
- 1Behavioural Neuroscience Division, Florey Institute of Neuroscience and Mental Health, University of Melbourne, Parkville, VIC, Australia
- 2Department of Anatomy and Neuroscience, University of Melbourne, Parkville, VIC, Australia
Huntington’s disease (HD) is an autosomal dominant disorder caused by a tandem repeat expansion encoding an expanded tract of glutamines in the huntingtin protein. HD is progressive and manifests as psychiatric symptoms (including depression), cognitive deficits (culminating in dementia), and motor abnormalities (including chorea). Having reached the twentieth anniversary of the discovery of the “genetic stutter” which causes HD, we still lack sophisticated insight into why so many HD patients exhibit affective disorders such as depression at very early stages, prior to overt appearance of motor deficits. In this review, we will focus on depression as the major psychiatric manifestation of HD, discuss potential mechanisms of pathogenesis identified from animal models, and compare depression in HD patients with that of the wider gene-negative population. The discovery of depressive-like behaviors as well as cellular and molecular correlates of depression in transgenic HD mice has added strong support to the hypothesis that the HD mutation adds significantly to the genetic load for depression. A key question is whether HD-associated depression differs from that in the general population. Whilst preclinical studies, clinical data, and treatment responses suggest striking similarities, there are also some apparent differences. We discuss various molecular and cellular mechanisms which may contribute to depression in HD, and whether they may generalize to other depressive disorders. The autosomal dominant nature of HD and the existence of models with excellent construct validity provide a unique opportunity to understand the pathogenesis of depression and associated gene-environment interactions. Thus, understanding the pathogenesis of depression in HD may not only facilitate tailored therapeutic approaches for HD sufferers, but may also translate to the clinical depression which devastates the lives of so many people.
Introduction
Huntington’s disease (HD) is a progressive neurodegenerative disorder that affects mood, cognition, and movement. HD provides a unique opportunity to study pathogenesis from its earliest stages, as the genetic cause of this autosomal dominant disease, with its array of divergent symptoms, is known. HD is caused by an abnormal expansion mutation of a tract of CAG trinucleotide tandem repeats close to the 5′ end of the huntingtin gene on chromosome 4 (The Huntington’s Disease Collaborative Research Group, 1993). HD was first described by the physician George Huntington in 1872, and our understanding of the mechanisms underlying HD has increased exponentially in the 20 years since the tandem repeat expansion in huntingtin was found to be the causative gene mutation. Despite this, a cure for this disease remains elusive. In light of this, much attention has been focused on symptom management. As a late onset disease, alleviation of especially early symptoms can greatly lengthen and improve the largely normal and productive periods of patients’ lives. HD presents with a classic triad of relentlessly progressive symptoms. Diagnosis is based on the appearance of motor symptoms, which most commonly appear in patients in the fourth or fifth decade of life. Some rare cases of juvenile onset are seen, with motor symptoms manifesting during or prior to teenage years; however juvenile onset HD, which is caused by exceptionally long CAG repeat expansions, accounts for only about 5% of all HD cases (Nance and Myers, 2001). Completing the triad are cognitive and psychiatric symptoms, which can arise years, often decades, prior to the inception of motor symptoms. There are also a plethora of non-CNS, peripheral symptoms, although it is not yet clear whether specific symptoms originate from cellular dysfunctions in central and/or peripheral tissues (van der Burg et al., 2009).
Psychiatric symptoms abound in HD (Leroi et al., 2002), including psychosis (Lovestone et al., 1996), irritability, anxiety, apathy (van Duijn et al., 2007), and obsessive and compulsive symptoms (Beglinger et al., 2008; van Duijn et al., 2008). However, depression is among the most prevalent of psychiatric symptoms, with a lifetime prevalence of major depression reported to be up to and over 50% in patients; moreover, depression is often diagnosed years if not decades prior motor symptom onset (Shiwach, 1994; Naarding et al., 2001; Paulsen et al., 2001, 2005; Duff et al., 2007; van Duijn et al., 2008; Gargiulo et al., 2009). This occurrence is hugely disproportionate when compared to lifetime prevalence of major depression in the general population, which has been found to be around 15% (Hasin et al., 2005; Kessler et al., 2005).
More than merely prevalent, depression has been rated by patients as one of the most debilitating symptoms of HD, affecting perceived quality of life more so than motor or even cognitive aspects of the disease (Ho et al., 2009). Depression increases the risk of suicide (Jensen et al., 1993; Harris and Barraclough, 1998; Cavanagh et al., 2003). In HD, almost 30% of patients attempt suicide at least once and death due to suicide among HD patients is four times that of the normal populace – a similar rate of death by suicide to that of those suffering from affective disorders (Farrer, 1986; Inskip et al., 1998). The presence of depression also leads to negative collateral effects such as the hastening of cognitive decline (Nehl et al., 2001; Smith et al., 2012). Whilst there is currently no cure available for HD, depression is largely seen as treatable. However, understanding of the etiology and pathophysiology of this early-onset symptom is required to allow early detection of depression and to apply the most suitable treatments.
Huntington’s disease is the most notorious of a group of neurodegenerative diseases caused by an expansion of a CAG trinucleotide repeat encoding poly-glutamine (poly-Q). Other diseases in this group include dentatorubropallidoluysian atrophy (DRPLA), spinocerebellar ataxia type 3 (SCA-3), spinal bulbar muscular atrophy (SBMA), and spinocerebellar ataxia types 1, 2, 6, 7, and 17 (David et al., 1997; Kakizuka, 1997; Koshy and Zoghbi, 1997; Robitaille et al., 1997; Ross, 1997; Nakamura, 2001; Nakamura et al., 2001). SCA patients show depression prevalence of between 15 and 17% in a large study with 526 genetically confirmed and clinically affected patients (117 SCA1, 163 SCA2, 139 SCA3, and 107 SCA6) (Schmitz-Hübsch et al., 2011), a rate similar to the prevalence in the general population (∼15%) and certainly less than that seen in HD. Other members of this group are very rare and studies have not been done specifically examining rates of depression. The overrepresentation of depression in HD may be due to the neurological importance of the huntingtin protein, its ubiquitousness and the subsequent consequences of the mutation.
Whilst the link between the length of CAG expansion and speed of deterioration has been established, evidence from the literature does not support the idea that depression in HD is affected by the length of CAG repeats. In a study with a cohort of 79 HD patients, no correlations were observed between CAG repeat length and personality change, psychosis, depression, or non-specific alterations (Weigell-Weber et al., 1996). No relationship was discerned even after correcting for predicted age of neurological onset in neurologically asymptomatic patients (Berrios et al., 2001). A more recent study examining 72 HD patients found that whilst the number of CAG repeats associated negatively with the age of onset of psychiatric disorders, neither the probability of developing psychiatric disorders nor the severity of psychiatric symptoms was correlated with the number of CAG repeats (Vassos et al., 2008). It therefore seems that although higher repeat number is associated with faster disease progression as a whole, it is insufficient to induce depression.
Although the ultimate cause of depression in HD may be different from that of depression in the HD gene-negative population (which we will refer to forthwith as “clinical depression”), recent research has shown that there are certainly perceptible similarities in the symptoms and even physiological anomalies. These similarities are worth examining as they may hold clues to the etiology of depression in HD and may be informative in establishing how best to treat this debilitating affective disorder. Various aspects of HD pathogenesis, including disrupted transcription, trafficking, signaling, homeostasis, synaptic and neuronal function, have been recently reviewed (Milnerwood and Raymond, 2010; Raymond et al., 2011; Ross and Tabrizi, 2011; Nithianantharajah and Hannan, 2012). Here, we discuss potential molecular and cellular mechanisms specifically involved in the etiology of depression in HD. This includes evidence implicating the dysregulation of serotonergic signaling, alteration of hypothalamic-pituitary-adrenal (HPA)-axis activity, and disruption of BDNF expression and trafficking as key pathogenic processes.
Diagnosing Depression in HD
Notwithstanding evidence of overwhelming prevalence of psychiatric symptoms, treatment of psychopathologies in HD has been scantly examined. Studies of the efficacy of various drugs in treating these symptoms have been few and far between, mostly with small cohorts, often case reports of a single patient with no controls (Naarding et al., 2001). While conventional antidepressants have been applied in the treatment of depression in HD, the effectiveness of different types of antidepressants, doses, treatment durations, and other factors have not been thoroughly examined. Having a different origin of disease, i.e., the mutation in the huntingtin gene, it is possible that disparities exist in HD which may translate to more efficacious treatment strategies. For instance, a study has found that passivity is a risk factor to earlier disease onset versus patients who were more active (Trembath et al., 2010). This reflects findings in mouse models where transgenic mice raised in enriched cages which provided enhanced sensory stimulations were rescued from depressive-like behavioral phenotypes seen in mice raised in standard cages (Pang et al., 2009; Du et al., 2012; Renoir et al., 2013). This suggests enhanced engagement and participation may have benefits in the treatment of depression. Little has been done with regards to behavioral therapy in HD but a study has shown that remotivational therapy improving quality of life of HD patients (Sullivan et al., 2001). A recent study on the use of antidepressants in pre-onset HD gene carriers found that about 22% of 787 prodromal patients are already on antidepressants, much more than in the control group, with around 13% on antidepressants (Rowe et al., 2012). However, a consensus on the prevalence of depression in HD has yet to be reached. The inconsistency in rates of depression reported between different studies is due to a variety of factors such as small sample sizes, differences in methodologies utilized, lack of control groups, and differences in the disease stages of participants (van Duijn et al., 2007). In particular, the usage of different assessment tools [Unified Huntington’s Disease Rating Scale (UHDR), Diagnostics and Statistics Manual of Mental Disorders (DSM), Neuropsychiatric Inventory (NPI), Beck Depression Inventory-II (BDI-II), Hamilton Rating Scale for Depression (Ham-D), Hospital Anxiety and Depression Scale (HADS), Depression Intensity Scale Circles (DISCs)] have resulted in very disparate estimations of the prevalence of depression in HD, ranging from 33 to 69%.
This large variation highlights the endogenous differences between how depression manifests with and without the HD gene mutation, causing the use of tools not tailored for depression in HD to produce inconsistent results. For example, standard rating scales for clinical depression contain items relating to symptoms of HD that are present regardless of the presence or absence of depression and therefore may skew the diagnosis. Weight loss and sleep disturbances are items included in many diagnostic tools as symptomatic of depression even though they are common symptoms in HD patients without depression. Therefore these symptoms may well be nothing but red herrings if included for diagnosis of depression in HD. Scales that contain less “somatic” items, such as the HADS and the DISCs were better able to accurately identify depression in HD (De Souza et al., 2010). Comparison between the BDI-II and the Ham-D found that the items best able to discriminate depression in HD patients, other than depressed mood, which had high correlations [correlation coefficient (r) = 0.834 for the BDI and 0.917 for the Ham-D], are those items measuring thoughts and attitudes around mood such as “discouraged about the future” (r = 0.653) and “satisfaction in life” (r = 0.64). Weight loss (r = 0.193 for BDI and 0.128 for Ham-D), loss of appetite (r = 0.319 for BDI, 0.297 for Ham-D), and other vegetative symptoms associated with clinical depression were, on the other hand, poor indicators of depression in HD (Rickards et al., 2011). Depression is notoriously heterogeneous in its presentation and these findings suggest possible dominance of a different cluster of symptoms in HD compared to most clinical depression patients. Accurate discriminators of depression in HD are required for better screening, diagnosis, and the gaging of treatment efficacy; areas that have not so far been systematically studied. Whether or not the differences reflected in the screening tools point to underlying pathophysiological divergence between depression in HD and clinical depression require further investigations. Besides the obvious consequences of depression itself, another important reason for the accurate and early detection of depression in HD is that depression has the negative effects of hastening cognitive decline and correlates with poorer cognitive performance in prodromal patients (Nehl et al., 2001; Smith et al., 2012), suggesting that earlier intervention can prolong the functional years of a patient’s life.
