 Amin Daei Sorkhabi1,2†
Amin Daei Sorkhabi1,2† Aila Sarkesh1,2†
Aila Sarkesh1,2† Hossein Saeedi2Faroogh Marofi3Mahnaz Ghaebi4
Hossein Saeedi2Faroogh Marofi3Mahnaz Ghaebi4 Nicola Silvestris5
Nicola Silvestris5 Behzad Baradaran2,6*‡
Behzad Baradaran2,6*‡ Oronzo Brunetti7‡
Oronzo Brunetti7‡- 1Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
- 2Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
- 3Department of Hematology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
- 4Cancer Gene Therapy Research Center (CGRC), Zanjan University of Medical Sciences, Zanjan, Iran
- 5Medical Oncology Unit, Department of Human Pathology "G. Barresi", University of Messina, Messina, Italy
- 6Neurosciences Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
- 7Medical Oncology Unit-Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Tumori “Giovanni Paolo II” of Bari, Bari, Italy
A high percentage of malignant gliomas are infected by human cytomegalovirus (HCMV), and the endogenous expression of HCMV genes and their products are found in these tumors. HCMV antigen expression and its implications in gliomagenesis have emerged as a promising target for adoptive cellular immunotherapy (ACT) strategies in glioblastoma multiforme (GB) patients. Since antigen-specific T cells in the tumor microenvironments lack efficient anti-tumor immune response due to the immunosuppressive nature of glioblastoma, CMV-specific ACT relies on in vitro expansion of CMV-specific CD8+ T cells employing immunodominant HCMV antigens. Given the fact that several hurdles remain to be conquered, recent clinical trials have outlined the feasibility of CMV-specific ACT prior to tumor recurrence with minimal adverse effects and a substantial improvement in median overall survival and progression-free survival. This review discusses the role of HCMV in gliomagenesis, disease prognosis, and recent breakthroughs in harnessing HCMV-induced immunogenicity in the GB tumor microenvironment to develop effective CMV-specific ACT.
Introduction
Glioblastoma multiforme (GB), also known as WHO‐grade IV glioma, is the most prevalent and lethal primary malignant tumor of the central nervous system (CNS), with a dismal prognosis despite substantial breakthroughs in disease treatment and the introduction of multimodal strategies combining surgery and chemoradiation (1–3). The limited efficacy of current therapeutic schemes is a cause of concern in the therapy of patients. The suboptimal “standard of care” comprises microsurgical tumor debulking, concomitant radiochemotherapy with temozolomide (TMZ), and consolidation TMZ chemotherapy, with a median survival of 15 months and a 5-year survival rate of less than 5%, underlining the imperative need for alternative therapies (4, 5).
Several immunosuppressive mechanisms that characterize the immunological milieu of GB have been investigated in an effort to improve GB treatment strategies. Nevertheless, emerging immunotherapeutic interventions aimed at stimulating specific immune responses targeting solid tumors have provided patients with GB a glimpse of hope over the last decade. At present, cancer immunotherapy may be broadly classified into four categories: i. monoclonal antibodies (mAb) against inhibitory immune checkpoint molecules, ii. oncolytic virotherapy, iii. adoptive cellular therapy (ACT), and iv. cellular vaccine therapy (6). ACT is a highly personalized therapeutic approach that involves a patient’s T cells to deliberately redirect them to induce anti-tumor immune responses, rather than relying on the cytotoxic processes of conventional therapies, over which GB cells are refractory (7). However, the primary issue in this process is to overcome dysfunctional T cells, which are characterized by a loss of effector function, notably impaired cytotoxicity, and reduced secretion of inflammatory cytokines such as interleukin-2 (IL-2), tumor necrosis factor-α (TNF-α), and/or interferon-γ (IFN-γ) (8, 9).
Virus-associated malignancies are viable targets for the ACT, which leverage viral antigens expressed by cancer cells to redirect virus-specific T lymphocytes to eradicate tumor cells. There is a plethora of evidence that human cytomegalovirus (HCMV) antigens are abundantly expressed in GB tissue but not in normal brain tissue and potentially contribute to gliomagenesis (10). However, since the 80-90% prevalence of HCMV in healthy adults does not correspond to the prevalence of GB (1 out of 30,000), HCMV is not labeled as a classic oncogenic virus (11, 12). Thereby, HCMV does not directly induce malignant cellular transformation, but rather contributes to GB pathogenesis via “oncomodulation” of host cellular pathways (13). In this context, HCMV is not eliminated from the host body after primary infection, and any potential immunosuppressive condition, notably GB-associated immunodeficiency, reactivates this virus, leading to the activation of oncogenic pathways (14). These insights open up a prospective avenue for stimulating dysfunctional CMV-specific T cells to elicit competent anti-tumor immune responses in order to overcome the heterogeneous and antigen-escaping nature of GB. In this review, we will discuss the implications of HCMV in GB pathogenesis and prognosis, as well as recent advances of CMV-specific T cell therapy in GB patients.
HCMV Gliomagenesis
HCMV has been implicated in gliomagenesis through a variety of distinctive mechanisms, which are classified into six major hallmarks of cancer, including tumor proliferation and invasion, inhibition of tumor cell apoptosis, autophagy, promotion of angiogenesis, tumor-associated immunodeficiency, and stemness induction (15) (Figure 1).
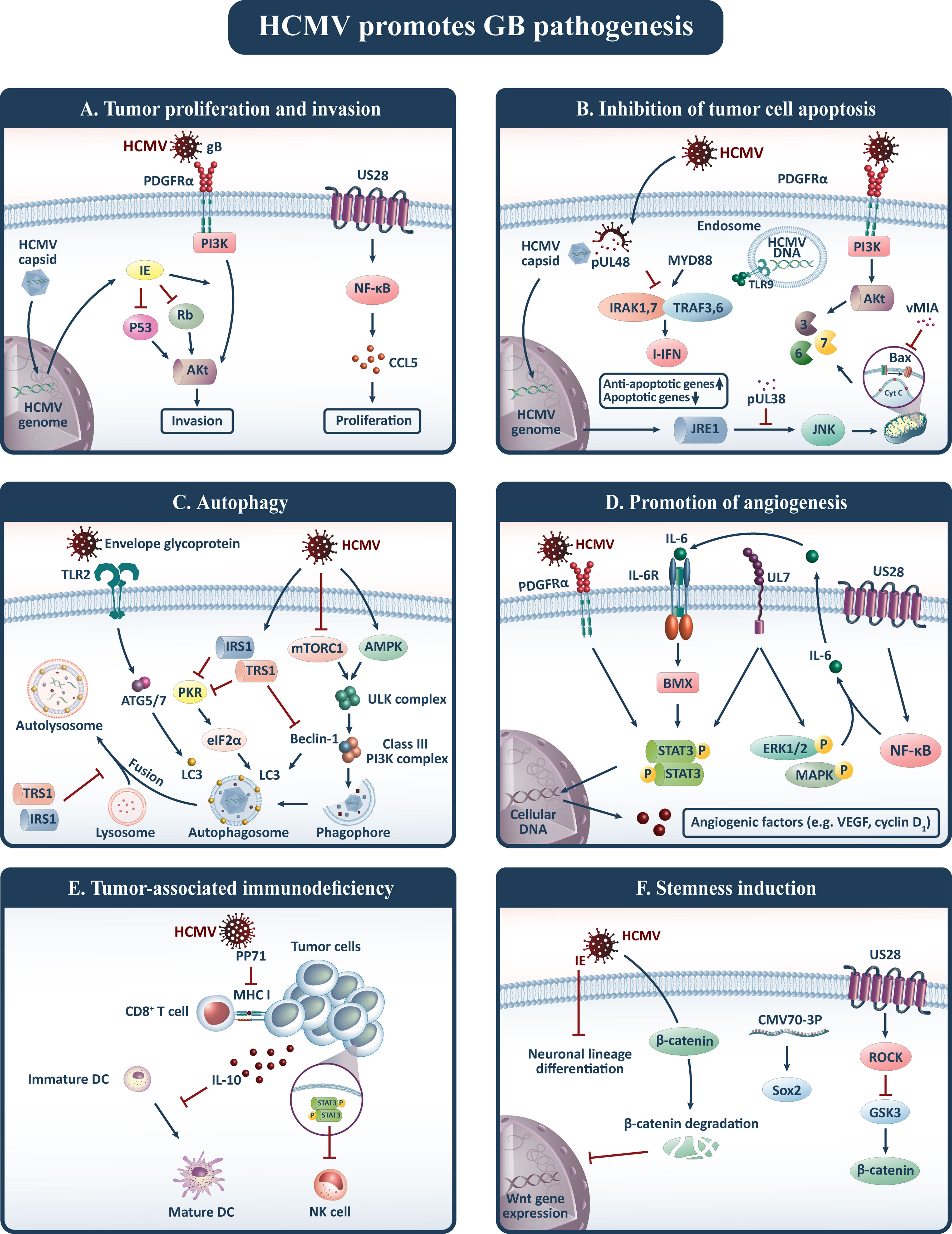
Figure 1 HCMV contributes to gliomagenesis and promotes six oncogenic pathways including: (A) tumor proliferation and invasion, (B) inhibition of tumor cell apoptosis, (C) autophagy, (D) promotion of angiogenesis, (E) tumor-associated immunodeficiency, and (F) stemness induction. Akt, Protein kinase B; CCL5, Chemokine (C-C motif) ligand 5; DC, Dendritic cell; HCMV, Human cytomegalovirus; IE, Immediate-early protein; I-IFN, type I interferon (I-IFN); IL, Interleukin; JNK, c-Jun N-terminal kinase; LC3, Light chain 3; MHC, Major histocompatibility complex; NF-κB, Nuclear factor-kappa B; NK, Natural killer; PDGFRα, Platelet-derived growth factor receptor alpha; PI3K, Phosphatidylinositol-3-kinase; Rb, Retinoblastoma; SOX2, SRY-Box 2; STAT3, Signal transducer and activator of transcription 3; TLRs, Toll-like receptors; TRAF3, TNF receptor-associated factor; Treg, Regulatory T cell; VEGF, Vascular endothelial growth factor; vMIA, viral mitochondria-localized inhibitor of apoptosis.
Tumor Proliferation and Invasion
Cell proliferation and invasion in human cancers are among the major areas of concern, leading to uncontrolled disease activity and fatal tumor cell invasion to the surrounding brain tissues, rendering it unresponsive to treatment interventions such as complete surgical resection. Hence explicating the mechanisms associated with tumor cell proliferation and invasion is critical in order to develop effective treatment approaches. HCMV generates oncomodulatory proteins that interact with tumor cell pathways, promoting tumorigenesis and tumor invasion. In this regard, the HCMV immediate-early (IE) proteins, which include IE1-72 or IE2-86, are an essential viral transcriptional activators that have been discovered to be expressed in more than 90% of glioma tumors (16). In particular, the HCMV-IE1-72 protein along with IE2-86 influences human glioblastoma growth by inactivating the p53 and the retinoblastoma (Rb) family of tumor suppressor proteins, resulting in cell cycle progression and apoptosis blockade while also actively engaging in cell cycle arrest to facilitate viral replication (17–20). However, it has been shown that these paradoxical cell cycle transformation mechanisms may enhance cellular proliferation, DNA synthesis, and entry into the cell cycle (21, 22). Furthermore, HCMV glycoprotein B (gB) is the viral abundant envelope glycoprotein that functions in the same way as the genuine ligand, platelet-derived growth factor (PDGF), by binding to the receptor tyrosine kinase (RTK)PDGFR-alpha (PDGFRα) to promote viral cellular entry (23). It has been demonstrated that gB is endogenously overexpressed in GB samples and contributes to the persistent phosphorylation of PDGFRα, protein kinase B (Akt), and Src. As a result, it triggers downstream RTK signaling of an essential oncogenic phosphatidylinositol-3-kinase (PI3K)-Akt axis, which is sufficient to enhance glial precursor proliferation and drive gliomagenesis (24, 25). Another HCMV-encoded protein that contributes to virus latency, cell-to-cell dissemination, immune evasion, and angioproliferative signaling is the chemokine receptor US28, which is extensively expressed in GB samples (26, 27). It has been proposed that US28 activates the hypoxia-inducible factor-1/pyruvate kinase M2 (HIF-1/PKM2) feedforward loop via Gαq-, CaMKII-, and Akt/mTOR-dependent pathways, directing proliferation, angiogenesis, and metabolic reprogramming in GB cells, and that US28 knockdown reverses proliferation in these cells (28). In agreement with these findings, in vivo animal studies established that nanobody-mediated US28 silencing restricts the tumor development of oncogenic glioma cells (29). These findings substantiate the notion that US28 is implicated in HCMV-mediated oncomodulation. Furthermore, immunofluorescence studies suggest that US28 upregulates phosphorylated signal transducer and activator of transcription 3 (p-STAT3) and endothelial nitric oxide synthase (e-NOS) that contribute to invasion of GB cells by upregulating pro-invasive factors including extracellular-signal-regulated kinase 1/2 (ERK1/2), focal adhesion kinase (FAK), and Src (30–32). FAK is an important player of integrin-dependent cell signaling that controls cell adhesion and migration, which are essential for metastasis. US28 constitutively activates FAK via phospholipase C- (PLC-), reducing cell adhesion and hence enhancing GB migration (33, 34).
Inhibition of Tumor Cell Apoptosis
Upon infection, one of the most important underlying mechanisms of viral persistence and replication in cells is the expression of proteins that restrict virus-mediated apoptosis. Studies have shown that HCMV enhances the expression of anti-apoptotic activating transcription factor 5 (ATF5), which is extensively upregulated in glioma cells and contributes to their survival (35). ATF5 tends to bind to an ATF5-specific regulatory element in the B-cell lymphoma/leukemia-2 (Bcl-2) P2 promoter to enhance Bcl-2 expression (36). This enhances anti-apoptotic Bcl-2 expression while decreasing apoptotic Bcl-2-associated X (Bax) protein expression, indicating that HCMV has an anti-apoptotic effect on glioma cells (37). Likewise, HCMV IE86 has been shown to impede apoptotic pathways in glioma cells through promoting heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP A2/B1)-mediated alternative splicing of Bcl-x, resulting in reduced Bcl-xS/Bcl-xL ratio (38). As well, HCMV-driven Bcl-2 and viral mitochondria-localized inhibitor of apoptosis (vMIA) are endowed with the ability to impede mitochondrial outer membrane permeabilization via interaction with Bax and adenine nucleotide translocase, as well as redirection of these proteins to other subcellular locations. As a result, infected cells’ proapoptotic potential is drastically reduced (39, 40). On the other hand, the vMIA interferes with the enzymatic activity of the inner membrane mitochondrial protein ATP synthasome, resulting in impaired phosphate transport and hence decreased mitochondrial ATP synthesis (41). As a consequence, since apoptosis is an energy-requiring process, bioenergy depletion inhibits it.
