Downregulation of Transcriptional Activity, Increased Inflammation, and Damage in the Placenta Following in utero Zika Virus Infection Is Associated With Adverse Pregnancy Outcomes
 Patrick S. Creisher1
Patrick S. Creisher1  Jun Lei2
Jun Lei2  Morgan L. Sherer1 Amanda Dziedzic1 Anne E. Jedlicka1
Morgan L. Sherer1 Amanda Dziedzic1 Anne E. Jedlicka1  Harish Narasimhan1 Anna Chudnovets2 Ariana D. Campbell1 Anguo Liu2 Andrew Pekosz1 Irina Burd2*
Harish Narasimhan1 Anna Chudnovets2 Ariana D. Campbell1 Anguo Liu2 Andrew Pekosz1 Irina Burd2*  Sabra L. Klein1*
Sabra L. Klein1*- 1W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, United States
- 2Department of Gynecology and Obstetrics, Integrated Research Center for Fetal Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Zika virus (ZIKV) infection during pregnancy causes serious adverse outcomes to the developing fetus, including fetal loss and birth defects known as congenital Zika syndrome (CZS). The mechanism by which ZIKV infection causes these adverse outcomes, and specifically the interplay between the maternal immune response and ZIKV replication has yet to be fully elucidated. Using an immunocompetent mouse model of transplacental ZIKV transmission and adverse pregnancy outcomes, we have previously shown that Asian lineage ZIKV disrupts placental morphology and induces elevated secretion of IL-1β. In the current manuscript, we characterized placental damage and inflammation during in utero African lineage ZIKV infection. Within 48 h after ZIKV infection at embryonic day 10, viral RNA was detected in placentas and fetuses from ZIKA infected dams, which corresponded with placental damage and reduced fetal viability as compared with mock infected dams. Dams infected with ZIKV had reduced proportions of trophoblasts and endothelial cells and disrupted placental morphology compared to mock infected dams. While placental IL-1β was increased in the placenta, but not the spleen, within 3 h post infection, this was not caused by activation of the NLRP3 inflammasome. Using bulk mRNAseq from placentas of ZIKV and mock infected dams, ZIKV infection caused profound downregulation of the transcriptional activity of genes that may underly tissue morphology, neurological development, metabolism, cell signaling and inflammation, illustrating that in utero ZIKV infections causes disruption of pathways associated with CZS in our model.
Introduction
Pregnant women and their developing fetuses are at high risk for severe outcomes from a variety of viral, bacterial, and parasitic infections including the “TORCH” pathogens (Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus), which are grouped because of their propensity to induce congenital disease (1, 2). Viral infections, specifically, often cause adverse consequences during pregnancy for the mother, fetus, or both. Pregnant people are at higher risk than nonpregnant people for severe disease from many viruses, including Influenza A viruses and Hepatitis E virus, which may be due to immunological changes associated with pregnancy (1, 3–5). Viruses also can infect the placenta, fetus, or both to cause adverse consequences, such as loss of pregnancy, preterm birth, birth defects, and growth and neurodevelopmental delays in the fetus and newborn (2, 4).
Zika virus (ZIKV) is a single-stranded positive sense RNA virus in the family Flaviviridae that received international and scientific attention in 2015–2016, when the virus spread throughout the Americas causing microcephaly and other congenital malformations, such as problems with hearing, vision, and mobility, in the babies of infected pregnant people (6, 7). The causal connection between ZIKV infection during pregnancy and subsequent birth defects, which are collectively referred to as Congenital Zika Syndrome (CZS), was confirmed with the detection of ZIKV RNA in amniotic fluid of pregnant women carrying children with microcephaly and the isolation of ZIKV from the brain of an infant who died after birth (6, 8). Notably, multiple systematic reviews and meta-analyses performed after the end of the epidemic confirmed the association between ZIKV infection during pregnancy and CZS (9–12).
Despite the causal link between prenatal ZIKV infection and adverse pregnancy and neonatal outcomes, the mechanism by which ZIKV infection contributes to CZS remains unclear. To date, most studies suggest that viral replication in the fetal brain as well as immunopathology induced by maternal inflammation cause adverse outcomes (13, 14). Animal models of ZIKV infection during pregnancy [reviewed in (14)] provide insight into mechanisms of ZIKV pathogenesis. Mouse models are commonly utilized for studies of viral pathogenesis (15), and their short gestation period and large litter sizes provide a benefit for studies during pregnancy (16).
Since the 2016 ZIKV epidemic, we have investigated the pathogenesis of ZIKV infection during pregnancy after intrauterine infection in immunocompetent mice. Collectively, we have shown ZIKV infection during pregnancy causes placental pathology, reduced fetal viability, congenital malformations, and reduced cortical thickness that is induced by maternal inflammation rather than direct virus infection (17, 18). We have identified placental secretion of IL-1β as a cause of inflammation and placental dysfunction (18). Elevated IL-1β signaling during pregnancy damages the placenta by disrupting placental architecture, including distortion of the labyrinth layer and reduction in trophoblast invasion, which may contribute to adverse neonatal health consequences (18, 19). Canonically, IL-1β activation and secretion is dependent on activation of the NLRP3 [nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3] inflammasome (20), which is active in placental cells of humans and mice (21–23).
As the organ of the maternal-fetal interface, the placenta is necessary for appropriate fetal development, protection against insults (e.g., viruses), provision of nutrients, oxygen, and waste exchange, and enabling maternal-fetal endocrine cross-talk (2, 16, 24, 25). Microbes associated with congenital disease, including ZIKV, likely have mechanisms to overcome placental defenses, resulting in damage to the placenta and disrupted fetal development [reviewed in (2)]. In the current study, we sought to investigate the mechanisms of placental damage during ZIKV infection, by characterizing the cells and pathways that underly ZIKV-induced placental damage and adverse fetal outcomes.
Materials and Methods
Viruses and Cells
Zika virus (ZIKV) strain IB H 30656 (Nigeria, 1968) was purchased from the American Type Culture Collection (ATCC, # VR-1839). The Paraíba (Brazil, 2015) strain was kindly provided by Stephen Whitehead of the National Institute of Allergy and Infectious Diseases. All procedures for handling ZIKV were approved by the Institutional Biosafety Committee. Stocks of ZIKV strains were generated by infecting Vero E6 cells at an MOI of 0.001 50% tissue culture infectious doses (TCID50) per cell in DMEM (Sigma #D5796) supplemented with 2.5% Fetal Bovine Serum (FBS, Gibco #26140079), 1 mM sodium pyruvate (Sigma #S8636), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco #15149-122), and 2 mM l-glutamine (Gibco #25030-081). Approximately 96 h after infection, the supernatant fluids were collected, clarified by centrifugation (900 g for 10 min), and stored in aliquots at −80°C. ASC expressing RAW 264.7 (RAW-ASC) cells (InvivoGen #raw-asc) were grown in DMEM (Sigma #D5796) supplemented with 10% heat-inactivated FBS (Gibco #26140079), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco #26140079), 100 μg/ml Normocin™ (InvivoGen #ant-nr-1), and 10 μg/ml blasticidin (InvivoGen #ant-bl-05). RAW-ASC cells were plated on six well plates, primed with Pam3CSK4 (InvivoGen #tlrl-pms) at a concentration of 100 ng/ml for 3 h and then stimulated with 10 μM of Nigericin (InvivoGen #tlrl-nig) for 6 h as a positive control for inflammasome component westerns.
Experimental Mice
Timed-pregnant adult (2–3 months of age) CD-1 IGS mice were purchased from Charles River Laboratories (strain code 022). Animals arrived at embryonic day 9 (E9) and were housed in pairs in a specific-pathogen-free animal BSL2 facility at Johns Hopkins University with ad libitum access to food and water. Mice were acclimated for 24 h prior to experiments (26) and procedures for animal experiments were performed consistently at the same time of day.
ZIKV Infections
At E10, mice were anesthetized continuously with isoflurane mixed with oxygen and underwent mini-laparotomy in the lower abdomen for ZIKV injections. Animals received either 106 TCID50 units of ZIKV in 100 μl or 100 μl DMEM alone as previously described (18, 26). Briefly, the inoculum was divided equally into four injections delivered into the uterine myometrium, opposite the placenta and between the gestational sacs of the first five fetuses closest to the cervix of one uterine horn. The contralateral uterine horn was not manipulated. All placental experiments were performed with placentas from the horn that was either ZIKV or mock inoculated. Routine closure was performed after injections and dams were returned to cages for recovery. Investigators were not blinded to the treatment group allocation.
