Increased metal content in the TDP-43A315T transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis
 Theresa N. T. Dang1
Theresa N. T. Dang1  Nastasia K. H. Lim1
Nastasia K. H. Lim1  Alexandra Grubman1
Alexandra Grubman1  Qiao-Xin Li2
Qiao-Xin Li2  Irene Volitakis2
Irene Volitakis2  Anthony R. White1,2
Anthony R. White1,2  Peter J. Crouch1,2*
Peter J. Crouch1,2*- 1Department of Pathology, The University of Melbourne, VIC, Australia
- 2Florey Institute of Neuroscience and Mental Health, The University of Melbourne, VIC, Australia
Disrupted metal homeostasis is a consistent feature of neurodegenerative disease in humans and is recapitulated in mouse models of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis (ALS) and neuronal ceriod lipofuscinosis. While the definitive pathogenesis of neurodegenerative disease in humans remains to be fully elucidated, disease-like symptoms in the mouse models are all driven by the presence or over-expression of a putative pathogenic protein, indicating an in vivo relationship between expression of these proteins, disrupted metal homeostasis and the symptoms of neuronal failure. Recently it was established that mutant TAR DNA binding protein-43 (TDP-43) is associated with the development of frontotemporal lobar degeneration and ALS. Subsequent development of transgenic mice that express human TDP-43 carrying the disease-causing A315T mutation has provided new opportunity to study the underlying mechanisms of TDP-43-related neurodegenerative disease. We assessed the cognitive and locomotive phenotype of TDP-43A315T mice and their wild-type littermates and also assessed bulk metal content of brain and spinal cord tissues. Metal levels in the brain were not affected by the expression of mutant TDP-43, but zinc, copper, and manganese levels were all increased in the spinal cords of TDP-43A315T mice when compared to wild-type littermates. Performance of the TDP-43A315T mice in the Y-maze test for cognitive function was not significantly different to wild-type mice. By contrast, performance of the TDP-43A315T in the rotarod test for locomotive function was consistently worse than wild-type mice. These preliminary in vivo data are the first to show that expression of a disease-causing form of TDP-43 is sufficient to disrupt metal ion homeostasis in the central nervous system. Disrupted metal ion homeostasis in the spinal cord but not the brain may explain why the TDP-43A315T mice show symptoms of locomotive decline and not cognitive decline.
Introduction
Metals such as iron, copper and zinc are needed for normal cell function and alterations to the bio-availability of these metals can therefore have devastating consequences on the survival and functionality of cells in all parts of the body. This is particularly true for functionality of the central nervous system (CNS) because metal ions are also required for normal synaptic function (Tamano and Takeda, 2011). Underscoring the significance of maintaining metal homeostasis in the CNS, disrupted metal homeostasis is evident in many neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). In AD, iron, copper, and zinc accumulate within amyloid plaques (Connor et al., 1992; Lovell et al., 1998), in PD iron is increased within the substantia nigra (Sofic et al., 1988), and in ALS copper, zinc, manganese, and several other metals are all increased in the cerebrospinal fluid (Roos et al., 2013).
In addition to human tissue affected by neurodegenerative disease, animal models of these diseases also recapitulate elements of metal dyshomeostasis; AD model mice that over-express the amyloid-β precursor protein have decreased levels of copper and zinc in the brain relative to wild-type controls (Maynard et al., 2002) copper and iron are altered in the brains of PD model mice (Matusch et al., 2010; Ayton et al., 2013) and zinc and copper levels are altered in mutant SOD1 mouse models of ALS (Kiaei et al., 2004; Tokuda et al., 2013). The presence of disrupted metal homeostasis in animal models of neurodegenerative disease as well as the human condition has encouraged development of therapeutic strategies that aim to correct this imbalance (Badrick and Jones, 2011; Crouch and Barnham, 2012). However, confirming whether the loss of metal homeostasis in neurodegenerative disease represents a cause of the disease or a consequence of neuronal dysfunction still requires further investigation.
Frontotemporal lobar degeneration (FTLD) is a collective term for neurodegenerative diseases that involve degeneration of the frontal and temporal lobes of the brain (Pickering-Brown, 2010). It can affect individuals from the age of 40, and in the aged population, is one of the most common causes of dementia (Knopman et al., 2004). There are three main sub-types of FTLD based on their histology; dementia lacking distinct histology, FTLD with tau-positive inclusions, and FTLD with tau-negative ubiquitin-positive inclusions (FTLD-U). In 2006 it was established that cytoplasmic inclusions present in the brains of people with FTLD-U and in the spinal cords of people with ALS contained aggregates of the TAR DNA binding protein-43 (TDP-43) (Neumann et al., 2006). This provided the first molecular evidence to explain the comorbidity that is common to ALS and FTLD (Lomen-Hoerth et al., 2002) and also led several groups to propose that ALS and FTLD-U represent two ends of a spectrum of TDP-43-mediated neurodegenerative diseases that can collectively be referred to as TDP-43 proteinopathies (Liscic et al., 2008; Chen-Plotkin et al., 2010).
It is currently unclear whether disrupted metal homeostasis is present in TDP-43 proteinopathies. In vitro studies have implicated a role for metals in the aggregation of endogenous TDP-43 (Caragounis et al., 2010), and metal-based therapeutic strategies have the potential to prevent aberrant TDP-43 metabolism (Parker et al., 2012); however, there is a current paucity of in vivo data. This study utilizes the recently developed TDP-43A315T mouse model of FTLD/ALS (Wegorzewska et al., 2009) to assess whether the presence of a pathogenic form of TDP-43 affects metal ion homeostasis in vivo, and if present, to assess whether altered metal homeostasis may be associated with the symptoms of neuronal decline in an animal model of TDP-43 proteinopathy.
