Conserved gene signatures shared among MAPT mutations reveal defects in calcium signaling
 Miguel A. Minaya1
Miguel A. Minaya1  Sidhartha Mahali1
Sidhartha Mahali1  Abhirami K. Iyer1
Abhirami K. Iyer1  Abdallah M. Eteleeb1 Rita Martinez1 Guangming Huang1 John Budde1
Abdallah M. Eteleeb1 Rita Martinez1 Guangming Huang1 John Budde1  Sally Temple2 Alissa L. Nana3 William W. Seeley3 Salvatore Spina3 Lea T. Grinberg3,4
Sally Temple2 Alissa L. Nana3 William W. Seeley3 Salvatore Spina3 Lea T. Grinberg3,4  Oscar Harari1,5,6
Oscar Harari1,5,6  Celeste M. Karch1,5,6*
Celeste M. Karch1,5,6*- 1Department of Psychiatry, Washington University in St Louis, St Louis, MO, United States
- 2Neural Stem Cell Institute, Rensselaer, NY, United States
- 3Department of Neurology, UCSF Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, United States
- 4Department of Pathology, University of Sao Paulo, Sao Paulo, Brazil
- 5Hope Center for Neurological Disorders, Washington University in St Louis, St Louis, MO, United States
- 6NeuroGenomics and Informatics Center, Washington University in St Louis, St Louis, MO, United States
Introduction: More than 50 mutations in the MAPT gene result in heterogeneous forms of frontotemporal lobar dementia with tau inclusions (FTLD-Tau). However, early pathogenic events that lead to disease and the degree to which they are common across MAPT mutations remain poorly understood. The goal of this study is to determine whether there is a common molecular signature of FTLD-Tau.
Methods: We analyzed genes differentially expressed in induced pluripotent stem cell–derived neurons (iPSC-neurons) that represent the three major categories of MAPT mutations: splicing (IVS10 + 16), exon 10 (p.P301L), and C-terminal (p.R406W) compared with isogenic controls. The genes that were commonly differentially expressed in MAPT IVS10 + 16, p.P301L, and p.R406W neurons were enriched in trans-synaptic signaling, neuronal processes, and lysosomal function. Many of these pathways are sensitive to disruptions in calcium homeostasis. One gene, CALB1, was significantly reduced across the three MAPT mutant iPSC-neurons and in a mouse model of tau accumulation. We observed a significant reduction in calcium levels in MAPT mutant neurons compared with isogenic controls, pointing to a functional consequence of this disrupted gene expression. Finally, a subset of genes commonly differentially expressed across MAPT mutations were also dysregulated in brains from MAPT mutation carriers and to a lesser extent in brains from sporadic Alzheimer disease and progressive supranuclear palsy, suggesting that molecular signatures relevant to genetic and sporadic forms of tauopathy are captured in a dish. The results from this study demonstrate that iPSC-neurons capture molecular processes that occur in human brains and can be used to pinpoint common molecular pathways involving synaptic and lysosomal function and neuronal development, which may be regulated by disruptions in calcium homeostasis.
Background
Frontotemporal lobal degeneration with tau inclusions (FTLD-tau) encompasses a heterogenous group of disorders characterized by frontal and temporal lobar atrophy, neuronal loss and gliosis, and the accumulation of neurofibrillary tangles (NFTs) (Bodea et al., 2016). In a subset of cases, FTLD-Tau is caused by rare, dominantly inherited mutations in the microtubule associated protein tau (MAPT) gene (Pottier et al., 2016). Common genetic variation in the MAPT gene also contributes to sporadic forms of FTLD-Tau, including progressive supranuclear palsy (PSP) and corticobasal degeneration (Ferrari et al., 2014; Kouri et al., 2015; Steele et al., 2018).
MAPT is alternatively spliced and developmentally regulated in the central nervous system, resulting in six canonical tau isoforms, which differ based on the absence (0N) or presence of one or two N-terminal inclusions (1N, 2N, respectively) and three or four repeats in the microtubule binding region (3R or 4R, respectively) (Neve et al., 1986; Lee et al., 2001). In adult brains, there is an equal balance of 3R and 4R tau isoforms. More than 50 mutations in the MAPT gene are reported to cause FTLD-Tau (https://www.alzforum.org/mutations/mapt). These mutations fall into three major categories. First, located in the intronic region near the stem-loop domain, a subset of mutations alter MAPT splicing via inclusion of exon 10 (e.g., 3R<4R tau) or exclusion of exon 10 (e.g., 3R>4R tau). Second, missense mutations may occur within exon 10, such that the mutation is only present in a subset of MAPT isoforms (i.e. 4R tau). Finally, missense mutations may occur outside of the microtubule binding region, which leads to the production of mutant protein among all tau isoforms. We asked whether the heterogeneity in MAPT mutations drive common molecular mechanisms. To begin to address this question, we studied MAPT mutations that fall into these three major categories: MAPT IVS10 + 16, p.P301L and p.R406W, respectively.
Human cellular models of the brain derived from induced pluripotent stem cells (iPSC) have become an important tool for studying molecular and cellular markers that may initiate disease (Livesey, 2014; Iovino et al., 2015; Silva et al., 2016; Guo et al., 2017; Wray, 2017; Gonzalez et al., 2018; Jiang et al., 2018; Karch et al., 2018; Nakamura et al., 2019; Bowles et al., 2021; Lagomarsino et al., 2021). Here, we coupled human iPSC models with CRISPR/Cas9 genome editing technology to create a system that allows us to distinguish the molecular signatures associated with MAPT mutations and to begin to resolve the molecular phenotypes of tauopathy. Together, our findings uncover key changes in trans-synaptic signaling, lysosomal function, and calcium signaling shared across MAPT mutations.
Materials and methods
Patient consent
Skin punches were performed following written informed consent from the donor. The informed consent was approved by the Washington University School of Medicine and University of California San Francisco Institutional Review Board and Ethics Committee (IRB 201104178, 201306108 and 10-03946). The consent allows for use of tissue by all parties, commercial and academic, for the purposes of research but not for use in human therapy.
The Washington University and University of California San Francisco Institutional Review Boards reviewed the Neuropathology Cores (from whom the brains were obtained) operating protocols as well as this specific study and determined it was exempt from approval. Our participants provide this consent by signing the hospital’s autopsy form. If the participant does not provide future consent before death the DPOA or next of kin provide it after death. All data were analyzed anonymously.
iPSC lines
Human iPSC used in this study (Supplementary Table S1; Supplementary Figure S1) have been previously described (Karch et al., 2019). Briefly, iPSC lines were generated using non-integrating Sendai virus carrying OCT3/4, SOX2, KLF4, and cMYC (Life Technologies) (Takahashi and Yamanaka, 2006; Ban et al., 2011). iPSC lines were characterized for the following parameters using standard methods (Takahashi and Yamanaka, 2006): pluripotency markers by immunocytochemistry (ICC) and quantitative PCR (qPCR), spontaneous or TriDiff differentiation into the three germ layers by ICC and qPCR, assessment of chromosomal abnormalities by karyotyping, and MAPT mutation status was confirmed by Sanger sequencing (Supplementary Figure S1). To determine the impact of the MAPT mutant allele on molecular phenotypes, we used CRISPR/Cas9-edited isogenic controls in which the mutant allele was reverted to the wild-type (WT) allele in each of the donor iPSC lines as previously described (GIH36C2; F11362.1; F0510.2; Supplementary Table S1; Supplementary Figure S1) (Karch et al., 2019). Resulting edited lines were characterized as described above in addition to on- and off-target sequencing (Supplementary Figure S1). All iPSC lines used in this study carry the MAPT H1/H1 common haplotype.
Differentiation of iPSCs into cortical neurons
iPSCs were differentiated into cortical neurons using a two-step approach as previously described (Karch et al., 2019) (https://dx.doi.org/10.17504/protocols.io.p9kdr4w). iPSCs were plated at a density of 65,000 cells per well in neural induction media (StemCell Technologies) in a 96-well v-bottom plate to form neural aggregates and after 5 days, transferred into culture plates. The resulting neural rosettes were then isolated by enzymatic selection (Neural Rosette Selection Reagent; StemCell Technologies) and cultured as neural progenitor cells (NPCs). NPCs were differentiated in planar culture in neuronal maturation medium (neurobasal medium supplemented with B27, GDNF, BDNF, cAMP). Neurons typically arose within 1 week after plating, identified using immunocytochemistry for β-tubulin III (Tuj1). The cells continue to mature and were analyzed at 6 weeks.
RNA extraction, sequencing, and transcript quantification
iPSC-derived neurons were re-suspended in 200 µL of 50:1 homogenization solution: 1-Thioglycerol solution. After addition of 200 µL of Promega lysis buffer, the samples were transferred to the appropriate well of the Maxwell RSC cartridge. DNase solution was added to each cartridge. TapeStation 4200 System (Agilent Technologies) was used to perform quality control of the RNA concentration, purity, and degradation based on the estimated RNA integrity Number (RIN), and DV200 (Supplementary Table S1). Samples were sequenced by an Illumina HiSeq 4000 Systems Technology with a read length of 1 × 150 bp, and an average library size of 36.5 ± 12.2 million reads per sample.
Identity-by-Descent (IDB) (Browning and Browning, 2010) and FastQC (Andrews et al., 2012) analyses were performed to confirm sample identity. STAR (v.2.6.0) (Dobin et al., 2012) was used to align the RNA sequences to the human reference genome: GRCh38.p13 (hg38). The quality of RNA alignment was evaluated using sequencing metrics such as read distribution, ribosomal content, and alignment quality in Picard (v.2.8.2). The average percentage of unique mapped reads in the BAM files was 80.3% ± 3.62, and the average percentage of total mapped reads to GRCh38.p13 was 90.1% ± 5.12 (Supplementary Table S1). IGV (Integrative Genomics Viewer) (Thorvaldsdottir et al., 2013) was used with the reference Human Genome (hg38) to visualize mutation containing reads and their absence in samples edited using CRISPR/Cas9 protocols (isogenic controls).
Salmon (v. 0.11.3) (Patro et al., 2017) was used to quantify the expression of the genes annotated within the human reference genome used in this project (GRCh38.p13). Protein coding genes were selected for downstream analyses.
Principal component and differential expression analyses
Principal component analyses (PCA) were performed based on 19,957 protein coding genes using regularized-logarithm transformation (rlog) counts. Differential gene expression was performed using the DESeq2 (v.1.22.2) R package (Love et al., 2014). PCA and differential gene expression analyses were performed independently for each set of MAPT mutations and isogenic controls. Each MAPT mutation and its isogenic control were considered independent cohorts due to their shared genetic background. As such, the relationship across the three MAPT mutation sets was evaluated using the MetaVolcanoR R package (v1.10.0) (Prada et al., 2021). The meta-analysis included those genes that were differentially expressed (p <0.05) in the same direction across the three cohorts (n = 275 genes). A meta-volcano plot summarizing the gene fold change of the MAPT IVS10 + 16, p.P301L, and p.R406W datasets was generated using a Random Effect Model (REM) estimation. PCA and Volcano plots were created for each comparison using the ggplot2 R package (v3.3.6) (Wickham, 2016).
Pathway enrichment and network analyses
ToppGene (Chen et al., 2009) and Enrichr (Chen et al., 2013; Kuleshov et al., 2016; Xie et al., 2021) were used to identify pathways in which differentially expressed genes are enriched. Gene ontologies (GOs) related to molecular function, biological process and cellular component were selected based on two criteria: i) p ≤ 0.05 and ii) number of query genes associated with each GO > 1. Gene relationships including physical, predicted and genetic interactions, and gene networks including co-expression and co-localization were annotated using the geneMANIA prediction server (Warde-Farley et al., 2010).