Tissue Collection
Mice were euthanized by CO2 exposure followed by cardiac exsanguination at either 3, 6, or 48 h post infection (hpi), depending on the experiment. At the time of euthanasia, the total number of viable and nonviable fetuses was quantified for each pregnant dam. Fetal viability was determined as the percentage of fetuses within the inoculated uterine horn for ZIKV- and mock-infected dams that were viable. Small or discolored fetuses or the absence of a fetus at an implantation site were counted as nonviable. Maternal spleen as well as the uterine horn, placentas, and fetuses of the inoculated horn were collected with tissue either flash frozen in a dry ice–isopropanol bath or fixed in 4% paraformaldehyde (Fisher Scientific # AAJ19943K2). All animal studies were conducted under animal BSL2 containment.
Total RNA Extraction and qRT-PCR
Tissues were weighed and homogenized in 1 ml of QIAzol Lysis Reagent (QIAGEN #1023537) using Lysing Matrix D tubes (MP Biomedicals #6913100) in a MP Fast-prep 24 5G instrument. Total RNA was extracted from tissue samples using the RNeasy Lipid Tissue Mini Kit (QIAGEN #74804), according to the manufacturer's instructions. ZIKV RNA copies were determined by one-step qRT-PCR reaction using the QuantiTect Probe RT-PCR Kit (QIAGEN #20443) according to the manufacturer's protocol. The real-time PCR primers and probe for ZIKV RNA detection were the ZIKV 1162c set described previously (27), and the primer sequences and concentrations were as follows: Fwd (100 μM), 5′-CCGCTGCCCAACACAAG-3′; Rev (100 μM), 5′-CCACTAACGTTCTTTTGCAGACAT-3′; probe (25 μM), 5′-/56-FAM/AGCCTACCT/ZEN/TGACAAGC AATCAGACACTCAA/3IABkFQ/-3′ (Integrated DNA Technologies). ZIKV RNA copies were determined relative to a standard curve produced using serial 10-fold dilutions of ZIKV RNA isolated from ZIKV stocks with a known infectious virus titer.
Hematoxylin and Eosin Staining and Immunohistochemistry
At 48 hpi (E12), mice were euthanized and placentas were fixed overnight at 4°C in 4% PFA. The following day, placentas were washed five times with PBS and immersed in 30% sucrose until saturation. Using a Leica CM1950 cryostat, the specimens were cut at 20-μm thickness and mounted on positively charged slides (Fisher Scientific #12-550-15). Routine hematoxylin and eosin (H&E) staining was performed to evaluate the morphological change of the placentas. For immunohistochemical staining, antigen retrieval was performed by boiling in citrate buffer (pH 6, Vector Lab, H-3300-250) for 20 min. Slides were washed with PBS, which was followed by permeabilization in PBS solution containing 0.05% Triton X-100 and 10% normal goat serum (Invitrogen #50197Z) for 30 min. Placentas were incubated with mouse anti-flavivirus group antigen (1:100 ThermoFisher #MA1-7397), rabbit anti-vimentin (1:200, Abcam # ab92547), or rabbit anti-cytokeratin (1:200, Dako #Z0622) overnight at 4°C. The next day, sections were rinsed with PBS and then incubated with donkey anti-mouse (ThermoFisher #R37115) or donkey anti-rabbit (ThermoFisher #R37119) fluorescent secondary antibodies (ThermoFisher #R37115) diluted 1:500 for 3 h at room temperature. DAPI (Roche #10236276001) was applied for counterstaining, followed by mounting with Fluoromount-G (eBioscience #00-4958-02). Cell density of vimentin and cytokeratin positive cell quantification was performed using Image J (1.47v). For each placenta, six random images in labyrinth at the middle level (thickest) of placenta were taken, and the average fluorescent area calculated for that placenta. One placenta per dam was used and 3–4 dams per group were analyzed.
TUNEL Staining
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed with Click-iT™ Plus TUNEL Assay (ThermoFisher #C10617) per the manufacturer protocol. Placental slides were fixed with 4% paraformaldehyde in PBS for 15 min at 37°C, rinsed with PBS, and permeabilized with proteinase K solution for 15 min at RT followed by staining with TUNEL working solution for 60 min at 37°C. Specimens were mounted on glass slides using Fluoromount-G (eBioscience #00-4958-02). For each placenta, 10 random images in labyrinth at the middle level (thickest) of placenta were taken, and the average percentage TUNEL+ (TUNEL+ fluorescent signal divided by the DAPI fluorescent signal) given for that placenta. One placenta per dam was used and 3–4 dams per group were analyzed.
In situ Hybridization
In situ hybridization was performed using RNAscope Multiplex Fluorescent Reagent Kit v2 Assay (ACDBio #323136). Following the protocol provided by the manufacture, RNAscope® Probe-V-ZIKV (ACDBio #467771) was applied to detect the viral signal in fetuses. All the images were viewed using a Zeiss Axioplan 2 microscope (Jena, Germany). Images were taken using a Zeiss AxioCam MRM.
IL-1β ELISA
Flash frozen placentas were weighed and homogenized in a 1:10 weight per volume of PBS using Lysing Matrix D tubes (MP Biomedicals #6913100) in a MP Fast-prep 24 5G instrument. Placental homogenates were stored at −80 °C until analysis. IL1β in placental homogenates was measured by ELISA according to the manufacturer's protocol (Abcam #100704).
Western Blot
Flash frozen placentas were weighed and homogenized in 1× Cell Lysis Buffer (Cell Signaling Technology #9803) with 1× Protease Inhibitor cocktail (Sigma-Aldrich #P8340) and 10 mM sodium fluoride (Fisher Scientific #S299 100) (20 μl lysis buffer per mg tissue). Protein lysates were stored at −80°C until analysis. The protein concentration of each lysate was measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific #23225). For each sample, 25 μg of protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) on NuPAGE 4–12% Bis-Tris gels (Thermo Fisher Scientific #NP0329). The gel was blotted onto Immobilon-FL PVDF Membrane (Millipore #IPFL00010) and the membranes were blocked using a 1:1 mixture of 1× PBS/Tween-20 solution (Sigma-Aldrich #P3563) and Intercept blocking buffer (LI-COR Biosciences #927-70001) for 30 min at room temperature. The membranes were treated with primary antibody diluted in blocking solution overnight at 4°C on a rocker. The membranes were washed with PBS-Tween thrice (10 min per wash) and incubated in secondary antibody solutions for 60 min at room temperature on a rocker. The membranes were washed thrice (10 min per wash) in PBS-Tween and imaged on a ProteinSimple FluorChem Q imager. Individual bands were quantified using Image Studio software (LI-COR Biosciences; version 3.1.4). The signal from each band was normalized against the GAPDH signal as a loading control and graphed as arbitrary units. Primary antibodies included: rabbit anti-phospho-NF-κB p65 (Ser536) (Cell Signaling #3033), mouse anti-NFkB p65 (Thermo Fisher Scientific #33-9900), rabbit anti-NLRP3 (Thermo Fisher Scientific #MA5-32255), rat anti-caspase 1 (Thermo Fisher Scientific #14-9832-82), and mouse anti-GAPDH (Abcam #ab8245). Secondary antibodies included: goat anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific #A11001), donkey anti-rabbit Alexa Fluor Plus 647 (Thermo Fisher Scientific #A32795), and goat anti-rat Alex Fluor 647 (Thermo Fisher Scientific #A21247).
mRNAseq and Analysis
Quantitation of Total RNA was performed with the Qubit BR RNA Assay kit and Qubit Flex Fluorometer (Invitrogen/ThermoFisher), and quality assessment performed by RNA ScreenTape analysis on an Agilent TapeStation 2200. Barcoded libraries for mRNA-Seq were prepared from 100 ng Total RNA using the Tecan Universal Plus mRNA Seq Library kit with NuQuant, according to manufacturer's recommended protocol. Quality of libraries was assessed by High Sensitivity DNA Lab Chips on an Agilent BioAnalyzer 2100. Quantitation was performed with NuQuant reagent, and confirmed by Qubit High Sensitivity DNA assay, on Qubit 4 and Qubit Flex Fluorometers (Invitrogen/ThermoFisher). Libraries were diluted, and equimolar pools prepared, according to manufacturer's protocol for appropriate sequencer. An Illumina iSeq Sequencer with iSeq100 i1 reagent V2 300 cycle kit was used for final quality assessment of the library pool. For deep mRNA sequencing, a 200 cycle (2 × 100 bp) Illumina NovaSeq SP run was performed at Johns Hopkins Genomics, Genetic Resources Core Facility, RRID:SCR_018669.
iSeq and NovaSeq data files were uploaded to the Partek Server and analysis with Partek Flow NGS software, with RNA Toolkit, was performed as follows: pre-alignment QA/QC; trimming of reads; alignment to mm 39 Reference Index using STAR 2.7.8a; post- alignment QA/QC; quantification of gene counts to annotation model (Partek E/M, Ensembl Transcript Release 103); filter and normalization of gene counts; and, identification and comparison of differentially expressed genes with GSA (gene specific analysis). From resulting gene lists, clustering and biological interpretation was performed. Ingenuity Pathways Analysis software (Qiagen) was used to identify molecular networks of relevance, and the pathways and biological processes most significantly related in the data sets.
