 Yufen Xu1,2†
Yufen Xu1,2† Borong Liao
Borong Liao Baosheng Wang
Baosheng Wang Hanhan Xia
Hanhan Xia- 1College of Horticulture and Landscape Architecture, Zhongkai University of Agriculture and Engineering, Guangzhou, China
- 2Coconut Research Institute of Chinese Academy of Tropical Agricultural Sciences, Wenchang, China
- 3Department of Evolution, Ecology and Organismal Biology, University of California, Riverside, CA, United States
- 4Key Laboratory of Plant Resources Conservation and Sustainable Utilization, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
Chrysanthemum (Chrysanthemum morifolium Ramat) is an important floricultural crop and medicinal herb. Modern chrysanthemum cultivars have complex genetic backgrounds because of multiple cycles of hybridization, polyploidization, and prolonged cultivation. Understanding the genetic background and hybrid origin of modern chrysanthemum cultivars can provide pivotal information for chrysanthemum genetic improvement and breeding. By now, the origin of cultivated chrysanthemums remains unclear. In this study, 36 common chrysanthemum cultivars from across the world and multiple wild relatives were studied to identify the maternal donor of modern chrysanthemum. Chloroplast (cp) genomes of chrysanthemum cultivars were assembled and compared with those of the wild relatives. The structure of cp genomes was highly conserved among cultivars and wild relatives. Phylogenetic analyses based on the assembled cp genomes showed that all chrysanthemum cultivars grouped together and shared 64 substitutions that were distinct from those of their wild relatives. These results indicated that a diverged lineage of the genus Chrysanthemum, which was most likely an extinct or un-sampled species/population, provided a maternal source for modern cultivars. These findings provide important insights into the origin of chrysanthemum cultivars, and a source of valuable genetic markers for chrysanthemum breeding programs.
Introduction
Chrysanthemum (Chrysanthemum morifolium Ramat) is one of the most popular and economically important floricultural crops in the world, noted for its ornamental, nutritional, and medicinal values (Shih et al., 2011; Shinoyama et al., 2012). It was first cultivated in China approximately 1,600 years ago, then successively introduced to Japan, Europe, and North America (Chen, 1985, 2012; Shih et al., 2011). Modern chrysanthemum cultivars are mainly allohexaploids (2n = 6x = 54; Kumari et al., 2019). They have diverse floral morphologies, lignified stems, and can thrive in a wide range of habitats (e.g., urban, rural and farmland). The complex genetic and phenotypic variation of C. morifolium is thought to be the result of multiple cycles of hybridization, polyploidization, and artificial selection (Shih et al., 2011; Chen, 2012). Interspecific hybridization among modern chrysanthemums and their wild relatives, are still widely used for cultivar improvement (Kumari et al., 2019). The selection of parent species is a critical step for hybrid breeding. Currently, phenotypic traits are frequently used to guide the choice of germplasm to develop new chrysanthemum cultivars with novel appearances and improved stress tolerance (Kumari et al., 2019). However, hybrid progeny can exhibit extreme or “transgressive” traits relative to their progenitors (Rieseberg et al., 1999; Schwarzbach et al., 2001). Thus, hybridization between parent species selected according to phenotype may not produce offspring with the expected characters, e.g., a combination of the parents’ phenotypic characters. This may be an especially common problem for chrysanthemum, because C. morifolium cultivars and wild relatives have complex genetic backgrounds because of high ploidy levels and a long history of hybridization and artificial selection. Understanding the genetic background and hybrid origin of chrysanthemum cultivars can provide pivotal information for choosing germplasm resources, reduce the time-consuming process of artificial crossing, and ultimately facilitate genetic improvement and breeding programs (Lim et al., 2008; Kuligowska et al., 2016).
The origin of cultivated chrysanthemums has attracted great attention over the past decades. Previous studies based on morphological and genetic data show that cultivated chrysanthemums are derived from interspecific hybridization and polyploidization, involving Chrysanthemum indicum L., Chrysanthemum zawadskii Herbich, Chrysanthemum argyrophyllum Y. Ling, Chrysanthemum dichrum (C. Shih) H. Ohashi & Yonek, Chrysanthemum nankingense (Hand.-Mazz.) X.D. Cui, and C. vestitum (Hemsl.) Stapf (Chen, 1985, 2012; Dai et al., 1998, 2005; Fukai et al., 2003; Ma et al., 2020). Multiple lines of evidence point to C. indicum (2n = 4x = 36) and C. vestitum (2n = 6x = 54) as pivotal players in the origin of modern chrysanthemums. First, hybrids between C. indicum and C. vestitum show some phenotypic characters similar to modern chrysanthemums (Chen, 1985, 2012). Second, C. indicum is widely used for multiple purposes in Central China, where cultivated chrysanthemums most likely originated (Chen, 1985, 2012). Recently, cpDNA data has been used to investigate the origin of modern chrysanthemums (Ma et al., 2020; Qi et al., 2021). For example, Ma et al. (2020) investigated the phylogenetic relationships between chrysanthemum cultivars and wild relatives using cp genomes. They found that chrysanthemum cultivars formed a strongly supported clade on cpDNA tree and diverged from all wild species, indicating that the maternally inherited cp genome of modern chrysanthemums might be derived from an extinct progenitor (Ma et al., 2020). By sequencing two cpDNA fragments in multiple populations of wild Chrysanthemum species, Qi et al. (2021) found high cpDNA variation within species and suggested that the maternal progenitor of modern chrysanthemums could be an un-sampled population of wild species. Because these studies sampled only a small number of cultivated chrysanthemums (Ma et al., 2020; Qi et al., 2021), they may not capture all cpDNA variations in cultivars. Therefore, the maternal donor of modern chrysanthemums remains poorly understood.