Mouse model of tauopathy
To evaluate whether the genes differentially expressed in iPSC-derived neurons were altered in animal models of tauopathy, we analyzed the gene expression in the Tau-P301L mouse model of tauopathy and non-transgenic controls (Ramsden et al., 2005). Transcriptomic data from mice was obtained from the Mouse Dementia Network (Matarin et al., 2015). Gene expression across the timepoints (2-, 4-, 8-, and 18-months old mice) was normalized to mice at 2 months of age and plotted. Differential gene expression at 18 months of age was analyzed by unpaired t-tests to assess significance.
Drug target identification
To determine whether differentially expressed genes were associated with known drugs, we interrogated: (i) the WEB-based Gene SeT AnaLysis Toolkit (Liao et al., 2019), (ii) the Drug-Gene Interaction Database (Freshour et al., 2021), and (iii) the DrugBank (Wishart et al., 2018).
Calcium imaging
To measure calcium levels in iPSC-derived neurons, MAPT IVS10 + 16 mutation (GIH36C2) and isogenic controls (GIH36C2Δ1D01) were analyzed. NPCs were differentiated into cortical neurons as described above. After 22 days in culture, 2 × 105 neurons of each genotype were seeded into poly-L-ornithine and laminin-coated 96-well plate. Ca2+ levels in the iPSC-derived neurons were then measured using the Invitrogen™ Fluo-4 Direct™ Calcium Assay Kit (catalog number: F10471) following manufacturer’s instructions. Briefly, at 36 days in culture, growth medium was replaced with 50 μL per well Fluo-4 DirectTM calcium assay buffer and 50 μL per well of the 2x Fluo-4 DirectTM calcium reagent loading solution. The 96-well plate was then incubated at 37°C for 60 min, after which Fluo-4 fluorescence in intact cells directly proportional to cytoplasmic Ca2+ levels was measured using Synergy HTX multi-mode microplate reader (BioTek Instruments excitation at 494 nm and emission at 516 nm). Negative controls included Fluo-4 DirectTM calcium assay buffer plus reagent with no neurons and neurons without assay reagent. Fluo-4 staining in cells was imaged under a Nikon Eclipse 80i fluorescent microscope at 20x magnification. After measuring cytoplasmic Ca2+ levels, cells were lysed to break cellular and organelle membranes in 1% Triton X-100 and total Fluo-4 fluorescence intensities from cytoplasmic and intracellular Ca2+ stores were measured as described above.
Human brain datasets
To determine whether the differentially expressed genes in the iPSC-derived neurons capture molecular processes that occur specifically in primary tauopathies or that represent more general pathways associated with neurodegeneration, we analyzed gene expression in human brains with primary tauopathy (e.g., MAPT mutation carriers and progressive supranuclear palsy (PSP)), secondary tauopathy (e.g., Alzheimer disease (AD)), and FTLD with TDP-43 pathology (FTLD-TDP). Primary tauopathy datasets included: i) middle temporal gyrus from MAPT IVS10 + 16 mutation carriers (2 samples) and healthy controls (3 samples); ii) insular cortex from MAPT R406W carriers (2 samples) and healthy controls (2 samples) (Jiang et al., 2018); and iii) Temporal cortex from progressive supranuclear palsy (PSP) brains (82 samples) and healthy control brains (76 samples; syn6090813) (Allen et al., 2015; Allen et al., 2016). Secondary tauopathy datasets included temporal cortex from AD brains (84 samples) and healthy controls (76 samples) (Allen et al., 2015; Allen et al., 2016). To determine whether gene expression changes in iPSC-neuron models reflect a more general impact on neurodegenerative pathways, we examined gene expression profiles isolated from tissue of FTLD-TDP caused by rare mutations the GRN, C9ORF72 expansions, or from sporadic cases (Knight ADRC)(Li et al., 2018; Dube et al., 2019; Wani et al., 2021): i) parietal lobe from GRN mutation carriers (5 samples), ii) C9ORF72 expansion carriers (5 samples), iii) sporadic cases (8 samples), and (iv) healthy controls (16 samples). Differential gene expression analyses comparing controls and disease diagnosed brains were performed using gene expression measures and including as covariates sex, age-at-death, RNA integrity number (RIN), and brain tissue source.
Results
MAPT mutations are sufficient to induce global transcriptomic changes in human neurons
The goal of this study was to identify common genes and pathways that are downstream of MAPT mutations and candidate drivers of disease pathogenesis in FTLD-Tau (Figure 1). To address this goal, we studied a series of MAPT mutations that represent three major mutation types: MAPT IVS10 + 16, p.P301L and p.R406W (Figure 1; Supplementary Table S1). Protein coding genes obtained from RNA-sequencing data generated from iPSC–derived neurons carrying one of these three MAPT mutations together with isogenic controls were analyzed (Figure 2A). Among isogenic pairs, each of the MAPT mutations were sufficient to induce global transcriptomic changes in iPSC-neurons: 81.66% principal component 1 (PC1) for MAPT IVS10 + 16; 79.33% PC1 for MAPT p.P301L; and 57.28% PC1 for MAPT p.R406W (Figures 2B,D,F). PCA of the CRISPR/Cas9-engineered MAPT WT lines from independent donors reveal donor-dependent clustering (Supplementary Figure S2), suggesting that genetic background of the donor is the largest driver of transcriptomic variation which is consistent with prior reports (Kilpinen et al., 2017). Given that the genetic background remains conserved within the isogenic pairs, we treated each pair as a cohort and performed differential expression analyses to determine the impact of the presence of each mutant allele (Figures 2C,E,G; Supplemental Tables 2 and 3). Together, these findings illustrate that FTLD-causing MAPT mutations are sufficient to produce robust gene expression changes in neurons.
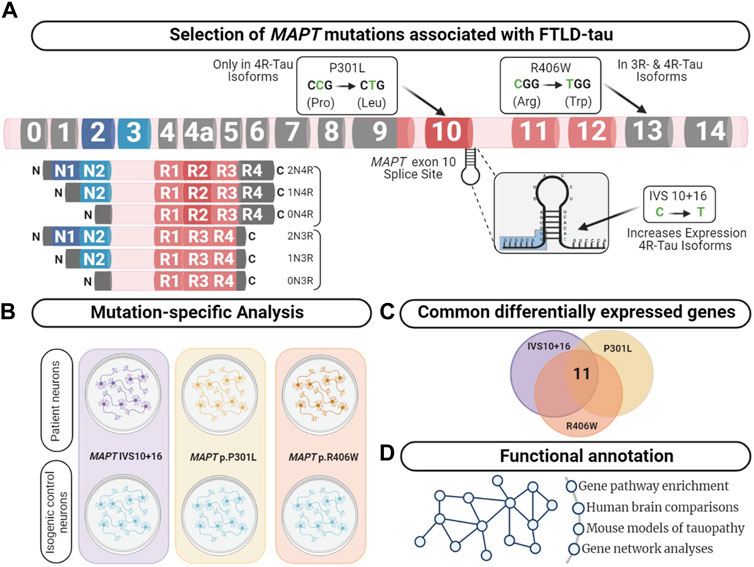
FIGURE 1. Integrative analysis to identify dysregulated pathways in FTLD-Tau. (A) MAPT gene annotated with the location of the mutations used in this study. Lower left panel displays the six major isoforms expressed in the central nervous system. (B) Comparison of human iPSC-neurons carrying the MAPT mutation specific and isogenic controls served as a discovery cohort to identify genes dysregulated across the three mutations. (C) Overlap analysis between all mutants compared with control. After multiple test corrections (BY-FDR ≤ 0.05), we identified 11 commonly differentially expressed genes. (D) Functional annotation was performed using the commonly differentially expressed genes.
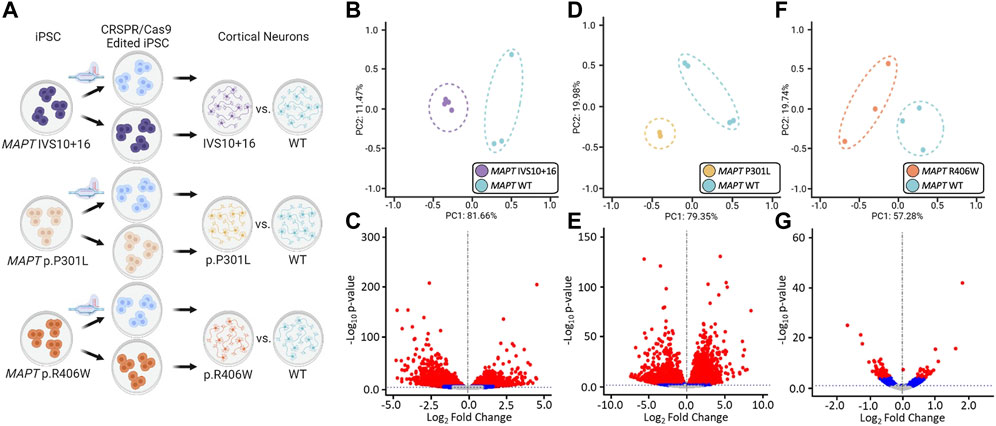
FIGURE 2. Global transcriptomic effects of MAPT IVS10 + 16, p.P301L, and p.R406W mutations. (A) Overview of iPSC differentiation into cortical neurons. (B–C) Principal component analyses and volcano plots obtained from MAPT IVS10 + 16 neurons compared to isogenic controls. (D–E) PCA and Volcano plots obtained from MAPT p.P301L neurons compared to isogenic controls. (F–G) PCA and Volcano plots obtained from MAPT p.R406W neurons compared to isogenic controls. PCA and Volcano plots were based on 19,957 protein-coding genes using regularized-logarithm transformation (rlog) counts. Volcano plots showing log2fold change between iPSC-derived neurons carrying MAPT mutations vs. isogenic controls, and the –log10 p-value for each gene. Red and blue dots within volcano plots represent, respectively, genes differentially expressed under adjusted-BY (BY-FDR) and unadjusted p-values (p ≤ 0.05).
MAPT mutations produce a shared gene expression signature in human neurons
Differential gene expression analyses within isogenic pairs illustrates that individual MAPT mutations produce global transcriptomic changes; thus, we sought to determine the extent of overlap in the differentially expressed genes among these three distinct MAPT mutation types (Figure 1C). We identified 11 differentially expressed genes across the three datasets (BY-FDR<0.05) (Figures 3A–D; Supplementary Table S4): CELSR1, CHRDL1, EFNB1, NOTCH1, CALB1, FOSL2, PLK2, PRICKLE2, ST8SIA3, NRP2, SPP1. Pathway analyses revealed that the 11 genes were enriched for i) trans-synaptic signaling pathways; ii) neuronal projection pathways; iii) lysosomal functions; and iv) calcium homeostasis (Figure 3E). These findings point to a common set of genes and pathways that are altered downstream of three distinct classes of MAPT mutations.
