Synaptic dysfunction in ALS and FTD: anatomical and molecular changes provide insights into mechanisms of disease
 Pauline A. Gelon
Pauline A. Gelon Paul A. Dutchak
Paul A. Dutchak Chantelle F. Sephton
Chantelle F. Sephton- Department of Psychiatry and Neuroscience, CERVO Brain Research Centre, Laval University, Quebec City, QC, Canada
Synaptic loss is a pathological feature of all neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). ALS is a disease of the cortical and spinal motor neurons resulting in fatal paralysis due to denervation of muscles. FTD is a form of dementia that primarily affects brain regions controlling cognition, language and behavior. Once classified as two distinct diseases, ALS and FTD are now considered as part of a common disease spectrum based on overlapping clinical, pathological and genetic evidence. At the cellular level, aggregation of common proteins and overlapping gene susceptibilities are shared in both ALS and FTD. Despite the convergence of these two fields of research, the underlying disease mechanisms remain elusive. However, recent discovers from ALS and FTD patient studies and models of ALS/FTD strongly suggests that synaptic dysfunction is an early event in the disease process and a unifying hallmark of these diseases. This review provides a summary of the reported anatomical and cellular changes that occur in cortical and spinal motor neurons in ALS and FTD tissues and models of disease. We also highlight studies that identify changes in the proteome and transcriptome of ALS and FTD models and provide a conceptual overview of the processes that contribute to synaptic dysfunction in these diseases. Due to space limitations and the vast number of publications in the ALS and FTD fields, many articles have not been discussed in this review. As such, this review focuses on the three most common shared mutations in ALS and FTD, the hexanucleuotide repeat expansion within intron 1 of chromosome 9 open reading frame 72 (C9ORF72), transactive response DNA binding protein 43 (TARDBP or TDP-43) and fused in sarcoma (FUS), with the intention of highlighting common pathways that promote synaptic dysfunction in the ALS-FTD disease spectrum.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal disease that stems from the degeneration of both upper cortical and lower spinal motor neurons resulting in dysfunction of voluntary muscles. In 1874, French neurologist Jean-Martin Charcot named the disease based on post-mortem observations that muscles lacked nourishment or were “atrophic” and the “lateral” regions of the spinal cord were hardened or had “sclerosis”. Our understanding of ALS has evolved from the simplistic view that the degeneration of motor neurons cause the disease, to the recognition that ALS is a multisystem neurodegenerative disease involving other neuron populations and regions in the brain and spinal cord, glial and immune cells as well as peripheral tissues, which contribute to the disease (Tortarolo et al., 2017; Grossman, 2019). ALS is a heterogenous disease regarding age of onset, site of onset, disease progression, as well as pathological characteristics. ALS patients are clinically classified as having classical or atypical ALS, based on the level of motor neuron involvement and pattern of onset (Grad et al., 2017). From the time an individual first experiences symptoms of ALS, their mean survival time is 3–5 years (Brown and Al-Chalabi, 2017). The global number of ALS cases per year is approximately 2.1–4.4 per 100,000 population (Chio et al., 2013; Xu et al., 2020) and the cumulative lifetime risk in the United States and Europe is one in 400 (Brown and Al-Chalabi, 2017). Sporadic ALS represents 90% of cases, with ~10% of ALS cases having a family history of the disease. In most cases, the mean age of onset for those who develop ALS is between the 55 and 75 years, with an average age of diagnosis at 55 years (Ingre et al., 2015). However, juvenile (<25 years old) and “young-onset” ALS cases (<45 years old) represent ~1% and 10% of all cases, respectively (Turner et al., 2012). In sporadic ALS the ratio of affected males to affected females is 2:1, and in familial ALS the ratio is approximately 1:1 (Brown and Al-Chalabi, 2017). Currently, there are no therapies or effective treatments for ALS, thus the need for a deeper understanding of this disease is greatly needed to develop targeted therapeutic strategies.
Frontotemporal dementia (FTD) was first described in 1892 by Arnold Pick as a form of dementia characterized by a progressive neuronal loss, primarily across the frontal and temporal lobes, which leads to changes in executive functions, personality, abnormal behaviors and language impairments (Kurz et al., 2014; Mulkey, 2019). Like ALS, FTD is also recognized as a heterogeneous clinical, pathological and genetic disorder and is broadly referred to as frontotemporal lobar degeneration (FTLD) due to the region-specific neuronal atrophy that occurs in the brain. Based on a patient’s clinical presentation, FTD is divided into three main subtypes that include behavioral variant (bvFTD), semantic dementia (svPPA), and progressive non-fluent aphasia (nfvPPA) (Olney et al., 2017; Devenney et al., 2019). From the time symptoms of FTD first appear, the mean survival time is 3 and 17 years (Olney et al., 2017; Devenney et al., 2019). The annual incidence of FTD is approximately 1.6–4.1 cases per 100,000 population per year (Onyike and Diehl-Schmid, 2013; Coyle-Gilchrist et al., 2016). Over half of FTD cases are sporadic, but ~40% have family histories of dementia, cognitive or motor disorder (Olney et al., 2017; Devenney et al., 2019). FTD is the most common type of dementia in individuals under the age of 65 with a mean age of onset at 52.8 years with the affected ratio of males to affected females being 14:3 (Ratnavalli et al., 2002). Similar to ALS, there are no effective treatments for FTD.
ALS and FTD are part of a common disease spectrum and share clinical, pathological and genetic features. In addition to well-characterized motor defects of ALS, ~50% of ALS patients have behavioral changes and cognitive abnormalities that are linked with degeneration of the frontal and temporal lobes, and are classified as ALS-FTD (Lomen-Hoerth et al., 2002). Among FTD subtypes, ~40% of patients with bvFTD develop motor neuron dysfunction and meet the criteria for motor neuron disease and are classified as FTD-MND (Lomen-Hoerth et al., 2002; Lipton et al., 2004). Moreover, ~50% of patients with MND develop varying forms of cognitive impairment (Lomen-Hoerth et al., 2003).
Landmark discoveries showing the RNA-binding protein, transactive response DNA binding protein 43 (TDP-43 or TARDBP), as the main component of ubiquitinated inclusions in both familial and sporadic forms of ALS and FTD helped establish new models of protein-aggregate dependent neuronal dysfunction (Arai et al., 2006; Neumann et al., 2006). Since this time, TDP-43 aggregation is now considered to be a general pathological marker for ALS and FTD (Saberi et al., 2015), as well as Alzheimer’s disease (Josephs et al., 2015), Parkinson’s disease (Nakashima-Yasuda et al., 2007), traumatic brain injuries (Johnson et al., 2011) and within the normal aging brain (Uchino et al., 2015). Nonetheless, the seminal discovery that TDP-43 mislocalization and aggregation occurs in both ALS and FTD brought forth novel research aimed at uncovering the overlapping mechanisms by which these diseases occur. Subsequently, fused in sarcoma (FUS) accumulation was identified in neurons and glia in familial ALS cases with gene mutations in FUS (Kwiatkowski et al., 2009; Vance et al., 2009). Following these studies, FUS aggregates were also identified in affected brain regions of patients with sporadic forms of FTD in the absence of FUS mutations (Neumann et al., 2009). In 2011, toxic accumulation of dipeptides repeat (DPR), which can be translated from a hexanucleotide repeat expansion of the GGGGCC (G4C2) nucleotides within intron 1 of chromosome 9 open reading frame 72 (C9ORF72), were identified in familial ALS and FTD cases with these gene expansions (DeJesus-Hernandez et al., 2011; Renton et al., 2011).
To date, there are at least 17 common genes that have been linked to the susceptibility of familial forms of ALS and FTD (Table 1) (Al-Saif et al., 2011; Belzil et al., 2013; Liscic et al., 2020; Anoar et al., 2021). Hexanucleotide repeat expansions within C9ORF72, mutations in TARDBP (or TDP-43) and FUS represent the majority of autosomal dominant mutations in familial ALS (Liscic et al., 2020; Anoar et al., 2021). Whereas C9ORF72, progranulin (GRN), and microtubule-associated protein tau (MAPT) represent the majority of autosomal dominant mutations found in familial FTD (Liscic et al., 2020; Anoar et al., 2021). Based on the most frequent overlapping genes and pathological protein deposits of ALS and FTLD, these diseases can be further classified as being ALS-TDP, ALS-FUS, ALS-C9 or FTLD-TDP, FTLD-FUS, FTLD-C9. While it is unlikely that a single gene is solely responsible for causing sporadic ALS and FTD, the data suggest that these pathologies are the result of alterations of multiple genes that are found in a common or related pathways, which collectively contribute to the neurodegenerative outcome (Liscic et al., 2020). Indeed, the common features of ALS and FTD reinforce this concept, but more work is needed in order to identify the primary mechanism(s) of disease.
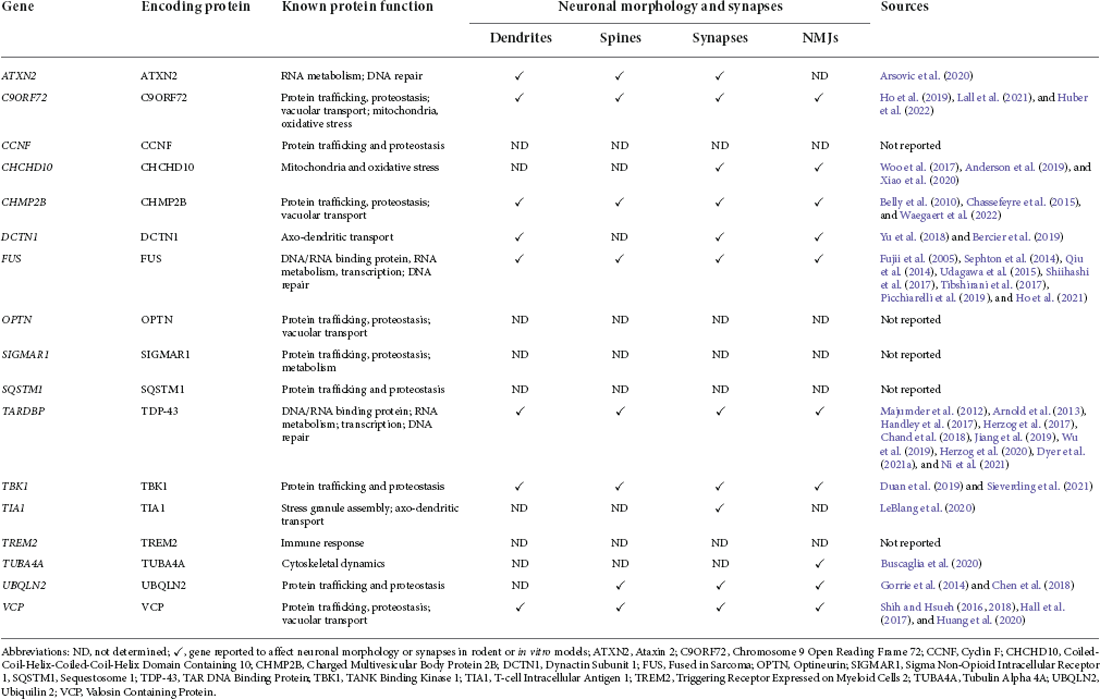
Table 1. ALS and FTD disease-associated gene mutations, their functions, and reported effects on neuromorphology and synapses.
While environmental and genetic risk factors are shown to be implicated in causing ALS and FTD, there is no consensus as to how these diseases originate (Al-Chalabi and Hardiman, 2013; Killin et al., 2016; Liscic et al., 2020). The identification of ALS and FTD-linked gene mutations in particular, have led to a multitude of studies that suggest mitochondrial function, cytoskeletal dynamics, protein homeostasis and axonal trafficking are implicated in causing these diseases (Abramzon et al., 2020; Castellanos-Montiel et al., 2020). Our understanding of the functional consequences caused by these defects are still ongoing, however, recent discovers made using in vivo and in vitro models of ALS/FTD strongly suggests that early synaptic dysfunction is a unifying hallmark of in these diseases. While cognitive impairments in ALS and motor defects in FTD have been reported in several early studies, the formal recognition of the convergent features of these diseases has only occurred recently, making the consolidation of the literature on ALS, ALS-FTD, and FTD-MND an inherent challenge. Here we provide a summary of the reported anatomical changes that occur in both cortical and spinal neurons in ALS, ALS-FTD, and FTD-MND. Additionally, we provide a summary of in vitro and in vivo models of ALS/FTD that report similar anatomical changes in motor neurons and their synapses. We describe recent work that identifies changes in the proteome and transcriptome in ALS/FTD models and provide a conceptual overview of the processes contributing to synaptic dysfunction in ALS and FTD. We also highlight recent work on C9ORF72, TDP-43, and FUS and their involvement in disrupting synaptic homeostasis.
Neuropathological Features of ALS and FTD
Neurons are highly complex cells with elaborate dendritic branches that contain numerous spines and an axon that can extend long distances to form synapses at its terminal bouton. Motor neurons form a complex network of circuits that enable voluntary and involuntary movements through innervation of muscles. Motor neurons are grouped into upper motor neurons (UMNs), also known as Betz cells, located in the motor cortex, and lower motor neurons (LMNs), located in the brainstem and spinal cord (Brown and Al-Chalabi, 2017). UMNs are responsible for integrating excitatory and inhibitory inputs from the cortex and translating them into signals that will initiate or inhibit voluntary movement. They form local synaptic connections within the cortex, as well as distal connections in the brainstem and spinal cord. The majority of UMN axons cross over the contralateral side in the brainstem and travel down the lateral corticospinal tract to form synapses directly with interneurons and lower motor neurons (LMNs), located in the anterior horn of the spinal cord. UMNs use the neurotransmitter, glutamate, to relay their signals and integrate excitatory and inhibitory inputs from the cortex to their synaptic targets. LMNs directly or indirectly innervate effector muscles at the neuromuscular junction (NMJ) via synaptic release of acetylcholine. Together, LMNs, muscle fibers and NMJs form a motor unit. LMNs include visceral motor neurons that contribute to both the sympathetic and parasympathetic functions of the autonomic nervous system and somatic motor neurons. Somatic motor neurons project their axons to skeletal muscles and are responsible for movement. They are further divided into alpha, beta and gamma motor neurons according to the muscle fiber they innervate within a specific muscle target (Zayia and Tadi, 2022).
The extent to which UMNs and LMNs are affected in ALS and FTD-MND can vary between individuals but the extent to which they are affected correlates with the type of motor dysfunction presented. When UMNs fail, there are signs of muscle stiffness and spasticity. When LMNs fail, muscle twitching or fasciculations occur as they degenerate and the muscles that they innervate atrophy (Brown and Al-Chalabi, 2017). In ALS, there is selective vulnerability of the alpha motor neurons. Among the first to degenerate are the fast fatiguing (FF) motor units, which are the largest motor units and innervate the fast-contracting and fatigable muscle fibers (FF type). This is followed by degeneration of the fast fatigue-resistant motor units, which innervate the fast-contracting and fatigue-resistant muscle fibers (FR type). Interestingly, the most resistant and least affected are the slow (S) motor units, which mainly innervate the slow contracting and fatigue-resistant fibers (S type) (Ragagnin et al., 2019). Collectively, it is the degeneration of the corticospinal tract and its motor units that leads to NMJ denervation, muscle atrophy, and loss of motor function. In classical ALS, which involves both UMNs and LMNs, facial muscles and extremities are mainly affected. Clinical subsets of ALS can be distinguished further by the anatomical location of disease onset. This includes bulbar onset where symptoms first appear in the muscles that control speech, mastication and swallowing due to onset of disease in motor neurons located in the brainstem. As well as limb or spinal onset, where symptoms initially present in the upper or lower limbs because of disease onset in UMNs and LMNs. Patients with bulbar onset have a much worse prognosis than those with spinal onset ALS, with an average survival time of <2 years following a diagnosis (Turner et al., 2010). Conversely, atypical forms of ALS, which involve either UMN or LMN, show spastic paraplegia, autoimmune disease, or demyelinating LMN disease, frequently with extended survival rates (Grad et al., 2017). Two other motor neuron diseases that should be mentioned are primary lateral sclerosis (PLS) with predominantly UMN involvement and comorbidities with FTD (Grace et al., 2011) and progressive muscular atrophy (PMA), which has predominant LMN involvement (Grad et al., 2017). Currently, these diseases are classified separately from ALS, however, it remains unclear if they are discrete disorders or variants of ALS and FTD-MND (Grad et al., 2017). In FTD-MND there is preferential degeneration of neurons in the frontal and temporal lobes, but there is also degeneration of UMNs and LMNs, which coincides with impaired motor function (Devenney et al., 2019). It is unclear why motor neurons are particularly vulnerable to degeneration in ALS and FTD-MND and it is equally unclear why some motor neurons are spared while other degenerate (Burrell et al., 2011; Nijssen et al., 2017).
The diagnosis of ALS, ALS-FTD, and FTD-MND is based on criteria that use clinical, electrophysiological and neuropathologic examination to assess motor and cognitive dysfunction. Diagnosing ALS is based on the El Escorial revised criteria (Brooks et al., 2000), to assess LMN and UMN degeneration and help rule out other diseases or processes involved in motor neuron dysfunction. Following the classification motor neuron disease, subsequent clinical criteria defines cognitive and behavioral dysfunctions that are used to diagnose ALS-FTD, based on criteria developed by Strong et al. (2009). These criteria require that an individual diagnosed with ALS meets at least two non-overlapping supportive diagnostic features from either the Neary and/or Hodge criteria for the diagnosis of FTD (Strong et al., 2009). The diagnosis of FTD using the Neary and Hodge criteria are defined by early decline in social and personal conduct, emotional blunting, loss of awareness and changes in verbal communication (Neary et al., 1998; Hodges and Miller, 2001). Following a diagnosis of FTD, clinical classification of FTD with motor neuron disease (FTD-MND) needs to meet the criteria of UMN and LMN dysfunction as defined by the El Escorial revised criteria (Geevasinga et al., 2016). However, due to the heterogeneity of symptoms experienced by individuals with ALS, ALS-FTD, and/or FTD-MND, it remains difficult to provide a definitive diagnosis of these diseases, especially in early disease stages when symptoms first appear.
There is growing evidence from ALS patients, FTD patients, and animal models that suggest synaptic dysfunction begins very early in the disease, before symptom onset and motor neuron death. Studies using electromyography (EMG), nerve conduction velocity (NCV), and transcranial magnetic stimulation (TMS) to examine the electrophysiological properties of LMNs show that ALS patients in the early stages of disease already have indications of corticospinal degeneration, loss of LMNs and altered excitability of surviving motor units (Dengler et al., 1990; Zanette et al., 2002; Vucic and Kiernan, 2006; Marchand-Pauvert et al., 2019). Comparable studies conducted in FTD-MND patients show similar abnormalities in central motor circuits (Alberici et al., 2008; Burrell et al., 2011; Coon et al., 2011) and can also be observed in FTD patients without clinical evidence of motor involvement (Alberici et al., 2008; Burrell et al., 2011; Coon et al., 2011). Investigation into the changes in corticospinal connectivity in ALS models indicate that excitability of motor neurons shifts from a hyperexcitable (Wainger et al., 2014) to a hypoexcitable state (Devlin et al., 2015; Naujock et al., 2016; Sommer et al., 2022) while NMJs are still functional (Martinez-Silva et al., 2018). The cause of changes in LMN excitability in ALS and FTD are under investigation, but may involve changes in synaptic inputs from UMNs (Pradhan and Bellingham, 2021). The idea that synaptic dysfunction occurs early in ALS, is further supported by post-mortem analysis of tissues from a 58-year-old ALS patient in the early stages of the disease who died unexpectedly (Fischer et al., 2004). This individual had muscle fibers that were grouped and angulated, which are consistent with acute and chronic denervation and reinnervation of the muscles by LMN synaptic terminals (Fischer et al., 2004), consistent with observations in pre-symptomatic ALS mouse models (Chand et al., 2018; Martineau et al., 2018). Importantly, there was little axon degeneration in the spinal cord, the corticospinal tract was intact with no degeneration of UMNs or LMNs and there was no activation of glial cells (Fischer et al., 2004). While degeneration of motor neurons is a defining pathological feature of ALS and FTD-MND, these findings strongly suggest that motor neuron loss and denervation of NMJs occurs late in the disease process.
