The Emerging Genetic Landscape of Hirschsprung Disease and Its Potential Clinical Applications
 Anwarul Karim
Anwarul Karim Clara Sze-Man Tang
Clara Sze-Man Tang Paul Kwong-Hang Tam
Paul Kwong-Hang Tam- 1Department of Surgery, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China
- 2Li Dak-Sum Research Center, The University of Hong Kong—Karolinska Institute Collaboration in Regenerative Medicine, Hong Kong, China
Hirschsprung disease (HSCR) is the leading cause of neonatal functional intestinal obstruction. It is a rare congenital disease with an incidence of one in 3,500–5,000 live births. HSCR is characterized by the absence of enteric ganglia in the distal colon, plausibly due to genetic defects perturbing the normal migration, proliferation, differentiation, and/or survival of the enteric neural crest cells as well as impaired interaction with the enteric progenitor cell niche. Early linkage analyses in Mendelian and syndromic forms of HSCR uncovered variants with large effects in major HSCR genes including RET, EDNRB, and their interacting partners in the same biological pathways. With the advances in genome-wide genotyping and next-generation sequencing technologies, there has been a remarkable progress in understanding of the genetic basis of HSCR in the past few years, with common and rare variants with small to moderate effects being uncovered. The discovery of new HSCR genes such as neuregulin and BACE2 as well as the deeper understanding of the roles and mechanisms of known HSCR genes provided solid evidence that many HSCR cases are in the form of complex polygenic/oligogenic disorder where rare variants act in the sensitized background of HSCR-associated common variants. This review summarizes the roadmap of genetic discoveries of HSCR from the earlier family-based linkage analyses to the recent population-based genome-wide analyses coupled with functional genomics, and how these discoveries facilitated our understanding of the genetic architecture of this complex disease and provide the foundation of clinical translation for precision and stratified medicine.
Introduction
Hirschsprung disease (HSCR), also known as colonic aganglionosis, is the leading cause of neonatal functional intestinal obstruction. It is a rare congenital developmental defect of the enteric nervous system (ENS) with a global incidence of 1 in 3,500–5,000 live births. The incidence of the disease varies widely among ethnic groups and is the highest among Asians (2.8/10,000 live births) (1, 2). The disease is characterized by the absence of enteric ganglia in the distal colon due to the failure of enteric neural crest cells (ENCCs) to fully colonize the hindgut during embryonic development. The incomplete innervation may result from any or a combination of genetic defects and environmental factors affecting migration, proliferation, differentiation, and survival of the ENCCs as well as impaired extracellular milieu or impaired interaction between the migrating neural crest-derived precursor cells and the mesenchymal microenvironment through which they migrate (3, 4).
HSCR is traditionally classified by the extent of aganglionosis. In short-segment HSCR (S-HSCR; 80% of cases), aganglionosis does not extend beyond the sigmoid region whereas in long-segment HSCR (L-HSCR; 20% of cases), it extends proximal to the sigmoid colon. Among the L-HSCR, if the aganglionosis extends to at least ileocecal valve, it is termed total colonic aganglionosis (TCA), which represents approximately 5% of all HSCR cases. The majority of HSCR cases (~70%; isolated HSCR) occur as isolated anomalies. Other cases are syndromic (syndromic HSCR) and often encountered with chromosomal aberrations and/or a range of other congenital malformations (1, 2, 5–8). The inheritance of HSCR is considered to be complex. It most commonly presents as sporadic forms (80–90%), which are more often S-HSCR and follow a multifactorial inheritance pattern. The remaining 10–20% of cases are familial and tend to be of L-HSCR/TCA with autosomal dominant inheritance (2, 9). HSCR also exhibits significant sex bias with a marked male preponderance. Especially for S-HSCR, the disease prevalence is four times higher in males than in females. Up till now, the only definitive treatment available for HSCR is surgery.
Over the past decades, through human genetic studies—beginning with the classical positional cloning, linkage analyses, and candidate gene screening—through genome-wide association studies (GWAS)—to the recent next-generation sequencing (NGS) studies, coupled with validation by functional genomic studies, many genetic variants and genes have been linked to HSCR. Majority of these genes belong to several key biological pathways that often crosstalk and have been shown to orchestrate the dynamic process of ENS development and HSCR pathogenesis, though some initially unsuspected candidate genes have also been uncovered by the bias-free approaches. These studies have provided solid evidence that most HSCR cases have complex (oligogenic/polygenic) genetic basis wherein multiple HSCR-associated common variants contribute to the genetic predisposition and modify the penetrance of rare damaging variants in disease-relevant genes and hence disease manifestation.
In this review, we explore the paradigm shift in genetic discoveries of HSCR from family-based, candidate gene studies to population-based, genome-wide analyses with the advent of genotyping and sequencing technologies and how these discoveries furthered our understanding of the genetic architecture of this complex disorder. This review primarily focuses on the emerging genetic landscape of rare coding variants, common regulatory variants, and the interplay between them as well as their differential contribution to HSCR and the disease subtypes, and how the new knowledge may pave the way for genome data to be incorporated as part of “routine” in the precision-medicine era.
Pre-GWAS ERA: Identification of Core HSCR Pathways Through Linkage Mapping and Candidate Gene Studies on Syndromic and Familial HSCR Cases
Genetic analyses to uncover the genes underlying HSCR began in late 1980s/early 1990s. Table 1 lists the main genes known to be involved in either syndromic or isolated HSCR. In the early days, analyses were primarily focused on the more Mendelian forms of syndromic and familial HSCR. These studies built upon the fundamental idea that disease causal variant and nearby genetic markers tend to be transmitted together due to linkage disequilibrium (LD). Such approach of positional cloning and linkage analysis have been applied to multiplex families where highly informative genetic markers are used to map the disease-associated loci of large effect. Once a locus is linked, a search for rare damaging mutations (i.e., variants with minor allele frequency <1% in general population) in candidate genes within the locus is ensued. This strategy remained very popular especially before the GWAS era. In fact, the two major HSCR genes—RET (in 10q11.2) and EDNRB (in 13q22.3)—representing the two core HSCR pathways as well as transcription factors underlying syndromic form of HSCR were initially discovered with this approach in the early 1990s (Table 1).
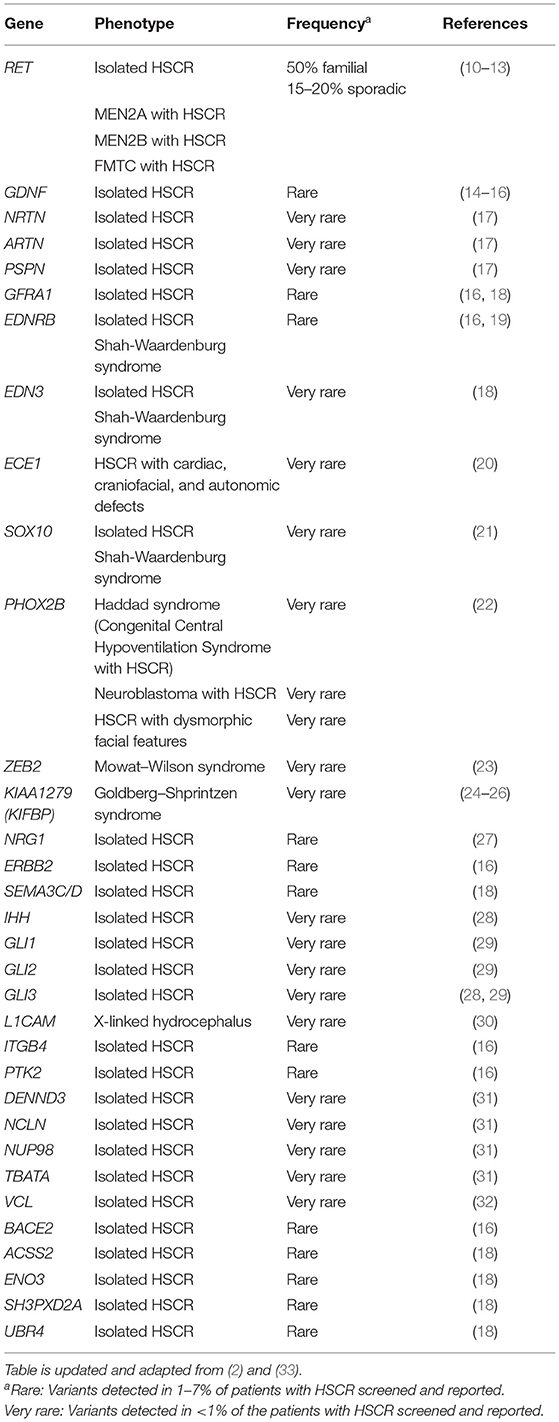
Table 1. Genes reported with rare damaging protein-altering variants in patients with HSCR.
RET Signaling
RET (Rearranged during Transfection) proto-oncogene encodes a transmembrane tyrosine-protein kinase receptor. It is activated by glial cell line-derived neurotrophic factor (GDNF) family ligands, such as GDNF, NRTN (neurturin), ARTN (artemin), PSPN (persephin), and co-receptors named GDNF-family receptor-α (GFRα1-4) (34). RET plays a pivotal role in both isolated and syndromic HSCR. The RET locus was first identified as a susceptibility locus for HSCR through linkage analyses in multiplex HSCR families (35, 36) facilitated by the finding of deletion of the proximal long arm of chromosome 10 in patients with isolated HSCR (37, 38) and the co-occurrence of HSCR and multiple endocrine neoplasia type 2 (MEN2) (39, 40). This was followed by the identification of numerous RET mutations, including missense, splicing variants, and short insertions and deletions (indels), across the whole spectrum of patients with HSCR occurring both as de novo and inherited events (10–13, 41–52). Damaging coding mutations in RET are identified in approximately 50% of familial and 15–20% of sporadic HSCR cases (1, 10–13), and it gradually becomes evident that rare coding variants in RET appear to play a less prominent role in sporadic and S-HSCR compared to the familial and L-HSCR (51, 53). Furthermore, somatic RET mutations have also been reported occasionally—occurring either as transmission of mosaic germline mutation from unaffected parent to affected patient or as somatic mutation in patients (54–57).