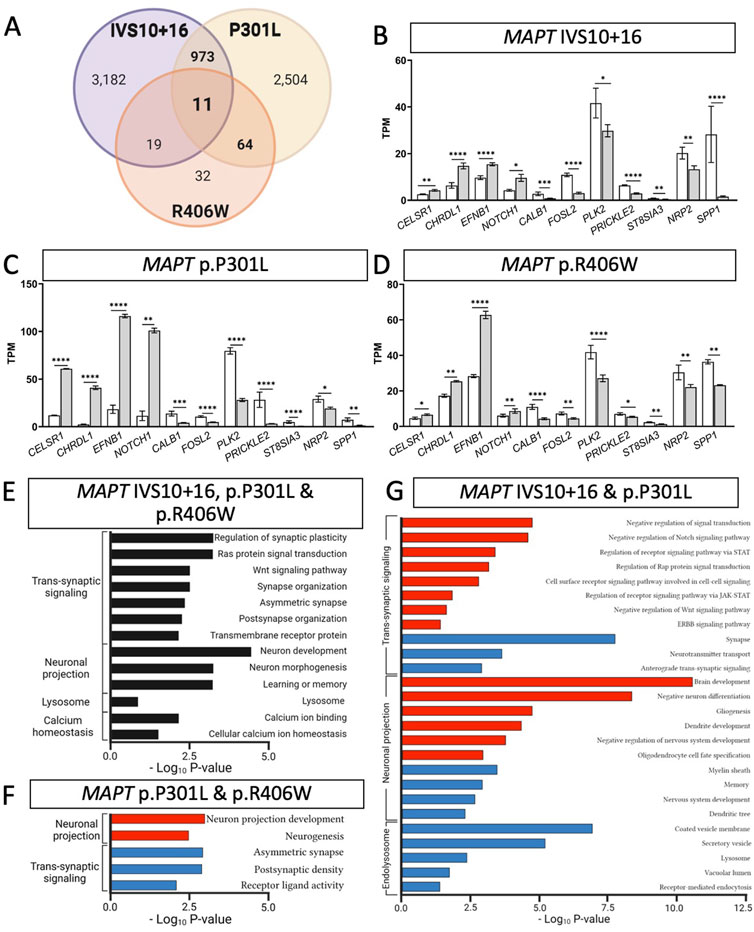
FIGURE 3. MAPT mutations result in common defects in synaptic signaling, neuronal projection, and lysosomal function. (A) Venn diagram presenting the differentially expressed genes common among iPSC-neurons carrying MAPT IVS10 + 16, p.P301L and p.R406W mutations (BY-FDR ≤ 0.05). (B–D) Normalized TPM expression of 11 genes shared between the three datasets (MAPT-IVS10 + 16, p.P301L and p.R406W; BY-FDR ≤ 0.05). (E) Bar graph showing the most significant pathways enriched among the 11 shared differentially genes (black bars). (F) Bar graph of the pathways enriched among the genes shared between MAPT p.P301L and p.R406W neurons. (G) Bar graph of the pathways enriched among the genes shared between MAPT IVS10 + 16 and p.P301L neurons. (F–G) Pathways from up-regulated genes (red bars). Pathways from down-regulated genes (blue bars).
Unique gene signatures
Beyond molecular signatures shared across the three MAPT mutations, we observed an imbalance in those genes shared between point mutations (MAPT p.P301L and p.R406W) and exon 10 mutations (MAPT IVS10 + 16 and p.P301L). We identified 64 genes that were shared between iPSC-neurons carrying the MAPT p.P301L and p.R406W mutations (BY-FDR<0.05; Figure 3A; Supplementary Table S5), while 973 genes were shared between iPSC-neurons carrying the MAPT IVS10 + 16 and p.P301L mutations (BY-FDR<0.05; Figure 3A; Supplementary Table S6). Thus, the number of differentially expressed genes associated with mutations located around the alternatively spliced exon 10 was 15-fold higher than the number of dysregulated genes associated with the MAPT p.P301L and p.R406W mutations.
Despite the different patterns in gene expression, pathway analyses were consistent with observations across all three MAPT mutations. The 64 dysregulated genes shared between the MAPT p.P301L and p.R406W neurons were enriched in pathways related to neurogenesis and trans-synaptic signaling (Figure 3F; Supplementary Table S7). Among MAPT IVS 10 + 16 and p.P301L (n = 973 genes), 409 up-regulated genes were enriched in pathways involved in the regulation of cell signaling: i) receptor signaling via JAK-STAT pathway; ii) Rap protein signal transduction; and iii) ERBB signaling pathway (Figure 3G; Supplementary Table S8). The 564 down-regulated genes were enriched in pathways involved in endolysosomal function: i) the coated vesicle membrane; ii) secretory vesicles; iii) lysosome; and iv) vacuolar lumen (Figure 3G; Supplementary Table S8).
A number of genes were found to be uniquely differentially expressed within each of the isogenic pairs, suggesting mutation-specific effects on gene expression (Figure 3A; Supplementary Table S3). The MAPT IVS10 + 16 mutation led to a significant increase in 1,804 unique genes, which are associated with synapse, learning, and vesicle-mediated transport (Supplementary Table S9), while the 1,746 uniquely down-regulated genes were associated with lysosome functions and apoptotic signaling (Supplementary Table S9). The MAPT p.P301L mutation produced 1,333 unique up-regulated genes that were associated with cell cycle processes (e.g., mitosis, meiosis, organellar fusion/division) and 1,645 unique down-regulated genes were associated with vesicle-mediated transport and neuron projections (Supplementary Table S9). Finally, the MAPT p.R406W mutation resulted in 217 uniquely up-regulated genes that were associated with lysosomal pathways, and the 174 uniquely down-regulated genes were associated with GABA receptor complex, neurotransmitter receptor activities, and synaptic signaling (Supplementary Table S9). Together, we demonstrate that many of these uniquely differentially expressed genes fall within pathways shared among all the MAPT mutations.
MAPT mutations lead to common genetic signatures that are associated with tau aggregation in mouse models of tauopathy
We sought to determine the extent to which the 11 commonly differentially expressed genes across MAPT mutations (Figure 3) were altered during disease course in the Tau-P301L mouse model of tauopathy. Using the Mouse Dementia Network (Matarin et al., 2015), we analyzed transcriptomic data generated from the cortex of WT and Tau-P301L mice collected at 2, 4, 8, and 18-months (Figure 4A). Compared with WT littermates, transgenic Tau-P301L mice develop tau aggregates beginning at 8 months of age (Ramsden et al., 2005). Among the 11 genes, six genes were differentially expressed at 18 months of age, when tau aggregation is most prominent (Figures 4B–G; Supplementary Figure S3): Celsr1, Chrdl1, Calb1, Plk2, Prickle2, and St8sia3. These six genes are highly related to one another and enriched in pathways related to neurodegeneration such as neurogenesis, behavior, learning, memory, and glutamatergic synapse (Figure 4H; Supplementary Table S10). At earlier timepoints when tau aggregation is beginning in the Tau-P301L mouse model, we observed statistical differences in expression of Efnb2 (8 months), Fosl2 (8 months), Calb1 (4 months), Nrp2 (8 months), Prickle2 (8 months), and St8sia3 (8 months). The Drug-Gene Interaction Database (Freshour et al., 2021) and DrugBank (Wishart et al., 2018) revealed that two genes, PLK2 and CALB1, are known targets of FDA approved drugs including tramadol, ethosuximide, levodopa, nicotine and oxcarbazepine, which are currently used to treat neurological symptoms (Supplementary Table S11). Together, these findings illustrate that MAPT mutations are sufficient to induce molecular changes in iPSC-neurons that are relevant to tau aggregation in vivo.
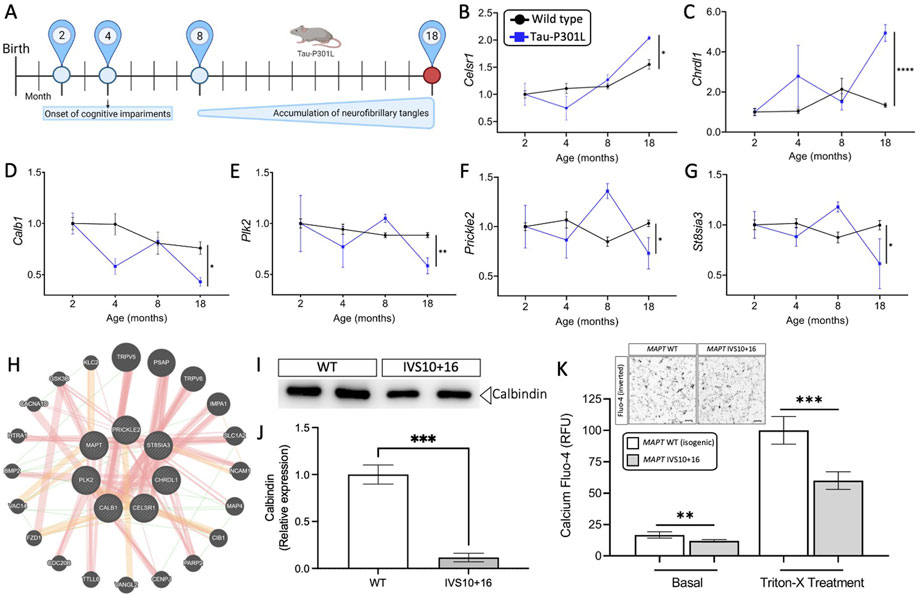
FIGURE 4. Genes altered in iPSC-neurons from MAPT mutation carriers are replicated in disease end-stage in the Tau-P301L tauopathy mouse model. (A) The expression of the 11 genes observed in iPSC-neurons were evaluated in the mouse model of tauopathy (Tau-P301L vs. wild type). Statistical comparisons reflect gene expression at disease end-stage (18 months old mice). (B–G) Expression of the six selected genes (2 up- and 4 down-regulated) in mice. Black circles, transgenic Tau-P301L mice; blue squares, non-transgenic control mice. Graphs show normalized gene expression relative to 2 months old mice. The genes presented were selected based on: (I) the expression of the genes in the human cell models and in mice followed the same direction, and (ii) the difference in the expression of each gene in 18-month old controls and transgenic Tau-P301L mice was significant. Statistical analyses (t-tests) were based on values normalized related to mice that were 2 months old. *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001. (H) Relatedness of the six selected genes dysregulated in human and mice, including the MAPT gene. The plot demonstrates the strong physical (orange nodes: 80.3%) and genetic interactions (green nodes 4.28%) between the query genes and genes have been related to pathways such as neurogenesis, behavior, learning or memory, and glutamatergic synapse. The size of the gene nodes is proportional to the degree to which the genes are related. Query genes are presented as striped dark grey balls and other selected genes subjected to interaction are presented as dark grey balls without stripes. Physical (orange; 80.3%), predicted (yellow; 9.48%), and genetic (green; 4.28%) interactions are displayed within the network. (I–K) Calcium is dysregulated in MAPT neurons. iPSC-neurons from MAPT IVS10 + 16 and isogenic control were cultured for 42 days and evaluated for calbindin 1 protein and intracellular calcium levels. (I) Immunoblot for calbindin 1 protein levels. (J) Quantification of calbindin 1 protein levels. (K) Inset, inverted fluorescent imaging representing the calcium concentration in iPSC-neurons treated with Fluo-4 Direct™ Calcium Assay Kit. Fluo4-Ca2+ binding dye relative fluorescence units (RFU) observed in iPSC-neurons carrying the IVS10 + 16 (grey bars) and isogenic controls (white bars) (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). Scale bar 20uM.