Materials and Methods
Materials
Rabbit antibody to TDP-43 was purchased from Proteintech (USA); rabbit antibodies to FLAG, histone H3, and GAPDH were obtained from Cell Signaling (Australia). All other chemicals were purchased from Sigma Aldrich (Australia) unless otherwise stated.
Animals
TDP-43A315T mice were purchased from The Jackson Laboratory Repository (Stock no. 010700; Bar Harbor, ME, USA). This mouse model was generated using the mouse prion promoter and a cDNA encoding human TARDBP with an A315T mutation and containing an N-terminal FLAG-tag (Wegorzewska et al., 2009). A colony of TDP-43A315T mice was maintained by breeding TDP-43A315T mice with non-transgenic C57/BL6 mice. Animals were group-housed under standard housing conditions with a 12 h light–dark cycle, and food and water ad libitum. All animals expressing the TDP-43A315T were confirmed via PCR according to the distributor’s protocol. Non-transgenic littermates not expressing the TDP-43A315T were used as wild-type controls. All animal protocols and procedures were approved by Melbourne Research Animal Ethics at The University of Melbourne, Australia. Only male mice were included in this study and the number of animals per group is stated in the figure legends.
Behavioral Testing
Locomotor activity was assessed using the rotarod assay (Turner et al., 2009); rod diameter 35 mm and elevation 200 mm. Mice were acclimatized to the rotarod by daily training for one week. Training involved three 5-min training runs per mouse per day. For each training run the mice were returned to the rotarod if they fell before the end of the 5-min period. Once the mice reached 5 weeks of age they were tested twice weekly. During testing, the rotarod was set to accelerate from 4 to 40 rpm over 5 min. Latency to fall was recorded in seconds. On each testing day each mouse was tested twice and only higher latency to fall score used for analysis.
Cognitive function was assessed using the Y-maze test as previously described (Hung et al., 2012) and performed at the age of 10 weeks. The floor of the maze was covered with sawdust which was replaced between each training and testing run in order to prevent residual odors that may affect performance. The walls of the maze were non-transparent and decorated internally so that each arm was visually unique. For training each mouse was allowed to explore only two arms of the maze (the novel arm was blocked with a non-transparent wall) for 10 min. At the end of training mice were removed from the maze for a period of 1 h before returning and being allowed to explore all 3 arms of the maze for 5 min. For training and testing the mice were placed into the maze at the same position in the starting arm of the maze. During testing the number of entries made to each arm of the maze was recorded and the percentage of entries into the novel third arm was calculated.
Tissue Collection and Preparation
Male mice (mean age of 12 weeks) were anesthetized by intraperitoneal injection of PBS (137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4, pH 7.4) supplemented with ketamine (20 mg/mL) and xylazine (4 mg/mL). Animals were then transcardially perfused with PBS containing 0.25% phosphatase inhibitor cocktail, 1% protease inhibitor (Roche) and 20 U/mL heparin. Perfused brain, spinal cord, liver, and quadriceps muscle were collected, frozen on dry ice and stored at -80°C until further processing.
For western blot analysis, enzyme-linked immunosorbent assay (ELISA) and protein oxidation detection, tissues were mechanically homogenized in 150–200 μL homogenizing buffer [PBS containing 1:100 protease inhibitor cocktail (Roche), 1:50 phosphatase inhibitor cocktail and 1:20 DNase (Roche)] before ultrasonication at 2 amp for 10 s. Homogenates were centrifuged at 18,000 g for 3 min at 4°C and the supernatant (soluble fraction) collected and the pellet (insoluble fraction) retained. Protein content was determined by the bicinchoninic acid assay (Thermo Scientific, USA). Fractions were stored at -80°C until further analysis.
Western Blot Analysis
Soluble tissue fractions were prepared in 4× loading buffer [250 mM Tris, 20% (v/v) glycerol, 8% (w/v) SDS, 2% (v/v) β-mercaptoethanol, 0.01% (w/v) bromophenol blue] and heated for 5 min at 95°C. Samples containing 10–30 μg protein were then loaded onto 4–12% NuPAGE Novex Bis–Tris Midi gels (Life Technologies) electrophoresed at 200 V for 40 min. Proteins were transferred onto PVDF membranes using the iBlot gel transfer device (Life Technologies) for 7 min according to the manufacturer’s instructions. Membranes were blocked with 4% (w/v) skim milk in PBS containing 0.05% (v/v) Tween-20 (PBST) followed by incubation with primary antibody (1:1000) overnight at 4°C. After washes in PBST, membranes were incubated with secondary antibody for 1 h at room temperature. Membranes were incubated with Western Lighting Ultra ECL (Perkin-Elmer) and imaged using the Fujifilm LAS-3000 Image reader. Blots were then stripped with 1% HCl for 15 min and reprobed for GAPDH or histone H3 as loading control. The optical density (OD) of bands was quantified using the ImageJ software and standardized to loading control (GAPDH/histone H3). The relative fold change for proteins in TDP-43A315T mouse tissue is expressed relative to wild-type mouse tissue.
Metal Analysis
Inductively coupled plasma mass spectrometry (ICP-MS) was used to measure bulk metal concentration in tissue samples as described elsewhere (Maynard et al., 2002). Briefly, perfused brain and spinal cord tissue samples were lyophilized then digested in HNO3 (65% Suprapur, Merck) overnight. Liver and quadriceps muscles were used as control non-CNS tissue. Tissues were heated at 90oC before the addition of H2O2 (30% Aristar, BDH). Samples were left to stand for ~30 min, before further heating at 70oC. The average reduced volume was determined and samples were further diluted with 1% HNO3. Measurements were made using an Agilent 7700 series ICP-MS instrument using a Helium Reaction Gas Cell and 200 ppb of Yttrium (Y89) as an internal control (ICP-MS-IS-MIX1-1, Accustandard). Results are expressed as micrograms of metal per gram of wet weight tissue (μg/g).
