Aurintricarboxylic Acid Suppresses Hepatitis B Virus Replication by Inhibition of RNase H Activity
 Meng Xu
Meng Xu Xiao Guan
Xiao Guan- 1Department of Pathology, The First Affiliated Hospital of China Medical University, Shenyang, China
- 2Division of Research and Development, Zhenjiang Meiruo Biotechnology Co., Ltd., National University Science and Technology Park, Zhenjiang, China
Background: Chronic hepatitis B virus (HBV) infection is a global health threat for which there is an urgent need to develop novel therapeutics.
Methods: Aurintricarboxylic acid (ATA) has been demonstrated with broad-spectrum antimicrobial and antiviral activities. In this study, we implemented ATA treatment in HBV-infected and HBV-transfected hepatocytes to uncover whether ATA inhibits HBV replication and its underlying mechanism.
Results: HBV DNA levels were significantly reduced, while viral proteins or transcripts were not altered. In contrast, ATA treatment did not further deteriorate HBV DNAs, viral proteins, and transcripts in hepatocytes transfected by an HBV RNase H dead mutant. Moreover, ATA showed an inhibitory effect on DNA synthesis in hepatocytes transfected with lamivudine-resistant HBV mutants.
Conclusions: ATA is a potent inhibitor of HBV replication by disrupting the RNase H activity of the viral polymerase.
Introduction
Being a high-risk factor for liver cirrhosis and hepatocellular carcinoma, hepatitis B virus (HBV) infection is a global public health issue affecting hundreds of millions of people (1). With a diameter of ~42 nm, HBV per se is an icosahedral nanosized particle, where the virion envelop contains HBV surface antigen (HBsAg), and the nucleocapsid encompasses HBV e-antigen (HBeAg) and core antigen (HBcAg) in complexes with viral polymerase (Pol) and genome (Figures 1A, B) (2). The HBV genome owns a partially double-stranded relaxed circular DNA (rcDNA) of ~3.2 kilobase (kb) pairs, one incomplete (+)-strand and the other complete (−)-strand in a covalent bond with Pol at the 5′-end (3).
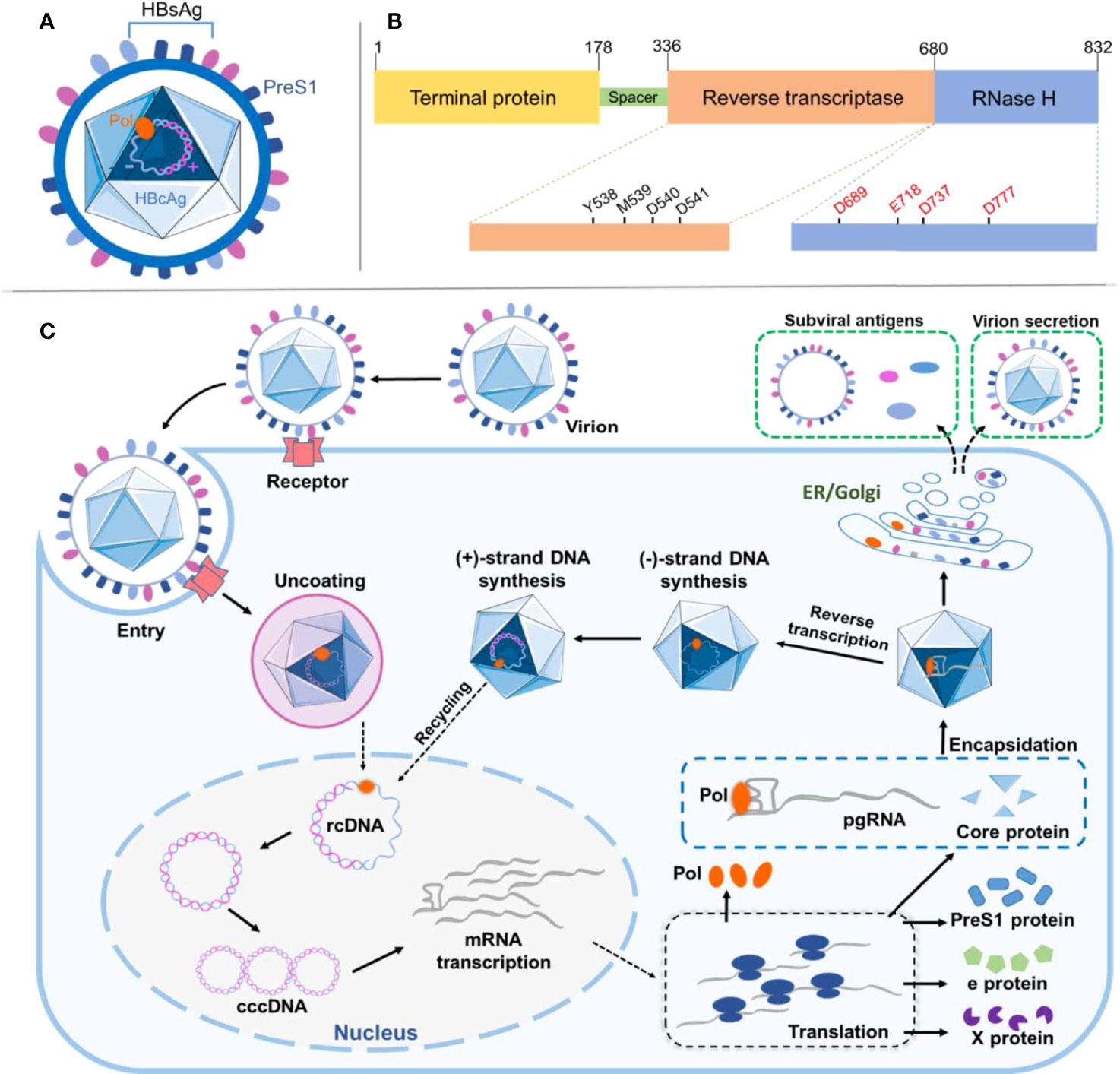
Figure 1 Viral structure and protein domain of HBV and its life cycle in the host. (A) Illustration of HBV particle with structural features as indicated and discussed in the text. (B) Depiction of HBV multifunctional domains where critical residues and motifs are indicated. (C) Schematic illustration showing HBV infection and replication in the host cell. All steps are as indicated and discussed in the text. HBV, hepatitis B virus.