Subsequent to the discovery of RET in HSCR pathogenesis, mutation screening was performed on interacting partners and other key members of RET signaling pathway with reference to known biological processes and animal models. Damaging rare variants in all four ligands of RET have been detected in patients with HSCR, yet RET still contributes to the vast majority of rare variants reported in genes in this core HSCR pathway (13–15, 17, 28, 51, 58–62). Mutations in these GDNF family ligands typically co-occurred with variants in other major HSCR genes, particularly in RET, except in a few instances (59, 61). As for those exceptions, the patients may also carry variants in other HSCR genes not yet discovered by that time. This is exemplified by a recent whole exome sequencing (WES) study by Sribudiani et al. (28) where the authors showed that a de novo in-frame deletion in GDNF was, together with rare inherited variants in IHH (Hedgehog signaling pathway, described later) and its mediator, GLI3, responsible for HSCR in a branch of a multigenerational HSCR family. Similarly for the co-receptors, no confirmed high-penetrant pathogenic variant has yet been described in patients except a marginal increase in burden of rare damaging protein-altering variants in GFRα1 that has been detected in a recent whole-genome sequencing (WGS) study on 443 East Asian S-HSCR cases compared to 493 controls (16, 18, 63). Altogether, these findings suggest that, in the majority of cases where variants are found in GDNF family ligands and co-receptors, they alone are not sufficient to cause HSCR and rather act as modifiers together with other risk variants (rare damaging or common regulatory risk variants) in a digenic/oligogenic pattern.
EDNRB Signaling
EDNRB (Endothelin receptor type B) gene encodes a non-specific G protein-coupled receptor for endothelins (EDN1, EDN2, and EDN3) (2). After synthesis, endothelins are converted into a shorter active form by endothelin converting enzyme (ECE-1) (2). Similar to RET, the contribution of EDNRB to the risk of HSCR was discovered by family-based studies. A HSCR susceptibility locus was first mapped to 13q22 by identity-by-descent and linkage analysis in a large, inbred, Mennonite kindred that manifested high incidence of HSCR and pigmentary disorders, the Waardenburg syndrome type 4 (WS4) (64). A follow-up study identified p.Trp276Cys mutation in EDNRB that showed incomplete penetrance and dosage effect in addition to being absent in some patients (65). This was followed by several reports of identification of EDNRB mutations in patients with HSCR and WS4 syndrome (11, 66–70). However, rare damaging variants of EDNRB account for only 3–7% of isolated HSCR cases (2).
Subsequent search for rare variants in genes in EDNRB pathway reported pathogenic mutations in EDN3 and ECE1 in patients with HSCR. In general, homozygous mutations of EDNRB and EDN3 are more commonly associated with WS4 and heterozygous mutations are more commonly associated with isolated HSCR (11, 66–73). Thus far, only one heterozygous ECE1 variant has been reported in a syndromic patient with HSCR (20).
Although rare damaging variants in EDNRB pathway genes are encountered in only a small fraction of patients with HSCR (~5%), they generally exhibit higher penetrance and confer higher risk than variants in RET signaling pathway genes. For example, in a WES study by Tilghman et al. on European HSCR cases and controls (18), pathogenic variants in EDNRB pathway genes (seven in EDNRB and one in EDN3) were exclusively present in HSCR cases. In addition, pathway-based odds ratio (OR) for EDNRB was considerably higher [OR = 69.03; 95% confidence interval (CI): 8.68–547.92] than that of the RET signaling pathway (OR = 16.03; 95% CI: 5.21–49.28). Consistently, EDNRB variants showed an overall higher risk than RET, for S-HSCR cases in East Asian WGS analysis (16).
Transcription Factors Critical for ENS Development
SOX10 encodes a transcription factor that is a key regulator of ENS development (74) and is implicated in Waardenburg syndrome with varying spectrum of features with or without HSCR (75–77). Although SOX10 variants were initially thought to cause HSCR as a part of WS4 (2), recent studies identified several isolated HSCR cases with SOX10 mutations—both in its coding region (21, 78) and in its enhancers (78–80).
PHOX2B also encodes a transcription factor required for normal development of the ENS (81). Genetic defects in PHOX2B have been described primarily with congenital central hypoventilation syndrome (CCHS) (82), which occur with HSCR in 15–20% of cases (83) and in some reports with neuroblastoma (84, 85). These strongly suggest a pathogenic role of PHOX2B in HSCR. In terms of rare coding variants, a de novo in-frame deletion in PHOX2B in a female patient with L-HSCR and other anomalies but without clinical manifestations of CCHS or neuroblastoma has been described (22). However, unlike SOX10, PHOX2B pathogenic variants in isolated HSCR are yet to be found. In addition to rare variants, common PHOX2B polymorphisms were shown to be moderately associated with HSCR in a Chinese population (86, 87) and interaction between RET and PHOX2B polymorphisms was demonstrated to significantly increase HSCR risk (88).
ZEB2, previously known as SIP1 or ZFHX1B, encodes a transcription factor that is crucial to direct the formation, migration, and specification of neural crest cells (89). Variants in this gene have been described with many patients with Mowat–Wilson syndrome, which includes HSCR in its phenotypic spectrum in approximately half of the cases (23). However, ZEB2 pathogenic variants have not been described in isolated HSCR yet. No enrichment for rare variants in ZEB2 was found in S-HSCR cases compared to controls, indicating that it is likely to be a very rare contributor to isolated HSCR (16).
Genes Related to Cytoskeleton
Homozygosity mapping followed by sequence analysis in a consanguineous family with multiple members having Goldberg–Shprintzen syndrome (GOSHS) identified homozygous truncating mutations in KIAA1279 (KIFBP) (24). HSCR has been reported in 64% of patients with GOSHS (25). Thus far, 16 different variants in this gene have been described in patients with GOSHS (25). KIAA1279 encodes for KIF-binding protein (KIFBP), which interacts with microtubule and actin filaments and plays a role in neurite outgrowth, neuronal development, and differentiation (90, 91). Mice null for this protein show delayed gut colonization by the neural crest-derived cells (92). KIFBP was shown to interact with several kinesins and SCG10 (a microtubule destabilizing protein), implicating the potential role of cytoskeleton/microtubule-related defects in HSCR pathogenesis (90). Related to this, it should also be noted that a slightly lower intensity of immunoreactivity of Microtubule-Associated Protein 5 (MAP5) was noted in the intestinal tissue of the aganglionic segment of patients with HSCR compared to the normoganglionic segment of the same patients or intestinal tissue of control individuals (93). However, aside from the KIAA1279 variants in patients with GOSHS, no variant has been reported in SCG10 (94) or other related genes yet in patients with HSCR.
The ERA of GWAS: Large Contribution of Common Variants Implicating New Disease Pathways and Emerging Genetic Landscape
Altogether, the rare damaging variants identified in RET and EDNRB pathways explain only a small fraction (<30%) of sporadic HSCR cases. The inability to detect rare broadly Mendelian variants in considerable proportions of patients, along with the variable expressivity and disease severity among carriers, implied that more complex genetic etiologies are likely to underlie HSCR. Other genetic factors or epistatic regulation of RET, either by variants within the locus or with other unlinked genes, must exist to explain the missing heritability.
Contribution of common variants to HSCR was first indicated by (i) the variable penetrance of a damaging RET missense mutation (RET c.1859G>C;p.Cys620Ser) under different haplotype backgrounds of common variants in a multiplex family (95) and (ii) subsequent reports of overrepresentation of certain RET haplotypes in patients with HSCR across populations (96–98). Later, a study by Emison et al. pinpointed a common functional RET intron 1 enhancer variant (RET+3; rs2435357 T/C) that largely increases risk of HSCR (OR~5). The risk allele (T) of RET+3, present in ~60% of European and ~80% of Asian cases, significantly reduces RET expression and confers higher risk for HSCR than rare alleles collectively (99). It is interesting to note that the risk allele frequency is substantially higher in the general population in East Asians than Europeans (47 vs. 23%), which could explain the higher incidence of HSCR in Asia (53). In contrast to rare damaging variants, contribution of this common enhancer variant is considerably higher in the major subtype of sporadic and isolated S-HSCR than the severe forms (53). Furthermore, another RET intron 1 risk variant (rs2506004 A/C), which is in near complete LD with rs2435357 (r2 = 0.99 in Asians and 1 in Europeans) and separated by only 217 base pairs from it, was also identified (100, 101). The risk allele (A) alters RET expression by interfering with the binding of transcription factors NXF, ARNT2, and SIM2 to this locus exemplifying that more than one variant may be relevant to disease pathogenesis in the same disease-associated haplotype (101). Altogether, these findings implied that common variants can predispose to HSCR in a low penetrance manner by modifying the phenotypic expression, which opened up a new area of genetic research on HSCR, including family-based and population-based association studies by detecting transmission disequilibrium of common single-nucleotide polymorphisms (SNP) from parents to proband and comparing frequencies of SNPs in cases vs. controls, respectively (53, 102–110). However, earlier association studies still relied on prior biological knowledge to detect association of variants in candidate region(s) and, with this candidate gene approach, no confident association beyond the RET locus could be detected or replicated.
The advances of high-throughput SNP array, assaying hundreds of thousands of SNPs simultaneously, permitted the transition to a hypothesis-free approach for a powerful detection of association in genome-wide scale, termed GWAS. Thus far, five array-based GWAS have been published on HSCR, which unraveled two novel, highly replicable GWAS significant loci—NRG1 and SEMA3C/D—in addition to RET (shown in Table 2) (102–108, 110). Transethnic meta-analysis of GWAS discovered universal as well as ancestry-specific risk alleles, highlighting the heterogeneity of the genetic architecture of this disease (111). Other common SNPs that were found with moderate associations in GWAS includes SNPs in or nearby PHOX2B, JAG1, and VRK2. Epistatic interaction within and between RET and other loci were discovered from these GWASs as well. Novel insights and pathways previously not linked to the genetic etiology of HSCR but aroused from findings of GWAS are summarized below.