Protein Oxidation Detection and MCP-1 Elisa
Oxidative stress and inflammation are associated with metal dyshomeostasis and neurodegeneration (Barnham et al., 2004; Molina-Holgado et al., 2007). To assess the oxidative and inflammatory status of the brain and spinal cord of the TDP-43A315T we measured levels of oxidized proteins and the inflammation marker monocyte chemoattractant protein 1 (MCP-1). Brain and spinal cord soluble and insoluble fractions were analyzed for oxidative modified proteins via the OxyBlot Protein Oxidation Detection Kit (Merck Millipore, Australia) according to the manufacturer’s instructions. The OxyBlot detects the carbonyl groups found on oxidized proteins. The OD of bands was quantified using the ImageJ software and values were standardized to the loading controls GAPDH and histone H3 for soluble and insoluble fractions, respectively. The relative fold change of oxidized proteins in TDP-43A315T mice is expressed relative to wild-type. Levels of MCP-1 were measured in brain, spinal cord, liver and quadriceps muscle using the Mouse CCL2/JE/MCP-1 DuoSet (R&D Systems, USA) according to the manufacturer’s instructions.
Statistical Analysis
All values are presented as mean ± SEM. All statistical analyses were performed using Graphpad Prism. Planned comparisons using two-tailed independent t-tests were used to analyze all data. Significance was set at p < 0.05.
Results
TDP-43A315T Mice Exhibit Locomotor Impairment but no Cognitive Deficit
Consistent with the original description of the TDP-43A315T mice (Wegorzewska et al., 2009), the FLAG-tagged TDP-43A315T was readily detected in the brain and spinal cord relative to the liver and quadriceps muscles, indicating that the mutant TDP-43 is mainly expressed within the CNS in this model (Figure 1A). When assessed using an antibody that detected both the FLAG-tagged mutant TDP-43 and the endogenous mouse TDP-43, expression of TDP-43 was ~7-fold higher in the transgenic animals compared to wild-type controls (Figure 1B).
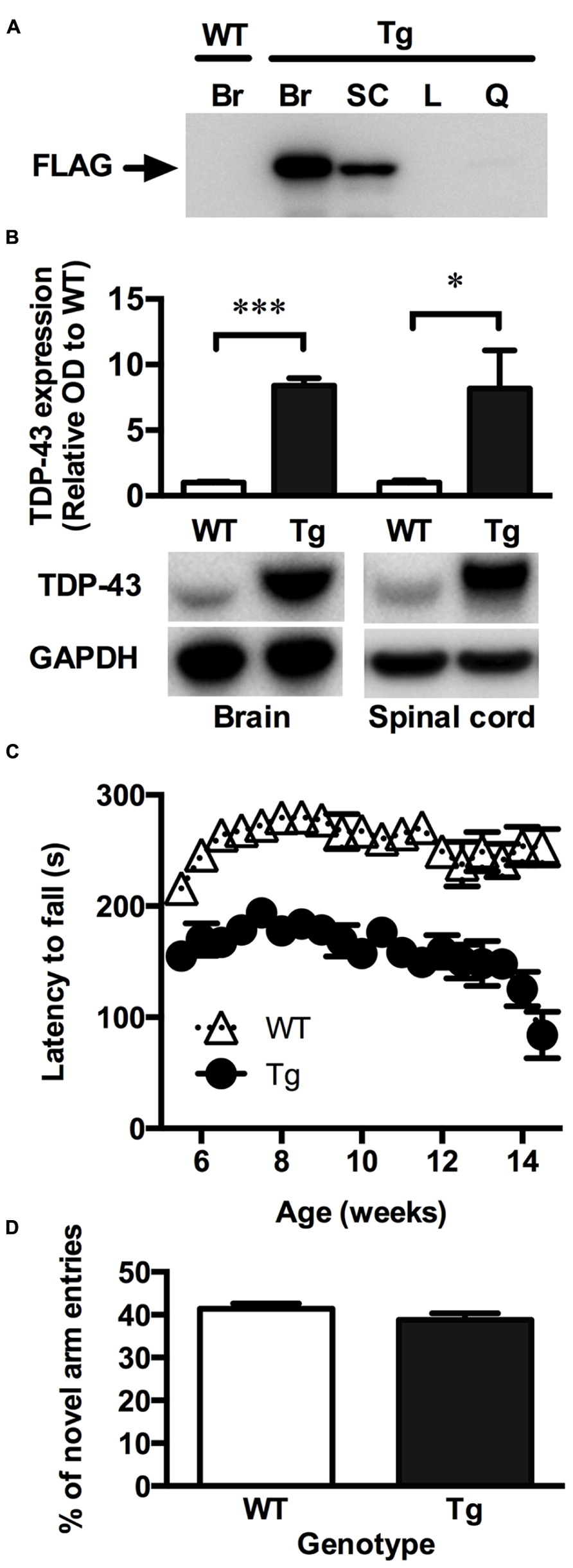
FIGURE 1. TDP-43A315T mice express mutant TDP-43 in the CNS and exhibit a locomotor impairment. (A) Representative western blot showing the absence of mutant FLAG-tagged TDP-43 (FLAG) expression in the brain of wild-type (WT) mice and the expression of FLAG-tagged TDP-43 in the brain (Br), and spinal cord (SC) but not liver (L) and quadriceps muscle (Q) of transgenic TDP-43A315T (Tg) mice. (B) Western blot analysis show a significant overexpression of total TDP-43 in the brain and spinal cord of Tg mice (n = 9) compared to WT (n = 9). ***p < 0.001; *p < 0.05. Blots shown are representative images. OD of bands were standardized to GAPDH and expressed relative to WT. (C) Tg mice exhibit a locomotor impairment compared to WT, scoring lower on the rotarod test than WT with a further decline in locomotor activity beginning at ~12 weeks. (D) No significant differences were detected in the number of novel arm entries of the Y-maze test between WT and Tg mice (p > 0.05).
