 Wenpan Dong
Wenpan Dong Chao Xu
Chao Xu Wenqing Li4†
Wenqing Li4† Xiaoman Xie
Xiaoman Xie Yizeng Lu
Yizeng Lu Yanlei Liu
Yanlei Liu Zhili Suo
Zhili Suo- 1State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 2Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Shandong Provincial Center of Forest Tree Germplasm Resources, Jinan, China
- 5Beijing Botanical Garden, Institute of Botany, Chinese Academy of Sciences, Beijing, China
Walnuts (Juglans of the Juglandaceae) are well-known economically important resource plants for the edible nuts, high-quality wood, and medicinal use, with a distribution from tropical to temperate zones and from Asia to Europe and Americas. There are about 21 species in Juglans. Classification of Juglans at section level is problematic, because the phylogenetic position of Juglans cinerea is disputable. Lacking morphological and DNA markers severely inhibited the development of related researches. In this study, the complete chloroplast genomes and two nuclear DNA regions (the internal transcribed spacer and ubiquitin ligase gene) of 10 representative taxa of Juglans were used for comparative genomic analyses in order to deepen the understanding on the application value of genetic information for inferring the phylogenetic relationship of the genus. The Juglans chloroplast genomes possessed the typical quadripartite structure of angiosperms, consisting of a pair of inverted repeat regions separated by a large single-copy region and a small single-copy region. All the 10 chloroplast genomes possessed 112 unique genes arranged in the same order, including 78 protein-coding, 30 tRNA, and 4 rRNA genes. A combined sequence data set from two nuclear DNA regions revealed that Juglans plants could be classified into three branches: (1) section Juglans, (2) section Cardiocaryon including J. cinerea which is closer to J. mandshurica, and (3) section Rhysocaryon. However, three branches with a different phylogenetic topology were recognized in Juglans using the complete chloroplast genome sequences: (1) section Juglans, (2) section Cardiocaryon, and (3) section Rhysocaryon plus J. cinerea. The molecular taxonomy of Juglans is almost compatible to the morphological taxonomy except J. cinerea (section Trachycaryon). Based on the complete chloroplast genome sequence data, the divergence time between section Juglans and section Cardiocaryon was 44.77 Mya, while section Rhysocaryon diverged from other sections in the genus Juglans was 47.61 Mya. Eleven of the 12 small inversions in the chloroplast genomes provided valuable phylogenetic information for classification of walnut plants at section and species levels. Our results are valuable for future studies on Juglans genetic diversity and will enhance the understanding on the phylogenetic evolution of Juglandaceae.
Introduction
Walnuts (Juglans L.) are well-known economically important resource trees for the edible nuts, high-quality wood, and medicinal use. Juglans, one of the eight living genera in the family Juglandaceae, has about 21 species in the world, with a distribution from tropical to temperate zones, and from Asia to Europe and Americas (Lu, 1982; Lu et al., 1999; APG III, 2009; Pei and Lu, 2011; Suo et al., 2012a; APG IV, 2016).
Juglans plants were classified into four sections according to morphology of leaf, floral, and fruit characteristics, i.e., section Rhysocaryon, section Cardiocaryon, section Trachycaryon, and section Juglans (syn. section Dioscaryon) (Dode, 1909; Manning, 1978; Lu et al., 1999). Manning (1978) and McGranahan and Leslie (1991) presented complete descriptions of the morphological variation, ecological distribution, and taxonomic treatment of the genus Juglans. Manchester (1987) considered that plants of the genus Juglans could be classified into three sections, and merged section Trachycaryon (Juglans cinerea) and section Cardiocaryon together according to the consistency of the walnut fossil characteristics.
Juglans section Juglans includes the two cultivated walnuts, J. regia with a distribution from southeastern Europe to China and the Himalayas, and J. sigillata distributed in Yunnan, Guizhou, Sichuan and Tibet of Southwest China. The nut of section Juglans is distinguished from that of related species by a dehiscent husk thin shell at fruit mature stage and narrow septum separating the kernel halves, all of which greatly facilitate kernel extraction (Dode, 1909; Manning, 1978; Lu et al., 1999; Martinez-Garcia et al., 2016). More than 300 walnut cultivars are documented under J. regia for producing edible nuts. J. regia has been cultivated for more than 6800 years (Beer et al., 2008; Joly and Visset, 2009; Pei and Lu, 2011). Section Trachycaryon, native to eastern North America, comprises a single species, J. cinerea L.. Section Rhysocaryon, endemic to North and South Americas (Stone et al., 2009; Suo et al., 2012a), consists of 16 taxa: J. australis, J. boliviana, J. californica, J. guatemalensis, J. jamaicensis, J. hindsii, J. hirsute, J. major, J. microcarpa, J. mollis, J. neotropica, J. nigra, J. olanchana, J. pyriformis, J. steyermarkii, and J. venezuelensis. Section Cardiocaryon contains three taxa native to East Asia: J. manshurica, J. cathayensis, and J. ailantifolia.
Molecular biological studies supported the sectional classification of Juglans based on morphological characteristics, except for section Trachycaryon represented by the single species J. cinerea. J. cinerea was placed within section Cardiocaryon when analysis was conducted using nuclear DNA sequence (the internal transcribed spacer, ITS), but within section Rhysocaryon when analysis was conducted using cpDNA sequences (NCS and matK) (Stanford et al., 2000; Aradhya et al., 2007). The phylogenetic position of J. cinerea is thus still problematic. Haplotype phylogeography suggested a geographical differentiation prior to the last glacial advance in eastern populations and separate postglacial migration paths for eastern and western populations when the detection was conducted using sequences from eight chloroplast DNA regions (Laricchia et al., 2015).
As a matter of fact, the plasticity of morphological traits is frequently observed due to influences from environmental conditions and different developmental stages. The internal transcribed spacer (ITS) sequence of nuclear ribosomal DNA and cpDNA fragments (rbcL, matK, and trnH–psbA) commonly recommended to use have only limited resolution in identifying closely related taxa in the Juglandaceae (Xiang et al., 2011; Dong et al., 2014, 2015; Suo et al., 2015). Thus, the phylogenetic relationship at section level in Juglans is still a challenging task, because of lacking morphological and DNA markers (Gunter et al., 1994; Cosmulescu and Botu, 2012; Ciarmiello et al., 2013; Suo et al., 2015). It is necessary to explore more genetic information for phylogenetic reconstruction of Juglans.
In recent years, the chloroplast genomes have been proven successfully to be more informative than cpDNA fragments in revealing phylogeny of land plants (Jansen et al., 2007; He et al., 2012; Suo et al., 2012b; Dong et al., 2016; Xu et al., 2017). Hu et al. (2017) used the complete chloroplast genome information to discuss genetic divergence of five Chinese Juglans taxa in comparison with the Fagaceae and the Betulaceae, the families closely related to the Juglandaceae. New nuclear DNA markers from the ubiquitin–proteasome system related DNA regions showed higher sensitivity and better resolution in detecting genetic diversity in genera Juglans and Lagerstroemia (Suo et al., 2015, 2016). The ubiquitin–proteasome system, which plays a key role in degradation of proteins, is imperative for maintaining the cellular homeostasis in eukaryotic cells (Ganoth et al., 2013; Marin, 2013). Furthermore, it has been reported that micro-structure mutations, such as small inversions, in chloroplast genomes may have a potential application value in the phylogenetic analysis of land plants (Kelchner and Wendel, 1996; Kim and Lee, 2005; Borsch and Quandt, 2009; Morrison, 2009).
The small size inversions (∼50 bp) are probably to be generated by intra-molecular recombination events (Ogihara et al., 1988; Hiratsuka et al., 1989). The possession of the same inversion is regarded as reliable evidence of shared ancestry (Jansen and Palmer, 1987; Doyle et al., 1992, 1996; Kim and Lee, 2004). The inverted repeats formed the stem structures and the small inversions formed the loops.
In this study, we report nine newly sequenced complete chloroplast genomes from Juglans (eight species and one cultivar). In addition, sequences from two nuclear DNA regions (ITS, and ubiquitin ligase gene), were also used to help resolving the genetic diversity in Juglans. The aims of our study are: (1) to upgrade the understanding on the application value in phylogenetic resolution of Juglans, (2) to provide more genetic resources for obtaining a better resolution on the phylogeny of the genus Juglans, and (3) to deepen the understanding on the genetic and evolutionary significance from the structural diversity of the chloroplast genomes.