There is evidence that alterations in dendritic branching and dendritic spines are an early pathological feature of ALS and FTD-MND. Indeed, dendritic length, complexity and branch thickness are indicators of a neuron’s health and connectivity (Kulkarni and Firestein, 2012; Kweon et al., 2017). Post-mortem Golgi staining and histological analysis of intact motor neurons from ALS and FTD-MND brain and spinal cord tissues show dendritic attrition and thinning of dendritic branches in the apical and basal dendrites of UMNs (Hammer et al., 1979; Horoupian et al., 1984; Ferrer et al., 1991; Genc et al., 2017) as well as LMNs of patients with motor defects (Kato et al., 1987). Consistent with the alterations in dendrites in ALS and ALS-FTD, synaptic integrity is also affected. In sporadic ALS, the pre-synaptic densities of UMNs are reported to be unchanged, however, the number of post-synaptic densities are significantly reduced (Genc et al., 2017; Henstridge et al., 2018). These findings are consistent with reports that pre-synaptic densities around the soma and proximal dendrites of the LMNs are significantly decreased in ALS spinal cord tissues (Sasaki and Maruyama, 1994). These observations support the hypothesis that ALS starts in the UMNs, descends to the LMNs and progresses to extra-motor brain regions (Braak et al., 2013; Brettschneider et al., 2013). In FTD and FTD-MND, synaptic loss, reduction in the number of spines (Liu et al., 1996; Ferrer, 1999; Lipton et al., 2001), and synaptic densities in the cortex have also been reported (Brun et al., 1995; Liu et al., 1996; Ferrer, 1999). Recent imaging techniques have been developed to study synaptic density in living FTD patients and support these histological findings. The use of [11C]UCB-J PET, a radio-ligand that selectively binds to synaptic vesicle protein 2A (SV2A), to assess the regional distribution of synaptic loss in patients with probable bvFTD and carriers of C9ORF72 hexanucleotide repeat expansions, showed that loss of synaptic densities occur in the frontotemporal region (Malpetti et al., 2021; Salmon et al., 2021). Importantly, this study showed that pre-symptomatic FTD-C9 patients had reduced synaptic density and that extensive synaptic loss was observed in patients with severe cognitive impairments (Malpetti et al., 2021). These findings are consistent with sporadic ALS patients that showed synaptic loss correlated with severity of cognitive impairments and not due to cortical atrophy (Henstridge et al., 2018). While dendritic spines of LMNs are not readily examined in ALS and FTD-MND patient studies, changes in LMN spines are found to occur in the pre-symptomatic stages of disease an ALS mouse model (Fogarty et al., 2020, 2021). Thus, characterization of LMN spines in ALS and FTD-MND may also be relevant to our understanding of corticospinal connectivity and disease. Collectively, these observations further support the view that dendritic attrition and synaptopathy are early markers of ALS and FTD and that synaptic defects occur in the corticospinal tracts prior to the clinical presentation of motor dysfunction, inflammation and motor neuron death (Figure 1).
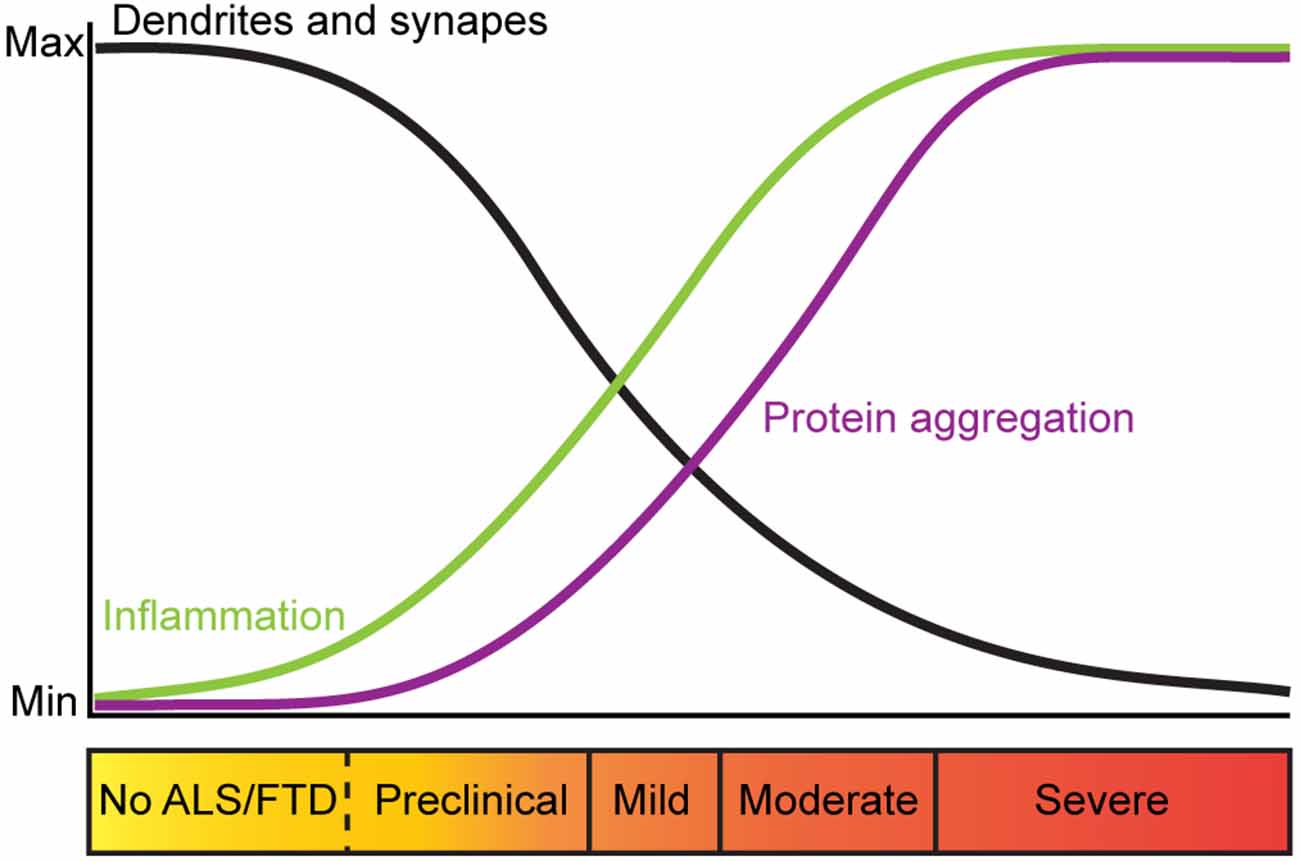
Figure 1. Trajectory of pathological changes and corresponding clinical presentation in ALS and FTD. Trajectory of changes in dendritic branches and synapses in ALS and FTD (black), inflammation (green) and protein aggregation (purple) relative to clinical symptoms of ALS and FTD. In ALS and FTD, dendritic branches and synapses are lost prior to clinical onset of disease, suggesting that perturbed neuronal morphology and synaptic maintenance are mechanisms that trigger synaptic loss and neurodegeneration associated with these diseases.
Genetic Links to Synaptic Dysfunction
Synaptopathy can occur due to changes including altered Ca2+ levels at synapses, altered protein expression within the synapse, and dysfunctional neurotransmitter cycling (Wishart et al., 2006; Lepeta et al., 2016; Fogarty, 2019). Concomitant with synaptopathy in ALS and FTD are abnormalities in the cytoskeleton, mitochondrial dysfunction and accumulation of misfolded proteins (Wishart et al., 2006; Sasaki and Iwata, 2007; Lepeta et al., 2016; Fogarty, 2019; Gao et al., 2019; Castellanos-Montiel et al., 2020). However, it is unclear how changes in these cellular processes trigger the cascade of events that lead to synaptic loss in ALS and FTD or whether changes at the synapse promote wide-spread neuronal dysfunction.
Motor neurons have complex dendritic branches and long axonal projections, which rely on the integrity of cytoskeletal proteins for maintaining structural stability and transport of mitochondria, proteins and RNA within a neuron (Konietzny et al., 2017; Theunissen et al., 2021). As such, cytoskeletal changes within motor neurons make them vulnerable to synaptic dysfunction and neurodegeneration. Several gene mutations that encode proteins implicated in cytoskeletal dynamics and axonal transport have been identified in familial ALS or FTD, including: dynactin subunit 1 (DCTN1) and tubulin alpha 4a (TUBA4A) (familial ALS and FTD) and kinesin family member 5A (KIF5A), myelin associated oligodendrocyte basic protein (MOBP) and profilin 1 (PFN1) (familial ALS) and MAPT (familial FTD) (Brown and Al-Chalabi, 2017; Olney et al., 2017; Devenney et al., 2019; Liscic et al., 2020; Theunissen et al., 2021). Genetic changes of these proteins reviewed in detail elsewhere, affect cytoskeleton dynamics, impair dendrite and axonal integrity, which would affect transport of mitochondria, proteins and RNA to the synapse (Castellanos-Montiel et al., 2020).
Mitochondria are critical for maintaining synaptic integrity. Mitochondria are situated within dendrites, at the base of dendritic spines and in the post-synaptic compartments to provide energy in the form of ATP, synthesis of glutamate, and modulate Ca2+ levels to regulate synaptic transmission (Waagepetersen et al., 2003; Li et al., 2004; Devine and Kittler, 2018; Giandomenico et al., 2022). The mitochondria produce more than 90% of neuronal ATP (Harris et al., 2012), which is required in many steps of the synaptic vesicle cycle (Chua et al., 2010; Sudhof and Rizo, 2011) and protein synthesis (Pontes et al., 2015). Failure of proper mitochondria trafficking, function and positioning in dendrites and synapses have been observed in ALS and FTD (Okamoto et al., 1990; Sasaki and Iwata, 2007) and may contribute to early synaptic loss in disease (Gao et al., 2019). The importance of proper mitochondrial function in synaptic regulation is supported by mutations in genes linked to mitochondrial function including: superoxide dismutase 1 (SOD1) for familial ALS and coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10), C9ORF72, FUS, and TARDBP for both familial ALS and FTD (Chaussenot et al., 2014; Williams et al., 2016; Lau et al., 2018; Smith et al., 2019; Liscic et al., 2020).
Due to the dynamic nature of the local synaptic environment, tight regulation of protein homeostasis can also impact several aspects of neuronal morphology and synaptic strength (Lottes and Cox, 2020; Giandomenico et al., 2022). As such, dendrites and synapses require some proteins to be transported from the cell body, however, they cannot rely on transport alone to maintain proteostasis (Lottes and Cox, 2020; Giandomenico et al., 2022). Instead, dendritic protein quality control systems, including free ribosomes and dendritic endoplasmic reticulum (ER) tubules, facilitate local translation (Lottes and Cox, 2020). Protein chaperones, endosomes, lysosomes, autophagosome and proteosomes that maintain proteostasis are also situation locally in dendrites (Lottes and Cox, 2020). Some proteins produced in the dendrites are then trafficked to activated synapses (Lottes and Cox, 2020). Within pre- and post-synaptic compartments several key elements such as free ribosomes and protein chaperones contribute to local proteostasis (Gorenberg and Chandra, 2017; Giandomenico et al., 2022). Altered proteostasis and accumulation of misfolded proteins caused by defects in protein synthesis and protein clearance pathways provides important insights into defects are underlying in ALS and FTD. Indeed, mutations in several genes linked with proteostasis and autophagy including charged multivesicular body protein 2B (CHMP2B), sequestosome 1 (SQSTM1/p62), ubiquilin-2 (UBQLN2), and valosin containing protein (VCP) are linked with familial ALS and FTD (Liscic et al., 2020). Integral to protein synthesis is trafficking and local translation of mRNA, which is another process that is altered in ALS and FTD and shown to essential in the maintenance of neuronal morphology and synapses. This is underscored by identified ALS and FTD mutations in genes that encode proteins involved in RNA metabolism including FUS, TARDBP, ataxin 2 (ATXN2), and T-cell intracellular antigen 1 (TIA1) (Ross et al., 2011; Takada, 2015; Brown and Al-Chalabi, 2017; Hofmann et al., 2019; Rubino et al., 2019). Based on these observations, there are strong indications that diverse mechanisms can contribute to synaptic dysfunction and biological distinctions of ALS and FTD (Figure 2).
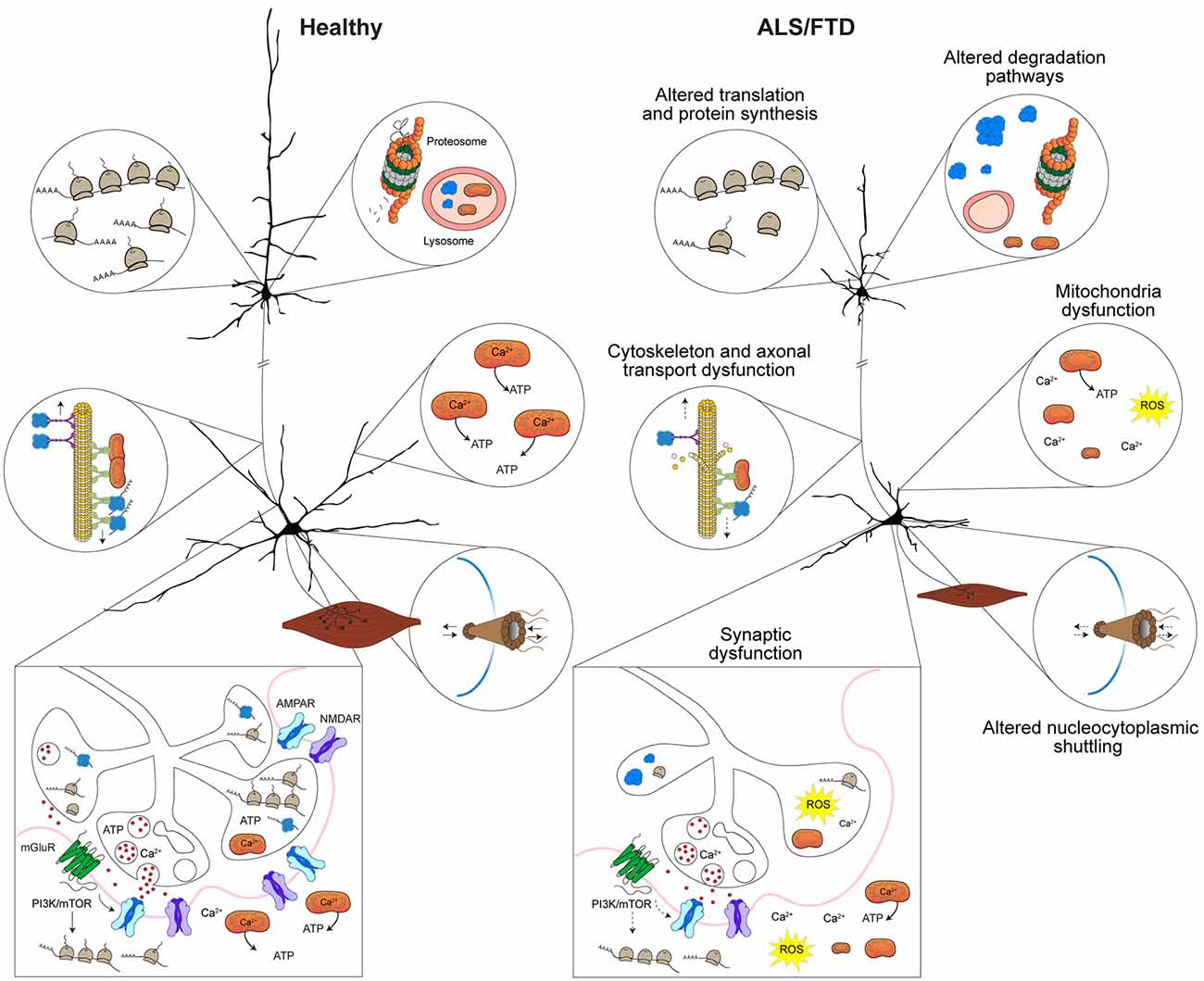
Figure 2. Mechanisms implicated in synaptic dysfunction in ALS and FTD. Altered proteostasis in ALS and FTD caused by misregulation of translation and protein synthesis affects several cellular processes including those implicated in neuronal morphology and synaptic homeostasis. Protein degradation pathways that involve the lysosome, autophagy, mitophagy, and the proteosome are dysfunctional in ALS and FTD result in toxic protein accumulation and lead to defects in neuronal function and synaptic communication. Cytoskeletal disruption or disarrangement that occur in ALS and FTD lead to structural instability of dendrites, axons and synapses and affect axonal transport of RNA granules, ribosomal subunits and organelles (i.e., mitochondria) to dendrites and synapse. In ALS and FTD, altered nucleocytoplasmic trafficking of proteins and altered physical properties of proteins (i.e., stress granule dynamics or liquid-liquid phase transitions) affect protein interactions and impact mRNA transport and local translation in axons, dendrites and synapses. Depletion or defective mitochondria within dendrites and at synapses that is observed in ALS and FTD affects Ca2+ levels and reduce ATP levels, which affects several processes including synaptic vesicle cycling and release, synaptic transmission, and local protein synthesis. Depicted upper and lower motor neurons were modified with permission from Sephton et al. (2014).
ALS/FTD Models of Disease
The identification of familial mutations linked with ALS and FTD have led to the development of in vivo and in vitro models of disease. These models have enabled more detailed biochemical investigation concerning how and when neuronal morphology and synaptic dysfunction occurs in relation to behavioral and pathological phenotypes. In this section, we discuss the known biological and pathological functions of C9ORF72, TDP-43, and FUS, which represent three major genes associated with ALS and FTD. Additionally, we discuss proteomic and transcriptomic findings that provide insight into mechanisms of synaptic dysfunction that occur in ALS and FTD.
C9ORF72: biological and pathological overview
The C9ORF72 locus is located on the reverse strand of chromosome locus 9p21.2. The gene includes two non-coding exons (1a and 1b) and 10 coding exons (from 2 to 11) that give rise to three coding variants. Alternative splicing of these RNA variants results in the production of two different protein isoforms: a 222-amino acid (aa) isoform (C9-short of 24 kDa) encoded by variant 1 and a 481-aa isoform (C9-long of 54 kDa) that is encoded by variant 2 and 3, which differ in their inclusion of the non-coding exon 1b or 1a, respectively (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012). The C9ORF72 human gene is highly conserved in primates and across different species, suggesting that C9ORF72 proteins have fundamental biological functions (Smeyers et al., 2021). C9ORF72 is ubiquitously expressed in all brain regions and the spinal cord (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Rizzu et al., 2016). In mice, the protein is localized in different cellular compartments during development and remains diffuse in the nucleus and soma in differentiated cells (Atkinson et al., 2015). The C9ORF72 protein can be found in biochemically purified synaptic fractions of brain homogenates (Atkinson et al., 2015) mainly in the pre-synaptic compartments (Frick et al., 2018), but also the post-synaptic compartment (Xiao et al., 2019). Analysis of C9ORF72 structure and its protein binding partners indicate that C9ORF72 acts as a guanine nucleotide exchange factor (GEF) for both Rab- and Rho-GTPases. It forms a complex with SMCR8 and WDR41 (Sellier et al., 2016; Sullivan et al., 2016) to act as a GDP/GTP exchange factor for RAB3, RAB8a, and RAB39b (Sellier et al., 2016; Sullivan et al., 2016; Frick et al., 2018), suggesting it has functions in membrane trafficking, synaptic transmission, cytoskeleton modulation, and autophagy. C9ORF72 also regulates actin dynamics (Sivadasan et al., 2016) and stress granules formation (Maharjan et al., 2017).