Gene dysregulation downstream of MAPT mutations impact calcium content
Next, we sought to explore the functional consequences of MAPT mutation-driven gene changes. Pathways associated with trans-synaptic signaling and lysosomal function were commonly altered by the three MAPT mutations and are regulated by calcium signaling (Figure 3E) (Lloyd-Evans and Waller-Evans, 2020). Among the 11 commonly differentially expressed genes (Figure 3), NOTCH1, PLK2, PRICKL2, and CALB1 are involved in calcium signaling. The CALB1 gene, which encodes the calbindin 1 protein and regulates Ca2+ entry into cells upon the stimulation of glutamate receptors (Noble et al., 2018), was significantly down-regulated in MAPT mutant iPSC-neurons (Figures 3B–D) and in 18-month old Tau-P301L mice when tau aggregation was present (Figure 4D). Thus, we hypothesized that reduced CALB1 expression leads to reduced intracellular calcium. To test this hypothesis, we verified that Calbindin 1 protein levels were significantly reduced in MAPT IVS10 + 16 neurons compared to isogenic controls (Figures 4I,J). We then measured calcium levels in iPSC-neurons from MAPT IVS10 + 16 and isogenic controls using Fluo-4 DirectTM, a cell-permeable fluorescent Ca2+ indicator. We observed a significant reduction in calcium levels under basal conditions, representing cytoplasmic calcium levels, in MAPT IVS10+16 iPSC-neurons compared with isogenic controls (Figure 4K). After treating with Triton-X to release intracellular calcium stores, we found that total calcium levels were also significantly reduced in MAPT IVS10+16 iPSC-neurons compared with isogenic controls (Figure 4K). Thus, we show that MAPT mutations are sufficient to disrupt calcium homeostasis in neurons.
Stem cell models capture tauopathy-relevant gene signatures
Leveraging isogenic iPSC lines to understand the contribution of a single allele to downstream phenotypes is a powerful system that when applied here has revealed gene signatures shared across MAPT mutations that also change during tau accumulation in mouse models of tauopathy. However, a limitation of this approach remains that iPSC-neurons are cultured in a dish and remain relatively immature. For example, iPSC-neurons predominantly express 0N3R tau (Patani et al., 2012; Sposito et al., 2015; Sato et al., 2018), while the adult brain expresses six tau isoforms (Hefti et al., 2018; Sato et al., 2018). Thus, we next sought to determine the extent to which the gene signatures we observe in iPSC-neurons from MAPT mutations are relevant to gene expression changes occurring in human brains with tauopathy and the extent to which these gene signatures are occurring across neurodegenerative diseases (Supplementary Table S12).
To determine the extent to which the iPSC-neuron model recapitulates gene signatures that occur in brains from MAPT mutation carriers, we analyzed transcriptomic datasets from MAPT IVS10 + 16 and MAPT p.R406W carrier brains compared with neuropathology free controls (Jiang et al., 2018). A meta-analysis of the three MAPT mutation pairs revealed that there are additional gene expression changes occurring commonly (Figure 5A); thus, for analyses of human brain datasets, we relaxed the p-value threshold and examined 275 genes (criteria: p <0.05 in single cohort analyses and Fisher’s exact FDR<0.05 in meta-analysis). Of the 275 genes changing in iPSC-neurons, we identified 114 genes in MAPT IVS10 + 16 brains and 141 genes in MAPT p.R406W brains (Figure 5B; Supplementary Table S12). The majority of the genes that are altered in MAPT carrier brains changed in the same direction as the iPSC-neuron model (Figure 5B), making gene signatures from the MAPT mutation carrier brains the most similar to the iPSC-neurons in hierarchical clustering analyses (Figure 5C). The genes that change as a function of the MAPT mutations in a dish and in disease pathology in the brains are commonly enriched in pathways related to trans-synaptic signaling, neuronal projects, and lysosomal function (Figures 5D, E; Supplemental Tables 13-14).
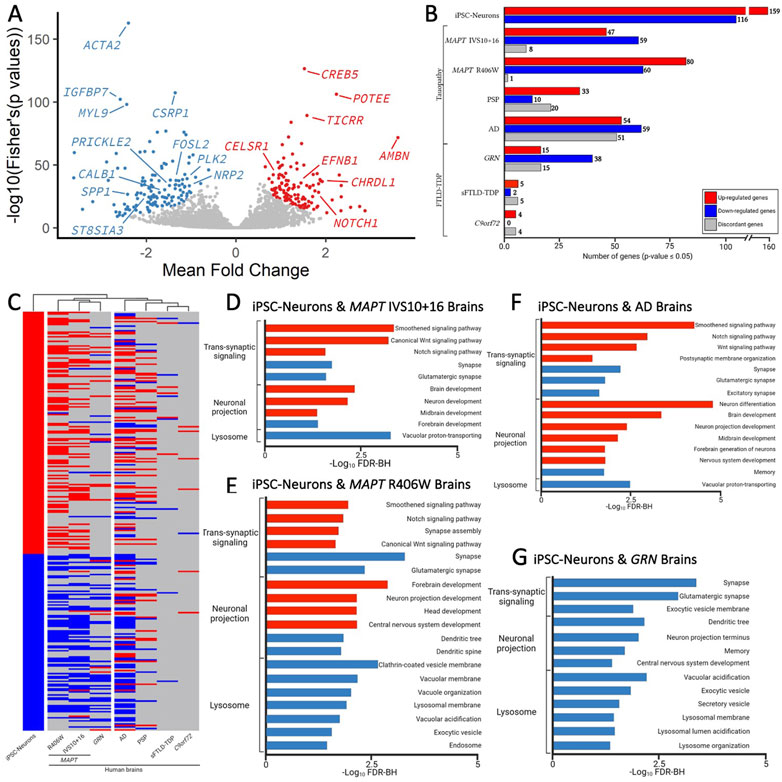
FIGURE 5. Genes changed in iPSC-neurons from MAPT mutations are also altered in brains from tauopathy patients. (A) Meta-Volcano plot identifies the common gene expression changes by MAPT IVS10 + 16, p.P301L and p.R406W. Red and blue dots represent up- and down-regulated genes, respectively. (B) Bar graph of the number of genes differentially expressed in iPSC-neurons from MAPT mutations (n = 275) and differentially expressed in brains from tauopathy patients (MAPT IVS10 + 16 and p.R406W carriers, PSP, and AD) and from FTLD-TDP (GRN mutation carriers, sporadic FTLD-TDP (sFTLD-TDP), and C9ORF72 expansion carriers). Up-regulated genes, red. Down-regulated genes, blue. Discordant genes (defined as p <0.05 and logFold change opposite of iPSC-neurons), gray. (C) Heatmap of gene expression of 275 genes in iPSC-neurons and human brains with hierarchical clustering revealing relatedness of cohorts. (D–F). Bar graphs showing the most significant pathways enriched shared between the iPSC-neurons and (D) MAPT IVS10 + 16 brains; (E) MAPT p.R406W brains; (F) sporadic AD brains; (G) GRN mutation carrier brains. Pathways from up-regulated genes (red); pathways from down-regulated genes (blue).
To determine the extent to which our genetic cellular model of primary tauopathy recapitulates molecular signatures of sporadic tauopathies, we analyzed transcriptomic data from AD, PSP, and control brains (Allen et al., 2016). We found that 63 of the 275 genes were differentially expressed in PSP brains, and 164 genes were differentially expressed in sporadic, late onset AD brains (Figure 5B; Supplementary Table S12). Interestingly, a large number of genes that were implicated in disease processes by the iPSC-neuronal model were changed in opposite directions in the PSP (n = 20) and AD brains (n = 51; Figure 5B), leading to a similar clustering of these brains with the iPSC-neuronal model but to a lesser extent than what we observe in brains from MAPT mutation carriers (Figure 5C). Again, the common genes were enriched in pathways related to trans-synaptic signaling, neuronal projection and lysosomal function (Figure 5F; Supplementary Tables S15–S16).
Synaptic dysfunction, neuronal projections, and lysosomal dysfunction have been implicated in many neurodegenerative diseases. Thus, we asked whether the gene signatures captured in the iPSC-neuronal model represents broader molecular changes in neurodegeneration. To address this question, we examined transcriptomic data from FTLD brains where the primary pathology is TDP-43 pathology (GRN, sporadic FTLD-TDP, and C9ORF72). There was minimal overlap between the genes changing in a dish as a function of the MAPT mutations and genes changing in sporadic FTLD-TDP (n = 8) and C9ORF72 expansion carrier brains (n = 12; Figures 5B,C; Supplementary Table S12). Interestingly, we observed substantial overlap between the iPSC-neuronal signatures and those occurring in GRN mutation carriers (n = 68; Figure 5B; Supplementary Table S12). Surprisingly, this led to brains from GRN mutation carriers clustering closely with brains from MAPT mutation carriers, appearing to be more similar in expression profile than sporadic tauopathies (Figure 5C). The genes were enriched in lysosomal function but also include trans-synaptic signaling and neuronal projections, all of which are consistent with proposed functions of the progranulin protein (Figure 5G; Supplementary Table S17) (Amin et al., 2022). Together, these analyses identified a set of gene signatures captured in stem cell models of MAPT mutations that also change in human brains with genetic and sporadic forms of tau pathology.
Discussion
The goal of this study was to identify commonly perturbed genes and pathways downstream of MAPT mutations and to define a core set of genes that drive disease pathogenesis in FTLD-Tau. MAPT mutations result in a range of clinical and neuropathological phenotypes (Whitwell et al., 2009; Ghetti et al., 2015; Moore et al., 2020). Here, we aimed to study three major classes of MAPT mutations that represent: splicing mutation (MAPT IVS10 + 16), 4R-expressing point mutation (MAPT p.P301L) and point mutation expressed in all isoforms (MAPT p.R406W). Our results suggest that these three mutations lead to a common series of events, causing the dysregulation of genes associated with pathways involved in synaptic, neuronal, and lysosomal function (Figure 6). Several of the differentially expressed genes were also altered in a mouse model of tauopathy, suggesting that these genes are relevant to disease pathogenesis and tau accumulation. A subset of genes were also found to be dysregulated in human brains from MAPT mutation carriers, AD, and PSP donors, illustrating that the molecular signatures we identified in iPSC-neurons are relevant to human disease. Together, this study demonstrates that iPSC-derived neurons capture molecular processes that occur in both mice and human brains, and can be used to model neurodegenerative diseases such as FTLD-Tau.
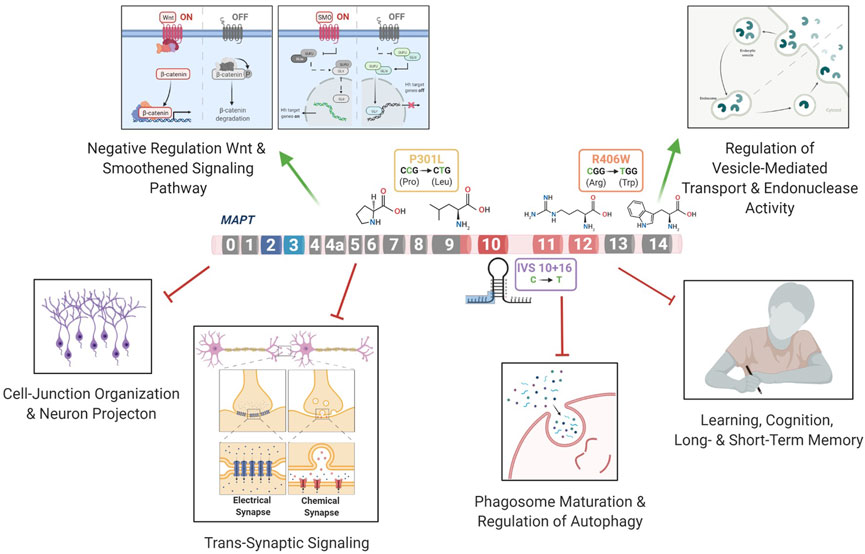
FIGURE 6. MAPT mutations lead to changes in the regulation of pathways related to FTLD-Tau.