Materials and Methods
Plant Materials and DNA Extraction
Fresh leaves were collected from the trees of J. nigra, J. major, and J. regia ‘Bokexiang’ growing in the Resources Nursery of the Forestry Bureau of Luoning County, Henan Province, China; J. sigillata, J. cathayensis, and J. hindsii from the Arboretum of the Forestry Academy of Yunnan Province, Kunming City, Yunnan Province, China; J. mandshurica growing in the Beijing Botanical Garden of the Chinese Academy of Sciences. J. regia from the plant of a natural population located in Taihang mountainous region of Yixian County, Hebei Province, China, and dried leaves of J. cinerea were taken from voucher specimen, 01816245, Chinese National Herbarium, collected May 5, 2006, at Sevier County, Tennessee, United States, No. 2274609 from PE Herbarium (Table 1). The (fresh) leaves from each accession were immediately dried with silica gel for further DNA extraction. Total genomic DNAs were extracted from each sample using the Plant Genomic DNA Kit (DP305) from Tiangen Biotech (Beijing) Co., Ltd., China.
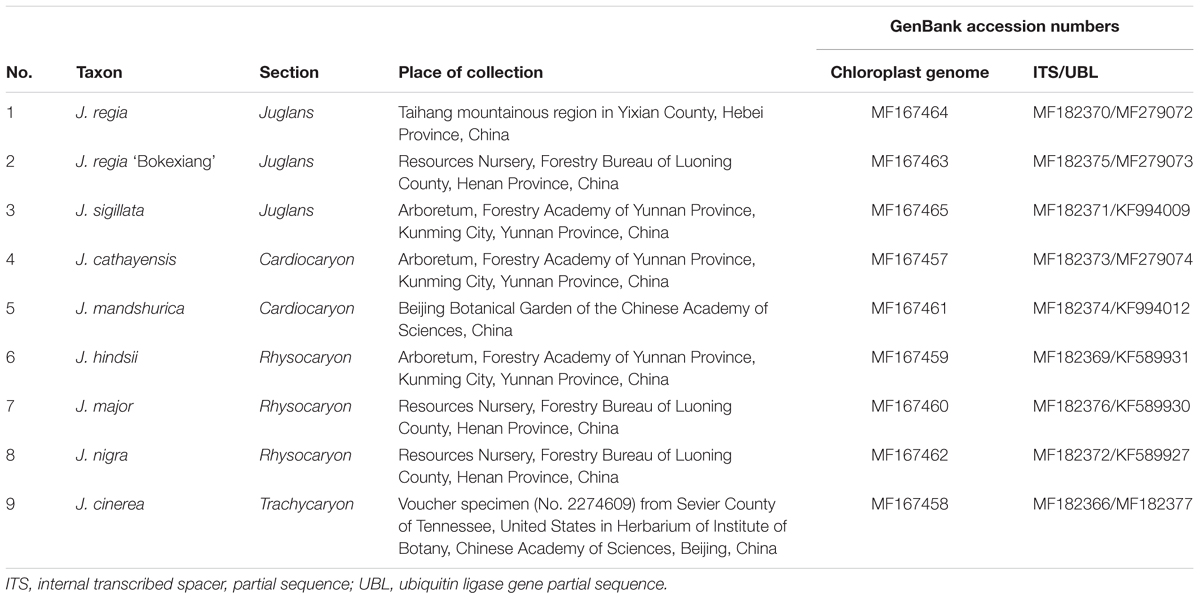
TABLE 1. Taxa of Juglans used in this study.
Chloroplast Genome Sequencing and Assembling
Four Juglans chloroplast genomes of J. regia, J. regia ‘Bokexiang’, J. sigillata and J. mandshurica were sequenced using the short-range PCR (Polymerase Chain Reaction) method reported by Dong et al. (2012, 2013). The PCR protocol was as follows: preheating at 94°C for 4.5 min, 34 cycles at 94°C for 50 s, annealing at 55°C for 40 s, and elongation at 72°C for 1.5 min, followed by a final extension at 72°C for 8 min. PCR amplification was performed in an Applied Biosystems VeritiTM 96-Well Thermal Cycler (Model#: 9902, made in Singapore). The amplified DNA fragments were sent to Shanghai Majorbio Bio-Pharm Technology Co., Ltd. (Beijing) for Sanger sequencing in both the forward and reverse directions using a 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, United States). The chloroplast DNA sequences were manually confirmed and assembled using Sequencher (v5.4) software.
Juglans cathayensis, J. cinerea, J. hindsii, J. major, and J. nigra (Supplementary Table S1) were sequenced using Illumina HiSeq 4000. Before sequencing, paired-end libraries with 300-bp insert size were constructed following the manufacturer’s protocol (Illumina Inc.). 303,763–1,744,889 mapped reads were obtained from 8,801,265–29,818,482 raw reads (Supplementary Table S1). The length of sequencing reads was 150 bp. The four junctions between the inverted repeat region (IRs) and the small single copy (SSC)/large single copy (LSC) region were checked by amplification using specific primers, followed by Sanger sequencing (Dong et al., 2013).
The high-throughput sequencing data were qualitatively assessed and assembled using SPAdes 3.6.1 (Bankevich et al., 2012). Using J. regia (KT963008) as a reference sequence, we selected chloroplast genome contigs using Blast method. The contigs of the chloroplast genome were assembled using Sequencher (v5.4) with default parameters and the gaps between contigs were filled in by amplification with PCR-based conventional Sanger sequencing using ABI 3730. The specific primers were designed based on the flanking sequences to bridge the gaps. After that, all reads were mapped to the spliced chloroplast genome sequence using Geneious 8.1 (Kearse et al., 2012) to avoid assembly errors.
Genome Annotation
Chloroplast genome annotation was performed using the Dual Organellar Genome Annotator (DOGMA) (Wyman et al., 2004). BLASTX and BLASTN searches were employed to accurately annotate the protein-encoding genes and to identify the locations of the ribosomal RNA (rRNA) and transfer RNA (tRNA) genes. Gene annotation information from other closely related plant species was also utilized for sure when the boundaries of the exons or introns could not be precisely determined because of the limited power of BLAST in chloroplast genome annotation. The chloroplast genome map was drawn using Genome Vx software (Conant and Wolfe, 2008). The nine chloroplast genomes newly sequenced in this study were deposited in GenBank (accession numbers MF167457-MF167465).
PCR Amplification of the Two Nuclear DNA Regions
The ITS sequences were amplified using the primer pair, ITS-u1 and ITS-u4, and following the PCR amplification conditions as reported by Cheng et al. (2016). The DNA sequence from the ubiquitin ligase gene region (UBE3) was amplified using the primer pair, H_UBE3_23f and H_UBE3_838r, and following the PCR amplification conditions as reported by Suo et al. (2015). The eleven ITS sequences and one ubiquitin ligase gene sequence were deposited in GenBank (accession numbers MF182366-MF182377). The ubiquitin ligase gene sequences of other samples used for comparative analysis in this study were downloaded from GenBank (accession numbers: KF994007-KF994018) (Suo et al., 2015). The DNA sequences of outgroups were also deposited in GenBank (Pterocarya stenoptera, MF182367 for ITS, KF994018 for UBE3; Cyclocarya paliurus, MF182368 for ITS, KF994017 for UBE3).
Sequence Divergence Analysis
The chloroplast genome sequences were aligned using MAFFT (Katoh and Standley, 2013) and were manually adjusted using Se-Al 2.0 (Rambaut, 1996). Variable and parsimony-informative base sites across the complete chloroplast genomes, the large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions of the chloroplast genomes were calculated using MEGA 6.0 software (Tamura et al., 2013). Sliding window analysis was conducted to generate nucleotide diversity (Pi) of the chloroplast genome using DnaSP (DNA Sequences Polymorphism version 5.10.01) software (Librado and Rozas, 2009). The step size was set to 200 bp, with a 600-bp window length. Repeating sequences were scanned over the complete chloroplast DNA sequences, species by species, using the REPuter program. Probable inversion regions associated with the repeated sequences were evaluated by detailed alignment and sequence similarity searches (Kurtz et al., 2001; Kim and Lee, 2005).
Phylogenetic Analysis
Maximum parsimony (MP) analyses were performed using PAUP v4b10 (Swofford, 2003). All characters were equally weighted, gaps were treated as missing, and character states were treated as unordered. Heuristic search was performed with MULPARS option, tree bisection-reconnection (TBR) branch swapping, and random stepwise addition with 1,000 replications. The Maximum likelihood (ML) analyses were conducted using RAxML 8.0 (Stamatakis, 2006). For ML analyses, the best-fit model, general time reversible (GTR)+G was used in all analysis as suggested with 1,000 bootstrap replicates.
Bayesian inference (BI) was conducted with Mrbayes v3.2 (Ronquist et al., 2012). The Markov chain Monte Carlo (MCMC) analysis was run for 2 × 5,000,000 generations. Trees were sampled at every 1,000 generations with the first 25% discarded as burn-in. The remaining trees were used to build a 50% majority-rule consensus tree. The stationarity was regarded to be reached when the average standard deviation of split frequencies remained below 0.01.