To investigate whether the TDP-43A315T mouse model recapitulate symptoms of ALS, the rotarod was used to assess locomotor activity. Lower scores on the rotarod are indicative of impaired locomotor function. TDP-43A315T mice consistently scored lower than wild-type littermates at all time-points examined (Figure 1C). The poor performance of TDP-43A315T mice on the rotarod decreased further towards the end of the study period, dropping sharply at ~12 weeks of age until end-stage at 14.2 ± 1.3 weeks. Unlike mutant SOD1 mouse models of ALS (Gurney et al., 1994; Wong et al., 1995) full hind-limb paralysis was not evident at end-stage in the TDP-43A315T mice. Accordingly, the TDP-43A315T mice maintained locomotor functionality through to end-stage (Figure 1C).
The Y-maze was used to examine whether the TDP-43A315T mice exhibited a cognitive deficit reminiscent of FTLD. Cognitive function was examined in males at 10 weeks of age to ensure that testing was assessed before the dramatic late stage decline in locomotor function. The number of entries into the novel arm of the Y-maze, indicative of short-term memory function, did not differ significantly between the TDP-43A315T mice and their age-matched wild-type littermates (p > 0.05; Figure 1D).
Increased Metal Levels in the Spinal Cord of TDP-43A315T Mice
Bulk metal analysis was performed using ICP-MS and levels of Na, Mg, Al, P, K, Ca, Ti, Mn, Fe, Cu, and Zn were quantified in the brain and spinal cord of TDP-43A315T and wild-type mice (Figure 2). Bulk levels of these metals were not significantly different in the brain between genotypes (p > 0.05; Figures 2A–K). However, Mn, Cu, and Zn were significantly increased in the spinal cord of TDP-43A315T mice when compared to wild-type controls by 19.8, 16.9, and 18.8%, respectively (p < 0.05; Figures 2H,J,K). Changes to Mn, Cu, and Zn observed in the spinal cord tissue appeared to be restricted to this tissue; as per the brain, these metals were not altered in the liver or quadriceps muscle (Figures 2H,J,K).
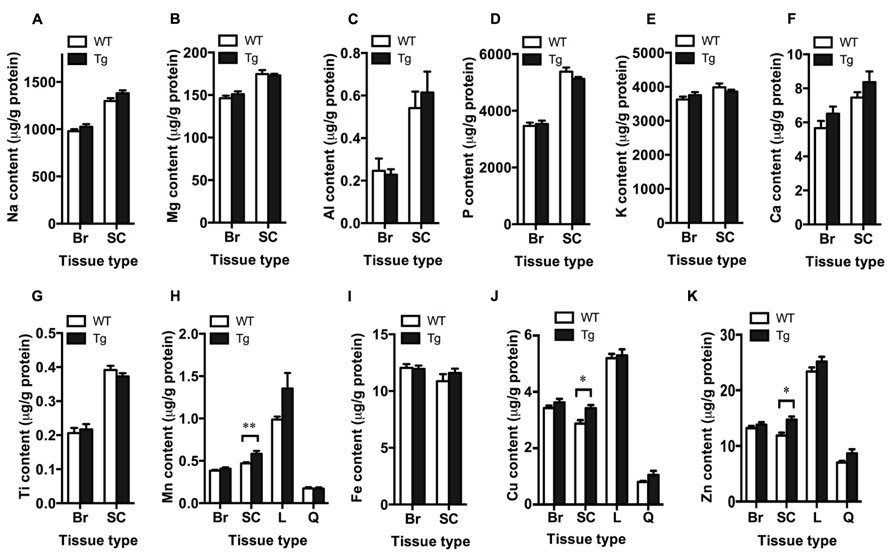
FIGURE 2. Increased metal content in the spinal cord of TDP-43A315T mice. ICP-MS analysis of bulk metal content in the brain (Br), spinal cord (SC), liver (L) and quadriceps muscle (Q) of TDP-43A315T mice (Tg), and wild-type litter mates (WT). (A–K) Planned comparisons revealed no significant differences in bulk levels of Na, Mg, Al, P, K, Ca, Ti, Mn, Fe, Cu, and Zn in the brain between genotyptes (n = 16 per genotype); however, (H) Mn, (J) Cu, and (K) Zn were significantly increased in the spinal cord of Tg mice compared to WT controls (n = 7 per genotype). **p < 0.01; *p < 0.5.
Increased Oxidative Stress in the Spinal Cord of TDP-43A315T Mice
The OxyBlot kit was used to detect the levels of oxidized proteins as a measure of oxidative stress in brain and spinal cord fractions. The level of oxidized proteins in the soluble brain and spinal cord fractions (containing mostly cytosolic proteins) of TDP-43A315T mice was not different from wild-type mice (Figure 3A). However, when assessing the insoluble fraction, containing mainly membrane and nuclear material, there was a significant 2.2-fold increase in the level of oxidized proteins in the spinal cord of TDP-43A315T mice when compared to wild-type (p < 0.05; Figure 3B). There were no differences in the insoluble brain fraction between genotypes (Figure 3B).