The three mutations located in distinct regions of the MAPT gene produced a common molecular signature of genes that were enriched for pathways involved in trans-synaptic signaling, neuronal projection and lysosomal regulation. Several pathways were related to functional changes described in FTLD. Wnt signaling has been previously shown to be associated with the MAPT IVS10 + 16 mutation (Harrison-Uy and Pleasure, 2012) and linked to several neurodegenerative disorders such as AD and FTLD (Korade and Mirnics, 2011; Rosen et al., 2011; Riise et al., 2015; Verheyen et al., 2018; Bottero et al., 2021). In addition, Notch signaling has been shown to be related to the microtubule stability within neurons (Bonini et al., 2013). The Ras signaling regulates basic cellular processes in the construction of neuronal networks, including neurogenesis, vesicular trafficking, or synaptic plasticity (Johnson and Chen, 2012; Qu et al., 2019). The synapse assembly has been associated with Alzheimer’s-type dementia (Clare et al., 2010). Anterograde trans-synaptic signaling; regulation of synaptic; and long-term synaptic potentiation have all been implicated in tauopathies (Purves et al., 2001; Gómez-Palacio-Schjetnan and Escobar, 2013; Liou et al., 2019). Additionally, several genes were enriched for pathways related to the loss of learning or memory and cognition (Rabinovici and Miller, 2010; Bott et al., 2014). Finally, we found that genes associated with dysregulation of the vesicle transport along microtubule pathway; phagosome maturation and autophagy were significantly reduced. This finding is consistent with a recent study of MAPT p.V337M-expressing cerebral organoids, where mutant organoids exhibited failure of protein homeostasis, a disruption of autophagy function, and loss of glutamatergic neurons (Bowles et al., 2021). Defects in endolysosomal pathways have been implicated in reduced clearance of protein and cell debris, which may contribute to neurodegeneration (Liu et al., 2012; Viegas et al., 2012; Gunawardena et al., 2014; Etchegaray et al., 2016; Kulkarni and Maday, 2018; Xu et al., 2021c). Thus, here, we identify a series of pathways that are common across different MAPT mutation types, suggesting that stem cell models capture defects observed in FTLD-Tau patients (Figure 6).
We report that several genes from the mutant iPSC-neurons were also altered at disease end-stage in the Tau-P301L mouse model of tauopathy: CALB1, PLK2, CELSR1, CHRDL1, PRICKLE2, and ST8SIA3. PLK2 (Polo like kinase 2) is associated with synaptic plasticity and prevention of cell death in neurodegenerative diseases (Kauselmann, 1999; Seeburg et al., 2005; Seeburg et al., 2008; Li et al., 2014; Weston et al., 2021). Modulating the activity of the PLK2 gene has been proposed as a therapeutic strategy for the treatment of Parkinson’s disease (Oueslati et al., 2013). CELSR1 (Cadherin EGF LAG seven-pass G-type receptor 1) encodes a receptor protein involved in cell adhesion and receptor-ligand interactions (Hadjantonakis et al., 1997). CELSR1 has been related to neurodevelopment and maintenance of the nervous system (Boutin et al., 2012), and mutations in this gene have been associated with neural tube defects (Robinson et al., 2012) and AD risk (Patel et al., 2019). CHRDL1 (Chordin-like 1) plays an important role in CNS development, learning, and promoting synaptic plasticity (Sun et al., 2007; Webb et al., 2012; Gao et al., 2013; Blanco-Suarez et al., 2018). PRICKLE2 (Prickle planar cell polarity protein 2) is an important cytoplasmic regulator of Wnt/PCP signaling (Katoh, 2005; Barrow, 2006; Vandervorst et al., 2019). Dysregulation of PRICKLE2 enhances the amyloid β (Aβ) plaque pathology and synaptic dysfunction in mice (Tao et al., 2011; Fujimura and Hatano, 2012). PRICKLE2 has been proposed to be a potential candidate for the diagnosis and treatment of AD (Sun et al., 2020). ST8SIA3 (alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 3) is involved in neurite growth, cell migration, and synaptic plasticity (Goodman et al., 1997; Lee et al., 1998; Eckhardt et al., 2000; Lin et al., 2019) and plays an important role in the development of Huntington’s disease, schizophrenia, and Parkinson’s disease (Belarbi et al., 2020; Moll et al., 2020). A strong physical interaction was observed between MAPT and CALB1, PLK2, CELSR1, CHRDL1, PRICKLE2, and ST8SIA3 and 20 additional genes. The glutamatergic synapse pathway was enriched among these genes. Impairments in glutamatergic circuits predispose GABAergic neurons to dysfunction (Ferrer, 1999; Bowie, 2008; Hughes et al., 2018; Benussi et al., 2019; Murley et al., 2020). Dysregulation of the glutamatergic system has been described in MAPT p.V337M cerebral organoids (Borroni et al., 2017; Borroni et al., 2018; Bowles et al., 2021) and MAPT p.R406W neurons (Jiang et al., 2018). Thus, our findings suggest that the glutamatergic synapse pathway is disrupted more commonly across MAPT mutations.
The absence of overlap in the five remaining genes, and the variability in the expression observed at 4 and 8 months of mouse disease may be driven by several factors. For example, gene expression profiles represent multiple cell-types in the brain which are not included in the iPSC-neuron model, which may mask neuronal-specific gene signatures. Additionally, species differences may also impact the gene signature profile.
CALB1 was found to be down regulated across the three MAPT mutations and reduced at disease end-stage in Tau-P301L mice. The CALB1 gene (Calbindin 1) regulates the calcium homeostasis in neurons (Noble et al., 2018), which plays a crucial role in neuronal development and memory performance (Sun et al., 2005; Soontornniyomkij et al., 2012; Kook et al., 2014; Goffigan-Holmes et al., 2018; Jung et al., 2020). Given the important role of CALB1 in neuronal calcium homeostasis, we measured calcium levels in the mutant and isogenic control neurons. We found that calcium levels were significantly reduced in MAPT mutant neurons compared with isogenic controls, supporting a dysregulation in calcium homeostasis. Calcium homeostasis is critical for the health and function of neurons and dysregulation of calcium leads to altered synaptic function, endolysosomal function, and neuronal development (Gleichmann and Mattson, 2011). This is in line with recent observations in genetically engineered iPSC expressing the MAPT IVS10 + 16 mutation, which showed disturbed intracellular calcium dynamics along with impaired neuronal activity (Britti et al., 2020; Kopach et al., 2021). The Drug-Gene Interaction Database (Freshour et al., 2021) and DrugBank (Wishart et al., 2018) revealed CALB1 and PLK2, also implicated in calcium regulation, are known targets of FDA-approved drugs including tramadol, ethosuximide, levodopa, nicotine and oxcarbazepine, which are currently utilized to treat neurological symptoms (Chen et al., 2015; Tambasco et al., 2018; Morana et al., 2020). Additional work is needed to evaluate calcium across these and other MAPT mutations. Thus, restoring calcium homeostasis may be a therapeutically viable approach for treating genetic forms of primary tauopathy.
A subset of the commonly differentially expressed genes from the MAPT mutant iPSC-neurons were found to be altered in human brains from MAPT IVS10 + 16 and p.R406W carriers. Brains from MAPT mutation carriers most closely recapitulated the gene signatures in a dish, but we also detected an overlap among sporadic tauopathies, including AD and PSP. Among the up-regulated genes, we observed an enrichment in pathways involving a negative regulation of: i) nervous system development; ii) neurogenesis; iii) neuron differentiation; iv) neuron projection development; and v) the canonical Wnt signaling pathway. Dysregulation of these pathways have been linked to FTLD patients (Rabinovici and Miller, 2010; Mann and Snowden, 2017; Yousef et al., 2017; Sobue et al., 2018; Bottero et al., 2021). Interestingly, we observed a broad reduction of gene enriched in lysosomal pathways. This is consistent with recent implications of lysosomal dysfunction in genetic and sporadic forms of FTLD-Tau (Polito et al., 2014; Caballero et al., 2018; Silva et al., 2019; Xu et al., 2019; Silva et al., 2020; Xu et al., 2021a; Bowles et al., 2021; Xu et al., 2021b; Caballero et al., 2021; Mahali et al., 2022; Silva et al., 2022). These gene signatures were largely specific for tauopathy; however, we identified a number of lysosomal genes that were altered in iPSC-neurons from MAPT mutations and in brains from GRN mutation carriers. GRN has been implicated in lysosomal function and neuronal integrity (Martens et al., 2012; Kao et al., 2017; Logan et al., 2021; Mohan et al., 2021; Simon et al., 2022). Thus, our stem cell model revealed several genes and pathways that are also altered in primary tauopathy patients.
Our results demonstrate the potential of iPSC technology to investigate disease mechanisms related to FTLD-Tau pathogenesis. A major challenge related to the iPSC technology and neuronal derivatives (iPSC-neurons) when modeling adult-onset neurodegenerative disease concerns capturing age-related phenotypes (Steg et al., 2021). While iPSC-neurons remain relatively immature and do not express the full complement of tau isoforms (Sposito et al., 2015; Sato et al., 2018), we are able to capture molecular signatures that change during disease course in mouse models of tauopathy and patient brains. Thus, there remains tremendous value in a combinatorial multiple model systems approach to identify key pathways that are affected early and remain relevant throughout the disease course.
Furthermore, the observation that different MAPT mutations may lead to a common pathophysiological mechanism has not been carefully studied. Thus, these findings have broader implications when considering therapeutic development and trial design. For example, drugs that target these common pathways may be effective across tauopathy patients. This study provides a framework for developing drugs targeting key dysregulated genes.
In conclusion, our approach provides a tractable system to identify genes altered directly by the MAPT-IVS10 + 16, p.P301L, and p.R406W mutations, which are relevant to tauopathies and that point to new therapeutic targets. The stem cell lines used in this research allowed for the identification of molecular drivers of disease, which could serve as a platform to identify new targets for drug development. Our iPSC-based cellular models have discovered a common gene signature which is enriched in dysregulated pathways involving synaptic connections, lysosome transport and neuronal development, and mechanisms that have been previously described to be altered in human brains and mouse models of tauopathy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Skin punches were performed following written informed consent from the donor. The informed consent was approved by the Washington University School of Medicine and University of California San Francisco Institutional Review Board and Ethics Committee (IRB 201104178, 201306108 and 10-03946). The consent allows for use of tissue by all parties, commercial and academic, for the purposes of research but not for use in human therapy. The patients/participants provided their written informed consent to participate in this study.
Author contributions
MM, SM, AI performed experiments, analyzed data and wrote the manuscript. RM, ST generated key reagents. JB, AE, and OH generated and QC composed data. CK designed the study, analyzed the data, supervised the study, wrote the manuscript and provided funding. All authors edited the manuscript.
Funding
This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the Neurogenomics and Informatics Center, and the Departments of Neurology and Psychiatry at Washington University School of Medicine. OH is an Archer Foundation Research Scientist. Confocal images were generated on a Zeiss LSM 880 Airyscan Confocal Microscope which was purchased with support from the Office of Research Infrastructure Programs (ORIP), a part of the NIH Office of the Director under grant OD021629. Funding provided by the National Institutes of Health (P30 AG066444, R01 AG056293, R56 NS110890, RF1 NS110890, U54 NS123985, R01 AG062359, R01 AG057777, K24 AG053435), Hope Center for Neurological Disorders (CK), Rainwater Charitable Organization (CK), Farrell Family Fund for Alzheimer’s Disease (CK), and UL1TR002345. The recruitment and clinical characterization of research participants at Washington University were supported by NIH P30AG066444 (JCM), P01AG03991 (JCM), and P01AG026276 (JCM). The UCSF Neurodegenerative Disease Brain Bank receives funding support from NIH grants P30AG062422, P01AG019724, U01AG057195, and U19AG063911, as well as the Rainwater Charitable Foundation and the Bluefield Project to Cure FTD. Diagrams were generated used BioRender.com.