Furthermore, the HCMV IE1 and IE2 proteins are both mutagenic, intervene with the function of the p53 and Rb tumor suppressors, foster the S-phase, and suppress apoptosis in infected cells (42, 43). In this respect, IE1 expression was shown to significantly downregulate p53 mRNA transcription in a human glioma cell line (44). These viral proteins also hinder apoptosis via the PI3K cellular pathway (45). Additionally, HCMV deubiquitinase suppresses the generation of anti-cancer type I interferons (I-IFNs), which contributes to the upregulation of anti-apoptotic genes and down-regulation of apoptosis-inducing genes, encouraging cells to surpass the G1-phase rapidly (46).
Autophagy
The catabolic program of autophagy is a self-defense mechanism that entails the provision of cellular material for lysosomal degradation to supply energy and macromolecules. This mechanism protects tumor cells from nutrient deprivation, provides the substrates essential for cell survival, and modulates apoptosis (47–49). Evidence suggests that several autophagy-related proteins display a proviral function, implying that HCMV is privileged to utilize components of the autophagic machinery for its advantage. In this context, the cross-talk between autophagy-initiating kinase ULK1 and the HCMV tegument protein pp28 during viral replication has been shown necessary for effective virus release (50). On the other hand, studies on human fibroblasts infected with HCMV have revealed that the virus can induce autophagy by lipidation of microtubule-associated protein 1 light chain 3 (LC3), a hallmark of autophagy (51). In addition, HCMV envelope glycoprotein stimulates toll-like receptor 2 (TLR2) that may contribute to autophagy induction (52).
During the infection of human fibroblasts, HCMV regulates autophagy in two opposed directions. In the early stages of infection, HCMV induces autophagy independent of de novo viral protein synthesis, as evidenced by an increment in the number of autophagosomes and autophagic influx, while in the late stages of infection (18-24h post-infection), HCMV suppresses autophagy via mechanisms reliant on de novo viral protein synthesis (53). TRS1 and IRS1, for example, are two anti-autophagic proteins encoded by HCMV that have been found to act as antagonists for the eukaryotic initiation factor 2α/protein kinase R (eIF2α/PKR) signaling pathway by binding to PKR and thus blocking eIF2α phosphorylation, which has a central role in autophagy induction. However, it has been suggested that the interplay between TRS1 and Beclin 1 is the dominant contributor in this anti-autophagic procedure (53–55).
Overall, HCMV infection necessitates a baseline level of autophagy as well as specific autophagic proteins in order to maintain effective viral morphogenesis. Thus, autophagy in HCMV-infected cells is characterized as an oncomodulator mechanism that enhances tumor cell viability while restricting viral infection in the late stages of infection.
Promotion of Angiogenesis
HCMV-infected tumor cells may rewire signals to adjacent neoplastic and endothelial cells, perpetuating autocrine and paracrine signaling that fosters tumor cell motility. Endothelial cell invasion and tumor cell neovascularization are involved in the malignant progression of glioma, and the HCMV is a central player in these pathways (56). In support of this notion, studies on a GB mouse model have suggested that murine CMV (MCMV) infection enhances intratumoral blood flow and angiogenesis, which has been linked to platelet-derived growth factor-D (PDGF-D) mediated pericyte recruitment (57). The expression of PDGF-D is further upregulated by nuclear factor-kappa B (NF-κB) signaling, which is mediated by CMV glycoprotein-induced phosphorylation of p56 (58, 59).
Furthermore, research has revealed that HCMV encodes UL7 protein, which serves homology to the N-terminal V-like domain of carcinoembryonic antigen-related cell adhesion molecule 1 (CEA-CAM1), a pro-angiogenic factor involved in signal transduction and cellular adhesion. Experiments on HCMV-infected endothelial cells evidenced that CEA-CAM1phosphorylates STAT3 and ERK1/2 MAP kinases and contributes to the secretion of pro-angiogenic factors such as IL-6. Thus, it provokes angiogenesis under physiological and pathological conditions and enhances the viability of endothelial cells by upregulating the anti-apoptotic factor survivin (60–62). Likewise, HCMV-infected cells are found to express the HCMV-driven US28 protein on their surface, which triggers the NF-κB pathway, directing the production of IL-6 and cyclooxygenase-2 (COX-2). Following that, IL-6 and COX-2 bind to their cognate receptors and induce STAT3-dependent VEGF and Cyclin D1 transcription, hence enhancing angiogenesis (26, 63, 64). Similarly, UL33 is another HCMV-encoded protein linked to an oncogenic signature that overlaps with US28-mediated signaling to some extent. It activates GB angiogenic pathways, including the IL-6-STAT3 axis, VEGF and COX-2 promoters, and hypoxia-induced factor-1 (HIF-1) (65). HIF-1 has been discovered to regulate anterior gradient protein 2 (AGR2) expression, which is involved in the adaptive hypoxia response to stimulate angiogenesis and tumor development in GB (66). Furthermore, HCMV-infected glioma stem cells (GSCs) release CMV IL-10, which interacts with monocytes to enhance CMV transcriptional activity and subverts them to a tumor-supportive M2 macrophage/microglia phenotype. As a result, it initiates a feed-forward loop toward glioma cells by boosting angiogenic VEGF production (30).
Another HCMV-encoded protein that stimulates angiogenesis in GB is phosphoprotein 71 (pp71), which is implicated in the activation of early viral gene expression (67). It has been suggested that pp71 is abundantly expressed in stem-like (CD133+) primary GB cells, especially in high-grade gliomas, and promotes stem cell factor expression via an NF-κB-dependent pathway (68, 69). In this way, stem cell factor binds to the c-kit receptor tyrosine kinase, triggering upregulation of endothelial progenitor cell migration and thereby accelerating angiogenesis in hypoxic settings (70). Additionally, the HCMV phosphoprotein65 (pp65) is ubiquitously expressed in 91% of GB tissues and has been observed to colocalize with endocan in the tumor cell cytoplasm (71). Endothelial cell-specific molecule-1 (ESM-1) or endocan is a proteoglycan biomarker of neoangiogenesis whose expression is regulated by VEGF in endothelial cells. The HCMV pp65 protein indirectly enhances endocan expression by modulating VEGF and cytokines such as TNF-α and IL-6, thereby enhancing glioma neovasculature (72).
Importantly, microRNA (miR)-217 and miR-199a-5p expressions are upregulated in HCMV-infected endothelial cells, which can suppress sirtuin 1 (SIRT1) and forkhead box O3 (FOXO3a) endogenously. This culminates in reinforced migration and tube formation of infected endothelial cells (73, 74). Similarly, miR-138 upregulates p-STAT3 protein expression through downregulating SIRT1 expression in HCMV-infected human umbilical vein endothelial cells (HUVECs), stimulating them to migrate and form tubes (75).
Taken together, HCMV promotes angiogenesis through multilateral interactions with stem cells and endothelial cells, resulting in upregulation of angiogenic factors, increased endothelial cell viability, and recruitment to form vessels required to supply tumor cells with adequate amounts of nutrients and oxygen.
Tumor-Associated Immunodeficiency
Infection with HCMV and its interplay with immune system components disrupt innate and adaptive immune pathways to evade viral elimination, resulting in compromised anti-tumor responses and antigen presentation. HCMV pp71 restrains the accumulation of major histocompatibility complex (MHC) class I in order to regulate protein trafficking and evade virus-specific CD8+ T cell recognition (76). Likewise, this viral tegument protein augments C-C motif chemokine ligand (CCL) 2/monocyte chemoattractant protein 1 (MCP1) through transcriptional upregulation, which has been attributed to glioma tumor grade and recurrence (77, 78). Also, HCMV infection tends to increase CCL5 expression, and since this ligand binds to and triggers the US28 pathway, it amplifies US28-induced GB invasiveness (31). On the other hand, US28-mediated recruitment of monocyte and macrophage increases in response to C-X3-C motif chemokine ligand (CX3CL)1/fractalkine elicited by GSC and tumor-associated macrophage (TAM) in the GB niche (34).
Along with monocyte chemotaxis to the tumor niche, HCMV-encoded IL-10, which appears to mimic the immunomodulatory effects of human IL-10, shifts monocyte polarization toward a deactivated pro-tumoral M2c phenotype, limiting CD4+ T cell activation and proliferation substantially (79). It hinders MHC class II and the co-stimulatory protein CD86 while raising the co-stimulatory inhibitory molecule B7-H1 (30). Additionally, this viral homolog amplifies its immunosuppressive effects in myeloid cells by upregulating human IL-10 and its positive regulator, tumor progression locus 2 (TPL2), as well as interacting with its receptor. This leads to an increase in heme oxygenase 1 (HO-1) expression, which is associated with immunosuppression (80).
The HCMV-induced immunosuppressive state in GB is exacerbated by additional mechanisms. In this context, research on CMV-infected mouse models has shown that CD4+Foxp3-IL-10+ Tregs contribute to an effective antiviral T cell response and viral clearance, whilst IL-10- and TGF-ß-dependent pathways have been shown to boost anti-inflammatory pathways in GB (81, 82). Thus, Treg-induced TGF-ß-mediated inactivation of the NKG2D receptor might enable tumor cells in GB patients to evade recognition by immune system cytolytic effector cells, resulting in tumor progression (83). Similarly, HCMV-encoded UL18, UL40, and UL142 are viral MHC class I homologs that enhance surface human leukocyte antigen (HLA)-E expression, which binds to the NK cell inhibitory receptor NKG2A/CD94 to resist NK lysis (84–86).
Taken together, HCMV triggers pathways that enable tumor cells to evade immune recognition while also interacting with immune cells, boosting immunosuppressive characteristics and compromising anti-tumor immune responses.
Stemness Induction
GSCs are neoplastic units with a high potential for unlimited self-renewal, multipotency, and tumor initiation, and they are appropriate hosts for persistent neurotropic HCMV infection as they lack DNA repair mechanisms, facilitate viral immune evasion, and have an extended life span (87). These stem cells endow GB tumors with chemoradioresistance and drive tumor recurrence (88).
Several preclinical studies have shed light on the cross-talk between HCMV infection and stemness induction in GB tumors. HCMV has shown strong tropism to GSCs with high expression of CD133 (30). Similarly, studies have shown that GSCs infected with HCMV outlive and contain greater amounts of viral DNA compared to glioma cell lines, which coincides with upregulation of stemness markers in GSCs (89). Of note, the stemness markers are expressed in GB tumors with interdependence on HCMV IE expression, rendering tumor-derived GSCs unable to differentiate into astrocytic or neuronal phenotypes, promoting GSCs features and therefore increasing tumor aggressiveness (90). It was suggested that HCMV IE1/IE2 is co-expressed with stemness markers including CD133, Nestin, and SRY-box 2 (Sox2) and is implicated in the miR-45-Sox2 axis, resulting in enhanced self-renewal of GSCs. Accordingly, IE1/IE2 knockdown by an RNA-i-based strategy limits Sox2 expression, tumorsphere formation, and induces apoptosis in HCMV positive GSCs (43). These findings provide credence to the notion that HCMV IE protein expression has a significant detrimental predictive role in GB tumors through induction of stemness properties (91).
Moreover, the HCMV-encoded CMV70-3P miRNA is expressed in GB tissues and upregulates Sox2 expression, which is linked to stemness induction. In this respect, studies have demonstrated that suppression of CMV70-3P impedes tumor migration and invasion, indicating that it is a promising target for GB therapy (92).
Expression of HCMV in GB
Beyond the proposed oncomodulatory mechanisms of HCMV implicated in gliomagenesis, the detection of HCMV in GB tissue has emerged as a debate with many incongruous and contradictory results since 2002. These are primarily attributed to methodological discrepancies; yet, these findings offer a viable path for translating these viral protein expressions into promising anti-HCMV therapies to approach GB (10). To this end, several studies have employed immunohistochemistry and in situ hybridization techniques to detect HCMV, which are capable of illuminating the site of virus particles and RNA accumulated within cancer tissues but are predisposed to user errors. Others have employed a sensitive nested polymerase chain reaction (PCR) approach that is sensitive enough to detect extremely low concentrations of viral DNA utilizing robust amplifying strategies (93–95). Despite this, recent PCR assays failed to identify CMV pp65, gB, and IE in GB patients’ peripheral blood or tumor samples (93, 96, 97). Other investigations, on the other hand, highlighted the significance of time in the test’s sensitivity. Herpesviral genomes inside cells become resistant to PCR amplification over time as a consequence of physical deterioration, reflecting that the most recent samples can be detected to express HCMV genes much more abundantly (98, 99). However, HCMV nucleic acid and immunodominant proteins such as pp65 and IE1-72 have been detected in a significant proportion of GB samples through in situ hybridization techniques (10, 16, 100–102). Others, using sensitive PCR, immunohistochemistry, and in situ hybridization, discovered that a significant proportion of GB samples contained HCMV antigen and DNA and that the expression rate may vary depending on glioma grade, with high-grade gliomas expressing viral antigens more than low-grade gliomas (95, 100, 103). In another study, researchers employed all three techniques to analyze HCMV expression in high-grade gliomas both retrospectively and prospectively, and though they found no significant expression of HCMV regardless of the approach except in low levels in three out of 18 plasma samples at baseline and only one in follow-up (104). In line with these studies, others in various investigations attempted alternative approaches like droplet digital PCR (ddPCR) and next-generation sequencing (NGS) technologies but were still unable to detect HCMV in GB samples (105, 106). The studies also provided insight on the possibility of false-positive results attributable to unidentified cross-reactivity among HCMV antibodies that bind to non-viral human proteins, as well as how varying concentrations of HCMV monoclonal antibodies might contribute to false-positive staining (104, 107). Taking all of the controversies into account, a recent systematic review of 81 studies recruiting 7024 GB samples and 2420 blood samples revealed that HCMV is expressed in 36% and 45.2% of samples, respectively (108). Importantly, none of the healthy surrounding brain tissues employed as a control counterpart were detected to express HCMV genes or their products, indicating a high level of protection against off-tumor cytotoxicity for potential CMV-based immunotherapeutics for GB patients (72, 109–112).