It is tempting to speculate, and indeed has been a predominant view, that depression in HD is a natural consequence to the reactive stress induced by the knowledge of being at risk of inheriting the disease and/or of positive genetic or clinical diagnosis of HD (Shiwach and Norbury, 1994). Whilst psychosocial stress undoubtedly adds to the psychological burden inducing depression (Codori et al., 2004; Larsson et al., 2006), it alone is now regarded as insufficient to fully account for the psychiatric co-morbidity. Prior to clinical onset, HD gene-positive carriers presented a higher current prevalence of major depression despite being ignorant of their gene status at the time of psychiatric assessment (Horowitz et al., 2001; Julien et al., 2007). That overrepresentation suggests that depression in HD patients is a behavioral manifestation of early neuropathology. Interestingly, the greater susceptibility of females to developing depression in the general population is also apparent within the HD population (Zielonka et al., 2012). These clinical aspects have been recapitulated by studies of the R6/1 transgenic and knock-in HdhQ111 mouse models (that are by definition unaware of their gene status) which report a female-specific depression-like behavioral phenotype (Pang et al., 2009; Pouladi et al., 2009; Du et al., 2012; Orvoen et al., 2012; Renoir et al., 2012), circumventing the caveat of psychosocial stress. Whilst sexual dimorphism has been reported in the CAG(n51) transgenic HD rats, the nature of the affective dysfunction in this model is less clear (Bode et al., 2008; Faure et al., 2011). Depressive-like behaviors have also been identified in the YAC transgenic HD mice (Pouladi et al., 2009), suggesting that it is a fundamental aspect of HD phenotypes. The study of other rodent models of HD might be relevant for the understanding of other specific psychiatric aspects of HD such as anxiety (Orvoen et al., 2012; Abada et al., 2013).
Although the pathophysiology leading to depression in HD arises from a different source than those of clinical depression, the successes of antidepressant treatments in ameliorating depression in HD, albeit mostly from case studies, suggest similarities in the underlying pathophysiology. Therefore it is important to take advantage of the extensive knowledge garnered in the study of clinical depression and to compare the two diseases in order to apply the most effective and tailored treatment for depression in HD.
The Serotonergic System
Symptomatic treatment of depression in HD and the efficacy of the range of antidepressant drugs often prescribed to HD patients have not been thoroughly examined. Case studies report benefits of selective serotonin reuptake inhibitors (SSRIs), selective noradrenergic reuptake inhibitors (SNRIs) (venlafaxine), atypical antipsychotics (olanzepine), monoamine oxidase inhibitors (MAOI), tetracyclic, and tricyclic antidepressants on small numbers of patients (Patel et al., 1996; Squitieri et al., 2001; Bonelli et al., 2003; Ciammola et al., 2009). A study of 26 HD patients with diagnoses of major depression treated with venlafaxine for 4 weeks showed significant improvement albeit a high rate of side effects such as irritability (Holl et al., 2010). In a more recent study by Rowe et al. (2012) of 787 prodromal HD participants, it was reported that 20% of prodromal patients were prescribed antidepressants with the vast majority using SSRIs (e.g., paroxetine, fluoxetine, and sertraline). However, the effectiveness SSRIs versus other types of antidepressants in diminishing the depression symptoms has yet to be specifically documented. Indeed the overall effectiveness of SSRI interventions in treating depression in HD has not been studied despite its frequent use.
Dysregulation of the serotonin (5-HT) signaling system has long been scrutinized in the field of clinical depression as an etiological cause (Castro et al., 1998; Yohrling et al., 2002). Compromising the 5-HT system affects its downstream elements; 5-HT upregulates the expression of cyclic adenosine monophosphate (cAMP) (Vaidya and Duman, 2001), which ultimately results in the activation of cAMP response element-binding protein (CREB). A loss of CREB signaling impairs adult hippocampal neurogenesis and disrupts normal hippocampal function (Jacobs et al., 2000; Urani et al., 2005), both of which have been proposed to be key pathologies of depression (Petersen et al., 2008; Lucassen et al., 2010). Based on findings from animal models, CREB signaling in HD is also disrupted, due to sequestration of its binding partner, CREB binding protein (CBP). Thus, this is a common molecular pathology that the disease shares with depression, and one which, along with the target gene BDNF as described below, is likely to mediate the deficits in hippocampal neurogenesis which is the cellular consistently observed in animal models of HD. Chronic treatment of different mouse models of HD with a variety of SSRIs have been found to rescue the neurogenesis deficits but there has yet to be a study demonstrating a possible rescue of neurogenesis through modulation of CREB signaling. That would be an interesting avenue of investigation for the future since compounds that increase phosphorylation of CREB and cAMP levels are reportedly associated with a rescue of cognitive deficits in the R6/1 transgenic mouse model (Giralt et al., 2013).
Serotonin reuptake inhibitors work in part by desensitizing 5-HT1A autoreceptors (Dawson et al., 2002; Rossi et al., 2008), which act to inhibit 5-HT production. Desensitization of these autoreceptors therefore increases 5-HT neurotransmission (Blier et al., 1998). It was also found that a single nucleotide polymorphism (SNP) on the promoter of 5-HT1A receptor which leads to the over-expression of 5-HT1A autoreceptors resulted in increased susceptibility to developing major depression in those with the mutation (Albert and Francois, 2010). As levels of 5-HT in the human brain can only be measured post-mortem, markers are used in patients to reflect 5-HT levels. One of these is measuring the primary metabolite of 5-HT, 5-hydroxyindoleacetic acid (5-HIAA) in the cerebrospinal fluid (CSF), which is deemed to reflect brain 5-HIAA levels (Wester et al., 1990). It has been found that 5-HIAA is reduced in the CSF of a sizeable proportion of patients with major depression, a finding replicated by post-mortem studies (Asberg, 1976; Asberg et al., 1976; van Praag and Plutchik, 1984; Roy et al., 1989). Reduced 5-HIAA levels have also been reported in HD patients, which is a clear indicator that there is a dysregulation of serotonin metabolism in HD (Caraceni et al., 1977; Jongen et al., 1980). Closely related, monoamine oxidase A, an enzyme that metabolizes 5-HT thereby decreasing the amount of available 5-HT, was elevated significantly in many brain regions of depressed patients (Meyer et al., 2006). This abnormal increase in metabolism of 5-HT has similarly been documented in the putamen and substantia nigra pars compacta of the basal ganglia, and in the pons of HD brains through quantitative enzyme radioautography (Richards et al., 2011). Interestingly, platelet monoamine oxidase activity was reported to be unchanged in HD patients with overt symptoms (Markianos et al., 2004) which suggests a dissociation between central and peripheral pathology in HD. Tryptophan, an essential amino acid from which serotonin is synthesized, has also been found to be reduced in a large proportion of patients with depression (Moller et al., 1983; Quintana, 1992). Experimentally induced tryptophan depletion via a special diet resulted in rapid relapse in remitted depression patients hours after ingestion of diet (Delgado et al., 1990). Plasma total and protein-bound tryptophan levels were found to be reduced in HD patients (Belendiuk et al., 1980) and while oral tryptophan administration increases blood 5-HT levels in healthy individuals, this response not observed in HD patients which is a further demonstration of a disease-associated impairment of 5-HT metabolism (Christofides et al., 2006). A recent study looking at the raphe nucleus, where 5-HT synthesis occurs, found a significant correlation between the level of depression in HD patients and stem raphe echogenicity using transcranial sonography (Krogias et al., 2011). This is similar to what is seen in clinical depression patients (Walter et al., 2007), further strengthening the evidence that an abnormality in 5-HT production may underlie both diseases. Recent work on compounds which influence tryptophan metabolism rescue neurodegeneration in the R6/2 mouse model of HD (Zwilling et al., 2011) but the effectiveness of such compounds to treat depressive behaviors have yet to be determined.
Besides changes to 5-HT levels, dysregulation of the receptors of 5-HT has also been implicated in depression. Major depression has been implicated with increased 5-HT1A autoreceptor density in the dorsal raphe nucleus (Stockmeier et al., 1998). Reduced post-synaptic 5-HT1A/2 receptor function has also been linked with depression with patients who had suffered major depression exhibiting global reduction in 5-HT1A and 5-HT2A receptor binding (Drevets et al., 1999, 2007; Messa et al., 2003; Moses-Kolko et al., 2008). This is reflected in animal models where genetic 5-HT1A receptor knock-out mice showed phenotypes of anxiety (Heisler et al., 1998; Parks et al., 1998; Ramboz et al., 1998) whereas over-expression of 5-HT1A receptor reduced anxiety-related behaviors (Kusserow et al., 2004). In post-mortem HD brains, reduced 5-HT1 receptor binding has been reported in the putamen, hippocampus (Cross et al., 1986), and in the basal ganglia and substantia nigra (Waeber and Palacios, 1989). Similar reductions in 5-HT1A receptor binding have been reported in the R6/2 transgenic mouse model, as well as a marked decrease in enzymatic activity of tryptophan hydroxylase (TPH) activity (the rate limiting enzyme for the biosynthesis of 5-HT) (Yohrling et al., 2002). The decrease in receptor binding is likely attributable to down-regulation of mRNA levels of various 5-HT receptors which is evident in R6/1 HD mice (Pang et al., 2009). All these culminate in abnormal physiological responses to the administration of 5-HT1A receptor agonist 8-OH-DPAT by R6/1 HD mice (Renoir et al., 2011a, 2012), thereby demonstrating how molecular neuropathology manifests as disease pathophysiology. Interestingly, chronic application of the SSRI sertraline was able to correct the hypersensitivity of the 5-HT1A autoreceptor in female R6/1 mice as well as depression-like phenotype (Renoir et al., 2012), suggesting an etiological link between disturbances to the 5-HT system with the HD-associated depression-like phenotype.
The HPA-Axis and Stress Responses
The HPA-axis is the major endocrine system responsible for stress adaptation. Stress activates the axis, resulting in the production of the stress hormone cortisol (corticosterone in rodents) (Papadimitriou and Priftis, 2009). Abnormally elevated HPA-axis activity is one of the most replicated biological findings in major depression (Pariante and Lightman, 2008; Stetler and Miller, 2011). Cortisol levels are usually tightly regulated because prolonged exposure to cortisol is damaging to the brain: reducing neurogenesis (Cameron and Gould, 1994; Wong and Herbert, 2006; Brummelte and Galea, 2010) and increasing apoptosis in the hippocampus (Sapolsky, 1986; Crochemore et al., 2005; Andrés et al., 2006; Liu et al., 2011) resulting in atrophy in the hippocampus of rats (Woolley et al., 1990) and monkeys (Sapolsky et al., 1990) as well as neuronal atrophy (Cerqueira et al., 2005a) and volume reduction in the prefrontal cortex (Cerqueira et al., 2005b). Dysregulation of the 5-HT neurotransmission has been linked with the HPA-axis abnormalities (Hery et al., 2000; Froger et al., 2004). The serotonin system is able to modulate anxiety and depression (Meaney et al., 1994; Ramboz et al., 1998; Harada et al., 2008; Brummett et al., 2012), possibly by regulating the HPA-axis through influencing glucocorticoid receptor (GR), whose activation is important in regulating the 5-HT system both in vivo and in vitro (Lanfumey et al., 2000; Erdeljan et al., 2001; Wang et al., 2009; Falkenberg and Rajeevan, 2010; Belay et al., 2011). For example, it has been found that serotonin neurotransmission is modulated by HPA-axis activity, with GR activation increasing 5-HT1A signaling (Hesen and Joels, 1996). Adrenalectomy reduced dentate gyrus neuronal morphology and this effect was reversed by using the 5-HT1A agonist ipsapirone (Huang et al., 1997). But sustained elevation of corticosteroid significantly reduced 5-HT1A receptor mRNA level as well as 5-HT1A binding density in the hippocampus and this is reversed by chronic administration of antidepressants (Lopez et al., 1998).