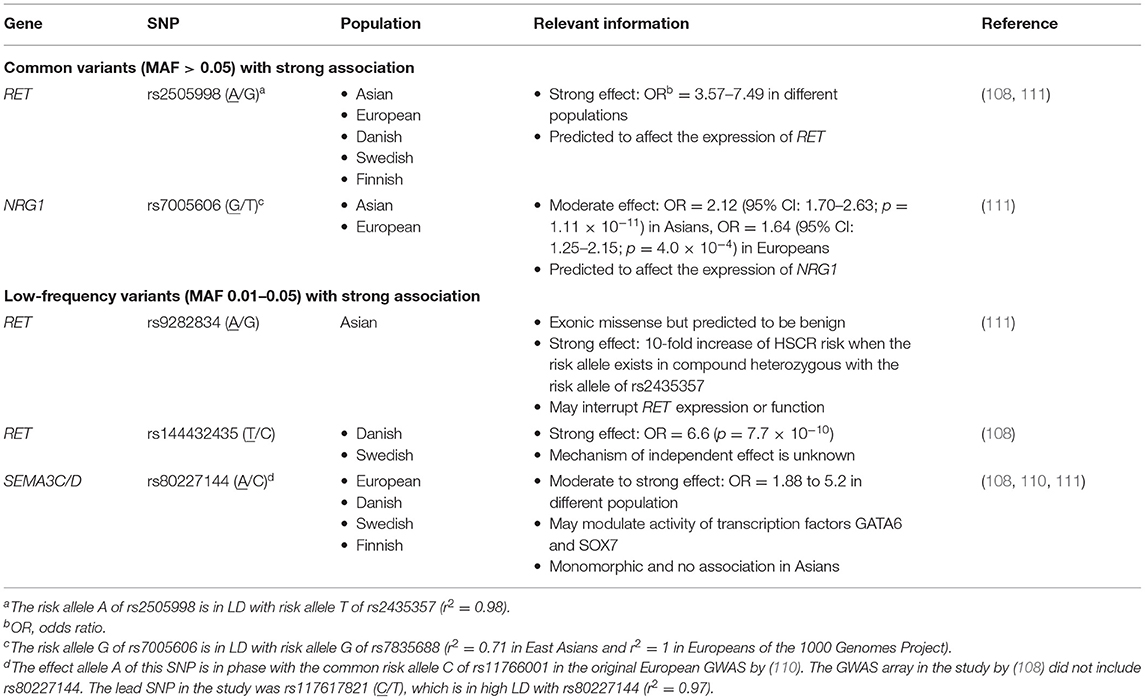
Table 2. Major GWAS loci associated with HSCR.
Neuregulin Signaling
Neuregulins (NRGs) are a family of growth factors that stimulate receptor tyrosine-protein kinase erbB (ERBB receptors) and are important regulators of neuronal migration and glia formation (112). Strong association of common SNPs (rs7835688 and rs16879552) lowering expression of NRG1 was first identified in a GWAS on Chinese population and was widely replicated across Asian studies (102, 104, 105, 113–115). Interestingly, the association was also reported in some Caucasian studies albeit with smaller and variable effect size (108, 110, 111). Following the GWAS discovery, patient-specific rare damaging coding variants were found in both Chinese and European sporadic HSCR cases. Functional analyses of these variants showed aberrant expression and uneven intracellular distribution of the mutant NRG1 proteins, proving that not only common variants but also rare coding variants in NRG1 can increase HSCR risk (27, 106). Besides NRG1, genome-wide copy number variation (CNV) analysis discovered the association of intronic deletions and duplications in NRG3, a paralog of NRG1, with HSCR on the same Chinese SNP array dataset (116). The recent population-based WGS study further identified a fourfold increase in the number of damaging rare variants in ERBB2, encoding the receptor for NRG1, in patients with HSCR compared to the controls (16). Altogether, these findings firmly established NRG-ERBB as one of the core HSCR pathways with significant contribution of both common and rare variants to disease pathogenesis.
Semaphorin Signaling
Semaphorins are extracellular signaling molecules that can directly bind to several receptor protein families particularly plexins and neuropilins. Semaphorins are important regulators of axon guidance and neural crest cell migration (117). Their involvement in HSCR pathogenesis has only been brought to focus with the identification of GWAS significant association signals within the intergenic region of SEMA3 gene cluster from the European and Danish GWASs (108, 110). Comparing the temporal and spatial localization with Ret in mouse ENS, SEMA3D and SEMA3C were considered to be the most likely candidate gene targets of the association in the cluster. Their roles in HSCR were further supported by the plausible interaction with RET such that co-knockdown of both sema3c/d and ret gave a more severe, aganglionic phenotype in zebrafish model. Recently, a twofold excess of rare deleterious variants in SEMA3C and SEMA3D was also observed in patients from a WES study (18).
Hedgehog and Notch Signaling
Hedgehog (Hh) signaling is mediated by transmembrane proteins Patched (PTCH1) and Smoothened (SMO) and the downstream effector GLI transcription factors. Notch receptors (NOTCH1 and NOTCH2) are transmembrane proteins activated by their ligands (DLL1 and DLL3). They are crucial regulators of ENS development (29), and key members of these pathways were suspected to be involved in HSCR pathogenesis. Using the Chinese GWAS data (102), a significant epistatic interaction between genes of these pathways (particularly driven by PTCH1 and DLL3 SNPs) conferring higher risk to HSCR was discovered (29). Functional analysis validated the interaction between the two signaling pathways and the Hedgehog/Notch interaction was shown to coordinate the proliferation and differentiation of ENCCs. Another study on an independent set of Chinese patients also suggested that common variants in PTCH1 may predispose to HSCR (118). Later, using targeted sequencing in 20 Chinese sporadic patients with HSCR devoid of RET coding sequence mutations, four rare coding variants in GLI1, GLI2, and GLI3 were identified, and these mutations were demonstrated to enhance GLI transcriptional activity (119). Finally, as mentioned earlier, inherited pathogenic variants of IHH and GLI3 genes, together with a de novo GDNF in-frame deletion, led to the manifestation of HSCR phenotype in one branch of a Dutch multiplex family, whereas the RET coding variant and intron-1 variant were responsible for the disease manifestation in another branch (28). These findings suggest that, although the reported variants in Hedgehog and Notch signaling pathway genes in patients with HSCR contribute to a small fraction of patients at present, more patients may be expected to be reported in the future especially with the increasing application of NGS and under oligogenic model harboring multiple variants in these genes.
Epistasis (Genetic Interaction) Is an Important Part of the Genetic Architecture of HSCR
Summarizing findings of GWAS studies, it becomes increasingly apparent that epistasis is an important component of the genetic architecture of HSCR, which contributes substantially to the modified penetrance of disease causal variants. In particular, the common RET enhancer variant (rs2435357) plays a pivotal role in epistatic interaction within and between HSCR genes and represents the main contributor to the sensitized genetic background of patients.
Within the RET locus, synergistic interaction between rs2435357 and other common/low-frequency/rare variants was observed. As reported in the transethnic meta-analysis, the low-frequency, Asian-specific missense variant (rs9282834) encoding RET D489N has no effect on HSCR risk alone (OR = 1.1); however, when it occurred in trans with rs2435357 (OR = 3.2), D489N increased risk of HSCR by at least 10-fold (OR = 16.7/26.7 in Chinese/Korean, respectively) (111). In addition, in the Chinese S-HSCR WGS study, the genetic effects of other rare RET coding mutations were largely modified to different degrees depending on the predicted pathogenicity in the presence of the common risk alleles. Again, no increase in disease risk was detected for individuals carrying only a single RET missense damaging or benign mutation; however, for individuals heterozygous with rs2435357, damaging missense RET mutations conferred ~5-fold increase in risk of HSCR on top of the effect of rs2435357. Similarly, for individuals with at least two high-risk alleles occurring in trans (i.e., rs2435357 TT or compound heterozygous for rs2435357 T allele and rs9282834 A allele), a non-damaging RET missense mutation also conferred a twofold increase in disease risk additionally. On the other hand, three cis-acting regulatory variants in three distinct enhancers of RET [rs2506030 (G/A), rs7069590 (T/C), and rs2435357 (T/C)] were shown to increase HSCR risk synergistically in patients of European ancestry (109). Functional studies have provided valuable biological insights underlying the genetic interaction. These risk alleles might disrupt the binding sites of RARB, GATA2, and SOX10, respectively, and their combined effect significantly dysregulated the expression of RET and other functionally related genes in the gene regulatory network through positive and negative feedback. These results altogether demonstrated how the RET enhancer allele may modify the penetrance of other coding variants or amplify its effect in conjunction with other enhancers to affect the phenotypic expression.
Moreover, the effect of variants in other HSCR genes may also depend on the RET genetic background. Interestingly, both of the new GWAS significant loci were shown to interact with RET. A genetic interaction between RET and NRG1 was observed (interaction p = 0.0095), in which the odds ratio increased by twofold for the RET rs2435357 risk genotype (TT) in the presence of NRG1 rs7835688 heterozygote. Such genetic interplay was later confirmed functionally by showing that Nrg1 inhibited the Gdnf-induced neuronal differentiation and Gdnf negatively regulated Nrg1 signaling by downregulating the expression of its receptor, ErbB2 (120). Likewise, the frequency of RET rs2435357 was higher for subjects with the SEMA3C/D rs12707682 risk allele, which is in line with the synergistic effects on gut innervation observed by co-knockdown of sema3c/d and ret in the zebrafish model. In mice, the reduced dosage of Ret or Ednrb, the second most mutated HSCR gene, independently yielded no obvious ENS phenotype, albeit combining the two oligogenic-null heterozygote models gave rise to aganglionic phenotype (121). In summary, these findings implied that the penetrance of mutations/functional variants in other HSCR susceptibility genes may also be modulated by RET variants and that a sensitized background of aberrant RET expression might be necessary for other genetic factors to act upon for disease manifestation (122).
Post-GWAS: Identification of HSCR-Associated Rare Variants and Genes by Unbiased Approach Using NGS Technology
The decreasing cost and hence increased adoption of NGS is expected to lead to a new era in genetic analysis of HSCR. WES and WGS studies investigating HSCR-associated rare variants in an unbiased manner are emerging and new biological insights not limited to differentiation, proliferation, and migration properties of the ENCCs begin to unveil.
Aberrant Extracellular Matrix (ECM) Composition Involving Focal Adhesion/ECM–Receptor Interaction
During the ENS development, ENCCs migrate from the neural tube and enter the foregut, and then migrate along the gastrointestinal tract in a rostrocaudal direction. The directed cell migration requires a coordinated interaction with the microenvironment including the ECM. Theoretically, genetic defects perturbing the interaction between cell and ECM may affect the migration and colonization of the ENCCs. In a recent WGS study on the severe forms of HSCR, genes with rare de novo, recessive or digenic variants in patients with L-HSCR/TCA were shown to be enriched for ECM–matrix receptor interaction (19). Furthermore, a significant enrichment of damaging rare variants in genes encoding cell-adhesion proteins, ITGB4 (Integrin beta-4) and PTK2/FAK (Focal adhesion kinase), was identified in an independent cohort of patients with S-HSCR (16). Integrins are a large family of cell surface receptors that connect ENCCs to the ECM. Upon activation, integrins undergo conformational change to recruit signaling molecules such as FAK and vinculin (VCL), a membrane-cytoskeletal protein in focal adhesion plaques, for the cell–matrix adhesions. Interestingly, a mutation in VCL (M209L) was also found in a Chinese patient with S-HSCR from integrative WES and transcriptomic analysis (32). Subsequent CRISPR/Cas9-mediated correction of the mutation in patient-specific induced pluripotent stem cells (iPSCs) efficiently rescued the differentiation and migration defects of the iPSC-derived ENCCs. In fact, abnormalities in the ECM composition in the affected bowel of patients with HSCR were noted decades ago (123). ECM proteins, such as laminins, collagens, tenascin, and fibronectin, were functionally shown to be involved in ENS development (124–126). The hypothesis that genetic defects affecting ECM composition underlie HSCR is further supported by the mouse models in which (1) loss of β1 integrin in ENCC resulted in colonic aganglionosis (127) and (2) lineage-specific upregulation of Col6a4 in Holstein mouse model increased total collagen VI protein levels in the ECM and resulted in slower migration of ENCCs and decreased extent of bowel colonization (128).