Although the detection of HCMV in GB samples has remained controversial, the inevitability of HCMV oncomodulatory pathways in gliomagenesis has raised the topic of the implications of HCMV infection for disease prognosis. A case-control study revealed that low-grade HCMV infection in GB patients is linked to prolonged survival, and HCMV infection was determined to be a negative prognostic indicator in GB patients (113). Further, the same author conducted a retrospective study and indicated that HCMV infection is a predictive marker and that the low-grade infection coincided with a median 20-month longer survival compared to high-grade infected GB patients (91). These findings, however, appear to be achieved in the exclusion of considering additional prognosticators, such as isocitrate dehydrogenase 1 (IDH-1) mutational status, O6-methylguanine methyltransferase (MGMT) methylation status, TMZ medication status, and so on. On the contrary, the majority of evaluations found no predictive significance for HCMV infection in GB patients (110, 114–116). Nonetheless, a recent meta-analysis of 7 studies and around 500 patients found no statistically significant impact of HCMV infection on GB prognosis (117).
CMV-Specific Adoptive T Cell Therapy
The expression of HCMV antigens in GB tissues but not in healthy brain tissues primes an intriguing avenue to exploit viral antigens to extend pre-existing antiviral immunity for GB therapy. It has led to a rigorous clinical evaluation of the safety and prospective therapeutic value of autologous CMV-specific T cell administration as a consolidative therapy for recurrent GB. To that aim, researchers employ in vitro stimulation to redirect polyfunctional CMV-specific CD8+ T cells to target immunodominant HCMV antigens and elicit an effective anti-tumor immune response (118).
Characteristics of CMV-Specific T Cells Derived From GB Patients
In healthy individuals, CMV-specific CD8+ and CD4+ T cells account for 10% and 9% of blood CD8+ and CD4+ T cells, respectively (119). Aside from the intact large T-cell response, the phenotype of CMV-specific T cells appears to be distinctive, as evidenced by their terminal differentiation state. In terminally differentiated CMV-specific CD8+ T cells, there is a high expression of NK receptor CD57, which is associated with immunosenescence, as well as cytokines IFNγ and TNF-α, and the cytotoxic molecules granzyme B and perforin. Additionally, these cells lack expression of co-stimulatory CD27 and CD28 molecules that contribute to expansion and activation of T cell receptor (TCR)-stimulated T cells (120).
Preoperative and postoperative flow cytometry analysis of blood from CMV-seropositive GB patients showed that CD4+CD57+ and CD4+CD28- T cells were more abundant than their control counterparts, which were found to have a negative impact on survival in these patients (121). Since the CD4+CD28- phenotype is specific to HCMV infection, it is postulated that HCMV antigens expressed in GB tissues persistently stimulate TCRs and drive the expansion of CD4+ T cells that express CD57 and subsequently lose the activation marker CD28, indicating defect proliferative capacity and senescence of these cells (121, 122). However, further analysis revealed that the precursor frequency of CMV-specific CD8+ T cells was within the range typically observed in healthy virus carriers, and a significant fraction of these cells were positive for the CD57 with CD27-CD57+ CMV-specific T cells being more frequent in GB patients compared to healthy virus carriers (123).
The CMV-specific T cells lack optimal cytotoxicity in the GB tumor microenvironment due to hypofunctionality in the production of multiple cytokines. In this respect, investigations have shown that exposure of GB patients’ peripheral blood to CMV-pp65 antigen does not lead to a substantial rise in IFNγ production (124). In the context of hypo/dysfunctionality of CMV-specific T cells in GB patients, it was suggested that the large proportion of CMV-specific T cells (60-70%) had attenuated polyfunctionality in expressing TNF-α, IFNγ, macrophage inflammatory protein-1β (MIP-1β), or CD107a (a marker for CD8+T-cell degranulation), which may be reconstituted to some extent with in vitro stimulation (123). Furthermore, CMV-specific CD8+ T cells isolated from resected GB tumor tissue disclosed the same disabilities in expressing IFNγ, TNF, IL-2, or CD107a and lacking expression of the marker for tissue-resident T cells, CD103. Likewise, tumor-infiltrating CMV-specific T cells express augmented levels of inhibitory receptors programmed cell death protein 1 (PD-1), T cell immunoglobulin and mucin-domain containing molecule-3 (TIM-3), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and lower levels of transcription factors T-bet, eomesodermin (EOMES), and lymphoid enhancer-binding factor 1 (LEF-1), while having a 4-fold lower frequency in tumor tissue compared to T cells circulating in peripheral blood (125). The overexpression of inhibitory receptors in CMV-specific T cells is potentially the basis for the inefficiency of these cells to respond to persistent antigen exposure, which is commonly referred to as immunological exhaustion (126).
CMV-Specific T Cell Manufacture for ACT
To begin the process of manufacturing polyfunctional CMV-specific T cells for the ACT, peripheral blood mononuclear cells (PBMCs) are harvested from peripheral blood through leukapheresis, resuspended in the growth medium, and subsequently stimulated with specified HLA class I and II-restricted peptide epitopes from HCMV antigens, including pp65. However, other approaches comprising CMV-pp65 alone, CMV-pp65 coupled with tumor-associated antigens (TAA), or even no antigen are applicable in this procedure. Thereafter, the peptide-pulsed cells are incubated and supplied with recombinant cytokines, including IL-2, which is introduced on day 0 and continued every 2 to 3 days. After 14 days of expansion, the sterility and microbiological tests, as well as the intracellular cytokine assays, are undertaken to qualify them for cryopreservation until future use. For adoptive transfer, T cells are thawed into clinical-grade normal saline and reinfused intravenously (123, 125, 127–129) (Figure 2).
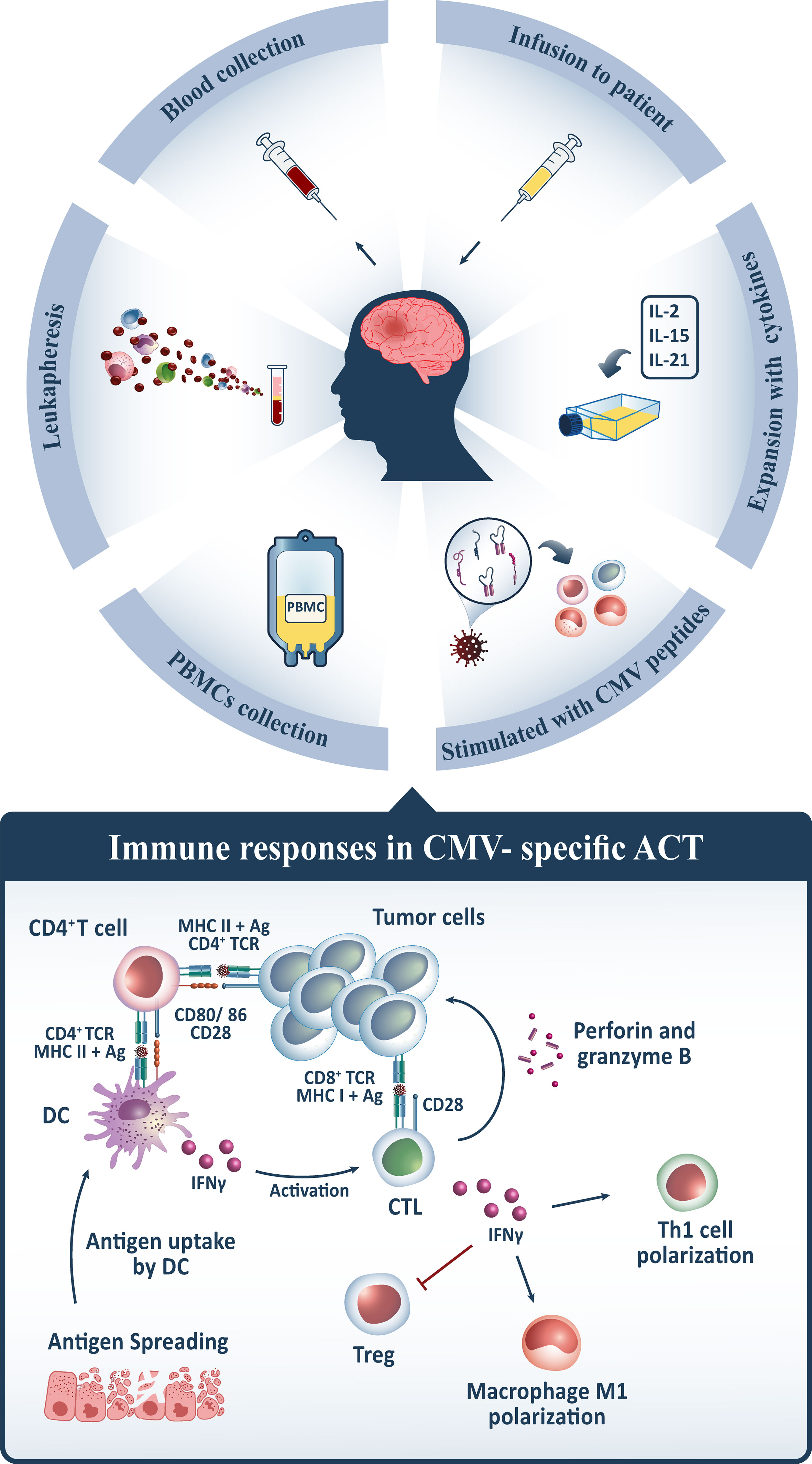
Figure 2 For CMV-specific ACT, PBMCs from GB patients’ peripheral blood are harvested, stimulated with CMV peptides, expanded in vitro by conditioning with multiple cytokines such as IL-2/IL-15/IL-21, and afterward reinfused intravenously. Following administration, CMV-specific CD8+ T cells traffic to tumor sites and recognize CMV antigens expressed on GB tumor cells, whilst interactions between CMV-specific CD4+ T cells and dendritic cells (DC) maintain its activation via IFN-γ release. CD8+ T cells release granzymes and perforin, which contribute to tumor cytolysis, and IFN-γ, which indirectly provokes other immune cells to elicit the desired anti-tumor immune response. It induces tumoricidal M1 polarization in macrophages, T helper1 (Th1) polarization, and inhibits regulatory T cells (Tregs) in the tumor microenvironment, which is associated with immunosuppression. Upon tumor cytolysis, tumor antigens are released, which are taken up by DCs and presented to cytotoxic T lymphocytes (CTLs), activating them and triggering a multi-antigen directed anti-tumor response.
Although the proposed procedure varies across trials, the key objective is to reconstitute polyfunctional CMV-specific T cells with adequate quantity, functional quality, high affinity, maturation, differentiation, and homing potential to elicit the desired clinical response in GB patients. In this respect, Luo et al. suggested an alternate cytokine cocktail consisting of IL-2, IL-15, and IL-21 to promote the expansion of high-affinity CMV-specific T cells with a homing phenotype (CCR6+ CXCR3+) and Th1 polarization (130). To elaborate, IL-2 is a commonly employed cytokine for T cell expansion that has concentration-dependent effects with some counter-productive consequences in the ACT setting (131, 132). IL-2 favors activation-induced cell death (AICD) and stimulates a variety of short-lived effector and long-lived memory T cell responses; however, it also contributes to the development, maintenance, and activity of Tregs, particularly the immunosuppressive phenotype, CD4+CD25+FOXP3+, which limits effector T cell response and proliferation (133, 134). To compensate for the negative impacts, the addition of IL-15 inhibits AICD and contributes to the development and persistence of long-lived memory CD8+ T cells. While the addition of IL-21 suppresses Tregs expansion and cooperates synergistically with IL-15 to boost antigen-specific CD8+ T cells expansion and IFNγ production, and both, unlike IL-2, induce suitable early-differentiated antigen-specific CD8+ T cells for immunotherapeutic applications (135–141). As well, the critical challenge in manufacturing methods is the migratory features and competency of CMV-specific T cells trafficking to the target region. Since healthy virus-naive individuals’ CMV-specific CD8+ T cells lack expression of CCR6, CXCR3, and CCR4, enhanced expression of CCR6 and CXCR3 via IL-2/IL-15/IL-21 conditioning might be a feasible approach to enhance CMV-specific CD8+ T cell trafficking to infection sites and eliciting anti-tumor secretions such as IFNγ and TNF-α (130).
Proposed Mechanism of CMV-Specific ACT
The purpose of contemporary immunotherapy is to enhance the function of cytotoxic T lymphocytes (CTLs) within the tumor microenvironment, boost CTL priming, and develop a long-term and effective anti-tumor immune response. To that aim, it is anticipated that after administration of CMV-specific T cells, these cells will traffic to tumor sites and interact with CMV-positive tumor cells to trigger cytolysis, therefore limiting tumor proliferation and progression. However, there are certain CMV-negative cells in the tumor tissue that may be targeted to some extent by ACT products. CD8+ T cells are a subset of lymphocytes that are specialized to recognize antigenic peptides presented by MHC class I molecules expressed by all tumor cell types; however, since persistent antigen exposure contributes to irreversible commitment to immunological exhaustion, CD4+ T cells are required to maintain the CD8+ T cell response and prevent exhaustion (142–145). CD4+ T cells interact with antigens and enable DCs to optimize antigen presentation and to deliver specific cytokines and co-stimulatory signals to encourage clonal expansion and differentiation of CD8+ T cells into an effector or memory T cell (146). Further, some DCs efficiently induce antigen cross-presentation of exogenous antigens on MHC class I molecules to activate CD8+ T cells through secretion of IFNγ (147). Following in vitro stimulation with immunodominant peptides, the CMV-specific CD8+ T cells reverse their competent cytolytic function to release several cytokines, including TNF-α and IFNγ that are involved in several anti-tumor immune responses. Upon antigen recognition, CTLs target tumor cells either directly through triggering synaptic exocytosis of cytotoxic granules comprising perforin and granzymes or indirectly through directing other immune components of tumor microenvironments toward tumor cells via secretion of cytokines, including IFNγ and TNF-α (148). IFNγ stimulates tumoricidal M1 macrophage polarization while also upregulating antigen-presenting machinery components such as MHC class I and II, and its synthesis and activities are amplified further by a positive feedback loop arising from inducing Th1 polarization. As well, increased IFNγ secretion suppresses Tregs in the tumor microenvironment (149). Of note, the potential role of CMV-specific ACT on T cell responses to other TAAs expressed in GB has been demonstrated (118). Following tumor cytolysis, CMV-specific ACT might have a bystander effect that extends T cell responses via epitope spreading. The antigen-presenting cells, including macrophages and DCs, engulf released antigens and cross-present them to T cells in order to prime CTLs, culminating in a multi-antigen directed immune response that targets a wide range of tumor cells (150) (Figure 2).