Studies have also described HPA-axis hyperactivity in HD patients. Earlier studies with small sample sizes found higher basal cortisol levels in moderate stage HD patients compared to controls at both morning (Leblhuber et al., 1995) and evening (Heuser et al., 1991). More recently, hyperactivity of the HPA-axis has been found in a small sample of clinically diagnosed, early stage HD patients compared to controls, with higher 24 h cortisol production, particularly in the morning and early afternoon periods (Aziz et al., 2009). Examining salivary cortisol in pre-symptomatic HD patients in comparison to controls, subtle but altered cortisol awakening responses were found, with higher cortisol concentration in patients at early morning periods, just after awakening (van Duijn et al., 2010). This indicates that HPA-axis dysregulation is an early pathophysiology in HD. However, another study with a comparatively large sample of patients found increased urine cortisol levels but only in moderate to late clinically diagnosed patients whereas pre-symptomatic and early stage patients did not display any differences in urine cortisol compared to 68 healthy controls (Björkqvist et al., 2006). But it is worthy to note that in this study, samples were collected in a narrow time window (14:00–17:00), when cortisol level is ebbing, leaving the possibility that the difference identified in the previous studies was masked in the early stage patients examined. Furthermore, analysis of patients was confounded by limitations to recruitment such as imbalance of gender (or indeed disregarding gender as a factor), differences in stages of disease progression as well as environmental confounders such as smoking, alcoholism, depression, and use of psychotropic medications.
Increased baseline corticosterone was initially found in R6/2 mice from an early age (Björkqvist et al., 2006). However, because of their rapid motor onset and deterioration, the R6/2 model is not an optimal one for examining pre-motor onset symptoms and pathophysiologies. Recently, it was found that at a pre-motor onset age, female R6/1 mice exhibit increased corticosterone release after physiological and pharmacological stresses whilst baseline levels did not differ between WT and R6/1 females (Du et al., 2012). This is more akin to what is seen in early, pre-motor symptomatic HD patients with little change in baseline cortisol levels, suggesting that in prodromal patients, differences may only become apparent when the HPA-axis is induced by stressors. Interestingly, the sex-dimorphic display of this physiological abnormality correlates with prior findings of depression-like behavioral phenotypes in pre-motor onset female R6/1 mice that were also absent in the males of the same age (Pang et al., 2009; Renoir et al., 2011b). Curiously, using pharmacological means as well as in vitro analysis, it was found that the source of the hyperactivity in the female R6/1 mice is the adrenal gland (Du et al., 2012). Studies examining the adrenal glands in HD have so far been scant. However, this finding raises the possibility of an early peripheral change which may influence central brain function. More research is required to examine the HPA-axis of HD patients in terms of its regulation and response to stress. Whilst adrenal hyperplasia has been found in victims of suicide (Szigethy et al., 1994) and a few studies reporting increased adrenal volume in depressed patients compared to controls, the lack of numbers and heterogeneity between studies makes it hard to draw useful conclusions about the role adrenal-specific pathology may play in the development of depression (Kessing et al., 2011).
BDNF and Neurotrophin Signaling
Depression is a complex affective disorder and is thought to result at least partially from an inability of the brain to make appropriate adaptations to environmental stressors, and may involve impaired neural plasticity (Duman et al., 2000; Duman, 2002; Manji et al., 2003; Czeh and Simon, 2005). This is evidenced by studies showing altered brain structures in depression subjects such as reduced cell number, cell density, and body size as well as reduced glial density in frontal cortical and hippocampal brain regions (Ongur et al., 1998; Cotter et al., 2001; Beasley et al., 2002; Rajkowska, 2002). These observations led to the hypothesis that a loss of neurotrophic factors, which are vital for the survival, development, and maintenance of neurons (Lewin and Barde, 1996; McAllister, 2001) as well as regulating synaptic and morphological plasticity (McAllister et al., 1999; Thoenen, 2000), is directly involved in the pathophysiology of depression and that its restitution may lie at the heart of successful treatment (Altar, 1999; Duman, 2004). Indeed, recently it was found that HD patients exhibited a significant decrease in peripheral BDNF gene expression (Krzyszton-Russjan et al., 2012). HD patients (Ciammola et al., 2007) and rodent models of HD (Strand et al., 2007) also show reduced levels of BDNF protein. Huntingtin has important roles in regulating both the transcription (Zuccato et al., 2001) and trafficking (DiFiglia et al., 1995; Gauthier et al., 2004) of BDNF, actions that are affected by the mutation in HD.
BDNF helps support the survival and integrity of existing neurons and encourage growth, migration, and differentiation of new neurons and synapses in the central as well as peripheral nervous systems (Cowansage et al., 2010). It also enhances neurogenesis in the hippocampus (Zigova et al., 1998; Benraiss et al., 2001; Pencea et al., 2001). The expression of BDNF is regulated via neuronal activity through calcium mediated mechanisms (Tabuchi, 2008) whilst its receptor tyrosine kinase-coupled receptor (TrkB), is also regulated in an activity-dependent manner (Nagappan and Lu, 2005). As compromised synaptic and structural plasticity is significantly associated with depression, the importance of BDNF in depression has therefore garnered considerable attention. Lower levels of BDNF in depression patients suggest a role of BDNF in the pathogenesis of depression (Yoshimura et al., 2010; Yoshida et al., 2012). Serum BDNF was found to be significantly reduced in antidepressant-naïve depression patients compared to those who were treated with antidepressants and there was a significant negative correlation between BDNF levels and the Hamilton Rating Scale for Depression (Shimizu et al., 2003). Hippocampal BDNF levels in post-mortem brains of depression patients were found to be higher in those treated with antidepressants at time of death (Chen et al., 2001; Karege et al., 2005). This suggests that antidepressants increase BDNF levels and may account, at least in part, for their potency. In animal models, a similar result was seen. It was found that early life stress lowers BDNF and alters stress sensitivity later in life (Cirulli et al., 2009), whereas increases in BDNF, either via social enrichment or direct infusion into the brain, was found to reduce susceptibility to depression-like behavior (Siuciak et al., 1997; Cirulli et al., 2010).
However, studies also found that increases in BDNF through communal nesting induced an anxiety phenotype by reducing latency to immobility and increasing immobility time in the forced-swim test (FST) (Branchi et al., 2006). Other studies examining the role of BDNF in depression have also found that heterozygous knock-out of BDNF did not alter anxiety profile on the elevated-plus maze (Montkowski and Holsboer, 1997; MacQueen et al., 2001; Gorski et al., 2003). BDNF heterozygotes or mice expressing the dominant negative TrkB receptor TrkB.T1, did not show any differences from WT mice in their performance on the FST, therefore showing that reduced BDNF signaling does not cause depression per se (Saarelainen et al., 2003). However, the fact that a lack of BDNF signaling negated the effects of antidepressant treatment argues that whilst it is not predictive of depression, it is integral for the potency of antidepressant action.
BDNF seems to mediate the positive effect of several classes of antidepressants including SSRIs, tricyclics, SNRI, and MAOI, which have been found to increase BDNF in serum, cortical astrocytes, prefrontal cortex, and hippocampus (Duman and Monteggia, 2006; Allaman et al., 2011; Molendijk et al., 2011). It is also upregulated by exercise and environmental enrichment, contributing to beneficial effects such as induction of cell survival, proliferation, and dendritic development, leading to augmented cognitive outcomes (Acheson et al., 1995; Cotman and Berchtold, 2002; Choi et al., 2009; Sun et al., 2010; Kazlauckas et al., 2011). Environmental enrichment and exercise (voluntary wheel running) have been found to rescue BDNF deficits in R6/1 HD mice as well as ameliorating affective, cognitive, and motor abnormalities (van Dellen et al., 2000; Spires et al., 2004; Pang et al., 2006, 2009; Nithianantharajah et al., 2008; Zajac et al., 2010; Renoir et al., 2013). A diet rich in omega-3 fatty acids, which has been shown to have positive effects on depression (Freeman, 2009), also increases BDNF levels in the hippocampus (Venna et al., 2009).
In BDNF-deficient mice, the behavioral effects of antidepressants were abolished (Saarelainen et al., 2003; Monteggia et al., 2004). TrkB T1 over-expressing transgenic mice, which show reduced TrkB activation in the brain, are resistant to the effects of antidepressants (Saarelainen et al., 2003), whereas over-expression of TrkB led to resistance to depression-like behavior, with SSRI administration unable to further increase this resistance (Koponen et al., 2005) indicating that TrkB signaling is required for the behavioral benefits of antidepressants. These findings suggest that BDNF may be a key molecule involved in various antidepressant treatment strategies. A possible mechanism mediating the antidepressant-induced increase of BDNF is the upregulated expression of CREB, a transcription factor that upregulates BDNF and TrkB (Nibuya et al., 1996). Interestingly, CREB-mediated transcription regulation requires the aid of CBP, whose expression is downregulated by mutant huntingtin (Kazantsev et al., 1999; Nucifora et al., 2001).
Antidepressants are widely used in the treatment of HD patients (Sackley et al., 2011). Recent studies suggest that chronic treatment with the SSRIs fluoxetine or sertraline increased hippocampal neurogenesis, ameliorated cognitive deficits, and depression-like behavioral symptoms in R6/1 mice (Grote et al., 2005; Renoir et al., 2012) and increased BDNF levels and neurogenesis in R6/2 mice (Peng et al., 2008). Chronic antidepressant treatment in depressed patients resulted in upregulation of CREB protein expression (Nibuya et al., 1996), CREB phosphorylation (Saarelainen et al., 2003), BDNF (Chen et al., 2001), and TrkB (Bayer et al., 2000) in the hippocampus. BDNF has been proposed to be a mediator of the effects of antidepressants (Koponen et al., 2005), by augmenting the survival and differentiation of adult-born neurons in the dentate gyrus (Groves, 2007). These results led to the hypothesis that depression in HD coincides with decreased activity in the serotonin-CREB-BDNF-TrkB pathway, resulting in cellular dysfunction and reduced neurogenesis in the hippocampus.
The link between HPA-axis, depression, and BDNF has been explored in rodent models of depression and given much attention. Social stress has been widely used as a useful model of depression (Henn and Vollmayr, 2005). Stressors such as forced immobilization (Smith et al., 1995) and social defeat (Pizarro et al., 2004) were found to decrease BDNF expression in the hippocampus and cortical and subcortical regions of rodent models. Induced elevation of corticosterone, mimicking the effect of stress, has also been associated with reduced levels of BDNF mRNA and protein in the hippocampus and frontal cortex of rodent models (Schaaf et al., 1997, 1998; Chao et al., 1998; Dwivedi et al., 2006). Adrenalectomy surgery caused an increase of BDNF in the hippocampus (Chao et al., 1998), whilst chronic GR activation reduces both CREB phosphorylation and BDNF expression (Focking et al., 2003). This suggests regulatory ability of glucocorticoids on BDNF expression. GR was also found to interact with the BDNF receptor TrkB and corticosterone reduces TrkB-GR interaction, causing reduced BDNF-triggered glutamate release and BDNF-stimulated PLC-γ (Numakawa et al., 2009). Thus, taken together, increased HPA-axis activity may initiate a chain reaction, leading to altered 5-HT signaling, reduced CREB-mediated transcription of BDNF and damage to the hippocampus and other brain regions, which in turn, reduces negative feedback on the HPA-axis in a negative cycle (Figure 1). The molecular and cellular processes may be impacted by environmental modulators, such as the cognitive stimulation and physical exercise induced by environmental enrichment. Complex gene–gene interactions and associated gene-environment interactions are presumably responsible for the variable incidence of depression both within HD patients (where each tandem repeat expansion mutation is embedded in a genome possessing a range of genetic modifiers) and the general population. Elucidation of this complexity at molecular, cellular, and systems levels will require a new generation of sophisticated animal models and clinical investigations.