To date, the hexanucleotide repeat G4C2 in intron 1 of C9ORF72 is the most frequent autosomal dominant mutation causing ALS and FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). Variation in length and expression levels of G4C2 repeats play a significant role in disease manifestation. For instance, the pathogenic length of >30 hexanucleotide repeats is found in ~10% of all ALS patients (Fratta et al., 2015). The characteristic pathological features in both ALS-C9 and FTLD-C9 cases is region-specific neuronal loss, gliosis, as well as accumulation of expanded transcripts in nuclear RNA foci and cytoplasmic TDP-43 aggregates (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Liu et al., 2019). Distinct pathologic mechanisms have been hypothesized for G4C2 expansion, including: (1) a loss-of-function due to haploinsufficiency of C9ORF72 protein expression; (2) a gain-of-function of the repeat RNA aggregates or RNA foci; and (3) aggregation of dipeptide repeats (DPRs), which are transcribed from G4C2 RNA repeats by repeat-associated non-ATG-initiated (RAN) translation (Ash et al., 2013; Mori et al., 2013; Mizielinska and Isaacs, 2014). Several metabolic, proteostasis and neuronal processes are known to be impaired in C9ORF72-mediated pathogenesis, including autophagy, lysosomal function, ubiquitin-proteasome system (UPS), unfolded protein response (UPR) and axonal transport (Alhindi et al., 2021; Nishimura and Arias, 2021; Smeyers et al., 2021), all of which are shown to be critical for cellular health, but also for the organization, structure, strength and function of the synapse.
C9ORF72: synaptic dysfunction in ALS/FTD models of disease
While some mouse models of C9orf72 ALS/FTD do not show any major phenotypes associated with the human disease (Batra and Lee, 2017), others display axon degeneration, denervation of NMJs and loss of motor neurons that correspond with changes in cognitive and motor function (Liu et al., 2016; Batra and Lee, 2017; Herranz-Martin et al., 2017; Riemslagh et al., 2021). Notably, global knockout mouse models of C9ORF72 do not develop motor deficits (Koppers et al., 2015; Atanasio et al., 2016; Sudria-Lopez et al., 2016; Dong et al., 2021), unlike models expressing G4C2 repeats. One study that characterized four unique mouse models of C9orf72 ALS/FTD, showed disease severity correlated with G4C2 repeat length and expression (Liu et al., 2016), consistent with findings in ALS/FTD-C9 patients. A single copy of C9-37 short repeats expressed in mice under control of a bacterial artificial chromosome (BAC) promoter caused no disease phenotype, whereas BAC-C9-36/29, which expressed short repeats with increased copies triggered mild disease progression (Liu et al., 2016). BAC-C9-500/32 and BAC-C9-500, with transgenes expressing copies containing a mix of ~500 and 32 repeats or ~500 repeats alone, showed a stronger decline in motor function, which corresponded with axonal degeneration, degeneration of UMN and LMN and denervation of NMJs (Liu et al., 2016). End-stage BAC-C9-500 and BAC-C9-500/32 mice that progressed more rapidly had extensive motor neuron loss in the lumbar spinal cord, along with antisense RNA foci and DPRs accumulation in ALS/FTD affected brain and spinal cord regions (Liu et al., 2016). These observations are consistent with another study that developed a mouse model overexpressing 102 G4C2 repeats mediated by adeno-associated virus (AAV) that found 102 G4C2 repeats generated DPR pathology, NMJ abnormalities, enhanced apoptosis of neuron populations, corresponding with deficits in gait and cognitive function (Herranz-Martin et al., 2017). Similarly, Thy1(GA)149-CFP transgenic mice with poly-GA inclusions within the brainstem and spinal cord had motor deficits in the absence of motor neuron loss (Schludi et al., 2017). Evidence points to DPR-mediated synaptic defects prior to cell death. Mobile GA aggregates found within neurites caused a reduction in SV2A (also known as SV2) and altered Ca2+ influx and synaptic vesicle release (Jensen et al., 2020). While the extent of the nucleotide expansion in C9ORF72 correlates with disease severity, further research into the mechanisms that contribute to C9ORF72 phenotypes will reveal novel therapeutic strategies.
The characterization of dendritic branches or spines have not been conducted in most C9orf72 ALS/FTD mouse models (Batra and Lee, 2017). However, in a microglia specific knockout mouse model of C9orf72, behavioral defects in these mice were attributed decreased dendritic branching in the motor cortex, which coincide with enhanced cortical synaptic pruning and decreased cortical synaptic proteins (Lall et al., 2021). In the same study, they found that C9orf72KO cortical neurons cultured in the absence of microglia did not have any changes in spine densities, however, in the presence of C9orf72KO microglia there was a reduction in synaptic densities and an increase in the number of pre-synaptic (vGlut1) and post-synaptic (PSD-95) markers within C9orf72KO microglia (Lall et al., 2021). These observations suggest that microglia have a role in dysfunctional synaptic pruning and synaptic loss observed in C9-ALS/FTD patients.
Findings from other in vitro models demonstrate a cell autonomous effect of C9orf72 on neuronal morphology and synapses. Primary hippocampal neurons from C9orf72KO mice have reduced dendritic arborization and decreased spine density (Ho et al., 2019). Furthermore, expression of the long isoform of human C9orf72, but not the short isoform, rescued the dendritic arborization phenotypes of C9orf72KO neurons through restoration of autophagy in these neurons (Ho et al., 2019). Overexpression of C9-RAN proteins in primary neurons resulted in RNA foci and DPR proteins as well as reduced dendritic branching (May et al., 2014; Zhang et al., 2014; Huber et al., 2022). The effects of poly-GA on these changes were attributed to impairments of the proteosome (May et al., 2014) and ER stress (Zhang et al., 2014). In addition to changes in dendritic branching, overexpression of 66 G4C2 mediated by AAV in primary hippocampal neurons resulted in fewer mature or mushroom-type spines, and increased numbers of stubby and thin spines (Huber et al., 2022). These morphological changes corresponded with hyperexcitation due to susceptibility of neurons to excitotoxicity (Huber et al., 2022), similar to reports from induced pluripotent stem cells (iPSCs)-derived motor neurons (Selvaraj et al., 2018; Shi et al., 2018). The hyperexcitation phenotype in neurons expressing C9ORF72 variants may be caused by the increased activity of N-methyl-D-aspartate (NMDA) or α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (Shi et al., 2018; Huber et al., 2022) and/or functional synaptic alterations due to impaired pre-synaptic vesicle dynamics (Perkins et al., 2021). In line with this, loss of C9orf72 in mouse brain lead to a post-synaptic upregulation of in AMPA receptor subunit, Gria1 (also known as GluR1 or GluA1) expression (Shi et al., 2018; Xiao et al., 2019) concomitant with a loss of Smrc8 expression and a decrease in Rab39b expression (Xiao et al., 2019). Changes in AMPA and NMDA receptor expression are also found in both patient-derived iPSCs and C9ORF72 patient post-mortem spinal cord tissues (Devlin et al., 2015; Selvaraj et al., 2018; Shi et al., 2018). These findings are consistent with C9ORF72 having both a loss and gain of function causing enhance neuronal sensitivity to excitotoxicity (Devlin et al., 2015; Selvaraj et al., 2018; Shi et al., 2018; Dong et al., 2021; Huber et al., 2022). Excitotoxicity of motor neurons due to over-stimulation of glutamate receptors was first proposed in the late 70s (Olney, 1978). Since then, experimental evidence has shown that major contributing factors to altered excitability and excitotoxicity of neurons in ALS/FTD are persistent activation of NMDA and AMPA receptors, due to reduced levels of astrocytic glutamate transporters and changes in AMPA and NMDA receptor composition and function, which lead to excess Na+ and Ca2+ influx and neuronal cell death (Van Den Bosch et al., 2006; King et al., 2016). Future studies will need to examine the mechanisms by which C9orf72 causes susceptibility of the neurons to glutamate-induced excitotoxicity, the susceptibility of different neuron populations and the interplay between different cell-types to better understand how this contributes to disease in ALS/FTLD-C9.
Mechanisms of synaptic dysfunction caused by C9ORF72
Comparative proteomic analysis of frontal cortical tissues from postmortem tissues of ALS, FTD, ALS-FTD, and ALS-C9 patients show these disease groups share a down-regulation of proteins associated with neurons and synapses and up-regulation of proteins associated with astrocytes and microglia (Umoh et al., 2018). Consistent with reports that C9ORF72 plays an important role in immune regulation (Burberry et al., 2016; O’Rourke et al., 2016; Sudria-Lopez et al., 2016), proteins associated with astrocytes and microglia were slightly enriched in ALS-C9 when compared to sporadic ALS (Umoh et al., 2018). Proteomic analysis of in vitro ALS/FTD-C9 models show that proteins involved in protein synthesis and protein degradation are predominantly affected (Hartmann et al., 2018; Lualdi et al., 2021). ALS-C9 patient-derived primary skin fibroblasts have altered expression of proteins involved in proteostasis including ribosomal proteins (RPL7 and RPL38), translation initiation and elongation factors (EIF3B, EIF4A1, EEF1A1, EEF1A2), members of the HSP90 family, proteasome 26S subunits and aminoacyl-tRNA synthetases (Lualdi et al., 2021). These findings are consistent with proteomic studies from primary mouse cortical neurons overexpressing poly-(PR)175, one of the five possible peptides produced via RAN translation, which show significant reduction in cytosolic ribosomal proteins that corresponded with a reduction in synaptic and axonal proteins (Hartmann et al., 2018). This study showed that poly-(GR)149, localized to the nucleolus and reduced ribosome levels and translation similar to poly-PR, suggesting that impaired ribosome biogenesis may cause changes to proteostasis and drive toxicity (Hartmann et al., 2018). In support of these findings, interactome analysis of poly-GR/PR revealed ribosomes and RNA-binding protein interactors (Hartmann et al., 2018), suggesting that sequestration of these factors by poly-GR/PR could somehow impair ribosome biogenesis and protein synthesis. The relevance of poly-GR/PR interactions with these proteins is not yet clear, however, ribosome biogenesis is critical for the production of functionally matured ribosomes that are important for dendritic growth and development (Slomnicki et al., 2016; Chau et al., 2018). These findings may be relevant for our understanding how C9ORF72 hexanucleotide repeat expansions can impact neuronal features. Other potentially relevant molecular pathways to neuronal morphology and synaptic function in ALS-C9 patient-derived primary skin fibroblasts include ER-phagosome crosstalk, vesicle trafficking, the Slit/Robo signaling pathway, glucose metabolism, mitochondrial transport and bioenergetics (Lualdi et al., 2021). Indeed, the role of C9ORF72 in mitochondria function has been previously discussed (Smith et al., 2019) and mitochondria defects and elevated levels of reactive oxygen species have been observed in C9ORF72 models (Dafinca et al., 2016; Onesto et al., 2016). However, the mechanism by which C9ORF72 variants cause these changes to occur and their impact on neuromorphology and synapses needs to be investigated further.
Consistent with proteomic discoveries, transcriptome profiles from C9ORF72 models show dysregulation of genes that are involved in vesicle transport pathways, endosomal trafficking, lysosomal function, proteostasis, as well as synaptic transmission and neural differentiation (Sareen et al., 2013; Prudencio et al., 2015; Dickson et al., 2019; Liu et al., 2020; Perkins et al., 2021; Szebenyi et al., 2021; Sommer et al., 2022). However, it is unclear if these changes are directly caused by C9ORF72 variants or by the secondary effects of other factors like TDP-43 nuclear depletion and protein aggregation (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Liu et al., 2019). To address the specific effects of C9ORF72, Liu et al. (2020) examined the transcriptome of cells without TDP-43 pathology from post-mortem human ALS/FTD-C9 brains. Their findings revealed that a hexanucleotide repeat expansion in C9ORF72 caused mild expression and splicing changes (Liu et al., 2020). The genes that showed expression differences were involved in synaptic vesicles fusion or formation, vesicle transport, endosomal trafficking, chaperone associated protein aggregation and DNA repair (Liu et al., 2020). The mechanism by which C9ORF72 causes changes in transcripts associated with these functions is not clear. One study examined mRNA stability in ALS-C9 fibroblasts and iPSCs and found that destabilization of ribosomal (RPL28, RPL38, RPS18) and mitochondrial transcripts (COX6B, COX6C-X1, COX5B, NDUFA1, NDUFA13) occurs, consistent with ALS/FTLD-C9 and sporadic ALS brain and spinal cord tissues (Tank et al., 2018). Pathways involving inflammation, focal adhesion and the cytoskeleton were significantly enriched among RNAs that had increased synthesis (Tank et al., 2018). The mechanism by which C9ORF72 destabilizes mRNA is not yet known, but may involve defects in nucleocytoplasmic transport of RNA or expanded C9ORF72 repeat sequestration of essential RBPs that regulate RNA stability (Ash et al., 2013; Mori et al., 2013; Mizielinska and Isaacs, 2014). Nevertheless, the impact of destabilizing ribosomal, mitochondria and other transcripts is likely to have a significant impact on a number of important cellular processes, including those involved in the maintenance of neuronal morphology and synaptic integrity.
TDP-43: biological and pathological overview
In humans, TARDBP is located at chromosomal locus 1p36.22 and is comprised of six exons, five of which encode a 43 kDa protein. The gene coding TDP-43 is highly conserved throughout evolution and is found in all higher eukaryotic species including Drosophila melanogaster, Xenopus laevis, and Caenorhabditis elegans (Wang et al., 2004; Ayala et al., 2005). TDP-43 is an RNA-binding protein that contains an N-terminal region, a nuclear localization signal (NLS), two RNA recognition motifs: RRM1 and RRM2, a nuclear export signal (NES), C-terminal low complexity domain composed of glutamine/asparagine-rich (Q/N) and glycine-rich regions (Dreyfuss et al., 1993; Ayala et al., 2008; King et al., 2012). TDP-43 is ubiquitously expressed and predominantly localized in the nucleus (Ayala et al., 2008). The protein is also found in cytoplasmic RNA granules, axons, dendrites as well as pre- and post-synapses (Majumder et al., 2012; Gopal et al., 2017; Khalfallah et al., 2018; Briese et al., 2020; Altman et al., 2021; Wong et al., 2021). Knockout of the TARDBP gene in mice is embryonic lethal and embryonic stem cells that do not express TDP-43 are cell-lethal (Sephton et al., 2010; Wu et al., 2010).
TDP-43 has roles in gene transcription and RNA metabolism, including pre-mRNA splicing, mRNA stability and mRNA transport, in addition to microRNA (miRNA) and long non-coding RNA (lncRNA) processing (Buratti and Baralle, 2010; Bjork et al., 2022). Using RNA immunoprecipitation assays nearly 30% of the transcriptome or more than 6,000 mRNA targets, have been identified to associate with TDP-43 (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011; Xiao et al., 2011). Further supporting the role of TDP-43 in regulating RNA metabolism, mass spectrometry studies of purified TDP-43 protein complexes show that it functionally associates with RNA splicing factors and proteins involved in RNA transport (Freibaum et al., 2010; Sephton et al., 2011). Analysis of TDP-43 and its interaction with proteins and DNA/RNA targets indicate that it is an important regulator of transcription and translation of genes involved in neuronal development and synaptic regulation (Sephton et al., 2012; Sephton and Yu, 2015; Alhindi et al., 2021; Bjork et al., 2022).
Cytoplasmic inclusion bodies that consist mainly of hyperphosphorylated and ubiquitinated TDP-43 are a common pathological feature observed in 95% of ALS and 50% of FTLD (Arai et al., 2006; Neumann et al., 2006; Chen-Plotkin et al., 2010). Following their discovery, missense mutations in the TARDBP gene were identified in both familial and sporadic cases of ALS (Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008). To date, over 50 TARDBP mutations from patients with sporadic and familial ALS have been identified (Lattante et al., 2013), accounting for ~5% of familial ALS and ~1% of sporadic cases (Chio et al., 2020). At least 9 TARDBP mutations have been identified in patients with FTD and FTD-MND (Borroni et al., 2009, 2010; Gitcho et al., 2009; Kovacs et al., 2009; Chio et al., 2010; Gelpi et al., 2014; Synofzik et al., 2014; Floris et al., 2015; Moreno et al., 2015; Caroppo et al., 2016). Among the TARDBP mutations identified, the A382T missense mutation is the most frequently identified in FTD patients (Chio et al., 2010; Synofzik et al., 2014; Floris et al., 2015; Caroppo et al., 2016).
In patients that have TARDBP mutations, TDP-43 aggregation is the most prominent histopathological feature (Chen-Plotkin et al., 2010). In ALS and FTD-MND, TDP-43 aggregates are found throughout affected regions of the cortical, brainstem and spinal cord, with the distribution of protein aggregates corresponding to the clinical features for each disorder (Geser et al., 2009). Many of the TDP-43 mutations are located within the glycine-rich C-terminal domain and promote aggregation of the protein (Johnson et al., 2009; Dewey et al., 2011). Proposed pathologic mechanisms of TARDBP mutations include altered physical properties related to stress granule formation, liquid-liquid phase transitions, and nucleocytoplasmic shuttling dynamics, which contribute to altered proteostasis, transcription, and translation that can impact a number of cellular functions (Loganathan et al., 2019; Bjork et al., 2022; Carey and Guo, 2022).
TDP-43: synaptic dysfunction in ALS/FTD models of disease
Several ALS/FTD mouse models of TDP-43 report that expression of ALS-linked mutations, cause brain atrophy, muscle denervation, motor neuron loss, and denervation of NMJs along with progressive motor impairments (McGoldrick et al., 2013; Picher-Martel et al., 2016; White et al., 2018; Ebstein et al., 2019; Huang et al., 2020; Alhindi et al., 2021). Mice overexpressing TDP-43Q331K or wild-type human TDP-43 (TDP-43WT), in the brain and spinal cord under control of the murine prion promoter (Prp), showed dose and age-dependent motor defects caused by mutant TDP-43, which corresponded with progressive motor axon degeneration, motor neuron death and denervation of NMJs (Arnold et al., 2013). Importantly, this study showed that these changes occurred in the absence of TDP-43 insoluble aggregates and nuclear loss of TDP-43 (Arnold et al., 2013). Further characterization of the TDP-43Q331K mice showed denervation of NMJs and impaired neurotransmission preceded defects in motor function and motor neuron loss (Chand et al., 2018). These findings corresponded with altered innervation of NMJs as well as the distribution of synaptic vesicle-related proteins at NMJs (Chand et al., 2018), indicative of ongoing synaptic remodeling. The observed phenotypes at the NMJ were found to be the result of defects in fusion and release of synaptic vesicles within the motor nerve terminal (Chand et al., 2018), consistent with the role of TDP-43 in stabilizing mRNAs required for synaptic vesicle release (Keihani et al., 2019).
Other reported changes in different TDP-43 mouse models involve alterations in dendritic branching and spines (Fogarty et al., 2016; Handley et al., 2017; Wu et al., 2019; Dyer et al., 2021a). Conditional knockout of TDP-43 in the mouse forebrain (TDP-43cKO) using the Camk2α promoter, showed that loss of TDP-43 in neurons caused dendritic attrition in layer V neurons UMN and loss of spines (Wu et al., 2019). These changes corresponded with brain atrophy, decreased long-term potentiation (LTP) in the hippocampus, and cognitive defects in 12-month-old mice, which were followed by motor defects (Wu et al., 2019). A hippocampal-specific knockout of TDP-43 showed similar changes to dendritic branches and spines in CA1 neurons along with decreased LTP and spatial memory, which were attributed to altered sortilin splicing and impaired brain-derived growth factor (BDNF) secretion (Tann et al., 2019). Reintroduction of sortilin or BDNF partially restored these phenotypes in TDP-43 knockout models (Tann et al., 2019). Thy1-YFP:Prp-TDP43A315T mice also showed significant reduction in dendritic branching and spine density in the layer V UMN prior to cognitive and motor dysfunction (Handley et al., 2017; Jiang et al., 2019). Morphological analysis of the spines in these mice revealed a significant impairment in the development of mature spines in the motor cortex and lowered synaptic transmission (Handley et al., 2017). Consistent with these findings, mice expressing TDP-43ΔNLS, a form of TDP-43 that is retained in the cytoplasm, in excitatory neurons under control of the Camk2α promoter, showed TDP-43ΔNLS, and not TDP-43WT expression, caused a significant loss in spine density in layer V UMN, with few mature spines and mostly thin spines (Dyer et al., 2021a). These changes corresponded with a decreased expression in AMPA receptor subunits, Gria1, Gria2, and Gria3, and NMDA receptor subunit 2A and hyperexcitability in layer V excitatory neurons of the motor cortex (Dyer et al., 2021b). While TDP-43 is shown to bind the mRNA of AMPA receptor subunits Gria2–4 (Sephton et al., 2011; Narayanan et al., 2013; Jiang et al., 2019), the mechanism by which cytoplasmic TDP-43 disrupts mRNA translation of ionic glutamate receptor subunits or other mRNAs related to changes in dendrites and spines remain undefined. Other mouse models of TDP-43 also exhibit changes in the excitability of motor neurons (Fogarty et al., 2016; Zhang et al., 2016), and the collective body of work suggests there are both extrinsic and intrinsic factors that contribute to these changes.