Neuronal Death Involving the BACE1–APP–BACE2 Pathway
Another novel finding from the NGS study was the stark excess of rare protein-altering variants in β-secretase 2 gene (BACE2) identified from patients with S-HSCR (16). BACE2 is a homolog of BACE1 encoding a protease that cleaves the amyloid precursor protein (APP) in the beta amyloid (Aβ) region and prevents its formation. Accumulation of Aβ induces neuronal death, representing the underlying cause of Alzheimer's disease. Using the iPSC platform, a patient-specific BACE2 rare variant was demonstrated to significantly reduce the APP processing activity of BACE2 and resulted in accumulation of Aβ and thereby apoptosis of enteric neurons. Similarly, correction of the mutation using a genome editing approach ameliorated the apoptotic phenotype. Together with the marginal association of common variants of PLD1 that may negatively regulate Aβ formation (16, 129, 130), these findings shed light on the important role of APP processing in HSCR pathogenesis.
Other New Findings From NGS Studies
Up till now, many other genes have been reported to be associated with HSCR from trio-based and case–control NGS studies, albeit these studies are more focused on the coding region. Functional characterization of all these genes remains a daunting task—even with the use of CRISPR/Cas9-mediated knockout or morpholino-mediated knockdown in zebrafish models in a relatively fast manner, not to mention elucidating the underlying disease mechanisms. Some of these new genes with functional support for their roles in ENS development include DENND3, NCLN, NUP98, and TBATA discovered from de novo mutation analysis of L-HSCR trios (31) and ACSS2, ENO3, SH3PXD2A, and UBR4 identified from the case–control WES study (Table 1) (18); however, how these genes predispose or cause HSCR remains to be explored. Compared to coding variants, identifying HSCR-associated functional non-coding variants from WGS studies is even more challenging and requires the development of novel analytical methods and integration of multi-omics data to facilitate their discoveries. A new framework, named MARVEL, has recently been developed and used together with functional analysis in human stem cells to identify several novel disease-associated regulatory elements, further highlighting two HSCR candidate genes—RASGEF1A and PIK3C2B (131).
Role of CNVs and Chromosomal Anomalies
Besides rare coding and common variants, CNVs and chromosomal abnormalities have also been frequently reported in HSCR cases. Chromosomal anomalies have been reported in up to 12% of patients with HSCR, although Down syndrome (trisomy 21) alone can be found in as much as 10% of HSCR cases, which increases HSCR risk by 50- to 100-fold (132, 133). In fact, these chromosomal anomalies have facilitated the discovery of several classical HSCR genes. CNVs that are beyond the resolution of conventional cytogenetics may contribute to the missing heritability of HSCR. These groups of CNVs are becoming more and more detectable with the advent of SNP arrays and NGS as well as a plethora of bioinformatic tools and algorithms for accurately detecting CNVs (134–137). Importantly, there appears to be a higher burden of rare CNVs in patients with HSCR compared to controls and larger CNVs in syndromic HSCR compared to isolated HSCR (116, 138, 139). Besides those affecting known HSCR genes (18, 19, 116, 139, 140), several other genic CNVs have also been detected in patients with HSCR lately, whose contribution to HSCR are yet to be functionally characterized—though many of these are also associated with other neurodevelopmental disorders (18). Among these, deletions in 16p11.2 appear to be particularly interesting as at least six patients with HSCR have been reported to have deletion in this locus (18, 141). However, there appears to be no common overlapping region deleted in all the reported patients with HSCR. Deletion and duplication of 16p11.2 are also implicated in intellectual disability and psychiatric disorders (142); however, none of the genes in this region have yet been characterized in animal models with respect to their roles in ENS. It is possible that one or more genes in this locus may be critical for ENS development and therefore deletion of their coding regions can result in haploinsufficiency and thus incomplete gut colonization. Alternatively, loss or disruption of their regulatory elements can lead to dysregulation of gene expression, and, when added up with the sensitized genetic background, can lead to HSCR.
Discussion
From the discoveries of HSCR-associated rare variants, common variants, and CNVs, it is evident that HSCR is a complex disease with involvement of multiple genes and pathways important for ENS development and function. Technological advances in genotyping and sequencing surmount the knowledge-intensive limitations of candidate gene approach and revolutionize genetic studies on HSCR, with disease-associated variants and genes being identified at an accelerated rate. In the past decade, the major genetic discoveries brought out by the hypothesis-free genome-wide approach have shed light into the genetic landscape and architecture of HSCR (Figure 1).
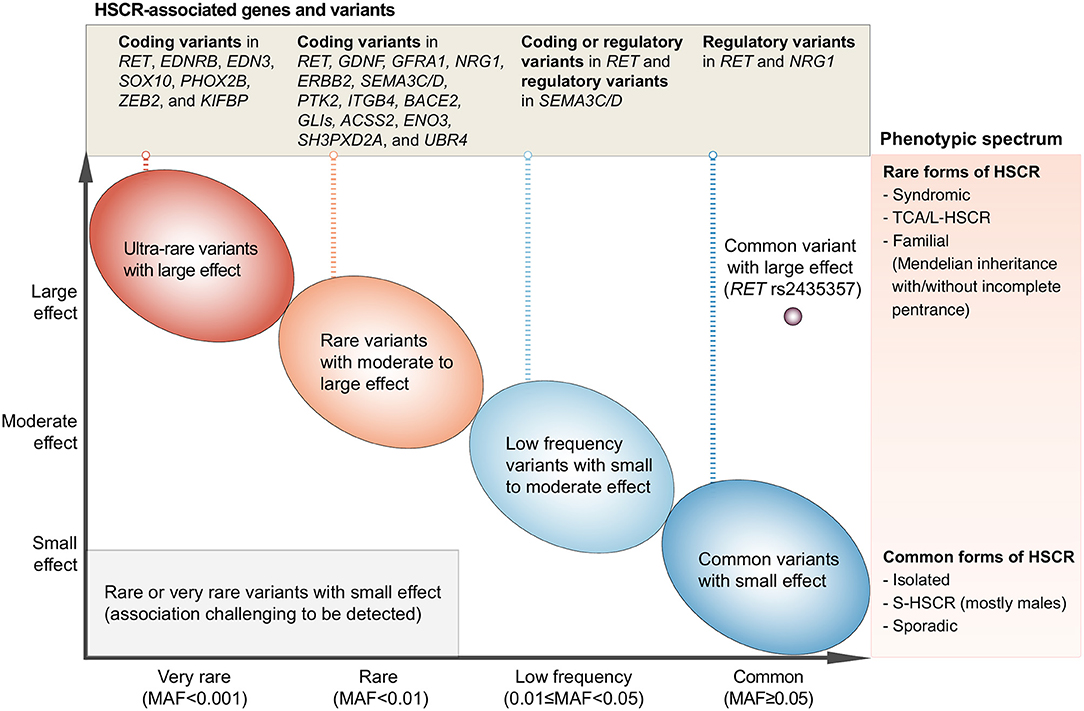
Figure 1. Current understanding of the genetic architecture of HSCR. The x-axis denotes the minor allele frequency (MAF) of variants and the y-axis denotes the effect size. HSCR-associated genetic variants range from variants with extremely low MAF and very large effect size (upper-left side of the plot) to the common/low-frequency variants with small to large effect size (right side of the plot). These variants have been reported in the HSCR loci shown in the upper panel and can cause HSCR with a wide spectrum of severity as denoted in the right panel. On one end of the phenotypic spectrum lies the syndromic HSCR and TCA/L-HSCR, which are the rarer and severe form of HSCR whereas on the other end lies the commonest form of HSCR, i.e., sporadic S-HSCR.
A varying degree of susceptibility to HSCR exists in the general population. This variation is largely due to the combinatorial effects of common, low-frequency, and rare inherited variants with increasing effect size (Figure 1; from right to left, from bottom to top). Indeed, the variable expressivity and phenotypic variability among carriers of the same genetic variant suggest a strong predisposition in the genetic background. The genetic findings reviewed here elegantly illustrate that common HSCR-associated variants, particularly in the major HSCR gene, RET, contribute not only additively but also synergistically to predisposition to HSCR. These common variants can provide a sensitized genetic background that modifies penetrance of rare disease-causing variants and, when the liability threshold is surpassed, will result in clinical manifestation. For example, a highly damaging ultra-rare loss-of-function variant in a key ENS gene (e.g., SOX10) is sufficient to cause the rarer form of the disease—i.e., L-HSCR/TCA—in a person with less sensitized genetic background. On the other hand, for a rare variant with moderate effect size, a highly sensitized genetic background is needed for the phenotypic expression of HSCR. Such joint and epistatic effects between and within regulatory and coding variants represent the integral component of the genetic architecture of HSCR, particularly for sporadic S-HSCR.
Another interesting point to note is that the biological pathways linked to HSCR are not independent of each other (Figure 2). Rather, there is considerable epistasis and crosstalk between these pathways both statistically and biologically, and a dynamic interaction of these pathways orchestrates the ENS development. A deeper understanding of the spatiotemporal crosstalk as well as the interaction between the migrating ENS precursor cells and the extracellular niche is warranted.