In this study, we performed comprehensive sampling in cultivars of C. morifolium. A total of 36 accessions were collected to represent common cultivars of different countries and to represent different flower morphologies. The whole cp genomes of chrysanthemum cultivars were assembled compared to those of their wild relatives to determine the origin of cultivated chrysanthemums. Phylogenetic analyses based on the assembled cp genomes elucidated the maternal origin of chrysanthemum cultivars. This study sheds light on the origin of modern chrysanthemums, and provides valuable genetic resources for the development of new cultivars.
Materials and Methods
Sampling, DNA Extraction, and Sequencing
Thirty-six common cultivars of C. morifolium with different origins and flower morphologies were collected, which included 11 Chinese traditional cultivars, nine Japanese cultivars, 10 Dutch cultivars, and six cultivars (referred as HD cultivars hereafter) recently developed by Houde Agricultural Technology (Guangzhou, China). Seven wild relatives of C. morifolium, and an outgroup species Ajania pacifica (Nakai) K.Bremer & Humphries (Anthemidinae, Asteraceae) were also sampled (Table 1; Figure 1). Seedlings from Houde Agricultural Technology were obtained for all cultivars and one wild species (Chrysanthemum nankingenese) for genome sequencing (Table 1). For other wild and outgroup species, the cp genome sequences from GenBank were downloaded with the following accession numbers: JN867589, MW539687, MH339742, MH165287, MN883841, NC037388, NC057203 (Table 1). Details of samples were provided in Table 1.
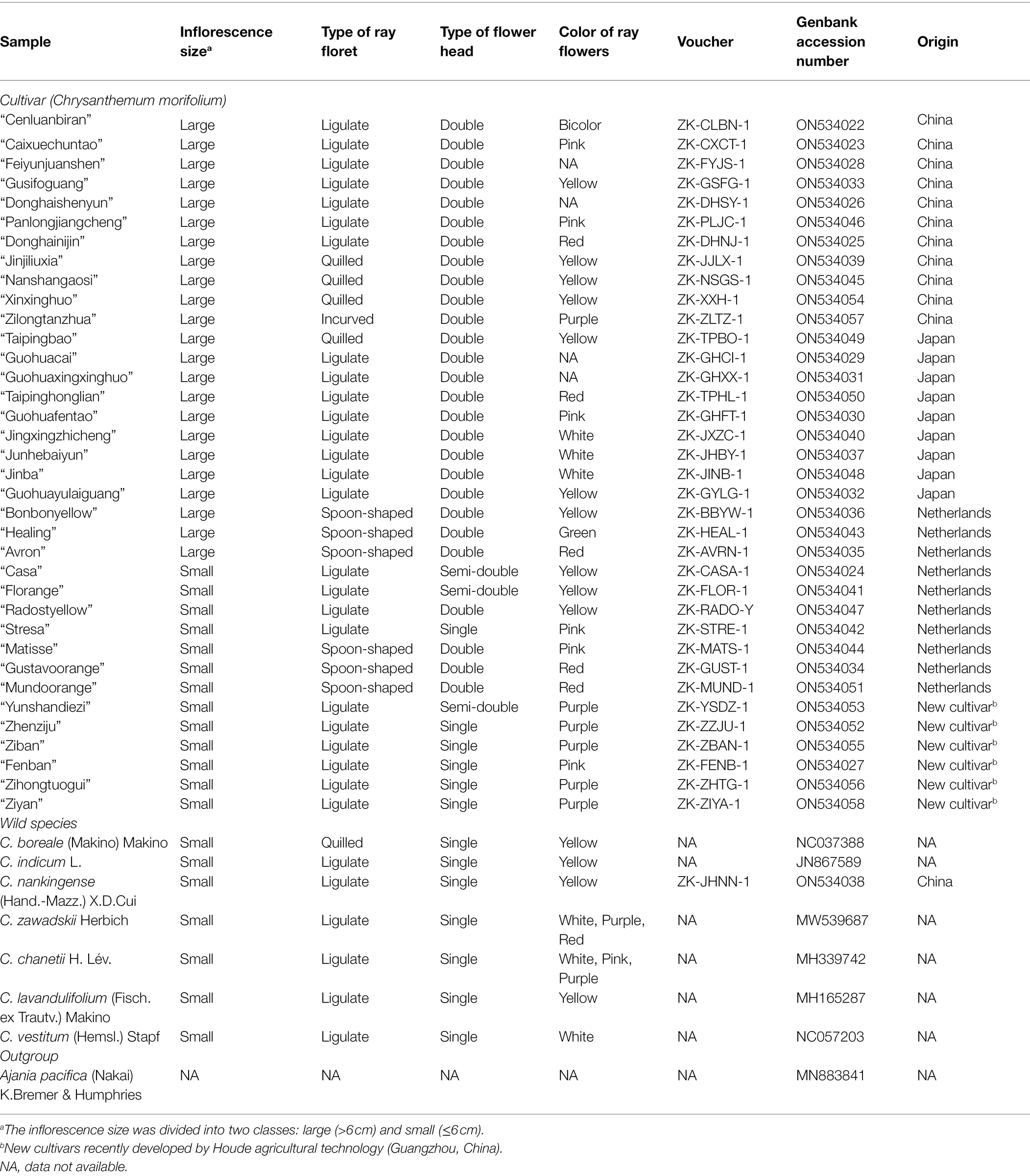
Table 1. Origin and flower characteristics of the 36 chrysanthemum cultivars and seven related wild species.