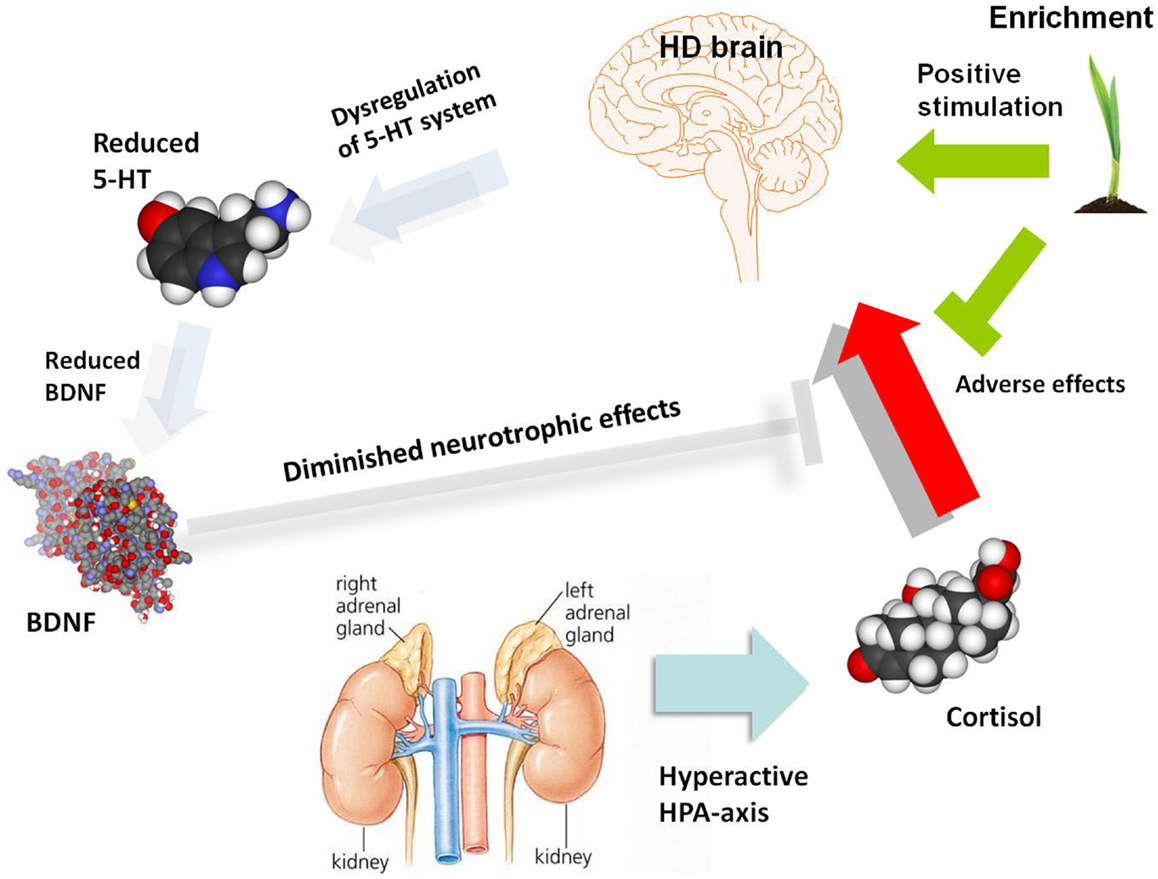
Figure 1. A schematic diagram illustrating potential molecular and cellular mediators of HD pathogenesis, with a focus on serotonergic and neurotrophin signaling as well as the HPA-dysfunction which may give rise to depression and associated symptoms. The role of environmental enrichment in ameliorating specific aspects of molecular, cellular, and systems dysfunction is also illustrated. There are clearly many other molecular and cellular candidates implicated in HD pathogenesis which are not shown in this diagram, which outlines a specific hypothesis regarding the etiology of depression in HD. 5-HT, serotonin; BDNF, brain-derived neurotrophic factor; HD, Huntington’s disease; HPA-axis, hypothalamic-pituitary-adrenal axis.
Summary
As a cure for HD remains elusive, effective strategies targeting symptom management are of great interest. This is especially pertinent with early onset psychiatric and cognitive symptoms, which tend to occur in the most productive years. Depression, being both early onset and having devastating consequences, is a priority target.
Collectively, HD shares certain hallmarks with a large portion of clinical depression patients: namely, disturbances of the 5-HT system, BDNF expression, and HPA-axis dysregulation. This is not perhaps surprising as the use of conventional antidepressants have shown to be equally efficacious in treating depression in HD. However, due to the relentless deterioration of the brain seen in HD, it is paramount to diagnose depression both early and accurately in the hope of stemming the decline with appropriate interventions. The inconsistent diagnostic results reflect the need for a tailored diagnostic tool for HD, taking into account many somatic symptoms such as weight loss which often seem to have little to do with depression. Furthermore, besides antidepressants such as SSRIs, other methods may be examined in detail as potential therapeutics. Strategies such as exercise interventions, occupational therapy, and other cognitive/behavioral treatments are under-utilized in HD and worthy of systematic investigation, both on their own and as adjuncts to pharmacological treatments.
Ultimately, the treatment of HD may require a sophisticated understanding of the pathogenesis of psychiatric, cognitive and motor symptoms, and the source of their heterogeneity in clinical populations. For example, it would be expected that genetic polymorphisms/mutations which predispose to depression in the general population might have additive effects on predisposition when combined with the HD mutation. Similarly, the kind of stressors which can trigger depression in genetically vulnerable individuals might be equally toxic for those who are HD gene-positive. A natural consequence of such detailed insights into pathogenesis could be pharmacogenomics, polypharmacy, and other tailored therapeutic approaches to improve the lives of those suffering from HD and depression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the support of NHMRC Project Grants and an ARC Future Fellowship FT3 (Anthony J. Hannan), as well as a University of Melbourne Research Scholarship (Xin Du). We thank past and present members of the Hannan Laboratory for useful discussions and data that have informed the writing of this manuscript.
References
Abada, Y. S., Schreiber, R., and Ellenbroek, B. (2013). Motor, emotional and cognitive deficits in adult BACHD mice: a model for Huntington’s disease. Behav. Brain Res. 238, 243–251. doi:10.1016/j.bbr.2012.10.039
Acheson, A., Conover, J. C., Fandl, J. P., DeChiara, T. M., Russell, M., Thadani, A., et al. (1995). A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 374, 450–453. doi:10.1038/374450a0
Albert, P. R., and Francois, B. L. (2010). Modifying 5-HT1A receptor gene expression as a new target for antidepressant therapy. Front. Neurosci. 4:35. doi:10.3389/fnins.2010.00035
Allaman, I., Fiumelli, H., Magistretti, P. J., and Martin, J. L. (2011). Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology (Berl.) 216, 75–84. doi:10.1007/s00213-011-2190-y
Altar, C. A. (1999). Neurotrophins and depression. Trends Pharmacol. Sci. 20, 59–61. doi:10.1016/S0165-6147(99)01309-7
Andrés, S., Cárdenas, S., Parra, C., Bravo, J., Greiner, M., Rojas, P., et al. (2006). Effects of long-term adrenalectomy on apoptosis and neuroprotection in the rat hippocampus. Endocrine 29, 299–307. doi:10.1385/ENDO:29:2:299
Asberg, M. (1976). Treatment of depression with tricyclic drugs – pharmacokinetic and pharmacodynamic aspects. Pharmakopsychiatr. Neuropsychopharmakol. 9, 18–26. doi:10.1055/s-0028-1094473
Asberg, M., Traskman, L., and Thoren, P. (1976). 5-HIAA in the cerebrospinal fluid. A biochemical suicide predictor? Arch. Gen. Psychiatry 33, 1193–1197. doi:10.1001/archpsyc.1976.01770100055005
Aziz, N. A., Pijl, H., Frolich, M., van der Graaf, A. W., Roelfsema, F., and Roos, R. A. (2009). Increased hypothalamic-pituitary-adrenal axis activity in Huntington’s disease. J. Clin. Endocrinol. Metab. 94, 1223–1228. doi:10.1210/jc.2008-2543
Bayer, T. A., Schramm, M., Feldmann, N., Knable, M. B., and Falkai, P. (2000). Antidepressant drug exposure is associated with mRNA levels of tyrosine receptor kinase B in major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 24, 881–888. doi:10.1016/S0278-5846(00)00115-9
Beasley, C. L., Cotter, D. R., and Everall, I. P. (2002). Density and distribution of white matter neurons in schizophrenia, bipolar disorder and major depressive disorder: no evidence for abnormalities of neuronal migration. Mol. Psychiatry 7, 564–570. doi:10.1038/sj.mp.4001038
Beglinger, L. J., Paulsen, J. S., Watson, D. B., Wang, C., Duff, K., Langbehn, D. R., et al. (2008). Obsessive and compulsive symptoms in prediagnosed Huntington’s disease. J. Clin. Psychiatry 69, 1758–1765. doi:10.4088/JCP.v69n1111
Belay, H., Burton, C. L., Lovic, V., Meaney, M. J., Sokolowski, M., and Fleming, A. S. (2011). Early adversity and serotonin transporter genotype interact with hippocampal glucocorticoid receptor mRNA expression, corticosterone, and behavior in adult male rats. Behav. Neurosci. 125, 150–160. doi:10.1037/a0022891
Belendiuk, K., Belendiuk, G. W., and Freedman, D. X. (1980). Blood monoamine metabolism in Huntington’s disease. Arch. Gen. Psychiatry 37, 325–332. doi:10.1001/archpsyc.1980.01780160095011
Benraiss, A., Chmielnicki, E., Lerner, K., Roh, D., and Goldman, S. A. (2001). Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J. Neurosci. 21, 6718–6731.