All sequence files and sample information have been deposited at NCBI Sequence Read Archive, NCBI BioProject: BioProject: accession number PRJNA797437.
Statistical Analysis
Fetal viability data were analyzed with a χ2 test. ZIKV RNA copies, TUNEL staining, IHC quantification, and Western Blot data were analyzed with unpaired two-tailed t-tests. IL-1β ELISA data were analyzed with two-way ANOVA with Bonferroni's post hoc for multiple comparisons. Data are presented as mean ± SEM. Mean differences were considered statistically significant at p < 0.05. Statistical analyses were performed using GraphPad Prism v9.2 (GraphPad Software).
Results
In utero Exposure to ZIKV Causes Placental Infection, Vertical Transmission, and Fetal Loss
Intrauterine infection of immunocompetent pregnant mice at E10 or E14 with either African or Asian lineage strains of ZIKV causes fetal infection and demise (18, 26). The use of a mouse-adapted ZIKV strain was hypothesized to cause a more robust phenotype (28). Therefore, the historic IB H 30,656 Nigeria 1968 (Nigeria ZIKV) strain, which has been passaged 21 times in suckling mice (29) was used. Consistent with previous studies using clinical isolates of South American ZIKV (18, 26), intrauterine infection at E10 with Nigeria ZIKV reduced fetal viability at 48 h post infection (hpi) compared with mock inoculated dams (Figure 1A).
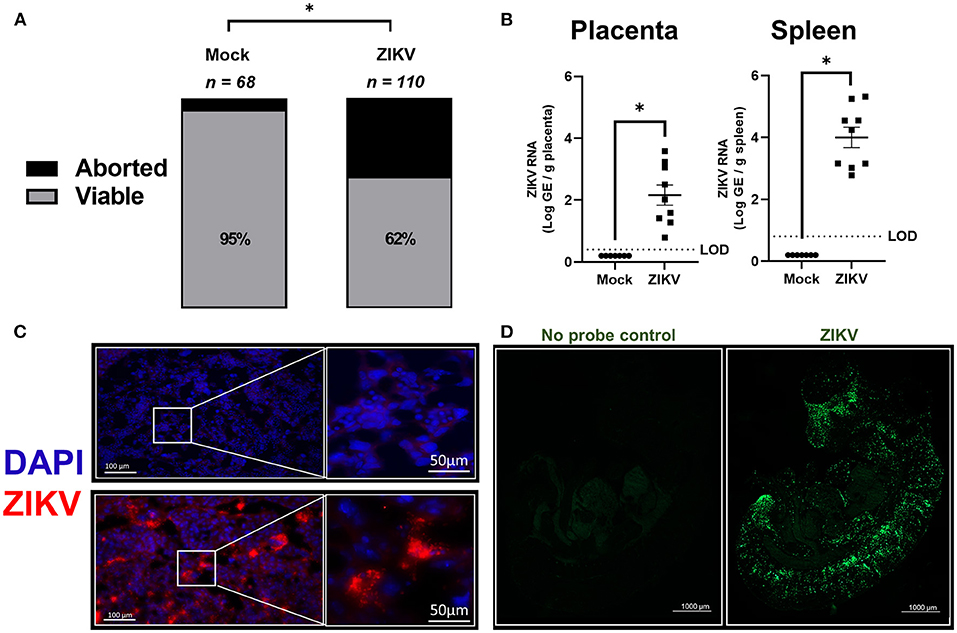
Figure 1. In utero exposure to ZIKV causes placental infection, vertical transmission, and fetal loss. At embryonic day (E) 10, pregnant mice underwent intrauterine injection of 106 TCID50 units of Nigeria ZIKV, Brazil ZIKV, or media (mock). (A) At 48 h post infection (hpi), dams were euthanized, and fetal viability was determined as the percentage of fetuses within the inoculated uterine horn (n = total number of fetuses from 11 to 16 dams per group from two independent replicates). (B) Nigeria ZIKV RNA copies were quantified from placenta or spleen collected 48 hpi. Data represent mean ± standard error of the mean from two independent replications (n = 7–9/group). (C) Placentas collected 48 hpi from either mock (upper panel) or Nigeria ZIKV (lower panel) infected dams were paraformaldehyde fixed and immunostained for ZIKV (red) and 4′6-diamidino-2-phenylindole (DAPI, blue) to label nuclei. Representative images taken at 20× magnification (left) and further zoomed 5.5 fold (right) are shown. (D) ZIKV RNA (green) was measured in fetuses of Brazil ZIKV infected dams by in situ hybridization (ISH) at 48 hpi. Samples were also imaged without RNA probe incubation as a control (left) to remove background and a representative image at 5× magnification is shown for fetuses of ZIKV infected dams (right). Significant differences (*p < 0.05) were determined by χ2 test (A) or unpaired two tailed t-test (B). Limit of detection (LOD) indicated with a dashed line. Scale bar: 100 or 50 μm (IHC, C), 1,000 μm (ISH, D).
ZIKV RNA copies were quantified at 48 hpi in placentas and spleens of dams infected with ZIKV at E10 (Figure 1B). ZIKV RNA was highly detectable in placentas and spleens of infected animals at 48 hpi (Figure 1B). ZIKV protein also was robustly detected in the labyrinth of placentas (Figure 1C, representative image) suggesting that infection may disrupt gas and nutrient exchange between mother and fetus.
Since Nigeria ZIKV has already been shown to productively infect fetuses, a 2015 Brazil clinical isolate (Brazil ZIKV) was used to infect pregnant dams at E10, with fetuses collected 48 hpi to measure the presence ZIKV RNA in the whole body by ISH. Consistent with previous studies using only quantitative PCR of fetal heads or bodies (21, 26, 28, 30), ZIKV RNA was predominantly localized to the developing fetal head and spinal cord (Figure 1D). Taken together with previously published studies (18, 26), these data illustrate that both African and Asian strains of ZIKV traverse the placenta and vertically infect fetuses with a tropism for the central nervous system, which is consistent with CZS.
ZIKV Infection Induces Placental Damage, Cell Death, and Disruption of Trophoblast and Endothelial Cell Layers
To further characterize the phenotypic placental damage caused by in utero ZIKV infection, placental morphology was analyzed (Figure 2A). Placentas of Nigeria ZIKV, collected at 48 hpi, showed significant damage, characterized by hemorrhage and mixing of maternal and fetal blood in the labyrinth as compared with placentas from mock infected dams (Figure 2A, highlighted in the lower panels), suggesting the loss of barrier function of trophoblast–endothelial cell layers. To define whether placental tissue damage included cell death, TUNEL staining in the labyrinth of placentas was performed, with the percentage of TUNEL positive cells quantified and reported as a proportion of DAPI+ cells (Figure 2B). The percentage of TUNEL+ cells in placentas of ZIKV infected dams was nearly double that of placentas of mock infected dams (Figure 2B, 3.3% in mock and 6.3% in ZIKV, p = 0.0186), indicating that ZIKV infection during pregnancy induces cell death in the placenta.
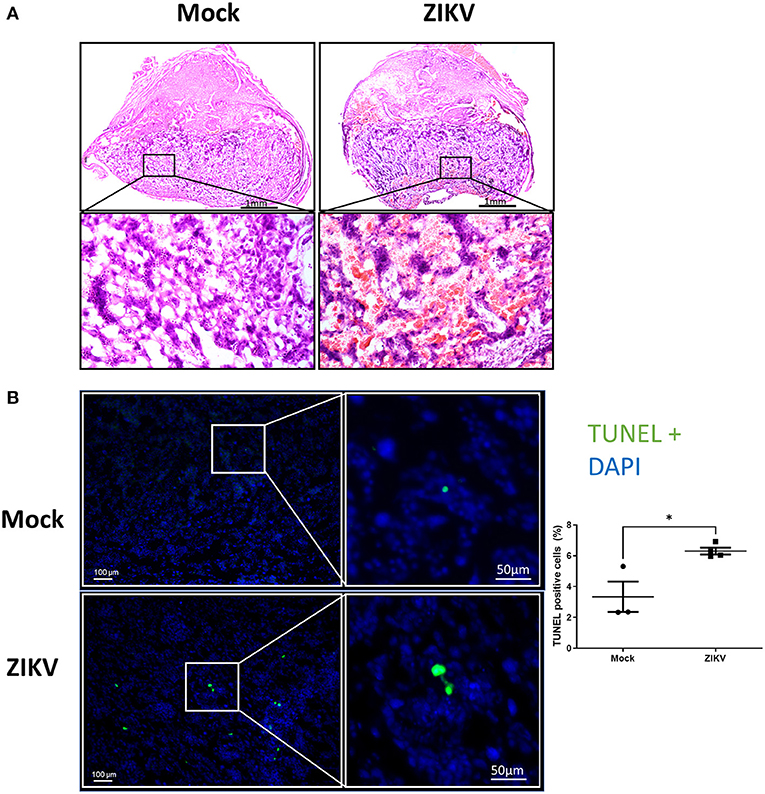
Figure 2. ZIKV infection induces placental damage and cell death. At E10, pregnant mice underwent intrauterine injection of 106 TCID50 units of Nigeria ZIKV or media (mock). Dams were euthanized at 48 hpi and placentas were paraformaldehyde-fixed. (A) Representative H and E images were taken at 5× magnification (upper panels) and 20× magnification (lower panels). (B) Representative TUNEL+ (green) stained placentas with DAPI to label nuclei taken at 100× magnification (left) and quantification of TUNEL+ cells as a percentage of DAPI+ cells. Data represent mean ± standard error of the mean (n = 3–4 dams/group, each dot indicates 1 placenta and is the mean quantification of 10 fields of view). Significant differences (*p < 0.05) were determined by unpaired two tailed t-test. Scale bar: 1 mm (HandE, A), 100 or 50 μm (TUNEL, B).
