis between TULP3 cargo and developmental models of Pkd1 mutants of varying severities could unmask either signal.
Lack of TULP3 in adult-onset models does not cause cystogenesis at 18 weeks. Cystogenesis in adult-onset Pkd1-Tulp3 double cko mice is fully suppressed at this stage. Intact kidney epithelial cilia in Tulp3 mutants argue for cilia-generated signaling rather than gross ciliary morphology defects in such suppression. Thus, it is highly likely that TULP3 traffics the ciliary component of CDCA (cCDCA) signal(s) predominantly in adult kidneys. Mild cystogenesis from Tulp3 deletion in adult-onset models occurs much later at 42 weeks, suggesting low CLCI activity of TULP3 cargoes only at older ages. Genetic epistasis between Tulp3 cargo mutants and Pkd1 mutants could therefore unmask cCDCA signal(s).
The complete lack of a TULP3 cargo using a conditional knockout strategy does not equate to Tulp3 cko that shows a selective loss of the corresponding TULP3 cargo from cilia alone without affecting the extraciliary pools. In certain cases, TULP3 cargoes that are selectively deficient in trafficking to cilia without affecting the functionality can be generated by mutating ciliary localizing signals targeted by TULP3 (e.g., for the GPCR cargo GPR161 (Hwang et al., 2021) or by targeting sequences that affect ciliary localization by TULP3 independent mechanisms (e.g., the RVxP motif for the lipidated protein ARL13B (Gigante et al., 2020), also a TULP3 cargo).
Although TULP3 loss does not affect ciliogenesis, complete lack of certain cargoes, such as ARL13B (Li et al., 2016; Seixas et al., 2016) or ARL13B-regulated INPP5E (Hakim et al., 2016), developmentally can cause ciliary disruption. These indirect effects of TULP3 cargoes on ciliary morphologies should be accounted for when performing genetic epistasis approaches between Pkd1 mutants and TULP3 cargo cko mutants.
Known TULP3 and IFT-A Cargoes as Potential CLCI and cCDCA Signals
Here we discuss TULP3-trafficked ciliary cargoes that might regulate both CLCI signal(s) independent of polycystins during development and cCDCA signal(s) repressed by polycystins in adult kidneys.
ARL13B
We and others recently showed that ARL13B is a TULP3 cargo in kidney epithelia in vivo (Hwang et al., 2019; Legue and Liem, 2019). ARL13B is a ciliary GTPase that regulates intraciliary trafficking of lipidated cargoes (Humbert et al., 2012). Inhibiting ARL13B would block multiple lipidated cargoes bound for cilia. In further support of the TULP3-regulated CLCI, developmental deletion of Arl13b causes mild fibrocystic kidney disease in mice models (Li et al., 2016; Seixas et al., 2016), similar to cilia loss. The zebrafish arl13b (scorpion) allele has nephric duct dilatation phenotypes, and analysis of phenotypic rescue using arl13b variants in this model suggests that ciliary localization is essential for in vivo function of ARL13B (Sun et al., 2004; Duldulao et al., 2009).
ARL13B Regulated Cargoes
ARL13B functions as a GEF for ARL3 (Gotthardt et al., 2015; Ivanova et al., 2017). ARL3GTP regulates the ciliary localization of farnesylated proteins (e.g., 5’ phosphatase INPP5E), and myristoylated proteins (e.g., NPHP3 and Cystin-1) by releasing them from their binding partners PDE6δ (Humbert et al., 2012) and UNC119B (Wright et al., 2011), respectively (Figure 6). The full list of ARL3-regulated lipidated cargoes in cilia is unknown. Residues flanking farnesylation site (CAAX box) regulate PDE6δ selectivity to cargoes (Fansa et al., 2016). The direct binding between TULP3 and certain cargoes, such as that between TULP3 and INPP5E (Humbert et al., 2012), could also factor in TULP3 mediated trafficking of these cargoes. Using IMCD3 Tulp3 ko cell lines, we recently showed that ARL13B and INPP5E were most affected, whereas NPHP3 and Cystin-1 were comparatively less affected (Palicharla et al., 2021). We also demonstrated a significant difference in the kinetics of loss of ARL13B and ARL3-dependent lipidated proteins from the Tulp3 cko kidney cilia: ARL13B is almost completely lost by P0, INPP5E by P5 and NPHP3 by P24. The percentage of ciliated cells and ciliary length in the collecting ducts was unchanged in Tulp3 cko mice (Hwang et al., 2019). The farnesylated protein LKB1, which is ARL3 independent for trafficking to cilia, is not TULP3 regulated. The effectiveness for complete depletion of INPP5E in cilia in Tulp3 cko might be related to direct binding between ARL13B and INPP5E and a requirement of such binding for effective ciliary retention of Inpp5e (Humbert et al., 2012; Qiu et al., 2021). NPHP3 is concentrated in the proximal ciliary inversin compartment by binding to NEK8 and ANKS6 that are required downstream of Inversin for NPHP3 localization (Bennett et al., 2020). Such binding might promote some retention of NPHP3 even in the absence of TULP3. Lack of Arl3 (Schrick et al., 2006) or ARL3-dependent lipidated proteins, Inpp5e and Nphp3 in developmental cko models (Wright et al., 2011; Humbert et al., 2012) or Cystin-1 mutants (Ricker et al., 2000; Hou et al., 2002; Omori et al., 2006), causes fibrocystic disease. Nonetheless, these cilia localization experiments in Tulp3 cko provide a potential road map for testing the most affected ARL13B regulated cargoes, such as INPP5E, as potential CLCI candidates.
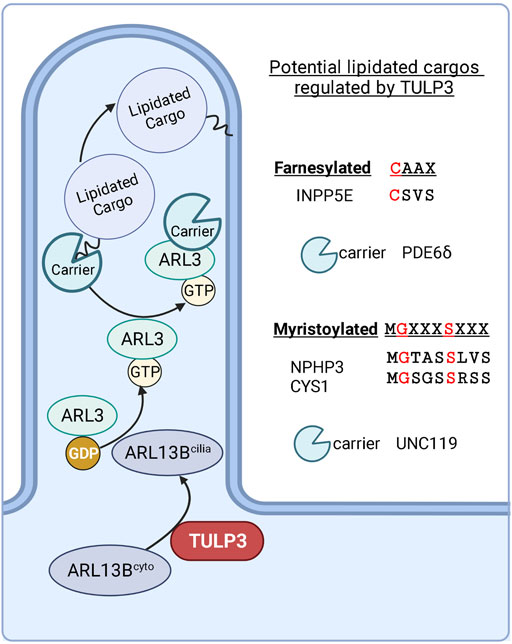
FIGURE 6. Potential lipidated cargoes of TULP3. TULP3 directs the trafficking of ARL13B to cilia. ARL13B regulates the release of farnesylated and myristoylated cargoes in the ciliary compartment via ARL3. Created in BioRender.
GPCRs
Multiple class A rhodopsin family GPCRs that are trafficked to cilia are TULP3 cargoes. One of these GPCRs, GPR161, is known to be ciliary in the IMCD3 cells (Mukhopadhyay et al., 2013) and expressed highly in the kidney CCD cells (Mehta et al., 2022). However, lack of GPR161 during kidney development does not cause cystogenesis (Hwang et al., 2019). Thus at least this GPCR is not a CLCI signal. Tolvaptan, an antagonist to the GPCR V2R, is the only FDA-approved drug to slow eGFR decline in patients (Torres et al., 2017; Torres et al., 2020). In addition to its prevailing basolateral plasma membrane localization, V2R was also reported to localize to the cilia (Raychowdhury et al., 2009; Sherpa et al., 2019). V2R is a Gαs coupled GPCR (Ausiello et al., 1987; Fenton et al., 2007), and Tolvaptan should prevent the cAMP level increase from Vasopressin. However, Tolvaptan treatment paradoxically increased cAMP levels in cilia in a V2R-independent manner (Sherpa et al., 2020), suggesting that such a nonspecific increase does not correspond to the role of V2R in the downstream signaling (Wang et al., 2005; Reif et al., 2011).