Estimation of Divergence Times
The BEAST v2.3.3 package (Bouckaert et al., 2014) was used to analyze the chloroplast genome dataset for assessment of Juglans divergence times using a relaxed molecular clock method (Drummond et al., 2006). We selected chloroplast genome dataset for divergence time analysis. For calibration, two constraints were used: (1) The age for the most recent common ancestor of the Juglandaceae was set to 79.9 Mya (71.2–96.4) and assigned a normal distribution (Xiang et al., 2014); (2) the Juglans crown group was set to age of 45 Mya (Manchester, 1987; Aradhya et al., 2007). We used an uncorrelated log-normal clock, a Yule tree prior, and a randomly generated starting tree. The data was assigned a GTR + I + G model of substitution. Runs were conducted for 500 million generations with parameters sampled every 5,000 steps. Tracer v.1.6 (Rambaut et al., 2014) was used to check convergence and stationarity, to determine the number of generations discarded as burn-in, and to confirm that effective sample size (ESS) values were over 200.
Results
Chloroplast Genome Features
The Juglans complete chloroplast genomes ranged from 159,714 (J. hopeiensis, GenBank accession no. KX671977) to 160,537 base pairs (bp) (J. regia voucher JREG20151001, GenBank accession no. KT870116) in length. All the chloroplast genomes possessed the typical quadripartite structure of angiosperms, consisting of a pair of the inverted repeat region (IRs: 26,023–26,039 bp) separated by a large single-copy region (LSC: 89,307–89,917 bp) and a small single-copy region (SSC: 18,352–18,429 bp) (Figure 1 and Table 2). All the 10 chloroplast genomes possessed 112 unique genes arranged in the same order, including 78 protein-coding, 30 tRNA, and 4 rRNA genes. GC content in each chloroplast genome is identically 36.1% (Supplementary Table S2).
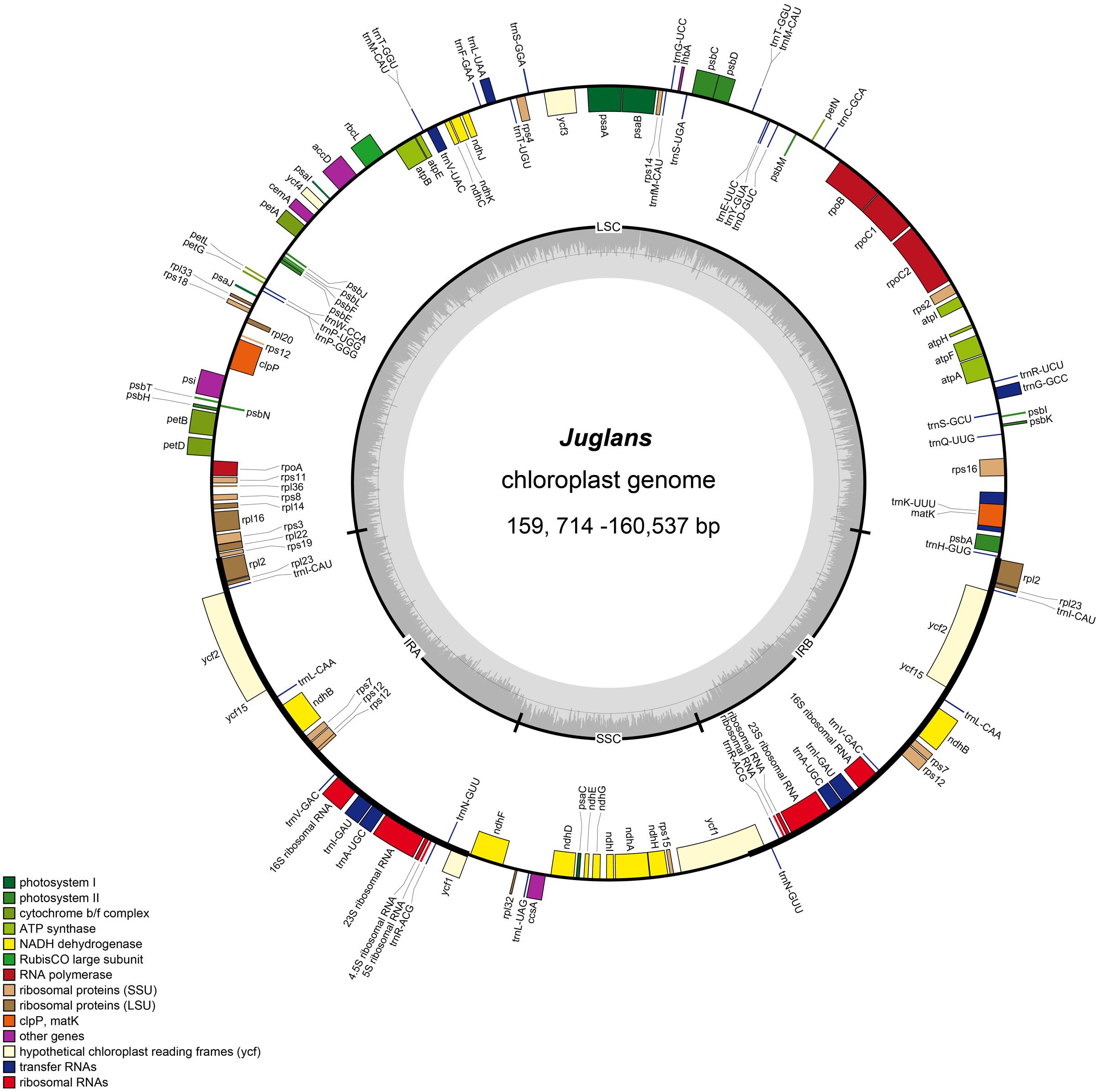
FIGURE 1. Gene map of Juglans chloroplast genome. The genes inside and outside of the circle are transcribed in the clockwise and counterclockwise directions, respectively. Genes belonging to different functional groups are shown in different colors. The thick lines indicate the extent of the inverted repeats (IRa and IRb) that separate the genomes into small single-copy (SSC) and large single-copy (LSC) regions.
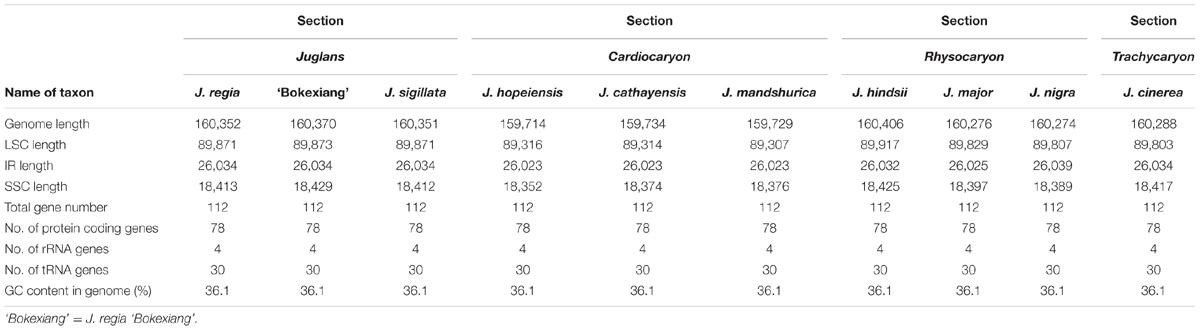
TABLE 2. Summary of complete chloroplast genome features of the 10 Juglans taxa.
Genome Sequence Divergence
The distribution of each single nucleotide polymorphic site (SNP) among the 10 Juglans chloroplast genomes is shown in Supplementary Table S3. There were 721 SNPs in LSC region, 268 in SSC region, and 30 in each of the IR regions.
At section level, the largest sequence divergence occurred between Juglans sections Cardiocaryon and Rhysocaryon, with the largest nucleotide substitution number (512 to 575) and the largest sequence distance (0.0032 to 0.0036). The smallest sequence divergence at section level was observed between sections Juglans and Cardiocaryon, with the lowest nucleotide substitution number (465 to 471) and the lowest sequence distance (0.0029 to 0.0030). The sequence divergence between sections Juglans and Rhysocaryon was observed to be intermediate, with the nucleotide substitution number ranged from 492 to 545, and the sequence distance ranged from 0.0031 to 0.0034 (Table 3).