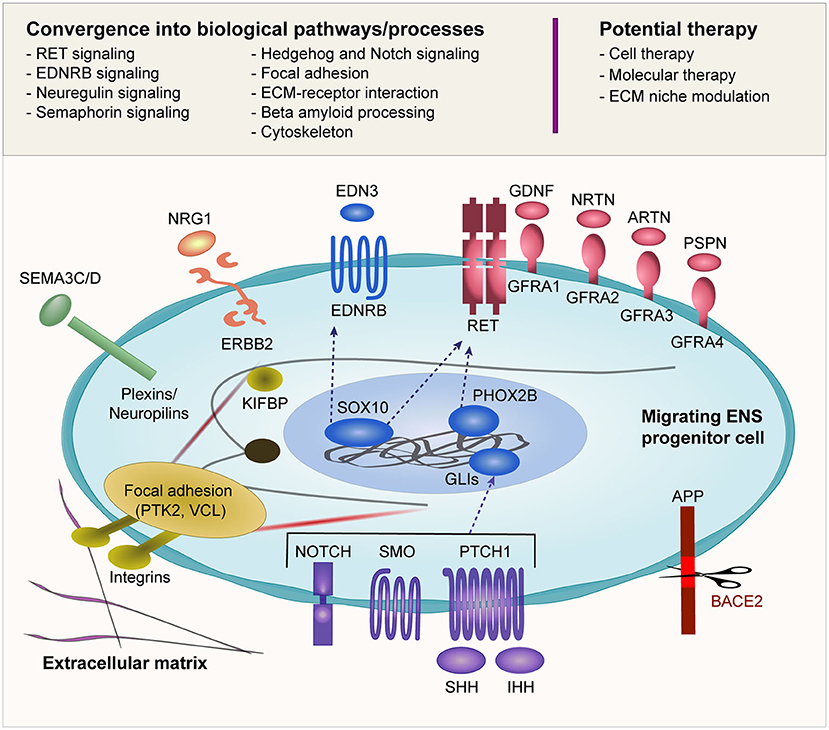
Figure 2. Biological pathways implicated in HSCR. Many of the reported HSCR-associated genes converge into several biological pathways as shown in this figure. The schematic diagram illustrates an ENCC migrating through the gut and its interaction with the surrounding environment including the ECM. The ENCC receives cues from the surrounding environment through different signaling molecules that regulate the cellular machinery as well as feedback interaction with the surrounding niche. The potential therapeutic approaches that are currently under investigation are also shown.
Despite the significant advancement in our understanding of HSCR genetics from the past three decades of research, the translation of these findings to clinical utility has yet to be established. Genetic risk prediction has potential clinical impact of assisting diagnosis, providing genetic counseling, informing treatment options, or predicting prognosis/complication/survival. Currently, there is no evidence-based consensus guideline for genetic testing of HSCR under clinical setting. The necessary first step for the clinical implementation of genetic risk prediction can be prioritized on the HSCR patient subgroups in which the phenotypic presentation is presumed to be caused by high penetrant pathogenic genetic variants and where the incremental benefits on clinical care and decision-making are optimal. Clinical genetic testing can first be considered in the case of syndromic presentation where a genetic diagnosis of a syndrome may lead to the discovery of additional anomalies in other organs and thereby enhance clinical management to improve patient care. Another potential application of clinical genetic testing can be in multiplex family where identification of segregating pathogenic variant may assist recurrence risk prediction and therefore allow genetic counseling. However, its clinical utility on non-syndromic and sporadic patients remains to be explored. Many genetic studies have demonstrated unequivocally that genetic variants of wide frequency spectrum (coding, regulatory, and CNV/chromosomal anomalies) can capture the genetic susceptibility to HSCR albeit with low to moderate discriminative power (18). Sophisticated methods combining polygenic risk of these genetic variants may leverage the performance of risk prediction, and such polygenic model may be applied for genetic testing of the major HSCR subgroup with high genetic heterogeneity. One powerful application of polygenic risk prediction is the potential ability to predict complications; however, up till now, it remains largely unknown if there exists any gene or variant significantly associated with prognosis or complication of HSCR (e.g., enterocolitis, severity of constipation, or incontinence following surgery). Further research should be encouraged to address these knowledge gaps. With the decreasing cost of NGS, WES and preferably WGS can be used in a research setting for genetic profiling. Whenever possible, parents should be included in genetic studies to facilitate the interpretation of variants. For each patient, comprehensive profiling of coding and non-coding variants, SNPs/Indels, and CNVs/chromosomal anomalies should be carried out. Together with other omics approaches (e.g., transcriptomics) and detailed phenotyping of the patients, more HSCR-associated candidate genes and biological pathways, and their differential contribution to the disease severity and complications can be discovered. In parallel, novel therapeutic options should be explored, e.g., stem cell therapy to regenerate the ENS, molecular therapy to modulate ENS formation or ECM niche manipulation, or correction of the culprit mutation with genome editing. While these alternative therapeutic approaches are still in the rudimentary stage, we envision a future where genetic testing would impact clinical care for most of the patients with HSCR by assisting diagnosis and clinical care, predicting complications, and, more importantly, providing alternative treatment options with much better clinical outcome.
Author Contributions
AK and CT wrote the manuscript. PT did the final critical review of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Theme-Based Research Scheme (Grant No. T12C-714/14-R) and Commissioned Paediatric Research at Hong Kong Children's Hospital (PR-HKU-1) to Paul Kwang-Hang Tam and General Research Fund (17113420) and Health Medical Research Fund (06171636) to Clara Sze-Man Tang.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. (2008) 45:1–14. doi: 10.1136/jmg.2007.053959
2. Tam PK, Garcia-Barcelo M. Genetic basis of Hirschsprung's disease. Pediatr Surg Int. (2009) 25:543–58. doi: 10.1007/s00383-009-2402-2
3. Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Transl Res.: the Journal of Laboratory and Clinical Medicine. (2013) 162:1–15. doi: 10.1016/j.trsl.2013.03.001
4. Henderson DJ, Copp AJ. Role of the extracellular matrix in neural crest cell migration. J Anat. (1997) 191:507–15. doi: 10.1046/j.1469-7580.1997.19140507.x
5. Pini Prato A, Rossi V, Mosconi M, Holm C, Lantieri F, Griseri P, et al. A prospective observational study of associated anomalies in Hirschsprung's disease. Orphanet J Rare Dis. (2013) 8:184. doi: 10.1186/1750-1172-8-184
6. Moore SW. Chromosomal and related Mendelian syndromes associated with Hirschsprung's disease. Pediatr Surg Int. (2012) 28:1045–58. doi: 10.1007/s00383-012-3175-6
7. Ryan ET, Ecker JL, Christakis NA, Folkman J. Hirschsprung's disease: Associated abnormalities and demography. J Pediatr Surg. (1992) 27:76–81. doi: 10.1016/0022-3468(92)90111-J
8. Karim A, Akter M, Aziz TT, Hoque M, Chowdhury TK, Imam MS, et al. Epidemiological characteristics of Hirschsprung's disease (HSCR): Results of a case series of fifty patients from Bangladesh. J Pediatr Surg. (2018) 53:1955–9. doi: 10.1016/j.jpedsurg.2017.12.029
9. Puri P, Nakamura H. Familial Hirschsprung's Disease. In: Puri P, editor. Hirschsprung's Disease and Allied Disorders. Cham: Springer International Publishing (2019). p. 115-9. doi: 10.1007/978-3-030-15647-3_6
10. Angrist M, Bolk S, Thiel B, Puffenberger EG, Hofstra RM, Buys CH, et al. Mutation analysis of the RET receptor tyrosine kinase in Hirschsprung disease. Hum Mol Genet. (1995) 4:821–30. doi: 10.1093/hmg/4.5.821
11. Garcia-Barcelo M, Sham MH, Lee WS, Lui VC, Chen BL, Wong KK, et al. Highly recurrent RET mutations and novel mutations in genes of the receptor tyrosine kinase and endothelin receptor B pathways in Chinese patients with sporadic Hirschsprung disease. Clin Chem. (2004) 50:93–100. doi: 10.1373/clinchem.2003.022061
12. Sancandi M, Ceccherini I, Costa M, Fava M, Chen B, Wu Y, et al. Incidence of RET mutations in patients with Hirschsprung's disease. J Pediatr Surg. (2000) 35:139–42. doi: 10.1016/S0022-3468(00)80031-7
13. Attie T, Pelet A, Edery P, Eng C, Mulligan LM, Amiel J, et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum Mol Genet. (1995) 4:1381–6. doi: 10.1093/hmg/4.8.1381
14. Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet. (1996) 14:341–4. doi: 10.1038/ng1196-341
15. Borghini S, Bocciardi R, Bonardi G, Matera I, Santamaria G, Ravazzolo R, et al. Hirschsprung associated GDNF mutations do not prevent RET activation. Eur J Hum Genet. (2002) 10:183–7. doi: 10.1038/sj.ejhg.5200785
16. Tang CS, Li P, Lai FP, Fu AX, Lau ST, So MT, et al. Identification of Genes Associated With Hirschsprung Disease, Based on Whole-Genome Sequence Analysis, and Potential Effects on Enteric Nervous System Development. Gastroenterology. (2018) 155:1908–22. doi: 10.1053/j.gastro.2018.09.012
17. Ruiz-Ferrer M, Torroglosa A, Luzon-Toro B, Fernandez RM, Antinolo G, Mulligan LM, et al. Novel mutations at RET ligand genes preventing receptor activation are associated to Hirschsprung's disease. J Mol Med. (Berlin, Germany). (2011) 89:471–80. doi: 10.1007/s00109-010-0714-2
18. Tilghman JM, Ling AY, Turner TN, Sosa MX, Krumm N, Chatterjee S, et al. Molecular Genetic Anatomy and Risk Profile of Hirschsprung's Disease. N Engl J Med. (2019) 380:1421–32. doi: 10.1056/NEJMoa1706594
19. Tang CS, Zhuang X, Lam WY, Ngan ES, Hsu JS, Michelle YU, et al. Uncovering the genetic lesions underlying the most severe form of Hirschsprung disease by whole-genome sequencing. Eur J Hum Genet: EJHG. (2018) 26:818–26. doi: 10.1038/s41431-018-0129-z
20. Hofstra RM, Valdenaire O, Arch E, Osinga J, Kroes H, Loffler BM, et al. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am J Hum Genet. (1999) 64:304–8. doi: 10.1086/302184
21. Sanchez-Mejias A, Watanabe Y. R MF, Lopez-Alonso M, Antinolo G, Bondurand N, et al. Involvement of SOX10 in the pathogenesis of Hirschsprung disease: report of a truncating mutation in an isolated patient. J Mol Med. (Berlin, Germany). (2010) 88:507–14. doi: 10.1007/s00109-010-0592-7