Berrios, G. E., Wagle, A. C., Marková, I. S., Wagle, S. A., Ho, L. W., Rubinsztein, D. C., et al. (2001). Psychiatric symptoms and CAG repeats in neurologically asymptomatic Huntington’s disease gene carriers. Psychiatry Res. 102, 217–225. doi:10.1016/S0165-1781(01)00257-8
Björkqvist, M., Petersén, A., Bacos, K., Isaacs, J., Norlén, P., Gil, J., et al. (2006). Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 15, 1713–1721. doi:10.1093/hmg/ddl094
Blier, P., Pineyro, G., el Mansari, M., Bergeron, R., and de Montigny, C. (1998). Role of somatodendritic 5-HT autoreceptors in modulating 5-HT neurotransmission. Ann. N. Y. Acad. Sci. 861, 204–216. doi:10.1111/j.1749-6632.1998.tb10192.x
Bode, F. J., Stephan, M., Suhling, H., Pabst, R., Straub, R. H., Raber, K. A., et al. (2008). Sex differences in a transgenic rat model of Huntington’s disease: decreased 17beta-estradiol levels correlate with reduced numbers of DARPP32+ neurons in males. Hum. Mol. Genet. 17, 2595–2609. doi:10.1093/hmg/ddn159
Bonelli, R. M., Mayr, B. M., Niederwieser, G., Reisecker, F., and Kapfhammer, H. P. (2003). Ziprasidone in Huntington’s disease: the first case reports. J. Psychopharmacol. (Oxford) 17, 459–460. doi:10.1177/0269881103174009
Branchi, I., D’Andrea, I., Sietzema, J., Fiore, M., Di Fausto, V., Aloe, L., et al. (2006). Early social enrichment augments adult hippocampal BDNF levels and survival of BrdU-positive cells while increasing anxiety- and “depression”-like behavior. J. Neurosci. Res. 83, 965–973. doi:10.1002/jnr.20789
Brummelte, S., and Galea, L. A. (2010). Chronic high corticosterone reduces neurogenesis in the dentate gyrus of adult male and female rats. Neuroscience 168, 680–690. doi:10.1016/j.neuroscience.2010.04.023
Brummett, B. H., Kuhn, C. M., Boyle, S. H., Babyak, M. A., Siegler, I. C., and Williams, R. B. (2012). Cortisol responses to emotional stress in men: association with a functional polymorphism in the 5HTR2C gene. Biol. Psychol. 89, 94–98. doi:10.1016/j.biopsycho.2011.09.013
Cameron, H. A., and Gould, E. (1994). Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61, 203–209. doi:10.1016/0306-4522(94)90224-0
Caraceni, T., Calderini, G., Consolazione, A., Riva, E., Algeri, S., Girotti, F., et al. (1977). Biochemical aspects of Huntington’s chorea. J. Neurol. Neurosurg. Psychiatr. 40, 581–587. doi:10.1136/jnnp.40.6.581
Castro, M. E., Pascual, J., Romon, T., Berciano, J., Figols, J., and Pazos, A. (1998). 5-HT1B receptor binding in degenerative movement disorders. Brain Res. 790, 323–328. doi:10.1016/S0006-8993(97)01566-7
Cavanagh, J. T., Carson, A. J., Sharpe, M., and Lawrie, S. M. (2003). Psychological autopsy studies of suicide: a systematic review. Psychol. Med. 33, 395–405. doi:10.1017/S0033291702006943
Cerqueira, J. J., Catania, C., Sotiropoulos, I., Schubert, M., Kalisch, R., Almeida, O. F., et al. (2005a). Corticosteroid status influences the volume of the rat cingulate cortex – a magnetic resonance imaging study. J. Psychiatr. Res. 39, 451–460. doi:10.1016/j.jpsychires.2005.01.003
Cerqueira, J. J., Pego, J. M., Taipa, R., Bessa, J. M., Almeida, O. F., and Sousa, N. (2005b). Morphological correlates of corticosteroid-induced changes in prefrontal cortex-dependent behaviors. J. Neurosci. 25, 7792–7800. doi:10.1523/JNEUROSCI.1598-05.2005
Chao, H. M., Sakai, R. R., Ma, L. Y., and McEwen, B. S. (1998). Adrenal steroid regulation of neurotrophic factor expression in the rat hippocampus. Endocrinology 139, 3112–3118. doi:10.1210/en.139.7.3112
Chen, B., Dowlatshahi, D., MacQueen, G., Wang, J. F., and Young, T. L. (2001). Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 50, 260–265. doi:10.1016/S0006-3223(01)01083-6
Choi, S. H., Li, Y., Parada, L. F., and Sisodia, S. S. (2009). Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Mol. Neurodegener. 4, 52. doi:10.1186/1750-1326-4-52
Christofides, J., Bridel, M., Egerton, M., Mackay, G. M., Forrest, C. M., Stoy, N., et al. (2006). Blood 5-hydroxytryptamine, 5-hydroxyindoleacetic acid and melatonin levels in patients with either Huntington’s disease or chronic brain injury. J. Neurochem. 97, 1078–1088. doi:10.1111/j.1471-4159.2006.03807.x
Ciammola, A., Sassone, J., Cannella, M., Calza, S., Poletti, B., Frati, L., et al. (2007). Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 574–577. doi:10.1002/ajmg.b.30501
Ciammola, A., Sassone, J., Colciago, C., Mencacci, N. E., Poletti, B., Ciarmiello, A., et al. (2009). Aripiprazole in the treatment of Huntington’s disease: a case series. Neuropsychiatr. Dis. Treat. 5, 1–4.
Cirulli, F., Berry, A., Bonsignore, L. T., Capone, F., D’Andrea, I., Aloe, L., et al. (2010). Early life influences on emotional reactivity: evidence that social enrichment has greater effects than handling on anxiety-like behaviors, neuroendocrine responses to stress and central BDNF levels. Neurosci. Biobehav. Rev. 34, 808–820. doi:10.1016/j.neubiorev.2010.02.008
Cirulli, F., Francia, N., Berry, A., Aloe, L., Alleva, E., and Suomi, S. J. (2009). Early life stress as a risk factor for mental health: role of neurotrophins from rodents to non-human primates. Neurosci. Biobehav. Rev. 33, 573–585. doi:10.1016/j.neubiorev.2008.09.001
Codori, A. M., Slavney, P. R., Rosenblatt, A., and Brandt, J. (2004). Prevalence of major depression one year after predictive testing for Huntington’s disease. Genet. Test. 8, 114–119. doi:10.1089/gte.2004.8.114
Cotman, C. W., and Berchtold, N. C. (2002). Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 25, 295–301. doi:10.1016/S0166-2236(02)02143-4
Cotter, D. R., Pariante, C. M., and Everall, I. P. (2001). Glial cell abnormalities in major psychiatric disorders: the evidence and implications. Brain Res. Bull. 55, 585–595. doi:10.1016/S0361-9230(01)00527-5
Cowansage, K. K., LeDoux, J. E., and Monfils, M. H. (2010). Brain-derived neurotrophic factor: a dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 3, 12–29. doi:10.2174/1874467211003010012
Crochemore, C., Lu, J., Wu, Y., Liposits, Z., Sousa, N., Holsboer, F., et al. (2005). Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatry 10, 790–798. doi:10.1038/sj.mp.4001679
Cross, A. J., Reynolds, G. P., Hewitt, L. M., and Slater, P. (1986). Brain serotonin receptors in Huntington’s disease. Neurochem. Int. 9, 431–435. doi:10.1016/0197-0186(86)90085-9
David, G., Abbas, N., Stevanin, G., Durr, A., Yvert, G., Cancel, G., et al. (1997). Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 17, 65–70. doi:10.1038/ng0997-65
Dawson, L. A., Nguyen, H. Q., Smith, D. L., and Schechter, L. E. (2002). Effect of chronic fluoxetine and WAY-100635 treatment on serotonergic neurotransmission in the frontal cortex. J. Psychopharmacol. (Oxford) 16, 145–152. doi:10.1177/026988110201600205
De Souza, J., Jones, L. A., and Rickards, H. (2010). Validation of self-report depression rating scales in Huntington’s disease. Mov. Disord. 25, 91–96. doi:10.1002/mds.22837
Delgado, P. L., Charney, D. S., Price, L. H., Aghajanian, G. K., Landis, H., and Heninger, G. R. (1990). Serotonin function and the mechanism of antidepressant action. Reversal of antidepressant-induced remission by rapid depletion of plasma tryptophan. Arch. Gen. Psychiatry 47, 411–418. doi:10.1001/archpsyc.1990.01810170011002
DiFiglia, M., Sapp, E., Chase, K., Schwarz, C., Meloni, A., Young, C., et al. (1995). Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14, 1075–1081. doi:10.1016/0896-6273(95)90346-1
Drevets, W. C., Frank, E., Price, J. C., Kupfer, D. J., Holt, D., Greer, P. J., et al. (1999). PET imaging of serotonin 1A receptor binding in depression. Biol. Psychiatry 46, 1375–1387. doi:10.1016/S0006-3223(99)00189-4
Drevets, W. C., Thase, M. E., Moses-Kolko, E. L., Price, J., Frank, E., Kupfer, D. J., et al. (2007). Serotonin-1A receptor imaging in recurrent depression: replication and literature review. Nucl. Med. Biol. 34, 865–877. doi:10.1016/j.nucmedbio.2007.06.008
Du, X., Leang, L., Mustafa, T., Renoir, T., Pang, T. Y., and Hannan, A. J. (2012). Environmental enrichment rescues female-specific hyperactivity of the hypothalamic-pituitary-adrenal axis in a model of Huntington’s disease. Transl. Psychiatry 2, e133. doi:10.1038/tp.2012.58
Duff, K., Paulsen, J. S., Beglinger, L. J., Langbehn, D. R., and Stout, J. C. (2007). Psychiatric symptoms in Huntington’s disease before diagnosis: the predict-HD study. Biol. Psychiatry 62, 1341–1346. doi:10.1016/j.biopsych.2006.11.034
Duman, R. S. (2002). Pathophysiology of depression: the concept of synaptic plasticity. Eur. Psychiatry 17(Suppl. 3), 306–310. doi:10.1016/S0924-9338(02)00654-5
Duman, R. S. (2004). Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med. 5, 11–25. doi:10.1385/NMM:5:1:011
Duman, R. S., Malberg, J., Nakagawa, S., and D’Sa, C. (2000). Neuronal plasticity and survival in mood disorders. Biol. Psychiatry 48, 732–739. doi:10.1016/S0006-3223(00)00935-5
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127. doi:10.1016/j.biopsych.2006.02.013
Dwivedi, Y., Rizavi, H. S., and Pandey, G. N. (2006). Antidepressants reverse corticosterone-mediated decrease in brain-derived neurotrophic factor expression: differential regulation of specific exons by antidepressants and corticosterone. Neuroscience 139, 1017–1029. doi:10.1016/j.neuroscience.2005.12.058
Erdeljan, P., MacDonald, J. F., and Matthews, S. G. (2001). Glucocorticoids and serotonin alter glucocorticoid receptor (GR) but not mineralocorticoid receptor (MR) mRNA levels in fetal mouse hippocampal neurons, in vitro. Brain Res. 896, 130–136. doi:10.1016/S0006-8993(01)02075-3
Falkenberg, V. R., and Rajeevan, M. S. (2010). Identification of a potential molecular link between the glucocorticoid and serotonergic signaling systems. J. Mol. Neurosci. 41, 322–327. doi:10.1007/s12031-009-9320-6
Farrer, L. A. (1986). Suicide and attempted suicide in Huntington disease: implications for preclinical testing of persons at risk. Am. J. Med. Genet. 24, 305–311. doi:10.1002/ajmg.1320240211
Faure, A., Höhn, S., Von Hörsten, S., Delatour, B., Raber, K., Le Blanc, P., et al. (2011). Altered emotional and motivational processing in the transgenic rat model for Huntington’s disease. Neurobiol. Learn. Mem. 95, 92–101. doi:10.1016/j.nlm.2010.11.010
Focking, M., Holker, I., and Trapp, T. (2003). Chronic glucocorticoid receptor activation impairs CREB transcriptional activity in clonal neurons. Biochem. Biophys. Res. Commun. 304, 720–723. doi:10.1016/S0006-291X(03)00665-X
Freeman, M. P. (2009). Omega-3 fatty acids in major depressive disorder. J. Clin. Psychiatry 70(Suppl. 5), 7–11. doi:10.4088/JCP.8157su1c.02
Froger, N., Palazzo, E., Boni, C., Hanoun, N., Saurini, F., Joubert, C., et al. (2004). Neurochemical and behavioral alterations in glucocorticoid receptor-impaired transgenic mice after chronic mild stress. J. Neurosci. 24, 2787–2796. doi:10.1523/JNEUROSCI.4132-03.2004
Gargiulo, M., Lejeune, S., Tanguy, M. L., Lahlou-Laforêt, K., Faudet, A., Cohen, D., et al. (2009). Long-term outcome of presymptomatic testing in Huntington disease. Eur. J. Hum. Genet. 17, 165–171. doi:10.1038/ejhg.2008.146
Gauthier, L. R., Charrin, B. C., Borrell-Pagès, M., Dompierre, J. P., Rangone, H., Cordelières, F. P., et al. (2004). Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138. doi:10.1016/j.cell.2004.06.018
Giralt, A., Saavedra, A., Carretón, O., Arumí, H., Tyebji, S., Alberch, J., et al. (2013). PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus. doi:10.1002/hipo.22128. [Epub ahead of print].
