
Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress

 , , , , , , and
, , , , , , and
Abstract
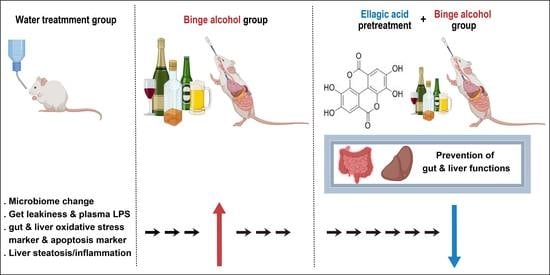
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
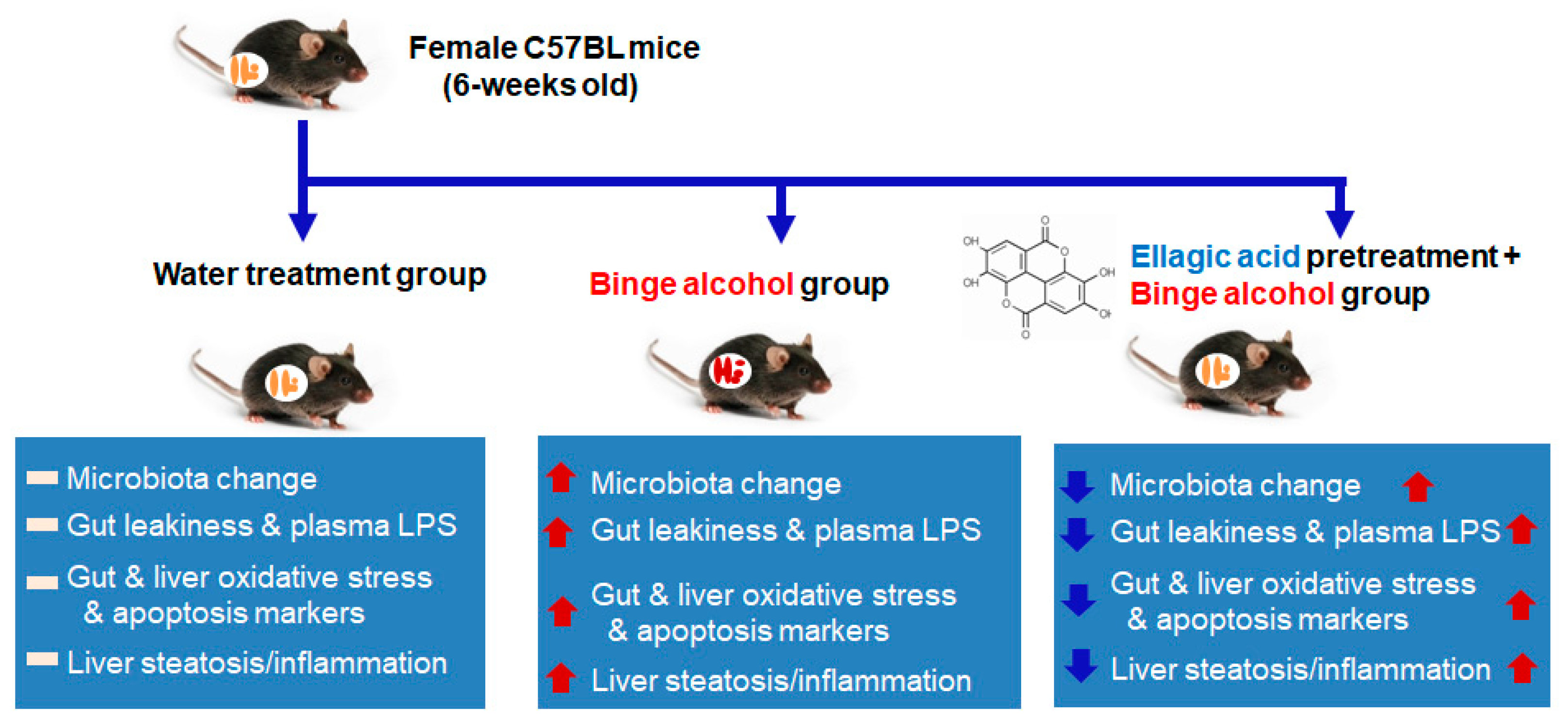
2.2. Animal Treatments
2.3. Histological Analysis and Plasma ALT Measurement
2.4. Endotoxin Assay
2.5. Determinations of Hepatic Triglyceride, Plasma Reactive Oxygen Species, and Blood Alcohol Concentration
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Immunoblot Analysis
2.8. Apoptosis Assay
2.9. Immunohistochemistry Analysis
2.10. Microbial 16S Sequencing and Bioinformatics
2.11. RNA Extraction and Quantitative Real-Time PCR
2.12. Statistical Analysis and Other Methods
3. Results
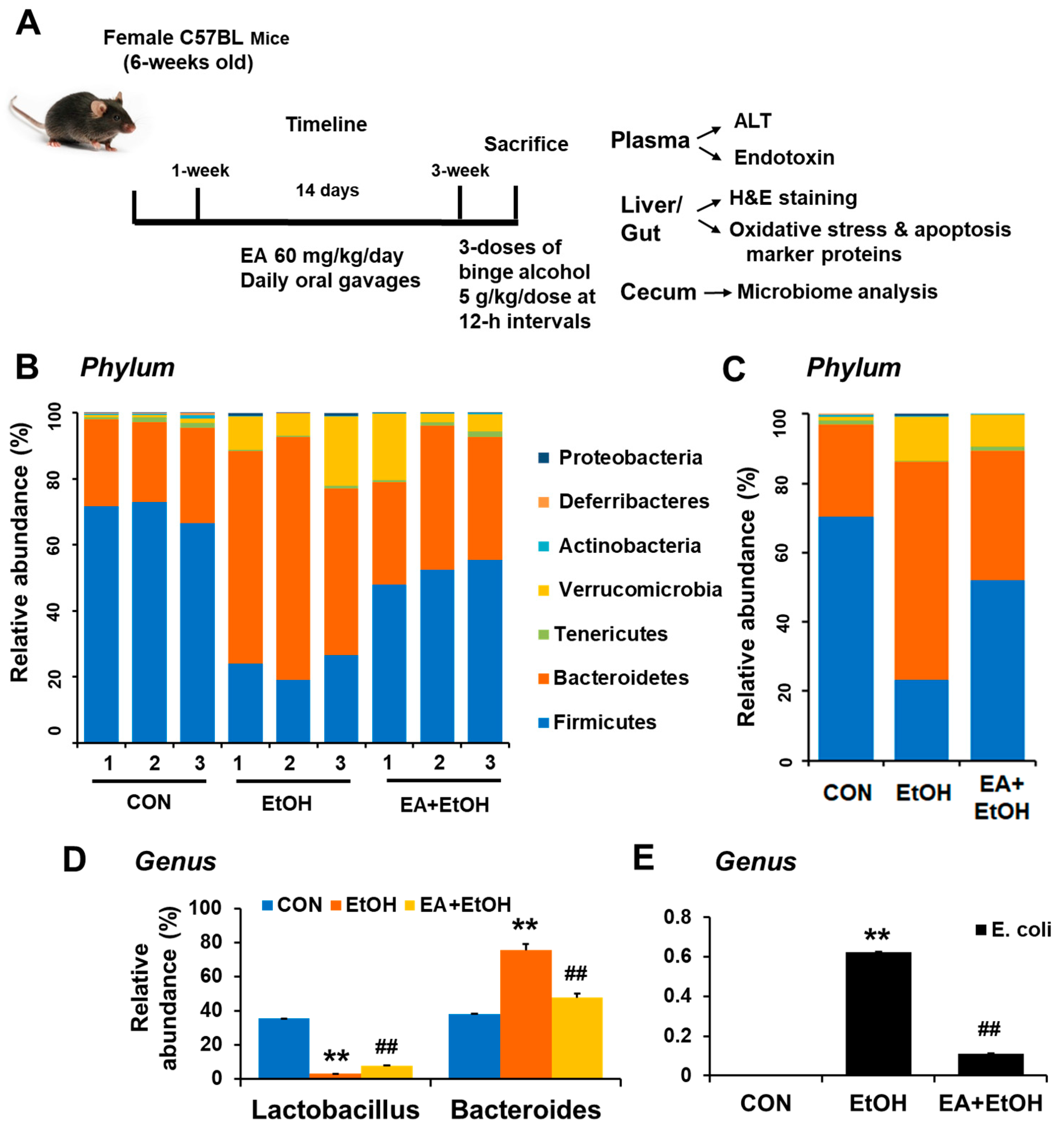
3.1. Ellagic Acid Pretreatment Prevents the Drastic Changes in Gut Microbiota in Binge Alcohol-Exposed Mice
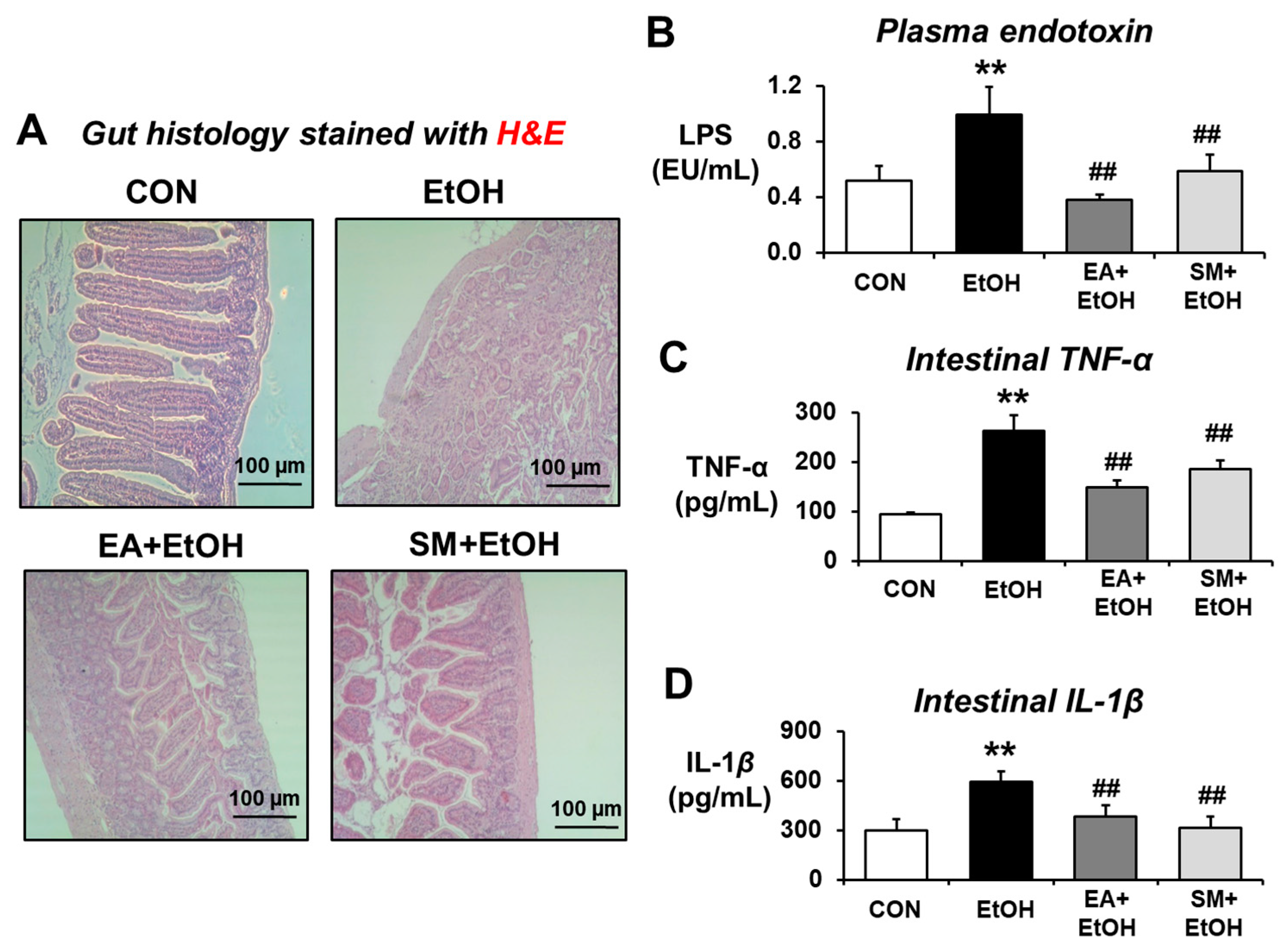
3.2. Ellagic Acid Pretreatment Averts the Increased Levels of Plasma Endotoxin and Intestinal TNF-α and IL-1β Proteins in Binge Alcohol-Exposed Mice
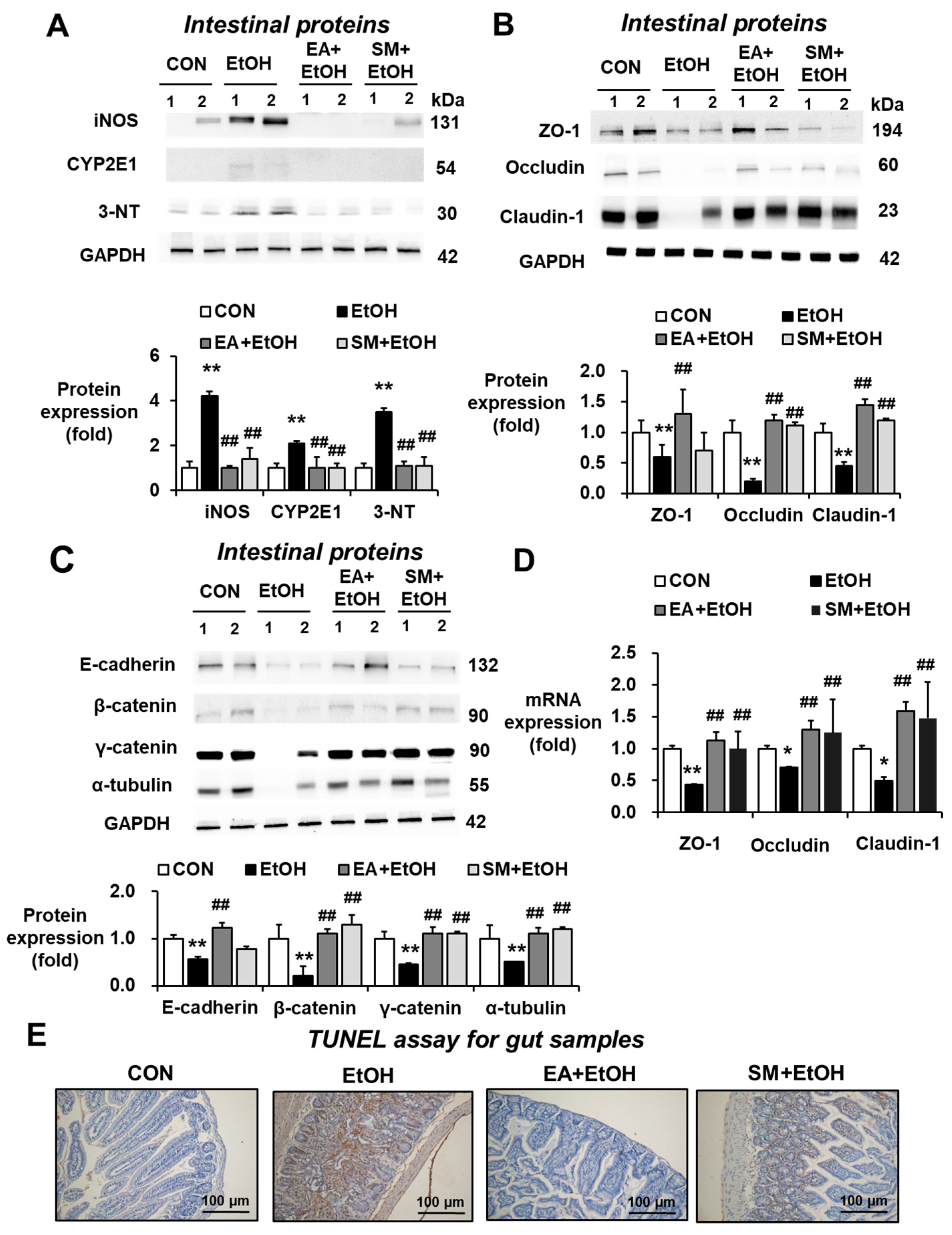
3.3. Ellagic Acid Pretreatment Reduced the Gut Oxidative Stress Markers in Binge Alcohol-Exposed Mice
3.4. Ellagic Acid Pretreatment Prevented Altered Levels of Gut Tight Junction, Adherent Junction, and Apoptosis Marker Proteins in Binge Alcohol-Exposed Mice
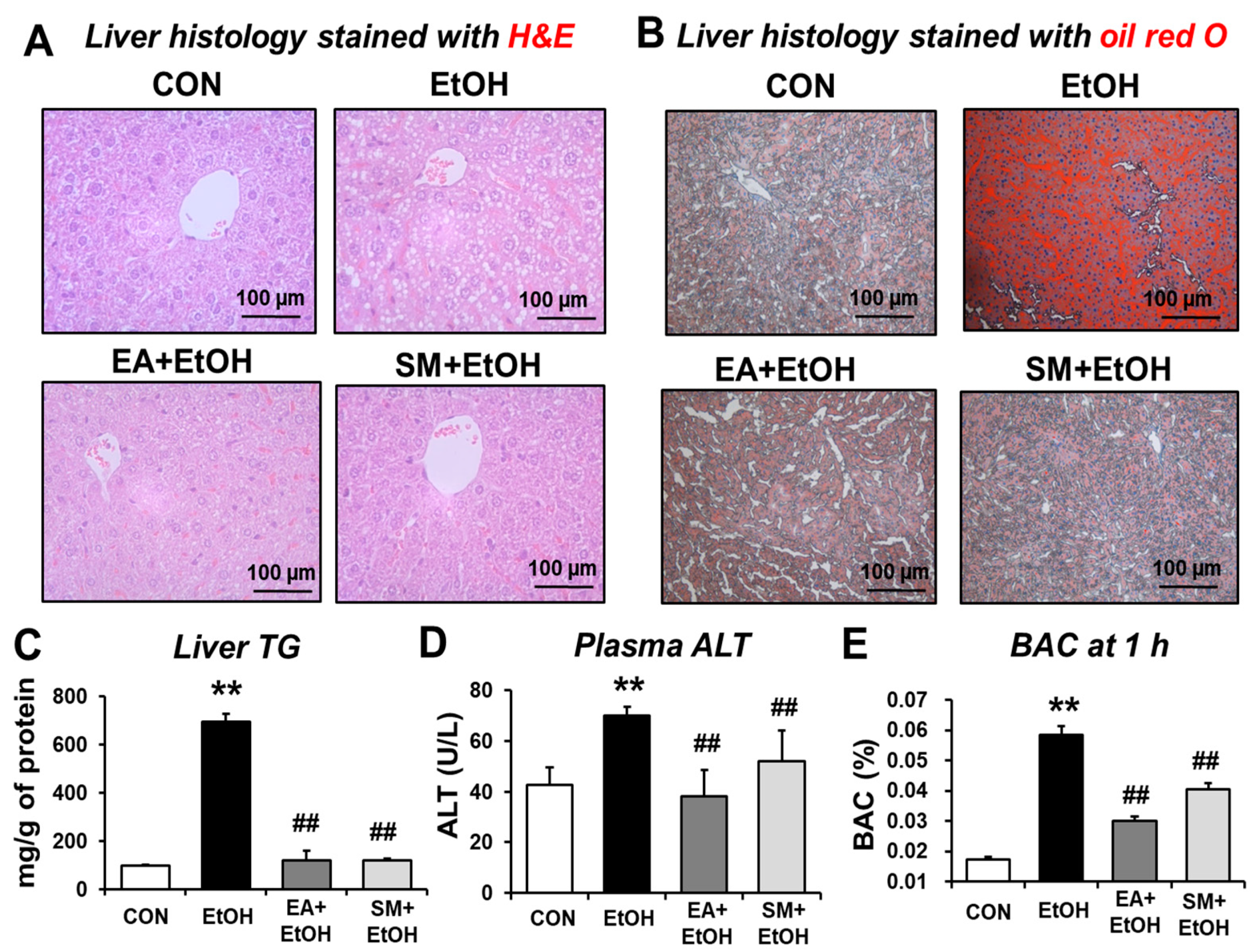
3.5. Ellagic Acid Pretreatment Prevented Alcohol-Mediated Hepatic Fat Accumulation, Plasma ALT, and Hepatic Triglyceride Levels
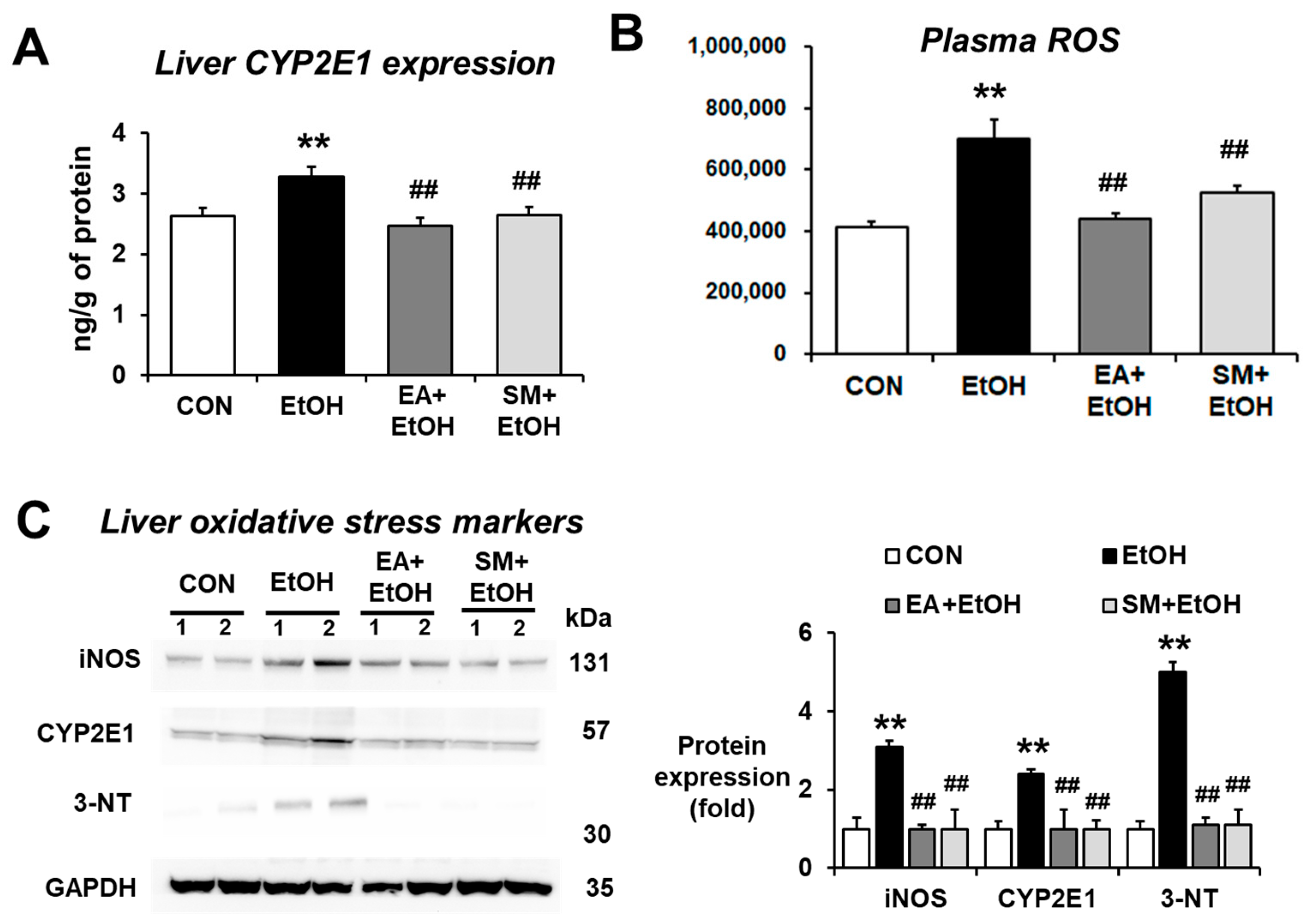
3.6. Ellagic Acid Pretreatment Attenuated the Hepatic Oxidative Stress Marker Proteins in Binge Alcohol-Exposed Mice
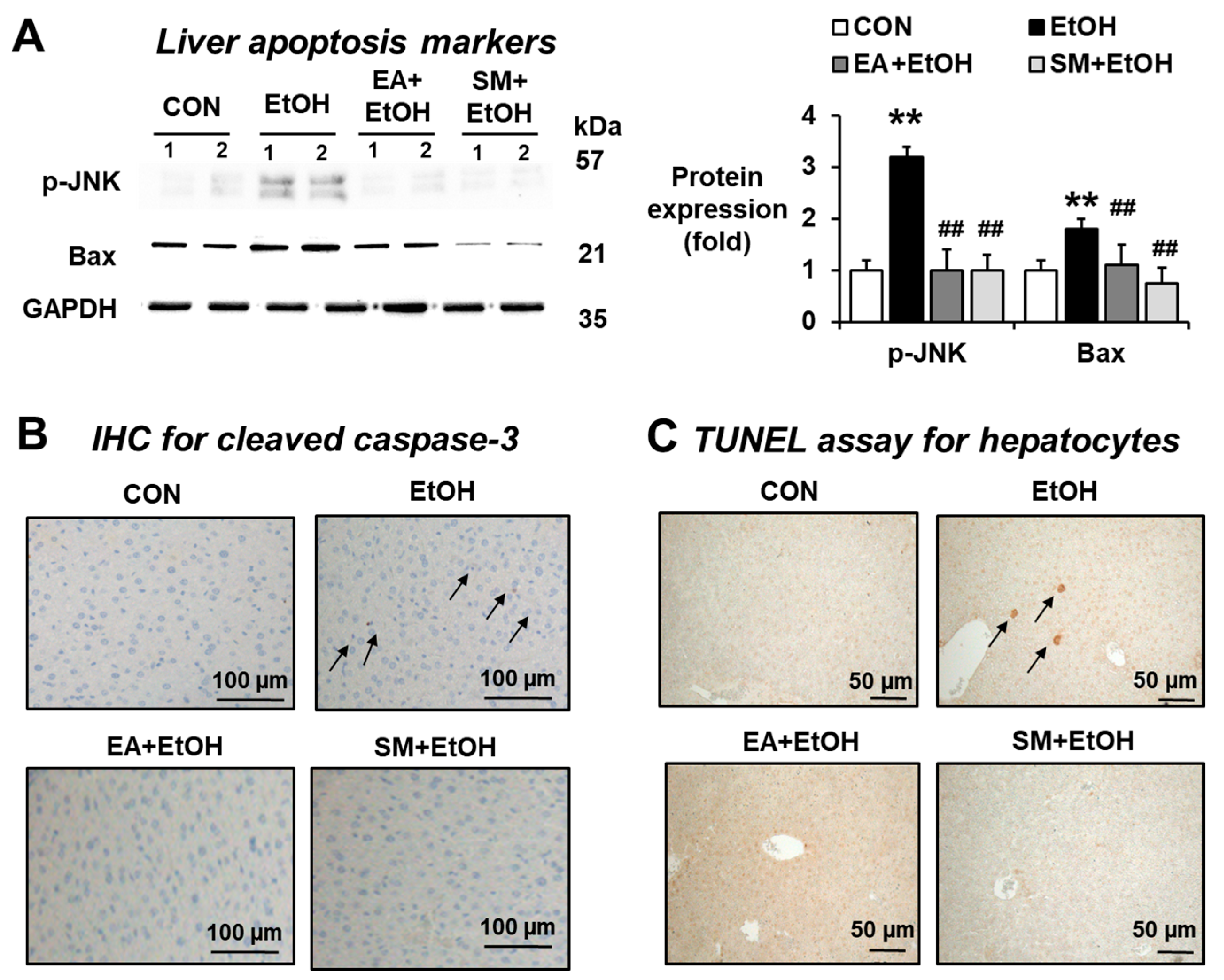