Preclinical and Clinical Findings of CMV-Specific ACT
In certain clinical settings, ACT employing genetically modified or unmodified antigen-specific T cells has resulted in long-term and promising clinical outcomes (151). Recent clinical trials have outlined the significance of CMV-specific ACT preceding GB tumor recurrence in being low-risk and offering enhanced progression-free survival and overall survival with compelling effective CMV-specific T cell response (Table 1).
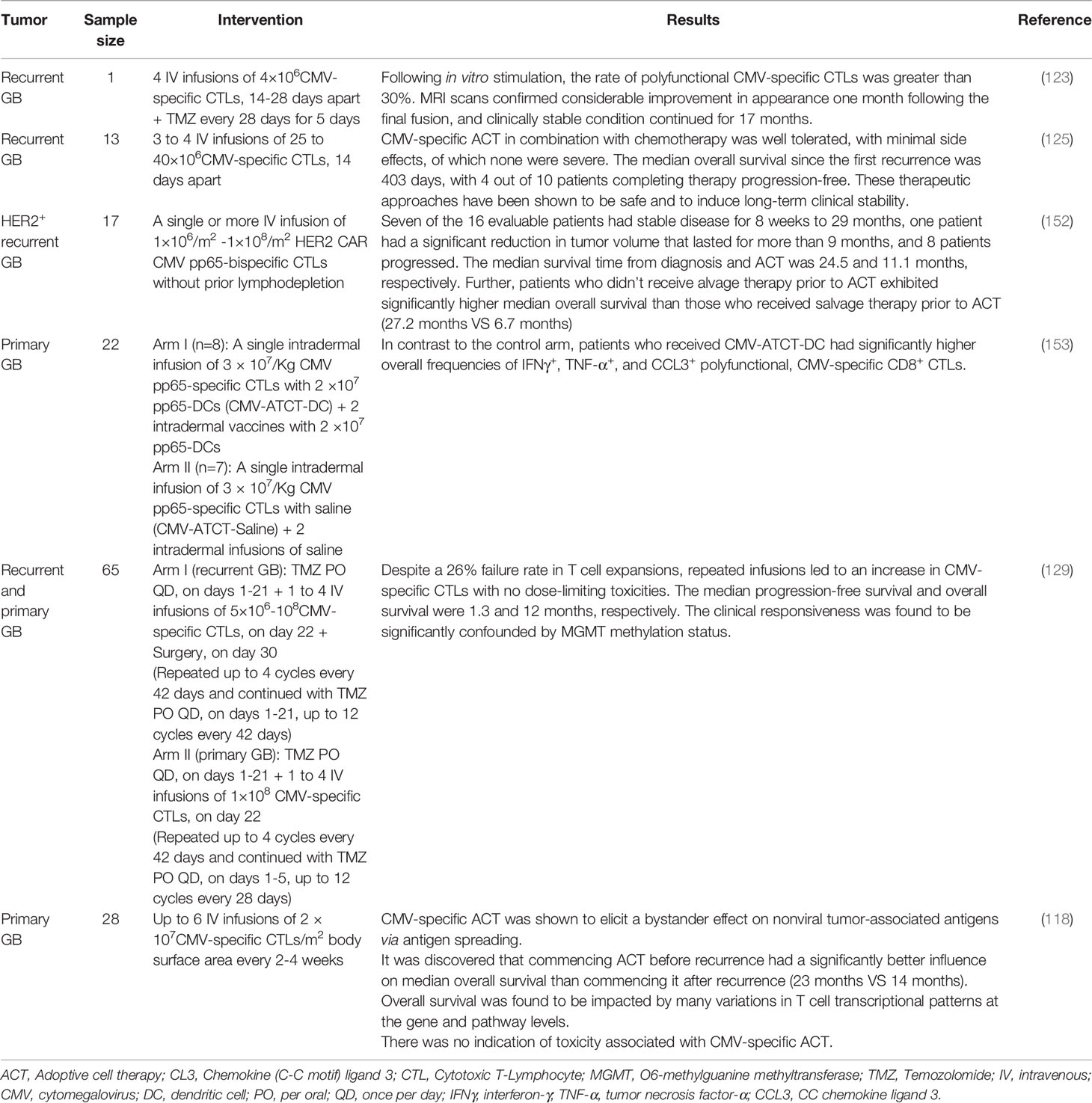
Table 1 Summary of clinical evidence on CMV-specific ACT.
Ghazi et al. discovered that in the peripheral blood of CMV-seropositive GB patients, the frequency of CMV-specific T cells, particularly those specific for the immunodominant pp65 antigen, was considerably lower than in healthy controls, implying the viability of polyclonal CMV-specific T cell generation via antigen-presenting cells (APCs) transduced with an adenoviral vector encoding IE1 and pp65 to kill CMV-positive target tissue (127). Further, Crough et al. administered four intravenous infusions of 40×106 CMV-specific CD8+ T cells to TMZ-treated patients at 14-28-day intervals in order to evaluate the polyfunctionality of these cells in vivo. It was found that in vitro stimulation of autologous CMV-specific CD8+ T cells induces a more than 20% rise in the proportion of polyfunctional virus-specific T cells, as well as a decrease in CD57+ antigen-specific T cells and an increase in CD27+ antigen-specific T cells. These led to a substantial decrease in the degree of enhancing tissue as evidenced by magnetic resonance imaging (MRI) scans and a marked improvement in other clinical parameters such as motor-neuron function (123).
For the first time in 2014, Schuessler et al. completed a phase 1 clinical trial in which they administered 3 to 4 infusions of 25 to 40×106 autologous CMV-specific T cells to 13 patients with recurrent GB, yielding the following findings: i, the clinical outcomes in these patients were found to be dose-independent of CMV-specific T cell infusion; ii, the clinical outcomes were unaffected by the detrimental consequences of concurrent standard therapeutic approaches; iii, CMV-specific ACT enhanced the median overall survival (>57 weeks), median progression-free survival (>35 weeks), and progression-free rate by 40% since the first recurrence, with only mild to moderate complications such as headaches or fatigue, none of which were serious (125). Conversely, Weathers et al. suggested that CMV-specific T cells, unlike CMV-negative T cells, exhibit anti-GB activity; despite the fact that seropositivity to CMV does not ensure tumor sensitivity to CMV-specific T cells, and failure in ex vivo expansion of CMV-specific T cells is highly attributable to the prior application of TMZ and other cytotoxic chemotherapies. Besides, they found that in spite of favorable enhancements in expressions of CD107a, TNF-α, IFNγ, IL-2, or effector memory markers (CD3+/CD8+/CCR7-/CD45RA-) during ex vivo expansions, there were no considerable alterations upon adoptive CMV-specific T cell transfer (129). Likewise, the stage of the disease impacts clinical responsiveness to ACT. In this regard, Smith et al. found that initiating adoptive CMV-specific T cell transfers prior to tumor recurrence, rather than thereafter, is associated with a 2.5 and 1.6-fold higher median progression-free survival (10 months vs 4 months) and median overall survival (23 months vs 14 months) since diagnosis, respectively (118).
Moreover, Nair et al. suggested in a preclinical study that autologous T cells stimulated with autologous DCs that were pulsed with CMV pp65 RNA could expand CMV pp65-specific T cells up to 10- to 20-fold, with a substantial rise in IFNγ+/CD4+ and IFNγ+/CD8+ cells (154). In line with these findings, Reap et al. performed a randomized clinical trial to assess the efficacy of combining DCs pulsed with CMV pp65 RNA with CMV pp65-specific T cells. They discovered that a single intravenous dose of 3×107 CMV pp65-specific T cells/kg in conjunction with three separate intradermal infusions of 2×107 DCs, as compared to a control group, had a significant influence on the frequency of polyfunctional CMV pp65-specific CD8+ T cells with enhanced capacity of simultaneous IFNγ, TNF-α, and CCL3 expression (153).
Since the heterogeneity of CMV pp65 antigen expression might be a major constraint in this therapeutic approach, some clinical trials have used genetically engineered CMV-specific T cells that express chimeric antigen receptors (CARs) to target GB-related surface proteins. Since GB tissues exhibit ineffective antigen presentation for T cell activation and express inhibitory surface ligands including PD-L1 and CTLA-4, the CAR construct incorporates co-stimulatory domains and the extracellular ligand-binding domain to bind the immunosuppressive signal and therefore decouple this binding from any immunosuppressive effects (155–157). To that aim, Ahmed et al. conducted a phase 1 clinical trial where they infused 1×106/m2-1×108/m2 pp65-specific T cells grafted with a second-generation human epidermal growth factor receptor 2 (HER2) CAR with a CD28.zeta signaling domain to 17 CMV seropositive patients with HER2+ recurrent GB who hadn’t undergone prior lymphodepletion. It resulted in partial clinical response and long-term disease stability in 1and 7 patients, respectively, as well as an increase in median overall survival (27 months since the first infusion). These findings pave the way for the development of CAR CMV bispecific immunotherapy in GB patients (152). However, further large-scale trials are warranted to develop an optimal protocol for incorporating adoptive transfer of CMV-specific T cells with appropriate genetic modifications, as well as DC vaccinations, to approach the GB patients’ treatment.
Influential Factors in Clinical Responsiveness to CMV-Specific ACT
Several cellular compositions and molecular characteristics of the tumor tissue have been established as having a substantial impact on clinical response to CMV-specific ACT in GB patients. As an instance, excessive PD-L1 expression in GB tissues has been identified as a significant pre-therapy marker that plays an essential part in ACT failure. According to Walker et al., long-term follow-up of patients who received CMV-specific ACT demonstrated that the frequency of PD-L1+ cells in tumor tissue prior to therapy correlated adversely with patient survival (158). It has been shown that the PD-1/PD-L1 axis contributes to CTL exhaustion and Treg augmentation, which protects tumor cells from CTL-mediated lysis (159). These findings call for more research into integrating anti-PD-L1 or genetically engineered T cells with PD-1 receptor blockade with CMV-specific ACT to maximize the clinical response of GB patients with predominant PD-L1 expression (160).
Furthermore, despite a paucity of evidence to indicate statistical relevance, additional cellular compositions such as CD3 and Sox2 have been linked to clinicopathological characteristics (158). Sox2 is a stemness marker involved in a variety of cellular activities, associated with maintaining the embryonic and pluripotent stem cell properties. It has been found to be overexpressed in high-grade gliomas, where it is linked to GSC generation and, therefore, poor prognosis (161–163). Sox2 expression is inversely correlated with CD3+ T cell infiltration, and patients with long-term survival had a relatively greater number of tumor-infiltrating CD3+ T cells (164).
MGMT, a cellular DNA repair enzyme that neutralizes the cytotoxic effects of alkylating drugs like TMZ, is another important biological component related to clinical response in GB patients (165). It has been demonstrated that MGMT promoter methylation is a mechanism of MGMT regulation in gliomas, which is a favorable predictor of progression-free survival and overall survival in TMZ-treated patients (166). In this respect, Weathers et al. revealed that GB patients with MGMT promoter-methylated tumors responded better to a combination of lymphodepleting dose-dense TMZ and CMV-specific ACT and that MGMT methylation status was a determining factor in radiographic outcomes (129). TMZ-induced lymphodepletion prior to ACT commencement exerts multilateral impact on the efficacy of ACT. Eliminating lymphocytes in the tumor microenvironment facilitates infused cells’ trafficking to the tumor site and their accessibility to various homeostatic cytokines, including IL-2, IL-7, and IL-15, while also reducing immunosuppressive cells that impair antigen-presentation. However, these outcomes are transient and will necessitate compensatory actions (167). Conversely, if immunotherapy is delivered shortly after or even during TMZ treatment, the immunosuppressive consequences will restrict immune cells from eliciting a robust anti-tumor response, signifying the timing of TMZ application for these patients. Nonetheless, large-scale immunological surveillance and clinical trials are warranted in order to optimize TMZ combination with CMV-based immunotherapeutic strategies (168).
Aside from MGMT promoter methylation and TMZ treatment, Walker et al. suggested that the presence of an IDH mutation is associated with a considerably better overall survival in GB patients (158). It was discovered that in IDH-mutated GB tumors, PD-L1 expression is downregulated relative to its IDH-wild-type counterpart, which correlated with greater T cell activation (169–171). On the other hand, IDH mutations were speculated to have immunomodulatory effects on both the natural course of disease and response to therapy, since IDH-mutated GB tumors exhibit low infiltration of TAMs, neutrophils, and myeloid cells, which is consistent with a survival benefit (172).
Moreover, Schuessler et al. and Smith et al. established that a signature of several genes in ACT products determines prolonged progression-free survival in patients with recurrent GB, including genes associated with T cell transcription factors, cytotoxic factors, checkpoint markers, and homing markers, as summarized in Table 2. The expression profile of these genes in ACT products contributes to CTL proliferation, cytotoxicity, chemotaxis, and interactions with multiple immunosuppressive and oncogenic pathways associated with tumor progression (118, 125).