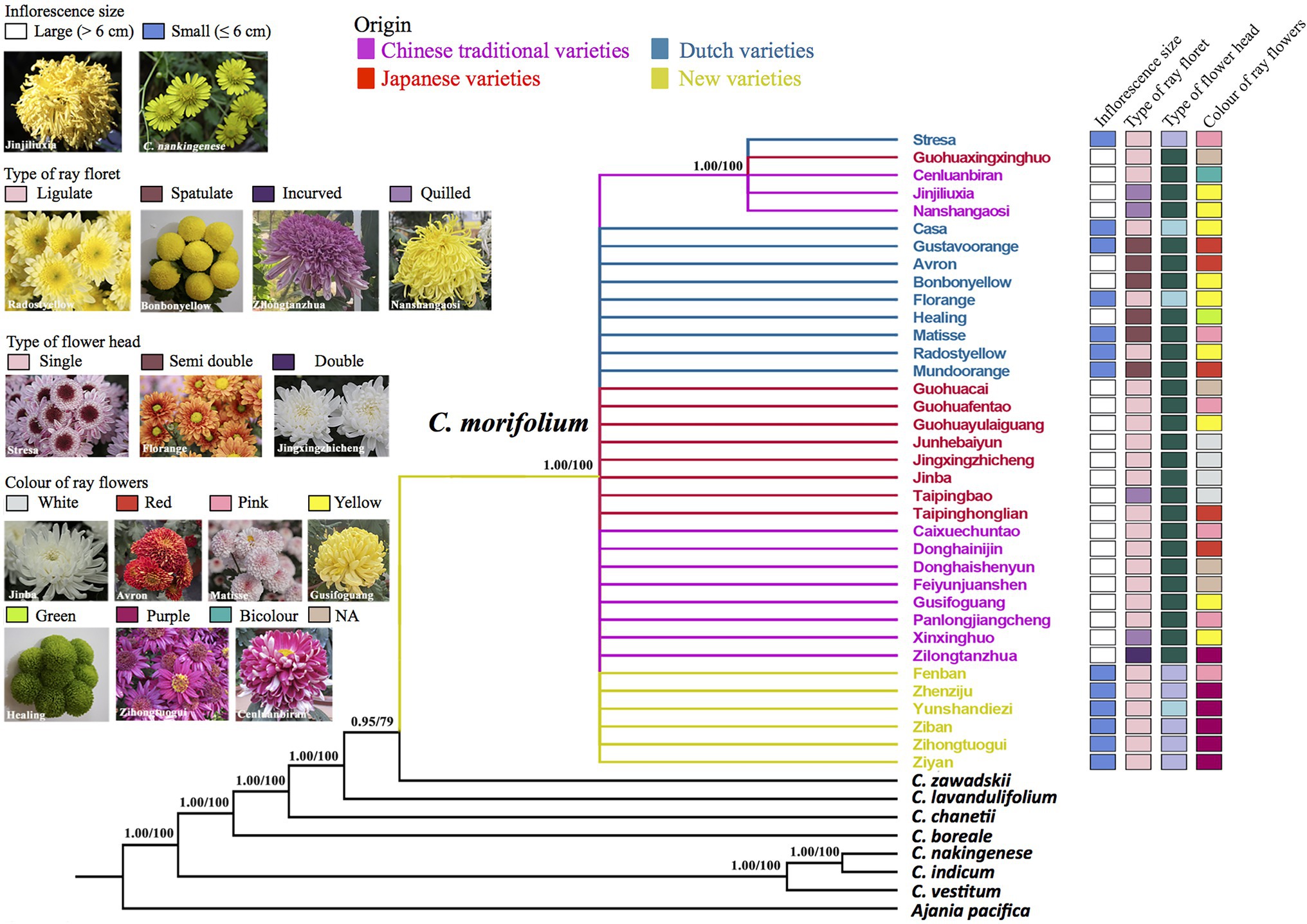
Figure 1. Phylogenetic relationships of chrysanthemums inferred from whole chloroplast genomes. Numbers above the branches are Bayesian posterior probability (PP) and likelihood bootsrap (BS) values. Branches with PP < 0.5 or BS < 50 are collapsed. The cultivar clades and names are indicated in different colors depending on their origin. Cultivar floral traits are showed on the right of the tree.
Total genomic DNA from 100 mg of fresh leaves was isolated using a DNeasy Plant MiniKit (Qiagen). DNA quality was examined by electrophoresis in 1% agarose, and DNA concentration was quantified with a BioPhotometer (Eppendorf, United States). High-quality DNA samples were sent to Novogene (Beijing, China) for library construction and sequencing. Genome sequencing was conducted on the Illumina HiSeq2500 platform to obtained 150 bp pair-end reads.
Chloroplast Genome Assembly and Annotation
De novo assembly of cp genomes for the 37 samples (36 cultivars and one wild species) was conducted using NOVOPlasty (Dierckxsens et al., 2017). For each sample, the cp genome was assembled by using 8–10 Gb of raw reads and a ribulose-bisphosphate carboxylase (RBCL) gene sequence from C. indicum (JN867589) as a seed sequence.