3.7. Ellagic Acid Pretreatment Prevented the Elevated Hepatic Apoptosis Marker Proteins in Binge Alcohol-Exposed Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sacks, J.J.; Gonzales, K.R.; Bouchery, E.E.; Tomedi, L.E.; Brewer, R.D. 2010 National and State Costs of Excessive Alcohol Consumption. Am. J. Prev. Med. 2015, 49, e73–e79. [Google Scholar] [CrossRef]
- Adachi, M.; Brenner, D.A. Clinical syndromes of alcoholic liver disease. Dig. Dis. 2005, 23, 255–263. [Google Scholar] [CrossRef]
- Tilg, H.; Day, C.P. Management strategies in alcoholic liver disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 24–34. [Google Scholar] [CrossRef]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Ambade, A.; Kodys, K.; Catalano, D.; Ward, D.V.; Szabo, G. Alcohol-related changes in the intestinal microbiome influence neutrophil infiltration, inflammation and steatosis in early alcoholic hepatitis in mice. PLoS ONE 2017, 12, e0174544. [Google Scholar]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Ballway, J.W.; Song, B.J. Translational approaches with antioxidant phytochemicals against alcohol-mediated oxidative stress, gut dysbiosis, intestinal barrier dysfunction, and fatty liver disease. Antioxidants 2021, 10, 384. [Google Scholar] [CrossRef]
- Wang, H.J.; Zakhari, S.; Jung, M.K. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J. Gastroenterol. 2010, 16, 1304–1313. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef]
- Bode, C.; Kugler, V.; Bode, J.C. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Singal, A.K.; Kamath, P.S.; Gores, G.J.; Shah, V.H. Alcoholic hepatitis: Current challenges and future directions. Clin. Gastroenterol. Hepatol. 2014, 12, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Nino, W.R.; Zazueta, C. Ellagic acid: Pharmacological activities and molecular mechanisms involved in liver protection. Pharmacol. Res. 2015, 97, 84–103. [Google Scholar] [CrossRef]