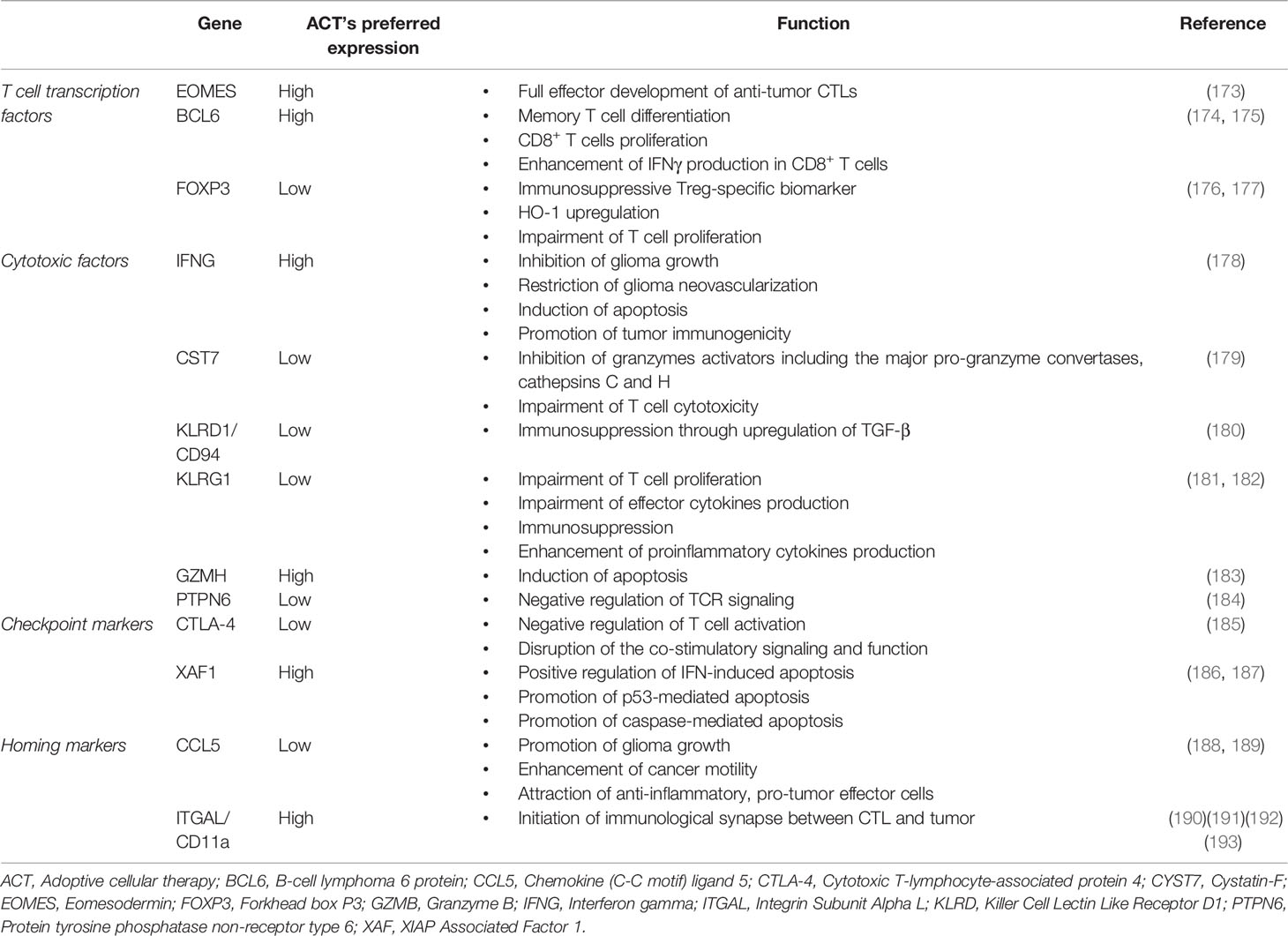
Table 2 The genetic profile of ACT products and their association with antitumor immune response.
Taken together, there is still a significant necessity to elucidate key factors in clinical responsiveness to CMV-specific ACT, which is acknowledged as an essential milestone toward developing more effective therapeutic approaches and eliminating the obstacles that contribute to treatment failure.
Conclusion
The most demanding aspect of developing immunotherapeutic approaches for solid tumors is identifying ideal tumor-specific antigens that can be targeted by immunotherapy while avoiding off-tumor toxicities and adverse effects. Like many difficult to treat solid cancers, GB tumors lack expression of antigenic epitopes and induce immunological exhaustion, both of which contribute to the tumor’s low immunogenicity and failure in immunotherapies. Hence, even minor evidence of tumor-specific antigen represents a ray of light for the development of effective antigen-directed immunotherapies. In this way, despite its controversial aspect, the concept of HCMV genes and their products expressions in GB tissues has attracted the interest of researchers seeking to translate it into an effective immunotherapeutic method for GB therapy. Particularly, studies have emphasized the exclusive expression of these antigens in GB tissues rather than adjacent healthy non-tumor tissues, indicating that CMV-specific immunotherapies outperform other methods owing to the lack of off-tumor CNS toxicities in this strategy. Several strategies, including antivirals, CMV antigen-pulsed DC vaccination, and CMV-specific ACT, have been evaluated in clinical trials, with encouraging indications of improved overall survival and progression-free survival in GB patients with minimal adverse effects.
In this review, we discussed HCMV implications for gliomagenesis and outlined various oncogenic pathways that HCMV modulates. Afterward, we discussed various studies with contradictory findings that have established HCMV expression in gliomas and its linkage with disease prognosis. Finally, the features of pre-existing anti-HCMV immunity were described, as well as their stimulation to efficiently target GB tumors. In summary, CMV-specific lymphocytes in the tumor microenvironment exhibit evidence of immunological exhaustion and senescence, making them unable to elicit the desired anti-tumor response, due in part to their specific nature to terminally differentiate and in part to the immunosuppressive nature of GB. As a result, employing immunodominant antigens of HCMV and ex vivo pre-conditioning with cytokines to reconstitute their cytotoxic activity is an essential step forward in this therapy. Although early phase studies of CMV-specific ACT have demonstrated substantial improvements in the appearance of MRI scans and patients’ survival, these findings are still preliminary, and research is required to broaden the application of this strategy in clinical settings. Additionally, determining the appropriate target group of patients for this approach based on influential markers in clinical responsiveness and, on the other hand, combining CMV-specific ACT with DC-vaccination, CAR T cell therapy, and immune checkpoint inhibitors are of interest to investigate in future studies to develop an efficacious multi-antigen directed approach with minimal side effects.
Author Contributions
ADS and AS extracted the data, took the lead in writing the manuscript and designing the figures and tables. HS designed the figures. FM, MG, and NS reviewed and edited the manuscript. OB and BB supervised and helped to develop the research question. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol (2015) 17 Suppl 4:iv1–iv62. doi: 10.1093/neuonc/nov189
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
3. Hasanpour Segherlou Z, Nouri-Vaskeh M, Noroozi Guilandehi S, Baghbanzadeh A, Zand R, Baradaran B, et al. GDF-15: Diagnostic, Prognostic, and Therapeutic Significance in Glioblastoma Multiforme. J Cell Physiol (2021) 236:(8):5564–5581. doi: 10.1002/jcp.30289
4. Davis ME. Epidemiology and Overview of Gliomas. Semin Oncol Nurs (2018) 34:420–9. doi: 10.1016/j.soncn.2018.10.001
5. Weller M, Van Den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO Guidelines on the Diagnosis and Treatment of Diffuse Gliomas of Adulthood. Nat Rev Clin Oncol (2021) 18:170–86. doi: 10.1038/s41571-020-00447-z
6. Wang J, Shen F, Yao Y, Wang LL, Zhu Y, Hu J. Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma. Front Oncol (2020) 10:59. doi: 10.3389/fonc.2020.00059
7. Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, et al. Intratumoral Heterogeneity: Pathways to Treatment Resistance and Relapse in Human Glioblastoma. Ann Oncol (2017) 28:1448–56. doi: 10.1093/annonc/mdx169
8. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and Are Functionally Impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
9. Mohme M, Schliffke S, Maire CL, Rünger A, Glau L, Mende KC, et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-Infiltrating Lymphocytes. Clin Cancer Res (2018) 24:4187–200. doi: 10.1158/1078-0432.CCR-17-2617
10. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res (2002) 62:3347–50.
11. Cannon MJ, Schmid DS, Hyde TB. Review of Cytomegalovirus Seroprevalence and Demographic Characteristics Associated With Infection. Rev Med Virol (2010) 20:202–13. doi: 10.1002/rmv.655
12. Mihelson N, Mcgavern DB. Viral Control of Glioblastoma. Viruses (2021) 13(7):1264. doi: 10.3390/v13071264
13. Hochhalter C, Carr C, O’neill B, Ware M, Strong M. The Association Between Human Cytomegalovirus and Glioblastomas: A Review. Neuroimmunol Neuroinflamm (2017) 4:96. doi: 10.20517/2347-8659.2017.10
14. La Rosa C, Diamond DJ. The Immune Response to Human CMV. Future Virol (2012) 7:279–93. doi: 10.2217/fvl.12.8
15. Joseph GP, Mcdermott R, Baryshnikova MA, Cobbs CS, Ulasov IV. Cytomegalovirus as an Oncomodulatory Agent in the Progression of Glioma. Cancer Lett (2017) 384:79–85. doi: 10.1016/j.canlet.2016.10.022
16. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, Mclendon RE, et al. Sensitive Detection of Human Cytomegalovirus in Tumors and Peripheral Blood of Patients Diagnosed With Glioblastoma. Neuro Oncol (2008) 10:10–8. doi: 10.1215/15228517-2007-035
17. Boldogh I, Abubakar S, Albrecht T. Activation of Proto-Oncogenes: An Immediate Early Event in Human Cytomegalovirus Infection. Science (1990) 247:561–4. doi: 10.1126/science.1689075
18. Hagemeier C, Caswell R, Hayhurst G, Sinclair J, Kouzarides T. Functional Interaction Between the HCMV IE2 Transactivator and the Retinoblastoma Protein. EMBO J (1994) 13:2897–903. doi: 10.1002/j.1460-2075.1994.tb06584.x
19. Zhu H, Shen Y, Shenk T. Human Cytomegalovirus IE1 and IE2 Proteins Block Apoptosis. J Virol (1995) 69:7960–70. doi: 10.1128/jvi.69.12.7960-7970.1995
20. Poma EE, Kowalik TF, Zhu L, Sinclair JH, Huang ES. The Human Cytomegalovirus IE1-72 Protein Interacts With the Cellular P107 Protein and Relieves P107-Mediated Transcriptional Repression of an E2F-Responsive Promoter. J Virol (1996) 70:7867–77. doi: 10.1128/jvi.70.11.7867-7877.1996
21. Castillo JP, Yurochko AD, Kowalik TF. Role of Human Cytomegalovirus Immediate-Early Proteins in Cell Growth Control. J Virol (2000) 74:8028–37. doi: 10.1128/JVI.74.17.8028-8037.2000
22. Cobbs CS, Soroceanu L, Denham S, Zhang W, Kraus MH. Modulation of Oncogenic Phenotype in Human Glioma Cells by Cytomegalovirus IE1-Mediated Mitogenicity. Cancer Res (2008) 68:724–30. doi: 10.1158/0008-5472.CAN-07-2291
23. Soroceanu L, Akhavan A, Cobbs CS. Platelet-Derived Growth Factor-Alpha Receptor Activation Is Required for Human Cytomegalovirus Infection. Nature (2008) 455:391–5. doi: 10.1038/nature07209
24. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, Vandenberg S, et al. PDGFR Alpha-Positive B Cells Are Neural Stem Cells in the Adult SVZ That Form Glioma-Like Growths in Response to Increased PDGF Signaling. Neuron (2006) 51:187–99. doi: 10.1016/j.neuron.2006.06.012
25. Cobbs C, Khan S, Matlaf L, Mcallister S, Zider A, Yount G, et al. HCMV Glycoprotein B Is Expressed in Primary Glioblastomas and Enhances Growth and Invasiveness via PDGFR-Alpha Activation. Oncotarget (2014) 5:1091–100. doi: 10.18632/oncotarget.1787
26. Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, Fraile-Ramos A, et al. HCMV-Encoded Chemokine Receptor US28 Mediates Proliferative Signaling Through the IL-6-STAT3 Axis. Sci Signal (2010) 3:ra58. doi: 10.1126/scisignal.2001180
27. Humby MS, O'connor CM. Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J Virol (2015) 90:2959–70. doi: 10.1128/JVI.02507-15
28. De Wit RH, Mujić-Delić A, Van Senten JR, Fraile-Ramos A, Siderius M, Smit MJ. Human Cytomegalovirus Encoded Chemokine Receptor US28 Activates the HIF-1α/PKM2 Axis in Glioblastoma Cells. Oncotarget (2016) 7:67966–85. doi: 10.18632/oncotarget.11817
29. Heukers R, Fan TS, De Wit RH, Van Senten JR, De Groof TWM, Bebelman MP, et al. The Constitutive Activity of the Virally Encoded Chemokine Receptor US28 Accelerates Glioblastoma Growth. Oncogene (2018) 37:4110–21. doi: 10.1038/s41388-018-0255-7
30. Dziurzynski K, Wei J, Qiao W, Hatiboglu MA, Kong LY, Wu A, et al. Glioma-Associated Cytomegalovirus Mediates Subversion of the Monocyte Lineage to a Tumor Propagating Phenotype. Clin Cancer Res (2011) 17:4642–9. doi: 10.1158/1078-0432.CCR-11-0414
31. Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, et al. Human Cytomegalovirus US28 Found in Glioblastoma Promotes an Invasive and Angiogenic Phenotype. Cancer Res (2011) 71:6643–53. doi: 10.1158/0008-5472.CAN-11-0744
32. Krishna BA, Miller WE, O'connor CM. US28: HCMV's Swiss Army Knife. Viruses (2018) 10(8):445. doi: 10.3390/v10080445
33. Miller WE, Zagorski WA, Brenneman JD, Avery D, Miller JL, O'connor CM. US28 Is a Potent Activator of Phospholipase C During HCMV Infection of Clinically Relevant Target Cells. PloS One (2012) 7:e50524. doi: 10.1371/journal.pone.0050524
34. Korbecki J, Gutowska I, Kojder I, Jeżewski D, Goschorska M, Łukomska A, et al. New Extracellular Factors in Glioblastoma Multiforme Development: Neurotensin, Growth Differentiation Factor-15, Sphingosine-1-Phosphate and Cytomegalovirus Infection. Oncotarget (2018) 9:7219–70. doi: 10.18632/oncotarget.24102
35. Sheng Z, Li L, Zhu LJ, Smith TW, Demers A, Ross AH, et al. A Genome-Wide RNA Interference Screen Reveals an Essential CREB3L2-ATF5-MCL1 Survival Pathway in Malignant Glioma With Therapeutic Implications. Nat Med (2010) 16:671–7. doi: 10.1038/nm.2158
36. Dluzen D, Li G, Tacelosky D, Moreau M, Liu DX. BCL-2 Is a Downstream Target of ATF5 That Mediates the Prosurvival Function of ATF5 in a Cell Type-Dependent Manner. J Biol Chem (2011) 286:7705–13. doi: 10.1074/jbc.M110.207639
37. Wang T, Qian D, Hu M, Li L, Zhang L, Chen H, et al. Human Cytomegalovirus Inhibits Apoptosis by Regulating the Activating Transcription Factor 5 Signaling Pathway in Human Malignant Glioma Cells. Oncol Lett (2014) 8:1051–7. doi: 10.3892/ol.2014.2264
38. Zhao R, Hu M, Liang S, Wang B, Yu B, Yang G, et al. IE86 Inhibits the Apoptosis and Promotes the Cell Proliferation of Glioma Cells via the hnRNP A2/B1-Mediated Alternative Splicing of Bcl-X. Int J Clin Exp Pathol (2019) 12:2775–85.
39. Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prévost MC, et al. Bcl-2 and Bax Modulate Adenine Nucleotide Translocase Activity. Cancer Res (2003) 63:541–6.
40. Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, et al. Cytomegalovirus Cell Death Suppressor vMIA Blocks Bax- But Not Bak-Mediated Apoptosis by Binding and Sequestering Bax at Mitochondria. Proc Natl Acad Sci USA (2004) 101:7988–93. doi: 10.1073/pnas.0401897101
41. Poncet D, Pauleau AL, Szabadkai G, Vozza A, Scholz SR, Le Bras M, et al. Cytopathic Effects of the Cytomegalovirus-Encoded Apoptosis Inhibitory Protein vMIA. J Cell Biol (2006) 174:985–96. doi: 10.1083/jcb.200604069
42. Shen Y, Zhu H, Shenk T. Human Cytomagalovirus IE1 and IE2 Proteins Are Mutagenic and Mediate "Hit-and-Run" Oncogenic Transformation in Cooperation With the Adenovirus E1A Proteins. Proc Natl Acad Sci USA (1997) 94:3341–5. doi: 10.1073/pnas.94.7.3341
43. Soroceanu L, Matlaf L, Khan S, Akhavan A, Singer E, Bezrookove V, et al. Cytomegalovirus Immediate-Early Proteins Promote Stemness Properties in Glioblastoma. Cancer Res (2015) 75:3065–76. doi: 10.1158/0008-5472.CAN-14-3307
44. Lee K, Jeon K, Kim JM, Kim VN, Choi DH, Kim SU, et al. Downregulation of GFAP, TSP-1, and P53 in Human Glioblastoma Cell Line, U373MG, by IE1 Protein From Human Cytomegalovirus. Glia (2005) 51:1–12. doi: 10.1002/glia.20179
45. Lukac DM, Alwine JC. Effects of Human Cytomegalovirus Major Immediate-Early Proteins in Controlling the Cell Cycle and Inhibiting Apoptosis: Studies With Ts13 Cells. J Virol (1999) 73:2825–31. doi: 10.1128/JVI.73.4.2825-2831.1999
46. Kumari P, Saha I, Narayanan A, Narayanan S, Takaoka A, Kumar NS, et al. Essential Role of HCMV Deubiquitinase in Promoting Oncogenesis by Targeting Anti-Viral Innate Immune Signaling Pathways. Cell Death Dis (2017) 8:e3078. doi: 10.1038/cddis.2017.461
47. Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy Is Activated in Colorectal Cancer Cells and Contributes to the Tolerance to Nutrient Deprivation. Cancer Res (2007) 67:9677–84. doi: 10.1158/0008-5472.CAN-07-1462
48. Mulcahy Levy JM, Thorburn A. Autophagy in Cancer: Moving From Understanding Mechanism to Improving Therapy Responses in Patients. Cell Death Differ (2020) 27:843–57. doi: 10.1038/s41418-019-0474-7
49. Shahverdi M, Hajiasgharzadeh K, Sorkhabi AD, Jafarlou M, Shojaee M, Jalili Tabrizi N, et al. The Regulatory Role of Autophagy-Related miRNAs in Lung Cancer Drug Resistance. BioMed Pharmacother (2022) 148:112735. doi: 10.1016/j.biopha.2022.112735
50. König P, Svrlanska A, Read C, Feichtinger S, Stamminger T. The Autophagy-Initiating Protein Kinase ULK1 Phosphorylates Human Cytomegalovirus Tegument Protein Pp28 and Regulates Efficient Virus Release. J Virol (2021) 95.
51. Mcfarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. Early Induction of Autophagy in Human Fibroblasts After Infection With Human Cytomegalovirus or Herpes Simplex Virus 1. J Virol (2011) 85:4212–21. doi: 10.1128/JVI.02435-10
52. Lussignol M, Esclatine A. “Chapter 2 - Cytomegalovirus and Autophagy”. In: Hayat MA, editor. Immunology. Academic Press (2018). p. 9–21.
53. Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, et al. The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction With Beclin 1. J Virol (2012) 86:2571–84. doi: 10.1128/JVI.05746-11
54. Lussignol M, Esclatine A. “Chapter 8 - Modulation of Autophagy by Herpesvirus Proteins”. In: Hayat MA, editor. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Amsterdam: Academic Press (2015). p. 145–58.
55. Humeau J, Leduc M, Cerrato G, Loos F, Kepp O, Kroemer G. Phosphorylation of Eukaryotic Initiation Factor-2α (Eif2α) in Autophagy. Cell Death Dis (2020) 11:433. doi: 10.1038/s41419-020-2642-6
56. Soroceanu L, Cobbs CS. Is HCMV a Tumor Promoter? Virus Res (2011) 157:193–203. doi: 10.1016/j.virusres.2010.10.026
57. Krenzlin H, Behera P, Lorenz V, Passaro C, Zdioruk M, Nowicki MO, et al. Cytomegalovirus Promotes Murine Glioblastoma Growth via Pericyte Recruitment and Angiogenesis. J Clin Invest (2019) 129:1671–83. doi: 10.1172/JCI123375
58. Yurochko AD, Hwang ES, Rasmussen L, Keay S, Pereira L, Huang ES. The Human Cytomegalovirus UL55 (Gb) and UL75 (Gh) Glycoprotein Ligands Initiate the Rapid Activation of Sp1 and NF-kappaB During Infection. J Virol (1997) 71:5051–9. doi: 10.1128/jvi.71.7.5051-5059.1997
59. Satoh J. Molecular Network of ChIP-Seq-Based NF-κb P65 Target Genes Involves Diverse Immune Functions Relevant to the Immunopathogenesis of Multiple Sclerosis. Mult Scler Relat Disord (2014) 3:94–106. doi: 10.1016/j.msard.2013.04.005
60. Botto S, Streblow DN, Defilippis V, White L, Kreklywich CN, Smith PP, et al. IL-6 in Human Cytomegalovirus Secretome Promotes Angiogenesis and Survival of Endothelial Cells Through the Stimulation of Survivin. Blood (2011) 117:352–61. doi: 10.1182/blood-2010-06-291245
61. Macmaniman JD, Meuser A, Botto S, Smith PP, Liu F, Jarvis MA, et al. Human Cytomegalovirus-Encoded Pul7 Is a Novel CEACAM1-Like Molecule Responsible for Promotion of Angiogenesis. mBio (2014) 5:e02035. doi: 10.1128/mBio.02035-14
62. Shomali N, Suliman Maashi M, Baradaran B, Daei Sorkhabi A, Sarkesh A, Mohammadi H, et al. Dysregulation of Survivin-Targeting microRNAs in Autoimmune Diseases: New Perspectives for Novel Therapies. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.839945
63. Maussang D, Verzijl D, Van Walsum M, Leurs R, Holl J, Pleskoff O, et al. Human Cytomegalovirus-Encoded Chemokine Receptor US28 Promotes Tumorigenesis. Proc Natl Acad Sci USA (2006) 103:13068–73. doi: 10.1073/pnas.0604433103
64. Maussang D, Langemeijer E, Fitzsimons CP, Stigter-Van Walsum M, Dijkman R, Borg MK, et al. The Human Cytomegalovirus-Encoded Chemokine Receptor US28 Promotes Angiogenesis and Tumor Formation via Cyclooxygenase-2. Cancer Res (2009) 69:2861–9. doi: 10.1158/0008-5472.CAN-08-2487
65. Van Senten JR, Bebelman MP, Fan TS, Heukers R, Bergkamp ND, Van Gasselt P, et al. The Human Cytomegalovirus-Encoded G Protein-Coupled Receptor UL33 Exhibits Oncomodulatory Properties. J Biol Chem (2019) 294:16297–308. doi: 10.1074/jbc.RA119.007796
66. Hong XY, Wang J, Li Z. AGR2 Expression Is Regulated by HIF-1 and Contributes to Growth and Angiogenesis of Glioblastoma. Cell Biochem Biophys (2013) 67:1487–95. doi: 10.1007/s12013-013-9650-4
67. Hwang J, Kalejta RF. Proteasome-Dependent, Ubiquitin-Independent Degradation of Daxx by the Viral Pp71 Protein in Human Cytomegalovirus-Infected Cells. Virology (2007) 367:334–8. doi: 10.1016/j.virol.2007.05.037
68. Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, et al. Neuronal and Glioma-Derived Stem Cell Factor Induces Angiogenesis Within the Brain. Cancer Cell (2006) 9:287–300. doi: 10.1016/j.ccr.2006.03.003
69. Matlaf LA, Harkins LE, Bezrookove V, Cobbs CS, Soroceanu L. Cytomegalovirus Pp71 Protein Is Expressed in Human Glioblastoma and Promotes Pro-Angiogenic Signaling by Activation of Stem Cell Factor. PloS One (2013) 8:e68176. doi: 10.1371/journal.pone.0068176
70. Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M. Stem Cell Factor/C-Kit Signaling Promotes the Survival, Migration, and Capillary Tube Formation of Human Umbilical Vein Endothelial Cells. J Biol Chem (2004) 279:18600–7. doi: 10.1074/jbc.M311643200
71. Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The Detection of CMV Pp65 and IE1 in Glioblastoma Multiforme. J Neurooncol (2011) 103:231–8. doi: 10.1007/s11060-010-0383-6
72. Xing Y, Wang Y, Wang S, Wang X, Fan D, Zhou D, et al. Human Cytomegalovirus Infection Contributes to Glioma Disease Progression via Upregulating Endocan Expression. Trans Res (2016) 177:113–26. doi: 10.1016/j.trsl.2016.06.008
73. Zhang S, Liu L, Wang R, Tuo H, Guo Y, Yi L, et al. MicroRNA-217 Promotes Angiogenesis of Human Cytomegalovirus-Infected Endothelial Cells Through Downregulation of SIRT1 and FOXO3A. PloS One (2013) 8:e83620. doi: 10.1371/journal.pone.0083620
74. Zhang S, Liu L, Wang R, Tuo H, Guo Y, Yi L, et al. MiR-199a-5p Promotes Migration and Tube Formation of Human Cytomegalovirus-Infected Endothelial Cells Through Downregulation of SIRT1 and eNOS. Arch Virol (2013) 158:2443–52. doi: 10.1007/s00705-013-1744-1
75. Zhang S, Liu L, Wang R, Tuo H, Guo Y, Yi L, et al. miR-138 Promotes Migration and Tube Formation of Human Cytomegalovirus-Infected Endothelial Cells Through the SIRT1/p-STAT3 Pathway. Arch Virol (2017) 162:2695–704. doi: 10.1007/s00705-017-3423-0
76. Trgovcich J, Cebulla C, Zimmerman P, Sedmak DD. Human Cytomegalovirus Protein Pp71 Disrupts Major Histocompatibility Complex Class I Cell Surface Expression. J Virol (2006) 80:951–63. doi: 10.1128/JVI.80.2.951-963.2006
77. Naing Z, Webel R, Hamilton S, Schmeiser C, Scott G, Marschall M, et al. Stimulatory Effects of Human Cytomegalovirus Tegument Protein Pp71 Lead to Increased Expression of CCL2 (Monocyte Chemotactic Protein-1) During Infection. J Gen Virol (2015) 96:1855–62. doi: 10.1099/vir.0.000101
78. Yang Q, Zhang J, Zhang X, Miao L, Zhang W, Jiang Z, et al. C-C Motif Chemokine Ligand 2/C-C Receptor 2 Is Associated With Glioma Recurrence and Poor Survival. Exp Ther Med (2021) 21:564. doi: 10.3892/etm.2021.9996
79. Avdic S, Cao JZ, Mcsharry BP, Clancy LE, Brown R, Steain M, et al. Human Cytomegalovirus Interleukin-10 Polarizes Monocytes Toward a Deactivated M2c Phenotype to Repress Host Immune Responses. J Virol (2013) 87:10273–82. doi: 10.1128/JVI.00912-13
80. Avdic S, Mcsharry BP, Steain M, Poole E, Sinclair J, Abendroth A, et al. Human Cytomegalovirus-Encoded Human Interleukin-10 (IL-10) Homolog Amplifies Its Immunomodulatory Potential by Upregulating Human IL-10 in Monocytes. J Virol (2016) 90:3819–27. doi: 10.1128/JVI.03066-15
81. Jost NH, Abel S, Hutzler M, Sparwasser T, Zimmermann A, Roers A, et al. Regulatory T Cells and T-Cell-Derived IL-10 Interfere With Effective Anti-Cytomegalovirus Immune Response. Immunol Cell Biol (2014) 92:860–71. doi: 10.1038/icb.2014.62
82. Li Z, Liu X, Guo R, Wang P. CD4(+)Foxp3(-) Type 1 Regulatory T Cells in Glioblastoma Multiforme Suppress T Cell Responses Through Multiple Pathways and Are Regulated by Tumor-Associated Macrophages. Int J Biochem Cell Biol (2016) 81:1–9. doi: 10.1016/j.biocel.2016.09.013
83. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-Beta Downregulates the Activating Receptor NKG2D on NK Cells and CD8+ T Cells in Glioma Patients. Neuro Oncol (2010) 12:7–13. doi: 10.1093/neuonc/nop009
84. Beck S, Barrell BG. Human Cytomegalovirus Encodes a Glycoprotein Homologous to MHC Class-I Antigens. Nature (1988) 331:269–72. doi: 10.1038/331269a0
85. Tomasec P, Braud VM, Rickards C, Powell MB, Mcsharry BP, Gadola S, et al. Surface Expression of HLA-E, an Inhibitor of Natural Killer Cells, Enhanced by Human Cytomegalovirus Gpul40. Science (2000) 1031 287(5455):1031. doi: 10.1126/science.287.5455.1031
86. Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, et al. Human Cytomegalovirus Encodes an MHC Class I-Like Molecule (UL142) That Functions to Inhibit NK Cell Lysis. J Immunol (2005) 175:7457–65. doi: 10.4049/jimmunol.175.11.7457
87. Tu SM. Stem Cell Theory of Cancer: Implications of a Viral Etiology in Certain Malignancies. Cancers (Basel) (2021) 13(11):2738. doi: 10.3390/cancers13112738
88. Alves ALV, Gomes INF, Carloni AC, Rosa MN, Da Silva LS, Evangelista AF, et al. Role of Glioblastoma Stem Cells in Cancer Therapeutic Resistance: A Perspective on Antineoplastic Agents From Natural Sources and Chemical Derivatives. Stem Cell Res Ther (2021) 12:206. doi: 10.1186/s13287-021-02231-x
89. Fiallos E, Judkins J, Matlaf L, Prichard M, Dittmer D, Cobbs C, et al. Human Cytomegalovirus Gene Expression in Long-Term Infected Glioma Stem Cells. PloS One (2014) 9:e116178. doi: 10.1371/journal.pone.0116178
90. Fornara O, Bartek J Jr., Rahbar A, Odeberg J, Khan Z, Peredo I, et al. Cytomegalovirus Infection Induces a Stem Cell Phenotype in Human Primary Glioblastoma Cells: Prognostic Significance and Biological Impact. Cell Death Differ (2016) 23:261–9. doi: 10.1038/cdd.2015.91
91. Rahbar A, Orrego A, Peredo I, Dzabic M, Wolmer-Solberg N, Strååt K, et al. Human Cytomegalovirus Infection Levels in Glioblastoma Multiforme Are of Prognostic Value for Survival. J Clin Virol (2013) 57:36–42. doi: 10.1016/j.jcv.2012.12.018
92. Ulasov IV, Kaverina NV, Ghosh D, Baryshnikova MA, Kadagidze ZG, Karseladze AI, et al. CMV70-3p miRNA Contributes to the CMV Mediated Glioma Stemness and Represents a Target for Glioma Experimental Therapy. Oncotarget (2017) 8:25989–99. doi: 10.18632/oncotarget.11175
93. Lau SK, Chen YY, Chen WG, Diamond DJ, Mamelak AN, Zaia JA, et al. Lack of Association of Cytomegalovirus With Human Brain Tumors. Mod Pathol (2005) 18:838–43. doi: 10.1038/modpathol.3800352