Upon infection, the specific binding between HBsAg pre-S1 in the HBV and sodium (Na+) taurocholate cotransporting polypeptide (NTCP) on the basolateral membrane of hepatocytes facilitates the receptor-mediated viral entry into the host, although other entry routes may also exist (Figure 1C) (4). The endocytotic virus is unenveloped and dissembled, followed by transportation of rcDNA into the nucleus and its complete ligation to form covalently closed circular DNA (cccDNA) (5). The cccDNA is further transcribed and processed to produce the pregenomic (pg) and precore viral RNAs for later translation of various viral proteins in the cytoplasm, including HBsAg, Pol, x proteins, and HBcAg (6). The core protein (HBc Ag) encapsidates pgRNA to form a nucleocapsid, where the Pol protein reverse-transcribes pgRNA to produce (−)-strand DNA. The Pol protein digests the pgRNA in the RNA·DNA hybrid through its RNase-H activity and synthesizes (+)-strand DNA to generate rcDNA in the nucleocapsid. The nucleocapsid is secreted as a virion after acquiring surface proteins (7). Interestingly, a point mutation (D737V) of Pol could abrogate its RNase H activity resulting in RNA·DNA hybrid in nucleocapsids (8). The vaccination against HBsAg has long proven valid in protecting vaccinees from HBV infection; however, once infected, due to HBV first forming covalently closed circular DNA (cccDNA) in the hepatocyte nucleus, a viral center molecule essential for HBV infection and persistence transcribes viral RNAs, including a replicative RNA intermediate called pgRNA, which encodes two viral proteins, HBV core (HBc) and HBV Pol. In general, cccDNA can persist throughout the life span of the quiescent hepatocytes without affecting their viability; therefore, viral elimination is ineffective. Currently, for chronic hepatitis B treatment, antiviral medications comprise of interferons aiming for serological clearance of HBsAg and nucleos(t)ides for inhibition of HBV Pol, showing limited success but developing substantial side effect and drug resistance (9). For example, lamivudine (also known as 3TC) is a cytidine dideoxynucleoside derivative and a potent inhibitor of reverse transcriptase in HBV replication. However, lamivudine treatment needs to be orally administered for a long period; otherwise, relapse occurs, causing genotype mutation and susceptibility loss (10).
Aurintricarboxylic acid (ATA) and its analogs have been shown to possess a broad spectrum of antiviral activity against a diversity of RNA/DNA viruses, including HIV (11), severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) (12), and Zika virus (13). Lately, it has also been proposed as a potential inhibitor for SARS-CoV-2 infection (14). In solutions, ATA self-assembles to form a group of polymers with heterogeneous sizes, and their antiviral activity varies in proportion to the degree of polymerization and the ensuing molecular weight (15, 16). The mechanism regarding antiviral actions of ATA may be twofold: one is to thwart the binding between viral particles and the host cell surface (11, 17) and the other is to interfere with protein–nucleic acid interaction in the viral replication (16, 18). However, no research on the potential inhibition of HBV by ATA has been yet reported. Herein, through this study, we investigate the antiviral activity of ATA against HBV and the underlying mechanism of its action by employing lamivudine-resistant HBV mutants.
Materials and Methods
Compounds
ATA was purchased from Sigma-Aldrich (St. Louis, MO, USA). ATA was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) as a 100-mM stock solution and stored at −20°C.
Cell Culture and Transfection
HepaRG cells purchased from Thermo Fisher (Waltham, MA, USA; catalog no. HPRGC10) were cultured in William’s medium E (Invitrogen, Carlsbad, CA, USA) supplemented with 2 mM of L-glutamine, 200 units/ml of penicillin, 200 μg/ml of streptomycin, 10% fetal bovine serum (FBS), 5 μg/ml of insulin, 20 ng/ml of epidermal growth factor, 50 μM of hydrocortisone (Sigma-Aldrich), and 2% DMSO. Huh-7 or HepG2.2.15 cells purchased from ATCC Cell Bank (Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle medium with 10 mM of HEPES, 200 units/ml of penicillin, 200 μg/ml of streptomycin, and 10% FBS. Plasmids were transfected into Huh-7 or HepaRG cells with Fugene 6 (Roche, Shanghai, China) or Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Hepatitis B Virus Infection
A 50-fold concentrated culture supernatant of HepG2.2.15 cells was used to prepare an infectious inoculum. The inoculum was prepared from freshly collected supernatants by precipitating viral particles in the presence of 6% polyethylene glycol 8000 (PEG8k). The virion pellets were resuspended in phosphate-buffered saline containing 25% FBS. Differentiated HepaRG cells (with 1 × 107 genome equivalent copies of HBV and 1 × 105 HepaRG cells were inoculated) were incubated with the concentrated infectious inoculum as reported for primary human hepatocytes and HepaRG cells (19). The secreted HBeAg, HBV DNA, and RNA were determined on the indicated days after infection.
Native Agarose Gel Electrophoresis Assay
Native agarose gel electrophoresis (NAGE) assays were performed as described previously (20). In brief, intact nucleocapsid particles were separated from crude extracts of HBV-replicating cells using agarose gel electrophoresis. Nucleocapsid particles within the gel were then denatured under alkaline conditions and transferred onto nitrocellulose membranes (Roche). Nucleocapsid DNA and core proteins were detected using Southern and Western blotting with a double-stranded HBV DNA probe spanning the whole viral genome and an anti-core antibody, respectively.
Expression Vectors
The HBV replicon or RNase H-defective replicon plasmid was described previously (8). Probe labeling and the development of Southern blotting signals were performed using an AlkPhos direct labeling system (Amersham, Little Chalfont, UK).