- Cho, Y.E.; Song, B.J. Pomegranate prevents binge alcohol-induced gut leakiness and hepatic inflammation by suppressing oxidative and nitrative stress. Redox Biol. 2018, 18, 266–278. [Google Scholar] [CrossRef]
- Cho, Y.E.; Yu, L.R.; Abdelmegeed, M.A.; Yoo, S.H.; Song, B.J. Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol-induced gut leakiness and liver injury. J. Hepatol. 2018, 69, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Kim, D.K.; Seo, W.; Gao, B.; Yoo, S.H.; Song, B.J. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome P450-2E1-mediated oxidative and nitrative stress. Hepatology 2021, 73, 2180–2195. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Mezey, E.; Hardwick, J.P.; Salem, N.; Clemens, D.L.; Song, B.J. Increased ethanol-inducible cytochrome P450-2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatol. Commun. 2017, 1, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.E.; Lee, M.H.; Song, B.J. Neuronal cell death and degeneration through increased nitroxidative stress and tau phosphorylation in HIV-1 transgenic rats. PLoS ONE 2017, 12, e0169945. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Hwang, B.B.; Chang, M.H.; Lee, J.H.; Heo, W.; Kim, J.K.; Pan, J.H.; Kim, Y.J. Kim, J.H. The edible insect Gryllus bimaculatus protects against gut-derived inflammatory responses and liver damage in nice after acute alcohol exposure. Nutrients 2019, 11, 857. [Google Scholar]
- Chen, P.; Starkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelson, D.R.; Gu, M.; Shellito, J.E.; Molina, P.E.; Taylor, C.M.; Luo, M.; Welsh, D.A. Intestinal microbial products from alcohol-fed mice contribute to intestinal permeability and peripheral immune activation. Alcohol Clin. Exp. Res. 2019, 43, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederbaum, A.I. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig. Dis. 2010, 28, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Morgan, K.; French, S.W.; Morgan, T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology 2002, 36, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, X.; Zhou, R.; Cederbaum, A. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem. Biophys. Res. Commun. 2010, 402, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Armoni, M.; Harel, C.; Kamieli, E.; Pessin, J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat induced obesity and IR. Am. J. Physiol. Endocrinol. Metabol. 2012, 302, E532–E539. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.-H.; Jang, S.; Gonzalez, F.J.; Song, B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic hepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG clinical guideline: Alcoholic liver disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef]
- Szabo, G. Clinical trial design for alcoholic hepatitis. Semin. Liver Dis. 2017, 37, 332–342. [Google Scholar] [CrossRef]
- Singal, A.K.; Shah, V.H. Current trials and novel therapeutic targets for alcoholic hepatitis. J. Hepatol. 2019, 70, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [Green Version]
- Song, B.J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.J. Preventive effects of indole-3-carbinol against alcohol-induced liver injury in mice via antioxidant, anti-inflammatory, and anti-apoptotic mechanisms: Role of gut-liver-adipose tissue axis. J. Nutr. Biochem. 2018, 55, 12–25. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Greenwel, P.; Cederbaum, A.I. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology 1999, 30, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef] [PubMed]