The cp genomes were annotated using GeSeq (Tillich et al., 2017) and Sequin was used for proofreading.1 The size, length of structural division, and gene content of the cp genome were measured according to the annotation results. The GC content of cp genomes was calculated using MEGA X (Kumar et al., 2018). The chloroplast genome map was obtained using OGDRAW (Greiner et al., 2019).
Comparative Analysis of the cp Genomes
The cp genome of C. indicum (JN867589) was used as a reference to align and compare the Chrysanthemum cp genomes using mVISTA (Frazer et al., 2004), and genome collinear analysis was performed with the Mauve tool (Darling et al., 2004). The expansion and contraction of IR boundaries were analyzed using IRscope (Amiryousefi et al., 2018).
The repeat regions on cp genome were annotated using REPuter (Kurtz et al., 2001). Forward, reverse, complementary, and palindromic repeat sequences with a hamming distance of 3 and minimum repeat size of 30 bp were searched. MISA (Beier et al., 2017) was also used to identify simple sequence repeats. The minimum number of repeats was set to 10, 6, 5, 5, 5, and 5 for mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide, respectively.
Phylogenetic Analyses
Phylogenetic analyses were performed using the Maximum Likelihood (ML) and Bayesian inference (BI) methods. For both ML and BI analyses, Jmodeltest v2.1.10 (Posada, 2008) was used to look for the best substitution model. The model GTR + GAMMA was chosen as the best model for ML and BI analyses. ML analysis was conducted in RAxML v8.2.12 (Stamatakis, 2014). The best ML tree was selected from 1,000 fast bootstrap replicates. BI analysis was conducted in MrBayes v3.2.7a (Ronquist et al., 2012). Markov Chain Monte Carlo (MCMC) runs were performed for 10 million generations with a sampling frequency of 1,000 generations. The temperature of the exchange chain was set to 0.2, and the “burninfrac” was set to 0.25, indicating that 2,500 burn-in samples were removed from the initial operation. The strict consequence tree and posterior probability (PP) were calculated from the remaining 7,500 trees. We chose A. pacifica as an outgroup for phylogenetic analyses because of its close relationship to the genus Chrysanthemum (Zhao et al., 2010; Liu et al., 2012; Ma et al., 2020).
Phenotypic Analyses
After flower opening, flower traits were measured according to the handbook of UPOV (International Union for the Protection of New Varieties of Plants) and the DUS (Distinctness, Uniformity and Stability) test guidelines issued by the Ministry of Agriculture of the People’s Republic of China. Floral traits were classified according to four morphological indexes: inflorescence size, type of ray floret, type of flower head, and color of ray flowers. The inflorescence size was divided into two classes: large (>6 cm) and small (≤6 cm). The shape of the ray floret was divided into five types: ligulate, spatulate, quilled, incurved, and spoon-shaped. The flower head was divided into four types: without ray florets, single, semi-double, and double. Using visual analysis as in the UPOV and DUS guidelines, color of ray flowers was divided into eight categories: white, yellow, red, purple, pink, green, bicolor, and intermediate color.
Results
Characterization of cp Genomes
In this study, cp genomes of 36 cultivars of C. morifolium and the wild species, C. nankingenese, were assembled. The length, number of annotated genes, and GC content for each genome was presented in Supplementary Table S1. All cp genomes consisted of a large single copy region (LSC), a small single copy region (SSC), and two inverted repeats (IR; Figure 2). The total length of cp genomes ranged from 151,058 to 151,096 bp for the 36 cultivars, which included 82,856–82,858 bp LSC, 18,294 bp SSC, and 24,954–24,972 bp IR (Supplementary Table S1). The length of the C. nankingenese cp genome was 150,967 bp, which included 82,740 bp, 18,311 bp, and 24,958 bp of LSC, SSC, and IR regions, respectively (Supplementary Table S1). For all assembled cp genomes, 110 unique genes arranged in the same order were annotated, which consisted of 79 protein-coding genes, 27 tRNAs, and four rRNAs (Supplementary Table S1). The GC content of all cp genomes was 37.5% (Supplementary Table S1). The assembled cp genomes were deposited in GenBank (Table 1).