Quantitative PCR and Reverse Transcription PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and treated twice with RNase-free DNase I (20, 21). Total RNA (1 μg) was used for reverse transcription, and the viral transcripts were quantified as described previously (20, 21). The results were normalized against the amplification results for internal control [i.e., glyceraldehyde 3-phosphate dehydrogenase (GAPDH)]. The primers used to detect HBV RNA were forward 5′-CGGAAATATACATCGTTTCCAT-3′ and reverse 5′-AAGAGTCCTCTTATGTAAGACCTT-3′, and the GAPDH primers were forward 5′-TGCACCACCAACTGCTTAGC-3′ and reverse 5′-GGCATGGACTGTGGTCATGAG-3′. RcDNA purification from either secreted virion or cytoplasm nucleocapsids was performed as previously described (22). Total RNA was extracted from cells using TRIzol according to the manufacturer’s instructions. Purified RNA (1 μg) was treated with DNase I (Invitrogen), immediately primed with oligo(dT) or random primers, and reverse transcribed using Superscript III reverse transcriptase (Invitrogen). Real-time PCR was performed using the ΔΔCt method.
Enzyme-Linked Immunosorbent Assay
HBeAg ELISA was performed as previously described (23).
Statistical Analysis
Statistical analysis was performed using Prism 6.0 (GraphPad software). Unpaired, two-tailed Student’s t-test was used for statistical comparison between groups, unless otherwise specifically mentioned.
Results
ATA Inhibits Hepatitis B Virus Replication
To examine the possible inhibition of HBV replication by ATA, we first used HepaRG cells for HBV infection since these cells express high levels of HBV receptor NTCP (4), and we treated HBV-infected HepaRG cells with different concentrations of ATA and so measured the HBeAg content. In this experiment, HepaRG cells were infected with HBV virions for 14 days in the absence or the presence of ATA at 5 (low) or 10 (high) μM, and viral loads (secreted HBeAg) in the supernatant were measured by HBe ELISA at different time points. As a result (Figure 2A), ATA inhibited HBV replication in the HepaRG cells in a dose-dependent manner. ATA continuously suppressed the HBeAg content, and on Day 14, 5 and 10 μg/ml of ATA reduced the HBeAg level to 63.4% and 27.2% of that with no treatment, respectively. Therefore, ATA had an inhibitory effect on HBV replication in HepaRG cells.
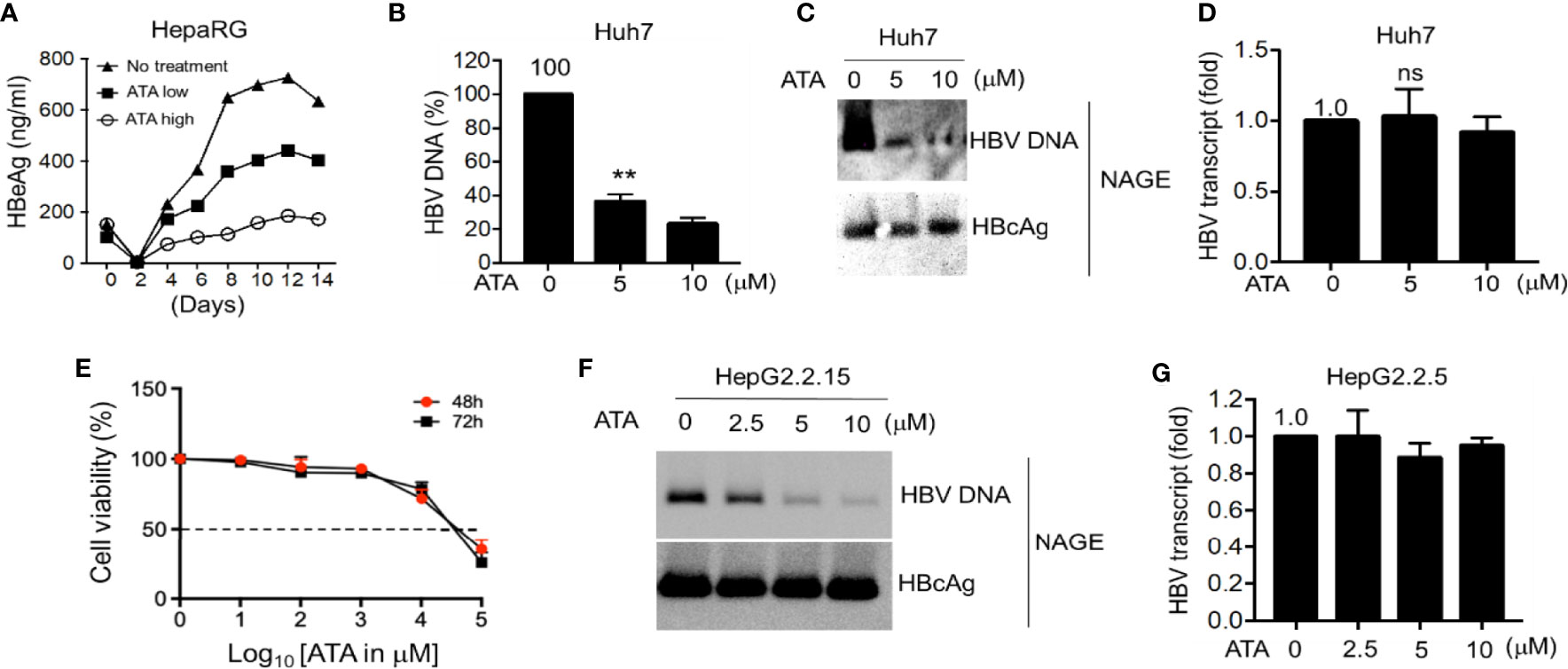
Figure 2 ATA inhibits HBV replication in hepatocytes. (A) Differentiated HepaRG cells were infected with HBV virions for 14 days in the presence or absence of ATA at 5 (low) or 10 (high) μM, and viral production was measured by HBe ELISA at the time points as indicated. (B) Huh-7 cells were transfected with an HBV replicon plasmid (HBV1.5). At 24 h after transfection, cells were treated with or without ATA for 48 h, and HBV DNA was extracted from secreted HBV virions in the culture medium and measured by qPCR using HBV-specific primers. (C) HBV nucleocapsids in cell lysates in (B) were estimated using the NAGE assay followed by Southern or Western blotting with HBV DNA probes or anti-HBc antibody (NAGE assay). (D) Total RNA in (B) was extracted, and RT-qPCR was performed to measure HBV transcripts. (E) Huh-7 cells (96-well plate format, 5 × 104 cells/well, triplicates) were treated with the indicated doses. Cell proliferation assays were performed at 48 and 72 h post-treatment. Dotted line indicates the 50% toxicity. Data are expressed as mean ± SD from three triplicates. (F) HBV nucleocapsids in HepG2.2.15 cells were estimated using the NAGE assay followed by Southern or Western blotting with HBV DNA probes or anti-HBc antibody. (G) Total RNA in (F) was extracted, and RT-qPCR was performed to measure HBV transcripts against GAPDH. **p < 0.01; ns, not significant (two-tailed, unpaired Student’s t-test). Data are mean ± SEM of three independent experiments. All Western blotting data are representative of three independent experiments. ATA, aurintricarboxylic acid; HBV, hepatitis B virus; NAGE, native agarose gel electrophoresis.