94. Poltermann S, Schlehofer B, Steindorf K, Schnitzler P, Geletneky K, Schlehofer JR. Lack of Association of Herpesviruses With Brain Tumors. J Neurovirol (2006) 12:90–9. doi: 10.1080/13550280600654573
95. Bhattacharjee B, Renzette N, Kowalik TF. Genetic Analysis of Cytomegalovirus in Malignant Gliomas. J Virol (2012) 86:6815–24. doi: 10.1128/JVI.00015-12
96. Priel E, Wohl A, Teperberg M, Nass D, Cohen ZR. Human Cytomegalovirus Viral Load in Tumor and Peripheral Blood Samples of Patients With Malignant Gliomas. J Clin Neurosci (2015) 22:326–30. doi: 10.1016/j.jocn.2014.06.099
97. Garcia-Martinez A, Alenda C, Irles E, Ochoa E, Quintanar T, Rodriguez-Lescure A, et al. Lack of Cytomegalovirus Detection in Human Glioma. Virol J (2017) 14:216. doi: 10.1186/s12985-017-0885-3
98. Millhouse S, Su YH, Zhang X, Wang X, Song BP, Zhu L, et al. Evidence That Herpes Simplex Virus DNA Derived From Quiescently Infected Cells In Vitro, and Latently Infected Cells In Vivo, Is Physically Damaged. J Neurovirol (2010) 16:384–98. doi: 10.3109/13550284.2010.515651
99. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant Association of Multiple Human Cytomegalovirus Genomic Loci With Glioblastoma Multiforme Samples. J Virol (2012) 86:854–64. doi: 10.1128/JVI.06097-11
100. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of Human Cytomegalovirus in Different Histological Types of Gliomas. Acta Neuropathol (2008) 116:79–86. doi: 10.1007/s00401-008-0359-1
101. Shamran HA, Kadhim HS, Hussain AR, Kareem A, Taub DD, Price RL, et al. Detection of Human Cytomegalovirus in Different Histopathological Types of Glioma in Iraqi Patients. BioMed Res Int (2015) 2015:642652. doi: 10.1155/2015/642652
102. Wakefield A, Pignata A, Ghazi A, Ashoori A, Hegde M, Landi D, et al. Is CMV a Target in Pediatric Glioblastoma? Expression of CMV Proteins, Pp65 and IE1-72 and CMV Nucleic Acids in a Cohort of Pediatric Glioblastoma Patients. J Neurooncol (2015) 125:307–15. doi: 10.1007/s11060-015-1905-z
103. Huang R, Qian D, Hu M, Zhang X, Song J, Li L, et al. Association Between Human Cytomegalovirus Infection and Histone Acetylation Level in Various Histological Types of Glioma. Oncol Lett (2015) 10:2812–20. doi: 10.3892/ol.2015.3638
104. Holdhoff M, Guner G, Rodriguez FJ, Hicks JL, Zheng Q, Forman MS, et al. Absence of Cytomegalovirus in Glioblastoma and Other High-Grade Gliomas by Real-Time PCR, Immunohistochemistry, and In Situ Hybridization. Clin Cancer Res (2017) 23:3150–7. doi: 10.1158/1078-0432.CCR-16-1490
105. Lin CT, Leibovitch EC, Almira-Suarez MI, Jacobson S. Human Herpesvirus Multiplex ddPCR Detection in Brain Tissue From Low- and High-Grade Astrocytoma Cases and Controls. Infect Agent Cancer (2016) 11:32. doi: 10.1186/s13027-016-0081-x
106. Strong MJ, Blanchard ET, Lin Z, Morris CA, Baddoo M, Taylor CM, et al. A Comprehensive Next Generation Sequencing-Based Virome Assessment in Brain Tissue Suggests No Major Virus - Tumor Association. Acta Neuropathol Commun (2016) 4:71. doi: 10.1186/s40478-016-0338-z
107. Yamashita Y, Ito Y, Isomura H, Takemura N, Okamoto A, Motomura K, et al. Lack of Presence of the Human Cytomegalovirus in Human Glioblastoma. Mod Pathol (2014) 27:922–9. doi: 10.1038/modpathol.2013.219
108. Peredo-Harvey I, Rahbar A, Söderberg-Nauclér C. Presence of the Human Cytomegalovirus in Glioblastomas-A Systematic Review. Cancers (Basel) (2021) 13(20):5051. doi: 10.3390/cancers13205051
109. Ahani N, Nikravesh A, Shirkoohi R, Karimi Arzenani M, Rokouei M, Alipour Eskandani M. Detection of Human Cytomegalovirus in Glioma Tumor Tissues. Comp Clin Pathol (2014) 23:1321–30. doi: 10.1007/s00580-013-1783-8
110. Ding D, Han S, Wang Z, Guo Z, Wu A. Does the Existence of HCMV Components Predict Poor Prognosis in Glioma? J Neurooncol (2014) 116:515–22. doi: 10.1007/s11060-013-1350-9
111. Stangherlin LM, Castro FL, Medeiros RS, Guerra JM, Kimura LM, Shirata NK, et al. Human Cytomegalovirus DNA Quantification and Gene Expression in Gliomas of Different Grades. PloS One (2016) 11:e0159604. doi: 10.1371/journal.pone.0159604
112. Yuan Z, Ye X, Zhu L, Zhang N, An Z, Zheng WJ. Virome Assembly and Annotation in Brain Tissue Based on Next-Generation Sequencing. Cancer Med (2020) 9:6776–90. doi: 10.1002/cam4.3325
113. Rahbar A, Stragliotto G, Orrego A, Peredo I, Taher C, Willems J, et al. Low Levels of Human Cytomegalovirus Infection in Glioblastoma Multiforme Associates With Patient Survival; -a Case-Control Study. Herpesviridae (2012) 3:3. doi: 10.1186/2042-4280-3-3
114. Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, et al. Effects of Valganciclovir as an Add-on Therapy in Patients With Cytomegalovirus-Positive Glioblastoma: A Randomized, Double-Blind, Hypothesis-Generating Study. Int J Cancer (2013) 133:1204–13. doi: 10.1002/ijc.28111
115. Yang CF, Ho HL, Lin SC, Hsu CY, Ho DM. Detection of Human Cytomegalovirus in Glioblastoma Among Taiwanese Subjects. PloS One (2017) 12:e0179366. doi: 10.1371/journal.pone.0179366
116. Han S, Wang PF, Xing YX, Song HW, Yao K, Lin ZX. Human Cytomegalovirus (HCMV) Infection was Not Correlated With Overall Survival in Glioblastomas. Neoplasma (2018) 65:431–5. doi: 10.4149/neo_2018_170124N59
117. Cai Z, Yang S, Li X, Chen F, Li W. Viral Infection and Glioma: A Meta-Analysis of Prognosis. BMC Cancer (2020) 20:549. doi: 10.1186/s12885-020-06796-3
118. Smith C, Lineburg KE, Martins JP, Ambalathingal GR, Neller MA, Morrison B, et al. Autologous CMV-Specific T Cells Are a Safe Adjuvant Immunotherapy for Primary Glioblastoma Multiforme. J Clin Invest (2020) 130:6041–53. doi: 10.1172/JCI138649
119. Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, et al. Broadly Targeted Human Cytomegalovirus-Specific CD4+ and CD8+ T Cells Dominate the Memory Compartments of Exposed Subjects. J Exp Med (2005) 202:673–85. doi: 10.1084/jem.20050882
120. Van Den Berg SPH, Pardieck IN, Lanfermeijer J, Sauce D, Klenerman P, Van Baarle D, et al. The Hallmarks of CMV-Specific CD8 T-Cell Differentiation. Med Microbiol Immunol (2019) 208:365–73. doi: 10.1007/s00430-019-00608-7
121. Fornara O, Odeberg J, Wolmer Solberg N, Tammik C, Skarman P, Peredo I, et al. Poor Survival in Glioblastoma Patients Is Associated With Early Signs of Immunosenescence in the CD4 T-Cell Compartment After Surgery. Oncoimmunology (2015) 4:e1036211. doi: 10.1080/2162402X.2015.1036211
122. Van Leeuwen EM, Remmerswaal EB, Heemskerk MH, Ten Berge IJ, Van Lier RA. Strong Selection of Virus-Specific Cytotoxic CD4+ T-Cell Clones During Primary Human Cytomegalovirus Infection. Blood (2006) 108:3121–7. doi: 10.1182/blood-2006-03-006809
123. Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex Vivo Functional Analysis, Expansion and Adoptive Transfer of Cytomegalovirus-Specific T-Cells in Patients With Glioblastoma Multiforme. Immunol Cell Biol (2012) 90:872–80. doi: 10.1038/icb.2012.19
124. Liu Z, Poiret T, Meng Q, Rao M, Von Landenberg A, Schoutrop E, et al. Epstein-Barr Virus- and Cytomegalovirus-Specific Immune Response in Patients With Brain Cancer. J Transl Med (2018) 16:182. doi: 10.1186/s12967-018-1557-9
125. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-Cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res (2014) 74:3466–76. doi: 10.1158/0008-5472.CAN-14-0296
126. Mirzaei R, Sarkar S, Yong VW. T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends Immunol (2017) 38:104–15. doi: 10.1016/j.it.2016.11.005
127. Ghazi A, Ashoori A, Hanley PJ, Brawley VS, Shaffer DR, Kew Y, et al. Generation of Polyclonal CMV-Specific T Cells for the Adoptive Immunotherapy of Glioblastoma. J Immunother (2012) 35:159–68. doi: 10.1097/CJI.0b013e318247642f
128. Smith C, Beagley L, Rehan S, Neller MA, Crooks P, Solomon M, et al. Autologous Adoptive T-Cell Therapy for Recurrent or Drug-Resistant Cytomegalovirus Complications in Solid Organ Transplant Recipients: A Single-Arm Open-Label Phase I Clinical Trial. Clin Infect Dis (2019) 68:632–40. doi: 10.1093/cid/ciy549
129. Weathers SP, Penas-Prado M, Pei BL, Ling X, Kassab C, Banerjee P, et al. Glioblastoma-Mediated Immune Dysfunction Limits CMV-Specific T Cells and Therapeutic Responses: Results From a Phase I/II Trial. Clin Cancer Res (2020) 26:3565–77. doi: 10.1158/1078-0432.CCR-20-0176
130. Luo XH, Meng Q, Liu Z, Paraschoudi G. Generation of High-Affinity CMV-Specific T Cells for Adoptive Immunotherapy Using IL-2, IL-15, and IL-21. Clin Immunol (2020) 217:108456. doi: 10.1016/j.clim.2020.108456
131. Ghaffari S, Torabi-Rahvar M, Omidkhoda A, Ahmadbeigi N. Impact of Various Culture Conditions on Ex Vivo Expansion of Polyclonal T Cells for Adoptive Immunotherapy. Apmis (2019) 127:737–45. doi: 10.1111/apm.12981
132. Sarkesh A, Sorkhabi AD, Ahmadi H, Abdolmohammadi-Vahid S, Parhizkar F, Yousefi M, et al. Allogeneic Lymphocytes Immunotherapy in Female Infertility: Lessons Learned and the Road Ahead. Life Sci (2022) 120503. doi: 10.1016/j.lfs.2022.120503
133. Kalia V, Sarkar S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front Immunol (2018) 29879. doi: 10.3389/fimmu.2018.02987
134. Ross SH, Cantrell DA. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu Rev Immunol (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
135. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in Regulating CD8+ T Cell Expansion and Function. J Exp Med (2005) 201:139–48. doi: 10.1084/jem.20041057
136. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 Instruct the Generation of Human Memory Stem T Cells From Naive Precursors. Blood (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
137. Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. IL15 and T-Cell Stemness in T-Cell-Based Cancer Immunotherapy. Cancer Res (2015) 75:5187–93. doi: 10.1158/0008-5472.CAN-15-1498
138. Chen Y, Yu F, Jiang Y, Chen J, Wu K, Chen X, et al. Adoptive Transfer of Interleukin-21-Stimulated Human CD8+ T Memory Stem Cells Efficiently Inhibits Tumor Growth. J Immunother (2018) 41:274–83. doi: 10.1097/CJI.0000000000000229
139. Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, et al. Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front Immunol (2019) 10:263. doi: 10.3389/fimmu.2019.00263
140. Bonte S, De Munter S, Billiet L, Goetgeluk G, Ingels J, Jansen H, et al. In Vitro OP9-DL1 Co-Culture and Subsequent Maturation in the Presence of IL-21 Generates Tumor Antigen-Specific T Cells With a Favorable Less-Differentiated Phenotype and Enhanced Functionality. Oncoimmunology (2021) 10:1954800. doi: 10.1080/2162402X.2021.1954800
141. Chamucero-Millares JA, Bernal-Estévez DA, Parra-López CA. Usefulness of IL-21, IL-7, and IL-15 Conditioned Media for Expansion of Antigen-Specific CD8+ T Cells From Healthy Donor-PBMCs Suitable for Immunotherapy. Cell Immunol (2021) 360:104257. doi: 10.1016/j.cellimm.2020.104257
142. Dranoff G. Cytokines in Cancer Pathogenesis and Cancer Therapy. Nat Rev Cancer (2004) 4:11–22. doi: 10.1038/nrc1252
143. Brooks DG, Mcgavern DB, Oldstone MB. Reprogramming of Antiviral T Cells Prevents Inactivation and Restores T Cell Activity During Persistent Viral Infection. J Clin Invest (2006) 116:1675–85. doi: 10.1172/JCI26856
144. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive Loss of Memory T Cell Potential and Commitment to Exhaustion During Chronic Viral Infection. J Virol (2012) 86:8161–70. doi: 10.1128/JVI.00889-12
145. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T Cell Help in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
146. Laidlaw BJ, Craft JE, Kaech SM. The Multifaceted Role of CD4(+) T Cells in CD8(+) T Cell Memory. Nat Rev Immunol (2016) 16:102–11. doi: 10.1038/nri.2015.10
147. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
148. Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front Immunol (2018) 9:14. doi: 10.3389/fimmu.2018.00014
149. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in Tumor Progression and Regression: A Review. biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
150. Brossart P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin Cancer Res (2020) 26:4442–7. doi: 10.1158/1078-0432.CCR-20-0305
151. Kiyotani K, Toyoshima Y, Nakamura Y. Personalized Immunotherapy in Cancer Precision Medicine. Cancer Biol Med (2021) 18(4):955–65. doi: 10.20892/j.issn.2095-3941.2021.0032
152. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
153. Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ, et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res (2018) 78:256–64. doi: 10.1158/0008-5472.CAN-17-0469
154. Nair SK, De Leon G, Boczkowski D, Schmittling R, Xie W, Staats J, et al. Recognition and Killing of Autologous, Primary Glioblastoma Tumor Cells by Human Cytomegalovirus Pp65-Specific Cytotoxic T Cells. Clin Cancer Res (2014) 20:2684–94. doi: 10.1158/1078-0432.CCR-13-3268
155. Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-Related Molecule B7-H1 by Glioma Cells: A Potential Mechanism of Immune Paralysis. Cancer Res (2003) 63:7462–7.