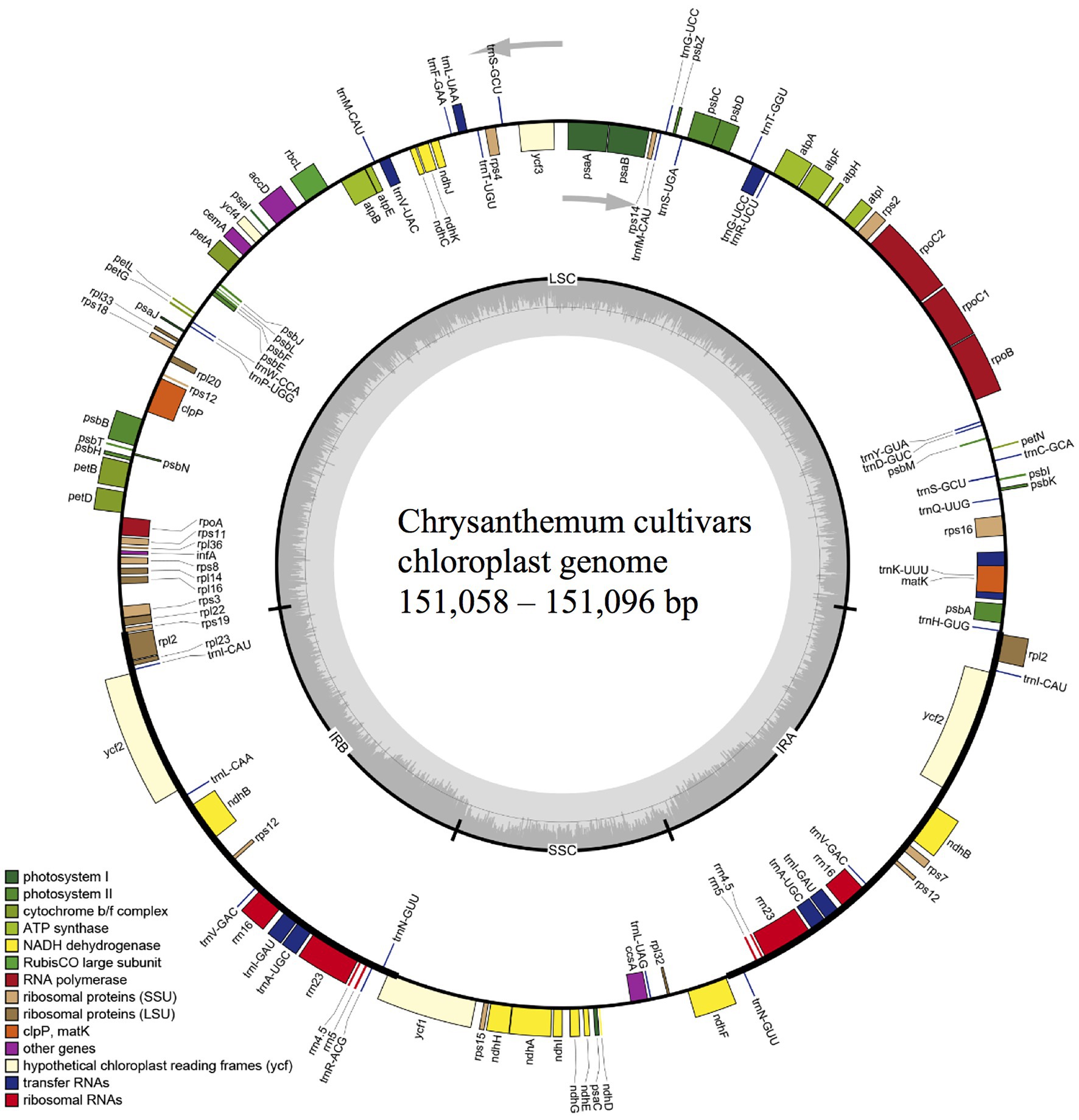
Figure 2. Chloroplast genome map of 36 chrysanthemum cultivars. Genes transcribed clockwise and counter-clockwise are showed inside and outside the circle, respectively. Genes belonging to different functional groups are color-coded. Dark gray in the inner circle corresponds to GC content. LSC, large single copy region; SSC, small single copy region; IRA and IRB, inverted repeats.
Structure of cp Genomes
The distribution of genes in the IR and SC boundaries for the 36 C. morifolium cultivars and C. nankingenese was examined to explore the potential expansion and contraction of the IR boundary. The positions of IR and SC boundaries were conserved among the assembled cp genomes. The boundary between LSC and IRb was located within the rps19 gene (Supplementary Figure S1). The length of the portions of the rps19 gene in the LSC and IRb regions were 218 and 61 bp, respectively. The boundary between SSC and IRb occurred within the ycf1 gene, with 558 and 4,457 bp (556 and 4,462 bp in C. nankingenese) of the gene located in the SSC and IRb regions, respectively (Supplementary Figure S1). The junction of SSC and IRa occurred 66 and 49 bp away from the ndhF gene (located in the SSC region) in the cp genomes of modern chrysanthemum cultivars and C. nankingenese, respectively (Supplementary Figure S1). The boundary between LSC and IRa was located between the rpl2 gene and the trnH gene. The trnH gene in the LSC region was 8 bp away from the boundary, while that of the rpl2 gene in IRa region was further away (Supplementary Figure S1). All assembled cp genomes were highly similar to the reference sequence (C. indicum) based on the results of mVISTA (Supplementary Figure S2). The coding regions were more conserved than the non-coding regions, and the most variable coding region was the ycf1 gene (Supplementary Figure S2).
Repeat Sequences of the cp Genome
We detected 43 dispersed repeats in the C. morifolium cultivar, “Fenban,” and 37 repeats in all the other cultivars (Supplementary Table S2). These repeats were either forward or palindromic. In C. nankingenese, 34 dispersed repeats, including 17 forward, 16 palindromic, and one complementary repeat, were identified (Supplementary Table S2). In addition to dispersed repeats, 41 SSRs in the cp genomes of C. morifolium cultivars, including 37 mononucleotide, one dinucleotide, and two trinucleotide SSRs, were detected (Supplementary Table S2). In C. nankingenese, 43 mononucleotides and two dinucleotide SSRs were found (Supplementary Table S2). There were no SSRs with a repeat unit longer than three nucleotides (e.g., tetranucleotide) in the cp genomes of C. morifolium cultivars and C. nankingenese.