To further characterize the damage to the placenta, changes to the trophoblast-endothelial cell barrier, which separates maternal and fetal blood in the labyrinth of the murine placenta (16, 25), was evaluated. Using cytokeratin (trophoblasts, Figure 3A) and vimentin (endothelial cells, Figure 3B) staining of placentas collected at 48 hpi, there was a significant loss of trophoblasts in placentas of ZIKV infected dams as indicated by a 51.5% reduction (p = 0.0458) in cytokeratin positive-area compared with mock placentas (Figure 3A, representative image and quantification). Similarly, disruption of the endothelial cell layer was observed, which could be seen morphologically by the loss of the streak-like vessel integrity seen in placentas of mock inoculated dams (Figure 3B, representative image). ZIKV infection caused a 34% decrease in vimentin-positive area compared to mock infected placentas (Figure 3B). Together, these data suggest that in utero ZIKV infection damages the placenta, including the placental labyrinth zone and disrupts both the trophoblast and endothelial cell layers of the maternal-fetal barrier. Disruption of the morphology of the labyrinth zone can compromise oxygen, nutrient, and waste exchange and impact fetal growth and viability (24, 25).
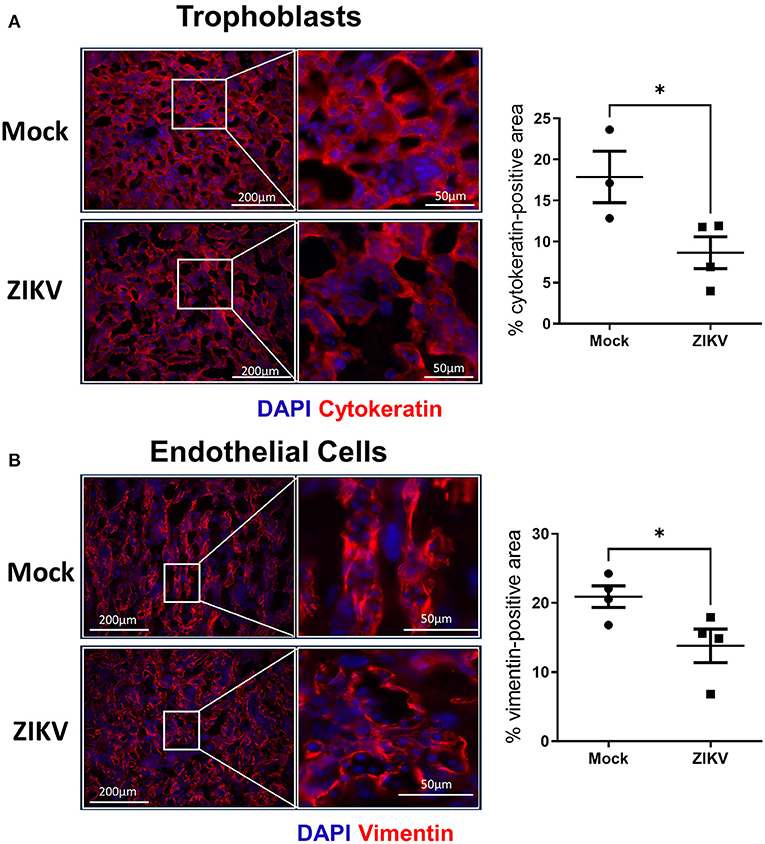
Figure 3. ZIKV infection disrupts the trophoblast and endothelial layer of the labyrinth zone. At E10, pregnant mice underwent intrauterine injection of 106 TCID50 units of Nigeria ZIKV or media (mock). Dams were euthanized at 48 hpi, and placentas were paraformaldehyde-fixed and immunostained for cytokeratin (A, red) to mark trophoblasts or vimentin (B, red) to mark endothelial cells and DAPI (blue) to label nuclei. Representative images taken at 20× magnification (left) and further zoomed (3.7- and 2.4- fold for trophoblasts and endothelial cells, respectively, right) are shown. Quantification of the percentage positive area for each marker is shown. Data represent mean ± standard error of the mean (n = 3–4 dams/group, each dot indicates 1 placenta and is the mean quantification of 6 fields of view). Significant differences (*p < 0.05) were determined by unpaired two tailed t-test. Scale bar: 200 μm.
Increased Placental IL-1β in the Absence of NLRP3 Inflammasome Upregulation
Previous studies have shown that IL-1β signaling during pregnancy induces placental damage and disruption of the placental labyrinth (19). To test the hypothesis that IL-1β underlies placental damage following infection with African lineage mouse adapted ZIKV, pregnant dams were infected at E10 with Nigeria ZIKV and placentas and spleens were collected at either 3, 6, or 48 hpi to measure IL-1β in tissue homogenates (Figures 4A,B). In placentas from ZIKV infected dams, IL-1β was increased at 3 hpi to levels consistent with systemic IL-1β treatment (19), and returned to baseline levels that were equivalent to placentas from mock inoculated dams by 6 hpi (Figure 4A) and 48 hpi (data not shown). No significant change in IL-1β concentrations in the spleen was observed with ZIKV infection (Figure 4B), suggesting that the increased placental IL-1β does not originate from systemic circulation.