22. Fernandez RM, Mathieu Y, Luzon-Toro B, Nunez-Torres R, Gonzalez-Meneses A, Antinolo G, et al. Contributions of PHOX2B in the pathogenesis of Hirschsprung disease. PLoS ONE. (2013) 8:e54043. doi: 10.1371/journal.pone.0054043
23. Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet J Rare Dis. (2007) 2:42. doi: 10.1186/1750-1172-2-42
24. Brooks AS, Bertoli-Avella AM, Burzynski GM, Breedveld GJ, Osinga J, Boven LG, et al. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am J Hum Genet. (2005) 77:120–6. doi: 10.1086/431244
25. Ozyavuz Cubuk P. Goldberg-Shprintzen Syndrome Associated with a Novel Variant in the KIFBP Gene. Molecular Syndromology. (2021):1–4. doi: 10.1159/000514531
26. Dafsari HS, Byrne S, Lin JP, Pitt M, Jongbloed JD, Flinter F, et al. Goldberg-Shprintzen megacolon syndrome with associated sensory motor axonal neuropathy. Am J Med Genet Part A. (2015) 167:1300–4. doi: 10.1002/ajmg.a.36873
27. Tang CS, Ngan ES, Tang WK, So MT, Cheng G, Miao XP, et al. Mutations in the NRG1 gene are associated with Hirschsprung disease. Hum Genet. (2012) 131:67–76. doi: 10.1007/s00439-011-1035-4
28. Sribudiani Y, Chauhan RK, Alves MM, Petrova L, Brosens E, Harrison C, et al. Identification of Variants in RET and IHH Pathway Members in a Large Family With History of Hirschsprung Disease. Gastroenterology. (2018) 155:118–29. doi: 10.1053/j.gastro.2018.03.034
29. Ngan ES, Garcia-Barcelo MM, Yip BH, Poon HC, Lau ST, Kwok CK, et al. Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J Clin Invest. (2011) 121:3467–78. doi: 10.1172/JCI43737
30. Takenouchi T, Nakazawa M, Kanemura Y, Shimozato S, Yamasaki M, Takahashi T, et al. Hydrocephalus with Hirschsprung disease: severe end of X-linked hydrocephalus spectrum. Am J Med Genet Part A. (2012) 158A:812–5. doi: 10.1002/ajmg.a.35245
31. Gui H, Schriemer D, Cheng WW, Chauhan RK, Antinolo G, Berrios C, et al. Whole exome sequencing coupled with unbiased functional analysis reveals new Hirschsprung disease genes. Genome Biol. (2017) 18:48. doi: 10.1186/s13059-017-1174-6
32. Lai FP, Lau ST, Wong JK, Gui H, Wang RX, Zhou T, et al. Correction of Hirschsprung-associated mutations in human induced pluripotent stem cells via clustered regularly interspaced short palindromic repeats/Cas9, restores neural crest cell function. Gastroenterology. (2017) 153:139–53. doi: 10.1053/j.gastro.2017.03.014
33. Tam PKH, Tang CSM, Garcia-Barceló M-M. Genetics of Hirschsprung's Disease. In: Puri P, editor. Hirschsprung's Disease and Allied Disorders. Cham: Springer International Publishing (2019). p. 121-31. doi: 10.1007/978-3-030-15647-3_7
34. Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature reviews Neuroscience. (2002) 3:383–94. doi: 10.1038/nrn812
35. Angrist M, Kauffman E, Slaugenhaupt SA, Matise TC, Puffenberger EG, Washington SS, et al. A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10. Nat Genet. (1993) 4:351–6. doi: 10.1038/ng0893-351
36. Lyonnet S, Bolino A, Pelet A, Abel L, Nihoul-Fékété C, Briard ML, et al. A gene for Hirschsprung disease maps to the proximal long arm of chromosome 10. Nat Genet. (1993) 4:346–50. doi: 10.1038/ng0893-346
37. Martucciello G, Bicocchi MP, Dodero P, Lerone M, Silengo Cirillo M, Puliti A, et al. Total colonic aganglionosis associated with interstitial deletion of the long arm of chromosome 10. Pediatr Surg Int. (1992) 7:308–10. doi: 10.1007/BF00183991
38. Fewtrell MS, Tam PK, Thomson AH, Fitchett M, Currie J, Huson SM, et al. Hirschsprung's disease associated with a deletion of chromosome 10 (q112q212): a further link with the neurocristopathies?. J Med Genet. (1994) 31:325–7. doi: 10.1136/jmg.31.4.325
39. Mahaffey SM, Martin LW, McAdams AJ, Ryckman FC, Torres M. Multiple endocrine neoplasia type II B with symptoms suggesting Hirschsprung's disease: a case report. J Pediatr Surg. (1990) 25:101–3. doi: 10.1016/S0022-3468(05)80172-1
40. Verdy M, Weber AM, Roy CC, Morin CL, Cadotte M, Brochu P. Hirschsprung's disease in a family with multiple endocrine neoplasia type 2. J Pediatr Gastroenterol Nutr. (1982) 1:603–7. doi: 10.1097/00005176-198212000-00027
41. Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung's disease. Nature. (1994) 367:377–8. doi: 10.1038/367377a0
42. Edery P, Lyonnet S, Mulligan LM, Pelet A, Dow E, Abel L, et al. Mutations of the RET proto-oncogene in Hirschsprung's disease. Nature. (1994) 367:378–80. doi: 10.1038/367378a0
43. Yin L, Barone V, Seri M, Bolino A, Bocciardi R, Ceccherini I, et al. Heterogeneity and Low Detection Rate of RET Mutations in Hirschsprung Disease. Eur J Hum Genet. (1994) 2:272–80. doi: 10.1159/000472371
44. Seri M, Yin L, Barone V, Bolino A, Celli I, Bocciardi R, et al. Frequency of RET mutations in long- and short-segment Hirschsprung disease. Hum Mutat. (1997) 9:243–9.
45. Svensson PJ, Molander ML, Eng C, Anvret M, Nordenskjold A. Low frequency of RET mutations in Hirschsprung disease in Sweden. Clin Genet. (1998) 54:39–44. doi: 10.1111/j.1399-0004.1998.tb03691.x
46. Fitze G, Cramer J, Ziegler A, Schierz M, Schreiber M, Kuhlisch E, et al. Association between c135G/A genotype and RET proto-oncogene germline mutations and phenotype of Hirschsprung's disease. Lancet. (2002) 359:1200–5. doi: 10.1016/S0140-6736(02)08218-1
47. Ruiz-Ferrer M, Fernandez RM, Antinolo G, Lopez-Alonso M, Eng C, Borrego S, et al. complex additive model of inheritance for Hirschsprung disease is supported by both RET mutations and predisposing RET haplotypes. Genet Med. (2006) 8:704–10.
48. Nunez-Torres R, Fernandez RM, Acosta MJ, Enguix-Riego Mdel V, Marba M, Carlos de, Agustin J, et al. Comprehensive analysis of RET common and rare variants in a series of Spanish Hirschsprung patients confirms a synergistic effect of both kinds of events. BMC Med Genet. (2011) 12:138. doi: 10.1186/1471-2350-12-138
49. So MT, Leon TY, Cheng G, Tang CS, Miao XP, Cornes BK, et al. RET mutational spectrum in Hirschsprung disease: evaluation of 601 Chinese patients. PLoS ONE. (2011) 6:e28986. doi: 10.1371/journal.pone.0028986
50. Carter TC, Kay DM, Browne ML, Liu A, Romitti PA, Kuehn D, et al. Hirschsprung's disease and variants in genes that regulate enteric neural crest cell proliferation, migration and differentiation. J Hum Genet. (2012) 57:485–93. doi: 10.1038/jhg.2012.54
51. Hofstra RM, Wu Y, Stulp RP, Elfferich P, Osinga J, Maas SM, et al. RET and GDNF gene scanning in Hirschsprung patients using two dual denaturing gel systems. Human mutation. (2000) 15:418–29. doi: 10.1002/(SICI)1098-1004(200005)15:5<418::AID-HUMU3>3.0.CO;2-2
52. Garcia-Barcelo M, Ganster RW, Lui VC, Leon TY, So MT, Lau AM, et al. TTF-1 and RET promoter SNPs: regulation of RET transcription in Hirschsprung's disease. Hum Mol Genet. (2005) 14:191–204. doi: 10.1093/hmg/ddi015
53. Emison ES, Garcia-Barcelo M, Grice EA, Lantieri F, Amiel J, Burzynski G, et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am J Hum Genet. (2010) 87:60–74. doi: 10.1016/j.ajhg.2010.06.007
54. Jiang Q, Wang Y, Li Q, Zhang Z, Xiao P, Wang H, et al. Sequence characterization of RET in 117 Chinese Hirschsprung disease families identifies a large burden of de novo and parental mosaic mutations. Orphanet J Rare Dis. (2019) 14:237. doi: 10.1186/s13023-019-1194-2
55. Jiang Q, Liu F, Miao C, Li Q, Zhang Z, Xiao P, et al. RET somatic mutations are underrecognized in Hirschsprung disease. Genet Med. (2018) 20:770–7. doi: 10.1038/gim.2017.178
56. Muller CM, Haase MG, Kemnitz I, Fitze G. Genetic mosaicism of a frameshift mutation in the RET gene in a family with Hirschsprung disease. Gene. (2014) 541:51–4. doi: 10.1016/j.gene.2014.02.027
57. Moore SW, Zaahl MG. Tissue specific somatic mutations and aganglionosis in Hirschsprung's disease. J Pediatr Surg. (2014) 49:258–61. doi: 10.1016/j.jpedsurg.2013.11.035
58. Eketjall S, Ibanez CF. Functional characterization of mutations in the GDNF gene of patients with Hirschsprung disease. Hum Mol Genet. (2002) 11:325–9. doi: 10.1093/hmg/11.3.325
59. Ivanchuk SM, Myers SM, Eng C, Mulligan LM. De novo mutation of GDNF, ligand for the RET/GDNFR-alpha receptor complex, in Hirschsprung disease. Hum Mol Genet. (1996) 5:2023–6. doi: 10.1093/hmg/5.12.2023
60. Doray B, Salomon R, Amiel J, Pelet A, Touraine R, Billaud M, et al. Mutation of the RET ligand, neurturin, supports multigenic inheritance in Hirschsprung disease. Hum Mol Genet. (1998) 7:1449–52. doi: 10.1093/hmg/7.9.1449
61. Salomon R, Attie T, Pelet A, Bidaud C, Eng C, Amiel J, et al. Germline mutations of the RET ligand GDNF are not sufficient to cause Hirschsprung disease. Nat Genet. (1996) 14:345–7. doi: 10.1038/ng1196-345
62. Miao X, Leon TY, Ngan ES, So MT, Yuan ZW, Lui VC, et al. Reduced RET expression in gut tissue of individuals carrying risk alleles of Hirschsprung's disease. Hum Mol Genet. (2010) 19:1461–7. doi: 10.1093/hmg/ddq020