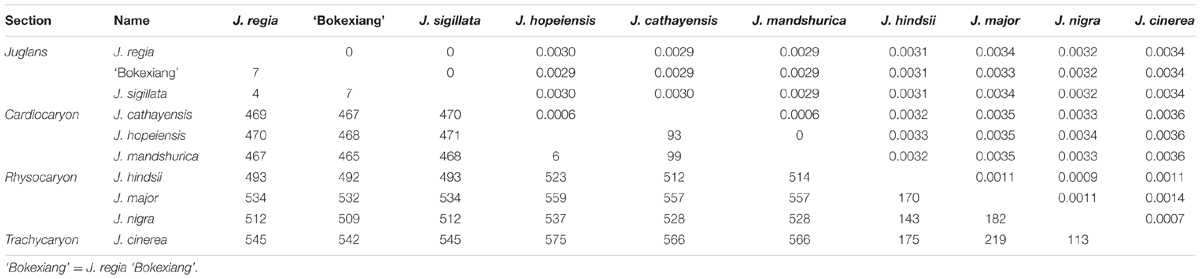
TABLE 3. Numbers of the pairwise nucleotide substitutions (the lower triangle) and sequence distance (the upper triangle) between the 10 complete cp genomes representing four sections within genus Juglans.
The lowest within-section-divergence was observed in section Juglans, with the lowest nucleotide substitutions of 4 to 7, and the lowest sequence distance of zero. The largest within-section-divergence was found in section Rhysocaryon, with the largest nucleotide substitutions of 143 to 182, and the largest sequence distance of 0.0009 to 0.0011. The intermediate within-section-divergence was observed in section Cardiocaryon (Table 3).
At taxon level, the largest sequence divergence was observed between J. cinerea and J. hopeiensis, with the highest nucleotide substitution number (575) and the highest sequence distance (0.0036). The lowest divergence was between J. cinerea and J. nigra of section Rhysocaryon, with the lowest nucleotide substitution number (113) and the lowest sequence distance (0.0007) (Table 3). J. hopeiensis is classified in section Cardiocaryon and is closer to J. mandshurica.
Six hyper-variable regions (Pi > 0.01) were uncovered among the sampled Juglans taxa. They are three intergenic spacers (rpoB-trnC, trnT-psbD, and psbE-petL) from the LSC region, and two gene regions (ycf1b and ycf1a) and one intron of ndhA from SSC region (Figure 2).
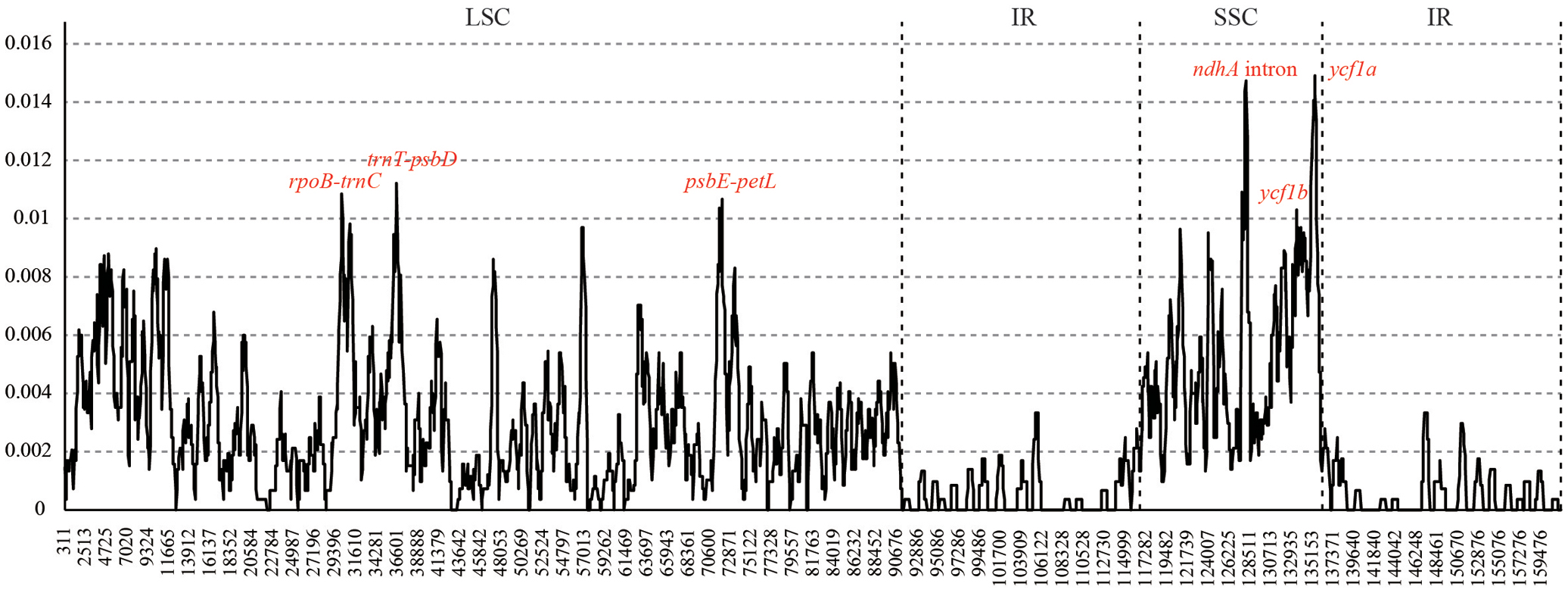
FIGURE 2. Sliding window analysis of the whole chloroplast genomes of 10 Juglans species. (window length: 600 bp, step size: 50 bp). X-axis: position of the midpoint of a window, Y-axis: nucleotide diversity of each window.
Small Inversions
It has been reported that each small inversion is commonly associated with a hairpin secondary structure in the chloroplast genomes (Kim and Lee, 2005). In this study, a total of 12 small inversions were uncovered based on the sequence alignment of the 10 complete chloroplast genomes representing the four sections of the genus Juglans, of which nine small inversions were located in LSC region, two in IR region, and one in SSC region. Eleven of the 12 small inversions were seen in intergenic spacers, and one of them was in ycf1 gene region of the chloroplast genomes (Table 4).
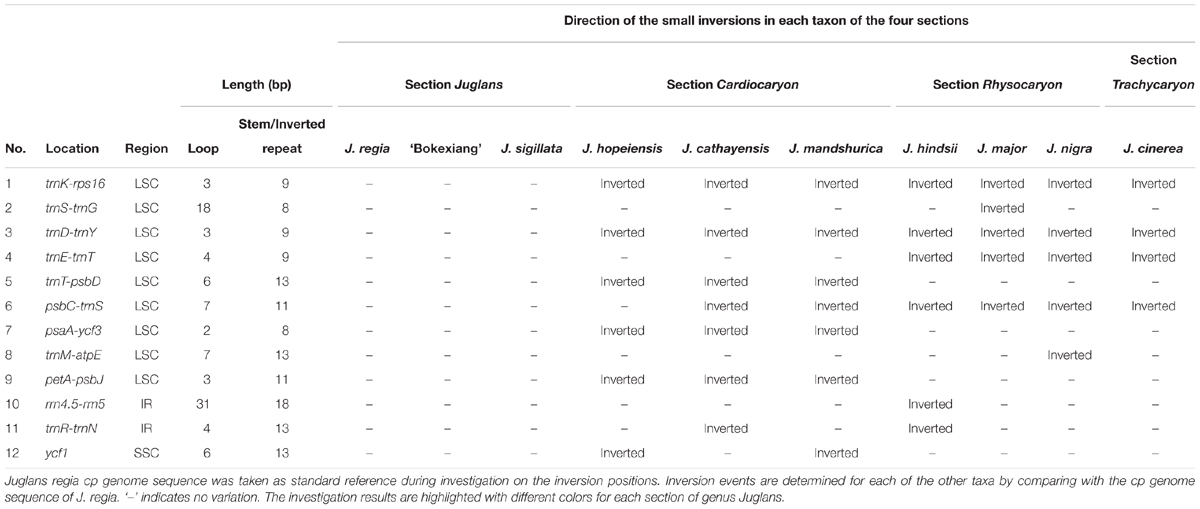
TABLE 4. The location and length of 12 small inversions.
Each of the small inversions from trnK-rps16 or trnD-trnY only occurred in section Juglans. The two small inversions from trnT-trnD, psbA-ycf3 and petA-psbJ only occurred in section Cardiocaryon. The small inversion from trnE-trnT occurred within section Rhysocaryon and section Trachycaryon (including a single species J. cinerea). Each small inversion from trnS-trnG, trnM-atpE, or rrn4.5-rrn5 occurred only in certain taxon within sect. Rhysocaryon. The small inversion from trnE-trnT was only observed in both section Juglans and section Cardiocaryon.