Consistent with mouse models, in vitro studies show dysregulation of TDP-43 causes defects in dendritic growth, maturation of spines and synaptic transmission (Majumder et al., 2012; Han et al., 2013; Herzog et al., 2017, 2020; Tibshirani et al., 2017; Jiang et al., 2019; Ni et al., 2021). Depletion of TDP-43 from primary hippocampal and cortical neurons caused a significant decrease in dendritic branches (Han et al., 2013; Herzog et al., 2017, 2020). In contrast, another report showed that TDP-43 depletion from primary hippocampal neurons resulted in an increase in the number of matured spines without affecting neuronal morphology (Majumder et al., 2012). These changes corresponded with increased levels of the active form of Rac family small GTPase 1 (Rac1), a known positive regulator of spinogenesis (Wiens et al., 2005), clustering of AMPA receptors on the dendritic surface as well as increased individual current amplitude and average amplitude of mEPSCs (Majumder et al., 2012). Changes in Rac1, GTP-Rac1, and Gria1 at the surface of the hippocampal neurons caused by TDP-43 knockdown were rescued with a Rac1 inhibitor or co-expression with wild-type TDP-43 (Majumder et al., 2012). The effect of TDP-43 depletion in dissociated hippocampal neurons differs from work using organotypic rat hippocampal slice cultures that showed TDP-43 knockdown reduced the spine density and function of mature synapses (Ni et al., 2021). Importantly, the effect on synapses caused by TDP-43 depletion were rescued by wildtype TDP-43, but not by ALS/FTLD-associated mutants (Ni et al., 2021). The discrepancies between these studies require further investigation, but may involve differences in developmental time-points, cell-types and expression levels of TDP-43. Nonetheless, these studies collectively point to important roles for TDP-43 in the development and maintenance of neuromorphology and synapses.
Indeed, the expression of ALS/FTD-associated TDP-43 mutants in primary neuron cultures provides evidence that TDP-43 variants negatively impact neuronal morphology. TDP-43WT, M337V or A315T mutants caused a significant decrease in dendritic branches (Herzog et al., 2020). These phenotypes could be partially rescued by overexpression of a constitutively active forms of calcium/calmodulin-dependent protein kinase IV (CaMKIV) and cAMP-response element binding protein (CREB) (Herzog et al., 2020), which are known regulators of dendritic growth (Redmond et al., 2002). Further examination of the TDP-43A315T mutant, showed a reduction in dendritic spines of pyramidal cortical neurons corresponded with decreased localization of AMPA receptor subunit Gria1 to synapses and decreased spine densities (Jiang et al., 2019). The functional consequences of these changes were reduced generation of action potentials in TDP-43A315T-expressing pyramidal neurons (Jiang et al., 2019). While AMPA and NMDA receptors are implicated in dendritic architecture and spines of neurons (Inglis et al., 2002; Espinosa et al., 2009), the mechanism by which TDP-43 alters the expression of AMPA and NMDA receptor subunits is still not known. Additionally, it is still not known how misregulation of other TDP-43 mRNA targets implicated in mitochondria transport, morphology and function, cytoskeleton dynamics, axonal transport, proteostasis and autophagy (Oberstadt et al., 2018; Gao et al., 2019; Wood et al., 2021) would impact neuronal integrity and synaptic function.
Dendritic attrition due to loss of intermediate and terminal branches are also observed in primary motor neurons expressing hTDP-43G348C, but not in primary motor neurons expressing hTDP-43WT (Tibshirani et al., 2017). These changes corresponded with a reduction in subunits of the chromatin remodeling complex, neuronal Brahma-related gene 1 (Brg1)-associated factor complex (nBAF) expression (Tibshirani et al., 2017). Dendritic defects were attenuated by co-expression of the critical nBAF subunit, Brg1 (Tibshirani et al., 2017). Importantly, this complex is shown to be important for neuronal differentiation, dendritic extension and synaptic function (Wu et al., 2007) and nBAF subunits are depleted in spinal motor neurons of sporadic ALS and ALS-C9 cases (Tibshirani et al., 2017). The different effects of TDP-43 variants vs. wild-type TDP-43 expression on neuromorphology is a potentially important observation and may provide clues to vulnerabilities of specific neuron populations and the molecular pathways involved.
Mechanisms of synaptic dysfunction caused by TDP-43
TDP-43 regulates diverse cellular processes including those involved in cytoskeletal dynamics, mitochondrial function, energy metabolism, axonal transport, signaling pathways and proteostasis (Iridoy et al., 2018; Oberstadt et al., 2018; Umoh et al., 2018; Wood et al., 2021; Bjork et al., 2022). Unbiased proteomic analysis of postmortem frontal cortical tissues from individuals with ALS and FTD revealed a correlation between TDP-43 pathology and cognitive dysfunction and significant changes in proteins associated with RNA-binding (HNRNPA1, MATR3, and PFN1 and TDP-43 itself), synaptic transmission (MAP1B, MAP1A, DCTN1, discs Large MAGUK Scaffold Protein 4: DLG4/PSD-95, Tubulins: TUBB4B, TUBA1A), mitochondria (NADH:ubiquinone oxidoreductase core subunits: NDUFV1, NDUFS1, NDUFA2, NDUFA8, ATP Synthase F1 Subunit Alpha: ATP5A1), astrocytes (Glial fibrillary acidic protein: GFAP), and microglia (Moesin: MSN) (Umoh et al., 2018). Similar changes were also found in post-mortem spinal cords from individuals with ALS-TDP and ubiquitin-positive FTLD (FTLD-U) (Iridoy et al., 2018). The mechanistic causes for changes protein expression caused by TDP-43 dysregulation and their relationship to synaptic dysfunction and disease require further investigation.
Studies that have examined the pre-symptomatic effects of TDP-43 in animal models provide insight into potential mechanisms by which TDP-43 initiate synaptic dysfunction. Transcriptomic analysis of pre-symptomatic and symptomatic TDP-43 mouse models demonstrate that significant changes in pre-mRNA splicing and RNA expression occur mostly in symptomatic mice (White et al., 2018; Gordon et al., 2019; Wu et al., 2019; Huang et al., 2020). Transcriptome analysis of forebrain TDP-43cKO mice showed fewer changes in gene expression from pre-symptomatic (3-month-old) compared to symptomatic (12-month-old) mice, the latter had a greater number of down-regulated genes (Wu et al., 2019). Glial-specific markers, Gfap, serpin peptidase inhibitor clade A member 3 (Serpina3a), and complement components (C4a/C4b), were constitutively up-regulated in both pre-symptomatic and symptomatic mice, whereas down-regulated genes in pre-symptomatic TDP-43cKO mice encoded proteins involved in synaptic, endosome, and autophagosome functions (Wu et al., 2019). The authors demonstrated that down-regulation of Dlg3/Psd-95 and the encoding synapse associated protein (SAP102) protein, were reduced in the cortex and synaptosomes of TDP-43cKO mice in an age-dependent manner. Consistent with SAP10 role in regulating synaptic plasticity through NMDA receptor recycling (Chen et al., 2012), down-stream mediators of the NMDA receptor, CaMKIV, NMDA receptor subunit NR2b (or Grin2B), and phospho-Erk1/2, are also found to be altered in TDP-43cKO mice (Wu et al., 2019). Several of the signaling proteins identified by Chen et al. (2012), are shown to activate CREB (Shaywitz and Greenberg, 1999), a transcription factor well known for regulating dendritic branching (Redmond et al., 2002). The implications of TDP-43 on the CREB signaling cascade are further supported with the identification of CREB2 and upstream activators of CREB as punitive TDP-43 RNA targets (Herzog et al., 2020). Collectively, these studies highlight the possible role of TDP-43 in regulating mRNAs involved in the NMDA receptor/CREB signaling cascade, but the direct and indirect role of TDP-43 in regulating this pathway and the implications for neuronal morphology and synaptic function require more detailed investigation.
In vitro evidence shows that the effects of TDP-43 on dendrites and synapses are mediated through its interactions with RNA. TDP-43 truncation mutants or point mutations in the RRM1 and RRM2 regions, which lack RNA binding, do not cause defects in dendritic growth (Herzog et al., 2017) and are not able to rescue synaptic defects caused by TDP-43 depletion (Ni et al., 2021). Moreover, the role of TDP-43 in RNA trafficking and translation regulation are shown to be important in maintaining neuronal morphology and synaptic integrity (Majumder et al., 2016; Tank et al., 2018; Briese et al., 2020; Altman et al., 2021). For example, TDP-43 is involved in Rac1 transport and translation in dendrites (Majumder et al., 2016), whereas depletion of TDP-43 alters Rac1 mRNA transport within neurons, which in turn affects Rac1 expression (Chu et al., 2019). Activation of the Rac1 pathway stabilizes dendritic spines and recruits AMPA receptors to the postsynaptic membrane, enhancing excitatory synaptic transmission (Wiens et al., 2005; Majumder et al., 2012). The previously mentioned changes in AMPA expression at dendritic spines in TDP-43 disease models are consistent with a potential reduction in Rac1-mediated AMPA recruitment to spines. There is also evidence that shows TDP-43 affects protein expression at the level of translation prior to global misregulation of gene and protein networks (Briese et al., 2020; Altman et al., 2021). Axonal accumulation of TDP-43ΔNLS was found to cause NMJ disruption through inhibition of local protein synthesis (Altman et al., 2021). Specifically, TDP-43ΔNLS accumulation promoted Ras-GAP SH3-domain-binding protein 1 (G3BP1)-positive ribonucleoprotein (RNP) assembly and mRNA sequestration within RNPs, which lead to a reduction in nuclear-encoded mitochondrial proteins and respiratory chain complex proteins by suppressing protein translation (Altman et al., 2021). The three reported nuclear-encoded mitochondrial genes, ATP5A1, Cox4i1, and Ndufa4, found to be affected by TDP-43, had increased mRNA abundance but decreased protein expression (Altman et al., 2021). These findings suggest that TDP-43ΔNLS sequestration of mRNA leads to deleterious effects on protein synthesis. Additionally, depletion of TDP-43 in primary mouse motor neurons can lead to translation repression and defects in in mRNA transport (Briese et al., 2020). Loss of TDP-43 led to defects in the axonal localization of transcripts encoding components of the cytoskeleton, the translational machinery and transcripts involved in mitochondrial energy metabolism (Briese et al., 2020). In the somatodendritic compartment of TDP-43 knockdown motor neurons, there was an upregulation of transcripts associated with neuron projection and dendrite development, synapses and cytoskeleton (Briese et al., 2020). Unique to axons, was the down-regulation of transcripts encoding translation initiation factors (eukaryotic initiation factor 4A-II: Eif4a2) and ribosomal proteins (40 S ribosomal proteins S8 and S3: Rps8 and Rps3, respectively) (Briese et al., 2020). These changes corresponded with a reduced capacity for protein production in axons of TDP-43 depleted motor neurons (Briese et al., 2020). Transcripts down-regulated in both the somatodendritic and axon compartments were associated with mitochondrial function and energy production, which corresponded with a significant decrease in the number of functional mitochondria in axons of TDP-43 depleted motor neurons (Briese et al., 2020). These findings demonstrate pathological TDP-43 interferes with multiple mitochondrial pathways, including bioenergetics, as well as biogenesis, quality control, transport and fission and fusion dynamics (Gao et al., 2019). Alterations in transcript and protein levels in TDP-43 models of ALS/FTD may involve several aspects of RNA metabolism including RNA trafficking, stability or translation regulation or possibly by its direct association with translation machinery (Russo et al., 2017; Chu et al., 2019; Charif et al., 2020; Sidibe et al., 2021). Therefore, further investigation is required to understand the mechanisms by TDP-43 causes changes in mRNA and protein levels and the temporal impact of these changes on neuronal morphology and synaptic function.
FUS: biological and pathological overview
Human FUS is located at the chromosomal locus 16p11.2 and is comprised of 15 exons, which encode a 53 kDa protein that migrates on SDS-PAGE between ~68–75 kDa. The gene encoding FUS is found in all higher eukaryotic species with homologs expressed in Drosophila melanogaster, Xenopus laevis, and Caenorhabditis elegans. Historically, FUS has also been called translated in liposarcoma (TLS) and is a member of the FET family of RNA-binding proteins that include Ewing sarcoma breakpoint region 1 (EWS) and TATA-box binding protein associated factor 15 (TAF15) (Tan and Manley, 2009). FUS contains a disordered N-terminal serine-tyrosine-glycine-glutamine (SYGQ)-rich region, followed by an arginine-glycine-glycine (RGG1) box in the middle of the protein, a central conserved RRM, and a zinc finger (ZnF) domain, that is flanked by RGG2 and RGG3 boxes and a proline-tyrosine nuclear localization sequence (PY-NLS) at the extreme C-terminal end (Lerga et al., 2001; Iko et al., 2004; Bentmann et al., 2012). FUS is ubiquitously expressed, predominantly localized in the nucleus of cells, and undergoes nucleocytoplasmic shuttling (Zinszner et al., 1997; Tsai et al., 2022). In the cytoplasm, FUS is observed within RNA granules and diffusely in non-RNA granules within the axons, dendrites, and synapses, prominently pre-synaptic within UMN and LMN (So et al., 2018; Deshpande et al., 2019; Sahadevan et al., 2021).
The nuclear functions of FUS include the regulation of transcription, splicing and DNA damage repair (Hoell et al., 2011; Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012; Schwartz et al., 2012; Mastrocola et al., 2013), whereas the cytoplasm functions include numerous roles in RNA transport and stability, miRNA processing, and translation regulation (Fujii and Takumi, 2005; Morlando et al., 2012; Udagawa et al., 2015; Kapeli et al., 2016; Kamelgarn et al., 2018; Sevigny et al., 2020). FUS can bind several thousand RNAs at coding, non-coding and 5’- and 3’-UTR regions (Hoell et al., 2011; Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012), mediated through its RRM, ZnF domain and three RGG boxes (Burd and Dreyfuss, 1994; Lerga et al., 2001; Iko et al., 2004; Ozdilek et al., 2017). In mouse brain, ~6,300 genes contain both TDP-43 and FUS binding sites, with overlapping binding sites in ~2,700 genes (Lagier-Tourenne et al., 2012). Functional analysis of FUS, its protein partners and DNA/RNA targets indicate that it is an important regulator of transcription and translation of genes that encode proteins that contribute to diverse cellular processes (Ishigaki et al., 2012; Lagier-Tourenne et al., 2012; Rogelj et al., 2012; Mastrocola et al., 2013; Sephton and Yu, 2015).
Over 50 mutations have been identified in the FUS gene that account for approximately 5–10% of familial ALS and ~0.4% of sporadic ALS (Kwiatkowski et al., 2009; Vance et al., 2009). Only a few FUS mutations have been identified in familial FTD (Van Langenhove et al., 2010; Huey et al., 2012). ALS and FTD-linked FUS mutations result in pathological aggregation of FUS, with varying levels of ubiquitination, in the cytoplasm and nucleus of neurons and glia throughout the affected regions of the cortex, brainstem and spinal cord (Kwiatkowski et al., 2009; Munoz et al., 2009; Vance et al., 2009; Suzuki et al., 2012). In contrast, most FTLD cases with FUS pathology (FTLD-FUS) show no evidence of mutations in the FUS gene (Seelaar et al., 2010; Nicolas et al., 2022). These cases usually present as bvFTD or FTD-MND and tend to effect younger individuals having more rapid disease progress (Perry et al., 2017). The majority of FUS mutations that have been identified are located within the C-terminal PY-NLS and are shown to affect the subcellular distribution of the protein (Bentmann et al., 2013). Mutations in the FUS 3’UTR have also been identified in familial ALS that increase FUS expression (Sabatelli et al., 2013). The proposed pathologic mechanisms of FUS misregulation involve mislocalization of the protein to the cytoplasm due to mutations in the PY-NLS. These defects alter physical properties of the protein involving aberrant stress granule dynamics and liquid-liquid phase transitions, which contribute to altered proteostasis, transcription and translation that impair dendrites and spine development (Sephton and Yu, 2015; Shang and Huang, 2016; Alhindi et al., 2021; Carey and Guo, 2022).
FUS: synaptic dysfunction in ALS/FTD models of disease
Several mouse models of ALS/FTD-FUS display similar aspects of the human disease, including changes to neuronal morphology and synapses, as well as cognitive and motor defects (McGoldrick et al., 2013; Picher-Martel et al., 2016; Alhindi et al., 2021). Global transgenic mice expressing low levels human wild-type FUS (FUSWT) or FUSR521G under Cre-inducible control of the CAG promoter, developed deficits in motor function, NMJ denervation and inflammation without the loss of motor neurons, degeneration of motor axons or FUS aggregation (Sephton et al., 2014). Importantly, only FUSR521G mice had dendritic attrition of the layer VI-V UMN and LMN and reduced mature spines of layer VI-V UMN, which corresponded with cognitive and motor impairments (Sephton et al., 2014). Characterization of early synaptic changes in pre-symptomatic FUSWT mice showed mitochondrial abnormalities in the pre-synaptic motor nerve terminals lead to smaller NMJs, loss of synaptic vesicles and synaptophysin (Syp) expression in the absence of motor neuron loss (So et al., 2018). Changes in mitochondrial abundance have also been observed in ALS/FTD-FUS patients and other disease models (Tradewell et al., 2012; Deng et al., 2015; Tsai et al., 2020), which may occur due to aberrant interactions with mRNA or protein interactions that lead to global changes in protein synthesis (Nakaya and Maragkakis, 2018; Tsai et al., 2020; Salam et al., 2021). Future studies will need to focus on understand the mechanisms by which FUSWT and FUS variants affect mitochondrial function in ALS/FTD-FUS disease models.
Other studies that examine the cell-autonomous effect of FUS variants show that restricted expression of FUS in the brain and spinal cord of mice is sufficient to promote dendritic attrition of UMN and LMN, alterations of dendritic spines and NMJs as well as behavior impairments (Qiu et al., 2014; Ho et al., 2021). Consistent with a loss of corticospinal connectivity observed in ALS and FTD-MND patients (Whitwell et al., 2011; Renga, 2022), Prp-FUSR521C mice had loss of dendritic branching and mature spines in UMN and a corresponding reduction in the number and size of presynaptic terminals in spinal motor neurons (Qiu et al., 2014). Brain atrophy and age-dependent cognitive defects in Prp-FUSR514G transgenic mice were also found to cause decreased dendritic spine density and LTP in the hippocampus of these mice (Ho et al., 2021). Consistent with synaptic defects caused by mislocalization of FUS variants to the cytoplasm, mouse models expressing FUSΔNLS, a FUS mutant that is retained in the cytoplasm, have reduced dendritic branching and spine densities, that correspond with behavioral impairments (Shiihashi et al., 2017; Picchiarelli et al., 2019). (KI) mice heterozygous for FusΔNLS/+ mice had endplate denervation, reduced surface of area and reduced neuromuscular transmission of NMJs by 1 month of age (Picchiarelli et al., 2019); defects that preceded motor phenotypes in these mice by 6 months of age (Scekic-Zahirovic et al., 2016). Mice expressing FUSΔNLS under the Thy1 promoter are shown to have FTD-associated cognitive defects including hyperactivity, social interactional deficits, and impaired fear memory retrieval, with decreased dendritic spine and synaptic density in the frontal cortex (Shiihashi et al., 2017). Importantly, the changes in neuronal morphology and synapses of are shown to precede neuronal loss (Shiihashi et al., 2017). Changes to synapses corresponded with significant decreases in mRNA levels for AMPA receptor subunits FUS knockin Gria3 and Gria4 (also known as GluR3 or GluA3 and GluR4 or GluA4, respectively) (Shiihashi et al., 2017). The functional consequence of reduced synaptic densities in Thy1-FUSΔNLS mice was a significant reduction in the frequency, but not the amplitude, of mEPSCc in layer V pyramidal neurons (Shiihashi et al., 2017). These results indicate that the function of each excitatory synaptic input is intact, but the total number of excitatory synaptic inputs is reduced in these mice. The mechanism by which FUS causes changes to the AMPA receptor subunits were not assessed in this study. However, mRNA transport was found to be impaired by cytoplasmic FUS aggregates found within the frontal cortex and hippocampus of Thy1-FUSΔNLS mice (Shiihashi et al., 2017), consistent with other studies that show cytoplasmic FUS aggregates alter translation through aberrant sequestration of RNA (Kamelgarn et al., 2018; Lopez-Erauskin et al., 2018). FUS depletion specifically in the hippocampus of mice causes delayed dendritic spine maturation and cognitive defects including hyperactivity, disinhibition and changes in social interactions (Udagawa et al., 2015). While global knockout of FUS leads to postnatal lethality (Hicks et al., 2000; Scekic-Zahirovic et al., 2016), conditional knockout of FUS in motor neurons is not sufficient to cause motor neuron death (Scekic-Zahirovic et al., 2016; Sharma et al., 2016). The effect of depleting FUS on motor neuron morphology was not examined in these studies. Collectively, these findings suggest that FUS misregulation can have both a loss- and gain-of-function effect on neuronal morphology and synapses.