- McKim, S.E.; Gabele, E.; Isayama, F.; Lambert, J.C.; Tucker, L.M.; Wheeler, M.D.; Connor, H.D.; Mason, R.P.; Doll, M.A.; Hein, D.W.; et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: Studies with knockout mice. Gastroenterology 2003, 125, 1834–1844. [Google Scholar] [CrossRef]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, D.; Wang, X.; Ward, S.C.; Cederbaum, A.I. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic. Biol. Med. 2010, 49, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rusyn, I.; Yin, M.; Gabele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig. 2000, 106, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Woodcroft, K.J.; Hafner, M.S.; Novak, R.F. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology 2002, 35, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Midde, N.M.; Miller, D.D.; Chauhan, S.; Kumar, A.; Kumar, S. Diallyl sulfide: Potential use in novel therapeutic interventions in alcohol, drugs, and disease mediated cellular toxicity by targeting cytochrome P450 2E1. Curr. Drug Metab. 2015, 16, 486–503. [Google Scholar] [CrossRef] [Green Version]
- Yoshigae, Y.; Sridar, C.; Kent, U.M.; Hollenberg, P.F. The inactivation of human CYP2E1 by phenethyl isothiocyanate, a naturally occurring chemopreventive agent, and its oxidative bioactivation. Drug Metab. Dispos. 2013, 41, 858–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, C.; Xing, G.; Qi, X.; Ren, J. Resveratrol downregulates Cyp2e1 and attenuates chemically induced hepatocarcinogenesis in SD Rats. J. Toxicol. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Bedada, S.K.; Neerati, P. Resveratrol pretreatment affects CYP2E1 activity of chlorzoxazone in healthy human volunteers. Phytother. Res. 2016, 30, 463–468. [Google Scholar] [CrossRef]