The small inversion from psbC-trnS occurred in section Juglans, including J. hopeiensis which is regarded as a natural hybrid between J. mandshurica and J. regia (molecular evidences from our experiments will be published in another article in detail). The small inversion in ycf1 occurred in either J. mandshurica or J. hopeiensis. The 4 bp small inversion of trnR-trnN occurred simultaneously in part of taxa in section Cardiocaryon and section Rhysocaryon, showing no phylogenetic implication (Table 4).
Phylogenetic Analysis
Phylogenetic analysis was conducted using each of the four sequence data sets: the complete chloroplast genome, LSC, SSC, or IR regions (Figure 3). The chloroplast genome sequences of Corylus chinensis (GenBank accession No. KX814336, Betulaceae), Ostrya rehderiana (GenBank accession No. KT454094, Betulaceae), Carpinus putoensis (GenBank accession No. KX695124, Betulaceae), Cyclocarya paliurus (GenBank accession No. KY246947, Juglandaceae), and Annamocarya sinensis (GenBank accession No. KX703001, Juglandaceae) were used as outgroups (Figure 3).
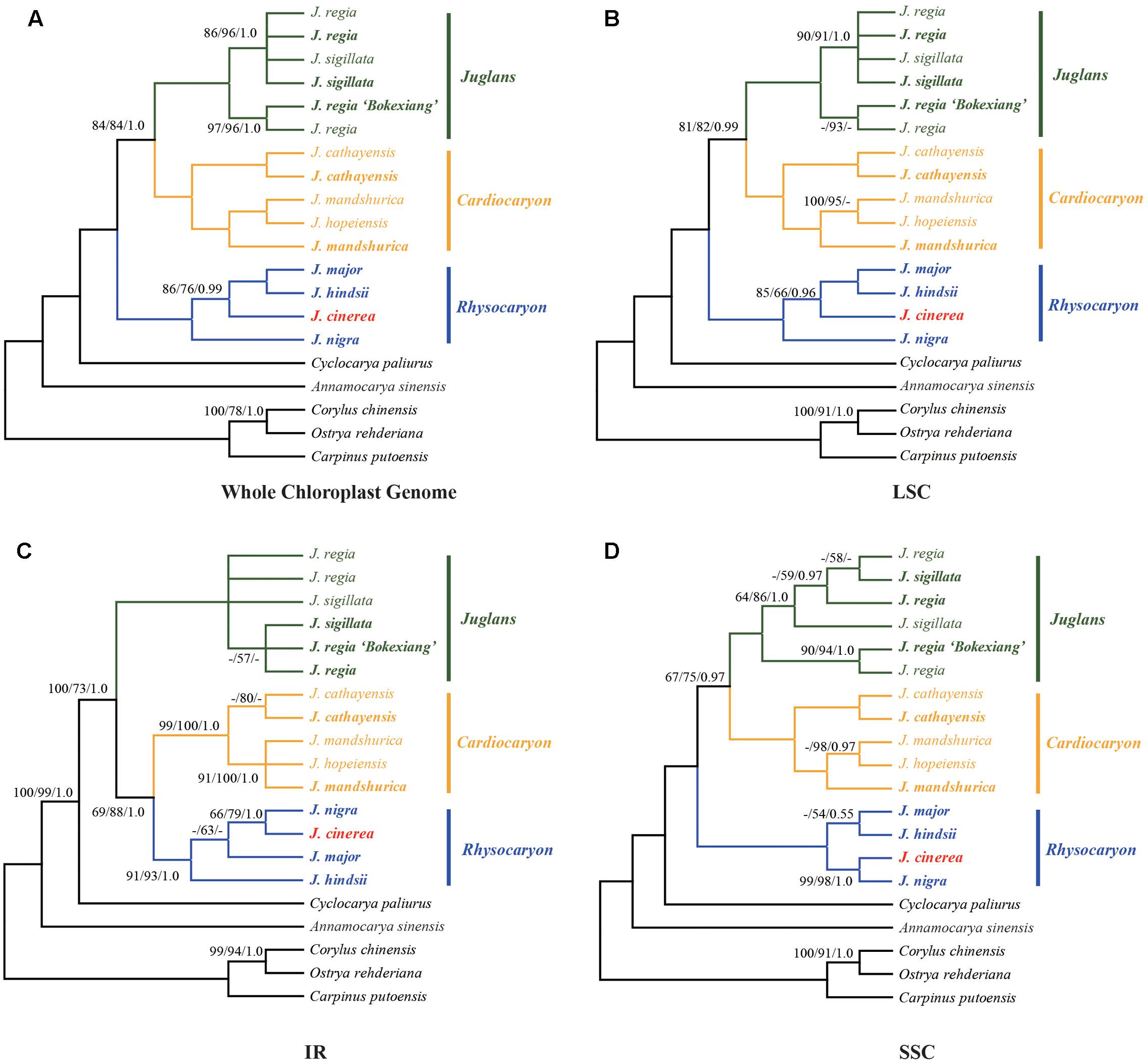
FIGURE 3. Phylogenetic relationships of Juglans inferred from maximum parsimony (MP), Bayesian inference (BI), and maximum likelihood (ML) analyses of different chloroplast genome data partitions. (A) Whole chloroplast genome. (B) LSC region. (C) IR region. (D) SSC region. Numbers near nodes indicate the MP bootstrap values (left) for each clade present in the 50% majority-rule consensus, ML bootstrap values (middle), and Bayesian posterior probability (right). Both MP and ML bootstrap support values = 100 and Bayesian posterior probability = 1.0 are not given at the nodes.
A combined sequence data set of a 724 bp length ITS sequence alignment with a 753 bp length ubiquitin ligase gene sequence alignment was used for the phylogenetic analyses. A total of 1,477 bp length nuclear DNA sequence alignment was used (Figure 4).
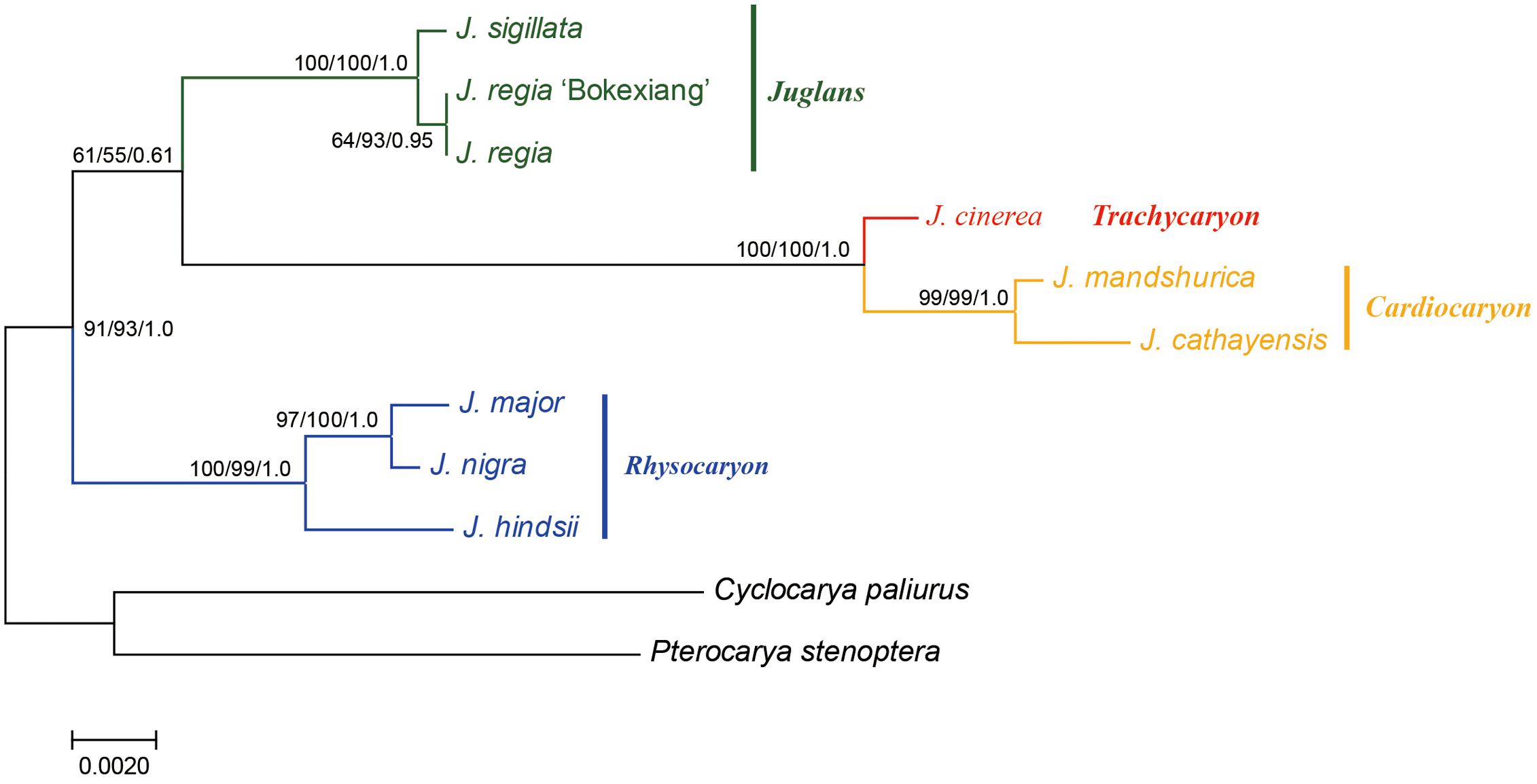
FIGURE 4. Phylogeny of Juglans based on ITS and ubiquitin ligase gene. Numbers near nodes indicate the MP bootstrap values (left) for each clade present in the 50% majority-rule consensus, ML bootstrap values (middle), and Bayesian posterior probability (right).