Dysregulation of FUS in vitro causes defects in dendritic growth, maturation of spines, and synaptic transmission (Fujii et al., 2005; Fujii and Takumi, 2005; Udagawa et al., 2015; Tibshirani et al., 2017; Qiu et al., 2021). Early work by Fujii et. al. established FUS as an important regulator of dendritic branching, synaptic maturation, and that FUS localizes within dendrites and synapses of primary hippocampal neurons in response to metabatropic glutamate receptor 5 (mGluR5) stimulation (Fujii and Takumi, 2005; Fujii et al., 2005). Moreover, FUSKO neurons have altered dendritic branching and immature dendritic spines (Fujii et al., 2005), down-regulation of Gria1 and AMPA receptor surface expression and reduced miniature EPSC amplitudes (Udagawa et al., 2015). Morphological changes in FUSKO neurons also corresponded with an increased RNA content in the dendrites (Fujii et al., 2005), consistent with the previously discussed functions of FUS in mRNA transport, stability and translation regulation (Fujii and Takumi, 2005; Morlando et al., 2012; Udagawa et al., 2015; Kapeli et al., 2016; Kamelgarn et al., 2018; Sevigny et al., 2020). Importantly, the ability of FUS to traffic within dendrites and synapses was dependant on intact microtubules and actin filaments as determined by the addition of cytochalasin B or nocodazole, inhibitors of the assembly of actin filaments or microtubules, respectively (Fujii et al., 2005).
Expression of ALS/FTD-associated FUS mutants in primary neurons shows that FUS dysregulation has an impact on dendritic branches and spines (Shiihashi et al., 2017; Tibshirani et al., 2017; Qiu et al., 2021). The defects in dendritic growth, branch points, and dendritic length in primary cortical neurons expressing FUSR521C were attributed to splicing defects of BDNF mRNA caused by aberrant FUSR521C binding to the 5’ splice junction sequences Bdnf mRNA (Qiu et al., 2021). Adding BDNF to these cultures only partially ameliorated defects in dendritic branching (Qiu et al., 2021), suggesting that other factors are contributing to these defects. hFUSΔNLS expression in primary cortical neurons, but not in cultures expressing hFUSWT, show similar defects in dendritic branching, but these changes were attributed to decreased mRNA trafficking and global protein synthesis in dendrites (Shiihashi et al., 2017). Dendritic attrition is also observed in primary motor neurons expressing hFUSR521G, but not in neurons expressing hFUSWT (Tibshirani et al., 2017). In this model, the primary branches closest to the soma of the motor neurons expressing hFUSR521G were spared, however, intermediate and terminal branches were significantly reduced, along with the average number of processes per neuron and total dendrite length (Tibshirani et al., 2017). These changes corresponded with a reduction in nuclear Brg1 expression (Tibshirani et al., 2017), which is an identified interactor of FUS (Chesi et al., 2013). The relevance between FUS dysregulation and expression and function of the nBAF complex remains to be fully elucidated, but the observed changes in nBAF subunits in sporadic ALS and ALS-C9 cases (Tibshirani et al., 2017) suggests important involvement in the disease. Collectively, these studies underscore the impact of transcription and translation and their potential impact on the function and structure of neurons.
Mechanisms of synaptic dysfunction caused by FUS
Findings from ALS/FTD-FUS patient and disease models show that FUS regulates diverse cellular processes including those involved in synaptic transmission, signaling cascades, cytoskeleton dynamics mitochondrial function and energy metabolism (Schweitzer et al., 2006; Martins-De-Souza et al., 2012; Garone et al., 2020). Unbiased proteomic analysis of frontal cortical tissues from postmortem tissues of FTLD-FUS and controls showed altered expression of proteins associated with synaptic communication (syntaxin subunits: STX1A, STX1B, and STXBP1, synapsin 3: SYN3, synaptotagmin I: SYT1) and those involved in cellular transport pathways (clathrin-associated adaptor proteins: AP1B1, AP2A1, AP2A2, AP2B1) (Martins-De-Souza et al., 2012). These findings were consistent with previously discussed changes in synaptic connectivity observed in FTLD and ALS (Henstridge et al., 2018; Malpetti et al., 2021; Salmon et al., 2021). Additionally, FTLD-FUS tissues had significant changes in proteins involved in signaling pathways (calcium-related proteins, CAMK2A, protein phosphatase 3, catalytic subunit, alpha isozyme: PPP3CA and neurochondrin: NCDN) (Martins-De-Souza et al., 2012). Other proteins with altered expression are implicated in cellular structure and cytoskeleton assembly including subunits of tubulin (TUBB1, TUBB2A, TUBB2C, and TUBA4B), dynamin 2 (DYN2) and neural growth regulator 1 (NEGR1) (Martins-De-Souza et al., 2012). Changed FTLD-FUS proteomic profiles are also associated with energy metabolism and mitochondrial function (glutamate dehydrogenase 2: GLUD2, glucose-6-phosphaste isomerase: GPI, malate dehydrogenase 1 MDH1 and NDUFS1) that are implicated in disease (Schweitzer et al., 2006; Martins-De-Souza et al., 2012). Similar changes were found in FUSP525L and FUSWT human iPSC lines differentiated into ventral spinal motor neurons (Garone et al., 2020). Genome-wide transcriptome analysis of FUS models reflect proteomic changes observed in patients along with a wide-range of altered cellular processes (Nakaya and Maragkakis, 2018; De Santis et al., 2019; Garone et al., 2020; Humphrey et al., 2020; Tsai et al., 2020; Ho et al., 2021). While these changes are likely to impact neuronal function and synaptic connectivity, the mechanistic causes for global changes in protein and gene expression by FUS dysregulation and their relationship to synaptic dysfunction are not known.
ALS/FTD-FUS models provide evidence that misregulation of RNA translation and protein synthesis by FUS underly alterations in neuromorphology and synapses (Sephton et al., 2014; Lopez-Erauskin et al., 2018). Genome-wide expression analysis of spinal cords from pre-symptomatic FUSR521G mice showed no significant changes in pre-mRNA splicing or mRNA expression but had altered protein synthesis at synapses that corresponded with dendritic attrition and fewer mature spines (Sephton et al., 2014). FUSR521C and R521H variants are shown to repress nascent protein production in the processes of primary hippocampal cultures (Lopez-Erauskin et al., 2018) and in axon terminals (Murakami et al., 2015). In vitro studies also show that FUS variants alter protein expression levels without significant changes in mRNA expression levels (Nakaya and Maragkakis, 2018; De Santis et al., 2019; Garone et al., 2020; Tsai et al., 2020). Expression of FUS variants in immortalized cell-lines, or in neurons differentiated from mouse embryonic stem cells, caused a decreased expression of mitochondrial encoded proteins without significant changes in overall translation or mRNA expression levels (Nakaya and Maragkakis, 2018; Tsai et al., 2020). FUS variants were found to sequester mRNAs encoding mitochondrial respiratory chain complex (RCC) proteins, causing a reduction in encoded proteins and disruption of mitochondrial networks (Tsai et al., 2020). The impact of these changes on neuronal functions were not assessed. Similarly, proteomic and transcriptomic profiles of isogenic pairs of FUSP525L and FUSWT human iPSC lines differentiated into ventral spinal cord populations showed no correlation between transcript and protein levels, effects that are attributed to selective binding of FUS mutants to the 3’UTRs of target mRNAs (De Santis et al., 2019; Garone et al., 2020). Misregulation of RNA translation and protein synthesis by FUS are shown to occur through different mechanisms including: defects in mRNA trafficking due to aberrant sequestration of RNA in granules or dysfunctional liquid-liquid phase transitions (Birsa et al., 2021), repression of translation through interactions with mRNA or polyribosomes (Udagawa et al., 2015; De Santis et al., 2019; Sevigny et al., 2020), and/or regulation of mRNA stability through non-sense mediated decay (NMD) (Kamelgarn et al., 2018; Ho et al., 2021). Future studies will need to investigate how reported changes in RNA translation and protein synthesis occur in these models and the impact on synaptic function.
FUS is shown to mediate the transport and stabilization of mRNAs important for synaptic function (Fujii and Takumi, 2005; Udagawa et al., 2015; Yokoi et al., 2017). Knocking down FUS in primary neurons leads to fewer mature dendritic spines, reduced miniature EPSC amplitude, and down-regulation of Gria1 expression and AMPA receptor surface expression (Udagawa et al., 2015). FUS was shown to regulate Gria1 mRNA stability at the 3’UTR and that changes in Gria1 expression were shown to occur through destabilization of Gria1 mRNA (Udagawa et al., 2015). Reintroduction of Gria1 in FUS knockdown mice partially restored behavioral impairments and in neurons restored synaptic dysfunction (Udagawa et al., 2015), demonstrating that FUS-mediated Gria1 expression is an important component for proper synaptic transmission. FUS and ELAV-like proteins were also shown to cooperatively control SynGAP mRNA stability in a 3’UTR length-dependent manner, resulting in a decrease expression of SynGAP α2 (Yokoi et al., 2017). Abnormal spine maturation and FTLD-like behavioral deficits in FUS-knockout mice were partially restored by expression of SynGAPα2 (Yokoi et al., 2017). While these findings provide evidence that FUS destabilization of specific mRNAs can impact synaptic function, it remains unclear whether other mRNA targets of FUS are also contributing to synaptic abnormalities caused by FUS misregulation. To address this question, FusΔNLS/+ mice were used to identify changes in synaptic RNAs (Sahadevan et al., 2021). FusΔNLS/+ mice had an increase in 485 RNAs associated with synaptic function and a decrease in 136 synaptic RNAs associated with cytoskeletal organization and RNA metabolism (Sahadevan et al., 2021). Interestingly, the majority of misregulated synaptic mRNAs in FusΔNLS/+ mice were not identified as synaptic mRNA targets of endogenous mouse FUS (Sahadevan et al., 2021), findings that could reflect the aberrant binding of FUS variants, but not wild-type FUS, to the 3’UTR region of mRNAs (Hoell et al., 2011; Garone et al., 2021). Among those identified as FUS targets, mRNAs with increased stability had FUS binding sites within exonic regions, while those with decreased stability had a higher proportion binding sites within 3’UTRs (Sahadevan et al., 2021). While the alterations in the synaptic RNA profiles in FusΔNLS/+ mice are likely to affect synaptic homeostasis, the direct implicates for these changes require further investigation. Collectively, these studies show that FUS dysfunction leads to alterations in protein translation and expression, however, the detailed mechanisms by which these changes alter neuronal function are not yet known.
Discussion
The clinical, pathological and genetic overlap between ALS and FTD has provided insights into mechanisms of disease, particularly as they relate to changes in neuronal morphology and synaptic dysfunction. Analysis of patient samples and ALS/FTD models strongly suggest that synaptic dysfunction occurs early in the disease process, before any significant neurodegeneration or cell death. Recent discoveries from proteomic and transcriptomic studies underscore the diverse cellular pathways and biological processes involved in synaptic dysfunction that contribute to the neurodegenerative pathology. Moreover, these studies highlight converging mechanisms of synaptic dysfunction in ALS and FTD. The identification of reliable biomarkers from body fluids or the development of new tools to image synaptic changes in patients may prove useful for early detection and diagnosis. Moreover, concentrated efforts to investigate the pre-symptomatic and early-symptomatic biochemical mechanisms leading to synaptic dysfunction will undoubtedly benefit our understanding of the disease and future therapeutic approaches.
The current body of work examining ALS/FTD-linked C9ORF72, TDP-43, and FUS variants, strongly implicates their involvement in synaptic dysfunction. The emphasis for future studies should be placed on examining the effects of these variants in both pre-symptomatic and symptomatic animal models to better understand the stages of disease progression and their contribution to changes in corticospinal connectivity and loss of synapses. Additionally, more work is needed to better understand the role of non-cell autonomous contributions of other cell types in maintaining dendritic spines and synapses. The current body of work from ALS/FTD-C9 patients and models suggest that C9ORF72 has several important roles in the soma and in distal regions if the neuron. To date, there are no studies that have analyzed the effects of C9ORF72 on axonal or synaptic proteomes and transcriptomes, which may provide additional insights into the mechanisms by which C9ORF72 causes early synaptic dysfunction. TDP-43 and FUS are important regulators of many cellular processes that are implicated in neuromorphology and synaptic homoeostasis through their RNA-binding capabilities. More work is needed to better understand and delineate the primary and secondary mechanisms by which dysregulation of TDP-43 and FUS initiate synaptic loss in both gain-of-function and loss-of-function models.
Author Contributions
CS: manuscript conceptualization, writing, and figure composition. PD: manuscript writing and editing. PG: literature review of manuscript and Table 1. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Brain Canada Future Leader’s Award (to CS), ALS Canada–Brain Canada Hudson Translational Team Grant (to CS), Natural Sciences and Engineering Research Council of Canada Grants RGPIN-2020-06376 and DGECR-2020-00060 (to CS), RGPIN-2018-06227 and DGECR-2018-00093 (to PD), and Fonds de Recherche du Québec Santé (to PD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14:42. doi: 10.3389/fnins.2020.00042
Al-Chalabi, A., and Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628. doi: 10.1038/nrneurol.2013.203
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Alberici, A., Bonato, C., Calabria, M., Agosti, C., Zanetti, O., Miniussi, C., et al. (2008). The contribution of TMS to frontotemporal dementia variants. Acta Neurol. Scand. 118, 275–280. doi: 10.1111/j.1600-0404.2008.01017.x
Alhindi, A., Boehm, I., and Chaytow, H. (2021). Small junction, big problems: neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J. Anat. . [Online ahead of print]. doi: 10.1111/joa.13463
Altman, T., Ionescu, A., Ibraheem, A., Priesmann, D., Gradus-Pery, T., Farberov, L., et al. (2021). Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat. Commun. 12:6914. doi: 10.1038/s41467-021-27221-8
Anderson, C. J., Bredvik, K., Burstein, S. R., Davis, C., Meadows, S. M., Dash, J., et al. (2019). ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 138, 103–121. doi: 10.1007/s00401-019-01989-y
Anoar, S., Woodling, N. S., and Niccoli, T. (2021). Mitochondria dysfunction in frontotemporal dementia/amyotrophic lateral sclerosis: lessons from Drosophila models. Front. Neurosci. 15:786076. doi: 10.3389/fnins.2021.786076
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Arnold, E. S., Ling, S. C., Huelga, S. C., Lagier-Tourenne, C., Polymenidou, M., Ditsworth, D., et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. U S A 110, E736–E745. doi: 10.1073/pnas.1222809110
Arsovic, A., Halbach, M. V., Canet-Pons, J., Esen-Sehir, D., Doring, C., Freudenberg, F., et al. (2020). Mouse ataxin-2 expansion downregulates CamKII and other calcium signaling factors, impairing granule—Purkinje neuron synaptic strength. Int. J. Mol. Sci. 21:6673. doi: 10.3390/ijms21186673
Ash, P. E., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atanasio, A., Decman, V., White, D., Ramos, M., Ikiz, B., Lee, H. C., et al. (2016). C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci. Rep. 6:23204. doi: 10.1038/srep23204
Atkinson, R. A., Fernandez-Martos, C. M., Atkin, J. D., Vickers, J. C., and King, A. E. (2015). C9ORF72 expression and cellular localization over mouse development. Acta Neuropathol. Commun. 3:59. doi: 10.1186/s40478-015-0238-7
Ayala, Y. M., Pantano, S., D’ambrogio, A., Buratti, E., Brindisi, A., Marchetti, C., et al. (2005). Human, Drosophila and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 348, 575–588. doi: 10.1016/j.jmb.2005.02.038
Ayala, Y. M., Zago, P., D’ambrogio, A., Xu, Y. F., Petrucelli, L., Buratti, E., et al. (2008). Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785. doi: 10.1242/jcs.038950
Batra, R., and Lee, C. W. (2017). Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/frontotemporal dementia. Front. Cell. Neurosci. 11:196. doi: 10.3389/fncel.2017.00196
Belly, A., Bodon, G., Blot, B., Bouron, A., Sadoul, R., and Goldberg, Y. (2010). CHMP2B mutants linked to frontotemporal dementia impair maturation of dendritic spines. J. Cell Sci. 123, 2943–2954. doi: 10.1242/jcs.068817
Belzil, V. V., Daoud, H., Camu, W., Strong, M. J., Dion, P. A., Rouleau, G. A., et al. (2013). Genetic analysis of SIGMAR1 as a cause of familial ALS with dementia. Eur. J. Hum. Genet. 21, 237–239. doi: 10.1038/ejhg.2012.135