- Domitrovic, R.; Cvijanovic, O.; Pernjak-Pugel, E.; Skoda, M.; Mikelic, L.; Crncevic-Orlic, Z. Berberine exerts nephroprotective effect against cisplatin-induced kidney damage through inhibition of oxidative/nitrosative stress, inflammation, autophagy and apoptosis. Food Chem. Toxicol. 2013, 62, 397–406. [Google Scholar] [CrossRef]
- Lee, S.E.; Koh, H.; Joo, D.J.; Nedumaran, B.; Jeon, H.J.; Park, C.S.; Harris, R.A.; Kim, Y.D. Induction of SIRT1 by melatonin improves alcohol-mediated oxidative liver injury by disrupting the CRBN-YY1-CYP2E1 signaling pathway. J. Pineal Res. 2020, 68, e12638. [Google Scholar] [CrossRef]
- Stice, C.P.; Xia, H.; Wang, X.D. Tomato lycopene prevention of alcoholic fatty liver disease and hepatocellular carcinoma development. Chronic Dis. Transl. Med. 2018, 4, 211–224. [Google Scholar] [CrossRef]
- Wilson, T.; Lewis, M.J.; Cha, K.L.; Gold, B. The effect of ellagic acid on xenobiotic metabolism by cytochrome P-450IIE1 and nitrosodimethylamine mutagenicity. Cancer Lett. 1992, 61, 129–134. [Google Scholar] [CrossRef]
- Brandon-Warner, E.; Sugg, J.A.; Schrum, L.W.; McKillop, I.H. Silibinin inhibits ethanol metabolism and ethanol-dependent cell proliferation in an in vitro model of hepatocellular carcinoma. Cancer Lett. 2010, 291, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, Y.T.; Wu, J.H.; Huang, C.C.; Peng, H.C.; Chen, Y.L.; Yang, S.C.; Chang, S.T. Protective effect of Acacia confusa bark extract and its active compound gallic acid against carbon tetrachloride-induced chronic liver injury in rats. Food Chem. Toxicol. 2009, 47, 1385–1392. [Google Scholar] [CrossRef]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.J. Preventive effects of dietary walnuts on high-fat-induced hepatic fat accumulation, oxidative stress and apoptosis in mice. J. Nutr. Biochem. 