We next explored ATA-mediated antiviral activity in HBV-transfected liver cells. Hepatocyte-derived cellular carcinoma Huh-7 cells were transfected with HBV replicon vectors for 24 h and further treated with or without ATA for 48 h before viral DNA in the supernatant containing the secreted virions was determined using qPCR (Figure 2B). The results showed that the treatment of ATA at 5 and 10 μM sustained the HBV DNA levels at 36.5% ± 4.2% and 23.4% ± 3.5% of that with no treatment, respectively, indicating a significant reduction of HBV production in ATA-treated hepatocytes (both p < 0.01). Moreover, in the cell lysates of different incubations as mentioned above, HBV nucleocapsids were estimated using NAGE assay, and total RNA was extracted to measure HBV transcripts using RT-qPCR. Levels of nucleocapsid-associated DNA (ncDNA) and cytoplasmic nucleocapsid core protein (i.e., HBcAg) were determined using HBV DNA probes and HBcAg antibody, respectively. In NAGE assays, the purified HBV particles were directly loaded onto a gel during electrophoresis, and thus ncDNA and HBV core (HBc) protein could be assessed using Southern and Western blotting, respectively, by HBV probes and an anti-HBV core protein antibody. As shown in Figure 2C, ATA treatments significantly lessened HBV DNA levels, whereas the HBcAg levels with or without ATA treatment were indistinguishable. This result revealed that the ATA had an inhibitory effect on viral DNAs but not on viral transcripts. This finding was confirmed when measuring HBV transcript levels using RT-qPCR. As shown in Figure 2D, HBV transcript from treatment with 5 or 10 μM of ATA displayed (1.03 ± 0.20)-fold or (0.92 ± 0.11)-fold of that with no treatment, showing no noticeable difference (p > 0.05). In addition, we also obtained the IC50 values of Huh-7 cells treated with ATA at different periods, which read 39.8 and 31.6 mM at times 48 and 72 h, respectively (Figure 2E).
We further used HBV stably expressing hepatocytes (HepG2.2.15 cells) to reaffirm the above findings with regard to the inhibitory machinery of ATA on HBV replication. At first, we applied NAGE assays to determine ncDNA and HBcAg levels in the presence or absence of ATA, and the results are shown in Figure 2F. It was consistently observed that ATA had a dose-dependent inhibition on ncDNA synthesis but not on HBcAg expression in HepG2.2.15 cells. Moreover, HBV transcripts became (1.00 ± 0.14)-, (0.89 ± 0.08)-, and (0.95 ± 0.04)-fold in the presence of 2.5, 5, and 10 μM of ATA (Figure 2G), respectively, compared to that with no treatment (all p > 0.05), confirming that ATA treatment did not impair HBV transcripts.
ATA Inhibits Hepatitis B Virus Pol RNase H Activity
Given the results insofar and the fact that ATA has been long regarded as a general inhibitor of nucleases, we postulated that ATA might inhibit HBV replication by blocking its Pol RNase H activity. To this end, a mutant HBV replicon plasmid was constructed where it expressed Pol protein with a point mutation D737V. This single amino acid replacement in the RNase H domain of the Pol protein causes the loss of its RNase H activity while maintaining DNA Pol activity, resulting in that HBV replication stalled at RNA/DNA hybrids in the nucleocapsids (8). We transfected Huh-7 cells with wild-type HBV or Pol RNase H dead mutant and further treated without or with ATA at different dosages. Results are shown in Figure 3A. While ATA had an inhibitory effect on wild-type HBV DNA levels but not on HBcAg levels, corroborating the results in Figure 1C, ATA treatment did not show any impact on HBV DNA levels in the RNase H dead mutant.
