 Fei Xiong
Fei Xiong Qiao Zhou
Qiao Zhou Xiaobo Huang3*
Xiaobo Huang3* Peng Cao
Peng Cao Yi Wang
Yi Wang- 1Department of Gastroenterology, Sichuan Academy of Medical Science and Sichuan Provincial People’s Hospital, Chengdu, China
- 2Department of Rheumatology and Immunology, Sichuan Academy of Medical Science and Sichuan Provincial People’s Hospital, Chengdu, China
- 3Department of Critical Care Medicine, Sichuan Academy of Medical Science and Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 4Department of Pharmacy, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Ferroptosis relies on iron, and ferroptotic cell death is triggered when the balance of the oxidation-reduction system is disrupted by excessive lipid peroxide accumulation. A close relationship between ferroptosis and nonalcoholic steatohepatitis (NASH) is formed by phospholipid peroxidation substrates, bioactive iron, and reactive oxygen species (ROS) neutralization systems. Recent studies into ferroptosis during NASH development might reveal NASH pathogenesis and drug targets. Our review summarizes NASH pathogenesis from the perspective of ferroptosis mechanisms. Further, we discuss the relationship between mitochondrial dysfunction, ferroptosis, and NASH. Finally, potential pharmacological therapies directed to ferroptosis in NASH are hypothesized.
Introduction
Nonalcoholic steatohepatitis (NASH) is a severe form of nonalcoholic fatty liver disease (NAFLD). Its histological characteristics are similar to those of alcoholic hepatitis, including macro-vesicular steatosis, hepatocyte ballooning, and necroinflammation, with or without fibrosis (Brunt et al., 1999; Matteoni et al., 1999; Kleiner et al., 2005). NASH may lead to cirrhosis in as many as 20% of patients (Matteoni et al., 1999) or to primary hepatocellular carcinoma (Angulo and Diehl, 2015; Wang et al., 2021). To date, lifestyle intervention is the immediate intervention for NASH (without fibrosis) (Diehl and Day, 2017), and pharmacological therapies for NASH are relatively rare, partly because NASH pathogenesis is too complex to be elucidated.
The liver is essential for regulating iron balance (Chen et al., 2022). Transferring receptor 1 (TFR1) and SLC39A14 deliver iron into hepatocytes and participate in many physiological and metabolic processes. Excess iron is stored as ferritin, and ferroportin (FPN) is critical for the elimination of iron. Hepcidin, ring finger protein 217, and a protein that regulates iron form a regulatory network that balances iron levels.
Similarly, some evidence suggests that iron plays a role in the progression of NASH (Bonkovsky et al., 1999; Facchini and Stoohs, 2002; Valenti et al., 2010; Nelson et al., 2011). In NASH patients, an increase in the haemochromatosis mutation rate is associated with an elevation in the hepatic iron level. More importantly, increased hepatic iron appears to be associated with liver fibrosis in NASH patients. Notably, iron is a crucial factor in insulin sensitivity. These studies imply that ferroptosis, a programmed cell death that relied on iron homeostasis, may play a role in the pathogenesis of NASH.
In this review, we focus on the potential mechanism of ferroptosis involved in the development of NASH and summarize some possible targets related to ferroptosis that may be important to NASH therapy.
Ferroptosis, mitochondria, and nonalcoholic steatohepatitis
In 2012, Dixon, Lemberg (Dixon et al., 2012) discovered a unique mode of death mediated by erastin in cancerous cells. They also found that certain iron chelators inhibited this unusual death modality and concluded that the death modality relied on iron; therefore, they called this form of death “ferroptosis.” They further found that the target of erastin was the cystine-glutamate transporter XC− and that ferroptosis was triggered after cystine-glutamate transporter XC− activity was inhibited by erastin. Cysteine deprivation (Gao et al., 2015) or suppression of cystine-glutamate transporter XC− disrupt glutathione (GSH) synthesis. GSH is an important antioxidant, and its depletion results in glutathione peroxidase 4 (GPX4) deactivation. Under normal conditions, hydroperoxides formed by PUFAs are reduced by GPX4. Inactivation of GPX4 leads to the accumulation of lipid peroxide. Bioactive iron triggers lipid peroxidation amplifies the generation of free oxygen radical species via the Fenton reaction and intensifies the dysregulation of oxidative lipid metabolism. Overall, the balance of the oxidation-reduction system in the cell is disrupted during ferroptosis. The mechanism map suggests a kind of strong connection between ferroptosis and diseases (Stockwell et al., 2017).
Because iron metabolism is closely associated with the liver, it is not surprising that ferroptosis has been recently found to play an essential role in hepatic injury (Wang et al., 2017; Chen et al., 2022). More interestingly, Minoru Tanaka et al. found ferroptosis was the first type of cell death via the choline-deficient, methionine-supplemented diet model and mixed lineage kinase domain-like protein (MLKL) knockout mice. The research hints ferroptosis is an important player in NAFLD conversion to NASH (Tsurusaki et al., 2019). Notably, three characteristics of NASH suggest ferroptosis is involved in the progression of NASH:
1) Lipid accumulation is the basis of hepatic steatosis. Excess lipid accumulation induces oxidative stress (Chen et al., 2020). Some reports have reported a high level of PUFAs in plasma samples taken from patients with NASH (Loomba et al., 2015), and PUFA intake increases the risk of NAFLD (Xie et al., 2021). These lines of evidence imply that PUFAs profoundly impact NASH development. The substrates for polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) are necessary for ferroptosis (Lei et al., 2022). Oxidized phospholipid-induced inflammation promotes the progression of NASH (Sun et al., 2020). In summary, this body of evidence suggests that hepatocytes in NASH patients are likely to be enriched with PUFA-PLs, creating a precondition for ferroptosis.