Polycystins
An hypomorphic Tulp3 mutant (K407I) shows cystogenesis in the embryonic kidney, but cilia are maintained (Legue and Liem, 2019). Ciliary disruption (Lin et al., 2003; Jonassen et al., 2008; San Agustin et al., 2016) or loss of ARL13B (Li et al., 2016; Seixas et al., 2016) or that of INPP5E (Hakim et al., 2016) does not cause cystogenesis during embryogenesis. Thus, the effects of the hypomorphic mutant are supposedly not only from lack of ARL13B/INPP5E trafficking to cilia. Polycystins themselves are also regulated in ciliary localization by TULP3 (Kim et al., 2014; Badgandi et al., 2017; Hwang et al., 2019), and PC2 is also partially reduced in ciliary levels in kidney epithelial cilia in the Tulp3 K407I mutant (Legue and Liem, 2019), which might partly explain this conundrum. PC2 trafficking to cilia is also regulated by the RVxP motif at its N-terminal end (Geng et al., 2006) while PC1 trafficking to cilia may be regulated by a similar RVxP motif at its C-terminal end (Ward et al., 2011; Su et al., 2015). The lack of the RVxP motif reduces proximity between TULP3 and PC2 (Hwang et al., 2019) but the coordination between TULP3 and the RVxP motif in the trafficking of PC2 is unclear. The Pkd2lrm4 (E442G) mutant in the first extracellular loop is unable to localize to cilia despite an intact RVxP (Walker et al., 2019), suggesting that the RVxP motif alone is not sufficient to cause a ciliary localization (Pearring et al., 2017). TULP3’s role in regulating polycystins abundance in cilia should not affect genetic epistasis analyses between potential TULP3 cargoes and polycystins to unravel cCDCA/CLCI signals.
Adenylyl Cyclases
Both AC5 (Wang et al., 2018) and AC6 (Rees et al., 2014) deletion individually reduces renal cyclic AMP and cyst growth in an orthologous mouse model of polycystic kidney disease, suggesting their role in ADPKD. A recent omics-level study of cAMP signaling components in kidney tissue showed that AC6 and AC5 are predominantly present in collecting ducts and less strong in proximal tubules (Mehta et al., 2022). AC5/6 are cilia localized. The Igarashi laboratory showed that AC5/6 are in a complex with protein A-kinase anchoring protein 150 (AKAP150) and protein kinase A (Choi et al., 2011). The authors proposed that PC2 interacts with the AC5/6 complex through its carboxy terminus. A cAMP-specific member of the phosphodiesterase family, PDE4C, is also located in renal primary cilia and interacts with the AKAP150 complex. An IFT-A core subunit IFT144 mutant (Ift144dmhd) shows reduced AC3 in the neural tube cilia (Liem et al., 2012). AC3 is one of the predominant ACs in the neurulation (Somatilaka et al., 2020). Whether such a role of IFT144 in AC3 trafficking encompasses other ciliary ACs and affects kidney expressed AC5 and AC6 is currently unknown. Besides, whether such a role of IFT144 in AC trafficking involves TULP3 interactions with the core subunits of IFT-A is also unknown. Recent optogenetic and/or chemogenetic techniques for manipulating the ciliary cAMP (Hansen et al., 2020; Truong et al., 2021) could be pivotal in determining if ciliary cAMP signaling plays a role in initiating cystogenesis.
Concluding Statement
What could be the molecular output propagated in cilia by CLCI and cCDCA signals? Based on our results showing early depletion of ARL13B and INPP5E from Tulp3 cko kidney epithelial cilia (Hwang et al., 2019; Palicharla et al., 2021), they are strong contenders for CLCI signals. INPP5E is a 5′phosphatase that generates PI(4)P from PI(4,5)P2 (Kisseleva et al., 2000). If INPP5E is the CLCI signal, by generating a PI(4)P exclusive ciliary membrane domain distinct from the plasma membrane rich in PI(4,5)P2, it could regulate ciliary components. The CLCI signal could be activating a transcription factor in cilia that is regulatable by the ciliary microenvironment. Such an example is seen in Hh pathway where GLI2 and GLI3 transcription factors are modified in cilia upon Hh addition (Haycraft et al., 2005). Some of the Gli-similar proteins (Lichti-Kaiser et al., 2012), at least two of which, GLIS3 (Kang et al., 2009) and GLIS2 (Attanasio et al., 2007), are ciliary, and show mild cystic changes perinatally upon deletion (Kang et al., 2009) or in the sensitized background of partial Kif3a loss (Lu et al., 2016). The cCDCA signal could similarly be activating a transcription factor in cilia that is regulatable by ciliary cAMP. An important feature of such a transcription factor would be that it localizes to cilia (in addition to the nucleus) but its deletion would not cause adult-onset cysts. Another feature would be its regulation of cell proliferation, like GLI2 in Hh-induced proliferation of cerebellar granule cells by CyclinD1/N-Myc (Yin et al., 2020). Nonetheless, understanding how ciliary signals transduce and amplify downstream cellular effects could provide important leads to understanding cystogenesis.
Author Contributions
FQ and SM had the idea for the article. FQ, RW, and SM performed the literature search and drafted the article. AM, VP, and S-HH critically read and revised the work. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Work in the FQ and SM laboratories is funded by the National Institutes of Health (R01DK128089). This work was also supported by Polycystic Kidney Disease Research Resource Consortium U54DK126114 (FQ) and PKD Foundation fellowship grants (215F19a to RW and 214F19a to VP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anvarian, Z., Mykytyn, K., Mukhopadhyay, S., Pedersen, L. B., and Christensen, S. T. (2019). Cellular Signalling by Primary Cilia in Development, Organ Function and Disease. Nat. Rev. Nephrol. 15, 199–219. doi:10.1038/s41581-019-0116-9
Anyatonwu, G. I., Estrada, M., Tian, X., Somlo, S., and Ehrlich, B. E. (2007). Regulation of Ryanodine Receptor-dependent Calcium Signaling by Polycystin-2. Proc. Natl. Acad. Sci. U.S.A. 104, 6454–6459. doi:10.1073/pnas.0610324104