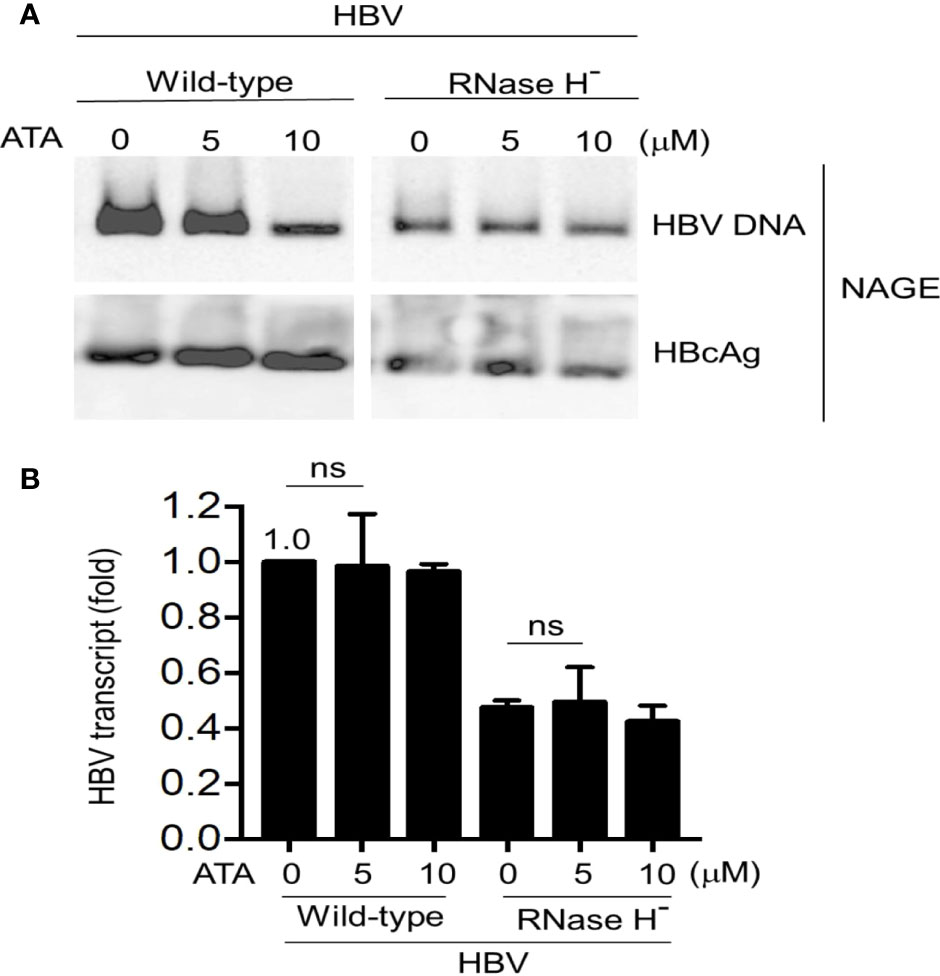
Figure 3 ATA does not affect the Pol RNase H mutant HBV replication in hepatocytes. Huh-7 cells were transfected with an RNase H-defective HBV replicon plasmid (HBV1.5 ΔRNase H). At 24 h after transfection, cells were treated with or without ATA for 48 h, and HBV nucleocapsids in cell lysates were estimated (A) using the NAGE assay followed by Southern or Western blotting with HBV DNA probes or anti-HBc antibody (NAGE assay). (B) Total RNA was extracted in panel (A) and HBV transcripts were measured by RT-qPCR normalized by GAPDH. ns, not significant (two-tailed, unpaired Student’s t-test). Data are mean ± SEM of three independent experiments. All Western blotting data are representative of three independent experiments. ATA, aurintricarboxylic acid; Pol, polymerase; HBV, hepatitis B virus; NAGE, native agarose gel electrophoresis.
In parallel, total RNA extracted from cell lysates (wild-type HBV or D737V mutant transfected Huh-7 cells) without or with ATA treatment was measured for viral transcript levels (Figure 3B). In wild-type HBV transfected cells, treatment with 5 and 10 μM of ATA did not inhibit viral transcripts, as HBV transcript levels remain (0.98 ± 0.19)-fold (p > 0.05) and (0.96 ± 0.03)-fold (p > 0.05), respectively, compared to that without ATA treatment. In contrast, viral transcript in D737V HBV mutant transfected cells was reduced to (0.47 ± 0.03)-fold of that in wild-type HBV transfected counterpart (p < 0.001), verifying the halted viral replication in this mutant-transfected host. Nevertheless, 5 or 10 μM of ATA treatment did not deteriorate viral transcripts in D737V HBV mutant transfected cells, validating that ATA treatment did not impair HBV transcripts. Put together, no noticeable inhibitory effect on viral transcripts of wild-type or RNase H-defective mutant HBV was induced by ATA treatment. Therefore, ATA inhibits HBV replication by disrupting viral Pol RNase H activity and triggering HBV stalls at RNA/DNA hybrids, evincing a potential new reagent to treat HBV infection.
ATA Inhibits Viral Replication of Drug-Resistant Mutant Hepatitis B Virus
Since ATA showed a distinctive mechanism to inhibit HBV Pol RNase H activity, we next determined whether the lamivudine-resistant HBV could be inhibited by ATA. In the following experiments, we transfected YIDD, YVDD, and YMDD (wild-type) HBV into hepatocytes, and we treated the cells without or with ATA or lamivudine. The YMDD motif is highly conserved in the Pol of the retroviruses, where M552I and M552V mutations were found predominant in long-term lamivudine-treated HBV patients who developed drug resistance (24). Results are shown in Figure 4A. It demonstrated that while both ATA and lamivudine significantly inhibited wild-type HBV ncDNA levels, ATA (but not lamivudine) treatment substantially lowered viral DNA levels in YIDD and YVDD mutants. At the same time, ATA or lamivudine treatment did not alter the levels of HBcAg proteins. Furthermore, HBV transcripts were intact in the presence of ATA or lamivudine at the dosages applied (Figure 4B). Taken together, ATA could inhibit lamivudine-resistant HBV mutants, suggesting a potent reagent against HBV genotype mutations.