2) The available evidence suggests that hepatic iron is involved in NASH. In physiological conditions, FPN is mainly expressed in the cytomembrane of Kupffer cells and hepatocytes around the portal vein (Drakesmith et al., 2015). Wang F et al. have shown FPN plays an important role in iron mobilization of hepatocyte and iron storage in macrophage in hepatocyte-specific FPN1 deletion mice (Zhang et al., 2012). Prolonged inflammation can produce more interleukin-6 (IL-6), and the binding of IL-6 to its receptor activates JAK/STAT pathway, resulting in STAT3 phosphorylation. Phosphorylated STAT3 enters the cell nucleus and activates the Hepcidin gene expression via locating the binding site in the Hepcidin promoter region (Camaschella et al., 2020). The combination of Hepcidin and FPN results in the change of the spatial configuration of FPN and leads to FPN degradation via ubiquitination and internalization (Billesbølle et al., 2020). Low expression of FPN inhibits iron release in macrophages and iron absorption in the duodenum (Donovan et al., 2005). There is evidence that IL-6 increases in NASH patients (Wieckowska et al., 2008) and STAT3 activation plays an important role in the fibrosis of NASH (Zhao et al., 2021). The molecular mechanism suggests that hepatic iron is associated with the development of NASH in a variety of pathological processes. Insulin resistance (Mendler et al., 1999) is directly associated with hepatic iron levels, and lower blood sugar decreases the iron concentration and inhibits iron transport. Other researchers have found a correlation between the hepatic iron level and the mutation rate of the haemochromatosis gene (HFE) (Bonkovsky et al., 1999). As a significant symbol in NASH development, an increase in hepatic iron has been related to the degree of hepatic fibrosis (George et al., 1998; Valenti et al., 2010). More importantly, elevated citrate has been detected in NAFLD; citrate indirectly produces free radicals via the Fenton reaction (van de Wier et al., 2013). The Fenton reaction can amplify the number of free oxygen radical species. Notably, bioactive iron is an essential component of ferroptosis, suggesting that ferroptosis promotes the progression of NASH via the Fenton reaction.
3) The cellular antioxidant ability is impaired. As mentioned above, the antioxidant ability in NASH is damaged or exhausted. The depletion of mGSH (Serviddio et al., 2008) is only one of many influences, and a lack of a variety of antioxidants has been found in NASH patients (Baskol et al., 2007), indicating that the ROS neutralization system in NASH was damaged. In recent years, researchers found that loss of Nrf2 is the key factor for the development of NASH (Xu et al., 2019). Nrf2 inhibits fatty acid biosynthesis by down-regulating ATP-citrate lyase, acetyl-CoA carboxylase 1, fatty acid synthase et al. (Yates et al., 2009; Kitteringham et al., 2010; Wu and Klaassen, 2011). The inhibiting action of Nrf2 on fatty acid biosynthesis might be attributed to Nrf2-mediated inhibition of the nuclear receptor liver X receptor–α gene (LXR-α) (Kay et al., 2011). More importantly, as a reducing agent, NADPH is necessary for biocatalysis and biotransformation of oxidized GSH by glutathione reductase. Nrf2 regulates NADPH biosynthesis with the help of malic enzyme and isocitrate dehydrogenase (Thimmulappa et al., 2002). Low expression of Nrf2 does not only promote the synthesis of fatty acids but also leads to the loss of reduced glutathione. Nrf−/− mice also show a correlation between Nrf2 and immunoreactions (Itoh et al., 2004). As indicated above, the inhibition of the cystine-glutamate transporter XC− induces ferroptosis. Studies have shown that cystine deprivation and glutaminolysis regulate ferroptosis (Gao et al., 2015). These studies reveal that ferroptosis is accompanied by the depletion of GSH. GSH is the primary antioxidant in the mitochondrial repair system, and its depletion shows that the cellular antioxidant ability is diminished after ferroptosis.
In summary, considering phospholipid peroxidation substrates, bioactive iron, and antioxidant system inhibition, ferroptosis may play an essential role in NASH development. Some indirect evidence supports this supposition; for example, haem oxygenase-1 blocked the progression of steatohepatitis (Wang et al., 2010). Moreover, recent research showed that haem oxygenase-1 induced ferroptosis mediated via mitochondrial factors (Wang et al., 2016). Further, the human genetic study brought home this point. Xingguo Liu et al. found that in two patients with mitochondrial DNA depletion syndrome (MDS), DGUOK mutant hepatocyte-like cells and hepatocyte organoids were more susceptible to iron overload-induced ferroptosis (Guo et al., 2021).