The sequence data set from either the whole chloroplast genome or SSC region provided the best and almost identical resolution in the phylogenetic analyses with high bootstrap support value in comparison with the sequence data set from each of the rest two chloroplast regions (LSC and IR regions). Generally, the walnut taxa could be separated into three branches by the chloroplast DNA sequence data sets: (1) section Juglans, (2) section Cardiocaryon, and (3) section Rhysocaryon including J. cinerea which is closer to J. nigra (Figure 3). However, the combined sequence data set from the two nuclear DNA regions revealed a different phylogenetic topology of three branches in Juglans: (1) section Juglans, (2) section Cardiocaryon plus section Trachycaryon (J. cinerea) which is closer to J. mandshurica, and (3) section Rhysocaryon (Figure 4). J. hopeiensis which was classified in section Cardiocaryon showed a closer relationship with J. mandshurica. Based on the complete chloroplast genome sequence data, the divergence time between section Juglans and section Cardiocaryon was 44.77 Mya. The divergence time of section Rhysocaryon from other sections in the genus Juglans was 47.61 Mya (Figure 5).
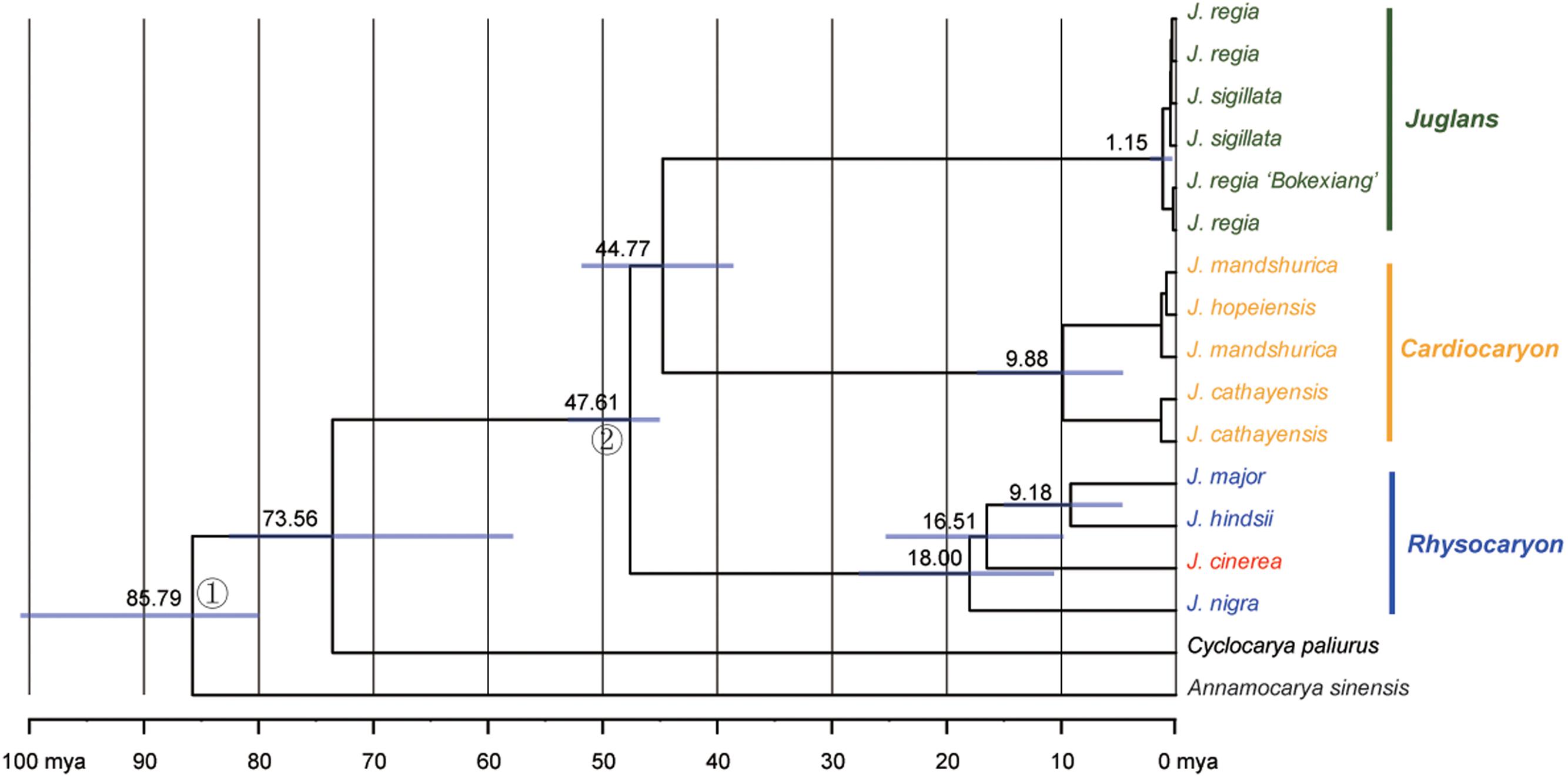
FIGURE 5. Chronogram obtained for Juglans under a Bayesian relaxed-clock approach based on the chloroplast genome dataset. Gray boxes indicate 95% confidence intervals on nodal ages.
Discussion
Structure, Size and Phylogenetic Information of Small Inversions
Large inversions are well characterized in the chloroplast genomes of various plant families/genera and the sequence data have been used to determine angiosperm lineages from the genus to phylum level (Jansen and Palmer, 1987; Milligan et al., 1989; Raubeson and Jansen, 1992; Hupfer et al., 2000; Kim and Lee, 2005). In contrast, limited plant groups were studied involving small inversions. For examples, a four base inversion associated with a hairpin secondary structure occurs within the rpl16 intron of the chloroplast genomes of some members of the genus Chusquea and related bamboo species (Poaceae) (Kelchner and Wendel, 1996). Sixteen small inversions ranging from 5 to 50 bp occurred in chloroplast genomes of phylogenetically distantly related groups of land plants including Poaceae, Fabaceae, and Solanaceae (Kim and Lee, 2005).
Within a single genus, Kim and Lee (2005) selected nine species of Jasminum (Oleaceae) to document the occurrence of the small inversions in closely related species. A 11 bp small inversion associated with a 19 bp inverted repeat was uncovered within the trnL-F non-coding regions in the chloroplast genome of Jasminum (Kim and Lee, 2005). In this study, 12 small inversions (loops) ranging from 2 to 31 bp in length were detected in chloroplast genomes of Juglans (Juglandaceae), they were associated with the inverted repeating sequences ranging from 8 to 18 bp in length. This indicated that there are large differences in the occurrence and distribution of small inversion between plant families. Our study further verified that small inversions are valuable genetic source for phylogenetic researches within a single genus, because more than 83.3% of them were found to be phylogenetically informative in revealing the genetic variations of Juglans plants at section and taxa levels.
In this study, a 4 bp small inversion in trnR-trnN intergenic spacer has no phylogenetic significance, indicating that careful analysis is necessary before use of small inversions.
Phylogeny of Juglans
In this study, we explored and analyzed new genetic information by sequencing the complete chloroplast genomes and two nuclear DNA regions (ITS, and ubiquitin ligase gene) of nine Juglans taxa representing the four sections of the genus Juglans previously published based on morphological characteristics. The phylogenetic analyses were conducted using three algorithms, MP, ML, and BI methods. No significant difference was found among the algorithms.
According to analysis based on DNA sequences from two nuclear gene regions, the monotypic section Trachycaryon (J. cinerea) is a sister to the section Cardiocaryon. These two sections (Trachycaryon and Cardiocaryon) together further formed one branch which is a sister group to section Juglans or section Rhysocaryon. Sectional level divergence occurred in Juglans around 44.77 to 47.61 Mya in Eocene. These are similar to the previous reports (Aradhya et al., 2007).