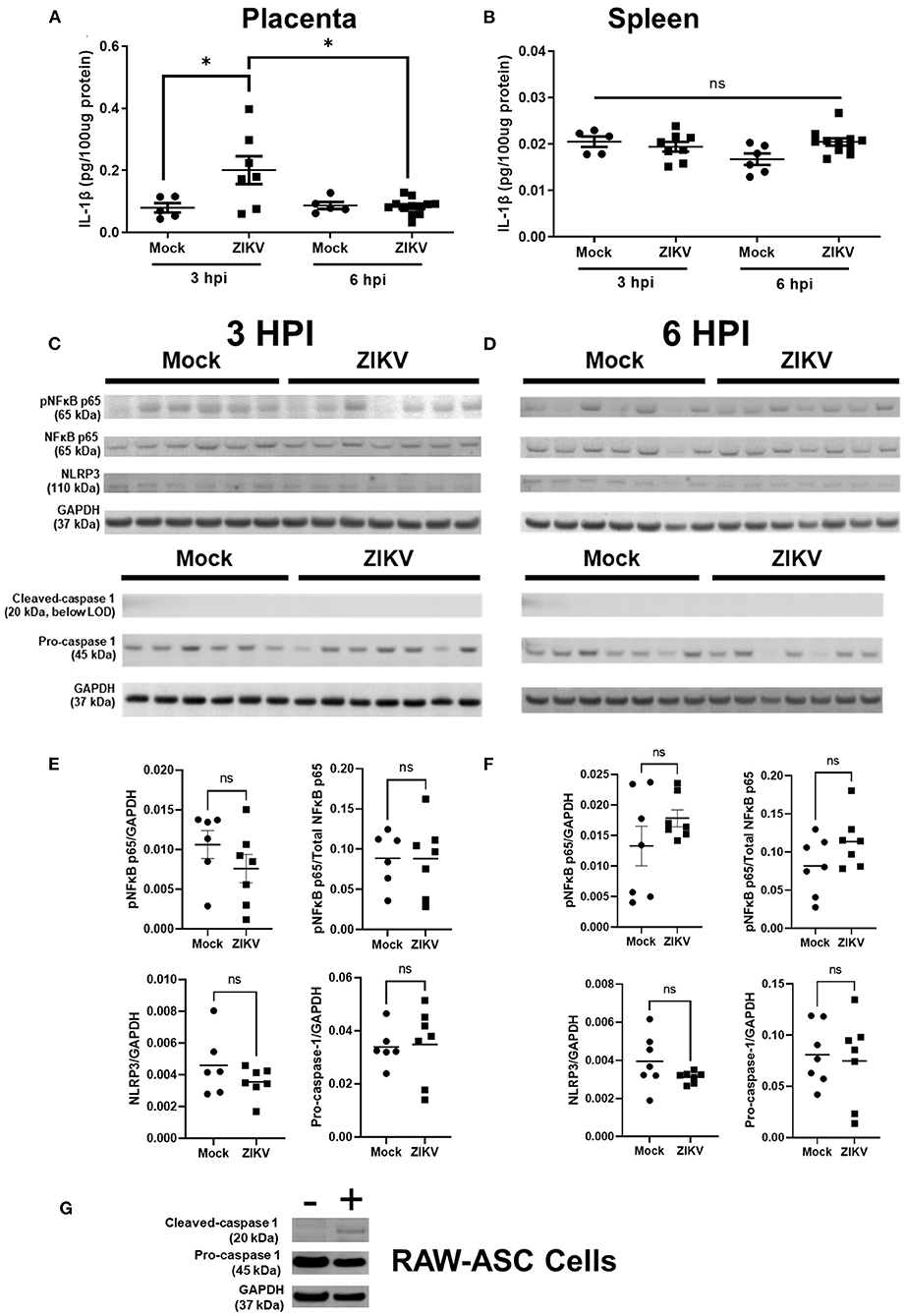
Figure 4. Intrauterine ZIKV infection transiently increases the IL-1β concentration but does not induce NLRP3 inflammasome upregulation or activation in placenta. At E10, pregnant mice underwent intrauterine injection of 106 TCID50 units of Nigeria ZIKV or media (mock). Placentas (A,C–F) and spleens (B) were collected at 3 hpi (C,E) or 6 hpi (D,F), homogenized, and analyzed by ELISA (A,B) or Western blot for phosphorylated (p)NFκB p65, total NFκB p65, and NLRP3 or cleaved-caspase 1 (20 kDa cleavage fragment) and pro-caspase 1. GAPDH is shown as the loading control. Placentas from two independent experiments were analyzed together to avoid gel to gel variability (C,D). Quantified protein expression (E,F) of pNFκB p65, NLRP3, and pro-caspase 1 in the placenta is shown as the fluorescence signal for each protein normalized to GAPDH. Phosphorylation of NFκB p65 is also shown as a proportion of total NFκB p65 per placenta. As a positive control for caspase-1 expression, RAW-ASC cells were primed and stimulated and cell lysate collected for Western blot analysis (G) Data represent mean ± standard error of the mean [n =5–12/group (ELISA), 6–7/group (WB)]. Significant differences were determined (*p < 0.05; ns, not significant) were determined by unpaired two tailed t-test (WB) or two-way ANOVA with Bonferroni post hoc test (ELISA).
IL-1β expression and release is canonically dependent upon the activation of inflammasomes, including the NLRP3 inflammasome (20), and both murine and human placental cells have the capacity to activate the NLRP3 inflammasome to release IL-1β (22, 23). Induction of NF-κB is a key component of the priming step of inflammasome activation (31). We hypothesized that the NLRP3 inflammasome would be activated by ZIKV infection of the placenta and might underlie elevated placental secretion of IL-1β. Using Nigeria ZIKV, pregnant dams were infected at E10 and euthanized at 3 hpi during peak placental IL-1β secretion and at 6 hpi, after IL-1β returned to baseline. Protein expression of components of the NLRP3 inflammasome cascade, including phosphorylated (at Ser536) and total-NF-κB p65, NLRP3 and caspase 1 (Figures 4C,D for blots, Figures 4E,F for quantification) were quantified in placental homogenates, relative to GAPDH (Figures 4E,F).
Protein expression of pNF-κB p65 in the placenta did not differ between ZIKV and mock infected dams at either timepoint (Figures 4C,E for 3 hpi and Figures 4D,F for 6 hpi). Placental NLRP3 expression in ZIKV compared to mock infected dams also did not differ at either timepoint (Figures 4C,E for 3 hpi and Figures 4D,F for 6 hpi). Together with the expression of pNF-κB p65, this is evidence that intrauterine ZIKV infection is not inducing NLRP3 inflammasome priming in the placenta at 3 or 6 hpi. Cleavage of pro-caspase 1 occurs upon assembly of inflammasomes, including the NLRP3 inflammasome, activating caspase 1 to process downstream products including IL-1beta, IL18, and Gasdermin D (32, 33). Measurement of cleaved caspase 1 is used as an indicator of inflammasome activation (19, 34, 35). Protein expression of the 20 kDa cleaved caspase 1 subunit was below the level of detection in placentas of both mock and ZIKV infected dams, while pro-caspase 1 was detectable and did not differ between ZIKV and mock infected dams (Figures 4C,E for 3 hpi and Figures 4D,F for 6 hpi). To ensure that the antibody utilized for measuring cleaved caspase-1 was functional, caspase-1 was measured in ASC expressing RAW 264.7 (RAW-ASC) cells, a mouse macrophage cell line capable of activating the NLRP3 inflammasome and illustrated robust expression (Figure 4G).
Taken together, these data indicate that the NLPR3 inflammasome is not activated in the placenta in response to intrauterine Nigeria ZIKV infection.
ZIKV Infection Downregulates Transcriptional Activity in the Placenta, Including Genes Associated With Cellular Function and Development
To transcriptionally evaluate the role of ZIKV infection and its contribution to the observed placental damage and disruption of substrate exchange in the placental labyrinth, we performed bulk mRNA sequencing of placentas from Nigeria ZIKV and mock inoculated dams collected 48 hpi. Utilizing standard cutoffs of a p-value ≤ 0.05 and fold change −2 < or >2, there were 928 differentially expressed genes between placentas of ZIKV and mock inoculated dams. Of these 928 genes, the majority (794) were downregulated in response to ZIKV infection (Figure 5A), revealing that ZIKV induces broad downregulation of host gene expression in the placenta during infection. To further characterize the effects of ZIKV infection on placental function, expression results of the 928 differentially expressed genes were imported into QIAGEN Ingenuity Pathway Analysis (IPA) to evaluate the interactions and associations of genes up or downregulated in the placenta during ZIKV infection (Figures 5B–F). The top five networks of diseases and functions containing genes differentially expressed in the placenta during ZIKV infection were “Cellular Development, Gene Expression, Tissue Morphology (Figure 5B)”, “Cell Signaling, Cell-To-Cell Signaling and Interaction, Nucleic Acid Metabolism (Figure 5C)”, “Cell-To-Cell Signaling and Interaction, Molecular Transport, Small Molecule Biochemistry (Figure 5D)”, “Endocrine System Disorders, Gastrointestinal Disease, Immunological Disease (Figure 5E)”, and “Digestive System Development and Function, Neurological Disease, Ophthalmic Disease (Figure 5F)”. Broadly, this network analysis further showcases the downregulation of host genes in the placenta by ZIKV infection (Figures 5B–F, green indicates significant downregulation). Genes associated with tissue morphology (Figure 5B) were downregulated by ZIKV infection between 2.056- and 1827.577-fold (fold change of specific genes indicated by shade) supporting the role of ZIKV in the morphological changes seen in the labyrinth of the placenta (Figures 2A, 3B). Genes associated with key cellular functions, including gene expression (Figure 5B), cellular signaling (Figures 5C,D), nucleic acid metabolism (Figure 5C), and molecular transport (Figure 5D) were downregulated between 2.004- and 947.01-fold by ZIKV infection, potentially causing cellular stress and the tissue damage observed histologically in placental slices (Figures 2, 3). Expression of genes associated with endocrine, gastrointestinal, immunological, neurological, and ophthalmic development and diseases were downregulated between 2.021- and 1348.931- fold by ZIKV infection (Figures 5E,F), consistent with evidence associating placental neurodevelopment gene expression with child neurobehavior (36, 37). Placental and fetal development is a tightly regulated process (16, 38), and downregulated gene expression changes induced in the placenta by ZIKV infection may have long-term consequences on the fetus.