Some reports have shown that NASH usually exhausts mitochondrial glutathione (mGSH) by increasing the mitochondrial cholesterol level (Serviddio et al., 2008; Josekutty et al., 2013). GSH plays a crucial role in the antioxidative system, and its function is facilitated by its reduction and conjugation (Forman et al., 2009). The depletion of mGSH suggests that mitochondria have lost antioxidative capacity and that ROS levels have increased. Hence, mGSH is at least part of the second hit in the 2-s hit theory of NASH. GSH levels are generally lower in NAFLD patients than in healthy people (Kumar et al., 2013). Although the details of mGSH transport have not been thoroughly explained (Ribas et al., 2014), a lower level of cellular GSH exacerbates mGSH depletion; that is, changes in cellular GSH charge and concentration affect mGSH levels. Data have shown that glutamate is usually increased in NAFLD patients (Gaggini et al., 2018); therefore, a decrease in GSH may partly contribute to increased glutaminolysis (Du et al., 2020). Moreover, some researchers have indicated that the cysteine level is increased in NAFLD patients (Kalhan et al., 2011). In summary, impaired mitochondria indicate damage to the ROS neutralization system.
Hence, glutamine is a critical factor in ferroptosis, and glutaminolysis regulates ferroptosis (Gao et al., 2015); α-ketoglutarate is a product of glutaminolysis and is part of the tricarboxylic acid cycle; therefore, mitochondria and ferroptosis seem to be linked (He et al., 2020). As mentioned above, mitochondrial dysfunction is accompanied by NASH development. Abnormal mitochondrial ultrastructure has also been observed in ferroptosis (Dixon et al., 2012). Similar to that in NASH, a change in mitochondrial morphology has been found in hepatocytes undergoing ferroptosis (Wang et al., 2017). Mitochondria are the primary sources of ROS (Murphy, 2009); therefore, mitochondria likely produce ROS, increasing lipid peroxidation. During NASH development, the depletion of mGSH implies that a decreased GSH level (Serviddio et al., 2008) is a result of mGSH exhaustion in mitochondria but not factors outside mitochondria. mGSH exhaustion-related ferroptosis is reported in programmed cell death of cardiomyocyte (Jang et al., 2021). RSL3 induced ferroptosis of cardiomyocytes via the accumulation of adrenoyl-phosphatidylethamines (PEs). The decrease of dicarboxylate carrier (DIC) and oxoglutarate carrier (OGC) leads to the depletion of mGSH and increase ROS. The interaction of mGSH and GPX4 regulates the process of ferroptosis with the help of the accumulation of PEs. The depletion of mGSH suggested that NASH may have a similar mechanism in NASH development.
Interestingly, the research showed the inhibition of DIC and OGC has a significant effect on mitochondrial membrane potential which was reduced by 14% less in PSA&BMA-treated cells (PSA, DIC inhibitor, and BMA, OGC inhibitor) than in the untreated cells (Jang et al., 2021). Earlier research also reported mitochondrial membrane potential changes in ferroptosis (Gao et al., 2019). The evolutionary formation of the membrane potential indicates that modification of membrane permeability—the transformation of membrane permeability—is most likely exacerbated by mGSH depletion. In the cellular defense mechanism, GPX4 is the most critical antioxidase; research has shown that lipid peroxidation induced by GPX4 is not evident in mitochondria (Angeli et al., 2014). However, further studies showed that GPX4 in mitochondria plays a defender in ferroptosis after dihydroorotate dehydrogenase (DHODH) is deactivated (Zhu et al., 2020; Mao et al., 2021). DHODH is located on the outside surface of the inner mitochondrial membrane. It can reduce coenzyme Q (CoQ) to ubiquinol (CoQH2). CoQ is a critical component in the electron transport chain. CoQH2 is the reduced form of CoQ; as an antioxidant, CoQH2 captures free radicals produced via lipid peroxidation. Hence, when the expression level of GPX4 is low, DHODH is expressed to prevent ferroptosis (Figure 1). Paradoxically, a recent study showed the benefit of using an inhibitor of DHODH to treat NAFLD (Zhu et al., 2020). Notably, the work concentrated on inflammatory cells, not hepatic cells or Kupffer cells. In summary, mitochondria are likely to regulate ferroptosis by inhibiting the ROS neutralization system.
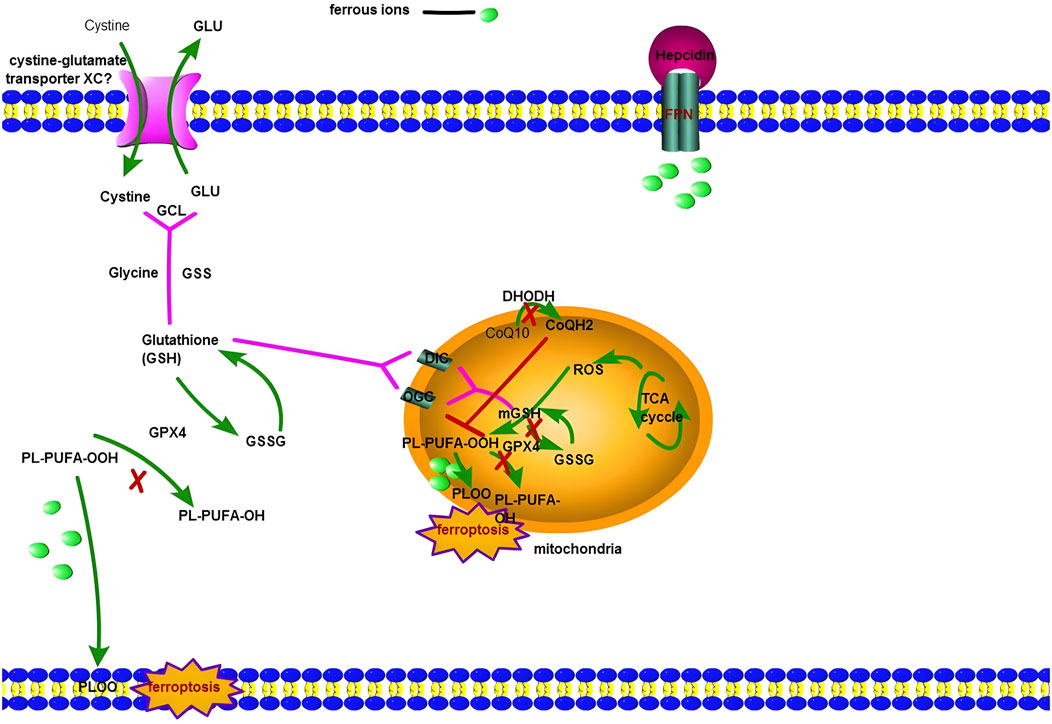