Arac, D., Boucard, A. A., Bolliger, M. F., Nguyen, J., Soltis, S. M., Südhof, T. C., et al. (2012). A Novel Evolutionarily Conserved Domain of Cell-Adhesion GPCRs Mediates Autoproteolysis. EMBO J. 31, 1364–1378. doi:10.1038/emboj.2012.26
Attanasio, M., Uhlenhaut, N. H., Sousa, V. H., O'Toole, J. F., Otto, E., Anlag, K., et al. (2007). Loss of GLIS2 Causes Nephronophthisis in Humans and Mice by Increased Apoptosis and Fibrosis. Nat. Genet. 39, 1018–1024. doi:10.1038/ng2072
Ausiello, D. A., Skorecki, K. L., Verkman, A. S., and Bonventre, J. V. (1987). Vasopressin Signaling in Kidney Cells. Kidney Int. 31, 521–529. doi:10.1038/ki.1987.31
Badgandi, H. B., Hwang, S.-h., Shimada, I. S., Loriot, E., and Mukhopadhyay, S. (2017). Tubby Family Proteins are Adapters for Ciliary Trafficking of Integral Membrane Proteins. J. Cell Biol. 216, 743–760. doi:10.1083/jcb.201607095
Behal, R. H., Miller, M. S., Qin, H., Lucker, B. F., Jones, A., and Cole, D. G. (2012). Subunit Interactions and Organization of the Chlamydomonas Reinhardtii Intraflagellar Transport Complex A Proteins. J. Biol. Chem. 287, 11689–11703. doi:10.1074/jbc.m111.287102
Bennett, H. W., Gustavsson, A.-K., Bayas, C. A., Petrov, P. N., Mooney, N., Moerner, W. E., et al. (2020). Novel Fibrillar Structure in the Inversin Compartment of Primary Cilia Revealed by 3D Single-Molecule Superresolution Microscopy. MBoC 31, 619–639. doi:10.1091/mbc.e19-09-0499
Bergmann, C., Guay-Woodford, L. M., Harris, P. C., Horie, S., Peters, D. J. M., and Torres, V. E. (2018). Polycystic Kidney Disease. Nat. Rev. Dis. Prim. 4, 50. doi:10.1038/s41572-018-0047-y
Boletta, A., and Germino, G. G. (2003). Role of Polycystins in Renal Tubulogenesis. Trends Cell Biol. 13, 484–492. doi:10.1016/s0962-8924(03)00169-7
Boletta, A. (2009). Emerging Evidence of a Link between the Polycystins and the mTOR Pathways. Pathogenetics 2, 6. doi:10.1186/1755-8417-2-6
Boulter, C., Mulroy, S., Webb, S., Fleming, S., Brindle, K., and Sandford, R. (2001). Cardiovascular, Skeletal, and Renal Defects in Mice with a Targeted Disruption of the Pkd1 Gene. Proc. Natl. Acad. Sci. U.S.A. 98, 12174–12179. doi:10.1073/pnas.211191098
Cai, Y., Fedeles, S. V., Dong, K., Anyatonwu, G., Onoe, T., Mitobe, M., et al. (2014). Altered Trafficking and Stability of Polycystins Underlie Polycystic Kidney Disease. J. Clin. Invest. 124, 5129–5144. doi:10.1172/jci67273
Cantiello, H. F. (2004). Regulation of Calcium Signaling by Polycystin-2. Am. J. Physiol. Renal Physiol. 286, F1012–F1029. doi:10.1152/ajprenal.00181.2003
Chapin, H. C., and Caplan, M. J. (2010). The Cell Biology of Polycystic Kidney Disease. J. Cell Biol. 191, 701–710. doi:10.1083/jcb.201006173
Chapin, H. C., Rajendran, V., and Caplan, M. J. (2010). Polycystin-1 Surface Localization Is Stimulated by Polycystin-2 and Cleavage at the G Protein-Coupled Receptor Proteolytic Site. Mol. Biol. Cell 21, 4338–4348. doi:10.1091/mbc.e10-05-0407
Choi, Y.-H., Suzuki, A., Hajarnis, S., Ma, Z., Chapin, H. C., Caplan, M. J., et al. (2011). Polycystin-2 and Phosphodiesterase 4C are Components of a Ciliary A-Kinase Anchoring Protein Complex that is Disrupted in Cystic Kidney Diseases. Proc. Natl. Acad. Sci. U.S.A. 108, 10679–10684. doi:10.1073/pnas.1016214108
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y. R., and Reiter, J. F. (2005). Vertebrate Smoothened Functions at the Primary Cilium. Nature 437, 1018–1021. doi:10.1038/nature04117
Davenport, J. R., Watts, A. J., Roper, V. C., Croyle, M. J., Van Groen, T., Wyss, J. M., et al. (2007). Disruption of Intraflagellar Transport in Adult Mice Leads to Obesity and Slow-Onset Cystic Kidney Disease. Curr. Biol. 17, 1586–1594. doi:10.1016/j.cub.2007.08.034
Delling, M., Decaen, P. G., Doerner, J. F., Febvay, S., and Clapham, D. E. (2013). Primary Cilia are Specialized Calcium Signalling Organelles. Nature 504, 311–314. doi:10.1038/nature12833
Delling, M., Indzhykulian, A. A., Liu, X., Li, Y., Xie, T., Corey, D. P., et al. (2016). Primary Cilia are Not Calcium-Responsive Mechanosensors. Nature 531, 656–660. doi:10.1038/nature17426
Delmas, P. (2004). Polycystins: From Mechanosensation to Gene Regulation. Cell 118, 145–148. doi:10.1016/j.cell.2004.07.007
Devane, J., Ott, E., Olinger, E. G., Epting, D., Decker, E., Friedrich, A., et al. (2022). Progressive Liver, Kidney, and Heart Degeneration in Children and Adults Affected by TULP3 Mutations. Am. J. Hum. Genet. 109, 928. doi:10.1016/j.ajhg.2022.03.015
Distefano, G., Boca, M., Rowe, I., Wodarczyk, C., Ma, L., Piontek, K. B., et al. (2009). Polycystin-1 Regulates Extracellular Signal-Regulated Kinase-dependent Phosphorylation of Tuberin to Control Cell Size through mTOR and its Downstream Effectors S6K and 4EBP1. Mol. Cell Biol. 29, 2359–2371. doi:10.1128/mcb.01259-08
Duldulao, N. A., Lee, S., and Sun, Z. (2009). Cilia Localization is Essential for In Vivo Functions of the Joubert Syndrome Protein Arl13b/Scorpion. Development 136, 4033–4042. doi:10.1242/dev.036350
Fansa, E. K., Kösling, S. K., Zent, E., Wittinghofer, A., and Ismail, S. (2016). PDE6δ-mediated Sorting of INPP5E into the Cilium is Determined by Cargo-Carrier Affinity. Nat. Commun. 7, 11366. doi:10.1038/ncomms11366
Fenton, R. A., Brønd, L., Nielsen, S., and Praetorius, J. (2007). Cellular and Subcellular Distribution of the Type-2 Vasopressin Receptor in the Kidney. Am. J. Physiol. Renal Physiol. 293, F748–F760. doi:10.1152/ajprenal.00316.2006
Fu, W., Wang, L., Kim, S., Li, J., and Dynlacht, B. D. (2016). Role for the IFT-A Complex in Selective Transport to the Primary Cilium. Cell Rep. 17, 1505–1517. doi:10.1016/j.celrep.2016.10.018
Gabow, P. A. (1993). Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 329, 332–342. doi:10.1056/nejm199307293290508
Gainullin, V. G., Hopp, K., Ward, C. J., Hommerding, C. J., and Harris, P. C. (2015). Polycystin-1 Maturation Requires Polycystin-2 in a Dose-dependent Manner. J. Clin. Invest. 125, 607–620. doi:10.1172/jci76972
Geng, L., Okuhara, D., Yu, Z., Tian, X., Cai, Y., Shibazaki, S., et al. (2006). Polycystin-2 Traffics to Cilia Independently of Polycystin-1 by Using an N-Terminal RVxP Motif. J. Cell Sci. 119, 1383–1395. doi:10.1242/jcs.02818