Bentmann, E., Haass, C., and Dormann, D. (2013). Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 280, 4348–4370. doi: 10.1111/febs.12287
Bentmann, E., Neumann, M., Tahirovic, S., Rodde, R., Dormann, D., Haass, C., et al. (2012). Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094. doi: 10.1074/jbc.M111.328757
Bercier, V., Hubbard, J. M., Fidelin, K., Duroure, K., Auer, T. O., Revenu, C., et al. (2019). Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol. Neurodegener. 14:27. doi: 10.1186/s13024-019-0327-3
Birsa, N., Ule, A. M., Garone, M. G., Tsang, B., Mattedi, F., Chong, P. A., et al. (2021). FUS-ALS mutants alter FMRP phase separation equilibrium and impair protein translation. Sci. Adv. 7:eabf8660. doi: 10.1126/sciadv.abf8660
Bjork, R. T., Mortimore, N. P., Loganathan, S., and Zarnescu, D. C. (2022). Dysregulation of translation in tdp-43 proteinopathies: deficits in the RNA supply chain and local protein production. Front. Neurosci. 16:840357. doi: 10.3389/fnins.2022.840357
Borroni, B., Archetti, S., Del Bo, R., Papetti, A., Buratti, E., Bonvicini, C., et al. (2010). TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features and disease course. Rejuvenation Res. 13, 509–517. doi: 10.1089/rej.2010.1017
Borroni, B., Bonvicini, C., Alberici, A., Buratti, E., Agosti, C., Archetti, S., et al. (2009). Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 30, E974–E983. doi: 10.1002/humu.21100
Braak, H., Brettschneider, J., Ludolph, A. C., Lee, V. M., Trojanowski, J. Q., Del Tredici, K., et al. (2013). Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. doi: 10.1038/nrneurol.2013.221
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Briese, M., Saal-Bauernschubert, L., Luningschror, P., Moradi, M., Dombert, B., Surrey, V., et al. (2020). Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 8:116. doi: 10.1186/s40478-020-00987-6
Brooks, B. R., Miller, R. G., Swash, M., Munsat, T. L., and World Federation of Neurology Research Group on Motor Neuron, D. (2000). El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Brown, R. H. Jr., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377:1602. doi: 10.1056/NEJMc1710379
Brun, A., Liu, X., and Erikson, C. (1995). Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration 4, 171–177. doi: 10.1006/neur.1995.0021
Buscaglia, G., Northington, K. R., Moore, J. K., and Bates, E. A. (2020). Reduced TUBA1A tubulin causes defects in trafficking and impaired adult motor behavior. eNeuro 7:ENEURO.0045-20.2020. doi: 10.1523/ENEURO.0045-20.2020
Buratti, E., and Baralle, F. E. (2010). The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429. doi: 10.4161/rna.7.4.12205
Burberry, A., Suzuki, N., Wang, J. Y., Moccia, R., Mordes, D. A., Stewart, M. H., et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8:347ra93. doi: 10.1126/scitranslmed.aaf6038
Burd, C. G., and Dreyfuss, G. (1994). Conserved structures and diversity of functions of RNA-binding proteins. Science 265, 615–621. doi: 10.1126/science.8036511
Burrell, J. R., Kiernan, M. C., Vucic, S., and Hodges, J. R. (2011). Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594. doi: 10.1093/brain/awr195
Carey, J. L., and Guo, L. (2022). Liquid-liquid phase separation of TDP-43 and FUS in physiology and pathology of neurodegenerative diseases. Front. Mol. Biosci. 9:826719. doi: 10.3389/fmolb.2022.826719
Caroppo, P., Camuzat, A., Guillot-Noel, L., Thomas-Anterion, C., Couratier, P., Wong, T. H., et al. (2016). Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol. Genet. 2:e80. doi: 10.1212/NXG.0000000000000080
Castellanos-Montiel, M. J., Chaineau, M., and Durcan, T. M. (2020). The neglected genes of ALS: cytoskeletal dynamics impact synaptic degeneration in ALS. Front. Cell. Neurosci. 14:594975. doi: 10.3389/fncel.2020.594975
Chand, K. K., Lee, K. M., Lee, J. D., Qiu, H., Willis, E. F., Lavidis, N. A., et al. (2018). Defects in synaptic transmission at the neuromuscular junction precede motor deficits in a TDP-43(Q331K) transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 32, 2676–2689. doi: 10.1096/fj.201700835R
Charif, S. E., Luchelli, L., Vila, A., Blaustein, M., and Igaz, L. M. (2020). Cytoplasmic expression of the ALS/FTD-related protein TDP-43 decreases global translation both in vitro and in vivo. Front. Cell. Neurosci. 14:594561. doi: 10.3389/fncel.2020.594561
Chassefeyre, R., Martinez-Hernandez, J., Bertaso, F., Bouquier, N., Blot, B., Laporte, M., et al. (2015). Regulation of postsynaptic function by the dementia-related ESCRT-III subunit CHMP2B. J. Neurosci. 35, 3155–3173. doi: 10.1523/JNEUROSCI.0586-14.2015
Chau, K. F., Shannon, M. L., Fame, R. M., Fonseca, E., Mullan, H., Johnson, M. B., et al. (2018). Downregulation of ribosome biogenesis during early forebrain development. eLife 7:e36998. doi: 10.7554/eLife.36998
Chaussenot, A., Le Ber, I., Ait-El-Mkadem, S., Camuzat, A., De Septenville, A., Bannwarth, S., et al. (2014). Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 35:e12884. doi: 10.1016/j.neurobiolaging.2014.07.022
Chen, B. S., Gray, J. A., Sanz-Clemente, A., Wei, Z., Thomas, E. V., Nicoll, R. A., et al. (2012). SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2, 1120–1128. doi: 10.1016/j.celrep.2012.09.024
Chen, T., Huang, B., Shi, X., Gao, L., and Huang, C. (2018). Mutant UBQLN2P497H in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol. Commun. 6:122. doi: 10.1186/s40478-018-0627-9
Chen-Plotkin, A. S., Lee, V. M., and Trojanowski, J. Q. (2010). TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 6, 211–220. doi: 10.1038/nrneurol.2010.18
Chesi, A., Staahl, B. T., Jovicic, A., Couthouis, J., Fasolino, M., Raphael, A. R., et al. (2013). Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat. Neurosci. 16, 851–855. doi: 10.1038/nn.3412
Chio, A., Calvo, A., Moglia, C., Restagno, G., Ossola, I., Brunetti, M., et al. (2010). Amyotrophic lateral sclerosis-frontotemporal lobar dementia in 3 families with p.Ala382Thr TARDBP mutations. Arch. Neurol. 67, 1002–1009. doi: 10.1001/archneurol.2010.173
Chio, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41, 118–130. doi: 10.1159/000351153
Chio, A., Mazzini, L., and Mora, G. (2020). Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 167:107986. doi: 10.1016/j.neuropharm.2020.107986
Chu, J. F., Majumder, P., Chatterjee, B., Huang, S. L., and Shen, C. J. (2019). TDP-43 regulates coupled dendritic mRNA transport-translation processes in co-operation with FMRP and Staufen1. Cell Rep. 29, e63118–e63133. doi: 10.1016/j.celrep.2019.10.061
Chua, J. J., Kindler, S., Boyken, J., and Jahn, R. (2010). The architecture of an excitatory synapse. J. Cell Sci. 123, 819–823. doi: 10.1242/jcs.052696
Coon, E. A., Sorenson, E. J., Whitwell, J. L., Knopman, D. S., and Josephs, K. A. (2011). Predicting survival in frontotemporal dementia with motor neuron disease. Neurology 76, 1886–1893. doi: 10.1212/WNL.0b013e31821d767b
Coyle-Gilchrist, I. T., Dick, K. M., Patterson, K., Vazquez Rodriquez, P., Wehmann, E., Wilcox, A., et al. (2016). Prevalence, characteristics and survival of frontotemporal lobar degeneration syndromes. Neurology 86, 1736–1743. doi: 10.1212/WNL.0000000000002638
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
De Santis, R., Alfano, V., De Turris, V., Colantoni, A., Santini, L., Garone, M. G., et al. (2019). Mutant FUS and ELAVL4 (HuD) aberrant crosstalk in amyotrophic lateral sclerosis. Cell Rep. 27, e53818–e53831. doi: 10.1016/j.celrep.2019.05.085
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, J., Yang, M., Chen, Y., Chen, X., Liu, J., Sun, S., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 11:e1005357. doi: 10.1371/journal.pgen.1005357
Dengler, R., Konstanzer, A., Kuther, G., Hesse, S., Wolf, W., Struppler, A., et al. (1990). Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve 13, 545–550. doi: 10.1002/mus.880130612
Deshpande, D., Higelin, J., Schoen, M., Vomhof, T., Boeckers, T. M., Demestre, M., et al. (2019). Synaptic FUS localization during motoneuron development and its accumulation in human ALS synapses. Front. Cell. Neurosci. 13:256. doi: 10.3389/fncel.2019.00256
Devenney, E. M., Ahmed, R. M., and Hodges, J. R. (2019). Frontotemporal dementia. Handb. Clin. Neurol. 167, 279–299. doi: 10.1016/B978-0-12-804766-8.00015-7
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Devlin, A. C., Burr, K., Borooah, S., Foster, J. D., Cleary, E. M., Geti, I., et al. (2015). Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 6:5999. doi: 10.1038/ncomms6999
Dewey, C. M., Cenik, B., Sephton, C. F., Dries, D. R., Mayer, P., 3rd, Good, S. K., et al. (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31, 1098–1108. doi: 10.1128/MCB.01279-10
Dickson, D. W., Baker, M. C., Jackson, J. L., DeJesus-Hernandez, M., Finch, N. A., Tian, S., et al. (2019). Extensive transcriptomic study emphasizes importance of vesicular transport in C9orf72 expansion carriers. Acta Neuropathol. Commun. 7:150. doi: 10.1186/s40478-019-0797-0
Dong, W., Ma, Y., Guan, F., Zhang, X., Chen, W., Zhang, L., et al. (2021). Ablation of C9orf72 together with excitotoxicity induces ALS in rats. FEBS J. 288, 1712–1723. doi: 10.1111/febs.15501
Dreyfuss, G., Matunis, M. J., Pinol-Roma, S., and Burd, C. G. (1993). hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem. 62, 289–321. doi: 10.1146/annurev.bi.62.070193.001445
Duan, W., Guo, M., Yi, L., Zhang, J., Bi, Y., Liu, Y., et al. (2019). Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging (Albany NY) 11, 2457–2476. doi: 10.18632/aging.101936
Dyer, M. S., Woodhouse, A., and Blizzard, C. A. (2021a). Cytoplasmic human TDP-43 mislocalization induces widespread dendritic spine loss in mouse upper motor neurons. Brain Sci. 11:883. doi: 10.3390/brainsci11070883
Dyer, M. S., Reale, L. A., Lewis, K. E., Walker, A. K., Dickson, T. C., Woodhouse, A., et al. (2021b). Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 157, 1300–1315. doi: 10.1111/jnc.15214
Ebstein, S. Y., Yagudayeva, I., and Shneider, N. A. (2019). Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep. 26, 364–373. doi: 10.1016/j.celrep.2018.12.045
Espinosa, J. S., Wheeler, D. G., Tsien, R. W., and Luo, L. (2009). Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron 62, 205–217. doi: 10.1016/j.neuron.2009.03.006
Ferrer, I. (1999). Neurons and their dendrites in frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 10, 55–60. doi: 10.1159/000051214
Ferrer, I., Roig, C., Espino, A., Peiro, G., and Matias Guiu, X. (1991). Dementia of frontal lobe type and motor neuron disease. A golgi study of the frontal cortex. J. Neurol. Neurosurg. Psychiatry 54, 932–934. doi: 10.1136/jnnp.54.10.932
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Floris, G., Borghero, G., Cannas, A., Di Stefano, F., Murru, M. R., Corongiu, D., et al. (2015). Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J. Neurol. 262, 375–384. doi: 10.1007/s00415-014-7575-5
Fogarty, M. J. (2019). Amyotrophic lateral sclerosis as a synaptopathy. Neural Regen. Res. 14, 189–192. doi: 10.4103/1673-5374.244782
Fogarty, M. J., Klenowski, P. M., Lee, J. D., Drieberg-Thompson, J. R., Bartlett, S. E., Ngo, S. T., et al. (2016). Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 6:37968. doi: 10.1038/srep37968
Fogarty, M. J., Mu, E. W. H., Lavidis, N. A., Noakes, P. G., and Bellingham, M. C. (2020). Size-dependent vulnerability of lumbar motor neuron dendritic degeneration in SOD1(G93A) mice. Anat. Rec. (Hoboken) 303, 1455–1471. doi: 10.1002/ar.24255
Fogarty, M. J., Mu, E. W. H., Lavidis, N. A., Noakes, P. G., and Bellingham, M. C. (2021). Size-dependent dendritic maladaptations of hypoglossal motor neurons in SOD1(G93A) mice. Anat. Rec. (Hoboken) 304, 1562–1581. doi: 10.1002/ar.24542
Fratta, P., Polke, J. M., Newcombe, J., Mizielinska, S., Lashley, T., Poulter, M., et al. (2015). Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiol. Aging 36, e1546–e1547. doi: 10.1016/j.neurobiolaging.2014.07.037
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6:72. doi: 10.1186/s40478-018-0579-0
Fujii, R., Okabe, S., Urushido, T., Inoue, K., Yoshimura, A., Tachibana, T., et al. (2005). The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 15, 587–593. doi: 10.1016/j.cub.2005.01.058
Fujii, R., and Takumi, T. (2005). TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. doi: 10.1242/jcs.02692
Gao, J., Wang, L., Yan, T., Perry, G., and Wang, X. (2019). TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 100:103396. doi: 10.1016/j.mcn.2019.103396
Garone, M. G., Alfano, V., Salvatori, B., Braccia, C., Peruzzi, G., Colantoni, A., et al. (2020). Proteomics analysis of FUS mutant human motoneurons reveals altered regulation of cytoskeleton and other ALS-linked proteins via 3’UTR binding. Sci. Rep. 10:11827. doi: 10.1038/s41598-020-68794-6
Garone, M. G., Birsa, N., Rosito, M., Salaris, F., Mochi, M., De Turris, V., et al. (2021). ALS-related FUS mutations alter axon growth in motoneurons and affect HuD/ELAVL4 and FMRP activity. Commun. Biol. 4:1025. doi: 10.1038/s42003-021-02538-8
Geevasinga, N., Loy, C. T., Menon, P., De Carvalho, M., Swash, M., Schrooten, M., et al. (2016). Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: a systematic review using individual patient data. Clin. Neurophysiol. 127, 2684–2691. doi: 10.1016/j.clinph.2016.04.005
Gelpi, E., Van Der Zee, J., Turon Estrada, A., Van Broeckhoven, C., and Sanchez-Valle, R. (2014). TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol. Appl. Neurobiol. 40, 225–230. doi: 10.1111/nan.12063
Genc, B., Jara, J. H., Lagrimas, A. K., Pytel, P., Roos, R. P., Mesulam, M. M., et al. (2017). Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 7:41765. doi: 10.1038/srep41765
Geser, F., Martinez-Lage, M., Robinson, J., Uryu, K., Neumann, M., Brandmeir, N. J., et al. (2009). Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch. Neurol. 66, 180–189. doi: 10.1001/archneurol.2008.558
Giandomenico, S. L., Alvarez-Castelao, B., and Schuman, E. M. (2022). Proteostatic regulation in neuronal compartments. Trends Neurosci. 45, 41–52. doi: 10.1016/j.tins.2021.08.002
Gijselinck, I., Van Langenhove, T., Van Der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
Gitcho, M. A., Bigio, E. H., Mishra, M., Johnson, N., Weintraub, S., Mesulam, M., et al. (2009). TARDBP 3’-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 118, 633–645. doi: 10.1007/s00401-009-0571-7
Gopal, P. P., Nirschl, J. J., Klinman, E., and Holzbaur, E. L. (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. U S A 114, E2466–E2475. doi: 10.1073/pnas.1614462114
Gordon, D., Dafinca, R., Scaber, J., Alegre-Abarrategui, J., Farrimond, L., Scott, C., et al. (2019). Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol Dis 121, 148–162. doi: 10.1016/j.nbd.2018.09.024
Gorenberg, E. L., and Chandra, S. S. (2017). The role of co-chaperones in synaptic proteostasis and neurodegenerative disease. Front. Neurosci. 11:248. doi: 10.3389/fnins.2017.00248
Gorrie, G. H., Fecto, F., Radzicki, D., Weiss, C., Shi, Y., Dong, H., et al. (2014). Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc. Natl. Acad. Sci. U S A 111, 14524–14529. doi: 10.1073/pnas.1405741111
Grace, G. M., Orange, J. B., Rowe, A., Findlater, K., Freedman, M., Strong, M. J., et al. (2011). Neuropsychological functioning in PLS: a comparison with ALS. Can. J. Neurol. Sci. 38, 88–97. doi: 10.1017/S0317167100120803
Grad, L. I., Rouleau, G. A., Ravits, J., and Cashman, N. R. (2017). Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb. Perspect. Med. 7:a024117. doi: 10.1101/cshperspect.a024117
Grossman, M. (2019). Amyotrophic lateral sclerosis - a multisystem neurodegenerative disorder. Nat. Rev. Neurol. 15, 5–6. doi: 10.1038/s41582-018-0103-y
Hall, C. E., Yao, Z., Choi, M., Tyzack, G. E., Serio, A., Luisier, R., et al. (2017). Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. 19, 1739–1749. doi: 10.1016/j.celrep.2017.05.024
Hammer, R. P. Jr., Tomiyasu, U., and Scheibel, A. B. (1979). Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp. Neurol. 63, 336–346. doi: 10.1016/0014-4886(79)90129-8
Han, J. H., Yu, T. H., Ryu, H. H., Jun, M. H., Ban, B. K., Jang, D. J., et al. (2013). ALS/FTLD-linked TDP-43 regulates neurite morphology and cell survival in differentiated neurons. Exp. Cell Res. 319, 1998–2005. doi: 10.1016/j.yexcr.2013.05.025
Handley, E. E., Pitman, K. A., Dawkins, E., Young, K. M., Clark, R. M., Jiang, T. C., et al. (2017). Synapse dysfunction of layer V pyramidal neurons precedes neurodegeneration in a mouse model of TDP-43 proteinopathies. Cereb. Cortex 27, 3630–3647. doi: 10.1093/cercor/bhw185
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hartmann, H., Hornburg, D., Czuppa, M., Bader, J., Michaelsen, M., Farny, D., et al. (2018). Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 1:e201800070. doi: 10.26508/lsa.201800070
Henstridge, C. M., Sideris, D. I., Carroll, E., Rotariu, S., Salomonsson, S., Tzioras, M., et al. (2018). Synapse loss in the prefrontal cortex is associated with cognitive decline in amyotrophic lateral sclerosis. Acta Neuropathol. 135, 213–226. doi: 10.1007/s00401-017-1797-4
Herranz-Martin, S., Chandran, J., Lewis, K., Mulcahy, P., Higginbottom, A., Walker, C., et al. (2017). Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis. Model. Mech. 10, 859–868. doi: 10.1242/dmm.029892
Herzog, J. J., Deshpande, M., Shapiro, L., Rodal, A. A., and Paradis, S. (2017). TDP-43 misexpression causes defects in dendritic growth. Sci. Rep. 7:15656. doi: 10.1038/s41598-017-15914-4