The phylogenetic topology obtained using molecular data is generally identical with the tree topology obtained using morphological data, except for the placement of J. cinerea (Manning, 1978). In Juglans, the phylogenetic position of J. cinerea has been controversial (Aradhya et al., 2007; Laricchia et al., 2015). In this study, phylogenetic analysis based on the chloroplast genome sequences showed that J. cinerea was positioned within section Rhysocaryon. The closer relationship between J. cinerea and J. nigra is identical with their current geographical occurrence. The chloroplast genome data do not support the isolated position of the monotypic section Trachycaryon (J. cinerea) based on morphological characters.
The distributional ranges of the Tertiary fossils of butternuts (J. cinerea) and black walnuts (J. nigra) do not overlap except in the northwestern parts of the United States around 40° N latitude, strongly suggesting that they may have evolved independently as suggested by Hills et al. (1974).
Sequences from eight different regions of the chloroplast genome in 197 trees in J. cinerea sampled from their distribution area revealed 10 haplotypes (Laricchia et al., 2015). The phylogenetic incongruence for J. cinerea based on nuclear DNA sequences and/or chloroplast genome sequences might be potentially caused by hybridization. Juglans section Trachycaryon based on morphological characteristics was supported by neither nuclear nor chloroplast DNA sequences. This is still a mystery at current stage and will be a key point for us to challenge in future phylogenetic studies of the genus Juglans (Aradhya et al., 2007). Further study by sampling more individual trees/populations of J. cinerea, members from section Rhysocaryon and section Cardiocaryon and utilizing potential information from the whole nuclear genome sequence of J. regia (Martinez-Garcia et al., 2016) will be meaningful.
Juglans hopeiensis was shown maternally belonging to the same chloroplast lineage with J. mandshurica in section Cardiocaryon in this study. This result is identical with the previous studies which suggested that J. hopeiensis is an inter-specific hybrid between J. mandshurica and J. regia (Rehder, 1940; Lu et al., 1999; Wu et al., 2000; Aradhya et al., 2007). Further study is necessary for a more clear elucidation involving the origin of J. hopeiensis.
Conclusion
This study reports the comparative genomic analysis results of nine Juglans chloroplast genome sequences with detailed gene annotation. More than 83.3% of the small inversions in the chloroplast genomes provided valuable genetic information for phylogenetic researches at taxon and section levels in Juglans. All of the Juglans taxa were discriminated completely with high bootstrap support values. The molecular taxonomy of Juglans is almost compatible to the currently accepted morphological taxonomy except J. cinerea (section Trachycaryon). The existence of the monotypic section Trachycaryon (J. cinerea) based on morphological characteristics was supported by neither nuclear nor chloroplast DNA sequences. The systematic position of J. cinerea shifted from a member of J. section Cardiocaryon based on the combined nuclear DNA sequence data set to a member of the section Rhysocaryon based on the chloroplast genome sequence data set. Further studies centering J. cinerea by sampling more samples will be helpful for clarifying the phylogenetic placement of J. cinerea. Sectional level divergence time of Juglans was 44.77 to 47.61 Mya in Eocene. These results obtained in this study are valuable for future researches on global Juglans genetic diversity and will enhance our understanding of the phylogenetic evolution of the Juglandaceae.
Author Contributions
WD conceived and designed the experiments, performed the experiments, conducted the chloroplast genome assembling, analyzed the data, wrote the paper, prepared figures, and/or tables, reviewed drafts of the paper. CX performed the experiments, analyzed the data, wrote the paper, reviewed drafts of the paper. WL, YiL, and XX conceived and designed the experiments, contributed reagents/materials/analysis tools, wrote the paper, reviewed drafts of the paper. YaL performed the experiments, prepared figures and/or tables. XJ wrote the paper, reviewed drafts of the paper. ZS conceived and designed the experiments, performed the experiments, analyzed the data, contributed reagents/materials/analysis tools, wrote the paper, reviewed drafts of the paper.
Funding
The study was financially supported by “Collection, Conservation, and Evaluation of Forest Tree Germplasm Resources” (LKZ201496-1-3) of Shandong Provincial Agricultural Elite Varieties Project, the joint projects No. 70009C1036 and 70009C1020, the National Natural Science Foundation of China (No. 30972412), and the National Forest Genetic Resources Platform (2005DKA21003).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors thank Runquan Dong, Huzhi Xu, Chenqian Wang, Fengmei Li, Peng Wang, Wenyu Ma, Zhihong Ding, and Weiwei Gao for help in field investigation and helpful discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01148/full#supplementary-material
References
APG III (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161, 105–121. doi: 10.1016/j.jep.2015.05.035
APG IV (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1016/j.jep.2015.05.035
Aradhya, M. K., Potter, D., Gao, F., and Simon, C. J. (2007). Molecular phylogeny of Juglans (Juglandaceae): a biogeographic perspective. Tree Genet. Genomes 3, 363–378. doi: 10.1007/s11295-006-0078-5
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beer, R., Kaiser, F., Schmidt, K., Ammann, B., Carraro, G., Grisa, E., et al. (2008). Vegetation history of the walnut forests in Kyrgyzstan (Central Asia): natural or anthropogenic origin? Quat. Sci. Rev. 27, 621–632. doi: 10.1016/j.quascirev.2007.11.012
Borsch, T., and Quandt, D. (2009). Mutational dynamics and phylogenetic utility of non-coding chloroplast DNA. Plant Syst. Evol. 282, 169–199. doi: 10.1007/s00606-009-0210-8
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Cheng, T., Xu, C., Lei, L., Li, C., Zhang, Y., and Zhou, S. (2016). Barcoding the kingdom Plantae: new PCR primers for ITS regions of plants with improved universality and specificity. Mol. Ecol. Resour. 16, 138–149. doi: 10.1111/1755-0998.12438
Ciarmiello, L. F., Pontecorvo, G., Piccirillo, P., Luca, A. D., Carillo, P., Kafantaris, I., et al. (2013). Use of nuclear and mitochondrial single nucleotide polymorphisms to characterize English walnut (Juglans regia L.) genotypes. Plant Mol. Biol. Rep. 31, 1116–1130. doi: 10.1007/s11105-013-0575-2
Conant, G. C., and Wolfe, K. H. (2008). GenomeVx: simple web-based creation of editable circular chromosome maps. Bioinformatics 24, 861–862. doi: 10.1093/bioinformatics/btm598
Cosmulescu, S., and Botu, M. (2012). Walnut biodiversity in south-western Romania-resource for perspective cultivars. Pak. J. Bot. 44, 307–311.
Dode, L. A. (1909). Contribution to the study of the genus Juglans (English translation by Cuendett, R.E). Bull. Soc. Dendrol. Fr. 11, 22–90.