Gigante, E. D., Taylor, M. R., Ivanova, A. A., Kahn, R. A., and Caspary, T. (2020). ARL13B Regulates Sonic Hedgehog Signaling from outside Primary Cilia. Elife 9, e50434. doi:10.7554/eLife.50434
Goetz, S. C., and Anderson, K. V. (2010). The Primary Cilium: A Signalling Centre during Vertebrate Development. Nat. Rev. Genet. 11, 331–344. doi:10.1038/nrg2774
Gotthardt, K., Lokaj, M., Koerner, C., Falk, N., Giessl, A., and Wittinghofer, A. (2015). A G-Protein Activation Cascade from Arl13B to Arl3 and Implications for Ciliary Targeting of Lipidated Proteins. Elife 4, e11859. doi:10.7554/eLife.11859
Grieben, M., Pike, A. C. W., Shintre, C. A., Venturi, E., El-Ajouz, S., Tessitore, A., et al. (2017). Structure of the Polycystic Kidney Disease TRP Channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 24, 114–122. doi:10.1038/nsmb.3343
Grimes, D. T., Keynton, J. L., Buenavista, M. T., Jin, X., Patel, S. H., Kyosuke, S., et al. (2016). Genetic Analysis Reveals a Hierarchy of Interactions between Polycystin-Encoding Genes and Genes Controlling Cilia Function during Left-Right Determination. PLoS Genet. 12, e1006070. doi:10.1371/journal.pgen.1006070
Ha, K., Nobuhara, M., Wang, Q., Walker, R. V., Qian, F., Schartner, C., et al. (2020). The Heteromeric PC-1/PC-2 Polycystin Complex is Activated by the PC-1 N-Terminus. Elife 9, e60684. doi:10.7554/eLife.60684
Hakim, S., Dyson, J. M., Feeney, S. J., Davies, E. M., Sriratana, A., Koenig, M. N., et al. (2016). Inpp5e suppresses polycystic kidney disease via inhibition of PI3K/Akt-dependent mTORC1 signaling. Hum. Mol. Genet. 25, 2295–2313. doi:10.1093/hmg/ddw097
Hama, T., and Park, F. (2016). Heterotrimeric G Protein Signaling in Polycystic Kidney Disease. Physiol. Genomics 48, 429–445. doi:10.1152/physiolgenomics.00027.2016
Hansen, J. N., Kaiser, F., Klausen, C., Stüven, B., Chong, R., Bönigk, W., et al. (2020). Nanobody-directed Targeting of Optogenetic Tools to Study Signaling in the Primary Cilium. Elife 9, e57907. doi:10.7554/eLife.57907
Hardy, E., and Tsiokas, L. (2020). Polycystins as Components of Large Multiprotein Complexes of Polycystin Interactors. Cell. Signal. 72, 109640. doi:10.1016/j.cellsig.2020.109640
Harris, P. C., and Torres, V. E. (2009). Polycystic Kidney Disease. Annu. Rev. Med. 60, 321–337. doi:10.1146/annurev.med.60.101707.125712
Haycraft, C. J., Banizs, B., Aydin-Son, Y., Zhang, Q., Michaud, E. J., and Yoder, B. K. (2005). Gli2 and Gli3 Localize to Cilia and Require the Intraflagellar Transport Protein Polaris for Processing and Function. PLoS Genet. 1, e53. doi:10.1371/journal.pgen.0010053
Hirano, T., Katoh, Y., and Nakayama, K. (2017). Intraflagellar Transport-A Complex Mediates Ciliary Entry and Retrograde Trafficking of Ciliary G Protein-Coupled Receptors. Mol. Biol. Cell 28, 429–439. doi:10.1091/mbc.e16-11-0813
Hogan, M. C., Manganelli, L., Woollard, J. R., Masyuk, A. I., Masyuk, T. V., Tammachote, R., et al. (2009). Characterization of PKD Protein-Positive Exosome-like Vesicles. J. Am. Soc. Nephrol. 20, 278–288. doi:10.1681/asn.2008060564
Hou, X., Mrug, M., Yoder, B. K., Lefkowitz, E. J., Kremmidiotis, G., D’Eustachio, P., et al. (2002). Cystin, a Novel Cilia-Associated Protein, Is Disrupted in the Cpk Mouse Model of Polycystic Kidney Disease. J. Clin. Invest. 109, 533–540. doi:10.1172/jci0214099
Hu, J., and Harris, P. C. (2020). Regulation of Polycystin Expression, Maturation and Trafficking. Cell. Signal. 72, 109630. doi:10.1016/j.cellsig.2020.109630
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., and Anderson, K. V. (2003). Hedgehog Signalling in the Mouse Requires Intraflagellar Transport Proteins. Nature 426, 83–87. doi:10.1038/nature02061
Hughes, J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millán, J. L., et al. (1995). The Polycystic Kidney Disease 1 (PKD1) Gene Encodes a Novel Protein with Multiple Cell Recognition Domains. Nat. Genet. 10, 151–160. doi:10.1038/ng0695-151
Humbert, M. C., Weihbrecht, K., Searby, C. C., Li, Y., Pope, R. M., Sheffield, V. C., et al. (2012). ARL13B, PDE6D, and CEP164 Form a Functional Network for INPP5E Ciliary Targeting. Proc. Natl. Acad. Sci. U.S.A. 109, 19691–19696. doi:10.1073/pnas.1210916109
Hwang, S.-H., Somatilaka, B. N., Badgandi, H., Palicharla, V. R., Walker, R., Shelton, J. M., et al. (2019). Tulp3 Regulates Renal Cystogenesis by Trafficking of Cystoproteins to Cilia. Curr. Biol. 29, 790–802. doi:10.1016/j.cub.2019.01.047
Hwang, S. H., Somatilaka, B. N., White, K., and Mukhopadhyay, S. (2021). Ciliary and Extraciliary Gpr161 Pools Repress Hedgehog Signaling in a Tissue-specific Manner. Elife 10, e67121. doi:10.7554/eLife.67121
Igarashi, P., and Somlo, S. (2002). Genetics and Pathogenesis of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 13, 2384–2398. doi:10.1097/01.asn.0000028643.17901.42
Iomini, C., Babaev-Khaimov, V., Sassaroli, M., and Piperno, G. (2001). Protein Particles in Chlamydomonas Flagella Undergo a Transport Cycle Consisting of Four Phases. J. Cell Biol. 153, 13–24. doi:10.1083/jcb.153.1.13
Iomini, C., Li, L., Esparza, J. M., and Dutcher, S. K. (2009). Retrograde Intraflagellar Transport Mutants Identify Complex A Proteins with Multiple Genetic Interactions in Chlamydomonas Reinhardtii. Genetics 183, 885–896. doi:10.1534/genetics.109.101915
Ivanova, A. A., Caspary, T., Seyfried, N. T., Duong, D. M., West, A. B., Liu, Z., et al. (2017). Biochemical Characterization of Purified Mammalian ARL13B Protein Indicates that it is an Atypical GTPase and ARL3 Guanine Nucleotide Exchange Factor (GEF). J. Biol. Chem. 292, 11091–11108. doi:10.1074/jbc.m117.784025
Jonassen, J. A., San Agustin, J., Follit, J. A., and Pazour, G. J. (2008). Deletion of IFT20 in the Mouse Kidney Causes Misorientation of the Mitotic Spindle and Cystic Kidney Disease. J. Cell Biol. 183, 377–384. doi:10.1083/jcb.200808137
Kakade, V. R., Tao, S., Rajagopal, M., Zhou, X., Li, X., Yu, A. S. L., et al. (2016). A cAMP and CREB-Mediated Feed-Forward Mechanism Regulates GSK3βin Polycystic Kidney Disease. J. Mol. Cell Biol. 8, 464–476. doi:10.1093/jmcb/mjw022