Herzog, J. J., Xu, W., Deshpande, M., Rahman, R., Suib, H., Rodal, A. A., et al. (2020). TDP-43 dysfunction restricts dendritic complexity by inhibiting CREB activation and altering gene expression. Proc. Natl. Acad. Sci. U S A 117, 11760–11769. doi: 10.1073/pnas.1917038117
Hicks, G. G., Singh, N., Nashabi, A., Mai, S., Bozek, G., Klewes, L., et al. (2000). Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat. Genet. 24, 175–179. doi: 10.1038/72842
Ho, W. Y., Agrawal, I., Tyan, S. H., Sanford, E., Chang, W. T., Lim, K., et al. (2021). Dysfunction in nonsense-mediated decay, protein homeostasis, mitochondrial function and brain connectivity in ALS-FUS mice with cognitive deficits. Acta Neuropathol. Commun. 9:9. doi: 10.1186/s40478-020-01111-4
Ho, W. Y., Tai, Y. K., Chang, J. C., Liang, J., Tyan, S. H., Chen, S., et al. (2019). The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 15, 827–842. doi: 10.1080/15548627.2019.1569441
Hodges, J. R., and Miller, B. (2001). The classification, genetics and neuropathology of frontotemporal dementia. Introduction to the special topic papers: part I. Neurocase 7, 31–35. doi: 10.1093/neucas/7.1.31
Hoell, J. I., Larsson, E., Runge, S., Nusbaum, J. D., Duggimpudi, S., Farazi, T. A., et al. (2011). RNA targets of wild-type and mutant FET family proteins. Nat. Struct. Mol. Biol. 18, 1428–1431. doi: 10.1038/nsmb.2163
Hofmann, J. W., Seeley, W. W., and Huang, E. J. (2019). RNA binding proteins and the pathogenesis of frontotemporal lobar degeneration. Annu. Rev. Pathol. 14, 469–495. doi: 10.1146/annurev-pathmechdis-012418-012955
Horoupian, D. S., Thal, L., Katzman, R., Terry, R. D., Davies, P., Hirano, A., et al. (1984). Dementia and motor neuron disease: morphometric, biochemical and Golgi studies. Ann. Neurol. 16, 305–313. doi: 10.1002/ana.410160306
Huang, S. L., Wu, L. S., Lee, M., Chang, C. W., Cheng, W. C., Fang, Y. S., et al. (2020). A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 8:3. doi: 10.1186/s40478-020-0881-5
Huber, N., Hoffmann, D., Giniatullina, R., Rostalski, H., Leskela, S., Takalo, M., et al. (2022). C9orf72 hexanucleotide repeat expansion leads to altered neuronal and dendritic spine morphology and synaptic dysfunction. Neurobiol. Dis. 162:105584. doi: 10.1016/j.nbd.2021.105584
Huey, E. D., Ferrari, R., Moreno, J. H., Jensen, C., Morris, C. M., Potocnik, F., et al. (2012). FUS and TDP43 genetic variability in FTD and CBS. Neurobiol. Aging 33:1016. doi: 10.1016/j.neurobiolaging.2011.08.004
Humphrey, J., Birsa, N., Milioto, C., Mclaughlin, M., Ule, A. M., Robaldo, D., et al. (2020). FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 48, 6889–6905. doi: 10.1093/nar/gkaa410
Iko, Y., Kodama, T. S., Kasai, N., Oyama, T., Morita, E. H., Muto, T., et al. (2004). Domain architectures and characterization of an RNA-binding protein, TLS. J. Biol. Chem. 279, 44834–44840. doi: 10.1074/jbc.M408552200
Inglis, F. M., Crockett, R., Korada, S., Abraham, W. C., Hollmann, M., Kalb, R. G., et al. (2002). The AMPA receptor subunit GluR1 regulates dendritic architecture of motor neurons. J. Neurosci. 22, 8042–8051. doi: 10.1523/JNEUROSCI.22-18-08042.2002
Ingre, C., Roos, P. M., Piehl, F., Kamel, F., and Fang, F. (2015). Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 7, 181–193. doi: 10.2147/CLEP.S37505
Iridoy, M. O., Zubiri, I., Zelaya, M. V., Martinez, L., Ausin, K., Lachen-Montes, M., et al. (2018). Neuroanatomical quantitative proteomics reveals common pathogenic biological routes between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int. J. Mol. Sci. 20:4. doi: 10.3390/ijms20010004
Ishigaki, S., Masuda, A., Fujioka, Y., Iguchi, Y., Katsuno, M., Shibata, A., et al. (2012). Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2:529. doi: 10.1038/srep00529
Jensen, B. K., Schuldi, M. H., Mcavoy, K., Russell, K. A., Boehringer, A., Curran, B. M., et al. (2020). Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72-ALS/FTD is rescued by SV2 replenishment. EMBO Mol. Med. 12:e10722. doi: 10.15252/emmm.201910722
Jiang, T., Handley, E., Brizuela, M., Dawkins, E., Lewis, K. E. A., Clark, R. M., et al. (2019). Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model. Mech. 12:dmm038109. doi: 10.1242/dmm.038109
Johnson, B. S., Snead, D., Lee, J. J., Mccaffery, J. M., Shorter, J., Gitler, A. D., et al. (2009). TDP-43 is intrinsically aggregation-prone and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.M109.010264
Johnson, V. E., Stewart, W., Trojanowski, J. Q., and Smith, D. H. (2011). Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 122, 715–726. doi: 10.1007/s00401-011-0909-9
Josephs, K. A., Whitwell, J. L., Tosakulwong, N., Weigand, S. D., Murray, M. E., Liesinger, A. M., et al. (2015). TAR DNA-binding protein 43 and pathological subtype of Alzheimer’s disease impact clinical features. Ann. Neurol. 78, 697–709. doi: 10.1002/ana.24493
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kamelgarn, M., Chen, J., Kuang, L., Jin, H., Kasarskis, E. J., Zhu, H., et al. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U S A 115, E11904–E11913. doi: 10.1073/pnas.1810413115
Kapeli, K., Pratt, G. A., Vu, A. Q., Hutt, K. R., Martinez, F. J., Sundararaman, B., et al. (2016). Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun. 7:12143. doi: 10.1038/ncomms12143
Kato, T., Hirano, A., and Donnenfeld, H. (1987). A Golgi study of the large anterior horn cells of the lumbar cords in normal spinal cords and in amyotrophic lateral sclerosis. Acta Neuropathol. 75, 34–40. doi: 10.1007/BF00686790
Keihani, S., Kluever, V., Mandad, S., Bansal, V., Rahman, R., Fritsch, E., et al. (2019). The long noncoding RNA neuroLNC regulates presynaptic activity by interacting with the neurodegeneration-associated protein TDP-43. Sci. Adv. 5:eaay2670. doi: 10.1126/sciadv.aay2670
Khalfallah, Y., Kuta, R., Grasmuck, C., Prat, A., Durham, H. D., Vande Velde, C., et al. (2018). TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 8:7551. doi: 10.1038/s41598-018-25767-0
Killin, L. O., Starr, J. M., Shiue, I. J., and Russ, T. C. (2016). Environmental risk factors for dementia: a systematic review. BMC Geriatr. 16:175. doi: 10.1186/s12877-016-0342-y
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. doi: 10.1016/j.brainres.2012.01.016
King, A. E., Woodhouse, A., Kirkcaldie, M. T., and Vickers, J. C. (2016). Excitotoxicity in ALS: overstimulation, or overreaction? Exp. Neurol. 275, 162–171. doi: 10.1016/j.expneurol.2015.09.019
Konietzny, A., Bar, J., and Mikhaylova, M. (2017). Dendritic actin cytoskeleton: structure, functions and regulations. Front. Cell. Neurosci. 11:147. doi: 10.3389/fncel.2017.00147
Koppers, M., Blokhuis, A. M., Westeneng, H. J., Terpstra, M. L., Zundel, C. A., Vieira De Sa, R., et al. (2015). C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438. doi: 10.1002/ana.24453
Kovacs, G. G., Murrell, J. R., Horvath, S., Haraszti, L., Majtenyi, K., Molnar, M. J., et al. (2009). TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy and chorea. Mov. Disord. 24, 1843–1847. doi: 10.1002/mds.22697
Kulkarni, V. A., and Firestein, B. L. (2012). The dendritic tree and brain disorders. Mol. Cell. Neurosci. 50, 10–20. doi: 10.1016/j.mcn.2012.03.005
Kurz, A., Kurz, C., Ellis, K., and Lautenschlager, N. T. (2014). What is frontotemporal dementia? Maturitas 79, 216–219. doi: 10.1016/j.maturitas.2014.07.001
Kweon, J. H., Kim, S., and Lee, S. B. (2017). The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep. 50, 5–11. doi: 10.5483/bmbrep.2017.50.1.131
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lagier-Tourenne, C., Polymenidou, M., Hutt, K. R., Vu, A. Q., Baughn, M., Huelga, S. C., et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 15, 1488–1497. doi: 10.1038/nn.3230
Lall, D., Lorenzini, I., Mota, T. A., Bell, S., Mahan, T. E., Ulrich, J. D., et al. (2021). C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 109, e82275–e82291. doi: 10.1016/j.neuron.2021.05.020
Lattante, S., Rouleau, G. A., and Kabashi, E. (2013). TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826. doi: 10.1002/humu.22319
Lau, D. H. W., Hartopp, N., Welsh, N. J., Mueller, S., Glennon, E. B., Morotz, G. M., et al. (2018). Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 9:327. doi: 10.1038/s41419-017-0022-7
LeBlang, C. J., Medalla, M., Nicoletti, N. W., Hays, E. C., Zhao, J., Shattuck, J., et al. (2020). Reduction of the RNA binding protein TIA1 exacerbates neuroinflammation in tauopathy. Front. Neurosci. 14:285. doi: 10.3389/fnins.2020.00285
Lepeta, K., Lourenco, M. V., Schweitzer, B. C., Martino Adami, P. V., Banerjee, P., Catuara-Solarz, S., et al. (2016). Synaptopathies: synaptic dysfunction in neurological disorders - a review from students to students. J. Neurochem. 138, 785–805. doi: 10.1111/jnc.13713
Lerga, A., Hallier, M., Delva, L., Orvain, C., Gallais, I., Marie, J., et al. (2001). Identification of an RNA binding specificity for the potential splicing factor TLS. J. Biol. Chem. 276, 6807–6816. doi: 10.1074/jbc.M008304200
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lipton, A. M., Cullum, C. M., Satumtira, S., Sontag, E., Hynan, L. S., White, C. L., et al. (2001). Contribution of asymmetric synapse loss to lateralizing clinical deficits in frontotemporal dementias. Arch. Neurol. 58, 1233–1239. doi: 10.1001/archneur.58.8.1233
Lipton, A. M., White, C. L., 3rd, and Bigio, E. H. (2004). Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 108, 379–385. doi: 10.1007/s00401-004-0900-9
Liscic, R. M., Alberici, A., Cairns, N. J., Romano, M., and Buratti, E. (2020). From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 15:31. doi: 10.1186/s13024-020-00373-9
Liu, X., Erikson, C., and Brun, A. (1996). Cortical synaptic changes and gliosis in normal aging, Alzheimer’s disease and frontal lobe degeneration. Dementia 7, 128–134. doi: 10.1159/000106867
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Liu, E. Y., Russ, J., Cali, C. P., Phan, J. M., Amlie-Wolf, A., Lee, E. B., et al. (2019). Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 27, e61409–e61421. doi: 10.1016/j.celrep.2019.04.003
Liu, E. Y., Russ, J., and Lee, E. B. (2020). Neuronal transcriptome from C9orf72 repeat expanded human tissue is associated with loss of C9orf72 function. Free Neuropathol. 1:23. doi: 10.17879/freeneuropathology-2020-2911
Loganathan, S., Lehmkuhl, E. M., Eck, R. J., and Zarnescu, D. C. (2019). To be or not to be‥.toxic-is RNA association With TDP-43 complexes deleterious or protective in neurodegeneration? Front. Mol. Biosci. 6:154. doi: 10.3389/fmolb.2019.00154
Lomen-Hoerth, C., Anderson, T., and Miller, B. (2002). The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. doi: 10.1212/wnl.59.7.1077
Lomen-Hoerth, C., Murphy, J., Langmore, S., Kramer, J. H., Olney, R. K., Miller, B., et al. (2003). Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 60, 1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d
Lopez-Erauskin, J., Tadokoro, T., Baughn, M. W., Myers, B., Mcalonis-Downes, M., Chillon-Marinas, C., et al. (2018). ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100, e7816–e7830. doi: 10.1016/j.neuron.2018.09.044
Lottes, E. N., and Cox, D. N. (2020). Homeostatic roles of the proteostasis network in dendrites. Front. Cell. Neurosci. 14:264. doi: 10.3389/fncel.2020.00264
Lualdi, M., Shafique, A., Pedrini, E., Pieroni, L., Greco, V., Castagnola, M., et al. (2021). C9ORF72 repeat expansion affects the proteome of primary skin fibroblasts in ALS. Int. J. Mol. Sci. 22:10385. doi: 10.3390/ijms221910385
Maharjan, N., Kunzli, C., Buthey, K., and Saxena, S. (2017). C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol. Neurobiol. 54, 3062–3077. doi: 10.1007/s12035-016-9850-1
Majumder, P., Chen, Y. T., Bose, J. K., Wu, C. C., Cheng, W. C., Cheng, S. J., et al. (2012). TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol. 124, 231–245. doi: 10.1007/s00401-012-1006-4
Majumder, P., Chu, J. F., Chatterjee, B., Swamy, K. B., and Shen, C. J. (2016). Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol. 132, 721–738. doi: 10.1007/s00401-016-1603-8
Malpetti, M., Holland, N., Jones, P. S., Ye, R., Cope, T. E., Fryer, T. D., et al. (2021). Synaptic density in carriers of C9orf72 mutations: a [(11) C]UCB-J PET study. Ann. Clin. Transl. Neurol. 8, 1515–1523. doi: 10.1002/acn3.51407
Marchand-Pauvert, V., Peyre, I., Lackmy-Vallee, A., Querin, G., Bede, P., Lacomblez, L., et al. (2019). Absence of hyperexcitability of spinal motoneurons in patients with amyotrophic lateral sclerosis. J. Physiol. 597, 5445–5467. doi: 10.1113/JP278117
Martineau, E., Di Polo, A., Vande Velde, C., and Robitaille, R. (2018). Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 7:e41973. doi: 10.7554/eLife.41973
Martinez-Silva, M. L., Imhoff-Manuel, R. D., Sharma, A., Heckman, C. J., Shneider, N. A., Roselli, F., et al. (2018). Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. eLife 7:e30955. doi: 10.7554/eLife.30955
Martins-De-Souza, D., Guest, P. C., Mann, D. M., Roeber, S., Rahmoune, H., Bauder, C., et al. (2012). Proteomic analysis identifies dysfunction in cellular transport, energy and protein metabolism in different brain regions of atypical frontotemporal lobar degeneration. J. Proteome Res. 11, 2533–2543. doi: 10.1021/pr2012279
Mastrocola, A. S., Kim, S. H., Trinh, A. T., Rodenkirch, L. A., and Tibbetts, R. S. (2013). The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 288, 24731–24741. doi: 10.1074/jbc.M113.497974
May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503. doi: 10.1007/s00401-014-1329-4
McGoldrick, P., Joyce, P. I., Fisher, E. M., and Greensmith, L. (2013). Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1832, 1421–1436. doi: 10.1016/j.bbadis.2013.03.012
Mizielinska, S., and Isaacs, A. M. (2014). C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function? Curr. Opin. Neurol. 27, 515–523. doi: 10.1097/WCO.0000000000000130
Moreno, F., Rabinovici, G. D., Karydas, A., Miller, Z., Hsu, S. C., Legati, A., et al. (2015). A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol. Commun. 3:19. doi: 10.1186/s40478-015-0190-6
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morlando, M., Dini Modigliani, S., Torrelli, G., Rosa, A., Di Carlo, V., Caffarelli, E., et al. (2012). FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. EMBO J. 31, 4502–4510. doi: 10.1038/emboj.2012.319
Mulkey, M. (2019). Understanding frontotemporal disease progression and management strategies. Nurs. Clin. North Am. 54, 437–448. doi: 10.1016/j.cnur.2019.04.011
Munoz, D. G., Neumann, M., Kusaka, H., Yokota, O., Ishihara, K., Terada, S., et al. (2009). FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 118, 617–627. doi: 10.1007/s00401-009-0598-9
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Nakashima-Yasuda, H., Uryu, K., Robinson, J., Xie, S. X., Hurtig, H., Duda, J. E., et al. (2007). Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 114, 221–229. doi: 10.1007/s00401-007-0261-2
Nakaya, T., and Maragkakis, M. (2018). Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 8:15575. doi: 10.1038/s41598-018-33964-0
Narayanan, R. K., Mangelsdorf, M., Panwar, A., Butler, T. J., Noakes, P. G., Wallace, R. H., et al. (2013). Identification of RNA bound to the TDP-43 ribonucleoprotein complex in the adult mouse brain. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 252–260. doi: 10.3109/21678421.2012.734520
Naujock, M., Stanslowsky, N., Bufler, S., Naumann, M., Reinhardt, P., Sterneckert, J., et al. (2016). 4-aminopyridine induced activity rescues hypoexcitable motor neurons from amyotrophic lateral sclerosis patient-derived induced pluripotent stem cells. Stem Cells 34, 1563–1575. doi: 10.1002/stem.2354
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/wnl.51.6.1546
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., Mackenzie, I. R., et al. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. doi: 10.1093/brain/awp214
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Ni, J., Ren, Y., Su, T., Zhou, J., Fu, C., Lu, Y., et al. (2021). Loss of TDP-43 function underlies hippocampal and cortical synaptic deficits in TDP-43 proteinopathies. Mol. Psychiatry. . [Online ahead of print]. doi: 10.1038/s41380-021-01346-0
Nicolas, G., Sevigny, M., Lecoquierre, F., Marguet, F., Deschenes, A., Del Pelaez, M. C., et al. (2022). A postzygotic de novo NCDN mutation identified in a sporadic FTLD patient results in neurochondrin haploinsufficiency and altered FUS granule dynamics. Acta Neuropathol. Commun. 10:20. doi: 10.1186/s40478-022-01314-x
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885. doi: 10.1007/s00401-017-1708-8
Nishimura, A. L., and Arias, N. (2021). Synaptopathy mechanisms in ALS caused by C9orf72 repeat expansion. Front. Cell. Neurosci. 15:660693. doi: 10.3389/fncel.2021.660693
O’Rourke, J. G., Bogdanik, L., Yanez, A., Lall, D., Wolf, A. J., Muhammad, A. K., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Oberstadt, M., Classen, J., Arendt, T., and Holzer, M. (2018). TDP-43 and cytoskeletal proteins in ALS. Mol. Neurobiol. 55, 3143–3151. doi: 10.1007/s12035-017-0543-1
Okamoto, K., Hirai, S., Shoji, M., Senoh, Y., and Yamazaki, T. (1990). Axonal swellings in the corticospinal tracts in amyotrophic lateral sclerosis. Acta Neuropathol. 80, 222–226. doi: 10.1007/BF00308929
Olney, J. W. (1978). “Neurotoxicity of excitatory amino acids,” in Kainic Acid as a Tool in Neurobiology, eds E. G. McGeer, J. W. Olney, and P. L. McGeer (New York: Raven Press), 95–121.