FIGURE 1. The figure shows ferroptosis involves both the cytoplasm and mitochondria. Cystine-glutamate transporter XC− is responsible for the transfer of Cystine into the macrophage (or hepatocytes around the portal vein), then Cysteine takes part in the process of Glutathione biosynthesis. GPX4 regulates the reduced state (GSH) and oxidized state (GSSH). Meanwhile, PL-PUFA-OOH is converted to PL-PUFA-OH by GPX4. When prolonged inflammation can produce more interleukin-6 (IL-6), and the binding of IL-6 to its receptor activates hepatocyte’s JAK/STAT pathway, resulting in STAT3 phosphorylation. Phosphorylated STAT3 enters the hepatocyte nucleus and activates the Hepcidin gene expression by locating the binding site in the Hepcidin promoter region. A large amount of Hepcidin protein is finally secreted out of the cells. It results in the combination of Hepcidin and FPN and leads to FPN degradation via ubiquitination and internalization, so macrophages (or hepatocytes around the portal vein) accumulate more iron. When GPX4 is deactivated, the Fenton reaction can initiate ferroptosis in the cytoplasm. For NASH patients, there exists another mechanism: ferroptosis in mitochondria. Mitochondria have two kinds of antioxidase: GPX4 and DHODH. DHODH is located on the outside surface of the inner mitochondrial membrane. It can reduce coenzyme Q (CoQ) to ubiquinol (CoQH2). CoQ is a critical component in the electron transport chain. CoQH2 is the reduced form of CoQ; as an antioxidant, CoQH2 captures free radicals produced via lipid peroxidation. Hence, ferroptosis will start successfully in mitochondrial if DHODH and GPX4 are both deactivated.
Potential drug targets of nonalcoholic steatohepatitis related to ferroptosis
To date, pharmacologic therapies for NASH have shown limited efficacy. As an antioxidant, vitamin E, which is used to treat NASH, has led to inconsistent results. A randomized control trial (RCT) showed that vitamin E or vitamin C reduced fibrosis scores (which was significant, p = 0.002) (Harrison et al., 2003). However, in another study, vitamin E treatment failed to reduce fibrosis scores (p = 0.24) (Sanyal et al., 2010). A meta-analysis revealed that trials with vitamin E indicated high heterogeneity (Musso et al., 2010). Some insulin sensitizers have shown a similar pattern (Musso et al., 2010). Furthermore, long-term use of vitamin E has a potential risk and large doses of vitamin E may increase all-cause mortality (Miller et al., 2005). In addition, vitamin E treatment may increase the risk of prostate cancer (Klein et al., 2011). More importantly, most trial periods are too short to maintain data accuracy, and these trials typically do not include crucial clinical outcomes, such as liver cirrhosis or liver cancer. Even the curative effects of GLP-1 receptor agonists are unclear (Newsome et al., 2021), indicating the need for additional data on GLP-1 receptor agonist trials.
Furthermore, GLP-1 receptor agonists are used only for patients with NASH and diabetes, and the scope of treatment is limited. In summary, the mechanism of insulin resistance is unlikely to be sufficiently similar to those of potential drug targets for NASH; therefore, we need to change our minds: ferroptosis may be a more analogous system. That is, based on the mechanism of ferroptosis, potential pharmacologic therapies can be classified into three attributional categories: regulating substrates for phospholipid peroxidation, iron chelating, and repairing the ROS neutralization system.
Substrates of phospholipid peroxidation can be regulated; for example, statins are a choice for regulating PUFAs (Nozue et al., 2013). However, data on statins as therapeutic drugs are insufficient to prove their benefit for NASH patients (Tomasiewicz et al, 2018). Paradoxically, omega-3 polyunsaturated fatty acids are beneficial to NAFLD patient (Masterton et al., 2010). However, PUFA-PLs are also found in normal cells. It is essential to maintain certain physiological functions, such as the cell membrane fluidity structure of the cytomembrane; therefore, PUFA-PLs may be necessary for hepatic cells. In addition, considering the mechanism, cells’ oxidation-reduction system is imbalanced during ferroptosis. When the imbalanced oxidation-reduction system cannot be corrected, the simple reduction in PUFAs may not be enough to inhibit ferroptosis. Therefore, regulating substrates for phospholipid peroxidation is not a key factor in ferroptosis.