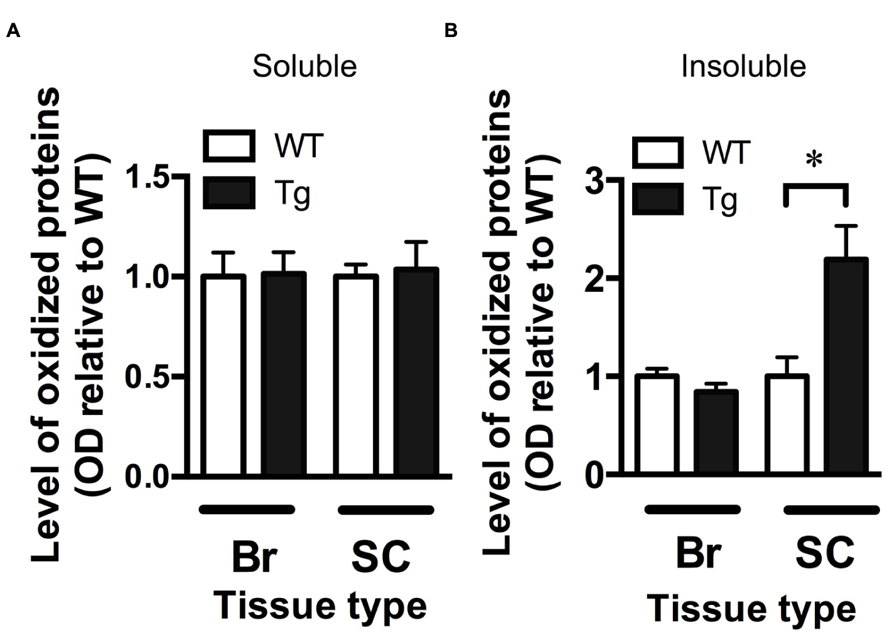
FIGURE 3. Increased oxidative stress in the spinal cord of TDP-43A315Tmice. (A) The level of oxidized proteins as determined by OxyBlot is unchanged in the soluble fraction (containing cytosolic proteins) of the brain (Br) and spinal cord (SC) of TDP-43A315T mice (Tg) compared to wild-type littermates (WT). (B) Levels of oxidized proteins were significantly increased in spinal cord insoluble fraction (containing membrane and nuclear material) of Tg compared to WT; *p < 0.05; n = 8 per genotype. No changes were detected in brain insoluble factions.
Increased Markers of Inflammation in the Brain and Spinal Cord of TDP-43A315T Mice
Monocyte chemoattractant protein-1 is involved in microglia-mediated inflammatory processes in the CNS and can be used as an indicator of increased inflammation (Deshmane et al., 2009). Levels of MCP-1, as detected by ELISA, were significantly increased in the brain and spinal cord of TDP-43A315T mice by 1.7- and 1.4-fold, respectively (p < 0.05; Figure 4).
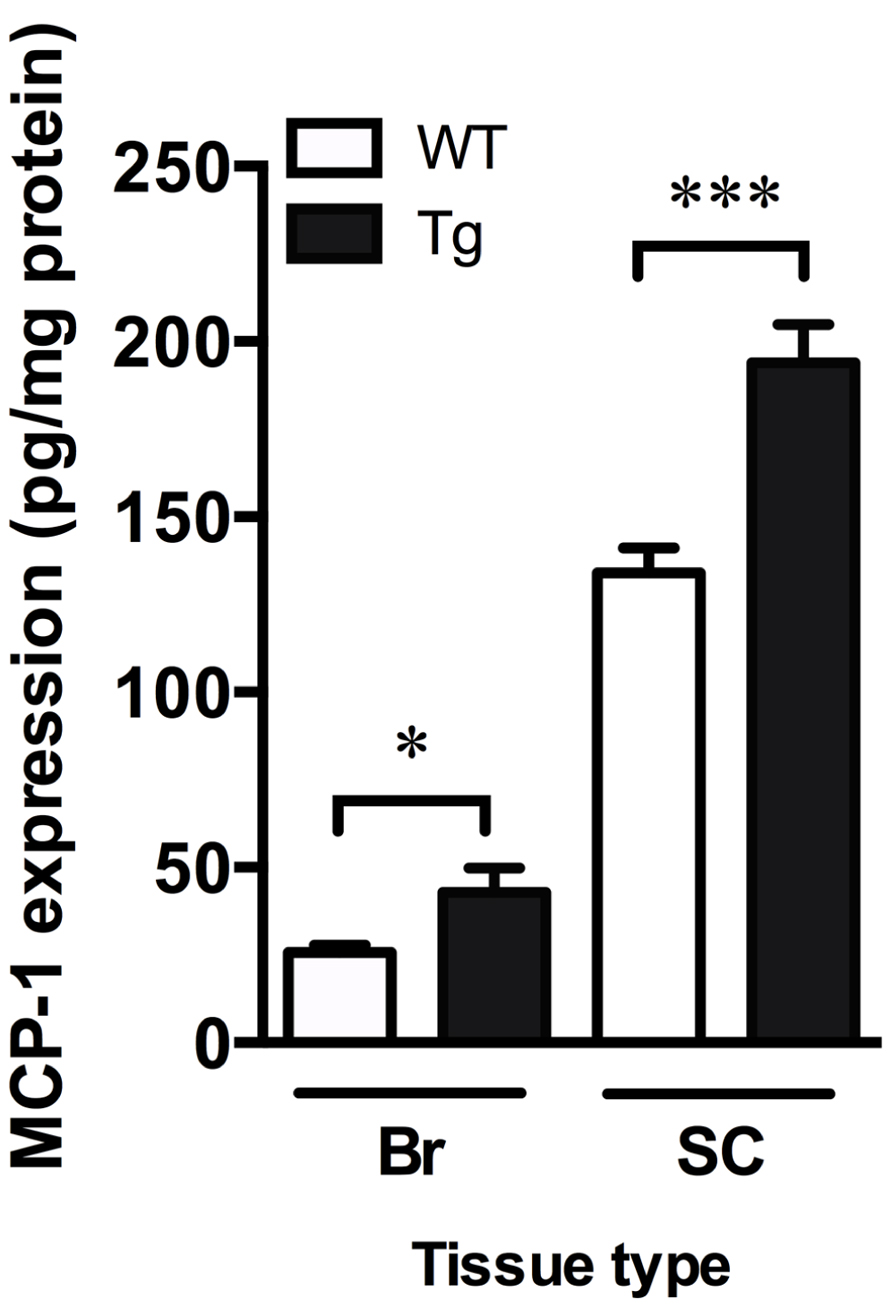
FIGURE 4. Increased markers of inflammation in TDP-43A315T mice. MCP-1 ELISA show a significant increase in MCP-1 content in the brain (Br) and spinal cord (SC) of TDP-43A315T mice (Tg) compared to wild-type (WT). ***p < 0.001; *p < 0.05. n = 9 per genotype for brain; n = 7 per genotype for spinal cord.