Olney, N. T., Spina, S., and Miller, B. L. (2017). Frontotemporal dementia. Neurol. Clin. 35, 339–374. doi: 10.1016/j.ncl.2017.01.008
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4:47. doi: 10.1186/s40478-016-0316-5
Onyike, C. U., and Diehl-Schmid, J. (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 25, 130–137. doi: 10.3109/09540261.2013.776523
Ozdilek, B. A., Thompson, V. F., Ahmed, N. S., White, C. I., Batey, R. T., Schwartz, J. C., et al. (2017). Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic. Acids Res. 45, 7984–7996. doi: 10.1093/nar/gkx460
Perkins, E. M., Burr, K., Banerjee, P., Mehta, A. R., Dando, O., Selvaraj, B. T., et al. (2021). Altered network properties in C9ORF72 repeat expansion cortical neurons are due to synaptic dysfunction. Mol. Neurodegener. 16:13. doi: 10.1186/s13024-021-00433-8
Perry, D. C., Brown, J. A., Possin, K. L., Datta, S., Trujillo, A., Radke, A., et al. (2017). Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 140, 3329–3345. doi: 10.1093/brain/awx254
Picchiarelli, G., Demestre, M., Zuko, A., Been, M., Higelin, J., Dieterle, S., et al. (2019). FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 22, 1793–1805. doi: 10.1038/s41593-019-0498-9
Picher-Martel, V., Valdmanis, P. N., Gould, P. V., Julien, J. P., and Dupre, N. (2016). From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 4:70. doi: 10.1186/s40478-016-0340-5
Polymenidou, M., Lagier-Tourenne, C., Hutt, K. R., Huelga, S. C., Moran, J., Liang, T. Y., et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468. doi: 10.1038/nn.2779
Pontes, M. H., Sevostyanova, A., and Groisman, E. A. (2015). When too much ATP is bad for protein synthesis. J. Mol. Biol. 427, 2586–2594. doi: 10.1016/j.jmb.2015.06.021
Pradhan, J., and Bellingham, M. C. (2021). Neurophysiological mechanisms underlying cortical hyper-excitability in amyotrophic lateral sclerosis: a review. Brain Sci. 11:549. doi: 10.3390/brainsci11050549
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Qiu, H., Lee, S., Shang, Y., Wang, W. Y., Au, K. F., Kamiya, S., et al. (2014). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 124, 981–999. doi: 10.1172/JCI72723
Qiu, H., Lee, S., Shang, Y., Wang, W. Y., Au, K. F., Kamiya, S., et al. (2021). ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 131:e149564. doi: 10.1172/JCI149564
Ragagnin, A. M. G., Shadfar, S., Vidal, M., Jamali, M. S., and Atkin, J. D. (2019). Motor neuron susceptibility in ALS/FTD. Front. Neurosci. 13:532. doi: 10.3389/fnins.2019.00532
Ratnavalli, E., Brayne, C., Dawson, K., and Hodges, J. R. (2002). The prevalence of frontotemporal dementia. Neurology 58, 1615–1621. doi: 10.1212/wnl.58.11.1615
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010. doi: 10.1016/s0896-6273(02)00737-7
Renga, V. (2022). Brain connectivity and network analysis in amyotrophic lateral sclerosis. Neurol. Res. Int. 2022:1838682. doi: 10.1155/2022/1838682
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riemslagh, F. W., Van Der Toorn, E. C., Verhagen, R. F. M., Maas, A., Bosman, L. W. J., Hukema, R. K., et al. (2021). Inducible expression of human C9ORF72 36x G4C2 hexanucleotide repeats is sufficient to cause RAN translation and rapid muscular atrophy in mice. Dis. Model. Mech. 14:dmm044842. doi: 10.1242/dmm.044842
Rizzu, P., Blauwendraat, C., Heetveld, S., Lynes, E. M., Castillo-Lizardo, M., Dhingra, A., et al. (2016). C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 4:37. doi: 10.1186/s40478-016-0306-7
Rogelj, B., Easton, L. E., Bogu, G. K., Stanton, L. W., Rot, G., Curk, T., et al. (2012). Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2:603. doi: 10.1038/srep00603
Ross, O. A., Rutherford, N. J., Baker, M., Soto-Ortolaza, A. I., Carrasquillo, M. M., DeJesus-Hernandez, M., et al. (2011). Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20, 3207–3212. doi: 10.1093/hmg/ddr227
Rubino, E., Mancini, C., Boschi, S., Ferrero, P., Ferrone, M., Bianca, S., et al. (2019). ATXN2 intermediate repeat expansions influence the clinical phenotype in frontotemporal dementia. Neurobiol. Aging 73:e7231. doi: 10.1016/j.neurobiolaging.2018.09.009
Russo, A., Scardigli, R., La Regina, F., Murray, M. E., Romano, N., Dickson, D. W., et al. (2017). Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum. Mol. Genet. 26, 1407–1418. doi: 10.1093/hmg/ddx035
Rutherford, N. J., Zhang, Y. J., Baker, M., Gass, J. M., Finch, N. A., Xu, Y. F., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4:e1000193. doi: 10.1371/journal.pgen.1000193
Sabatelli, M., Moncada, A., Conte, A., Lattante, S., Marangi, G., Luigetti, M., et al. (2013). Mutations in the 3’ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4748–4755. doi: 10.1093/hmg/ddt328
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi: 10.1016/j.ncl.2015.07.012
Sahadevan, S., Hembach, K. M., Tantardini, E., Perez-Berlanga, M., Hruska-Plochan, M., Megat, S., et al. (2021). Synaptic FUS accumulation triggers early misregulation of synaptic RNAs in a mouse model of ALS. Nat. Commun. 12:3027. doi: 10.1038/s41467-021-23188-8
Salam, S., Tacconelli, S., Smith, B. N., Mitchell, J. C., Glennon, E., Nikolaou, N., et al. (2021). Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 11:13613. doi: 10.1038/s41598-021-93189-6
Salmon, E., Bahri, M. A., Plenevaux, A., Becker, G., Seret, A., Delhaye, E., et al. (2021). In vivo exploration of synaptic projections in frontotemporal dementia. Sci. Rep. 11:16092. doi: 10.1038/s41598-021-95499-1
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Sasaki, S., and Iwata, M. (2007). Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66, 10–16. doi: 10.1097/nen.0b013e31802c396b
Sasaki, S., and Maruyama, S. (1994). Decreased synaptophysin immunoreactivity of the anterior horns in motor neuron disease. Acta Neuropathol. 87, 125–128. doi: 10.1007/BF00296180
Scekic-Zahirovic, J., Sendscheid, O., El Oussini, H., Jambeau, M., Sun, Y., Mersmann, S., et al. (2016). Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 35, 1077–1097. doi: 10.15252/embj.201592559
Schludi, M. H., Becker, L., Garrett, L., Gendron, T. F., Zhou, Q., Schreiber, F., et al. (2017). Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 134, 241–254. doi: 10.1007/s00401-017-1711-0
Schwartz, J. C., Ebmeier, C. C., Podell, E. R., Heimiller, J., Taatjes, D. J., Cech, T. R., et al. (2012). FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 26, 2690–2695. doi: 10.1101/gad.204602.112
Schweitzer, K., Decker, E., Zhu, L., Miller, R. E., Mirra, S. S., Spina, S., et al. (2006). Aberrantly regulated proteins in frontotemporal dementia. Biochem. Biophys. Res. Commun. 348, 465–472. doi: 10.1016/j.bbrc.2006.07.113
Seelaar, H., Klijnsma, K. Y., De Koning, I., Van Der Lugt, A., Chiu, W. Z., Azmani, A., et al. (2010). Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J. Neurol. 257, 747–753. doi: 10.1007/s00415-009-5404-z
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9:347. doi: 10.1038/s41467-017-02729-0
Sephton, C. F., Cenik, B., Cenik, B. K., Herz, J., and Yu, G. (2012). TDP-43 in central nervous system development and function: clues to TDP-43-associated neurodegeneration. Biol. Chem. 393, 589–594. doi: 10.1515/hsz-2012-0115
Sephton, C. F., Cenik, C., Kucukural, A., Dammer, E. B., Cenik, B., Han, Y., et al. (2011). Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 286, 1204–1215. doi: 10.1074/jbc.M110.190884
Sephton, C. F., Good, S. K., Atkin, S., Dewey, C. M., Mayer, P., 3rd, Herz, J., et al. (2010). TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 285, 6826–6834. doi: 10.1074/jbc.M109.061846
Sephton, C. F., Tang, A. A., Kulkarni, A., West, J., Brooks, M., Stubblefield, J. J., et al. (2014). Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc. Natl. Acad. Sci. U S A 111, E4769–E4778. doi: 10.1073/pnas.1406162111
Sephton, C. F., and Yu, G. (2015). The function of RNA-binding proteins at the synapse: implications for neurodegeneration. Cell. Mol. Life Sci. 72, 3621–3635. doi: 10.1007/s00018-015-1943-x
Sevigny, M., Bourdeau Julien, I., Venkatasubramani, J. P., Hui, J. B., Dutchak, P. A., Sephton, C. F., et al. (2020). FUS contributes to mTOR-dependent inhibition of translation. J. Biol. Chem. 295, 18459–18473. doi: 10.1074/jbc.RA120.013801
Shang, Y., and Huang, E. J. (2016). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. doi: 10.1016/j.brainres.2016.03.036
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7:10465. doi: 10.1038/ncomms10465
Shaywitz, A. J., and Greenberg, M. E. (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861. doi: 10.1146/annurev.biochem.68.1.821
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shih, Y.-T., and Hsueh, Y.-P. (2016). VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat. Commun. 7:11020. doi: 10.1038/ncomms11020
Shih, Y.-T., and Hsueh, Y.-P. (2018). The involvement of endoplasmic reticulum formation and protein synthesis efficiency in VCP- and ATL1-related neurological disorders. J. Biomed. Sci. 25:2. doi: 10.1186/s12929-017-0403-3
Shiihashi, G., Ito, D., Arai, I., Kobayashi, Y., Hayashi, K., Otsuka, S., et al. (2017). Dendritic homeostasis disruption in a novel frontotemporal dementia mouse model expressing cytoplasmic fused in sarcoma. EBioMedicine 24, 102–115. doi: 10.1016/j.ebiom.2017.09.005
Sidibe, H., Khalfallah, Y., Xiao, S., Gomez, N. B., Fakim, H., Tank, E. M. H., et al. (2021). TDP-43 stabilizes G3BP1 mRNA: relevance to amyotrophic lateral sclerosis/frontotemporal dementia. Brain 144, 3461–3476. doi: 10.1093/brain/awab217
Sivadasan, R., Hornburg, D., Drepper, C., Frank, N., Jablonka, S., Hansel, A., et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618. doi: 10.1038/nn.4407
Sieverding, K., Ulmer, J., Bruno, C., Satoh, T., Tsao, W., Freischmidt, A., et al. (2021). Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43(G298S) transgenic mice. Exp. Neurol. 335:113496. doi: 10.1016/j.expneurol.2020.113496
Slomnicki, L. P., Pietrzak, M., Vashishta, A., Jones, J., Lynch, N., Elliot, S., et al. (2016). Requirement of neuronal ribosome synthesis for growth and maintenance of the dendritic tree. J. Biol. Chem. 291, 5721–5739. doi: 10.1074/jbc.M115.682161
Smeyers, J., Banchi, E. G., and Latouche, M. (2021). C9ORF72: what it is, what it does and why it matters. Front. Cell. Neurosci. 15:661447. doi: 10.3389/fncel.2021.661447
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933. doi: 10.1016/j.neulet.2017.06.052
So, E., Mitchell, J. C., Memmi, C., Chennell, G., Vizcay-Barrena, G., Allison, L., et al. (2018). Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 27, 463–474. doi: 10.1093/hmg/ddx415
Sommer, D., Rajkumar, S., Seidel, M., Aly, A., Ludolph, A., Ho, R., et al. (2022). Aging-dependent altered transcriptional programs underlie activity impairments in human C9orf72-mutant motor neurons. Front. Mol. Neurosci. 15:894230. doi: 10.3389/fnmol.2022.894230
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Strong, M. J., Grace, G. M., Freedman, M., Lomen-Hoerth, C., Woolley, S., Goldstein, L. H., et al. (2009). Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 131–146. doi: 10.1080/17482960802654364
Sudhof, T. C., and Rizo, J. (2011). Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 3:a005637. doi: 10.1101/cshperspect.a005637
Sudria-Lopez, E., Koppers, M., De Wit, M., Van Der Meer, C., Westeneng, H. J., Zundel, C. A., et al. (2016). Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 132, 145–147. doi: 10.1007/s00401-016-1581-x
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Suzuki, N., Kato, S., Kato, M., Warita, H., Mizuno, H., Kato, M., et al. (2012). FUS/TLS-immunoreactive neuronal and glial cell inclusions increase with disease duration in familial amyotrophic lateral sclerosis with an R521C FUS/TLS mutation. J. Neuropathol. Exp. Neurol. 71, 779–788. doi: 10.1097/NEN.0b013e318264f164
Synofzik, M., Born, C., Rominger, A., Lummel, N., Schols, L., Biskup, S., et al. (2014). Targeted high-throughput sequencing identifies a TARDBP mutation as a cause of early-onset FTD without motor neuron disease. Neurobiol. Aging 35, 1212.e1–1215.e1. doi: 10.1016/j.neurobiolaging.2013.10.092
Szebenyi, K., Wenger, L. M. D., Sun, Y., Dunn, A. W. E., Limegrover, C. A., Gibbons, G. M., et al. (2021). Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 24, 1542–1554. doi: 10.1038/s41593-021-00923-4
Takada, L. T. (2015). The genetics of monogenic frontotemporal dementia. Dement. Neuropsychol. 9, 219–229. doi: 10.1590/1980-57642015DN93000003
Tan, A. Y., and Manley, J. L. (2009). The TET family of proteins: functions and roles in disease. J. Mol. Cell Biol. 1, 82–92. doi: 10.1093/jmcb/mjp025
Tank, E. M., Figueroa-Romero, C., Hinder, L. M., Bedi, K., Archbold, H. C., Li, X., et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9:2845. doi: 10.1038/s41467-018-05049-z
Tann, J. Y., Wong, L. W., Sajikumar, S., and Ibanez, C. F. (2019). Abnormal TDP-43 function impairs activity-dependent BDNF secretion, synaptic plasticity and cognitive behavior through altered Sortilin splicing. EMBO J. 38:e100989. doi: 10.15252/embj.2018100989
Theunissen, F., West, P. K., Brennan, S., Petrovic, B., Hooshmand, K., Akkari, P. A., et al. (2021). New perspectives on cytoskeletal dysregulation and mitochondrial mislocalization in amyotrophic lateral sclerosis. Transl. Neurodegener. 10:46. doi: 10.1186/s40035-021-00272-z
Tibshirani, M., Zhao, B., Gentil, B. J., Minotti, S., Marques, C., Keith, J., et al. (2017). Dysregulation of chromatin remodelling complexes in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 4142–4152. doi: 10.1093/hmg/ddx301
Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., et al. (2011). Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458. doi: 10.1038/nn.2778
Tortarolo, M., Lo Coco, D., Veglianese, P., Vallarola, A., Giordana, M. T., Marcon, G., et al. (2017). Amyotrophic lateral sclerosis, a multisystem pathology: insights into the role of TNFα. Mediators Inflamm. 2017:2985051. doi: 10.1155/2017/2985051
Tradewell, M. L., Yu, Z., Tibshirani, M., Boulanger, M. C., Durham, H. D., Richard, S., et al. (2012). Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum. Mol. Genet. 21, 136–149. doi: 10.1093/hmg/ddr448
Tsai, Y. L., Coady, T. H., Lu, L., Zheng, D., Alland, I., Tian, B., et al. (2020). ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 34, 785–805. doi: 10.1101/gad.335836.119
Tsai, Y. L., Mu, Y. C., and Manley, J. L. (2022). Nuclear RNA transcript levels modulate nucleocytoplasmic distribution of ALS/FTD-associated protein FUS. Sci. Rep. 12:8180. doi: 10.1038/s41598-022-12098-4
Turner, M. R., Barnwell, J., Al-Chalabi, A., and Eisen, A. (2012). Young-onset amyotrophic lateral sclerosis: historical and other observations. Brain 135, 2883–2891. doi: 10.1093/brain/aws144
Turner, M. R., Scaber, J., Goodfellow, J. A., Lord, M. E., Marsden, R., and Talbot, K. (2010). The diagnostic pathway and prognosis in bulbar-onset amyotrophic lateral sclerosis. J. Neurol. Sci. 294, 81–85. doi: 10.1016/j.jns.2010.03.028
Uchino, A., Takao, M., Hatsuta, H., Sumikura, H., Nakano, Y., Nogami, A., et al. (2015). Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol. Commun. 3:35. doi: 10.1186/s40478-015-0215-1
Udagawa, T., Fujioka, Y., Tanaka, M., Honda, D., Yokoi, S., Riku, Y., et al. (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 6:7098. doi: 10.1038/ncomms8098
Umoh, M. E., Dammer, E. B., Dai, J., Duong, D. M., Lah, J. J., Levey, A. I., et al. (2018). A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. doi: 10.15252/emmm.201708202
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi: 10.1016/S1474-4422(08)70071-1
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Van Langenhove, T., Van Der Zee, J., Sleegers, K., Engelborghs, S., Vandenberghe, R., Gijselinck, I., et al. (2010). Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74, 366–371. doi: 10.1212/WNL.0b013e3181ccc732
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vucic, S., and Kiernan, M. C. (2006). Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. 117, 1458–1466. doi: 10.1016/j.clinph.2006.04.016
Waagepetersen, H. S., Sonnewald, U., and Schousboe, A. (2003). Compartmentation of glutamine, glutamate and GABA metabolism in neurons and astrocytes: functional implications. Neuroscientist 9, 398–403. doi: 10.1177/1073858403254006
Waegaert, R., Dirrig-Grosch, S., Liu, H., Boutry, M., Luan, P., Loeffler, J. P., et al. (2022). Alteration of the neuromuscular junction and modifications of muscle metabolism in response to neuron-restricted expression of the CHMP2B(intron5) mutant in a mouse model of ALS-FTD syndrome. Biomolecules 12:497. doi: 10.3390/biom12040497
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wang, H. Y., Wang, I. F., Bose, J., and Shen, C. K. J. (2004). Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83, 130–139. doi: 10.1016/s0888-7543(03)00214-3
White, M. A., Kim, E., Duffy, A., Adalbert, R., Phillips, B. U., Peters, O. M., et al. (2018). TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 21, 552–563. doi: 10.1038/s41593-018-0113-5
Whitwell, J. L., Weigand, S. D., Gunter, J. L., Boeve, B. F., Rademakers, R., Baker, M., et al. (2011). Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology 77, 393–398. doi: 10.1212/WNL.0b013e318227047f
Wiens, K. M., Lin, H., and Liao, D. (2005). Rac1 induces the clustering of AMPA receptors during spinogenesis. J. Neurosci. 25, 10627–10636. doi: 10.1523/JNEUROSCI.1947-05.2005
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. doi: 10.1038/ncomms11253
Wishart, T. M., Parson, S. H., and Gillingwater, T. H. (2006). Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 65, 733–739. doi: 10.1097/01.jnen.0000228202.35163.c4
Wong, C. E., Jin, L. W., Chu, Y. P., Wei, W. Y., Ho, P. C., and Tsai, K. J. (2021). TDP-43 proteinopathy impairs mRNP granule mediated postsynaptic translation and mRNA metabolism. Theranostics 11, 330–345. doi: 10.7150/thno.51004
Wood, A., Gurfinkel, Y., Polain, N., Lamont, W., and Lyn Rea, S. (2021). Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 22:4705. doi: 10.3390/ijms22094705
Woo, J.-A. A., Liu, T., Trotter, C., Fang, C. C., De Narvaez, E., Lepochat, P., et al. (2017). Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 8:15558. doi: 10.1038/ncomms15558
Wu, L. S., Cheng, W. C., Chen, C. Y., Wu, M. C., Wang, Y. C., Tseng, Y. H., et al. (2019). Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun. 7:50. doi: 10.1186/s40478-019-0674-x
Wu, L. S., Cheng, W. C., Hou, S. C., Yan, Y. T., Jiang, S. T., and Shen, C. K. (2010). TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48, 56–62. doi: 10.1002/dvg.20584
Wu, J. I., Lessard, J., Olave, I. A., Qiu, Z., Ghosh, A., Graef, I. A., et al. (2007). Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108. doi: 10.1016/j.neuron.2007.08.021
Xiao, S., Mckeever, P. M., Lau, A., and Robertson, J. (2019). Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol. Commun. 7:161. doi: 10.1186/s40478-019-0812-5
Xiao, S., Sanelli, T., Dib, S., Sheps, D., Findlater, J., Bilbao, J., et al. (2011). RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci. 47, 167–180. doi: 10.1016/j.mcn.2011.02.013
Xiao, Y., Zhang, J., Shu, X., Bai, L., Xu, W., Wang, A., et al. (2020). Loss of mitochondrial protein CHCHD10 in skeletal muscle causes neuromuscular junction impairment. Hum. Mol. Genet. 29, 1784–1796. doi: 10.1093/hmg/ddz154
Xu, L., Liu, T., Liu, L., Yao, X., Chen, L., Fan, D., et al. (2020). Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. 267, 944–953. doi: 10.1007/s00415-019-09652-y
Yokoi, S., Udagawa, T., Fujioka, Y., Honda, D., Okado, H., Watanabe, H., et al. (2017). 3’UTR length-dependent control of SynGAP Isoform α2 mRNA by FUS and ELAV-like proteins promotes dendritic spine maturation and cognitive function. Cell Rep. 20, 3071–3084. doi: 10.1016/j.celrep.2017.08.100
Yokoseki, A., Shiga, A., Tan, C. F., Tagawa, A., Kaneko, H., Koyama, A., et al. (2008). TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542. doi: 10.1002/ana.21392
Yu, J., Lai, C., Shim, H., Xie, C., Sun, L., Long, C.-X., et al. (2018). Genetic ablation of dynactin p150(Glued) in postnatal neurons causes preferential degeneration of spinal motor neurons in aged mice. Mol. Neurodegener. 13:10. doi: 10.1186/s13024-018-0242-z
Zanette, G., Tamburin, S., Manganotti, P., Refatti, N., Forgione, A., and Rizzuto, N. (2002). Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin. Neurophysiol. 113, 1688–1697. doi: 10.1016/s1388-2457(02)00288-2
Zhang, Y. J., Jansen-West, K., Xu, Y. F., Gendron, T. F., Bieniek, K. F., Lin, W. L., et al. (2014). Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 128, 505–524. doi: 10.1007/s00401-014-1336-5
Zhang, W., Zhang, L., Liang, B., Schroeder, D., Zhang, Z. W., Cox, G. A., et al. (2016). Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 19, 557–559. doi: 10.1038/nn.4257
Keywords: amyotrophic lateral sclerosis (ALS; Lou Gehrig Disease), frontotemporal dementia (FTD), motor neuron disease (MND), synapse, dendrite, C9ORF72, TDP-43 (TAR DNA binding protein 43), fused in sarcoma (FUS)
Citation: Gelon PA, Dutchak PA and Sephton CF (2022) Synaptic dysfunction in ALS and FTD: anatomical and molecular changes provide insights into mechanisms of disease. Front. Mol. Neurosci. 15:1000183. doi: 10.3389/fnmol.2022.1000183
Received: 21 July 2022; Accepted: 01 September 2022;
Published: 03 October 2022.
Edited by:
Matthew Livesey, The University of Sheffield, United KingdomReviewed by:
Alberto Catanese, University of Ulm, GermanyMatthew J. Fogarty, Mayo Clinic, United States
Copyright © 2022 Gelon, Dutchak and Sephton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chantelle F. Sephton, chantelle.sephton.1@ulaval.ca