Acknowledgments
We would like to thank the research subjects and their families who generously participated in this study. We thank Dr. Aimee Kao who generously provided the fibroblasts used to generate the GIH36C2 iPSC line (supported by the Rainwater Charitable Organization).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MS declared a past co-authorship with the authors SM, ST and CK to the handling editor.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1051494/full#supplementary-material
References
Allen, M., Carrasquillo, M. M., Funk, C., Heavner, B. D., Zou, F., Younkin, C. S., et al. (2016). Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3 (1), 160089. doi:10.1038/sdata.2016.89
Allen, M., Kachadoorian, M., Carrasquillo, M. M., Karhade, A., Manly, L., Burgess, J. D., et al. (2015). Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol. Genet. 1 (2), e15. doi:10.1212/NXG.000000000000001
Amin, S., Carling, G., and Gan, L. (2022). New insights and therapeutic opportunities for progranulin-deficient frontotemporal dementia. Curr. Opin. Neurobiol. 72, 131–139. doi:10.1016/j.conb.2021.10.001
Andrews, S., Krueger, F., Segonds-Pichon, A., Biggins, L., Krueger, C., and Wingett, S. (2012). FastQC: A quality control tool for high throughput sequence data. Cambridge, MA: Babraham Institute. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ ((Accessed).
Ban, H., Nishishita, N., Fusaki, N., Tabata, T., Saeki, K., Shikamura, M., et al. (2011). Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. U. S. A. 108 (34), 14234–14239. doi:10.1073/pnas.1103509108
Barrow, J. R. (2006). Wnt/PCP signaling: A veritable polar star in establishing patterns of polarity in embryonic tissues. Semin. Cell. Dev. Biol. 17 (2), 185–193. doi:10.1016/j.semcdb.2006.04.002
Belarbi, K., Cuvelier, E., Bonte, M.-A., Desplanque, M., Gressier, B., Devos, D., et al. (2020). Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol. Neurodegener. 15 (1), 59. doi:10.1186/s13024-020-00408-1
Benussi, A., Alberici, A., Buratti, E., Ghidoni, R., Gardoni, F., Di Luca, M., et al. (2019). Toward a glutamate hypothesis of frontotemporal dementia. Front. Neurosci. 13 (304), 304. doi:10.3389/fnins.2019.00304
Blanco-Suarez, E., Liu, T.-F., Kopelevich, A., and Allen, N. J. (2018). Astrocyte-secreted chordin-like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron 100 (5), 1116–1132. e13. doi:10.1016/j.neuron.2018.09.043
Bodea, L. G., Eckert, A., Ittner, L. M., Piguet, O., and Götz, J. (2016). Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J. Neurochem. 138 (S1), 71–94. doi:10.1111/jnc.13600
Bonini, S. A., Ferrari-Toninelli, G., Montinaro, M., and Memo, M. (2013). Notch signalling in adult neurons: A potential target for microtubule stabilization. Ther. Adv. Neurological Disord. 6 (6), 375–385. doi:10.1177/1756285613490051
Borroni, B., Stanic, J., Verpelli, C., Mellone, M., Bonomi, E., Alberici, A., et al. (2017). Anti-AMPA GluA3 antibodies in frontotemporal dementia: A new molecular target. Sci. Rep. 7 (1), 6723. doi:10.1038/s41598-017-06117-y
Borroni, B., Benussi, A., Premi, E., Alberici, A., Marcello, E., Gardoni, F., et al. (2018). Biological, neuroimaging, and neurophysiological markers in frontotemporal dementia: Three faces of the same coin. J. Alzheimers Dis. 62 (3), 1113–1123. doi:10.3233/JAD-170584
Bott, N. T., Radke, A., Stephens, M. L., and Kramer, J. H. (2014). Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener. Dis. Manag. 4 (6), 439–454. doi:10.2217/nmt.14.34
Bottero, V., Alrafati, F., Santiago, J. A., and Potashkin, J. A. (2021). Transcriptomic and network meta-analysis of frontotemporal dementias. Front. Mol. Neurosci. 14 (239), 747798. doi:10.3389/fnmol.2021.747798
Boutin, C., Goffinet, A. M., and Tissir, F. (2012). Celsr1-3 cadherins in PCP and brain development. Curr. Top. Dev. Biol. 101, 161–183. doi:10.1016/B978-0-12-394592-1.00010-7
Bowie, D. (2008). Ionotropic glutamate receptors & CNS disorders. CNS Neurol. Disord. Drug Targets 7 (2), 129–143. doi:10.2174/187152708784083821
Bowles, K. R., Silva, M. C., Whitney, K., Bertucci, T., Berlind, J. E., Lai, J. D., et al. (2021). ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell. 184 (17), 4547–4563. e17. doi:10.1016/j.cell.2021.07.003
Britti, E., Ros, J., Esteras, N., and Abramov, A. Y. (2020). Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell. Calcium 86, 102150. doi:10.1016/j.ceca.2019.102150
Browning, S. R., and Browning, B. L. (2010). High-resolution detection of identity by descent in unrelated individuals. Am. J. Hum. Genet. 86 (4), 526–539. doi:10.1016/j.ajhg.2010.02.021
Caballero, B., Wang, Y., Diaz, A., Tasset, I., Juste, Y. R., Stiller, B., et al. (2018). Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 17 (1), e12692. doi:10.1111/acel.12692
Caballero, B., Bourdenx, M., Luengo, E., Diaz, A., Sohn, P. D., Chen, X., et al. (2021). Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 12 (1), 2238. doi:10.1038/s41467-021-22501-9
Chen, J., Bardes, E. E., Aronow, B. J., and Jegga, A. G. (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37 (2), W305–W311. doi:10.1093/nar/gkp427
Chen, E. Y., Tan, C. M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G. V., et al. (2013). Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128. doi:10.1186/1471-2105-14-128
Chen, X., Mccue, H. V., Wong, S. Q., Kashyap, S. S., Kraemer, B. C., Barclay, J. W., et al. (2015). Erratum to: Ethosuximide ameliorates neurodegenerative disease phenotypes by modulating DAF-16/FOXO target gene expression. Mol. Neurodegener. 10 (1), 54. doi:10.1186/s13024-015-0051-6
Clare, R., King, V. G., Wirenfeldt, M., and Vinters, H. V. (2010). Synapse loss in dementias. J. Neurosci. Res. 88 (10), 2083–2090. doi:10.1002/jnr.22392
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2012). Star: Ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
Dube, U., Del-Aguila, J. L., Li, Z., Budde, J. P., Jiang, S., Hsu, S., et al. (2019). An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 22 (11), 1903–1912. doi:10.1038/s41593-019-0501-5
Eckhardt, M., Bukalo, O., Chazal, G., Wang, L., Goridis, C., Schachner, M., et al. (2000). Mice deficient in the polysialyltransferase ST8SiaIV/PST-1 allow discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity. J. Neurosci. 20 (14), 5234–5244. doi:10.1523/JNEUROSCI.20-14-05234.2000
Etchegaray, J. I., Elguero, E. J., Tran, J. A., Sinatra, V., Feany, M. B., and Mccall, K. (2016). Defective phagocytic corpse processing results in neurodegeneration and can Be rescued by TORC1 activation. J. Neurosci. 36 (11), 3170–3183. doi:10.1523/JNEUROSCI.1912-15.2016
Ferrari, R., Hernandez, D. G., Nalls, M. A., Rohrer, J. D., Ramasamy, A., Kwok, J. B., et al. (2014). Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurol. 13 (7), 686–699. doi:10.1016/S1474-4422(14)70065-1
Ferrer, I. (1999). Neurons and their dendrites in frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 10 (1), 55–60. doi:10.1159/000051214
Freshour, S. L., Kiwala, S., Cotto, K. C., Coffman, A. C., Mcmichael, J. F., Song, J. J., et al. (2021). Integration of the drug–gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49 (D1), D1144–D1151. doi:10.1093/nar/gkaa1084
Fujimura, L., and Hatano, M. (2012). “Role of Prickle1 and Prickle2 in neurite outgrowth in murine neuroblastoma cells,” in Methods in molecular Biology (New York: Springer), 173–185.
Gao, W.-L., Zhang, S.-Q., Zhang, H., Wan, B., and Yin, Z.-S. (2013). Chordin-like protein 1 promotes neuronal differentiation by inhibiting bone morphogenetic protein-4 in neural stem cells. Mol. Med. Rep. 7 (4), 1143–1148. doi:10.3892/mmr.2013.1310
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathology Appl. Neurobiol. 41 (1), 24–46. doi:10.1111/nan.12213
Gleichmann, M., and Mattson, M. P. (2011). Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal 14 (7), 1261–1273. doi:10.1089/ars.2010.3386
Goffigan-Holmes, J., Sanabria, D., Diaz, J., Flock, D., and Chavez-Valdez, R. (2018). Calbindin-1 expression in the Hippocampus following neonatal hypoxia-ischemia and therapeutic hypothermia and deficits in spatial memory. Dev. Neurosci. 40 (5-6), 508–522. doi:10.1159/000497056
Gómez-Palacio-Schjetnan, A., and Escobar, M. L. (2013). “Neurotrophins and synaptic plasticity,” in Neurogenesis and neural plasticity (Springer Berlin Heidelberg), 117–136.
Gonzalez, C., Armijo, E., Bravo-Alegria, J., Becerra-Calixto, A., Mays, C. E., and Soto, C. (2018). Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 23 (12), 2363–2374. doi:10.1038/s41380-018-0229-8
Goodman, C. S., Davis, G. W., and Zito, K. (1997). The many faces of fasciclin II: Genetic analysis reveals multiple roles for a cell adhesion molecule during the generation of neuronal specificity. Cold Spring Harb. Symp. Quant. Biol. 62, 479–491.
Gunawardena, S., Anderson, E., and White, J. (2014). Axonal transport and neurodegenerative disease: Vesicle-motor complex formation and their regulation. Degener. Neurological Neuromuscul. Dis. 4, 29.