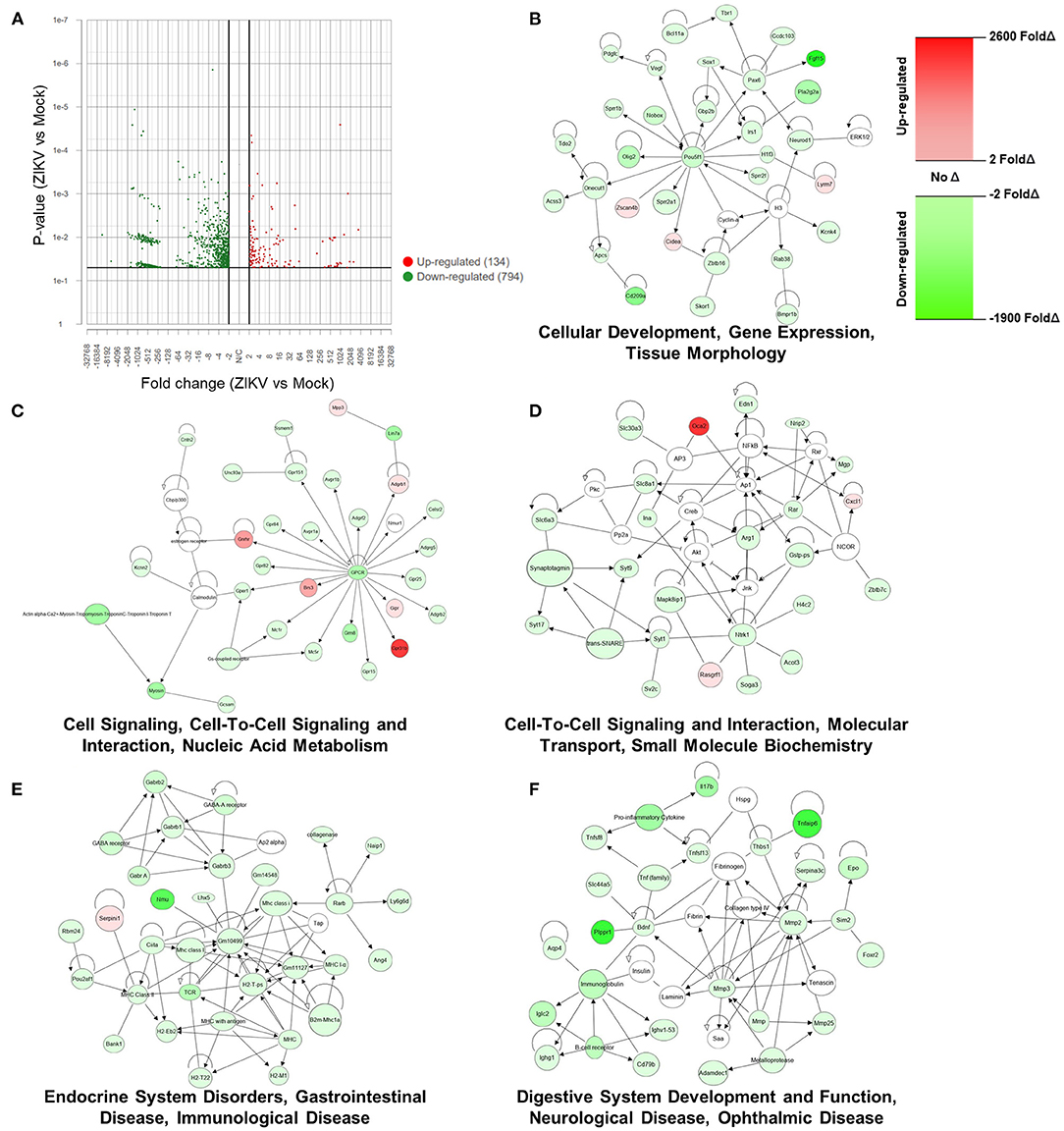
Figure 5. ZIKV infection downregulates host mRNA expression in the placenta, including genes associated with cellular function and development. At E10, pregnant mice underwent intrauterine injection of 106 TCID50 units of Nigeria ZIKV or media (mock). Dams were euthanized at 48 hpi, placentas collected, and RNA extracted. mRNA libraries for NGS were prepared with Tecan Genomics Universal Plus mRNA Seq kit and sequenced with a 200 cycle (2 × 100 bp) SP run on an Illumina NovaSeq 6,000 and analyzed in Partek® Flow® (A) and QIAGEN Ingenuity Pathway Analysis (B–F). A Volcano Plot showing the 928 genes that were differentially expressed between ZIKV and mock placentas using cut-offs of p-value ≤ 0.05 and fold change −2 < or >2. Red indicates upregulation and green indicates downregulation; individual dots indicate individual genes (A). The top five networks associated with the 928 differentially expressed genes, were identified using Ingenuity Pathway Analysis. Red indicates significant upregulation and green indicates downregulation with the shade indicating the magnitude of the fold change (B).
Discussion
Placental inflammation, dysfunction, and damage are major contributing factors to adverse perinatal outcomes during maternal infections (39–43). Intrauterine infection of dams with diverse ZIKVs cause productive infection of the placentas and fetuses, disrupt the placental architecture, and reduce fetal viability. Increased secretion of IL-1β, but not other proinflammatory cytokines, in the placenta causes adverse fetal outcomes in our model (18). In this study, we sought to further characterize the placental damage observed after intrauterine ZIKV infection, both transcriptionally as well as at cellular and tissue levels. Disruption of the maternal-fetal barrier, including cell death and significant reductions in both trophoblasts and endothelial cells within the labyrinth zone, were observed. ZIKV also induced dramatic downregulation of genes in the placenta, including those associated with cellular functions and tissue morphology, which may provide a potential transcriptional mechanism of the observed damage to both the placenta and fetal tissue.
Phylogenetic analyses indicate two major lineages of ZIKV, the Asian lineage and the African lineage, with viruses of the 2015–2017 epidemic clustering within the Asian lineage (44, 45). While lineage-specific mutations exist (45, 46), the contributions of these mutations to pathogenesis and development of CZS remains unresolved. Most studies indicate that African lineage strains are more virulent than Asian lineage strains in vivo (47–49). For example, African lineage viruses replicate to higher titers in fetal organs than do Asian lineage viruses in a porcine model (49). The molecular mechanisms that define these lineage-dependent differences remain unclear. In our model of intrauterine ZIKV infection of pregnant mice, both Asian (Brazil, 2015; Puerto Rico, 2015) and African (Nigeria, 1968) lineage viruses replicate in the placenta, induce placental damage, upregulate placental IL-1β secretion, and reduce fetal viability (18, 26). Additionally, both Asian and African strains vertically transmit, with ZIKV detectable in fetal tissues, including the brain and spinal cord, and cause cortical thinning, congenital malformations, and behavioral deficits in offspring (18, 26). Our data add to a growing body of literature suggesting that both Asian and African lineage viruses have the capacity to cause adverse perinatal outcomes.
We have shown previously that adverse perinatal outcomes, including disruption of the placental architecture, congenital malformations, and reduced fetal viability are caused by transient elevation of placental IL-1β, which is reversed by coadministration of an IL-1 receptor antagonist (18). Elevated IL-1β secretion occurs after intrauterine infection with either Asian [Brazil, 2015; (18)] or African (Nigeria, 1968) lineage viruses. Further, studies of intrauterine inflammation in the absence of replicating virus infection through intrauterine injection of lipopolysaccharide further reveal a role for IL-1β as a key mediator of perinatal brain injury and disrupted offspring neurodevelopment (19, 50–53). Consistent with these observations, treatment of dams with recombinant IL-1β at E14-E17 of pregnancy results distortion of the placental labyrinth structure, decreased numbers of mononuclear trophoblast giant cells, and reduced proportions of endothelial cells as compared to placentas from control dams, with fetal brains exhibiting evidence of reduced cortical neuronal morphology (19). Collectively, these data suggest that IL-1β is a significant mediator of placental dysfunction and adverse perinatal outcomes. The molecular mechanism of increased placental IL-1β secretion, however, has not been reported. Based on results from the current study, the NLRP3 inflammasome was not activated in response to ZIKV infection in the placenta. Evidence exists, however, for IL-1β activation and secretion via non-canonical pathways (54–57), which should be considered in future studies.
During gestation, the placenta is the site of nutrient, oxygen, and waste exchange between the mother and fetus. In mice, this occurs specifically in the placental labyrinth, a structure where maternal and fetal blood are separated by three layers of trophoblasts and one layer of endothelial cells (16, 24, 25, 58). Evidence from diverse models indicates that deficits in maternal-fetal substrate exchange cause adverse fetal outcomes, including intrauterine growth restrictions and abortion (38, 59–62). We and others have previously observed placental damage after ZIKV infection (18, 26, 30), a finding also seen in human term placentas (63). In the current study, we have further defined this damage as a significant loss of trophoblasts and endothelial cells within the maternal-fetal barrier, and identified transcriptional changes associated with the observed placental damage. Others have focused on transcriptionally profiling cells infected with ZIKV in vitro [reviewed in (64)] or have used human placental tissue at-term, well after ZIKV infection has been controlled (65, 66). In contrast, we utilized placentas collected during peak ZIKV replication (26) and found robust downregulation of genes, including those involved in cellular function and tissue modeling. As placental and fetal (including fetal neurodevelopment) is a highly regulated process (16, 24, 25, 38), these transcriptional changes may have long-lasting effects on the offspring, in addition to underpinning the observed placental damage.