Phylogeny Reconstruction
Maximum Likelihood and BI analyses of cp genomes produced similar trees (Figure 1). The genus Chrysanthemum was monophyletic. All 36 cultivars of C. morifolium formed a strongly supported group (BS = 100, PP = 1.00), and wild species of the genus Chrysanthemum were paraphyletic to this group (Figure 1). The relationships among cultivars were largely unresolved because of low genetic diversity on cpDNA data (mean pair-wise diversity = 2.0 × 10−6), with the exception of five cultivars (“Cenluanbiran,” “Guohuaxingxinghuo,” “Jinjiliuxia,” “Nanshangaosi,” and “Stresa”) that shared a mutation and formed a clade with strong support (BS = 100, PP = 1.00) on the cpDNA tree (Figure 1). The wild species, C. zawadskii, was most closely related to the clade made up of the C. morifolium cultivars, followed by Chrysanthemum lavandulifolium, Chrysanthemum chanetii, and Chrysanthemum boreale (Figure 1). This result is different from a recent phylogenetic analyses based on cp genomes, in which the C. lavandulifolium was sister to the cultivars (Ma et al., 2020). This difference could be due to high cpDNA variation in C. zawadskii, and different chlorotypes have been sequenced in these two studies (Genbank accession number, MW539687 vs. MG799556). Three wild species, namely, C. vestitum, C. indicum, and C. nankingenese grouped together, with the latter two more closely related to each other.
Phenotypic Characterization
The 36 C. morifolium cultivars showed very high morphological variation (Figure 1; Supplementary Figure S3; Table 1). All the Chinese traditional and the Japanese cultivars have large flowers and double flower heads; all the HD cultivars have small flowers and single (or semi-double) flower heads; and the Dutch cultivars have both large and small flowers, and three types of flower heads (single, semi-double, and double; Figure 1; Supplementary Figure S3; Table 1). There were four types of ray florets observed in the 36 cultivars (Figure 1; Supplementary Figure S3; Table 1). The ligulate and spoon types of ray florets were found in cultivars with large and small flowers, and ligulate was the most common type of ray floret (floret-type of 15 large flower and 10 small flower cultivars). The quilled and incurved types of ray floret were only found in four and one large flower cultivars, respectively. Flower color was successfully documented in 32 cultivars. The most common color was yellow (10 cultivars), followed by pink (6), purple (6), red (5), and white (3). The white and bicolors were rare and only produced by the “Cenluanbiran” and “Healing” cultivars, respectively.
Discussion
Maternal Genome Donor of Chrysanthemum Cultivars
Our phylogenetic analyses of cp genomes revealed that all chrysanthemum cultivars formed a strongly supported clade (BS = 100, BI = 1.00), and shared 64 substitutions that were distinct from wild species of the genus Chrysanthemum (Supplementary Data 1). This result is consistent with the suggestion that an extinct or un-sampled wild Chrysanthemum species served as the maternal donor of chrysanthemum cultivars (Ma et al., 2020; Qi et al., 2021). This conclusion, however, depends on the assumption that cpDNA variations of chrysanthemum cultivars and wild species have been well represented in phylogenetic analyses. Because there are more than 20,000 chrysanthemum cultivars all over the world, a comprehensive sampling with all cultivars is impossible. In this study, we collected 36 accessions to represent common cultivars of different countries and different flower morphologies. Our sample size is much larger than previous studies (12 accessions; Ma et al., 2020; Qi et al., 2021), and should comprise most, if not all, of the cpDNA variations in chrysanthemum cultivars. Domesticated chrysanthemum was thought to originate from multiple species, such as C. argyrophyllum, C. indicum, C. lavandulifolium, C. nankingense, C. vestitum, and C. zawadskii (Chen, 1985; Dai et al., 1998; Ma et al., 2020; Qi et al., 2021). A recent study revealed high cpDNA variations within two wild Chrysanthemum species, C. indicum and C. vestitum (Qi et al., 2021). Therefore, to elucidate whether the maternal parents of chrysanthemum cultivars were extinct or un-sampled, future studies should perform range-wild sampling of all wild species, and comparative analysis of whole cp genome sequences.
The genetic divergence between cultivars and wild species was slightly higher than that among wild species (0.0012 vs. 0.0010, p = 0.011, Wilcoxon rank sum test), which suggested that the maternal donor species diverged from the other wild species before the domestication of chrysanthemum. The divergent cp genomes of cultivars could also be explained by an increased rate of substitution caused by artificial selection for improving chrysanthemum ornamental value. However, in this case, we would expect that the high divergence between the cultivar lineages that experienced different artificial selection histories. In contrast, the divergence between cultivars was extremely low (mean pair-wise diversity = 2.0 × 10−6), regardless of their distinct floral morphologies. In addition, the chloroplast genome of plants is relatively conserved and contains genes involved in photosynthesis, transcription, and translation (Wicke et al., 2011), which were unlikely to be under directional selection during the breeding of Chrysanthemum.
Utility of cp Genomes for Phylogenetic and Population Genetic Analyses
Chloroplast genomes are haploid, maternally inherited, and structurally conserved in most flowering plants, making the cp genome an ideal genetic markers for tracking the evolution of plants at both high and low taxonomic levels (Xing and Liu, 2008; Rogalski et al., 2015; Daniell et al., 2016). By taking advantage of next-generation sequencing technologies and bioinformatics tools, the cp genomes can be assembled from whole genome sequencing data, avoiding the time-consuming processes of chloroplast isolation and purification (Dierckxsens et al., 2017; Freudenthal et al., 2020; Jin et al., 2020). Cp genomes are widely used to study evolutionary history of plants, and provide a high resolution tool for deciphering phylogenetic relationships between closely related species, such as those in the families Asteraceae (Vargas et al., 2017), Fabaceae (Koenen et al., 2020), Fagaceae (Zhou et al.), Polemoniaceae (Rose et al., 2021), and Saxifragaceae (Folk et al., 2017). The phylogeny of the genus Chrysanthemum is well resolved based on the complete cp genomes from the current and a previous study (Ma et al., 2020), which contrasts with the low resolution phylogeny based on a few cpDNA fragments (Zhao et al., 2003; Qi et al., 2021).