2016, 38, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Devipriya, N.; Sudheer, A.R.; Menon, V.P. Dose-response effect of ellagic acid on circulatory antioxidants and lipids during alcohol-induced toxicity in experimental rats. Fundam. Clin. Pharmacol. 2007, 21, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Devipriya, N.; Sudheer, A.R.; Vishwanathan, P.; Menon, V.P. Modulatory potential of ellagic acid, a natural plant polyphenol on altered lipid profile and lipid peroxidation status during alcohol-induced toxicity: A pathohistological study. J. Biochem. Mol. Toxicol. 2008, 22, 101–112. [Google Scholar] [CrossRef]
- Gonzalez-Sarrias, A.; Azorin-Ortuno, M.; Yanez-Gascon, M.J.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Dissimilar in vitro and in vivo effects of ellagic acid and its microbiota-derived metabolites, urolithins, on the cytochrome P450 1A1. J. Agric. Food Chem. 2009, 57, 5623–5632. [Google Scholar] [CrossRef] [PubMed]
- Devipriya, N.; Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Effect of ellagic acid, a natural polyphenol, on alcohol-induced prooxidant and antioxidant imbalance: A drug dose dependent study. Singap. Med. J. 2007, 48, 311–318. [Google Scholar]
- Devipriya, N.; Sudheer, A.R.; Srinivasan, M.; Menon, V.P. Effect of ellagic acid, a plant polyphenol, on fibrotic markers (MMPs and TIMPs) during alcohol-induced hepatotoxicity. Toxicol Mech. Methods 2007, 17, 349–356. [Google Scholar]
- Barch, D.H.; Rundhaugen, L.M.; Thomas, P.E.; Kardos, P.; Pillay, N.S. Dietary ellagic acid inhibits the enzymatic activity of CYP1A1 without altering hepatic concentrations of CYP1A1 or CYP1A1 mRNA. Biochem. Biophys. Res. Commun. 1994, 201, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Szaefer, H.; Jodynis-Liebert, J.; Cichocki, M.; Matuszewska, A.; Baer-Dubowska, W. Effect of naturally occurring plant phenolics on the induction of drug metabolizing enzymes by o-toluidine. Toxicology 2003, 186, 67–77. [Google Scholar] [CrossRef]