Infection with viruses, including influenza A viruses, Cytomegalovirus, and Hepatitis E virus, during pregnancy also result in maternal inflammation, transplacental and fetal infections, or both (1, 2, 13). There is a growing need to elucidate the role of virus replication vs. maternal inflammation as mediators of adverse perinatal outcomes from infections, including SARS-CoV2. Several studies illustrate SARS-CoV2 infection during pregnancy contributes to negative maternal and fetal outcomes, such as preterm birth, although presence of vertical transmission is debated (67–69). Animal models of viral infections during pregnancy will be integral for identifying the molecular mechanisms of adverse pregnancy outcomes.
Data Availability Statement
mRNAseq data have been deposited at NCBI Sequence Read Archive, NCBI BioProject accession number PRJNA797437. Other data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Johns Hopkins Animal Care and Use Committee.
Author Contributions
SK, IB, PC, JL, MS, AP, HN, and ACh conceptualized and designed the experiments. PC, JL, MS, HN, ACh, and IB performed animal experiments. PC, MS, HN, and AP grew and quantified viruses. PC, JL, ACh, AD, AJ, and ACa performed assays. PC, MS, AD, and ACa statistically analyzed and graphed data. PC and SK wrote the manuscript with input from all authors. All authors read and provided edits to drafts and approved the final submission.
Funding
Funding provided by a grant from the NIH/NICHD (R01HD097608; SK and IB), training grants from the NIH/NIAID (2T32AI007417-26; PC) as well as the Vivien Thomas Scholars Initiative at the Johns Hopkins University (ACa).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the expert animal care staff at Johns Hopkins for assistance with maintenance of ZIKV-infected dams in our facilities.
References
1. Vermillion MS, Klein SL. Pregnancy and infection: using disease pathogenesis to inform vaccine strategy. NPJ Vaccines. (2018) 3:6. doi: 10.1038/s41541-017-0042-4
2. Megli CJ, Coyne CB. Infections at the maternal-fetal interface: an overview of pathogenesis and defence. Nat Rev Microbiol. (2021) 20:67–82. doi: 10.1038/s41579-021-00610-y
3. Kourtis AP, Read JS, Jamieson DJ. Pregnancy and infection. N Engl J Med. (2014) 371:1077. doi: 10.1056/NEJMra1213566
4. Racicot K, Mor G. Risks associated with viral infections during pregnancy. J Clin Invest. (2017) 127:1591–9. doi: 10.1172/JCI87490
5. Abu-Raya B, Michalski C, Sadarangani M, Lavoie PM. Maternal immunological adaptation during normal pregnancy. Front Immunol. (2020) 11:575197. doi: 10.3389/fimmu.2020.575197
6. Arora HS. A to Z of zika virus: a comprehensive review for clinicians. Glob Pediatr Health. (2020) 7:2333794X20919595. doi: 10.1177/2333794X20919595
7. Maslow JN, Roberts CC. Zika virus: a brief history and review of its pathogenesis rediscovered. Methods Mol Biol. (2020) 2142:1–8. doi: 10.1007/978-1-0716-0581-3_1
8. Schuler-Faccini L, Ribeiro EM, Feitosa IM, Horovitz DD, Cavalcanti DP, Pessoa A, et al. Possible association between zika virus infection and microcephaly-Brazil, 2015. Morb Mortal Wkly Rep. (2016) 65:59–62. doi: 10.15585/mmwr.mm6503e2
9. Coelho AVC, Crovella S. Microcephaly prevalence in infants born to zika virus-infected women: a systematic review and meta-analysis. Int J Mol Sci. (2017) 18:1714. doi: 10.3390/ijms18081714
10. Nithiyanantham SF, Badawi A. Maternal infection with Zika virus and prevalence of congenital disorders in infants: systematic review and meta-analysis. Can J Public Health. (2019) 110:638–48. doi: 10.17269/s41997-019-00215-2
11. Gallo LG, Martinez-Cajas J, Peixoto HM, Pereira A, Carter JE, Mckeown S, et al. Another piece of the Zika puzzle: assessing the associated factors to microcephaly in a systematic review and meta-analysis. BMC Public Health. (2020) 20:827. doi: 10.1186/s12889-020-08946-5
12. Martins MM, Alves Da Cunha AJL, Robaina JR, Raymundo CE, Barbosa AP, Medronho RA. Fetal, neonatal, and infant outcomes associated with maternal Zika virus infection during pregnancy: a systematic review and meta-analysis. PLoS ONE. (2021) 16:e0246643. doi: 10.1371/journal.pone.0246643
13. Coyne CB, Lazear HM. Zika virus-reigniting the TORCH. Nat Rev Microbiol. (2016) 14:707–15. doi: 10.1038/nrmicro.2016.125
14. Narasimhan H, Chudnovets A, Burd I, Pekosz A, Klein SL. Animal models of congenital zika syndrome provide mechanistic insight into viral pathogenesis during pregnancy. PLoS Negl Trop Dis. (2020) 14:e0008707. doi: 10.1371/journal.pntd.0008707
15. Empey KM. Peebles, RS Jr, Janssen WJ. Mouse models of viral infection methods. Mol Biol. (2018) 1809:395–414. doi: 10.1007/978-1-4939-8570-8_26
16. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol. (2019) 4:eaat6114. doi: 10.1126/sciimmunol.aat6114
17. Szaba FM, Tighe M, Kummer LW, Lanzer KG, Ward JM, Lanthier P, et al. Zika virus infection in immunocompetent pregnant mice causes fetal damage and placental pathology in the absence of fetal infection. PLoS Pathog. (2018) 14:e1006994. doi: 10.1371/journal.ppat.1006994
18. Lei J, Vermillion MS, Jia B, Xie H, Xie L, Mclane MW, et al. (2019) IL-1 receptor antagonist therapy mitigates placental dysfunction and perinatal injury following Zika virus infection. JCI Insight 4. doi: 10.1172/jci.insight.122678
19. Chudnovets A, Lei J, Na Q, Dong J, Narasimhan H, Klein SL, et al. Dose-dependent structural and immunological changes in the placenta and fetal brain in response to systemic inflammation during pregnancy. Am J Reprod Immunol. (2020) 84:e13248. doi: 10.1111/aji.13248
20. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
21. C Weel I, Romao-Veiga M, Matias ML, Fioratti EG, Peracoli JC, et al. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J Reprod Immunol. (2017) 123:40–47. doi: 10.1016/j.jri.2017.09.002
22. Sano M, Shimazaki S, Kaneko Y, Karasawa T, Takahashi M, Ohkuchi A, et al. Palmitic acid activates NLRP3 inflammasome and induces placental inflammation during pregnancy in mice. J Reprod Dev. (2020) 66:241–8. doi: 10.1262/jrd.2020-007
23. Megli C, Morosky S, Rajasundaram D, Coyne CB. Inflammasome signaling in human placental trophoblasts regulates immune defense against Listeria monocytogenes infection. J Exp Med. (2021) 218. doi: 10.1084/jem.20200649
24. Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. (2010) 120:1016–25. doi: 10.1172/JCI41211
25. Bronson SL, Bale TL. The Placenta as a mediator of stress effects on neurodevelopmental reprogramming. Neuropsychopharmacology. (2016) 41:207–18. doi: 10.1038/npp.2015.231
26. Vermillion MS, Lei J, Shabi Y, Baxter VK, Crilly NP, Mclane M, et al. Intrauterine Zika virus infection of pregnant immunocompetent mice models transplacental transmission and adverse perinatal outcomes. Nat Commun. (2017) 8:14575. doi: 10.1038/ncomms14575
27. Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis. (2008) 14:1232–9. doi: 10.3201/eid1408.080287
28. Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger-Lucarelli J, Uccellini MB, et al. An immunocompetent mouse model of zika virus infection. Cell Host Microbe. (2018) 23:672–685.e676. doi: 10.1016/j.chom.2018.04.003
29. Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, et al. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis. (2012) 6:e1477. doi: 10.1371/journal.pntd.0001477
30. Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, et al. Zika Virus Infection during pregnancy in mice causes placental damage and fetal demise. Cell. (2016) 165:1081–91. doi: 10.1016/j.cell.2016.05.008
31. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. (2019) 20: 3328. doi: 10.3390/ijms20133328
32. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
33. Zhao C, Zhao W. NLRP3 inflammasome-a key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
34. Gross O. Measuring the inflammasome. Methods Mol Biol. (2012) 844:199–222. doi: 10.1007/978-1-61779-527-5_15