Iron chelators appear to be good choices for regulating ferroptosis. Brent Stockwell et al. discovered ferroptosis was inhibited via the intervention of certain iron chelators (Dixon et al., 2012). However, in a cohort study, the main clinical effects of iron accumulation in NASH patients were insignificant (Younossi et al., 1999). In this unselected cohort, certain unknown confounding factors were not eliminated, and the number of patients with NASH was lower than that of the other cohort. Therefore, further research is needed. In the past, targeted pharmacologic therapies for NASH usually focused on insulin resistance and antioxidants; fewer studies have been directed to hepatic iron as a drug target for NASH. Notably, no significant benefit has been obtained via phlebotomy (Adams et al., 2015); hepatic iron was not adequately removed. In the clinic, a chelating agent is a potential therapeutic choice. For example, deferoxamine is usually suitable for beta-thalassemia and other iron-overload conditions. Compared with phlebotomy, hepatic parenchymal cells can take up more iron. This means that deferoxamine can clear iron directly from hepatocytes. Indeed, the half-life of deferoxamine is short; the potential question is whether iron accumulates in mitochondria. In rat hepatic cells, the iron level in mitochondria was double that of the cytoplasm (Paul et al., 2017). Iron chelators may not be able to capture enough bioactive iron in mitochondria to benefit NASH patients; therefore, further physiological efficacy and safety data need to be generated in future relevant clinical studies of NASH.
Repairing the ROS neutralization system via GSH is a good choice in ferroptosis. A pilot study showed a benefit from biochemical tests for patients with NASH (Irie et al., 2016). However, similar to iron chelators, GSH levels can be exhausted in mitochondria (Serviddio et al., 2008). It is unclear whether supplementation with GSH can lead to mGSH level recovery. A method to transport GSH into mitochondria is also a challenge. Additional data on GSH effects are needed in the future to develop mGSH-targeted pharmacologic therapy for NASH. We now know that GPX4 and DHODH are two central defense systems against ferroptosis in mitochondria (Mao et al., 2021). As DHODH is depleted, the defense system increasingly relies on GPX4 and vice versa. Pharmaceutical inhibitors of DHODH are potential for cancer treatment; however, the same drug target is not useful for developing NASH therapies.
Interestingly, a DHODH inhibitor drug, vidofludimus, showed the potential to reverse hepatic steatosis and reduce inflammation (Zhu et al., 2020). However, the results of these studies do not seem to comport with the mechanism of ferroptosis. Immune cells were the targets in the study. However, one hypothesis suggests that DHODH inhibition upregulates the expression of GPX4 under certain conditions and thus inhibits ferroptosis. In addition, a recent study of the regulation of cancer immunity by ferroptosis sheds some light on that question. Weiping Zou, Weimin Wang et al. found CD8+ cells induce tumor ferroptosis by interferon gamma they produced (Wang et al., 2019). The research first discovered ferroptosis is a new mechanism of anti-tumor, and it has widely applied prospects in the immunotherapy of tumours. According to this line of thinking, In the development of NASH, vidofludimus may regulate hepatic cell ferroptosis via the inhibition of Immune cells. Additional research is needed to explore the relevant mechanism of DHODH action in ferroptosis.
Conclusion and perspectives
Importantly, although the pathogenesis of NASH is still debated, a large body of evidence shows that ferroptosis likely plays an essential role in NASH development. Evidence suggesting a relationship between ferroptosis and NASH has been mainly focused on three aspects: (Brunt et al., 1999) A high level of PUFAs is found in NASH patients, and PUFAs undergo oxidative phosphorylation, a process that induces inflammation. These characteristics suggest that PUFA-PLs play roles in NASH. (Kleiner et al., 2005) Iron participates in the development of NASH. Importantly, the Fenton reaction has been associated with the development of NASH; this reaction can amplify the number of free oxygen radical species involved in ferroptosis. (Matteoni et al., 1999) Evidence from a wide range of sources suggests that the balance of the oxidation-reduction system in hepatic cells is disrupted in NASH. Considering these three characteristics, we have discussed lipid-lowering agents, iron chelators, and GSH for use in drug therapies for NASH. Furthermore, we explored potential drug targets, such as bioactive iron in mitochondria and DHODH inhibitors.
As the research into ferroptosis in NASH advances, we believe significant progress will be made to show the future relationship between ferroptosis and NASH, providing new ideas and laying a foundation for identifying potential pharmacologic targets for NASH.
Interestingly, accumulating evidence suggests that mitochondria may play an important role in the progression of NASH and potentially in ferroptosis. Considerable evidence supports the idea that impaired mitochondrial function is involved in NASH, as indicated by abnormal morphology, gene expression, defective mitophagy, and depletion of mGSH. Similar evidence is found for ferroptosis. Thus, a strong association between NASH and ferroptosis seems possible. With intensive ongoing research, a better understanding of how mitochondria are involved in NASH and ferroptosis will contribute to resolving the debate on whether mitochondria regulate ferroptosis.