Guo, W., Fumagalli, L., Prior, R., and Van Den Bosch, L. (2017). Current advances and limitations in modeling ALS/FTD in a dish using induced pluripotent stem cells. Front. Neurosci. 11, 671. doi:10.3389/fnins.2017.00671
Hadjantonakis, A.-K., Sheward, W. J., Harmar, A. J., De Galan, L., Hoovers, J. M. N., and Little, P. F. R. (1997). Celsr1, a neural-specific gene encoding an unusual seven-pass transmembrane receptor, maps to mouse chromosome 15 and human chromosome 22qter. Genomics 45 (1), 97–104. doi:10.1006/geno.1997.4892
Harrison-Uy, S. J., and Pleasure, S. J. (2012). Wnt signaling and forebrain development. Cold Spring Harb. Perspect. Biol. 4 (7), a008094. doi:10.1101/cshperspect.a008094
Hefti, M. M., Farrell, K., Kim, S., Bowles, K. R., Fowkes, M. E., Raj, T., et al. (2018). High-resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS One 13 (4), e0195771. doi:10.1371/journal.pone.0195771
Hughes, L. E., Rittman, T., Robbins, T. W., and Rowe, J. B. (2018). Reorganization of cortical oscillatory dynamics underlying disinhibition in frontotemporal dementia. Brain 141 (8), 2486–2499. doi:10.1093/brain/awy176
Iovino, M., Agathou, S., Gonzalez-Rueda, A., Del Castillo Velasco-Herrera, M., Borroni, B., Alberici, A., et al. (2015). Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138 (11), 3345–3359. doi:10.1093/brain/awv222
Jiang, S., Wen, N., Li, Z., Dube, U., Del Aguila, J., Budde, J., et al. (2018). Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl. Psychiatry 8 (1), 265. doi:10.1038/s41398-018-0319-z
Johnson, D. S., and Chen, Y. H. (2012). Ras family of small GTPases in immunity and inflammation. Curr. Opin. Pharmacol. 12 (4), 458–463. doi:10.1016/j.coph.2012.02.003
Jung, E. M., Yoo, Y. M., Park, S. Y., Ahn, C., Jeon, B. H., Hong, E. J., et al. (2020). Calbindin-D(9k) is a novel risk gene for neurodegenerative disease. Cell. Physiol. Biochem. 54 (3), 438–456. doi:10.33594/000000229
Kao, A. W., McKay, A., Singh, P. P., Brunet, A., and Huang, E. J. (2017). Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci. 18 (6), 325–333. doi:10.1038/nrn.2017.36
Karch, C. M., Hernandez, D., Wang, J. C., Marsh, J., Hewitt, A. W., Hsu, S., et al. (2018). Human fibroblast and stem cell resource from the dominantly inherited alzheimer network. Alzheimers Res. Ther. 10 (1), 69. doi:10.1186/s13195-018-0400-0
Karch, C. M., Kao, A. W., Karydas, A., Onanuga, K., Martinez, R., Argouarch, A., et al. (2019). A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell. Rep. 13 (5), 939–955. doi:10.1016/j.stemcr.2019.09.006
Katoh, M. (2005). WNT/PCP signaling pathway and human cancer (review). Oncol. Rep. 14 (6), 1583–1588. doi:10.3892/or.14.6.1583
Kauselmann, G., Weiler, M., Wulff, P., JeSSberger, S., Konietzko, U., Scafidi, J., et al. (1999). The polo-like protein kinases Fnk and Snk associate with a Ca2+- and integrin-binding protein and are regulated dynamically with synaptic plasticity. EMBO J. 18 (20), 5528–5539. doi:10.1093/emboj/18.20.5528
Kilpinen, H., Goncalves, A., Leha, A., Afzal, V., Alasoo, K., Ashford, S., et al. (2017). Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546 (7658), 370–375. doi:10.1038/nature22403
Kook, S.-Y., Jeong, H., Kang, M. J., Park, R., Shin, H. J., Han, S.-H., et al. (2014). Crucial role of calbindin-D28k in the pathogenesis of Alzheimer’s disease mouse model. Cell. Death Differ. 21 (10), 1575–1587. doi:10.1038/cdd.2014.67
Kopach, O., Esteras, N., Wray, S., Abramov, A. Y., and Rusakov, D. A. (2021). Genetically engineered MAPT 10+16 mutation causes pathophysiological excitability of human iPSC-derived neurons related to 4R tau-induced dementia. Cell. Death Dis. 12 (8), 716. doi:10.1038/s41419-021-04007-w
Korade, Ž., and Mirnics, K. (2011). Wnt signaling as a potential therapeutic target for frontotemporal dementia. Neuron 71 (6), 955–957. doi:10.1016/j.neuron.2011.09.002
Kouri, N., Ross, O. A., Dombroski, B., Younkin, C. S., Serie, D. J., Soto-Ortolaza, A., et al. (2015). Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6, 7247. doi:10.1038/ncomms8247
Kuleshov, M. V., Jones, M. R., Rouillard, A. D., Fernandez, N. F., Duan, Q., Wang, Z., et al. (2016). Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. doi:10.1093/nar/gkw377
Kulkarni, V. V., and Maday, S. (2018). Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 78 (3), 298–310. doi:10.1002/dneu.22562
Lagomarsino, V. N., Pearse, R. V., Liu, L., Hsieh, Y.-C., Fernandez, M. A., Vinton, E. A., et al. (2021). Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron 109, 3402–3420.e9. doi:10.1016/j.neuron.2021.08.003
Lee, Y. C., Kim, Y. J., Lee, K. Y., Kim, K. S., Kim, B. U., Kim, H. N., et al. (1998). Cloning and expression of cDNA for a human Sia alpha 2,3Gal beta 1, 4GlcNA:alpha 2,8-sialyltransferase (hST8Sia III). Arch. Biochem. Biophys. 360 (1), 41–46. doi:10.1006/abbi.1998.0909
Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi:10.1146/annurev.neuro.24.1.1121
Li, J., Ma, W., Wang, P.-Y., Hurley, P. J., Bunz, F., and Hwang, P. M. (2014). Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free Radic. Biol. Med. 73, 270–277. doi:10.1016/j.freeradbiomed.2014.05.022
Li, Z., Del-Aguila, J. L., Dube, U., Budde, J., Martinez, R., Black, K., et al. (2018). Genetic variants associated with Alzheimer's disease confer different cerebral cortex cell-type population structure. Genome Med. 10 (1), 43. doi:10.1186/s13073-018-0551-4
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z., and Zhang, B. (2019). WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47 (W1), W199–W205. doi:10.1093/nar/gkz401
Lin, C.-Y., Lai, H.-L., Chen, H.-M., Siew, J.-J., Hsiao, C.-T., Chang, H.-C., et al. (2019). Functional roles of ST8SIA3-mediated sialylation of striatal dopamine D2 and adenosine A2A receptors. Transl. Psychiatry 9 (1), 209. doi:10.1038/s41398-019-0529-z
Liou, C. J., Tong, M., Vonsattel, J. P., and De La Monte, S. M. (2019). Altered brain expression of insulin and insulin-like growth factors in frontotemporal lobar degeneration: Another degenerative disease linked to dysregulation of insulin metabolic pathways. ASN Neuro 11, 1759091419839515. doi:10.1177/1759091419839515
Liu, X.-A., Rizzo, V., and Puthanveettil, S. (2012). Pathologies of axonal transport in neurodegenerative diseases. Transl. Neurosci. 3 (4), 355–372. doi:10.2478/s13380-012-0044-7
Livesey, F. J. (2014). Human stem cell models of dementia. Hum. Mol. Genet. 23 (R1), R35–R39. doi:10.1093/hmg/ddu302
Lloyd-Evans, E., and Waller-Evans, H. (2020). Lysosomal Ca(2+) homeostasis and signaling in health and disease. Cold Spring Harb. Perspect. Biol. 12 (6), a035311. doi:10.1101/cshperspect.a035311
Logan, T., Simon, M. J., Rana, A., Cherf, G. M., Srivastava, A., Davis, S. S., et al. (2021). Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell. 184 (18), 4651–4668 e25. doi:10.1016/j.cell.2021.08.002
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Mahali, S., Martinez, R., King, M., Verbeck, A., Harari, O., Benitez, B. A., et al. (2022). Defective proteostasis in induced pluripotent stem cell models of frontotemporal lobar degeneration. Transl. Psychiatry 12 (1), 508. doi:10.1038/s41398-022-02274-5
Mann, D. M. A., and Snowden, J. S. (2017). Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 27 (6), 723–736. doi:10.1111/bpa.12486
Martens, L. H., Zhang, J., Barmada, S. J., Zhou, P., Kamiya, S., Sun, B., et al. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. investigation 122, 3955–3959. doi:10.1172/JCI63113
Matarin, M., Salih, D. A., Yasvoina, M., Cummings, D. M., Guelfi, S., Liu, W., et al. (2015). A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell. Rep. 10 (4), 633–644. doi:10.1016/j.celrep.2014.12.041
Mohan, S., Sampognaro, P. J., Argouarch, A. R., Maynard, J. C., Welch, M., Patwardhan, A., et al. (2021). Processing of progranulin into granulins involves multiple lysosomal proteases and is affected in frontotemporal lobar degeneration. Mol. Neurodegener. 16 (1), 51. doi:10.1186/s13024-021-00472-1
Moll, T., Shaw, P. J., and Cooper-Knock, J. (2020). Disrupted glycosylation of lipids and proteins is a cause of neurodegeneration. Brain 143 (5), 1332–1340. doi:10.1093/brain/awz358
Moore, K. M., Nicholas, J., Grossman, M., Mcmillan, C. T., Irwin, D. J., Massimo, L., et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: An international retrospective cohort study. Lancet Neurology 19 (2), 145–156. doi:10.1016/S1474-4422(19)30394-1
Morana, P., Mucci, F., Baroni, S., Della Vecchia, A., Piccinni, A., Morana, B., et al. (2020). Effectiveness of clozapine, oxcarbazepine and rivastigmine combination in a bipolar disorder patient with initial cerebral atrophy. Clin. Case Rep. 8 (2), 254–257. doi:10.1002/ccr3.2462
Murley, A. G., Rouse, M. A., Jones, P. S., Ye, R., Hezemans, F. H., O’Callaghan, C., et al. (2020). GABA and glutamate deficits from frontotemporal lobar degeneration are associated with disinhibition. Brain 143 (11), 3449–3462. doi:10.1093/brain/awaa305
Nakamura, M., Shiozawa, S., Tsuboi, D., Amano, M., Watanabe, H., Maeda, S., et al. (2019). Pathological progression induced by the frontotemporal dementia-associated R406W tau mutation in patient-derived iPSCs. Stem Cell. Rep. 13 (4), 684–699. doi:10.1016/j.stemcr.2019.08.011
Neve, R. L., Harris, P., Kosik, K. S., Kurnit, D. M., and Donlon, T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387 (3), 271–280. doi:10.1016/0169-328x(86)90033-1
Noble, J. W., Almalki, R., Roe, S. M., Wagner, A., Duman, R., and Atack, J. R. (2018). The X-ray structure of human calbindin-D28K: An improved model. Acta Crystallogr. Sect. D. Struct. Biol. 74 (10), 1008–1014. doi:10.1107/S2059798318011610
Oueslati, A., Schneider, B. L., Aebischer, P., and Lashuel, H. A. (2013). Polo-like kinase 2 regulates selective autophagic -synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. 110 (41), E3945–E3954. doi:10.1073/pnas.1309991110
Patani, R., Lewis, P. A., Trabzuni, D., Puddifoot, C. A., Wyllie, D. J., Walker, R., et al. (2012). Investigating the utility of human embryonic stem cell-derived neurons to model ageing and neurodegenerative disease using whole-genome gene expression and splicing analysis. J. Neurochem. 122 (4), 738–751. doi:10.1111/j.1471-4159.2012.07825.x
Patel, D., Mez, J., Vardarajan, B. N., Staley, L., Chung, J., Zhang, X., et al. (2019). Association of rare coding mutations with alzheimer disease and other dementias among adults of European ancestry. JAMA Netw. Open 2 (3), e191350. doi:10.1001/jamanetworkopen.2019.1350
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14 (4), 417–419. doi:10.1038/nmeth.4197
Polito, V. A., Li, H., Martini-Stoica, H., Wang, B., Yang, L., Xu, Y., et al. (2014). Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 6 (9), 1142–1160. doi:10.15252/emmm.201303671
Pottier, C., Ravenscroft, T. A., Sanchez-Contreras, M., and Rademakers, R. (2016). Genetics of FTLD: Overview and what else we can expect from genetic studies. J. Neurochem. 138 (S1), 32–53. doi:10.1111/jnc.13622
Prada, C., Lima, D., and Nakaya, H. (2021). MetaVolcanoR: Gene expression meta-analysis visualization tool. R package version 1.6.0.