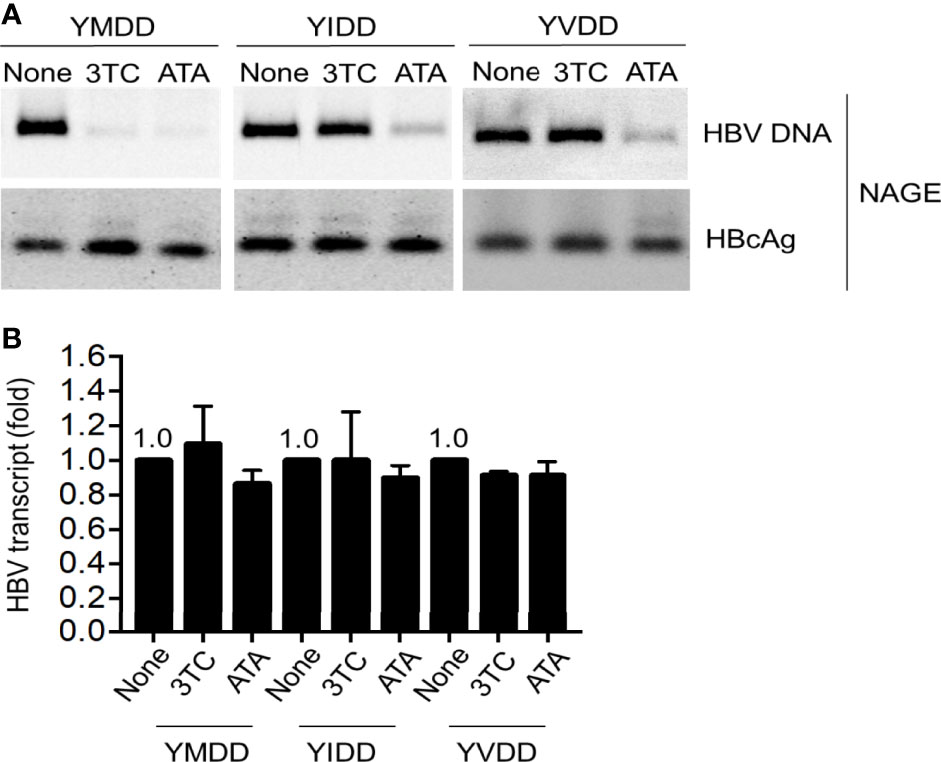
Figure 4 ATA inhibits lamivudine-resistant mutant HBV replication. Huh-7 cells were transfected with HBV replicon plasmids, as indicated. At 24 h after transfection, cells were treated with or without 3TC or ATA for 48 h, and HBV nucleocapsids in cell lysates were estimated (A) using the NAGE assay followed by Southern or Western blotting with HBV DNA probes or anti-HBc antibody (NAGE assay). (B) Total RNA was extracted in panel (A), and HBV transcripts were measured by RT-qPCR normalized by GAPDH. Data are mean ± SEM of three independent experiments. All Western blotting data are representative of three independent experiments. ATA, aurintricarboxylic acid; HBV, hepatitis B virus; NAGE, native agarose gel electrophoresis.
Discussions
In this study, we report for the very first time that ATA owns a potent inhibitory activity for HBV Pol-mediated DNA synthesis in both wild-type and lamivudine-resistant hepatocytes. ATA has been reported with inhibitory activities in a variety of DNA or RNA viruses and with minimized toxicity to a diversity of cells (25). Upon desolvation in aqueous solutions, ATA forms polymerized compounds of polyanionic aromatic structure and so becomes highly negatively charged. This feature promotes its binding to positively charged viral envelope glycoproteins, prohibiting viruses from docking onto cell membranes and further entering the host (11). In addition, ATA is a potent inhibitor of endonuclease activity and protein–nucleic acid interactions (18, 26). This inhibition might be attributed to polyanionic ATAs outcompeting nucleic acids in binding to the active sites of endonucleases, stopping the formation of protein–nucleic acid complexes (16).
HBV replication relies on the proper functioning of the encoded Pol, a multifunctional enzyme that comprises four domains: terminal protein (TP), spacer, reverse transcriptase (RT)/DNA Pol (HP), and RNase H (Figure 1B) (27). Importantly, HBV replication is governed by the RT to employ viral pgRNA template to synthesize (−)-strand DNA, followed by the RNase H cleavage of RNA/DNA heteroduplex, and the HP synthesis of (−)-strand DNA to (+)-strand DNA for further formation of rcDNA (28). In our study, we showed that in the absence of RNase H activity, ATA treatment did not further impair HBV transcripts or HBV DNA levels, suggesting that the drug target of ATA may reside in the RNase H domain rather than the RT domain. HBV D737V mutant was reported with the compromised capability of synthesizing truncated (−)-strand of viral DNA, but not of (+)-strand (8). Therefore, it was derived that ATA might not affect (−)-strand HBV DNA synthesis by retaining the DNA Pol activity of Pol. Instead, ATA could insult RNase H activity of Pol, leading to the formation of RNA/DNA hybrids and retardation of HBV replication. As a result, rcDNA synthesis was stuck. Further experimental outcomes using lamivudine-resistant mutants whose variations in the YMDD motif fall in the RT domain added that ATA inhibits HBV replications via targeting the RNase H domain.
In the RNase H domain of human HBV, a conserved “DEDD” motif together with residues R703 and R781 has been thought of as a critical catalytic site to fulfill the RNase H activity (29, 30). Through electrostatic interactions, polyanionic ATA, similar to clinically tried or approved nucleos(t)ides, outcompetes the binding of nucleic acids to restrain RNA cleavage in the RNA/DNA hybrids, thereby interrupting the HBV replication in the host to produce progeny virions. More intriguingly, polymerized ATA possesses a unique conjugation structure along their central carbon backbone as well as across the quinone methide moiety (Figure 5), casting a “magnetic” net to the catalyzing enzymes via much increased affinity, such as aromatic stacking, hydrogen bonding, and electronic interaction, finally blocking the nucleic acid attachment. More studies to understand the interplays between ATA and HBV Pol are still underway.