Gorski, J. A., Balogh, S. A., Wehner, J. M., and Jones, K. R. (2003). Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience 121, 341–354. doi:10.1016/S0306-4522(03)00426-3
Grote, H. E., Bull, N. D., Howard, M. L., van Dellen, A., Blakemore, C., Bartlett, P. F., et al. (2005). Cognitive disorders and neurogenesis deficits in Huntington’s disease mice are rescued by fluoxetine. Eur. J. Neurosci. 22, 2081–2088. doi:10.1111/j.1460-9568.2005.04365.x
Groves, J. O. (2007). Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 12, 1079–1088. doi:10.1038/sj.mp.4002075
Harada, K., Yamaji, T., and Matsuoka, N. (2008). Activation of the serotonin 5-HT2C receptor is involved in the enhanced anxiety in rats after single-prolonged stress. Pharmacol. Biochem. Behav. 89, 11–16. doi:10.1016/j.pbb.2007.10.016
Harris, E. C., and Barraclough, B. (1998). Excess mortality of mental disorder. Br. J. Psychiatry 173, 11–53. doi:10.1192/bjp.173.1.11
Hasin, D. S., Goodwin, R. D., Stinson, F. S., and Grant, B. F. (2005). Epidemiology of major depressive disorder: results from the National Epidemiologic Survey on Alcoholism and Related Conditions. Arch. Gen. Psychiatry 62, 1097–1106. doi:10.1001/archpsyc.62.10.1097
Heisler, L. K., Chu, H. M., Brennan, T. J., Danao, J. A., Bajwa, P., Parsons, L. H., et al. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. U.S.A. 95, 15049–15054. doi:10.1073/pnas.95.25.15049
Henn, F. A., and Vollmayr, B. (2005). Stress models of depression: forming genetically vulnerable strains. Neurosci. Biobehav. Rev. 29, 799–804. doi:10.1016/j.neubiorev.2005.03.019
Hery, M., Semont, A., Fache, M. P., Faudon, M., and Hery, F. (2000). The effects of serotonin on glucocorticoid receptor binding in rat raphe nuclei and hippocampal cells in culture. J. Neurochem. 74, 406–413. doi:10.1046/j.1471-4159.2000.0740406.x
Hesen, W., and Joels, M. (1996). Modulation of 5HT1A responsiveness in CA1 pyramidal neurons by in vivo activation of corticosteroid receptors. J. Neuroendocrinol. 8, 433–438. doi:10.1046/j.1365-2826.1996.04724.x
Heuser, I. J., Chase, T. N., and Mouradian, M. M. (1991). The limbic-hypothalamic-pituitary-adrenal axis in Huntington’s disease. Biol. Psychiatry 30, 943–952. doi:10.1016/0006-3223(91)90007-9
Ho, A. K., Gilbert, A. S., Mason, S. L., Goodman, A. O., and Barker, R. A. (2009). Health-related quality of life in Huntington’s disease: which factors matter most? Mov. Disord. 24, 574–578. doi:10.1002/mds.22412
Holl, A. K., Wilkinson, L., Painold, A., Holl, E. M., and Bonelli, R. M. (2010). Combating depression in Huntington’s disease: effective antidepressive treatment with venlafaxine XR. Int. Clin. Psychopharmacol. 25, 46–50. doi:10.1097/YIC.0b013e3283348018
Horowitz, M. J., Field, N. P., Zanko, A., Donnelly, E. F., Epstein, C., and Longo, F. (2001). Psychological impact of news of genetic risk for Huntington disease. Am. J. Med. Genet. 103, 188–192. doi:10.1002/ajmg.1538
Huang, J., Strafaci, J. A., and Azmitia, E. C. (1997). 5-HT1A receptor agonist reverses adrenalectomy-induced loss of granule neuronal morphology in the rat dentate gyrus. Neurochem. Res. 22, 1329–1337. doi:10.1023/A:1022062921438
Inskip, H. M., Harris, E. C., and Barraclough, B. (1998). Lifetime risk of suicide for affective disorder, alcoholism and schizophrenia. Br. J. Psychiatry 172, 35–37. doi:10.1192/bjp.172.1.35
Jacobs, B. L., van Praag, H., and Gage, F. H. (2000). Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol. Psychiatry 5, 262–269. doi:10.1038/sj.mp.4000712
Jensen, P., Sorensen, S. A., Fenger, K., and Bolwig, T. G. (1993). A study of psychiatric morbidity in patients with Huntington’s disease, their relatives, and controls. Admissions to psychiatric hospitals in Denmark from 1969 to 1991. Br. J. Psychiatry 163, 790–797. doi:10.1192/bjp.163.6.790
Jongen, P. J., Renier, W. O., and Gabreels, F. J. (1980). Seven cases of Huntington’s disease in childhood and levodopa induced improvement in the hypokinetic – rigid form. Clin. Neurol. Neurosurg. 82, 251–261. doi:10.1016/0303-8467(80)90017-7
Julien, C. L., Thompson, J. C., Wild, S., Yardumian, P., Snowden, J. S., Turner, G., et al. (2007). Psychiatric disorders in preclinical Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 78, 939–943. doi:10.1136/jnnp.2006.103309
Kakizuka, A. (1997). Degenerative ataxias: genetics, pathogenesis and animal models. Curr. Opin. Neurol. 10, 285–290. doi:10.1097/00019052-199708000-00002
Karege, F., Vaudan, G., Schwald, M., Perroud, N., and La Harpe, R. (2005). Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res. Mol. Brain Res. 136, 29–37. doi:10.1016/j.molbrainres.2004.12.020
Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D., and Housman, D. (1999). Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 96, 11404–11409. doi:10.1073/pnas.96.20.11404
Kazlauckas, V., Pagnussat, N., Mioranzza, S., Kalinine, E., Nunes, F., Pettenuzzo, L., et al. (2011). Enriched environment effects on behavior, memory and BDNF in low and high exploratory mice. Physiol. Behav. 102, 475–480. doi:10.1016/j.physbeh.2010.12.025
Kessing, L. V., Willer, I. S., and Knorr, U. (2011). Volume of the adrenal and pituitary glands in depression. Psychoneuroendocrinology 36, 19–27. doi:10.1016/j.psyneuen.2010.05.007
Kessler, R. C., Berglund, P., Demler, O., Jin, R., Merikangas, K. R., and Walters, E. E. (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 593–602. doi:10.1001/archpsyc.62.6.617
Koponen, E., Rantamaki, T., Voikar, V., Saarelainen, T., MacDonald, E., and Castren, E. (2005). Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell. Mol. Neurobiol. 25, 973–980. doi:10.1007/s10571-005-8468-z
Koshy, B. T., and Zoghbi, H. Y. (1997). The CAG/polyglutamine tract diseases: gene products and molecular pathogenesis. Brain Pathol. 7, 927–942. doi:10.1111/j.1750-3639.1997.tb00894.x
Krogias, C., Strassburger, K., Eyding, J., Gold, R., Norra, C., Juckel, G., et al. (2011). Depression in patients with Huntington disease correlates with alterations of the brain stem raphe depicted by transcranial sonography. J. Psychiatry Neurosci. 36, 187–194. doi:10.1503/jpn.100067
Krzyszton-Russjan, J., Zielonka, D., Jackiewicz, J., Kusmirek, S., Bubko, I., Klimberg, A., et al. (2012). A study of molecular changes relating to energy metabolism and cellular stress in people with Huntington’s disease: looking for biomarkers. J. Bioenerg. Biomembr. 45, 71–85.
Kusserow, H., Davies, B., Hörtnagl, H., Voigt, I., Stroh, T., Bert, B., et al. (2004). Reduced anxiety-related behaviour in transgenic mice overexpressing serotonin 1A receptors. Brain Res. Mol. Brain Res. 129, 104–116. doi:10.1016/j.molbrainres.2004.06.028
Lanfumey, L., Mannoury La Cour, C., Froger, N., and Hamon, M. (2000). 5-HT-HPA interactions in two models of transgenic mice relevant to major depression. Neurochem. Res. 25, 1199–1206. doi:10.1023/A:1007683810230
Larsson, M. U., Luszcz, M. A., Bui, T. H., and Wahlin, T. B. (2006). Depression and suicidal ideation after predictive testing for Huntington’s disease: a two-year follow-up study. J. Genet. Couns. 15, 361–374. doi:10.1007/s10897-006-9027-6
Leblhuber, F., Peichl, M., Neubauer, C., Reisecker, F., Steinparz, F. X., Windhager, E., et al. (1995). Serum dehydroepiandrosterone and cortisol measurements in Huntington’s chorea. J. Neurol. Sci. 132, 76–79. doi:10.1016/0022-510X(95)00114-H
Leroi, I., O’Hearn, E., Marsh, L., Lyketsos, C. G., Rosenblatt, A., Ross, C. A., et al. (2002). Psychopathology in patients with degenerative cerebellar diseases: a comparison to Huntington’s disease. Am. J. Psychiatry 159, 1306–1314. doi:10.1176/appi.ajp.159.8.1306
Lewin, G. R., and Barde, Y. A. (1996). Physiology of the neurotrophins. Annu. Rev. Neurosci. 19, 289–317. doi:10.1146/annurev.ne.19.030196.001445
Liu, B., Zhang, H., Xu, C., Yang, G., Tao, J., Huang, J., et al. (2011). Neuroprotective effects of icariin on corticosterone-induced apoptosis in primary cultured rat hippocampal neurons. Brain Res. 1375, 59–67. doi:10.1016/j.brainres.2010.12.053
Lopez, J. F., Chalmers, D. T., Little, K. Y., and Watson, S. J. (1998). A.E. Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol. Psychiatry 43, 547–573. doi:10.1016/S0006-3223(97)00484-8
Lovestone, S., Hodgson, S., Sham, P., Differ, A. M., and Levy, R. (1996). Familial psychiatric presentation of Huntington’s disease. J. Med. Genet. 33, 128–131. doi:10.1136/jmg.33.2.128
Lucassen, P. J., Meerlo, P., Naylor, A. S., van Dam, A. M., Dayer, A. G., Fuchs, E., et al. (2010). Regulation of adult neurogenesis by stress, sleep disruption, exercise and inflammation: implications for depression and antidepressant action. Eur. Neuropsychopharmacol. 20, 1–17. doi:10.1016/j.euroneuro.2009.08.003
MacQueen, G. M., Ramakrishnan, K., Croll, S. D., Siuciak, J. A., Yu, G., Young, L. T., et al. (2001). Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav. Neurosci. 115, 1145–1153. doi:10.1037/0735-7044.115.5.1145
Manji, H. K., Quiroz, J. A., Sporn, J., Payne, J. L., Denicoff, K., Gray, N. A., et al. (2003). Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol. Psychiatry 53, 707–742. doi:10.1016/S0006-3223(03)00117-3
Markianos, M., Panas, M., Kalfakis, N., and Vassilopoulos, D. (2004). Platelet monoamine oxidase activity in subjects tested for Huntington’s disease gene mutation. J. Neural Transm. 111, 475–483. doi:10.1007/s00702-003-0103-x
McAllister, A. K. (2001). Neurotrophins and neuronal differentiation in the central nervous system. Cell. Mol. Life Sci. 58, 1054–1060. doi:10.1007/PL00000920
McAllister, A. K., Katz, L. C., and Lo, D. C. (1999). Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci. 22, 295–318. doi:10.1146/annurev.neuro.22.1.295
Meaney, M. J., Diorio, J., Francis, D., LaRocque, S., O’Donnell, D., Smythe, J. W., et al. (1994). Environmental regulation of the development of glucocorticoid receptor systems in the rat forebrain. The role of serotonin. Ann. N. Y. Acad. Sci. 746, 260–273; discussion 89–93. doi:10.1111/j.1749-6632.1994.tb39243.x
Messa, C., Colombo, C., Moresco, R. M., Gobbo, C., Galli, L., Lucignani, G., et al. (2003). 5-HT(2A) receptor binding is reduced in drug-naive and unchanged in SSRI-responder depressed patients compared to healthy controls: a PET study. Psychopharmacology (Berl.) 167, 72–78.