Although single cpDNA fragments contain insufficient information to resolve the relationships for all species, concatenated analyses of multiple fragments may provide valuable information for species delimitation. Relatively high genetic variation in the intergenic (e.g., trnH-psbA) and the coding regions of ycf1 was found. Some of these regions are used as genetic markers for phylogenetic analyses in land plants (CBOL Plant Working Group, 2009; Dong et al., 2015; Bagheri et al., 2017), and may be promising DNA-barcode markers in the genus Chrysanthemum. Simple repeats in the cp genome (i.e., cpSSRs) are important genetic markers for population genetic analyses. The mutation rate in cpSSR regions has been estimated at 3.2 × 10−5–7.9 × 10−5 per site per year (Provan et al., 1999), thousands of times higher than elsewhere in the cp genome (1 × 10−9–3 × 10−9 per site per year; Wolfe et al., 1987). Thus, the cpSSRs found in this study have the potential to be used to detect genetic variations between recently diverged lineages, such as chrysanthemum cultivars or populations of wild chrysanthemum species. However, it worth noting that sequencing repetitive regions is technically challenging for next-generation sequencing (Treangen and Salzberg, 2011). New laboratory methods and novel computational tools will be required to accurately genotype the cpSSR regions in plants (Šarhanová et al., 2018).
Conclusion
We assembled 37 cp genomes of chrysanthemum cultivars and wild species. The structure of these cp genomes was highly conserved in the genus Chrysanthemum, with similar IR-SSC boundaries, number and order of genes, and content of repetitive elements. Phylogenetic analyses based on cpDNA data revealed a strongly supported clade formed by chrysanthemum cultivars, suggesting a lineage of the genus Chrysanthemum as well as its subsequent cultivars unidirectionally providing a maternal source in breeding programs for developing modern cultivars. The high divergence between the cp genomes of chrysanthemum cultivars and wild species indicates that the maternal parent might be an extinct or un-sampled species (or population). Moreover, the low cpDNA polymorphism in chrysanthemum cultivars suggests that either the maternal parent had very low cpDNA variation, or only a few individuals served as the maternal donor of modern cultivars. In addition to maternal origin information of chrysanthemum cultivars, this study provides cp genomic resources for developing genetic markers that can be used in phylogenetic and phylogeographic studies of the genus Chrysanthemum. Specially, we suggest that repetitive regions (e.g., cpSSR) with elevated mutation rates may contain enough genetic variation be used to delineate chrysanthemum cultivars and populations of wild relatives. This study sheds new light on the origin of chrysanthemum cultivars and provides a valuable genetic resource for the continued development of varieties.
Data Availability Statement
The data present in the study are deposited in the Genbank (https://www.ncbi.nlm.nih.gov/genbank/), accession number ON534022-ON534058.
Author Contributions
HX and BW designed this study. YX, BL, and HX collected samples. YX and BL analyzed the data. HX, BW, and KO wrote the original draft, HZ, FW, and HX supervised the project. All authors read and approved the final manuscript.
Funding
This work was supported by the Major Technological Innovation of Guangdong Province of China (no. 2020B020220009) and the National Natural Science Foundation of China (no. 32001244).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.923442/full#supplementary-material
Footnotes
References
Amiryousefi, A., Hyvonen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Bagheri, A., Maassoumi, A. A., Rahiminejad, M. R., Brassac, J., and Blattner, F. R. (2017). Molecular phylogeny and divergence times of Astragalus section Hymenostegis: an analysis of a rapidly diversifying species group in Fabaceae. Sci. Rep. 7:14033. doi: 10.1038/s41598-017-14614-3
Beier, S., Thiel, T., Muench, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
CBOL Plant Working Group (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U. S. A. 106, 12794–12797. doi: 10.1073/pnas.0905845106
Chen, J. Y. (1985). Studies on the origin of Chinese florist’s chrysanthemum. Acta Hortic. 167, 349–361.
Chen, J. Y. (2012). The Origin of Garden Chrysanthemum. Hefei: Anhui Science & Technology Publishing House.
Dai, S. L., Chen, J. Y., and Li, W. B. (1998). Application of RAPD analysis in the study on the origin of Chinese cultivated chrysanthemum. Acta Bot. Sin. 40, 1053–1059.
Dai, S. L., Wang, W. K., and Xu, Y. X. (2005). Phylogenetic relationship of Dendranthema (DC.) Des Moul. revealed by fluorescent in situ hybridization. J. Integr. Plant Biol. 47, 783–791. doi: 10.1111/j.1744-7909.2005.00068.x
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5:8348. doi: 10.1038/srep08348
Folk, R. A., Mandel, J. R., and Freudenstein, J. V. (2017). Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66, 320–337. doi: 10.1093/sysbio/syw083
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Freudenthal, J. A., Pfaff, S., Terhoeven, N., Korte, A., Ankenbrand, M. J., and Förster, F. (2020). The landscape of chloroplast genome assembly tools. bioRxiv, 665–869. doi: 10.1101/665869
Fukai, S., Wei, Z., and Goi, M. (2003). Dendranthema species as chrysanthemum genetic resources. Acta Hortic. 620, 223–230. doi: 10.17660/ActaHortic.2003.620.26
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Koenen, E. J. M., Ojeda, D. I., Steeves, R., Migliore, J., Bakker, F. T., Wieringa, J. J., et al. (2020). Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol. 225, 1355–1369. doi: 10.1111/nph.16290
Kuligowska, K., Lutken, H., and Mueller, R. (2016). Towards development of new ornamental plants: status and progress in wide hybridization. Planta 244, 1–17. doi: 10.1007/s00425-016-2493-7
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumari, S., Dhiman, S. R., and Gupta, Y. C. (2019). Advances in breeding of Chrysanthemum: a review. Int. J. Curr. Microbiol. App. Sci. 8, 1631–1643. doi: 10.20546/ijcmas.2019.808.193
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lim, K. B., Barba Gonzalez, R., Zhou, S., Ramanna, M. S., and Tuyl, J. (2008). “Interspecific hybridization in Lily (Lilium): taxonomic and commercial aspects of using species hybrids in breeding,” in Floriculture, Ornamental and Plant Biotechnology. 5, 138–145.