156. Bagley SJ, Desai AS, Linette GP, June CH, O'rourke DM. CAR T-Cell Therapy for Glioblastoma: Recent Clinical Advances and Future Challenges. Neuro Oncol (2018) 20:1429–38. doi: 10.1093/neuonc/noy032
157. Liu F, Huang J, Liu X, Cheng Q, Luo C, Liu Z. CTLA-4 Correlates With Immune and Clinical Characteristics of Glioma. Cancer Cell Int (2020) 20:7. doi: 10.1186/s12935-019-1085-6
158. Walker DG, Shakya R, Morrison B, Neller MA, Matthews KK, Nicholls J, et al. Impact of Pre-Therapy Glioblastoma Multiforme Microenvironment on Clinical Response to Autologous CMV-Specific T-Cell Therapy. Clin Transl Immunol (2019) 8:e01088. doi: 10.1002/cti2.1088
159. Chen RQ, Liu F, Qiu XY, Chen XQ. The Prognostic and Therapeutic Value of PD-L1 in Glioma. Front Pharmacol (2018) 9:1503. doi: 10.3389/fphar.2018.01503
160. Abdoli Shadbad M, Hemmat N, Khaze Shahgoli V, Derakhshani A, Baradaran F, Brunetti O. A Systematic Review on PD-1 Blockade and PD-1 Gene-Editing of CAR-T Cells for Glioma Therapy: From Deciphering to Personalized Medicine. Front Immunol (2022) 12:788211. doi: 10.3389/fimmu.2021.78821
161. Miconi G, Palumbo P, Dehcordi SR, La Torre C, Lombardi F, Evtoski Z, et al. Immunophenotypic Characterization of Human Glioblastoma Stem Cells: Correlation With Clinical Outcome. J Cell Biochem (2015) 116:864–76. doi: 10.1002/jcb.25043
162. Raysi Dehcordi S, Ricci A, Di Vitantonio H, De Paulis D, Luzzi S, Palumbo P, et al. Stemness Marker Detection in the Periphery of Glioblastoma and Ability of Glioblastoma to Generate Glioma Stem Cells: Clinical Correlations. World Neurosurg (2017) 105:895–905. doi: 10.1016/j.wneu.2017.05.099
163. Fang X, Huang Z, Zhai K, Huang Q, Tao W, Kim L, et al. Inhibiting DNA-PK Induces Glioma Stem Cell Differentiation and Sensitizes Glioblastoma to Radiation in Mice. Sci Transl Med (2021) (600):eabc7275. 13. doi: 10.1126/scitranslmed.abc7275
164. Robinson MH, Vasquez J, Kaushal A, Macdonald TJ, Velázquez Vega JE, Schniederjan M, et al. Subtype and Grade-Dependent Spatial Heterogeneity of T-Cell Infiltration in Pediatric Glioma. J Immunother Cancer (2020) 8(2):e001066. doi: 10.1136/jitc-2020-001066
165. Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, et al. Correlation of O6-Methylguanine Methyltransferase (MGMT) Promoter Methylation With Clinical Outcomes in Glioblastoma and Clinical Strategies to Modulate MGMT Activity. J Clin Oncol (2008) 26:4189–99. doi: 10.1200/JCO.2007.11.5964
166. Annavarapu S, Gogate A, Pham T, Davies K, Singh P, Robert N. Treatment Patterns and Outcomes for Patients With Newly Diagnosed Glioblastoma Multiforme: A Retrospective Cohort Study. CNS Oncol (2021), 10(3):Cns76. doi: 10.2217/cns-2021-0007
167. Bechman N, Maher J. Lymphodepletion Strategies to Potentiate Adoptive T-Cell Immunotherapy - What Are We Doing; Where Are We Going? Expert Opin Biol Ther (2021) 21:627–37. doi: 10.1080/14712598.2021.1857361
168. Hotchkiss KM, Sampson JH. Temozolomide Treatment Outcomes and Immunotherapy Efficacy in Brain Tumor. J Neuro-Oncol (2021) 151:55–62. doi: 10.1007/s11060-020-03598-2
169. Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, et al. Molecular and Clinical Characterization of PD-L1 Expression at Transcriptional Level via 976 Samples of Brain Glioma. Oncoimmunology (2016) 5:e1196310. doi: 10.1080/2162402X.2016.1196310
170. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational Burden, Immune Checkpoint Expression, and Mismatch Repair in Glioma: Implications for Immune Checkpoint Immunotherapy. Neuro Oncol (2017) 19:1047–57. doi: 10.1093/neuonc/nox026
171. Mu L, Long Y, Yang C, Jin L, Tao H, Ge H, et al. The IDH1 Mutation-Induced Oncometabolite, 2-Hydroxyglutarate, May Affect DNA Methylation and Expression of PD-L1 in Gliomas. Front Mol Neurosci (2018) 11:82. doi: 10.3389/fnmol.2018.00082
172. Friedrich M, Bunse L, Wick W, Platten M. Perspectives of Immunotherapy in Isocitrate Dehydrogenase-Mutant Gliomas. Curr Opin Oncol (2018) 30:368–74. doi: 10.1097/CCO.0000000000000478
173. Li J, He Y, Hao J, Ni L, Dong C. High Levels of Eomes Promote Exhaustion of Anti-Tumor CD8(+) T Cells. Front Immunol (2018) 2981 9. doi: 10.3389/fimmu.2018.02981
174. Ichii H, Sakamoto A, Kuroda Y, Tokuhisa T. Bcl6 Acts as an Amplifier for the Generation and Proliferative Capacity of Central Memory CD8+ T Cells. J Immunol (2004) 173:883–91. doi: 10.4049/jimmunol.173.2.883
175. Han J, Ma Y, Ma L, Tan D, Niu H, Bai C, et al. Id3 and Bcl6 Promote the Development of Long-Term Immune Memory Induced by Tuberculosis Subunit Vaccine. Vaccines (Basel) (2021) 9(2):126. doi: 10.3390/vaccines9020126
176. Yue Q, Zhang X, Ye HX, Wang Y, Du ZG, Yao Y, et al. The Prognostic Value of Foxp3+ Tumor-Infiltrating Lymphocytes in Patients With Glioblastoma. J Neurooncol (2014) 116:251–9. doi: 10.1007/s11060-013-1314-0
177. Guo XY, Zhang GH, Wang ZN, Duan H, Xie T, Liang L, et al. A Novel Foxp3-Related Immune Prognostic Signature for Glioblastoma Multiforme Based on Immunogenomic Profiling. Aging (Albany NY) (2021) 13:3501–17. doi: 10.18632/aging.202282
178. Kane A, Yang I. Interferon-Gamma in Brain Tumor Immunotherapy. Neurosurg Clin N Am (2010) 21:77–86. doi: 10.1016/j.nec.2009.08.011
179. Prunk M, Nanut MP, Sabotic J, Svajger U, Kos J. Increased Cystatin F Levels Correlate With Decreased Cytotoxicity of Cytotoxic T Cells. Radiol Oncol (2019) 53:57–68. doi: 10.2478/raon-2019-0007
180. He H, Yang P, Jiang L, Zhang J, Zhao C, Chen L, et al. Upregulation of CD94 on CD8+T Cells in Anterior Chamber-Associated Immune Deviation. BMC Immunol (2008) 9:53. doi: 10.1186/1471-2172-9-53
181. Henson SM, Akbar AN. KLRG1–more Than a Marker for T Cell Senescence. Age (Dordr) (2009) 31:285–91. doi: 10.1007/s11357-009-9100-9
182. Li L, Wan S, Tao K, Wang G, Zhao E. KLRG1 Restricts Memory T Cell Antitumor Immunity. Oncotarget (2016) 7:61670–8. doi: 10.18632/oncotarget.11430
183. Hou Q, Zhao T, Zhang H, Lu H, Zhang Q, Sun L, et al. Granzyme H Induces Apoptosis of Target Tumor Cells Characterized by DNA Fragmentation and Bid-Dependent Mitochondrial Damage. Mol Immunol (2008) 45:1044–55. doi: 10.1016/j.molimm.2007.07.032
184. Watson HA, Wehenkel S, Matthews J, Ager A. SHP-1: The Next Checkpoint Target for Cancer Immunotherapy? Biochem Soc Trans (2016) 44:356–62. doi: 10.1042/BST20150251
185. Huang J, Liu F, Liu Z, Tang H, Wu H, Gong Q, et al. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front Pharmacol (2017) 8:242. doi: 10.3389/fphar.2017.00242
186. Zou B, Chim CS, Pang R, Zeng H, Dai Y, Zhang R, et al. XIAP-Associated Factor 1 (XAF1), a Novel Target of P53, Enhances P53-Mediated Apoptosis via Post-Translational Modification. Mol Carcinog (2012) 51:422–32. doi: 10.1002/mc.20807
187. Jeong SI, Kim JW, Ko KP, Ryu BK, Lee MG, Kim HJ, et al. XAF1 Forms a Positive Feedback Loop With IRF-1 to Drive Apoptotic Stress Response and Suppress Tumorigenesis. Cell Death Dis (2018) 9:806. doi: 10.1038/s41419-018-0867-4
188. Kranjc MK, Novak M, Pestell RG, Lah TT. Cytokine CCL5 and Receptor CCR5 Axis in Glioblastoma Multiforme. Radiol Oncol (2019) 53:397–406. doi: 10.2478/raon-2019-0057
189. Novak M, Koprivnikar Krajnc M, Hrastar B, Breznik B, Majc B, Mlinar M, et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int J Mol Sci (2020) 21(12):4199. doi: 10.3390/ijms21124199
190. Reina M, Espel E. Role of LFA-1 and ICAM-1 in Cancer. Cancers (Basel) (2017) 9(11):153. doi: 10.20944/preprints201709.0146.v1
191. Cobbs CS. Evolving Evidence Implicates Cytomegalovirus as a Promoter of Malignant Glioma Pathogenesis. Herpesviridae (2011) 2:10. doi: 10.1186/2042-4280-2-10
192. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 Signaling in the Hematopoietic System Elicits Multicomponent Antitumor Immunity. Nat Med (2005) 11:1314–21. doi: 10.1038/nm1325
Keywords: immunotherapy, adoptive cellular therapy (ACT), CMV-specific T cell, cytomegalovirus (CMV), herpes virus, glioblastoma multiforme, glioma, brain tumor
Citation: Daei Sorkhabi A, Sarkesh A, Saeedi H, Marofi F, Ghaebi M, Silvestris N, Baradaran B and Brunetti O (2022) The Basis and Advances in Clinical Application of Cytomegalovirus-Specific Cytotoxic T Cell Immunotherapy for Glioblastoma Multiforme. Front. Oncol. 12:818447. doi: 10.3389/fonc.2022.818447
Received: 05 December 2021; Accepted: 24 March 2022;
Published: 19 April 2022.
Edited by:
Michel Salzet, Lille University of Science and Technology, FranceReviewed by:
Helene Castel, Université de Rouen, FranceThomas Daubon, UMR5095 Institut de Biochimie et Génétique Cellulaires (IBGC), France
Copyright © 2022 Daei Sorkhabi, Sarkesh, Saeedi, Marofi, Ghaebi, Silvestris, Baradaran and Brunetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Behzad Baradaran, baradaranb@tbzmed.ac.ir; Oronzo Brunetti, o.brunetti@oncologico.bari.it
†These authors have contributed equally to this work
‡These authors share last authorship