Meyer, J. H., Ginovart, N., Boovariwala, A., Sagrati, S., Hussey, D., Garcia, A., et al. (2006). Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 63, 1209–1216. doi:10.1001/archpsyc.63.11.1209
Milnerwood, A. J., and Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 33, 513–523. doi:10.1016/j.tins.2010.08.002
Molendijk, M. L., Bus, B. A., Spinhoven, P., Penninx, B. W., Kenis, G., Prickaerts, J., et al. (2011). Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol. Psychiatry 16, 1088–1095. doi:10.1038/mp.2010.98
Moller, S. E., Honore, P., and Larsen, O. B. (1983). Tryptophan and tyrosine ratios to neutral amino acids in endogenous depression. Relation to antidepressant response to amitriptyline and lithium + L-tryptophan. J. Affect. Disord. 5, 67–79. doi:10.1016/0165-0327(83)90038-1
Monteggia, L. M., Barrot, M., Powell, C. M., Berton, O., Galanis, V., Gemelli, T., et al. (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 101, 10827–10832. doi:10.1073/pnas.0402141101
Montkowski, A., and Holsboer, F. (1997). Intact spatial learning and memory in transgenic mice with reduced BDNF. Neuroreport 8, 779–782. doi:10.1097/00001756-199702100-00040
Moses-Kolko, E. L., Wisner, K. L., Price, J. C., Berga, S. L., Drevets, W. C., Hanusa, B. H., et al. (2008). Serotonin 1A receptor reductions in postpartum depression: a positron emission tomography study. Fertil. Steril. 89, 685–692. doi:10.1016/j.fertnstert.2007.03.059
Naarding, P., Kremer, H. P., and Zitman, F. G. (2001). Huntington’s disease: a review of the literature on prevalence and treatment of neuropsychiatric phenomena. Eur. Psychiatry 16, 439–445. doi:10.1016/S0924-9338(01)00604-6
Nagappan, G., and Lu, B. (2005). Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 28, 464–471. doi:10.1016/j.tins.2005.07.003
Nakamura, K. (2001). SCA17, a novel polyglutamine disease caused by the expansion of polyglutamine tracts in TATA-binding protein. Rinsho Shinkeigaku 41, 1123–1125.
Nakamura, K., Jeong, S. Y., Uchihara, T., Anno, M., Nagashima, K., Nagashima, T., et al. (2001). SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 10, 1441–1448. doi:10.1093/hmg/10.14.1441
Nance, M. A., and Myers, R. H. (2001). Juvenile onset Huntington’s disease – clinical and research perspectives. Ment. Retard Dev. Disabil. Res. Rev. 7, 153–157. doi:10.1002/mrdd.1022
Nehl, C., Ready, R. E., Hamilton, J., and Paulsen, J. S. (2001). Effects of depression on working memory in presymptomatic Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 13, 342–346. doi:10.1176/appi.neuropsych.13.3.342
Nibuya, M., Nestler, E. J., and Duman, R. S. (1996). Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J. Neurosci. 16, 2365–2372.
Nithianantharajah, J., Barkus, C., Murphy, M., and Hannan, A. J. (2008). Gene-environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol. Dis. 29, 490–504. doi:10.1016/j.nbd.2007.11.006
Nithianantharajah, J., and Hannan, A. J. (2012). Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington’s disease. Neuroscience. doi:10.1016/j.neuroscience.2012.05.043. [Epub ahead of print].
Nucifora, F. C. Jr., Sasaki, M., Peters, M. F., Huang, H., Cooper, J. K., Yamada, M., et al. (2001). Interference by Huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science 291, 2423–2428. doi:10.1126/science.1056784
Numakawa, T., Kumamaru, E., Adachi, N., Yagasaki, Y., Izumi, A., and Kunugi, H. (2009). Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. U.S.A. 106, 647–652. doi:10.1073/pnas.080088 8106
Ongur, D., Drevets, W. C., and Price, J. L. (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U.S.A. 95, 13290–13295. doi:10.1073/pnas.95.22.13290
Orvoen, S., Pla, P., Gardier, A. M., Saudou, F., and David, D. J. (2012). Huntington’s disease knock-in male mice show specific anxiety-like behaviour and altered neuronal maturation. Neurosci. Lett. 507, 127–132. doi:10.1016/j.neulet.2011.11.063
Pang, T. Y., Du, X., Zajac, M. S., Howard, M. L., and Hannan, A. J. (2009). Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 18, 753–766. doi:10.1093/hmg/ddn385
Pang, T. Y., Stam, N. C., Nithianantharajah, J., Howard, M. L., and Hannan, A. J. (2006). Differential effects of voluntary physical exercise on behavioral and brain-derived neurotrophic factor expression deficits in Huntington’s disease transgenic mice. Neuroscience 141, 569–584. doi:10.1016/j.neuroscience.2006.04.013
Papadimitriou, A., and Priftis, K. N. (2009). Regulation of the hypothalamic-pituitary-adrenal axis. Neuroimmunomodulation 16, 265–271. doi:10.1159/000216184
Pariante, C. M., and Lightman, S. L. (2008). The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 31, 464–468. doi:10.1016/j.tins.2008.06.006
Parks, C. L., Robinson, P. S., Sibille, E., Shenk, T., and Toth, M. (1998). Increased anxiety of mice lacking the serotonin1A receptor. Proc. Natl. Acad. Sci. U.S.A. 95, 10734–10739. doi:10.1073/pnas.95.18.10734
Patel, S. V., Tariot, P. N., and Asnis, J. (1996). L-Deprenyl augmentation of fluoxetine in a patient with Huntington’s disease. Ann. Clin. Psychiatry 8, 23–26. doi:10.3109/10401239609149087
Paulsen, J. S., Nehl, C., Hoth, K. F., Kanz, J. E., Benjamin, M., Conybeare, R., et al. (2005). Depression and stages of Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 17, 496–502. doi:10.1176/appi.neuropsych.17.4.496
Paulsen, J. S., Ready, R. E., Hamilton, J. M., Mega, M. S., and Cummings, J. L. (2001). Neuropsychiatric aspects of Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 71, 310–314. doi:10.1136/jnnp.71.3.310
Pencea, V., Bingaman, K. D., Wiegand, S. J., and Luskin, M. B. (2001). Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J. Neurosci. 21, 6706–6717.
Peng, Q., Masuda, N., Jiang, M., Li, Q., Zhao, M., Ross, C. A., et al. (2008). The antidepressant sertraline improves the phenotype, promotes neurogenesis and increases BDNF levels in the R6/2 Huntington’s disease mouse model. Exp. Neurol. 210, 154–163. doi:10.1016/j.expneurol.2007.10.015
Petersen, A., Wortwein, G., Gruber, S. H., and Mathe, A. A. (2008). Escitalopram reduces increased hippocampal cytogenesis in a genetic rat depression model. Neurosci. Lett. 436, 305–308. doi:10.1016/j.neulet.2008.03.035
Pizarro, J. M., Lumley, L. A., Medina, W., Robison, C. L., Chang, W. E., Alagappan, A., et al. (2004). Acute social defeat reduces neurotrophin expression in brain cortical and subcortical areas in mice. Brain Res. 1025, 10–20. doi:10.1016/j.brainres.2004.06.085
Pouladi, M. A., Graham, R. K., Karasinska, J. M., Xie, Y., Santos, R. D., Petersén, A., et al. (2009). Prevention of depressive behaviour in the YAC128 mouse model of Huntington disease by mutation at residue 586 of huntingtin. Brain 132, 919–932. doi:10.1093/brain/awp006
Quintana, J. (1992). Platelet serotonin and plasma tryptophan decreases in endogenous depression. Clinical, therapeutic, and biological correlations. J. Affect. Disord. 24, 55–62. doi:10.1016/0165-0327(92)90019-3
Rajkowska, G. (2002). Cell pathology in mood disorders. Semin. Clin. Neuropsychiatry 7, 281–292. doi:10.1053/scnp.2002.35228
Ramboz, S., Oosting, R., Amara, D. A., Kung, H. F., Blier, P., Mendelsohn, M., et al. (1998). Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc. Natl. Acad. Sci. U.S.A. 95, 14476–14481. doi:10.1073/pnas.95.24.14476
Raymond, L. A., Andre, V. M., Cepeda, C., Gladding, C. M., Milnerwood, A. J., and Levine, M. S. (2011). Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience 198, 252–273. doi:10.1016/j.neuroscience.2011.08.052
Renoir, T., Chevarin, C., Lanfumey, L., and Hannan, A. J. (2011a). Effect of enhanced voluntary physical exercise on brain levels of monoamines in Huntington disease mice. PLoS Curr. 3:RRN1281. doi:10.1371/currents.RRN1281
Renoir, T., Zajac, M. S., Du, X., Pang, T. Y., Leang, L., Chevarin, C., et al. (2011b). Sexually dimorphic serotonergic dysfunction in a mouse model of Huntington’s disease and depression. PLoS ONE 6:e22133. doi:10.1371/journal.pone.0022133
Renoir, T., Pang, T. Y., Mo, C., Chan, G., Chevarin, C., Lanfumey, L., et al. (2013). Differential effects of early environmental enrichment on emotionality related behaviours in Huntington’s disease transgenic mice. J. Physiol. (Lond.) 591, 41–55. doi:10.1113/jphysiol.2012.239798
Renoir, T., Pang, T. Y., Zajac, M. S., Chan, G., Du, X., Leang, L., et al. (2012). Treatment of depressive-like behaviour in Huntington’s disease mice by chronic sertraline and exercise. Br. J. Pharmacol. 165, 1375–1389. doi:10.1111/j.1476-5381.2011.01567.x
Richards, G., Messer, J., Waldvogel, H. J., Gibbons, H. M., Dragunow, M., Faull, R. L., et al. (2011). Up-regulation of the isoenzymes MAO-A and MAO-B in the human basal ganglia and pons in Huntington’s disease revealed by quantitative enzyme radioautography. Brain Res. 1370, 204–214. doi:10.1016/j.brainres.2010.11.020
Rickards, H., De Souza, J., Crooks, J., van Walsem, M. R., van Duijn, E., Landwehrmeyer, B., et al. (2011). Discriminant analysis of beck depression inventory and hamilton rating scale for depression in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 23, 399–402. doi:10.1176/appi.neuropsych.23.4.399
Robitaille, Y., Lopes-Cendes, I., Becher, M., Rouleau, G., and Clark, A. W. (1997). The neuropathology of CAG repeat diseases: review and update of genetic and molecular features. Brain Pathol. 7, 901–926. doi:10.1111/j.1750-3639.1997.tb00893.x
Ross, C. A. (1997). Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19, 1147–1150. doi:10.1016/S0896-6273(00)80405-5
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98. doi:10.1016/S1474-4422(10)70245-3
Rossi, D. V., Burke, T. F., McCasland, M., and Hensler, J. G. (2008). Serotonin-1A receptor function in the dorsal raphe nucleus following chronic administration of the selective serotonin reuptake inhibitor sertraline. J. Neurochem. 105, 1091–1099. doi:10.1111/j.1471-4159.2007.05201.x
Rowe, K. C., Paulsen, J. S., Langbehn, D. R., Wang, C., Mills, J., Beglinger, L. J., et al. (2012). Patterns of serotonergic antidepressant usage in prodromal Huntington disease. Psychiatry Res. 196, 309–314. doi:10.1016/j.psychres.2011.09.005
Roy, A., De Jong, J., and Linnoila, M. (1989). Cerebrospinal fluid monoamine metabolites and suicidal behavior in depressed patients. A 5-year follow-up study. Arch. Gen. Psychiatry 46, 609–612. doi:10.1001/archpsyc.1989.01810070035005
Saarelainen, T., Hendolin, P., Lucas, G., Koponen, E., Sairanen, M., MacDonald, E., et al. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 23, 349–357.
Sackley, C., Hoppitt, T. J., Calvert, M., Gill, P., Eaton, B., Yao, G., et al. (2011). Huntington’s disease: current epidemiology and pharmacological management in UK primary care. Neuroepidemiology 37, 216–221. doi:10.1159/000331912
Sapolsky, R. M. (1986). Glucocorticoid toxicity in the hippocampus. Temporal aspects of synergy with kainic acid. Neuroendocrinology 43, 440–444. doi:10.1159/000124561
Sapolsky, R. M., Uno, H., Rebert, C. S., and Finch, C. E. (1990). Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J. Neurosci. 10, 2897–2902.