- Parlesak, A.; Schafer, C.; Schutz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Nanji, A.A.; Khettry, U.; Sadrzadeh, S.M.H. Lactobacillus Feeding Reduces Endotoxemia and Severity of Experimental Alcoholic Liver (Disease). Exp. Biol. Med. 1994, 205, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic Analyses of Alcohol Induced Pathogenic Alterations in the Intestinal Microbiome and the Effect of Lactobacillus rhamnosus GG Treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef] [Green Version]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Jin, X.; Li, J.; Zhang, T.; Zhang, W.; Shi, W.; Fan, S.; Wang, X.; Wang, J.; Zhong, B.; et al. Effects of silymarin, glycyrrhizin, and oxymatrine on the pharmacokinetics of ribavirin and its major metabolite in rats. Phytother. Res. 2016, 30, 618–626. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-h.; Sim, Y.; Hwang, J.-h.; Kwun, I.-S.; Lim, J.-H.; Kim, J.; Kim, J.-I.; Baek, M.-C.; Akbar, M.; Seo, W.; et al. Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress. Antioxidants 2021, 10, 1386. https://doi.org/10.3390/antiox10091386
Kim D-h, Sim Y, Hwang J-h, Kwun I-S, Lim J-H, Kim J, Kim J-I, Baek M-C, Akbar M, Seo W, et al. Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress. Antioxidants. 2021; 10(9):1386. https://doi.org/10.3390/antiox10091386
Chicago/Turabian StyleKim, Dong-ha, Yejin Sim, Jin-hyeon Hwang, In-Sook Kwun, Jae-Hwan Lim, Jihoon Kim, Jee-In Kim, Moon-Chang Baek, Mohammed Akbar, Wonhyo Seo, and et al. 2021. "Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress" Antioxidants 10, no. 9: 1386. https://doi.org/10.3390/antiox10091386