35. Zheng Y, Liu Q, Wu Y, Ma L, Zhang Z, Liu T, et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. (2018) 37:e99347. doi: 10.15252/embj.201899347
36. Green BB, Kappil M, Lambertini L, Armstrong DA, Guerin DJ, Sharp AJ, et al. Expression of imprinted genes in placenta is associated with infant neurobehavioral development. Epigenetics. (2015) 10:834–41. doi: 10.1080/15592294.2015.1073880
37. Lester BM, Marsit CJ. Epigenetic mechanisms in the placenta related to infant neurodevelopment. Epigenomics. (2018) 10:321–33. doi: 10.2217/epi-2016-0171
38. Woods L, Perez-Garcia V, Hemberger M. Regulation of placental development and its impact on fetal growth-new insights from mouse models. Front Endocrinol. (2018) 9:570. doi: 10.3389/fendo.2018.00570
39. Redline RW. Placental inflammation. Semin Neonatol. (2004) 9:265–74. doi: 10.1016/j.siny.2003.09.005
40. Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum Reprod Update. (2006) 12:747–55. doi: 10.1093/humupd/dml016
41. Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, et al. Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol. (2018) 3:eaao1680. doi: 10.1126/sciimmunol.aao1680
42. Goldstein JA, Gallagher K, Beck C, Kumar R, Gernand AD. Maternal-fetal inflammation in the placenta and the developmental origins of health and disease. Front Immunol. (2020) 11:531543. doi: 10.3389/fimmu.2020.531543
43. Matoba N, Mestan KK, Collins JW Jr. Understanding Racial Disparities of Preterm Birth Through the Placenta. Clin Ther. (2021) 43:287–96. doi: 10.1016/j.clinthera.2020.12.013
44. Ye Q, Liu ZY, Han JF, Jiang T, Li XF, Qin CF. Genomic characterization and phylogenetic analysis of Zika virus circulating in the Americas. Infect Genet Evol. (2016) 43:43–9. doi: 10.1016/j.meegid.2016.05.004
45. Beaver JT, Lelutiu N, Habib R, Skountzou I. Evolution of two major zika virus lineages: implications for pathology, immune response, vaccine development. Front Immunol. (2018) 9:1640. doi: 10.3389/fimmu.2018.01640
46. Gong Z, Xu X, Han GZ. The Diversification of Zika Virus: Are There Two Distinct Lineages? Genome Biol Evol. (2017) 9:2940–5. doi: 10.1093/gbe/evx223
47. Tripathi S, Balasubramaniam VR, Brown JA, Mena I, Grant A, Bardina SV, et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. (2017) 13:e1006258. doi: 10.1371/journal.ppat.1006258
48. Smith DR, Sprague TR, Hollidge BS, Valdez SM, Padilla SL, Bellanca SA, et al. African and Asian zika virus isolates display phenotypic differences both in vitro and in vivo. Am J Trop Med Hyg. (2018) 98:432–44. doi: 10.4269/ajtmh.17-0685
49. Udenze D, Trus I, Berube N, Gerdts V, Karniychuk U. The African strain of Zika virus causes more severe in utero infection than Asian strain in a porcine fetal transmission model. Emerg Microbes Infect. (2019) 8:1098–107. doi: 10.1080/22221751.2019.1644967
50. Fan LW, Tien LT, Zheng B, Pang Y, Rhodes PG, Cai Z. Interleukin-1beta-induced brain injury and neurobehavioral dysfunctions in juvenile rats can be attenuated by alpha-phenyl-n-tert-butyl-nitrone. Neuroscience. (2010) 168:240–52. doi: 10.1016/j.neuroscience.2010.03.024
51. Leitner K, Al Shammary M, Mclane M, Johnston MV, Elovitz MA, Burd I. IL-1 receptor blockade prevents fetal cortical brain injury but not preterm birth in a mouse model of inflammation-induced preterm birth and perinatal brain injury. Am J Reprod Immunol. (2014) 71:418–26. doi: 10.1111/aji.12216
52. Rosenzweig JM, Lei J, Burd I. Interleukin-1 receptor blockade in perinatal brain injury. Front Pediatr. (2014) 2:108. doi: 10.3389/fped.2014.00108
53. Na Q, Chudnovets A, Liu J, Lee JY, Dong J, Shin N, et al. Placental Macrophages Demonstrate Sex-Specific Response to Intrauterine Inflammation and May Serve as a Marker of Perinatal Neuroinflammation. J Reprod Immunol. (2021) 147:103360. doi: 10.1016/j.jri.2021.103360
54. Black RA, Kronheim SR, Cantrell M, Deeley MC, March CJ, Prickett KS, et al. Generation of biologically active interleukin-1 beta by proteolytic cleavage of the inactive precursor. J Biol Chem. (1988) 263:9437–42. doi: 10.1016/S0021-9258(19)76559-4
55. Hazuda DJ, Strickler J, Kueppers F, Simon PL, Young PR. Processing of precursor interleukin 1 beta and inflammatory disease. J Biol Chem. (1990) 265:6318–22. doi: 10.1016/S0021-9258(19)39328-7
56. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
57. Pyrillou K, Burzynski LC, Clarke MCH. Alternative pathways of IL-1 activation, and its role in health and disease. Front Immunol. (2020) 11:613170. doi: 10.3389/fimmu.2020.613170
58. Fowden AL, Sibley CP. Placental phenotype and fetal growth. J Physiol. (2009) 587:3429. doi: 10.1113/jphysiol.2009.175968
59. Ware CB, Horowitz MC, Renshaw BR, Hunt JS, Liggitt D, Koblar SA, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. (1995) 121:1283–99. doi: 10.1242/dev.121.5.1283
60. Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci U S A. (1998) 95:5082–7. doi: 10.1073/pnas.95.9.5082
61. Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, et al. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol. (2003) 23:5043–55. doi: 10.1128/MCB.23.14.5043-5055.2003
62. Ueno M, Lee LK, Chhabra A, Kim YJ, Sasidharan R, Van Handel B, et al. c-Met-dependent multipotent labyrinth trophoblast progenitors establish placental exchange interface. Dev Cell. (2013) 27:373–86. doi: 10.1016/j.devcel.2013.10.019
63. Rabelo K, De Souza LJ, Salomao NG, Machado LN, Pereira PG, Portari EA, et al. Zika Induces Human Placental Damage and Inflammation. Front Immunol. (2020) 11:2146. doi: 10.3389/fimmu.2020.02146
64. Gratton R, Tricarico PM, Agrelli A, Colaco Da Silva HV, Coelho Bernardo L, Crovella S, et al. In vitro zika virus infection of human neural progenitor cells: meta-analysis of RNA-Seq assays. Microorganisms. (2020) 8:270. doi: 10.3390/microorganisms8020270
65. Lum FM, Narang V, Hue S, Chen J, Mcgovern N, Rajarethinam R, et al. Immunological observations and transcriptomic analysis of trimester-specific full-term placentas from three Zika virus-infected women. Clin Transl Immunology. (2019) 8:e01082. doi: 10.1002/cti2.1082
66. Amaral MS, Goulart E, Caires-Junior LC, Morales-Vicente DA, Soares-Schanoski A, Gomes RP, et al. Differential gene expression elicited by ZIKV infection in trophoblasts from congenital Zika syndrome discordant twins. PLoS Negl Trop Dis. (2020) 14:e0008424. doi: 10.1371/journal.pntd.0008424
67. Carrasco I, Munoz-Chapuli M, Vigil-Vazquez S, Aguilera-Alonso D, Hernandez C, Sanchez-Sanchez C, et al. SARS-COV-2 infection in pregnant women and newborns in a Spanish cohort (GESNEO-COVID) during the first wave. BMC Pregnancy Childbirth. (2021) 21:326. doi: 10.1186/s12884-021-03784-8
68. Norman M, Naver L, Soderling J, Ahlberg M, Hervius Askling H, Aronsson B, et al. Association of maternal SARS-CoV-2 infection in pregnancy with neonatal outcomes. JAMA. (2021) 325:2076–86. doi: 10.1001/jama.2021.5775
Keywords: flavivirus, inflammation, IL-1β, neurological development, trophoblasts
Citation: Creisher PS, Lei J, Sherer ML, Dziedzic A, Jedlicka AE, Narasimhan H, Chudnovets A, Campbell AD, Liu A, Pekosz A, Burd I and Klein SL (2022) Downregulation of Transcriptional Activity, Increased Inflammation, and Damage in the Placenta Following in utero Zika Virus Infection Is Associated With Adverse Pregnancy Outcomes. Front. Virol. 2:782906. doi: 10.3389/fviro.2022.782906
Received: 24 September 2021; Accepted: 24 January 2022;
Published: 22 February 2022.
Edited by:
Vikki M. Abrahams, Yale University, United StatesReviewed by:
Nardhy Gomez-Lopez, Wayne State University, United StatesSeth Guller, Yale University, United States
Copyright © 2022 Creisher, Lei, Sherer, Dziedzic, Jedlicka, Narasimhan, Chudnovets, Campbell, Liu, Pekosz, Burd and Klein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina Burd, iburd@jhmi.edu; Sabra L. Klein, sklein2@jhu.edu