Discussion
Metal dyshomeostasis is implicated in a number of neurodegenerative diseases (Sofic et al., 1988; Connor et al., 1992; Lovell et al., 1998), but whether metals play a role in the degeneration of neurons in TDP-43 associated forms of diseases such as FTLD-U and ALS is unknown. In this study, the TDP-43A315T mouse model of FTLD/ALS TDP-43 proteinopathy was used to examine potential metal changes in the CNS due to expression of a pathogenic form of mutant TDP-43. Consistent with previous studies (Wegorzewska et al., 2009), FLAG-tagged mutant TDP-43 was mainly expressed within the CNS of TDP-43A315T mice resulting in the overexpression of TDP-43 in the brain and spinal cord. These animals exhibited a locomotor impairment reminiscent of ALS. The TDP-43A315T mice performed poorly on the rotarod test and demonstrated a progressive loss of locomotor activity towards end-stage. This observation is consistent with those reported by Esmaeili and colleagues (2013). Full hind-limb paralysis was not evident prior to the premature death of the TDP-43A315T mice.
Bulk metal analysis of the spinal cord revealed a significant increase in the levels of manganese, Cu and Zn due to expression of mutant TDP-43 in the TDP-43A315T mice (Figure 2). These changes appear specific to the spinal cord tissue as manganese, copper and zinc were not altered in the brain, liver, or quadriceps muscle (Figure 2). Rotarod analysis of the TDP-43A315T mice revealed a clear deficit relative to wild-type mice, but the Y-maze assessment of the TDP-43A315T mice indicated expression of the mutant TDP-43 did not affect the cognitive function of these animals (Figure 1D). This apparent demarcation between symptoms of neuronal decline in the spinal cord compared to neuronal decline in the brain is despite the higher levels of the mutant TDP-43 expression in the brain compared to spinal cord (Figure 1B). Thus, the effects of mutant TDP-43 expression on metals in the spinal cord, but not the brain, may have contributed to the predominantly locomotor phenotype of the TDP-43A315T mice.
The mechanisms by which mutant TDP-43 could contribute to elevated levels of these metals in the spinal cord remain to be fully elucidated, and it is not yet clear whether the altered metal content of the spinal cord represents a cause or consequence of neuronal dysfunction. The data generated for levels of oxidized proteins (Figure 3), however, are in part consistent with the altered metal content representing a causative event. Despite no change to levels of oxidized proteins in the soluble fraction, the abundance of oxidized proteins in the insoluble fraction was elevated in the spinal cords of TDP-43A315T mice. Oxidative damage is evident in the spinal cords of ALS cases (Niebroj-Dobosz et al., 2004) as well as mutant SOD1 mouse models of the disease (Soon et al., 2011), and altered metal homeostasis has been associated with this oxidative damage. Most notably, the pro-oxidant toxic gain-of-function ascribed to mutant SOD1 in SOD1-associated cases of ALS is proposed to be the result of altered metallation of SOD1 (Beckman et al., 2001). The normal anti-oxidant activity of SOD1 requires equimolar binding of Zn and Cu, but disruptions to this metal stoichiometry, including disruptions caused by ALS-associated SOD1 mutations (Crow et al., 1997; Roberts et al., 2007) confer toxic pro-oxidant activity to the SOD1 (Estevez et al., 1999). Significantly, altered metallation of wild-type SOD1 also confers a pro-oxidant toxic gain-of-function (Estevez et al., 1999). It is possible therefore, that the mutant-TDP-43 induced disruption of metal homeostasis detected in the spinal cords of the TDP-43A315T mice contributed to a neurotoxic oxidative mechanism already proposed as a significant pathogenic event in ALS.
In contrast to oxidative damage, the data generated for levels of the inflammatory marker MCP-1 are less clear with respect to inflammation possibly contributing to the mutant TDP-43-induced phenotype of the TDP-43A315T mice. Levels of MCP-1 were elevated in the spinal cords of TDP-43A315T mice, and they were also elevated in the brain (Figure 4). Given the mice did not display an overt cognitive phenotype indicative of neuronal dysfunction in the brain, these data may suggest the inflammation present in the brains of the TDP-43A315T mice represents a relatively non-specific response to the over-expression of an exogenous protein. Alternatively, these data may represent a specific consequence of mutant TDP-43 expression, a possibility consistent with evidence for increased inflammation in ALS and FTLD (Galimberti et al., 2008; Letiembre et al., 2009; Papadimitriou et al., 2010). As discussed below, the absence of an overt cognitive phenotype in the TDP-43A315T mice may be due to the premature death of these animals preventing the opportunity for the manifestation of neuronal dysfunction in the brain.