63. Borrego S, Fernandez RM, Dziema H, Niess A, Lopez-Alonso M, Antinolo G, et al. Investigation of germline GFRA4 mutations and evaluation of the involvement of GFRA1, GFRA2, GFRA3, and GFRA4 sequence variants in Hirschsprung disease. J Med Genet. (2003) 40:e18. doi: 10.1136/jmg.40.3.e18
64. Puffenberger EG, Kauffman ER, Bolk S, Matise TC, Washington SS, Angrist M, et al. Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22. Hum Mol Genet. (1994) 3:1217–25. doi: 10.1093/hmg/3.8.1217
65. Puffenberger EG, Hosoda K, Washington SS, Nakao K. deWit D, Yanagisawa M, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell. (1994) 79:1257–66. doi: 10.1016/0092-8674(94)90016-7
66. Verheij JB, Kunze J, Osinga J, van Essen AJ, Hofstra RM, ABCD. syndrome is caused by a homozygous mutation in the EDNRB gene. Am J Med Genet. (2002) 108:223–5. doi: 10.1002/ajmg.10172
67. Sakai T, Nirasawa Y, Itoh Y, Wakizaka A. Japanese patients with sporadic Hirschsprung: mutation analysis of the receptor tyrosine kinase proto-oncogene, endothelin-B receptor, endothelin-3, glial cell line-derived neurotrophic factor and neurturin genes: a comparison with similar studies. Eur J Pediatr. (2000) 159:160–7. doi: 10.1007/s004310050043
68. Amiel J, Attie T, Jan D, Pelet A, Edery P, Bidaud C, et al. Heterozygous endothelin receptor B (EDNRB) mutations in isolated Hirschsprung disease. Hum Mol Genet. (1996) 5:355–7. doi: 10.1093/hmg/5.3.355
69. Bidaud C, Salomon R, Van Camp G, Pelet A, Attié T, Eng C, et al. Endothelin-3 Gene Mutations in Isolated and Syndromic Hirschsprung Disease. Eur J Hum Genet. (1997) 5:247–51. doi: 10.1159/000484771
70. Attie T, Till M, Pelet A, Amiel J, Edery P, Boutrand L, et al. Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung disease. Hum Mol Genet. (1995) 4:2407–9. doi: 10.1093/hmg/4.12.2407
71. Hofstra RM, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg EJ, Stulp RP, et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet. (1996) 12:445–7. doi: 10.1038/ng0496-445
72. Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet. (1996) 12:442–4. doi: 10.1038/ng0496-442
73. Svensson PJ, Von Tell D, Molander ML, Anvret M, Nordenskjold A, A. heterozygous frameshift mutation in the endothelin-3 (EDN-3) gene in isolated Hirschsprung's disease. Pediatr Res. (1999) 45:714–7. doi: 10.1203/00006450-199905010-00018
74. Bondurand N, Sham MH. The role of SOX10 during enteric nervous system development. Dev Biol. (2013) 382:330–43. doi: 10.1016/j.ydbio.2013.04.024
75. Burke EA, Reichard KE, Wolfe LA, Brooks BP, DiGiovanna JJ, Hadley DW, et al. A novel frameshift mutation in SOX10 causes Waardenburg syndrome with peripheral demyelinating neuropathy, visual impairment and the absence of Hirschsprung disease. Am J Med Genet A. (2020) 182:1278–83. doi: 10.1002/ajmg.a.61542
76. Jalilian N, Tabatabaiefar MA, Alimadadi H, Noori-Daloii MR. SOX10 mutation causes Waardenburg syndrome associated with distinctive phenotypic features in an Iranian family: A clue for phenotype-directed genetic analysis. Int J Pediatr Otorhinolaryngol. (2017) 96:122–6. doi: 10.1016/j.ijporl.2017.03.016
77. Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. (2010) 31:391–406. doi: 10.1002/humu.21211
78. Lecerf L, Kavo A, Ruiz-Ferrer M, Baral V, Watanabe Y, Chaoui A, et al. An impairment of long distance SOX10 regulatory elements underlies isolated Hirschsprung disease. Hum Mutat. (2014) 35:303–7. doi: 10.1002/humu.22499
79. Chan KK, Wong CK, Lui VC, Tam PK, Sham MH. Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: Differential effects on target gene regulation. J Cell Biochem. (2003) 90:573–85. doi: 10.1002/jcb.10656
80. Sham MH, Lui VC, Fu M, Chen B, Tam PK. SOX10 is abnormally expressed in aganglionic bowel of Hirschsprung's disease infants. Gut. (2001) 49:220–6. doi: 10.1136/gut.49.2.220
81. Elworthy S, Pinto JP, Pettifer A, Cancela ML, Kelsh RN. Phox2b function in the enteric nervous system is conserved in zebrafish and is sox10-dependent. Mech Dev. (2005) 122:659–69. doi: 10.1016/j.mod.2004.12.008
82. Bishara J, Keens TG, Perez IA. The genetics of congenital central hypoventilation syndrome: clinical implications. Appl Clin Genet. (2018) 11:135–44. doi: 10.2147/TACG.S140629
83. Sandoval RL, Zaconeta CM, Margotto PR, Cardoso MT, Franca EM, Medina CT, et al. Congenital central hypoventilation syndrome associated with Hirschsprung's Disease: case report and literature review. Rev Paul Pediatr. (2016) 34:374–8. doi: 10.1016/j.rppede.2015.10.009
84. Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, et al. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. (2008) 27:469–76. doi: 10.1038/sj.onc.1210659
85. Mosse YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. (2004) 75:727–30. doi: 10.1086/424530
86. Garcia-Barcelo M, Sham MH, Lui VC, Chen BL, Ott J, Tam PK. Association study of PHOX2B as a candidate gene for Hirschsprung's disease. Gut. (2003) 52:563–7. doi: 10.1136/gut.52.4.563
87. Zhao J, Zhu Y, Xie X, Yao Y, Zhang J, Zhang R, et al. Pleiotropic effect of common PHOX2B variants in Hirschsprung disease and neuroblastoma. Aging. (2019) 11:1252–61. doi: 10.18632/aging.101834
88. Miao X, Garcia-Barcelo MM, So MT, Leon TY, Lau DK, Liu TT, et al. Role of RET and PHOX2B gene polymorphisms in risk of Hirschsprung's disease in Chinese population. Gut. (2007) 56:736. doi: 10.1136/gut.2006.116145
89. Hegarty SV, Sullivan AM, O'Keeffe GW. Zeb2: A multifunctional regulator of nervous system development. Prog Neurobiol. (2015) 132:81–95. doi: 10.1016/j.pneurobio.2015.07.001
90. Alves MM, Burzynski G, Delalande JM, Osinga J, van der Goot A, Dolga AM, et al. KBP interacts with SCG10, linking Goldberg-Shprintzen syndrome to microtubule dynamics and neuronal differentiation. Hum Mol Genet. (2010) 19:3642–51. doi: 10.1093/hmg/ddq280
91. Drevillon L, Megarbane A, Demeer B, Matar C, Benit P, Briand-Suleau A, et al. KBP-cytoskeleton interactions underlie developmental anomalies in Goldberg-Shprintzen syndrome. Hum Mol Genet. (2013) 22:2387–99. doi: 10.1093/hmg/ddt083
92. Hirst CS, Stamp LA, Bergner AJ, Hao MM, Tran MX, Morgan JM, et al. Kif1bp loss in mice leads to defects in the peripheral and central nervous system and perinatal death. Sci Rep. (2017) 7:16676. doi: 10.1038/s41598-017-16965-3
93. Tam PK, Owen G. An immunohistochemical study of neuronal microtubule-associated proteins in Hirschsprung's disease. Hum Pathol. (1993) 24:424–31. doi: 10.1016/0046-8177(93)90092-U
94. Alves MM, Osinga J, Verheij JB, Metzger M, Eggen BJ, Hofstra RM. Mutations in SCG10 are not involved in Hirschsprung disease. PLoS ONE. (2010) 5:e15144. doi: 10.1371/journal.pone.0015144
95. Borrego S, Saez ME, Ruiz A, Gimm O, Lopez-Alonso M, Antinolo G, et al. Specific polymorphisms in the RET proto-oncogene are over-represented in patients with Hirschsprung disease and may represent loci modifying phenotypic expression. J Med Genet. (1999) 36:771–4. doi: 10.1136/jmg.36.10.771
96. Borrego S, Ruiz A, Saez ME, Gimm O, Gao X, Lopez-Alonso M, et al. RET genotypes comprising specific haplotypes of polymorphic variants predispose to isolated Hirschsprung disease. J Med Genet. (2000) 37:572–8. doi: 10.1136/jmg.37.8.572
97. Garcia-Barcelo MM, Sham MH, Lui VC, Chen BL, Song YQ, Lee WS, et al. Chinese patients with sporadic Hirschsprung's disease are predominantly represented by a single RET haplotype. J Med Genet. (2003) 40:e122. doi: 10.1136/jmg.40.11.e122
98. Sancandi M, Griseri P, Pesce B, Patrone G, Puppo F, Lerone M, et al. Single nucleotide polymorphic alleles in the 5' region of the RET proto-oncogene define a risk haplotype in Hirschsprung's disease. J Med Genet. (2003) 40:714–8. doi: 10.1136/jmg.40.9.714
99. Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, et al. A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature. (2005) 434:857–63. doi: 10.1038/nature03467
100. Burzynski GM, Nolte IM, Bronda A, Bos KK, Osinga J, Plaza Menacho I, et al. Identifying candidate Hirschsprung disease-associated RET variants. Am J Hum Genet. (2005) 76:850–8. doi: 10.1086/429589
101. Sribudiani Y, Metzger M, Osinga J, Rey A, Burns AJ, Thapar N, et al. Variants in RET associated with Hirschsprung's disease affect binding of transcription factors and gene expression. Gastroenterology. (2011) 140:572–82. doi: 10.1053/j.gastro.2010.10.044
102. Garcia-Barcelo MM, Tang CS, Ngan ES, Lui VC, Chen Y, So MT, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung's disease. Proc Natl Acad Sci U S A. (2009) 106:2694–9. doi: 10.1073/pnas.0809630105
103. Tang CS, Tang WK, So MT, Miao XP, Leung BM, Yip BH, et al. Fine mapping of the NRG1 Hirschsprung's disease locus. PLoS ONE. (2011) 6:e16181. doi: 10.1371/journal.pone.0016181
104. Phusantisampan T, Sangkhathat S, Phongdara A, Chiengkriwate P, Patrapinyokul S, Mahasirimongkol S. Association of genetic polymorphisms in the RET-protooncogene and NRG1 with Hirschsprung disease in Thai patients. J Hum Genet. (2012) 57:286–93. doi: 10.1038/jhg.2012.18