Author contributions
YW, PC, and XH contributed to the conception and design of the work. FX, QZ, and YW drafted the manuscript. FX and QZ prepared the figure. FX and QZ substantively revised the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (81802504, 81872207, 81903901), Sichuan Science and Technology Bureau (2019YFS0439, 2020JDJQ0067, 2020JDRC0118, 2021YJ0564, 2022YFH0005), the Science and Technology Innovation Project of Chengdu, China (No. 2021-YF05-00225-SN), a Sichuan Medical Association grant (No. Q19037), and a grant from Sichuan Provincial Health Commission (20PJ105).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, L. A., Crawford, D. H., Stuart, K., House, M. J., St Pierre, T. G., Webb, M., et al. (2015). The impact of phlebotomy in nonalcoholic fatty liver disease: a prospective, randomized, controlled trial. Hepatology 61 (5), 1555–1564. doi:10.1002/hep.27662
Angeli, J., Schneider, M., Proneth, B., Tyurina, Y., Tyurin, V., Hammond, V., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191. doi:10.1038/ncb3064
Angulo, P., and Diehl, A. M. (2015). Fibrosis in nonalcoholic fatty liver disease: mechanisms and clinical implications. Semin. Liver Dis. 35 (2), 132–145. doi:10.1055/s-0035-1550065
Baskol, G., Baskol, M., and Kocer, D. (2007). Oxidative stress and antioxidant defenses in serum of patients with non-alcoholic steatohepatitis. Clin. Biochem. 40 (11), 776–780. doi:10.1016/j.clinbiochem.2007.02.006
Billesbølle, C. B., Kretsch, R. C., Powers, A. S., Gonen, S., Schneider, S., Arvedson, T., et al. (2020). Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 586 (7831), 807–811. doi:10.1038/s41586-020-2668-z
Bonkovsky, H. L., Tortorelli, K., LeClair, P., Cobb, J., Lambrecht, R. W., Banner, B. F., et al. (1999). Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J. Hepatol. 31 (3), 421–429. doi:10.1016/s0168-8278(99)80032-4
Brunt, E. M., Di Bisceglie, A. M., Neuschwander-Tetri, B. A., and Bacon, B. R. (1999). Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 94 (9), 2467–2474. doi:10.1111/j.1572-0241.1999.01377.x
Camaschella, C., Silvestri, L., and Nai, A. (2020). Iron metabolism and iron disordersrevisited in the hepcidin era. Haematologica 105 (2), 260–272. doi:10.3324/haematol.2019.232124
Chen, J., Li, X., Ge, C., Min, J., and Wang, F. (2022). The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29 (3), 467–480. doi:10.1038/s41418-022-00941-0
Chen, Z., Tian, R., She, Z., Cai, J., and Li, H. (2020). Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 152, 116–141. doi:10.1016/j.freeradbiomed.2020.02.025
Diehl, A. M., and Day, C. (2017). Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 23377 (21), 2063–2072. doi:10.1056/NEJMra1503519
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Donovan, A., Pinkus, J. L., Pinkus, G. S., Zon, L. I., Robine, S., Andrews, N. C., et al. (2005). The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1 (3), 191–200. doi:10.1016/j.cmet.2005.01.003
Drakesmith, H., Ganz, T., and Nemeth, E. (2015). Ironing out ferroportin. Cell Metab. 22 (5), 777–787. doi:10.1016/j.cmet.2015.09.006
Du, K., Chitneni, S. K., Suzuki, A., Wang, Y., Henao, R., Hyun, J., et al. (2020). Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell. Mol. Gastroenterol. Hepatol. 10 (1), 1–21. doi:10.1016/j.jcmgh.2019.12.006
Facchini, F. S., and Stoohs, R. A. (2002). Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology 122 (4), 931–939. doi:10.1053/gast.2002.32403
Forman, H. J., Zhang, H., and Rinna, A. (2009). Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 30 (1-2), 1–12. doi:10.1016/j.mam.2008.08.006
Gaggini, M., Carli, F., Rosso, C., Buzzigoli, E., Marietti, M., Della Latta, V., et al. (2018). Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 67 (1), 145–158. doi:10.1002/hep.29465
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi:10.1016/j.molcel.2015.06.011
Gao, M., Yi, J., Zhu, J., Minikes, A. M., Monian, P., Thompson, C. B., et al. (2019). Role of mitochondria in ferroptosis. Mol. Cell 73 (2), 354–363. doi:10.1016/j.molcel.2018.10.042
George, D. K., Goldwurm, S., Macdonald, G. A., Cowley, L. L., Walker, N. I., Ward, P. J., et al. (1998). Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 114 (2), 311–318. doi:10.1016/s0016-5085(98)70482-2
Guo, J., He, X., Li, S., Wu, Y., Xiang, G., Bao, F., et al. (2021). A combined model of human iPSC-derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome. Adv. Sci. 8 (10), 2004680. doi:10.1002/advs.202004680
Harrison, S. A., Torgerson, S., Hayashi, P., Ward, J., and Schenker, S. (2003). Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 98 (11), 2485–2490. doi:10.1111/j.1572-0241.2003.08699.x
He, R., Du, S., Lei, T., Xie, X., and Wang, Y. (2020). Glycogen synthase kinase 3β in tumorigenesis and oncotherapy (Review). Oncol. Rep. 44 (6), 2373–2385. doi:10.3892/or.2020.7817
Irie, M., Sohda, T., Anan, A., Fukunaga, A., Takata, K., Tanaka, T., et al. (2016). Reduced glutathione suppresses oxidative stress in nonalcoholic fatty liver disease. Euroasian J. Hepatogastroenterol. 6 (1), 13–18. doi:10.5005/jp-journals-10018-1159
Itoh, K., Ishii, Y., Ishii, T., Shibata, T., Kawamoto, Y., Kelly, V., et al. (2004). Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12, 14)-prostaglandin j(2). Mol. Cell. Biol. 24 (1), 36–45. doi:10.1128/mcb.24.1.36-45.2004