The data presented in this study indicate increased metal levels in the spinal cords of the TDP-43A315T mice may have contributed to their locomotor impairment, while the lack of cognitive impairment may be due to the absence of metal dyshomeostasis in the brain. However, this study only performed bulk metal analysis whereby whole tissue homogenates were analyzed for total metal levels. Region specific changes to metals or the redistribution of metals in the brain may therefore have been undetected by this methodology. Alternate analytical techniques such as laser-ablation ICP-MS which allows for spatial resolution of metal concentrations within tissues (Hare et al., 2012) or liquid chromatography ICP-MS which allows resolution of metalloproteins prior to metal quantitation (Lothian et al., 2013) may therefore be required before excluding the possibility that expression of mutant TDP-43 caused relatively subtle changes to metals within the brains of the TDP-43A315T mice. In addition, it must be noted that the TDP-43A315T mouse model has recently been reported to die prematurely from gastrointestinal complications before the development of full ALS- and FTLD-like symptoms (Guo et al., 2012; Esmaeili and colleagues, 2013). Although we did not study these gastrointestinal complications, we did find that the TDP-43A315T mice died suddenly before the presence of full hind-limb paralysis. This raises the possibility that if these mice had not died from their gastrointestinal complications they may have gone on to exhibit symptoms of cognitive decline. Thus, the non-CNS related premature death of the TDP-43A315T mice may have been the only factor that limited the detection of potential alterations to brain metal homeostasis and/or cognitive impairment, especially if FTLD-like symptoms develop much later than ALS-like symptoms.
Overall, this preliminary study is the first to report altered metal content in the spinal cord of the TDP-43A315T mouse model of FTLD and ALS. The increase in metal content is associated with increased oxidative stress and inflammation in this tissue. The mechanism by which mutant TDP-43 can alter metal levels and the effect of these changes on the oxidative and inflammatory status of the CNS remains to be elucidated, and an analysis of these changes to spinal cord metal levels is needed to address their temporal relationship with the progressive phenotype of the TDP-43A315T mice. Nonetheless, the data from this study provide evidence to support the role of metal dyshomeostasis in neurodegenerative diseases, including TDP-43 proteinopathies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, the Brain Foundation, the Motor Neurone Disease Research Institution of Australia, and The University of Melbourne.
References
Ayton, S., Lei, P., Duce, J. A., Wong, B. X., Sedjahtera, A., Adlard, P. A., et al. (2013). Ceruloplasmin dysfunction and therapeutic potential for parkinson disease. Ann. Neurol. 73, 554–559. doi: 10.1002/ana.23817
Badrick, A. C., and Jones, C. E. (2011). Reorganizing metals: the use of chelating compounds as potential therapies for metal-related neurodegenerative disease. Curr. Top. Med. Chem. 11, 543–552. doi: 10.2174/156802611794785181
Barnham, K. J., Masters, C. L., and Bush, A. I. (2004). Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214. doi: 10.1038/nrd1330
Beckman, J. S., Estevez, A. G., Crow, J. P., and Barbeito, L. (2001). Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 24, S15–20. doi: 10.1016/S0166-2236(01)00004-2
Caragounis, A., Price, K. A., Soon, C. P., Filiz, G., Masters, C. L., Li, Q. X., et al. (2010). Zinc induces depletion and aggregation of endogenous TDP-43. Free Radic. Biol. Med. 48, 1152–1161. doi: 10.1016/j.freeradbiomed.2010.01.035
Chen-Plotkin, A. S., Lee, V. M., and Trojanowski, J. Q. (2010). TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220. doi: 10.1038/nrneurol.2010.18
Connor, J. R., Menzies, S. L., St Martin, S. M., and Mufson, E. J. (1992). A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res. 31, 75–83. doi: 10.1002/jnr.490310111
Crouch, P. J., and Barnham, K. J. (2012). Therapeutic redistribution of metal ions to treat Alzheimer’s disease. Acc. Chem. Res. 45, 1604–1611. doi: 10.1021/ar300074t
Crow, J. P., Sampson, J. B., Zhuang, Y., Thompson, J. A., and Beckman, J. S. (1997). Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 69, 1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x
Deshmane, S. L., Kremlev, S., Amini, S., and Sawaya, B. E. (2009). Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 29, 313–326. doi: 10.1089/jir.2008.0027
Esmaeili, M. A., Panahi, M., Yadav, S., Hennings, L., and Kiaei, M. (2013). Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int. J. Exp. Pathol. 94, 56–64. doi: 10.1111/iep.12006
Estevez, A. G., Crow, J. P., Sampson, J. B., Reiter, C., Zhuang, Y., Richardson, G. J., et al. (1999). Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 286, 2498–2500. doi: 10.1126/science.286.5449.2498
Galimberti, D., Venturelli, E., Fenoglio, C., Guidi, I., Villa, C., Bergamaschini, L., et al. (2008). Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degeneration. J. Neurol. 255, 539–544. doi: 10.1007/s00415-008-0737-6
Guo, Y., Wang, Q., Zhang, K., An, T., Shi, P., Li, Z., et al. (2012). HO-1 induction in motor cortex and intestinal dysfunction in TDP-43 A315T transgenic mice. Brain Res. 1460, 88–95. doi: 10.1016/j.brainres.2012.04.003
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Hare, D., Austin, C., and Doble, P. (2012). Quantification strategies for elemental imaging of biological samples using laser ablation-inductively coupled plasma-mass spectrometry. Analyst 137, 1527–1537. doi: 10.1039/c2an15792f