105. Kim JH, Cheong HS, Sul JH, Seo JM, Kim DY, Oh JT, et al. A genome-wide association study identifies potential susceptibility loci for Hirschsprung disease. PLoS ONE. (2014) 9:e110292. doi: 10.1371/journal.pone.0110292
106. Luzon-Toro B, Torroglosa A, Nunez-Torres R, Enguix-Riego MV, Fernandez RM, de Agustin JC, et al. Comprehensive analysis of NRG1 common and rare variants in Hirschsprung patients. PLoS ONE. (2012) 7:e36524. doi: 10.1371/journal.pone.0036524
107. Kapoor A, Jiang Q, Chatterjee S, Chakraborty P, Sosa MX, Berrios C, et al. Population variation in total genetic risk of Hirschsprung disease from common RET, SEMA3 and NRG1 susceptibility polymorphisms. Hum Mol Genet. (2015) 24:2997–3003. doi: 10.1093/hmg/ddv051
108. Fadista J, Lund M, Skotte L, Geller F, Nandakumar P, Chatterjee S, et al. Genome-wide association study of Hirschsprung disease detects a novel low-frequency variant at the RET locus. Eur J Hum Genet: EJHG. (2018) 26:561–9. doi: 10.1038/s41431-017-0053-7
109. Chatterjee S, Kapoor A, Akiyama JA, Auer DR, Lee D, Gabriel S, et al. Enhancer variants synergistically drive dysfunction of a gene regulatory network in hirschsprung disease. Cell. (2016) 167:355–68. doi: 10.1016/j.cell.2016.09.005
110. Jiang Q, Arnold S, Heanue T, Kilambi KP, Doan B, Kapoor A, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am J Hum Genet. (2015) 96:581–96. doi: 10.1016/j.ajhg.2015.02.014
111. Tang CS, Gui H, Kapoor A, Kim JH, Luzon-Toro B, Pelet A, et al. Trans-ethnic meta-analysis of genome-wide association studies for Hirschsprung disease. Hum Mol Genet. (2016) 25:5265–75. doi: 10.1093/hmg/ddw333
112. Rio C, Rieff HI Qi P, Khurana TS, Corfas G. Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron. (1997) 19:39–50. doi: 10.1016/S0896-6273(00)80346-3
113. Wei Z, Yu X, Wu W, Bai M, Lu Y, Song H, et al. Common variants of NRG1 and ITGB4 confer risk of Hirschsprung disease in Han Chinese population. J Pediatr Surg. (2020) 55:2758–65. doi: 10.1016/j.jpedsurg.2020.04.008
114. Yang D, Yang J, Li S, Jiang M, Cao G, Yang L, et al. Effects of RET, NRG1 and NRG3 polymorphisms in a Chinese population with Hirschsprung disease. Sci Rep. (2017) 7:43222. doi: 10.1038/srep43222
115. Zhang Y, Xie X, Zeng J, Wu Q, Zhang R, Zhu D, et al. Association of NRG1 and AUTS2 genetic polymorphisms with Hirschsprung disease in a South Chinese population. J Cell Mol Med. (2018) 22:2190–9. doi: 10.1111/jcmm.13498
116. Tang CS, Cheng G, So MT, Yip BH, Miao XP, Wong EH, et al. Genome-wide copy number analysis uncovers a new HSCR gene: NRG3. PLoS Genet. (2012) 8:e1002687. doi: 10.1371/journal.pgen.1002687
117. Alto LT, Terman JR. Semaphorins and their Signaling Mechanisms. Methods Mol Biol. (2017) 1493:1–25. doi: 10.1007/978-1-4939-6448-2_1
118. Wang Y, Wang J, Pan W, Zhou Y, Xiao Y, Zhou K, et al. Common genetic variations in Patched1 (PTCH1) gene and risk of hirschsprung disease in the Han Chinese population. PLoS ONE. (2013) 8:e75407. doi: 10.1371/journal.pone.0075407
119. Liu JA, Lai FP, Gui HS, Sham MH, Tam PK, Garcia-Barcelo MM, et al. Identification of GLI mutations in patients with hirschsprung disease that disrupt enteric nervous system development in mice. Gastroenterology. (2015) 149:1837–48. doi: 10.1053/j.gastro.2015.07.060
120. Gui H, Tang WK, So MT, Proitsi P, Sham PC, Tam PK, et al. RET and NRG1 interplay in Hirschsprung disease. Hum Genet. (2013) 132:591–600. doi: 10.1007/s00439-013-1272-9
121. Carrasquillo MM, McCallion AS, Puffenberger EG, Kashuk CS, Nouri N, Chakravarti A. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet. (2002) 32:237–44. doi: 10.1038/ng998
122. Brosens E, Burns AJ, Brooks AS, Matera I, Borrego S, Ceccherini I, et al. Genetics of enteric neuropathies. Dev Biol. (2016) 417:198–208. doi: 10.1016/j.ydbio.2016.07.008
123. Parikh DH, Tam PK, Van Velzen D, Edgar D. The extracellular matrix components, tenascin and fibronectin, in Hirschsprung's disease: an immunohistochemical study. J Pediatr Surg. (1994) 29:1302–6. doi: 10.1016/0022-3468(94)90101-5
124. Gao N, Hou P, Wang J, Zhou T, Wang D, Zhang Q, et al. Increased Fibronectin Impairs the Function of Excitatory/Inhibitory Synapses in Hirschsprung Disease. Cell Mol Neurobiol. (2020) 40:617–28. doi: 10.1007/s10571-019-00759-4
125. Akbareian SE, Nagy N, Steiger CE, Mably JD, Miller SA, Hotta R, et al. Enteric neural crest-derived cells promote their migration by modifying their microenvironment through tenascin-C production. Dev Biol. (2013) 382:446–56. doi: 10.1016/j.ydbio.2013.08.006
126. Fu M, Barlow-Anacker AJ, Kuruvilla KP, Bowlin GL, Seidel CW, Trainor PA, et al. 37/67-laminin receptor facilitates neural crest cell migration during enteric nervous system development. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. (2020) 34:10931–47. doi: 10.1096/fj.202000699R
127. Breau MA, Pietri T, Eder O, Blanche M, Brakebusch C, Fassler R, et al. Lack of beta1 integrins in enteric neural crest cells leads to a Hirschsprung-like phenotype. Development (Cambridge, England). (2006) 133:1725–34. doi: 10.1242/dev.02346
128. Soret R, Mennetrey M, Bergeron KF, Dariel A, Neunlist M, Grunder F, et al. A collagen VI-dependent pathogenic mechanism for Hirschsprung's disease. J Clin Invest. (2015) 125:4483–96. doi: 10.1172/JCI83178
129. Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, et al. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci U S A. (2006) 103:1941–6. doi: 10.1073/pnas.0510708103
130. Niu WB, Bai MR, Song HL, Lu YJ, Wu WJ, Gong YM. et al. Association of variants in PLD1, 3p241, and 10q1121 regions with hirschsprung's disease in Han Chinese population. Front Genet. (2020) 11:738. doi: 10.3389/fgene.2020.00738
131. Fu AX, Lui KN, Tang CS, Ng RK, Lai FP, Lau ST, et al. Whole-genome analysis of noncoding genetic variations identifies multiscale regulatory element perturbations associated with Hirschsprung disease. Genome Res. (2020) 30:1618–32. doi: 10.1101/gr.264473.120
132. Sergi CM, Caluseriu O, McColl H, Eisenstat DD. Hirschsprung's disease: clinical dysmorphology, genes, micro-RNAs, and future perspectives. Pediatr Res. (2017) 81:177–91. doi: 10.1038/pr.2016.202
133. Heuckeroth RO. Hirschsprung disease - integrating basic science and clinical medicine to improve outcomes. Nat Rev Gastroenterol Hepatol. (2018) 15:152–67. doi: 10.1038/nrgastro.2017.149
134. Zhuang X, Ye R, So MT, Lam WY, Karim A, Yu M, et al. A random forest-based framework for genotyping and accuracy assessment of copy number variations. NAR Genom Bioinform. (2020) 2:lqaa071. doi: 10.1093/nargab/lqaa071
135. Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet. (2015) 6:138.
136. Antaki D, Brandler WM, Sebat J. SV2: accurate structural variation genotyping and de novo mutation detection from whole genomes. Bioinformatics (Oxford, England). (2018) 34:1774–7. doi: 10.1093/bioinformatics/btx813
137. Moreno-Cabrera JM, Del Valle J, Castellanos E, Feliubadalo L, Pineda M, Brunet J, et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur J Hum Geneti: EJHG. (2020) 28:1645–55. doi: 10.1038/s41431-020-0675-z
138. Kuil L, MacKenzie KC, Tang CS, Windster JD, Le TL, Karim A, et al. Size matters: large copy number losses reveal novel Hirschsprung disease genes. medRxiv. (2020):2020.11.02.20221481. doi: 10.1101/2020.11.02.20221481
139. Lantieri F, Gimelli S, Viaggi C, Stathaki E, Malacarne M, Santamaria G, et al. Copy number variations in candidate genomic regions confirm genetic heterogeneity and parental bias in Hirschsprung disease. Orphanet J Rare Dis. (2019) 14:270. doi: 10.1186/s13023-019-1205-3
140. Jiang Q, Ho YY, Hao L, Nichols Berrios C, Chakravarti A. Copy number variants in candidate genes are genetic modifiers of Hirschsprung disease. PLoS ONE. (2011) 6:e21219. doi: 10.1371/journal.pone.0021219
141. Sampson MG, Coughlin CR, 2nd, Kaplan P, Conlin LK, Meyers KE, Zackai EH, et al. Evidence for a recurrent microdeletion at chromosome 16p11.2 associated with congenital anomalies of the kidney and urinary tract (CAKUT) and Hirschsprung disease. Am J Med Genet Part A. (2010) 152A:2618–22. doi: 10.1002/ajmg.a.33628
Keywords: Hirschsprung disease, aganglionosis, genetics, genetic architecture, rare variants, GWAS, next-generating sequencing, common variants
Citation: Karim A, Tang CS and Tam PKH (2021) The Emerging Genetic Landscape of Hirschsprung Disease and Its Potential Clinical Applications. Front. Pediatr. 9:638093. doi: 10.3389/fped.2021.638093
Received: 05 December 2020; Accepted: 02 July 2021;
Published: 05 August 2021.
Edited by:
Andrew S. Day, University of Otago, New ZealandReviewed by:
Victor Manuel Navas-López, Hospital Materno-Infantil, SpainMaria M. Alves, Erasmus University Medical Center, Netherlands
Copyright © 2021 Karim, Tang and Tam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul Kwong-Hang Tam, paultam@hku.hk; Clara Sze-Man Tang, claratang@hku.hk