Kang, H. S., Beak, J. Y., Kim, Y.-S., Herbert, R., and Jetten, A. M. (2009). Glis3 is Associated with Primary Cilia and Wwtr1/TAZ and Implicated in Polycystic Kidney Disease. Mol. Cell Biol. 29, 2556–2569. doi:10.1128/mcb.01620-08
Kim, J., Kato, M., and Beachy, P. A. (2009). Gli2 Trafficking Links Hedgehog-dependent Activation of Smoothened in the Primary Cilium to Transcriptional Activation in the Nucleus. Proc. Natl. Acad. Sci. U.S.A. 106, 21666–21671. doi:10.1073/pnas.0912180106
Kim, H., Xu, H., Yao, Q., Li, W., Huang, Q., Outeda, P., et al. (2014). Ciliary Membrane Proteins Traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent Mechanism. Nat. Commun. 5, 5482. doi:10.1038/ncomms6482
Kim, S., Nie, H., Nesin, V., Tran, U., Outeda, P., Bai, C.-X., et al. (2016). The Polycystin Complex Mediates Wnt/Ca2+ Signalling. Nat. Cell Biol. 18, 752–764. doi:10.1038/ncb3363
Kisseleva, M. V., Wilson, M. P., and Majerus, P. W. (2000). The Isolation and Characterization of a cDNA Encoding Phospholipid-specific Inositol Polyphosphate 5-phosphatase. J. Biol. Chem. 275, 20110–20116. doi:10.1074/jbc.m910119199
Kobayashi, T., Ishida, Y., Hirano, T., Katoh, Y., and Nakayama, K. (2021). Cooperation of the IFT-A Complex with the IFT-B Complex is Required for Ciliary Retrograde Protein Trafficking and GPCR Import. Mol. Biol. Cell 32, 45–56. doi:10.1091/mbc.e20-08-0556
Kopinke, D., Norris, A. M., and Mukhopadhyay, S. (2021). Developmental and Regenerative Paradigms of Cilia Regulated Hedgehog Signaling. Seminars Cell Dev. Biol. 110, 89–103. doi:10.1016/j.semcdb.2020.05.029
Köttgen, M., and Walz, G. (2005). Subcellular Localization and Trafficking of Polycystins. Pflugers Arch. Eur. J. Physiol. 451, 286–293. doi:10.1007/s00424-005-1417-3
Koulen, P., Cai, Y., Geng, L., Maeda, Y., Nishimura, S., Witzgall, R., et al. (2002). Polycystin-2 is an Intracellular Calcium Release Channel. Nat. Cell Biol. 4, 191–197. doi:10.1038/ncb754
Kurbegovic, A., Kim, H., Xu, H., Yu, S., Cruanès, J., Maser, R. L., et al. (2014). Novel Functional Complexity of Polycystin-1 by GPS Cleavage In Vivo: Role in Polycystic Kidney Disease. Mol. Cell Biol. 34, 3341–3353. doi:10.1128/mcb.00687-14
Lea, W. A., Mcgreal, K., Sharma, M., Parnell, S. C., Zelenchuk, L., Charlesworth, M. C., et al. (2020). Analysis of the Polycystin Complex (PCC) in Human Urinary Exosome-like Vesicles (ELVs). Sci. Rep. 10, 1500. doi:10.1038/s41598-020-58087-3
Lee, K., Boctor, S., Barisoni, L. M. C., and Gusella, G. L. (2015). Inactivation of Integrin-β1 Prevents the Development of Polycystic Kidney Disease after the Loss of Polycystin-1. J. Am. Soc. Nephrol. 26, 888–895. doi:10.1681/asn.2013111179
Legue, E., and Liem, K. F. (2019). Tulp3 is a Ciliary Trafficking Gene that Regulates Polycystic Kidney Disease. Curr. Biol. 29, 803–812. doi:10.1016/j.cub.2019.01.054
Li, Y., Tian, X., Ma, M., Jerman, S., Kong, S., Somlo, S., et al. (2016). Deletion of ADP Ribosylation Factor-like GTPase 13B Leads to Kidney Cysts. J. Am. Soc. Nephrol. 27, 3628–3638. doi:10.1681/asn.2015091004
Lichti-Kaiser, K., Zeruth, G., Kang, H. S., Vasanth, S., and Jetten, A. M. (2012). Gli-similar Proteins: Their Mechanisms of Action, Physiological Functions, and Roles in Disease. Vitam. Horm. 88, 141–171. doi:10.1016/b978-0-12-394622-5.00007-9
Liem, K. F., Ashe, A., He, M., Satir, P., Moran, J., Beier, D., et al. (2012). The IFT-A Complex Regulates Shh Signaling through Cilia Structure and Membrane Protein Trafficking. J. Cell Biol. 197, 789–800. doi:10.1083/jcb.201110049
Lin, F., Hiesberger, T., Cordes, K., Sinclair, A. M., Goldstein, L. S. B., Somlo, S., et al. (2003). Kidney-specific Inactivation of the KIF3A Subunit of Kinesin-II Inhibits Renal Ciliogenesis and Produces Polycystic Kidney Disease. Proc. Natl. Acad. Sci. U.S.A. 100, 5286–5291. doi:10.1073/pnas.0836980100
Lu, W., Peissel, B., Babakhanlou, H., Pavlova, A., Geng, L., Fan, X., et al. (1997). Perinatal Lethality with Kidney and Pancreas Defects in Mice with a Targetted Pkd1 Mutation. Nat. Genet. 17, 179–181. doi:10.1038/ng1097-179
Lu, D., Rauhauser, A., Li, B., Ren, C., Mcenery, K., Zhu, J., et al. (2016). Loss of Glis2/NPHP7 Causes Kidney Epithelial Cell Senescence and Suppresses Cyst Growth in the Kif3a Mouse Model of Cystic Kidney Disease. Kidney Int. 89, 1307–1323. doi:10.1016/j.kint.2016.03.006
Ma, M., Tian, X., Igarashi, P., Pazour, G. J., and Somlo, S. (2013). Loss of Cilia Suppresses Cyst Growth in Genetic Models of Autosomal Dominant Polycystic Kidney Disease. Nat. Genet. 45, 1004–1012. doi:10.1038/ng.2715
Ma, M., Gallagher, A. R., and Somlo, S. (2017). Ciliary Mechanisms of Cyst Formation in Polycystic Kidney Disease. Cold Spring Harb. Perspect. Biol. 9, a028209. doi:10.1101/cshperspect.a028209
Ma, M., Legué, E., Tian, X., Somlo, S., and Liem, K. F. (2019). Cell-Autonomous Hedgehog Signaling is Not Required for Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 30, 2103–2111. doi:10.1681/asn.2018121274
Ma, M. (2021). Cilia and Polycystic Kidney Disease. Seminars Cell Dev. Biol. 110, 139–148. doi:10.1016/j.semcdb.2020.05.003
Mehta, Y. R., Lewis, S. A., Leo, K. T., Chen, L., Park, E., Raghuram, V., et al. (2022). "ADPKD-omics": Determinants of Cyclic AMP Levels in Renal Epithelial Cells. Kidney Int. 101, 47–62. doi:10.1016/j.kint.2021.10.014
Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S. L., Veldhuisen, B., Saris, J. J., et al. (1996). PKD2, a Gene for Polycystic Kidney Disease that Encodes an Integral Membrane Protein. Science 272, 1339–1342. doi:10.1126/science.272.5266.1339
Mukhopadhyay, S., Wen, X., Chih, B., Nelson, C. D., Lane, W. S., Scales, S. J., et al. (2010). TULP3 Bridges the IFT-A Complex and Membrane Phosphoinositides to Promote Trafficking of G Protein-Coupled Receptors into Primary Cilia. Genes Dev. 24, 2180–2193. doi:10.1101/gad.1966210
Mukhopadhyay, S., Wen, X., Ratti, N., Loktev, A., Rangell, L., Scales, S. J., et al. (2013). The Ciliary G-Protein-Coupled Receptor Gpr161 Negatively Regulates the Sonic Hedgehog Pathway via cAMP Signaling. Cell 152, 210–223. doi:10.1016/j.cell.2012.12.026