Jang, S. C-D. X., Tyurina, Y. Y., St Croix, C. M., Kapralov, A. A., Tyurin, V. A., Bayır, H., et al. (2021). Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 45, 102021. doi:10.1016/j.redox.2021.102021
Josekutty, J., Iqbal, J., Iwawaki, T., Kohno, K., and Hussain, M. M. (2013). Microsomal triglyceride transfer protein inhibition induces endoplasmic reticulum stress and increases gene transcription via Ire1α/cJun to enhance plasma ALT/AST. J. Biol. Chem. 288 (20), 14372–14383. doi:10.1074/jbc.M113.459602
Kalhan, S. C., Guo, L., Edmison, J., Dasarathy, S., McCullough, A. J., Hanson, R. W., et al. (2011). Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 60 (3), 404–413. doi:10.1016/j.metabol.2010.03.006
Kay, H. Y., Hwang, S. J., Choi, H. S., Gilroy, R. K., Wan, Y. J., Kim, S. G., et al. (2011). Nrf2 inhibits LXRα-dependent hepatic lipogenesis by competing with FXR for acetylase binding. Antioxid. Redox Signal. 15 (8), 2135–2146. doi:10.1089/ars.2010.3834
Kitteringham, N. R., Walsh, J., Randle, L., Jenkins, R. E., Sison, R., Goldring, C. E., et al. (2010). Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J. Proteomics 73 (8), 1612–1631. doi:10.1016/j.jprot.2010.03.018
Klein, E. A., Thompson, I. M., Tangen, C. M., Crowley, J. J., Lucia, M. S., Goodman, P. J., et al. (2011). Vitamin E and the risk of prostate cancer: the selenium and vitamin E cancer prevention trial (SELECT). JAMA 306 (14), 1549–1556. doi:10.1001/jama.2011.1437
Kleiner, B. E., Van Natta, M., Behling, C., Contos, M. J., Cummings, O. W., Ferrell, L. D., et al. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41 (6), 1313–1321. doi:10.1002/hep.20701
Kumar, A., Sharma, A., Duseja, A., Das, A., Dhiman, R. K., Chawla, Y. K., et al. (2013). Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have higher oxidative stress in comparison to chronic viral hepatitis. J. Clin. Exp. Hepatol. 3 (1), 12–18. doi:10.1016/j.jceh.2012.10.009
Lei, G., Zhuang, L., and Gan, B. (2022). Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396. doi:10.1038/s41568-022-00459-0
Loomba, R., Quehenberger, O., Armando, A., and Dennis, E. (2015). Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J. Lipid Res. 56 (1), 185–192. doi:10.1194/jlr.P055640
Mao, C., Liu, X., Zhang, Y., Lei, G., Yan, Y., Lee, H., et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593 (7860), 586–590. doi:10.1038/s41586-021-03539-7
Masterton, G. S., Plevris, J. N., and Hayes, P. C. (2010). Review article: omega-3 fatty acids - a promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 31 (7), 679–692. doi:10.1111/j.1365-2036.2010.04230.x
Matteoni, C. A., Gramlich, T., Boparai, N., Liu, Y. C., and McCullough, A. J. (1999). Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 116 (6), 1413–1419. doi:10.1016/s0016-5085(99)70506-8
Mendler, M-H., Turlin, B., Moirand, R., Jouanolle, A-M., Sapey, T., Guyader, D., et al. (1999). Insulin resistance–associated hepatic iron overload. Gastroenterology 117 (5), 1155–1163. doi:10.1016/s0016-5085(99)70401-4
Miller, E. R., Pastor-Barriuso, R., Dalal, D., Riemersma, R. A., Appel, L. J., and Guallar, E. (2005). Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 142 (1), 37–46. doi:10.7326/0003-4819-142-1-200501040-00110
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417 (1), 1–13. doi:10.1042/BJ20081386
Musso, G., Gambino, R., Cassader, M., and Pagano, G. (2010). A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 52 (1), 79–104. doi:10.1002/hep.23623
Nelson, J. E., Brunt, E. M., Yeh, M. M., Kleiner, D. E., Unalp-Arida, A., Kowdley, K. V., et al. (2011). Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 53 (2), 448–457. doi:10.1002/hep.24038
Newsome, P. N., Buchholtz, K., Cusi, K., Linder, M., Okanoue, T., Ratziu, V., et al. (2021). A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N. Engl. J. Med. 384 (12), 1113–1124. doi:10.1056/NEJMoa2028395
Nozue, T., Yamamoto, S., Tohyama, S., Fukui, K., Umezawa, S., Onishi, Y., et al. (2013). Effects of statins on serum n-3 to n-6 polyunsaturated fatty acid ratios in patients with coronary artery disease. J. Cardiovasc. Pharmacol. Ther. 18, 320–326. doi:10.1177/1074248412473202
Paul, B. T., Manz, D. H., Torti, F. M., and Torti, S. V. (2017). Mitochondria and iron: current questions. Expert Rev. Hematol. 10 (1), 65–79. doi:10.1080/17474086.2016.1268047
Ribas, V., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2014). Glutathione and mitochondria. Front. Pharmacol. 5, 151. doi:10.3389/fphar.2014.00151
Sanyal, A. J., Chalasani, N., Kowdley, K. V., McCullough, A., Diehl, A. M., Bass, N. M., et al. (2010). Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 362 (18), 1675–1685. doi:10.1056/NEJMoa0907929
Serviddio, G., Bellanti, F., Tamborra, R., Rollo, T., Capitanio, N., Romano, A. D., et al. (2008). Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 57 (7), 957–965. doi:10.1136/gut.2007.147496
Stockwell, B. R., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., Fulda, S., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171 (2), 273–285. doi:10.1016/j.cell.2017.09.021
Sun, X., Seidman, J. S., Zhao, P., Troutman, T. D., Spann, N. J., Que, X., et al. (2020). Neutralization of oxidized phospholipids ameliorates non-alcoholic steatohepatitis. Cell Metab. 31 (1), 189–206. doi:10.1016/j.cmet.2019.10.014
Thimmulappa, R. K., Srisuma, S., Kensler, T. W., Yamamoto, M., and Biswal, S. (2002). Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 62 (18), 5196–5203.