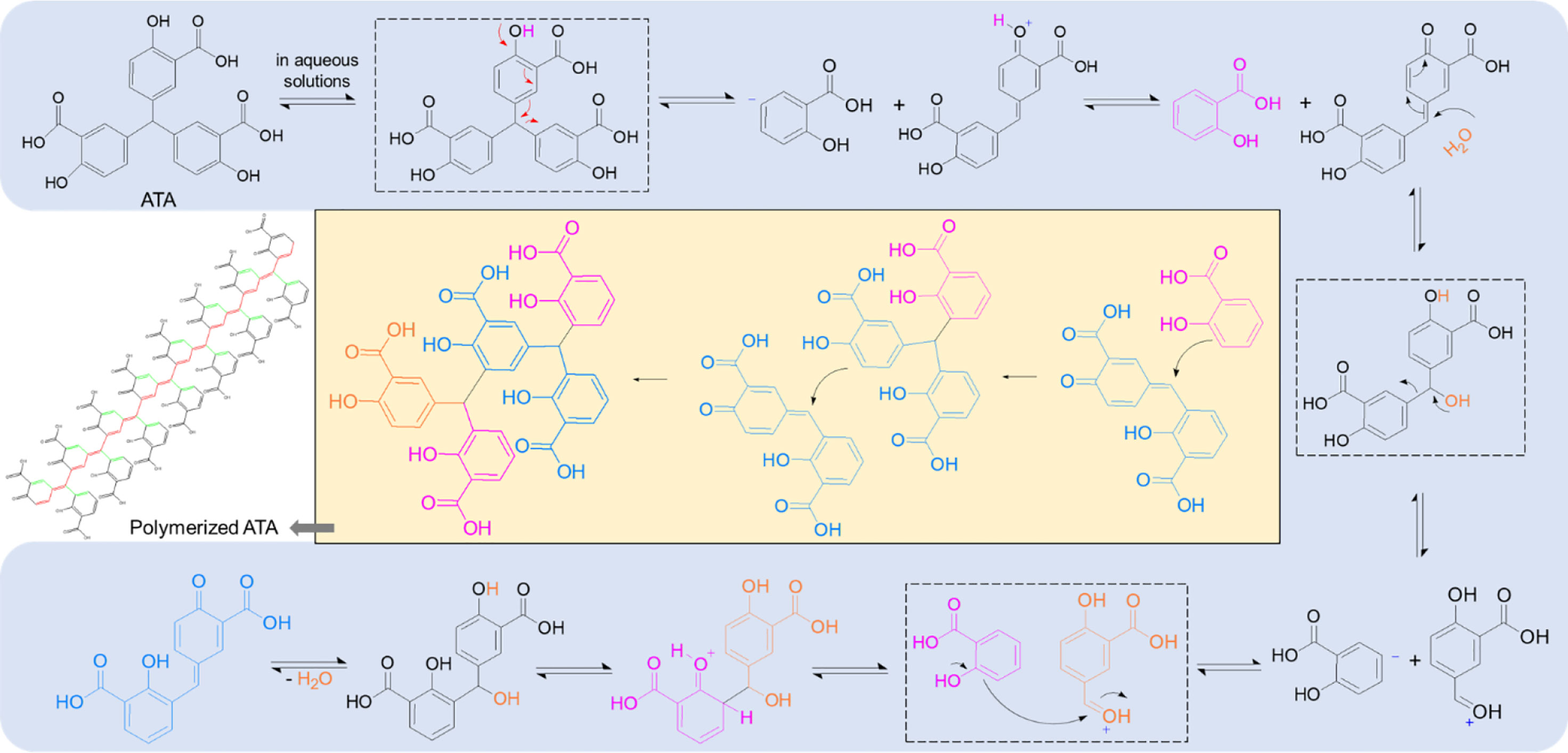
Figure 5 ATA polymerizes in the aqueous solutions and forms conjugation structures. The mechanism with regard to ATA polymer formation, indicating a conjugation structure along the unsaturated carbon backbone (red) as well as across the quinone methide moiety (green), shown in the polymerized ATA structure. ATA, aurintricarboxylic acid.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
XG conceived the idea and directed the research. MX and XG performed the experiments and analyzed the data. XG wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
Author XG was employed by Zhenjiang Meiruo Biotechnology Co., Ltd.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ATA, aurintricarboxylic acid; cccDNA, covalently closed circular DNA; HBc, HBV core; HBcAg, HBV core antigen; HBeAg, HBV e-antigen; HBsAg, HBV surface antigen; HBV, hepatitis B virus; IFN, interferon; NTCP, sodium (Na+) taurocholate cotransporting polypeptide; ncDNA, nucleocapsid-associated DNA; pgRNA, pregenomic RNA; Pol, polymerase; rcDNA, relaxed circular DNA; SARS-CoV, severe acute respiratory syndrome-associated coronavirus.
References
1. Dienstag JL. Hepatitis B Virus Infection. N Engl J Med (2008) 359:1486–500. doi: 10.1056/NEJMra0801644
2. Venkatakrishnan B, Zlotnick A. The Structural Biology of Hepatitis B Virus: Form and Function. Annu Rev Virol (2016) 3:429–51. doi: 10.1146/annurev-virology-110615-042238
3. Liang TJ. Hepatitis B: The Virus and Disease. Hepatology (2009) 49:S13–21. doi: 10.1002/hep.22881
4. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium Taurocholate Cotransporting Polypeptide Is a Functional Receptor for Human Hepatitis B and D Virus. Elife (2012) 1:e00049. doi: 10.7554/eLife.00049
5. Seto WK, Lo YR, Pawlotsky JM, Yuen MF. Chronic Hepatitis B Virus Infection. Lancet (2018) 392:2313–24. doi: 10.1016/S0140-6736(18)31865-8
6. Nassal M. HBV cccDNA: Viral Persistence Reservoir and Key Obstacle for a Cure of Chronic Hepatitis B. Gut (2015) 64:1972–84. doi: 10.1136/gutjnl-2015-309809
7. Grimm D, Thimme R, Blum HE. HBV Life Cycle and Novel Drug Targets. Hepatol Int (2011) 5:644–53. doi: 10.1007/s12072-011-9261-3
8. Radziwill G, Tucker W, Schaller H. Mutational Analysis of the Hepatitis B Virus P Gene Product: Domain Structure and RNase H Activity. J Virol (1990) 64:613–20. doi: 10.1128/jvi.64.2.613-620.1990
9. Lok AS, McMahon BJ, Brown RS, Wong JB, Ahmed AT, Farah W, et al. Antiviral Therapy for Chronic Hepatitis B Viral Infection in Adults: A Systematic Review and Meta-Analysis. Hepatology (2016) 63:284–306. doi: 10.1002/hep.28280
10. Dusheiko G. Lamivudine Treatment of Chronic Hepatitis B. Rev Med Virol (1998) 8:153–9. doi: 10.1002/(SICI)1099-1654(199807/09)8:3<153::AID-RMV228>3.0.CO;2-Y
11. Reymen D, Witvrouw M, Esté J, Neyts J, Schols D, Andrei G, et al. Mechanism of the Antiviral Activity of New Aurintricarboxylic Acid Analogues. Antiviral Chem Chemotherapy (1996) 7:142–52. doi: 10.1177/095632029600700304
12. He R, Adonov A, Traykova-Adonova M, Cao J, Cutts T, Grudesky E, et al. Potent and Selective Inhibition of SARS Coronavirus Replication by Aurintricarboxylic Acid. Biochem Biophys Res Commun (2004) 320:1199–203. doi: 10.1016/j.bbrc.2004.06.076
13. Park JG, Avila-Perez G, Madere F, Hilimire TA, Nogales A, Almazan F, et al. Potent Inhibition of Zika Virus Replication by Aurintricarboxylic Acid. Front Microbiol (2019) 10:718. doi: 10.3389/fmicb.2019.00718
14. Zhou H, Fang Y, Xu T, Ni WJ, Shen AZ, Meng XM. Potential Therapeutic Targets and Promising Drugs for Combating SARS-CoV-2. Br J Pharmacol (2020) 177:3147–61. doi: 10.1111/bph.15092
15. González RG, Blackburn BJ, Schleich T. Fractionation and Structural Elucidation of the Active Components of Aurintricarboxylic Acid, a Potent Inhibitor of Protein Nucleic Acid Interactions. Biochim Biophys Acta (BBA)-Nucleic Acids Protein Synthesis (1979) 562:534–45. doi: 10.1016/0005-2787(79)90116-3