Dong, W. P., Liu, H., Xu, C., Zuo, Y. J., Chen, Z. J., and Zhou, S. L. (2014). A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: a case study on ginsengs. BMC Genet. 15:138. doi: 10.1186/s12863-014-0138-z
Dong, W. P., Liu, J., Yu, J., Wang, L., and Zhou, S. L. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7:e35071. doi: 10.1371/journal.pone.0035071
Dong, W. P., Xu, C., Cheng, T., Lin, K., and Zhou, S. L. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Dong, W. P., Xu, C., Li, C. H., Sun, J. H., Zuo, Y. J., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Dong, W. P., Xu, C., Li, D. L., Jin, X. B., Lu, Q., and Suo, Z. L. (2016). Comparative analysis of the complete chloroplast genome sequences in psammophytic Haloxylon species (Amaranthaceae). PeerJ 4:e2699. doi: 10.7717/peerj.2699
Doyle, J. J., Davis, J. I., Soreng, R. J., Garvin, D., and Anderson, M. J. (1992). Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc. Natl. Acad. Sci. U.S.A. 89, 7722–7726. doi: 10.1073/pnas.89.16.7722
Doyle, J. J., Doyle, J. L., Ballenger, J. A., and Palmer, J. D. (1996). The distribution and phylogenetic significance of a 50-kb chloroplast DNA inversion in the flowering plant family Leguminosae. Mol. Phylogenet. Evol. 5, 429–438. doi: 10.1006/mpev.1996.0038
Drummond, A. J., Ho, S. Y., Phillips, M. J., and Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. doi: 10.1371/journal.pbio.0040088
Ganoth, A., Tsfadia, Y., and Wiener, R. (2013). Ubiquitin: molecular modeling and simulations. J. Mol. Graph. Model. 46, 29–40. doi: 10.1016/j.jmgm.2013.09.006
Gunter, L. E., Kochert, G., and Giannasi, D. E. (1994). Phylogenetic relationships of the Juglandaceae. Plant Syst. Evol. 192, 11–29. doi: 10.1007/BF00985904
He, L. X., Suo, Z. L., Zhang, C. H., Jin, X. B., Zhao, D. X., Zhao, X. Q., et al. (2012). Classification of Chinese medicinal tree peony cultivars based on chloroplast DNA sequences. AASRI Procedia 1, 344–352. doi: 10.1016/j.aasri.2012.06.053
Hills, L. V., Klovan, J. E., and Sweet, A. R. (1974). Juglans eocinerea n. sp., Beaufort Formation (Tertiary), southwestern Banks Inland, Arctic Canada. Can. J. Bot. 52, 65–90. doi: 10.1139/b74-011
Hiratsuka, J., Shimada, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M., et al. (1989). The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194. doi: 10.1007/BF02464880
Hu, Y., Woeste, K. E., and Zhao, P. (2017). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Hupfer, H., Swiatek, M., Hornung, S., Herrmann, R. G., Maier, R. M., Chiu, W. L., et al. (2000). Complete nucleotide sequence of the Oenothera elata plastid chromosome, representing plastome I of the five distinguishable Euoenothera plastomes. Mol. Gen. Genet. 263, 581–585. doi: 10.1007/pl00008686
Jansen, K. R., and Palmer, J. D. (1987). A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proc. Natl. Acad. Sci. U.S.A. 84, 5818–5822. doi: 10.1073/pnas.84.16.5818
Jansen, R. K., Cai, Z. Q., Raubeson, L. A., Daniell, H., dePamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Joly, C., and Visset, L. (2009). Evolution of vegetation landscapes since the Late Mesolithic on the French West Atlantic coast. Rev. Palaeobot. Palynol. 154, 124–179. doi: 10.1016/j.revpalbo.2008.12.011
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelchner, S. A., and Wendel, J. F. (1996). Hairpins create minute inversions in non-coding regions of chloroplast DNA. Curr. Genet. 30, 259–262. doi: 10.1007/s002940050130
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, K. J., and Lee, H. L. (2005). Wide spread occurrence of small inversions in the chloroplast genomes of land plants. Mol. Cells 19, 104–113.
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Laricchia, K. M., McCleary, T. S., Hoban, S. M., Borkowski, D., and Romero-Severson, J. (2015). Chloroplast haplotypes suggest preglacial differentiation and separate postglacial migration paths for the threatened North American forest tree Juglans cinerea L. Tree Genet. Genomes 11, 30. doi: 10.1007/s11295-015-0852-3
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lu, A. M., Stone, D. E., and Grauke, L. J. (1999). “Juglandaceae,” in Flora of China, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press), 277–285.
Manchester, S. R. (1987). The fossil history of the Juglandaceae. Monogr. Syst. Bot. Mo. Bot. Gard. 21, 1–137.
Manning, W. E. (1978). The classification within the Juglandaceae. Ann. Mo. Bot. Gard. 65, 1058–1087. doi: 10.2307/2398782
Marin, I. (2013). Evolution of plant HECT ubiquitin ligases. PLoS ONE 8:e68536. doi: 10.1371/journal.pone.0068536
Martinez-Garcia, P. J., Crepeau, M. W., Puiu, D., Gonzalez-Ibeas, D., Whalen, J., Stevens, K. A., et al. (2016). The walunut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 87, 507–532. doi: 10.1111/tpj.13207
McGranahan, G., and Leslie, C. (1991). “Walnuts (Juglans),” in Genetic Resources of Temperate Fruit and Nut Crops, eds J. N. Moore and J. R. Ballington Jr. (Wageningen: International Society for Horticultural Science), 907–951.
Milligan, B. G., Hampton, J. N., and Palmer, J. D. (1989). Dispersed repeats and structural reorganization in subclover chloroplast DNA. Mol. Biol. Evol. 6, 355–368.
Morrison, D. A. (2009). A framework for phylogenetic sequence alignment. Plant Syst. Evol. 282, 127–149. doi: 10.1007/s00606-008-0072-5
Ogihara, Y., Terachi, T., and Sasakuma, T. (1988). Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. U.S.A. 85, 8573–8577. doi: 10.1073/pnas.85.22.8573
Pei, D., and Lu, X. Z. (2011). Walnut Germplasm Resources in China (In Chinese). Beijing: China Forestry Publishing House, 1–208.
Rambaut, A., Suchard, M., Xie, D., and Drummond, A. (2014). Tracer v1. 6. Available at: http://beast.bio.ed.ac.uk/Tracer
Raubeson, L. A., and Jansen, R. K. (1992). Chloroplast DNA evidence on the ancient evolutionary split in vascular land plants. Science 255, 1697–1699. doi: 10.1126/science.255.5052.1697
Rehder, A. (1940). Manual of Cultivated Trees and Shrubs in North America. MacMillan, NY: The Macmillan Company.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. PMID: 16928733 doi: 10.1093/bioinformatics/btl446
Stanford, A. M., Harden, R., and Parks, C. R. (2000). Phylogeny and biogeography of Juglans (Juglandaceae) based on matK and ITS sequence data. Am. J. Bot. 87, 872–882. doi: 10.2307/2656895
Stone, D. E., Oh, S. H., Tripp, E. A., Rios, G. L. E., and Manos, P. S. (2009). Natural history distribution, phylogenetic relationships, and conservation of Central American black walnuts (Juglans section Rhysocaryon). J. Torrey Bot. Soc. 136, 1–25. doi: 10.3159/08-RA-036R.1
Suo, Z. L., Chen, L. N., Pei, D., Jin, X. B., and Zhang, H. J. (2015). A new nuclear DNA marker from ubiquitin ligase gene region for genetic diversity detection of walnut germplasm resources. Biotechnol. Rep. 5, 40–45. doi: 10.1016/j.btre.2014.11.003
Suo, Z. L., Li, W. Y., Jin, X. B., and Zhang, H. J. (2016). A new nuclear DNA marker revealing both microsatellite variations and single nucleotide polymorphic loci: a case study on classification of cultivars in Lagerstroemia indica L. J. Microb. Biochem. Technol. 8, 266–271. doi: 10.4172/1948-5948.1000296
Suo, Z. L., Pei, D., Ma, Q. G., and Jin, X. B. (2012a). Genetic formation of paradox hybrids (Juglans L.) revealed by nrDNA IGS8-ETS1 region. AASRI Procedia 1, 156–165. doi: 10.1016/j.aasri.2012.06.025
Suo, Z. L., Zhang, C. H., Zheng, Y. Q., He, L. X., Jin, X. B., Hou, B. X., et al. (2012b). Revealing genetic diversity of tree peonies at micro-evolution level with hyper-variable chloroplast markers and floral traits. Plant Cell Rep. 31, 2199–2213. doi: 10.1007/s00299-012-1330-0
Swofford, D. L. (2003). PAUP∗. Phylogenetic Analysis Using Parsimony (∗and Other Methods). Version4b10. Sunderland, MA: Sinauer.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Wu, Y. M., Pei, D., Xi, S. K., and Li, J. R. (2000). A study on the genetic relationship among species in Juglans L. using RAPD markers. Acta Hortic. Sin. 27, 17–22.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xiang, X.-G., Wang, W., Li, R.-Q., Lin, L., Liu, Y., Zhou, Z.-K., et al. (2014). Large-scale phylogenetic analyses reveal fagalean diversification promoted by the interplay of diaspores and environments in the Paleogene. Perspect. Plant Ecol. Evol. Syst. 16, 101–110. doi: 10.1016/j.ppees.2014.03.001
Xiang, X. G., Zhang, J. B., Lu, A. M., and Li, R. Q. (2011). Molecular identification on species in Juglandaceae: a tiered method. J. Syst. Evol. 49, 252–260. doi: 10.1111/j.1759-6831.2011.00116.x
Keywords: Juglans, complete chloroplast genome, small inversion, internal transcribed spacer, ubiquitin ligase gene, phylogeny
Citation: Dong W, Xu C, Li W, Xie X, Lu Y, Liu Y, Jin X and Suo Z (2017) Phylogenetic Resolution in Juglans Based on Complete Chloroplast Genomes and Nuclear DNA Sequences. Front. Plant Sci. 8:1148. doi: 10.3389/fpls.2017.01148
Received: 19 March 2017; Accepted: 15 June 2017;
Published: 30 June 2017.
Edited by:
Jose I. Hormaza, IHSM La Mayora (CSIC), SpainReviewed by:
Federico J. Albertazzi, University of Costa Rica, Costa RicaAureliano Bombarely, Virginia Tech, United States
Copyright © 2017 Dong, Xu, Li, Xie, Lu, Liu, Jin and Suo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhili Suo, zlsuo@ibcas.ac.cn
†These authors have contributed equally to this work.