Purves, D., Augustine, G., and Fitzpatrick, D. (2001). Neuroscience long-term synaptic potentiation. Neuroscience 2nd edition edn. Sunderland MA: Sinauer Associates.
Qu, L., Pan, C., He, S.-M., Lang, B., Gao, G.-D., Wang, X.-L., et al. (2019). The ras superfamily of small GTPases in non-neoplastic cerebral diseases. Front. Mol. Neurosci. 12 (121), 121. doi:10.3389/fnmol.2019.00121
Rabinovici, G. D., and Miller, B. L. (2010). Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24 (5), 375–398. doi:10.2165/11533100-000000000-00000
Ramsden, M., Kotilinek, L., Forster, C., Paulson, J., McGowan, E., SantaCruz, K., et al. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J. Neurosci. 25 (46), 10637–10647. the official journal of the Society for Neuroscience. doi:10.1523/JNEUROSCI.3279-05.2005
Riise, J., Plath, N., Pakkenberg, B., and Parachikova, A. (2015). Aberrant Wnt signaling pathway in medial temporal lobe structures of Alzheimer’s disease. J. Neural Transm. 122 (9), 1303–1318. doi:10.1007/s00702-015-1375-7
Robinson, A., Escuin, S., Doudney, K., Vekemans, M., Stevenson, R. E., Greene, N. D. E., et al. (2012). Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 33 (2), 440–447. doi:10.1002/humu.21662
Rosen, Y., EzraWexler, M., EricVersano, R., Coppola, G., Gao, F., Winden, D., et al. (2011). Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron 71 (6), 1030–1042. doi:10.1016/j.neuron.2011.07.021
Sato, C., Barthelemy, N. R., Mawuenyega, K. G., Patterson, B. W., Gordon, B. A., Jockel-Balsarotti, J., et al. (2018). Tau kinetics in neurons and the human central nervous system. Neuron 97 (6), 861–864. e7. doi:10.1016/j.neuron.2018.04.035
Seeburg, D. P., Pak, D., and Sheng, M. (2005). Polo-like kinases in the nervous system. Oncogene 24 (2), 292–298. doi:10.1038/sj.onc.1208277
Seeburg, D. P., Feliu-Mojer, M., Gaiottino, J., Pak, D. T. S., and Sheng, M. (2008). Critical role of CDK5 and polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron 58 (4), 571–583. doi:10.1016/j.neuron.2008.03.021
Silva, M. C., Cheng, C., Mair, W., Almeida, S., Fong, H., Biswas, M. H. U., et al. (2016). Human iPSC-derived neuronal model of tau-a152t frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell. Rep. 7 (3), 325–340. doi:10.1016/j.stemcr.2016.08.001
Silva, M. C., Ferguson, F. M., Cai, Q., Donovan, K. A., Nandi, G., Patnaik, D., et al. (2019). Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 8, e45457. doi:10.7554/eLife.45457
Silva, M. C., Nandi, G. A., Tentarelli, S., Gurrell, I. K., Jamier, T., Lucente, D., et al. (2020). Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat. Commun. 11 (1), 3258. doi:10.1038/s41467-020-16984-1
Silva, M. C., Nandi, G., Donovan, K. A., Cai, Q., Berry, B. C., Nowak, R. P., et al. (2022). Discovery and optimization of tau targeted protein degraders enabled by patient induced pluripotent stem cells-derived neuronal models of tauopathy. Front. Cell. Neurosci. 16, 801179. doi:10.3389/fncel.2022.801179
Simon, M. J., Logan, T., DeVos, S. L., and Di Paolo, G. (2022). Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell. Biol. doi:10.1016/j.tcb.2022.09.006
Sobue, G., Ishigaki, S., and Watanabe, H. (2018). Pathogenesis of frontotemporal lobar degeneration: Insights from loss of function theory and early involvement of the caudate nucleus. Front. Neurosci. 12 (473), 473. doi:10.3389/fnins.2018.00473
Soontornniyomkij, V., Risbrough, V. B., Young, J. W., Soontornniyomkij, B., Jeste, D. V., and Achim, C. L. (2012). Hippocampal calbindin-1 immunoreactivity correlate of recognition memory performance in aged mice. Neurosci. Lett. 516 (1), 161–165. doi:10.1016/j.neulet.2012.03.092
Sposito, T., Preza, E., Mahoney, C. J., Seto-Salvia, N., Ryan, N. S., Morris, H. R., et al. (2015). Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum. Mol. Genet. 24 (18), 5260–5269. doi:10.1093/hmg/ddv246
Steele, N. Z., Bright, A. R., Lee, S. E., Fong, J. C., Bonham, L. W., Karydas, A., et al. (2018). Frequency of frontotemporal dementia gene variants in C9ORF72, MAPT, and GRN in academic versus commercial laboratory cohorts. Adv. Genomics Genet. 8, 23–33. doi:10.2147/AGG.S164047
Steg, L. C., Shireby, G. L., Imm, J., Davies, J. P., Franklin, A., Flynn, R., et al. (2021). Novel epigenetic clock for fetal brain development predicts prenatal age for cellular stem cell models and derived neurons. Mol. Brain 14 (1), 98. doi:10.1186/s13041-021-00810-w
Sun, F., Jiang, F., Zhang, N., Li, H., Tian, W., and Liu, W. (2020). Upregulation of Prickle2 ameliorates alzheimer's disease-like pathology in a transgenic mouse model of alzheimer's disease. Front. Cell. Dev. Biol. 8, 565020. doi:10.3389/fcell.2020.565020
Sun, Z., Wang, H. B., Deng, Y. P., Lei, W. L., Xie, J. P., Meade, C. A., et al. (2005). Increased calbindin-D28k immunoreactivity in striatal projection neurons of R6/2 Huntington's disease transgenic mice. Neurobiol. Dis. 20 (3), 907–917. doi:10.1016/j.nbd.2005.05.023
Sun, M., Thomas, M. J., Herder, R., Bofenkamp, M. L., Selleck, S. B., and O'Connor, M. B. (2007). Presynaptic contributions of chordin to hippocampal plasticity and spatial learning. J. Neurosci. 27 (29), 7740–7750. doi:10.1523/JNEUROSCI.1604-07.2007
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126 (4), 663–676. doi:10.1016/j.cell.2006.07.024
Tambasco, N., Romoli, M., and Calabresi, P. (2018). Levodopa in Parkinson’s disease: Current status and future developments. Curr. Neuropharmacol. 16 (8), 1239–1252. doi:10.2174/1570159X15666170510143821
Tao, H., Manak, J. R., Sowers, L., Mei, X., Kiyonari, H., Abe, T., et al. (2011). Mutations in Prickle orthologs cause seizures in flies, mice, and humans. Am. J. Hum. Genet. 88 (2), 138–149. doi:10.1016/j.ajhg.2010.12.012
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform 14 (2), 178–192. doi:10.1093/bib/bbs017
Vandervorst, K., Dreyer, C. A., Konopelski, S. E., Lee, H., Ho, H.-Y. H., and Carraway, K. L. (2019). Wnt/PCP signaling contribution to carcinoma collective cell migration and metastasis. Cancer Res. 79 (8), 1719–1729. doi:10.1158/0008-5472.CAN-18-2757
Verheyen, A., Diels, A., Reumers, J., Van Hoorde, K., Van den Wyngaert, I., van Outryve d'Ydewalle, C., et al. (2018). Genetically engineered iPSC-derived FTDP-17 MAPT neurons display mutation-specific neurodegenerative and neurodevelopmental phenotypes. Stem Cell. Rep. 11 (2), 363–379. doi:10.1016/j.stemcr.2018.06.022
Viegas, M. S., Estronca, L. M. B. B., and Vieira, O. V. (2012). Comparison of the kinetics of maturation of phagosomes containing apoptotic cells and IgG-opsonized particles. PLoS ONE 7 (10), e48391. doi:10.1371/journal.pone.0048391
Wani, A., Zhu, J., Ulrich, J. D., Eteleeb, A., Sauerbeck, A. D., Reitz, S. J., et al. (2021). Neuronal VCP loss of function recapitulates FTLD-TDP pathology. Cell. Rep. 36 (3), 109399. doi:10.1016/j.celrep.2021.109399
Warde-Farley, D., Donaldson, S. L., Comes, O., Zuberi, K., Badrawi, R., Chao, P., et al. (2010). The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38 (2), W214–W220. doi:10.1093/nar/gkq537
Webb, R., TomMatarin, M., Gardner, C., JessicaKelberman, D., Hassan, H., Ang, W., et al. (2012). X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am. J. Hum. Genet. 90 (2), 247–259. doi:10.1016/j.ajhg.2011.12.019
Weston, L. J., Stackhouse, T. L., Spinelli, K. J., Boutros, S. W., Rose, E. P., Osterberg, V. R., et al. (2021). Genetic deletion of Polo-like kinase 2 reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. J. Biol. Chem. 296, 100273. doi:10.1016/j.jbc.2021.100273
Whitwell, J. L., Jack, C. R., Boeve, B. F., Senjem, M. L., Baker, M., Ivnik, R. J., et al. (2009). Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology 73 (13), 1058–1065. doi:10.1212/WNL.0b013e3181b9c8b9
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46 (D1), D1074–D1082. doi:10.1093/nar/gkx1037
Wray, S. (2017). Modeling tau pathology in human stem cell derived neurons. Brain Pathol. 27 (4), 525–529. doi:10.1111/bpa.12521
Xie, Z., Bailey, A., Kuleshov, M. V., Clarke, D. J. B., Evangelista, J. E., Jenkins, S. L., et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc. 1 (3), e90. doi:10.1002/cpz1.90
Xu, Y., Zhang, S., and Zheng, H. (2019). The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 15 (4), 583–598. doi:10.1080/15548627.2018.1532258
Xu, Y., Du, S., Marsh, J. A., Horie, K., Sato, C., Ballabio, A., et al. (2021a). TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol. Psychiatry 26 (10), 5925–5939. doi:10.1038/s41380-020-0738-0
Xu, Y., Propson, N. E., Du, S., Xiong, W., and Zheng, H. (2021b). Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. U. S. A. 118 (27), e2023418118. doi:10.1073/pnas.2023418118
Xu, Y., Propson, N. E., Du, S., Xiong, W., and Zheng, H. (2021c). Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. 118 (27), e2023418118. doi:10.1073/pnas.2023418118
Keywords: IPSC-derived neurons, frontotemporal dementia (FTD), MAPT mutations (tau), transcriptomics, calcium signaling
Citation: Minaya MA, Mahali S, Iyer AK, Eteleeb AM, Martinez R, Huang G, Budde J, Temple S, Nana AL, Seeley WW, Spina S, Grinberg LT, Harari O and Karch CM (2023) Conserved gene signatures shared among MAPT mutations reveal defects in calcium signaling. Front. Mol. Biosci. 10:1051494. doi: 10.3389/fmolb.2023.1051494
Received: 22 September 2022; Accepted: 13 January 2023;
Published: 09 February 2023.
Edited by:
Paul Seidler, University of Southern California, United StatesReviewed by:
Maria Catarina Silva, Massachusetts General Hospital and Harvard Medical School, United StatesLiviu-Gabriel Bodea, Queensland Brain Institute, The University of Queensland, Australia
Copyright © 2023 Minaya, Mahali, Iyer, Eteleeb, Martinez, Huang, Budde, Temple, Nana, Seeley, Spina, Grinberg, Harari and Karch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celeste M. Karch, karchc@wustl.edu