16. Gonzalez RG, Haxo RS, Schleich T. Mechanism of Action of Polymeric Aurintricarboxylic Acid, a Potent Inhibitor of Protein-Nucleic Acid Interactions. Biochemistry (1980) 19:4299–303. doi: 10.1021/bi00559a023
17. Cushman M, Wang P, Chang SH, Wild C, De Clercq E, Schols D, et al. Preparation and Anti-HIV Activities of Aurintricarboxylic Acid Fractions and Analogs: Direct Correlation of Antiviral Potency With Molecular Weight. J Med Chem (1991) 34:329–37. doi: 10.1021/jm00105a052
18. Shadrick WR, Mukherjee S, Hanson AM, Sweeney NL, Frick DN. Aurintricarboxylic Acid Modulates the Affinity of Hepatitis C Virus NS3 Helicase for Both Nucleic Acid and ATP. Biochemistry (2013) 52:6151–9. doi: 10.1021/bi4006495
19. Gripon P, Rumin S, Urban S, Le SJ, Glaise D, Cannie I, et al. Infection of a Human Hepatoma Cell Line by Hepatitis B Virus. Proc Natl Acad Sci USA (2002) 99:15655–60. doi: 10.1073/pnas.232137699
20. Liang G, Kitamura K, Wang Z, Liu G, Chowdhury S, Fu W, et al. RNA Editing of Hepatitis B Virus Transcripts by Activation-Induced Cytidine Deaminase. Proc Natl Acad Sci USA (2013) 110:2246–51. doi: 10.1073/pnas.1221921110
21. Liang G, Liu G, Kitamura K, Wang Z, Chowdhury S, Monjurul A, et al. TGF-β Suppression of HBV RNA Through AID-Dependent Recruitment of an RNA Exosome Complex. PloS Pathog (2015) 11:e1004780. doi: 10.1371/journal.ppat.1004780
22. Nguyen DH, Hu J. Reverse Transcriptase- and RNA Packaging Signal-Dependent Incorporation of APOBEC3G Into Hepatitis B Virus Nucleocapsids. J Virol (2008) 82:6852–61. doi: 10.1128/JVI.00465-08
23. Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science (2014) 343:1221–8. doi: 10.1126/science.1243462
24. Gutfreund KS, Williams M, George R, Bain VG, Ma MM, Yoshida EM, et al. Genotypic Succession of Mutations of the Hepatitis B Virus Polymerase Associated With Lamivudine Resistance. J Hepatol (2000) 33:469–75. doi: 10.1016/S0168-8278(00)80284-6
25. Balzarini J, Mitsuya H, De Clercq E, Broder S. Aurintricarboxylic Acid and Evans Blue Represent Two Different Classes of Anionic Compounds Which Selectively Inhibit the Cytopathogenicity of Human T-Cell Lymphotropic Virus Type III/Lymphadenopathy-Associated Virus. Biochem Biophys Res Commun (1986) 136:64–71. doi: 10.1016/0006-291X(86)90877-6
26. Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, et al. Characterization of Aurintricarboxylic Acid as a Potent Hepatitis C Virus Replicase Inhibitor. Antiviral Chem Chemother (2009) 20:19–36. doi: 10.3851/IMP1286
27. Tavis JE, Zoidis G, Meyers MJ, Murelli RP. Chemical Approaches to Inhibiting the Hepatitis B Virus Ribonuclease H. ACS Infect Dis (2019) 5:655–8. doi: 10.1021/acsinfecdis.8b00045
28. Jones SA, Hu J. Hepatitis B Virus Reverse Transcriptase: Diverse Functions as Classical and Emerging Targets for Antiviral Intervention. Emerg Microbes Infect (2013) 2:e56. doi: 10.1038/emi.2013.56
29. Ko C, Shin Y-C, Park W-J, Kim S, Kim J, Ryu W-S. Residues Arg703, Asp777, and Arg781 of the RNase H Domain of Hepatitis B Virus Polymerase Are Critical for Viral DNA Synthesis. J Virol (2014) 88:154–63. doi: 10.1128/JVI.01916-13
Keywords: hepatitis B virus, aurintricarboxylic acid, polymerase, lamivudine-resistant, RNase H
Citation: Xu M and Guan X (2022) Aurintricarboxylic Acid Suppresses Hepatitis B Virus Replication by Inhibition of RNase H Activity. Front.Virol. 2:861494. doi: 10.3389/fviro.2022.861494
Received: 09 February 2022; Accepted: 21 April 2022;
Published: 20 May 2022.
Edited by:
Carlos M. de Noronha, Albany Medical College, United StatesReviewed by:
Hussein Hassan Aly, National Institute of Infectious Diseases (NIID), JapanChristian K. Pfaller, Paul Ehrlich Institute, Germany
Copyright © 2022 Xu and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Guan, merrillbiotech@163.com