Mukhopadhyay, S., Badgandi, H. B., Hwang, S.-h., Somatilaka, B., Shimada, I. S., and Pal, K. (2017). Trafficking to the Primary Cilium Membrane. Mol. Biol. Cell 28, 233–239. doi:10.1091/mbc.e16-07-0505
Nauli, S. M., and Zhou, J. (2004). Polycystins and Mechanosensation in Renal and Nodal Cilia. Bioessays 26, 844–856. doi:10.1002/bies.20069
Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., et al. (2003). Polycystins 1 and 2 Mediate Mechanosensation in the Primary Cilium of Kidney Cells. Nat. Genet. 33, 129–137. doi:10.1038/ng1076
Nauli, S. M., Rossetti, S., Kolb, R. J., Alenghat, F. J., Consugar, M. B., Harris, P. C., et al. (2006). Loss of Polycystin-1 in Human Cyst-Lining Epithelia Leads to Ciliary Dysfunction. J. Am. Soc. Nephrol. 17, 1015–1025. doi:10.1681/asn.2005080830
Norman, R. X., Ko, H. W., Huang, V., Eun, C. M., Abler, L. L., Zhang, Z., et al. (2009). Tubby-like Protein 3 (TULP3) Regulates Patterning in the Mouse Embryo through Inhibition of Hedgehog Signaling. Hum. Mol. Genet. 18, 1740–1754. doi:10.1093/hmg/ddp113
Ocbina, P. J. R., and Anderson, K. V. (2008). Intraflagellar Transport, Cilia, and Mammalian Hedgehog Signaling: Analysis in Mouse Embryonic Fibroblasts. Dev. Dyn. 237, 2030–2038. doi:10.1002/dvdy.21551
Omori, S., Hida, M., Fujita, H., Takahashi, H., Tanimura, S., Kohno, M., et al. (2006). Extracellular Signal-Regulated Kinase Inhibition Slows Disease Progression in Mice with Polycystic Kidney Disease. J. Am. Soc. Nephrol. 17, 1604–1614. doi:10.1681/asn.2004090800
Padovano, V., Kuo, I. Y., Stavola, L. K., Aerni, H. R., Flaherty, B. J., Chapin, H. C., et al. (2017). The Polycystins Are Modulated by Cellular Oxygen-Sensing Pathways and Regulate Mitochondrial Function. Mol. Biol. Cell 28, 261–269. doi:10.1091/mbc.e16-08-0597
Padovano, V., Mistry, K., Merrick, D., Gresko, N., and Caplan, M. J. (2020). A Cut above (And below): Protein Cleavage in the Regulation of Polycystin Trafficking and Signaling. Cell. Signal. 72, 109634. doi:10.1016/j.cellsig.2020.109634
Palicharla, V. R., Hwang, S.-H., Somatilaka, B. N., Badgandi, H. B., Legué, E., Tran, V. M., et al. (2021). Interactions between TULP3 Tubby Domain Cargo Site and ARL13B Amphipathic Helix Promote Lipidated Protein Transport to Cilia. bioRxiv. doi:10.1101/2021.05.25.445488
Parnell, S. C., Magenheimer, B. S., Maser, R. L., Rankin, C. A., Smine, A., Okamoto, T., et al. (1998). The Polycystic Kidney Disease-1 Protein, Polycystin-1, Binds and Activates Heterotrimeric G-Proteins In Vitro. Biochem. Biophys. Res. Commun. 251, 625–631. doi:10.1006/bbrc.1998.9514
Parnell, S. C., Magenheimer, B. S., Maser, R. L., Pavlov, T. S., Havens, M. A., Hastings, M. L., et al. (2018). A Mutation Affecting Polycystin-1 Mediated Heterotrimeric G-Protein Signaling Causes PKD. Hum. Mol. Genet. 27, 3313–3324. doi:10.1093/hmg/ddy223
Pazour, G. J., San Agustin, J. T., Follit, J. A., Rosenbaum, J. L., and Witman, G. B. (2002). Polycystin-2 Localizes to Kidney Cilia and the Ciliary Level Is Elevated in Orpk Mice with Polycystic Kidney Disease. Curr. Biol. 12, R378–R380. doi:10.1016/s0960-9822(02)00877-1
Pazour, G. J. (2004). Intraflagellar Transport and Cilia-dependent Renal Disease: The Ciliary Hypothesis of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 15, 2528–2536. doi:10.1097/01.asn.0000141055.57643.e0
Pearring, J. N., San Agustin, J. T., Lobanova, E. S., Gabriel, C. J., Lieu, E. C., Monis, W. J., et al. (2017). Loss of Arf4 Causes Severe Degeneration of the Exocrine Pancreas but Not Cystic Kidney Disease or Retinal Degeneration. PLoS Genet. 13, e1006740. doi:10.1371/journal.pgen.1006740
Pema, M., Drusian, L., Chiaravalli, M., Castelli, M., Yao, Q., Ricciardi, S., et al. (2016). mTORC1-mediated Inhibition of Polycystin-1 Expression Drives Renal Cyst Formation in Tuberous Sclerosis Complex. Nat. Commun. 7, 10786. doi:10.1038/ncomms10786
Picariello, T., Brown, J. M., Hou, Y., Swank, G., Cochran, D. A., King, O. D., et al. (2019). A Global Analysis of IFT-A Function Reveals Specialization for Transport of Membrane-Associated Proteins into Cilia. J. Cell Sci., 132jcs220749. doi:10.1242/jcs.220749
Piontek, K. B., Huso, D. L., Grinberg, A., Liu, L., Bedja, D., Zhao, H., et al. (2004). A Functional Floxed Allele of Pkd1 that Can Be Conditionally Inactivated In Vivo. J. Am. Soc. Nephrol. 15, 3035–3043. doi:10.1097/01.asn.0000144204.01352.86
Piperno, G., Siuda, E., Henderson, S., Segil, M., Vaananen, H., and Sassaroli, M. (1998). Distinct Mutants of Retrograde Intraflagellar Transport (IFT) Share Similar Morphological and Molecular Defects. J. Cell Biol. 143, 1591–1601. doi:10.1083/jcb.143.6.1591
Pisitkun, T., Shen, R.-F., and Knepper, M. A. (2004). Identification and Proteomic Profiling of Exosomes in Human Urine. Proc. Natl. Acad. Sci. U.S.A. 101, 13368–13373. doi:10.1073/pnas.0403453101
Ponting, C. P., Hofmann, K., and Bork, P. (1999). A latrophilin/CL-1-like GPS Domain in Polycystin-1. Curr. Biol. 9, R585–R588. doi:10.1016/s0960-9822(99)80379-0
Praetorius, H. A., Frokiaer, J., Nielsen, S., and Spring, K. R. (2003). Bending the Primary Cilium Opens Ca2+-Sensitive Intermediate-Conductance K+ Channels in MDCK Cells. J. Membr. Biol. 191, 193–200. doi:10.1007/s00232-002-1055-z
Qian, F., Boletta, A., Bhunia, A. K., Xu, H., Liu, L., Ahrabi, A. K., et al. (2002). Cleavage of Polycystin-1 Requires the Receptor for Egg Jelly Domain and Is Disrupted by Human Autosomal-Dominant Polycystic Kidney Disease 1-associated Mutations. Proc. Natl. Acad. Sci. U.S.A. 99, 16981–16986. doi:10.1073/pnas.252484899
Qin, J., Lin, Y., Norman, R. X., Ko, H. W., and Eggenschwiler, J. T. (2011). Intraflagellar Transport Protein 122 Antagonizes Sonic Hedgehog Signaling and Controls Ciliary Localization of Pathway Components. Proc. Natl. Acad. Sci. U.S.A. 108, 1456–1461. doi:10.1073/pnas.1011410108
Qiu, H., Fujisawa, S., Nozaki, S., Katoh, Y., and Nakayama, K. (2021). Interaction of INPP5E with ARL13B is Essential for its Ciliary Membrane Retention but Dispensable for its Ciliary Entry. Biol. Open 10, bio057653. doi:10.1242/bio.057653