Tomasiewicz, K., Flisiak, R., Halota, W., Jaroszewicz, J., Lebensztejn, D., Lisik, W., et al. (2018). Recommendations for the management of non-alcoholic fatty liver disease (NAFLD). Clin. Exp. Hepatol. 4 (3), 153–157. doi:10.5114/ceh.2018.78118
Tsurusaki, S. T. Y., Koumura, T., Nakasone, M., Sakamoto, T., Matsuoka, M., Imai, H., et al. (2019). Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 10 (6), 449. doi:10.1038/s41419-019-1678-y
Valenti, L., Bugianesi, E., Dongiovanni, P., Galmozzi, E., Vanni, E., Canavesi, E., et al. (2010). HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 138 (3), 905–912. doi:10.1053/j.gastro.2009.11.013
van de Wier, B., Balk, J. M., Haenen, G. R., Giamouridis, D., Bakker, J. A., Bast, B. C., et al. (2013). Elevated citrate levels in non-alcoholic fatty liver disease: the potential of citrate to promote radical production. FEBS Lett. 587 (15), 2461–2466. doi:10.1016/j.febslet.2013.06.019
Wang, H., An, P., Xie, E., Wu, Q., Fang, X., Gao, H., et al. (2017). Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66 (2), 449–465. doi:10.1002/hep.29117
Wang, R., Wang, S., Wu, C. W., Wong, V. W., Chan, H. L., Farrell, G. C., et al. (2010). Heme oxygenase-1 protects against steatohepatitis in both cultured hepatocytes and mice. Gastroenterology 138 (2), 694–704.
Wang, W., Choi, J. E., Gijón, M., Kennedy, P. D., Johnson, J. K., Liao, P., et al. (2019). CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569 (7755), 270–274. doi:10.1038/s41586-019-1170-y
Wang, Y., Xie, Q., Tan, H., Liao, M., Zhu, S., Zheng, L. L., et al. (2021). Targeting cancer epigenetic pathways with small-molecule compounds: Therapeutic efficacy and combination therapies. Pharmacol. Res. 173, 105702. doi:10.1016/j.phrs.2021.105702
Wang, Y. Q., Chang, S. Y., Wu, Q., Gou, Y. J., Jia, L., Cui, Y. M., et al. (2016). The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308. doi:10.3389/fnagi.2016.00308
Wieckowska, A., Li, Z., Lopez, R., Zein, N. N., and Feldstein, A. E. (2008). Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 103 (6), 1372–1379. doi:10.1111/j.1572-0241.2007.01774.x
Wu, K. C., and Klaassen, C. D. (2011). Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 123 (2), 590–600. doi:10.1093/toxsci/kfr183
Xie, Y., Tian, H., Xiang, B., Li, D., Liu, J., Cai, Z., et al. (2021). Total polyunsaturated fatty acid intake and the risk of non-alcoholic fatty liver disease in Chinese han adults: a secondary analysis based on a case-control study. BMC Gastroenterol. 21 (1), 451. doi:10.1186/s12876-021-02039-2
Xu, D., Jeong, S., Qian, Y., Wu, H., Xia, Q., Kong, X., et al. (2019). The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 9, 1428. doi:10.3389/fphar.2018.01428
Yates, M. S., Dolan, P. M., Osburn, W. O., Shin, S., McCulloch, C. C., Silkworth, J. B., et al. (2009). Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 30 (6), 1024–1031. doi:10.1093/carcin/bgp100
Younossi, Z. M., Gramlich, T., Bacon, B. R., Matteoni, C. A., Boparai, N., O'Neill, R., et al. (1999). Hepatic iron and nonalcoholic fatty liver disease. Hepatology 30 (4), 847–850. doi:10.1002/hep.510300407
Zhang, Z., Guo, X., An, P., Tao, Y., and Wang, F. (2012). Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 56 (3), 961–971. doi:10.1002/hep.25746
Zhao, J., Yu, Y. R., and Qi, Y-F. (2021). STAT3: a key regulator in liver fibrosis. Ann. Hepatol. 21, 100224. doi:10.1016/j.aohep.2020.06.010
Keywords: ferroptosis, dihydroorotate dehydrogenase, mitochondrial glutathione, NASH, mitochondria
Citation: Xiong F, Zhou Q, Huang X, Cao P and Wang Y (2022) Ferroptosis plays a novel role in nonalcoholic steatohepatitis pathogenesis. Front. Pharmacol. 13:1055793. doi: 10.3389/fphar.2022.1055793
Received: 28 September 2022; Accepted: 23 November 2022;
Published: 02 December 2022.
Edited by:
Suleyman Kaplan, Ondokuz Mayıs University, TurkeyReviewed by:
Kiymet Kübra Tüfekci, Kastamonu University, TurkeyTomohide Kurahashi, Osaka Rosai Hospital, Japan
Copyright © 2022 Xiong, Zhou, Huang, Cao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaobo Huang, drhuangxb@163.com; Peng Cao, caopeng1989@hust.edu.cn; Yi Wang, w_yi2022@163.com
†These authors have contributed equally to this work