Liu, P. L., Wan, Q., Guo, Y. P., Yang, J., and Rao, G. Y. (2012). Phylogeny of the genus Chrysanthemum L.: evidence from single-copy nuclear gene and chloroplast DNA sequences. PLoS One 7:e48970. doi: 10.1371/journal.pone.0048970
Ma, Y. P., Zhao, L., Zhang, W. J., Zhang, Y. H., Xing, X., Duan, X. X., et al. (2020). Origins of cultivars of Chrysanthemum-evidence from the chloroplast genome and nuclear LFY gene. J. Syst. Evol. 58, 925–944. doi: 10.1111/jse.12682
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Provan, J., Soranzo, N., Wilson, N. J., Goldstein, D. B., and Powell, W. (1999). A low mutation rate for chloroplast microsatellites. Genetics 153, 943–947. doi: 10.1093/genetics/153.2.943
Qi, S., Twyford, A. D., Ding, J. Y., Borrell, J. S., Wang, L. Z., Ma, Y. P., et al. (2021). Natural interploidy hybridization among the key taxa involved in the origin of horticultural chrysanthemums. J. Syst. Evol. doi: 10.1111/jse.12810
Rieseberg, L. H., Archer, M. A., and Wayne, R. K. (1999). Transgressive segregation, adaptation and speciation. Heredity 83, 363–372. doi: 10.1038/sj.hdy.6886170
Rogalski, M., Vieira, L. d. N., Fraga, H. P., and Guerra, M. P. (2015). Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front. Plant Sci. 6:586. doi: 10.3389/fpls.2015.00586
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). Mrbayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rose, J. P., Toledo, C. A. P., Lemmon, E. M., Lemmon, A. R., and Sytsma, K. J. (2021). Out of sight, out of mind: widespread nuclear and plastid-nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Syst. Biol. 70, 162–180. doi: 10.1093/sysbio/syaa049
Šarhanová, P., Pfanzelt, S., Brandt, R., Himmelbach, A., and Blattner, F. R. (2018). SSR-seq: genotyping of microsatellites using next-generation sequencing reveals higher level of polymorphism as compared to traditional fragment size scoring. Ecol. Evol. 8, 10817–10833. doi: 10.1002/ece3.4533
Schwarzbach, A. E., Donovan, L. A., and Rieseberg, L. H. (2001). Transgressive character expression in a hybrid sunflower species. Am. J. Bot. 88, 270–277. doi: 10.2307/2657018
Shih, C., Christopher, J. H., and Michael, G. G. (2011). “Chrysanthemum,” in Flora of China. eds. Z. Y. Wu, P. H. Raven, and D. Y. Hong (Beijing, St. Louis: Science Press, Missouri Botanical Garden Press), 669–676.
Shinoyama, H., Aida, R., Ichikawa, H., Nomura, Y., and Mochizuki, A. (2012). Genetic engineering of chrysanthemum (Chrysanthemum morifolium): current progress and perspectives. Plant Biotechnol. 29, 323–337. doi: 10.5511/plantbiotechnology.12.0521a
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Treangen, T. J., and Salzberg, S. L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. doi: 10.1038/nrg3117
Vargas, O. M., Ortiz, E. M., and Simpson, B. B. (2017). Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 214, 1736–1750. doi: 10.1111/nph.14530
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Mueller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U. S. A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Xing, S.-C., and Liu, C. J. (2008). Progress in chloroplast genome analysis. Prog. Biochem. Biophys. 35, 21–28.
Zhao, H. B., Chen, F. D., Chen, S. M., Wu, G. S., and Guo, W. M. (2010). Molecular phylogeny of Chrysanthemum, Ajania and ITS allies (anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trnL-F IGS sequences. Plant Syst. Evol. 284, 153–169. doi: 10.1007/s00606-009-0242-0
Keywords: Chrysanthemum, chloroplast genome, maternal donor, genetic marker, phylogenetic analysis
Citation: Xu Y, Liao B, Ostevik KL, Zhou H, Wang F, Wang B and Xia H (2022) The Maternal Donor of Chrysanthemum Cultivars Revealed by Comparative Analysis of the Chloroplast Genome. Front. Plant Sci. 13:923442. doi: 10.3389/fpls.2022.923442
Edited by:
Xiaohua Jin, Institute of Botany (CAS), ChinaReviewed by:
Christiane M. Ritz, Senckenberg Museum of Natural History Görlitz, GermanyMohammad Nadeem, King Saud University, Saudi Arabia
Copyright © 2022 Xu, Liao, Ostevik, Zhou, Wang, Wang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanhan Xia, xiahanhan@zhku.edu.cn
†These authors have contributed equally to this work and share first authorship