Quidwai, T., Wang, J., Hall, E. A., Petriman, N. A., Leng, W., Kiesel, P., et al. (2021). A WDR35-dependent Coat Protein Complex Transports Ciliary Membrane Cargo Vesicles to Cilia. Elife 10, e69786. doi:10.7554/eLife.69786
Raychowdhury, M. K., Ramos, A. J., Zhang, P., Mclaughin, M., Dai, X.-Q., Chen, X.-Z., et al. (2009). Vasopressin Receptor-Mediated Functional Signaling Pathway in Primary Cilia of Renal Epithelial Cells. Am. J. Physiol. Renal Physiol. 296, F87–F97. doi:10.1152/ajprenal.90509.2008
Rees, S., Kittikulsuth, W., Roos, K., Strait, K. A., Van Hoek, A., and Kohan, D. E. (2014). Adenylyl Cyclase 6 Deficiency Ameliorates Polycystic Kidney Disease. J. Am. Soc. Nephrol. 25, 232–237. doi:10.1681/asn.2013010077
Reif, G. A., Yamaguchi, T., Nivens, E., Fujiki, H., Pinto, C. S., and Wallace, D. P. (2011). Tolvaptan Inhibits ERK-dependent Cell Proliferation, Cl− Secretion, and In Vitro Cyst Growth of Human ADPKD Cells Stimulated by Vasopressin. Am. J. Physiol. Ren. Physiol. 301, F1005–F1013. doi:10.1152/ajprenal.00243.2011
Ricker, J. L., Gattone, V. H., Calvet, J. P., and Rankin, C. A. (2000). Development of Autosomal Recessive Polycystic Kidney Disease in BALB/c-cpk/cpk Mice. J. Am. Soc. Nephrol. 11, 1837–1847. doi:10.1681/asn.v11101837
Rohatgi, R., Milenkovic, L., and Scott, M. P. (2007). Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 317, 372–376. doi:10.1126/science.1139740
Rosenbaum, J. L., and Witman, G. B. (2002). Intraflagellar Transport. Nat. Rev. Mol. Cell Biol. 3, 813–825. doi:10.1038/nrm952
San Agustin, J. T., Klena, N., Granath, K., Panigrahy, A., Stewart, E., Devine, W., et al. (2016). Genetic Link between Renal Birth Defects and Congenital Heart Disease. Nat. Commun. 7, 11103. doi:10.1038/ncomms11103
Schrick, J. J., Vogel, P., Abuin, A., Hampton, B., and Rice, D. S. (2006). ADP-ribosylation Factor-like 3 Is Involved in Kidney and Photoreceptor Development. Am. J. Pathol. 168, 1288–1298. doi:10.2353/ajpath.2006.050941
Seixas, C., Choi, S. Y., Polgar, N., Umberger, N. L., East, M. P., Zuo, X., et al. (2016). Arl13b and the Exocyst Interact Synergistically in Ciliogenesis. Mol. Biol. Cell 27, 308–320. doi:10.1091/mbc.e15-02-0061
Senum, S. R., Li, Y. M., Benson, K. A., Joli, G., Olinger, E., Lavu, S., et al. (2022). Monoallelic IFT140 Pathogenic Variants Are an Important Cause of the Autosomal Dominant Polycystic Kidney-Spectrum Phenotype. Am. J. Hum. Genet. 109, 136–156. doi:10.1016/j.ajhg.2021.11.016
Shen, P. S., Yang, X., Decaen, P. G., Liu, X., Bulkley, D., Clapham, D. E., et al. (2016). The Structure of the Polycystic Kidney Disease Channel PKD2 in Lipid Nanodiscs. Cell 167, 763–773. doi:10.1016/j.cell.2016.09.048
Sherpa, R. T., Mohieldin, A. M., Pala, R., Wachten, D., Ostrom, R. S., and Nauli, S. M. (2019). Sensory Primary Cilium is a Responsive cAMP Microdomain in Renal Epithelia. Sci. Rep. 9, 6523. doi:10.1038/s41598-019-43002-2
Sherpa, R. T., Mohieldin, A. M., Pala, R., Wachten, D., Ostrom, R. S., and Nauli, S. M. (2020). Author Correction: Sensory Primary Cilium is a Responsive cAMP Microdomain in Renal Epithelia. Sci. Rep. 10, 1731. doi:10.1038/s41598-020-58754-5
Shibazaki, S., Yu, Z., Nishio, S., Tian, X., Thomson, R. B., Mitobe, M., et al. (2008). Cyst Formation and Activation of the Extracellular Regulated Kinase Pathway after Kidney Specific Inactivation of Pkd1. Hum. Mol. Genet. 17, 1505–1516. doi:10.1093/hmg/ddn039
Shillingford, J. M., Murcia, N. S., Larson, C. H., Low, S. H., Hedgepeth, R., Brown, N., et al. (2006). The mTOR Pathway Is Regulated by Polycystin-1, and its Inhibition Reverses Renal Cystogenesis in Polycystic Kidney Disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5466–5471. doi:10.1073/pnas.0509694103
Somatilaka, B. N., Hwang, S.-H., Palicharla, V. R., White, K. A., Badgandi, H., Shelton, J. M., et al. (2020). Ankmy2 Prevents Smoothened-independent Hyperactivation of the Hedgehog Pathway via Cilia-Regulated Adenylyl Cyclase Signaling. Dev. Cell 54, 710–726. doi:10.1016/j.devcel.2020.06.034
Su, X., Wu, M., Yao, G., El-Jouni, W., Luo, C., Tabari, A., et al. (2015). Regulation of Polycystin-1 Ciliary Trafficking by Motifs at its C-Terminus and Polycystin-2 but Not by Cleavage at the GPS Site. J. Cell Sci. 128, 4063–4073. doi:10.1242/jcs.160556
Su, Q., Hu, F., Ge, X., Lei, J., Yu, S., Wang, T., et al. (2018). Structure of the Human PKD1-PKD2 Complex. Science 361, eaat9819. doi:10.1126/science.aat9819
Sun, Z., Amsterdam, A., Pazour, G. J., Cole, D. G., Miller, M. S., and Hopkins, N. (2004). A Genetic Screen in Zebrafish Identifies Cilia Genes as a Principal Cause of Cystic Kidney. Development 131, 4085–4093. doi:10.1242/dev.01240
Takahara, M., Katoh, Y., Nakamura, K., Hirano, T., Sugawa, M., Tsurumi, Y., et al. (2018). Ciliopathy-associated Mutations of IFT122 Impair Ciliary Protein Trafficking but Not Ciliogenesis. Hum. Mol. Genet. 27, 516–528. doi:10.1093/hmg/ddx421
The European Polycystic Kidney Disease Consortium (1994). The Polycystic Kidney Disease 1 Gene Encodes a 14 Kb Transcript and Lies within a Duplicated Region on Chromosome 16. Cell 78, 725–794. doi:10.1016/0092-8674(94)90137-6
Torres, V. E., and Harris, P. C. (2014). Strategies Targeting cAMP Signaling in the Treatment of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 25, 18–32. doi:10.1681/asn.2013040398
Torres, V. E., Wang, X., Qian, Q., Somlo, S., Harris, P. C., and Gattone, V. H. (2004). Effective Treatment of an Orthologous Model of Autosomal Dominant Polycystic Kidney Disease. Nat. Med. 10, 363–364. doi:10.1038/nm1004
Torres, V. E., Boletta, A., Chapman, A., Gattone, V., Pei, Y., Qian, Q., et al. (2010). Prospects for mTOR Inhibitor Use in Patients with Polycystic Kidney Disease and Hamartomatous Diseases. Clin. J. Am. Soc. Nephrol. 5, 1312–1329. doi:10.2215/cjn.01360210
Torres, V. E., Chapman, A. B., Devuyst, O., Gansevoort, R. T., Perrone, R. D., Koch, G., et al. (2017). Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 377, 1930–1942. doi:10.1056/nejmoa1710030