Schaaf, M. J., de Jong, J., de Kloet, E. R., and Vreugdenhil, E. (1998). Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 813, 112–120. doi:10.1016/S0006-8993(98)01010-5
Schaaf, M. J., Hoetelmans, R. W., de Kloet, E. R., and Vreugdenhil, E. (1997). Corticosterone regulates expression of BDNF and trkB but not NT-3 and trkC mRNA in the rat hippocampus. J. Neurosci. Res. 48, 334–341. doi:10.1002/(SICI)1097-4547(19970515)48:4<334::AID-JNR5>3.0.CO;2-C
Schmitz-Hübsch, T., Coudert, M., Tezenas du Montcel, S., Giunti, P., Labrum, R., Dürr, A., et al. (2011). Depression comorbidity in spinocerebellar ataxia. Mov. Disord. 26, 870–876. doi:10.1002/mds.23698
Shimizu, E., Hashimoto, K., Okamura, N., Koike, K., Komatsu, N., Kumakiri, C., et al. (2003). Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 54, 70–75. doi:10.1016/S0006-3223(03)00181-1
Shiwach, R. (1994). Psychopathology in Huntington’s disease patients. Acta Psychiatr. Scand. 90, 241–246. doi:10.1111/j.1600-0447.1994.tb01587.x
Shiwach, R. S., and Norbury, C. G. (1994). A controlled psychiatric study of individuals at risk for Huntington’s disease. Br. J. Psychiatry 165, 500–505. doi:10.1192/bjp.165.4.500
Siuciak, J. A., Lewis, D. R., Wiegand, S. J., and Lindsay, R. M. (1997). Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol. Biochem. Behav. 56, 131–137. doi:10.1016/S0091-3057(96)00169-4
Smith, M. A., Makino, S., Kvetnansky, R., and Post, R. M. (1995). Effects of stress on neurotrophic factor expression in the rat brain. Ann. N. Y. Acad. Sci. 771, 234–239. doi:10.1111/j.1749-6632.1995.tb44684.x
Smith, M. M., Mills, J. A., Epping, E. A., Westervelt, H. J., and Paulsen, J. S. (2012). Depressive symptom severity is related to poorer cognitive performance in prodromal Huntington disease. Neuropsychology 26, 664–669. doi:10.1037/a0029218
Spires, T. L., Grote, H. E., Varshney, N. K., Cordery, P. M., van Dellen, A., Blakemore, C., et al. (2004). Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J. Neurosci. 24, 2270–2276. doi:10.1523/JNEUROSCI.1658-03.2004
Squitieri, F., Cannella, M., Piorcellini, A., Brusa, L., Simonelli, M., and Ruggieri, S. (2001). Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 14, 69–72.
Stetler, C., and Miller, G. E. (2011). Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom. Med. 73, 114–126. doi:10.1097/PSY.0b013e31820ad12b
Stockmeier, C. A., Shapiro, L. A., Dilley, G. E., Kolli, T. N., Friedman, L., and Rajkowska, G. (1998). Increase in serotonin-1A autoreceptors in the midbrain of suicide victims with major depression-postmortem evidence for decreased serotonin activity. J. Neurosci. 18, 7394–7401.
Strand, A. D., Baquet, Z. C., Aragaki, A. K., Holmans, P., Yang, L., Cleren, C., et al. (2007). Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 27, 11758–11768. doi:10.1523/JNEUROSCI.2461-07.2007
Sullivan, F. R., Bird, E. D., Alpay, M., and Cha, J. H. (2001). Remotivation therapy and Huntington’s disease. J. Neurosci. Nurs. 33, 136–142. doi:10.1097/01376517-200106000-00005
Sun, H., Zhang, J., Zhang, L., Liu, H., Zhu, H., and Yang, Y. (2010). Environmental enrichment influences BDNF and NR1 levels in the hippocampus and restores cognitive impairment in chronic cerebral hypoperfused rats. Curr. Neurovasc. Res. 7, 268–280. doi:10.2174/156720210793180819
Szigethy, E., Conwell, Y., Forbes, N. T., Cox, C., and Caine, E. D. (1994). Adrenal weight and morphology in victims of completed suicide. Biol. Psychiatry 36, 374–380. doi:10.1016/0006-3223(94)91212-2
Tabuchi, A. (2008). Synaptic plasticity-regulated gene expression: a key event in the long-lasting changes of neuronal function. Biol. Pharm. Bull. 31, 327–335. doi:10.1248/bpb.31.327
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983. doi:10.1016/0092-8674(93)90585-E
Thoenen, H. (2000). Neurotrophins and activity-dependent plasticity. Prog. Brain Res. 128, 183–191. doi:10.1016/S0079-6123(00)28016-3
Trembath, M. K., Horton, Z. A., Tippett, L., Hogg, V., Collins, V. R., Churchyard, A., et al. (2010). A retrospective study of the impact of lifestyle on age at onset of Huntington disease. Mov. Disord. 25, 1444–1450. doi:10.1002/mds.23108
Urani, A., Chourbaji, S., and Gass, P. (2005). Mutant mouse models of depression: candidate genes and current mouse lines. Neurosci. Biobehav. Rev. 29, 805–828. doi:10.1016/j.neubiorev.2005.03.020
Vaidya, V. A., and Duman, R. S. (2001). Depresssion – emerging insights from neurobiology. Br. Med. Bull. 57, 61–79. doi:10.1093/bmb/57.1.61
van Dellen, A., Blakemore, C., Deacon, R., York, D., and Hannan, A. J. (2000). Delaying the onset of Huntington’s in mice. Nature 404, 721–722. doi:10.1038/35008142
van Duijn, E., Selis, M. A., Giltay, E. J., Zitman, F. G., Roos, R. A., van Pelt, H., et al. (2010). Hypothalamic-pituitary-adrenal axis functioning in Huntington’s disease mutation carriers compared with mutation-negative first-degree controls. Brain Res. Bull. 83, 232–237. doi:10.1016/j.brainresbull.2010.08.006
van der Burg, J. M., Bjorkqvist, M., and Brundin, P. (2009). Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 8, 765–774. doi:10.1016/S1474-4422(09)70178-4
van Duijn, E., Kingma, E. M., Timman, R., Zitman, F. G., Tibben, A., Roos, R. A., et al. (2008). Cross-sectional study on prevalences of psychiatric disorders in mutation carriers of Huntington’s disease compared with mutation-negative first-degree relatives. J. Clin. Psychiatry 69, 1804–1810. doi:10.4088/JCP.v69n1116
van Duijn, E., Kingma, E. M., and van der Mast, R. C. (2007). Psychopathology in verified Huntington’s disease gene carriers. J. Neuropsychiatry Clin. Neurosci. 19, 441–448. doi:10.1176/appi.neuropsych.19.4.441
van Praag, H. M., and Plutchik, R. (1984). Depression type and depression severity in relation to risk of violent suicide attempt. Psychiatry Res. 12, 333–338. doi:10.1016/0165-1781(84)90049-0
Vassos, E., Panas, M., Kladi, A., and Vassilopoulos, D. (2008). Effect of CAG repeat length on psychiatric disorders in Huntington’s disease. J. Psychiatr. Res. 42, 544–549. doi:10.1016/j.jpsychires.2007.05.008
Venna, V. R., Deplanque, D., Allet, C., Belarbi, K., Hamdane, M., and Bordet, R. (2009). PUFA induce antidepressant-like effects in parallel to structural and molecular changes in the hippocampus. Psychoneuroendocrinology 34, 199–211. doi:10.1016/j.psyneuen.2008.08.025
Waeber, C., and Palacios, J. M. (1989). Serotonin-1 receptor binding sites in the human basal ganglia are decreased in Huntington’s chorea but not in Parkinson’s disease: a quantitative in vitro autoradiography study. Neuroscience 32, 337–347. doi:10.1016/0306-4522(89)90082-1
Walter, U., Prudente-Morrissey, L., Herpertz, S. C., Benecke, R., and Hoeppner, J. (2007). Relationship of brainstem raphe echogenicity and clinical findings in depressive states. Psychiatry Res. 155, 67–73. doi:10.1016/j.pscychresns.2006.12.001
Wang, H. T., Han, F., and Shi, Y. X. (2009). Activity of the 5-HT1A receptor is involved in the alteration of glucocorticoid receptor in hippocampus and corticotropin-releasing factor in hypothalamus in SPS rats. Int. J. Mol. Med. 24, 227–231.
Weigell-Weber, M., Schmid, W., and Spiegel, R. (1996). Psychiatric symptoms and CAG expansion in Huntington’s disease. Am. J. Med. Genet. 67, 53–57. doi:10.1002/(SICI)1096-8628(19960216)67:1<53::AIDAJMG9>3.0.CO;2-T
Wester, P., Bergstrom, U., Eriksson, A., Gezelius, C., Hardy, J., and Winblad, B. (1990). Ventricular cerebrospinal fluid monoamine transmitter and metabolite concentrations reflect human brain neurochemistry in autopsy cases. J. Neurochem. 54, 1148–1156. doi:10.1111/j.1471-4159.1990.tb01942.x
Wong, E. Y., and Herbert, J. (2006). Raised circulating corticosterone inhibits neuronal differentiation of progenitor cells in the adult hippocampus. Neuroscience 137, 83–92. doi:10.1016/j.neuroscience.2005.08.073
Woolley, C. S., Gould, E., and McEwen, B. S. (1990). Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 531, 225–231. doi:10.1016/0006-8993(90)90778-A
Yohrling, I. G., Jiang, G. C., DeJohn, M. M., Robertson, D. J., Vrana, K. E., and Cha, J. H. (2002). Inhibition of tryptophan hydroxylase activity and decreased 5-HT1A receptor binding in a mouse model of Huntington’s disease. J. Neurochem. 82, 1416–1423. doi:10.1046/j.1471-4159.2002.01084.x
Yoshida, T., Ishikawa, M., Niitsu, T., Nakazato, M., Watanabe, H., Shiraishi, T., et al. (2012). Decreased serum levels of mature brain-derived neurotrophic factor (BDNF), but not its precursor proBDNF, in patients with major depressive disorder. PLoS ONE 7:e42676. doi:10.1371/journal.pone.0042676
Yoshimura, R., Umene-Nakano, W., Hoshuyama, T., Ikenouchi-Sugita, A., Hori, H., Katsuki, A., et al. (2010). Plasma levels of brain-derived neurotrophic factor and interleukin-6 in patients with dysthymic disorder: comparison with age- and sex-matched major depressed patients and healthy controls. Hum. Psychopharmacol. 25, 566–569. doi:10.1002/hup.1155
Zajac, M. S., Pang, T.Y., Wong, N., Weinrich, B., Leang, L.S., Craig, J. M., et al. (2010). Wheel running and environmental enrichment differentially modify exon-specific BDNF expression in the hippocampus of wild-type and pre-motor symptomatic male and female Huntington’s disease mice. Hippocampus 20, 621–636. doi:10.1002/hipo.20658
Zielonka, D., Marinus, J., Roos, R. A., De Michele, G., Di Donato, S., Putter, H., et al. (2012). The influence of gender on phenotype and disease progression in patients with Huntington’s disease. Parkinsonism Relat. Disord. 19, 192–197. doi:10.1016/j.parkreldis.2012.09.012
Zigova, T., Pencea, V., Wiegand, S. J., and Luskin, M. B. (1998). Intraventricular administration of BDNF increases the number of newly generated neurons in the adult olfactory bulb. Mol. Cell. Neurosci. 11, 234–245. doi:10.1006/mcne.1998.0684
Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., et al. (2001). Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293, 493–498. doi:10.1126/science.1059581
Keywords: Huntington’s disease, neurodegeneration, depression, psychiatric disorders, serotonin, BDNF, stress, polyglutamine disease
Citation: Du X, Pang TYC and Hannan AJ (2013) A tale of two maladies? Pathogenesis of depression with and without the Huntington’s disease gene mutation. Front. Neurol. 4:81. doi: 10.3389/fneur.2013.00081
Received: 18 April 2013; Accepted: 11 June 2013;
Published online: 09 July 2013.
Edited by:
Thomas M. Durcan, McGill University, CanadaCopyright: © 2013 Du, Pang and Hannan. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Xin Du and Anthony J. Hannan, Florey Institute of Neuroscience and Mental Health, Kenneth Myer Building, At Genetics Lane off Royal Parade, University of Melbourne, VIC 3010, Australia e-mail: xin.du@florey.edu.au; anthony.hannan@florey.edu.au