Hung, L. W., Villemagne, V. L., Cheng, L., Sherratt, N. A., Ayton, S., White, A. R., et al. (2012). The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 209, 837–854. doi: 10.1084/jem.20112285
Kiaei, M., Bush, A. I., Morrison, B. M., Morrison, J. H., Cherny, R. A., Volitakis, I., et al. (2004). Genetically decreased spinal cord copper concentration prolongs life in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 24, 7945–7950. doi: 10.1523/jneurosci.2000-04.2004
Knopman, D. S., Petersen, R. C., Edland, S. D., Cha, R. H., and Rocca, W. A. (2004). The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology 62, 506–508. doi: 10.1212/01.WNL.0000106827.39764.7E
Letiembre, M., Liu, Y., Walter, S., Hao, W., Pfander, T., Wrede, A., et al. (2009). Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol. Aging 30, 759–768. doi: 10.1016/j.neurobiolaging.2007.08.018
Liscic, R. M., Grinberg, L. T., Zidar, J., Gitcho, M. A., and Cairns, N. J. (2008). ALS and FTLD: two faces of TDP-43 proteinopathy. Eur. J. Neurol. 15, 772–780. doi: 10.1111/j.1468-1331.2008.02195.x
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi: 10.1212/WNL.59.7.1077
Lothian, A., Hare, D. J., Grimm, R., Ryan, T. M., Masters, C. L., and Roberts, B. R. (2013). Metalloproteomics: principles, challenges and applications to neurodegeneration. Front. Aging Neurosci. 5:35. doi: 10.3389/fnagi.2013.00035
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., and Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. doi: 10.1016/S0022-510X(98)00092-6
Matusch, A., Depboylu, C., Palm, C., Wu, B., Hoglinger, G. U., Schafer, M. K., et al. (2010). Cerebral bioimaging of Cu, Fe, Zn, and Mn in the MPTP mouse model of Parkinson’s disease using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). J. Am. Soc. Mass Spectrom. 21, 161–171. doi: 10.1016/j.jasms.2009.09.022
Maynard, C. J., Cappai, R., Volitakis, I., Cherny, R. A., White, A. R., Beyreuther, K., et al. (2002). Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676. doi: 10.1074/jbc.M204379200
Molina-Holgado, F., Hider, R. C., Gaeta, A., Williams, R., and Francis, P. (2007). Metals ions and neurodegeneration. Biometals 20, 639–654. doi: 10.1007/s10534-006-9033-z
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Niebroj-Dobosz, I., Dziewulska, D., and Kwiecinski, H. (2004). Oxidative damage to proteins in the spinal cord in amyotrophic lateral sclerosis (ALS). Folia Neuropathol. 42, 151–156.
Papadimitriou, D., Le Verche, V., Jacquier, A., Ikiz, B., Przedborski, S., and Re, D. B. (2010). Inflammation in ALS and SMA: sorting out the good from the evil. Neurobiol. Dis. 37, 493–502. doi: 10.1016/j.nbd.2009.10.005
Parker, S. J., Meyerowitz, J., James, J. L., Liddell, J. R., Nonaka, T., Hasegawa, M., et al. (2012). Inhibition of TDP-43 accumulation by bis(thiosemicarbazonato)-copper complexes. PLoS ONE 7:e42277. doi: 10.1371/journal.pone.0042277
Pickering-Brown, S. M. (2010). Review: recent progress in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 36, 4–16. doi: 10.1111/j.1365–2990.2009.01045.x
Roberts, B. R., Tainer, J. A., Getzoff, E. D., Malencik, D. A., Anderson, S. R., Bomben, V. C., et al. (2007). Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 373, 877–890. doi: 10.1016/j.jmb.2007.07.043
Roos, P. M., Vesterberg, O., Syversen, T., Flaten, T. P., and Nordberg, M. (2013). Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 151, 159–170. doi: 10.1007/s12011-012-9547-x
Sofic, E., Riederer, P., Heinsen, H., Beckmann, H., Reynolds, G. P., Hebenstreit, G., et al. (1988). Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural. Transm. 74, 199–205. doi: 10.1007/BF01244786
Soon, C. P., Donnelly, P. S., Turner, B. J., Hung, L. W., Crouch, P. J., Sherratt, N. A., et al. (2011). Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 286, 44035–44044. doi: 10.1074/jbc.M111.274407
Tamano, H., and Takeda, A. (2011). Dynamic action of neurometals at the synapse. Metallomics 3, 656–661. doi: 10.1039/c1mt00008j
Tokuda, E., Okawa, E., Watanabe, S., Ono, S. I., and Marklund, S. L. (2013). Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 54, 308–319. doi: 10.1016/j.nbd.2013.01.001
Turner, B. J., Parkinson, N. J., Davies, K. E., and Talbot, K. (2009). Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol. Dis. 34, 511–517. doi: 10.1016/j.nbd.2009.03.005
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 106, 18809–18814. doi: 10.1073/pnas.0908767106
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. doi: 10.1016/0896-6273(95)90259-7
Keywords: amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), TAR DNA binding protein-43 (TDP-43), copper (Cu), zinc (Zn), manganese (Mn), neurodegenerative disease
Citation: Dang TNT, Lim NKH, Grubman A, Li Q-X, Volitakis I, White AR and Crouch PJ (2014) Increased metal content in the TDP-43A315T transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Front. Aging Neurosci. 6:15. doi: 10.3389/fnagi.2014.00015
Received: 04 December 2013; Accepted: 24 January 2014;
Published online: 11 February 2014.
Edited by:
Paul Adlard, The Mental Health Research Institute, AustraliaReviewed by:
Xuechu Zhen, Shanghai Institute of Materia Medica–Chinese Academy of Sciences, ChinaBlaine Russell Roberts, Florey Institute of Neuroscience and Mental Health, Australia
Copyright © 2014 Dang, Lim, Grubman, Li, Volitakis, White and Crouch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Crouch, Department of Pathology, The University of Melbourne, Parkville, VIC 3010, Australia e-mail: pjcrouch@unimelb.edu.au