Torres, V. E., Chapman, A. B., Devuyst, O., Gansevoort, R. T., Perrone, R. D., Lee, J., et al. (2020). Multicenter Study of Long-Term Safety of Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 16, 48–58. doi:10.2215/cjn.10250620
Trudel, M., Yao, Q., and Qian, F. (2016). The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells 5, 3. doi:10.3390/cells5010003
Truong, M. E., Bilekova, S., Choksi, S. P., Li, W., Bugaj, L. J., Xu, K., et al. (2021). Vertebrate Cells Differentially Interpret Ciliary and Extraciliary cAMP. Cell 184, 2911. doi:10.1016/j.cell.2021.04.002
Vien, T. N., Wang, J., Ng, L. C. T., Cao, E., and Decaen, P. G. (2020). Molecular Dysregulation of Ciliary Polycystin-2 Channels Caused by Variants in the TOP Domain. Proc. Natl. Acad. Sci. U.S.A. 117, 10329–10338. doi:10.1073/pnas.1920777117
Walker, R. V., Keynton, J. L., Grimes, D. T., Sreekumar, V., Williams, D. J., Esapa, C., et al. (2019). Ciliary Exclusion of Polycystin-2 Promotes Kidney Cystogenesis in an Autosomal Dominant Polycystic Kidney Disease Model. Nat. Commun. 10, 4072. doi:10.1038/s41467-019-12067-y
Wang, X., Gattone, V., Harris, P. C., and Torres, V. E. (2005). Effectiveness of Vasopressin V2 Receptor Antagonists OPC-31260 and OPC-41061 on Polycystic Kidney Disease Development in the PCK Rat. J. Am. Soc. Nephrol. 16, 846–851. doi:10.1681/asn.2004121090
Wang, J., Kaletsky, R., Silva, M., Williams, A., Haas, L. A., Androwski, R. J., et al. (2015). Cell-Specific Transcriptional Profiling of Ciliated Sensory Neurons Reveals Regulators of Behavior and Extracellular Vesicle Biogenesis. Curr. Biol. 25, 3232–3238. doi:10.1016/j.cub.2015.10.057
Wang, Q., Cobo-Stark, P., Patel, V., Somlo, S., Han, P.-L., and Igarashi, P. (2018). Adenylyl Cyclase 5 Deficiency Reduces Renal Cyclic AMP and Cyst Growth in an Orthologous Mouse Model of Polycystic Kidney Disease. Kidney Int. 93, 403–415. doi:10.1016/j.kint.2017.08.005
Wang, Z., Ng, C., Liu, X., Wang, Y., Li, B., Kashyap, P., et al. (2019). The Ion Channel Function of Polycystin-1 in the Polycystin-1/polycystin-2 Complex. EMBO Rep. 20, e48336. doi:10.15252/embr.201948336
Wang, W., Silva, L. M., Wang, H. H., Kavanaugh, M. A., Pottorf, T. S., Allard, B. A., et al. (2022). Ttc21b Deficiency Attenuates Autosomal Dominant Polycystic Kidney Disease in a Kidney Tubular- and Maturation-Dependent Manner. Kidney Int. S0085-2538 (22), 00387–00388. doi:10.1016/j.kint.2022.04.034
Ward, H. H., Brown-Glaberman, U., Wang, J., Morita, Y., Alper, S. L., Bedrick, E. J., et al. (2011). A Conserved Signal and GTPase Complex Are Required for the Ciliary Transport of Polycystin-1. Mol. Biol. Cell 22, 3289–3305. doi:10.1091/mbc.e11-01-0082
Wheatley, D. N. (2005). Landmarks in the First Hundred Years of Primary (9+0) Cilium Research. Cell Biol. Int. 29, 333–339. doi:10.1016/j.cellbi.2005.03.001
Wright, K. J., Baye, L. M., Olivier-Mason, A., Mukhopadhyay, S., Sang, L., Kwong, M., et al. (2011). An ARL3-UNC119-RP2 GTPase Cycle Targets Myristoylated NPHP3 to the Primary Cilium. Genes Dev. 25, 2347–2360. doi:10.1101/gad.173443.111
Yin, W.-C., Satkunendran, T., Mo, R., Morrissy, S., Zhang, X., Huang, E. S., et al. (2020). Dual Regulatory Functions of SUFU and Targetome of GLI2 in SHH Subgroup Medulloblastoma. Dev. Cell 52, 132. doi:10.1016/j.devcel.2019.12.014
Yoder, B. K., Hou, X., and Guay-Woodford, L. M. (2002). The Polycystic Kidney Disease Proteins, Polycystin-1, Polycystin-2, Polaris, and Cystin, Are Co-localized in Renal Cilia. J. Am. Soc. Nephrol. 13, 2508–2516. doi:10.1097/01.asn.0000029587.47950.25
Yoshiba, S., Shiratori, H., Kuo, I. Y., Kawasumi, A., Shinohara, K., Nonaka, S., et al. (2012). Cilia at the Node of Mouse Embryos Sense Fluid Flow for Left-Right Determination via Pkd2. Science 338, 226–231. doi:10.1126/science.1222538
Yu, S., Hackmann, K., Gao, J., He, X., Piontek, K., García-González, M. A., et al. (2007). Essential Role of Cleavage of Polycystin-1 at G Protein-Coupled Receptor Proteolytic Site for Kidney Tubular Structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18688–18693. doi:10.1073/pnas.0708217104
Yu, Y., Ulbrich, M. H., Li, M.-H., Buraei, Z., Chen, X.-Z., Ong, A. C. M., et al. (2009). Structural and Molecular Basis of the Assembly of the TRPP2/PKD1 Complex. Proc. Natl. Acad. Sci. U.S.A. 106, 11558–11563. doi:10.1073/pnas.0903684106
Yu, S. S., Wang, E., Chiang, C. Y., Cheng, P. H., Yeh, Y. S., Wu, Y. Y., et al. (2022). Large Deletion of Wdr19 in Developing Renal Tubules Disrupts Primary Ciliogenesis, Leading to Polycystic Kidney Disease in Mice. J. Pathology 257, 5–16. doi:10.1002/path.5863
Zhang, X. M., Ramalho-Santos, M., and Mcmahon, A. P. (2001). Smoothened Mutants Reveal Redundant Roles for Shh and Ihh Signaling Including Regulation of L/R Asymmetry by the Mouse Node. Cell 105, 781–792. doi:10.1016/s0092-8674(01)00385-3
Zhang, B., Tran, U., and Wessely, O. (2018). Polycystin 1 Loss of Function is Directly Linked to an Imbalance in G-Protein Signaling in the Kidney. Development 145, dev158931. doi:10.1242/dev.158931
Zhang, C., Balbo, B., Ma, M., Zhao, J., Tian, X., Kluger, Y., et al. (2021). Cyclin-Dependent Kinase 1 Activity is a Driver of Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 32, 41–51. doi:10.1681/asn.2020040511
Keywords: polycystic kidney disease, cystogenesis, primary cilia, polycystin 1 and 2, tubby-like protein 3, cilia-dependent cyst activation, cilia-localized cyst inhibition, intraflagellar transport
Citation: Walker RV, Maranto A, Palicharla VR, Hwang S-H, Mukhopadhyay S and Qian F (2022) Cilia-Localized Counterregulatory Signals as Drivers of Renal Cystogenesis. Front. Mol. Biosci. 9:936070. doi: 10.3389/fmolb.2022.936070
Received: 04 May 2022; Accepted: 30 May 2022;
Published: 23 June 2022.
Edited by:
Annarita Di Mise, University of Bari Aldo Moro, ItalyReviewed by:
Jong Hoon Park, Sookmyung Women’s University, South KoreaMiriam Zacchia, University of Campania Luigi Vanvitelli, Italy
Copyright © 2022 Walker, Maranto, Palicharla, Hwang, Mukhopadhyay and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Qian, fqian@som.umaryland.edu