Merging the Multi-Target Effects of Phytochemicals in Neurodegeneration: From Oxidative Stress to Protein Aggregation and Inflammation

 , ,
, ,
Abstract
:1. Introduction
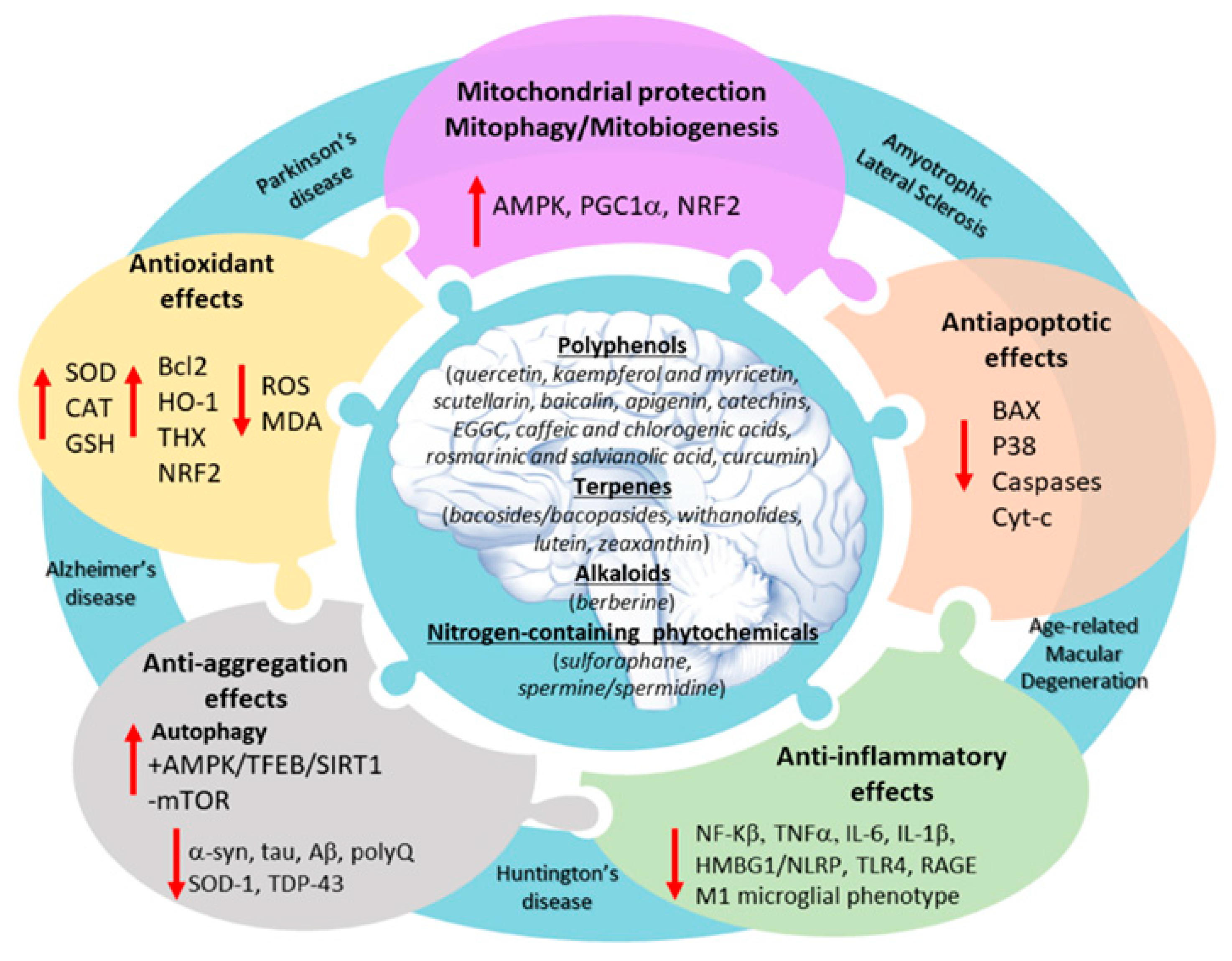
2. An Overview of Neuroprotective Phytochemical Classes
2.1. Polyphenols
2.2. Terpenes
2.3. Alkaloids
2.4. Other Nitrogen-Containing Phytochemicals
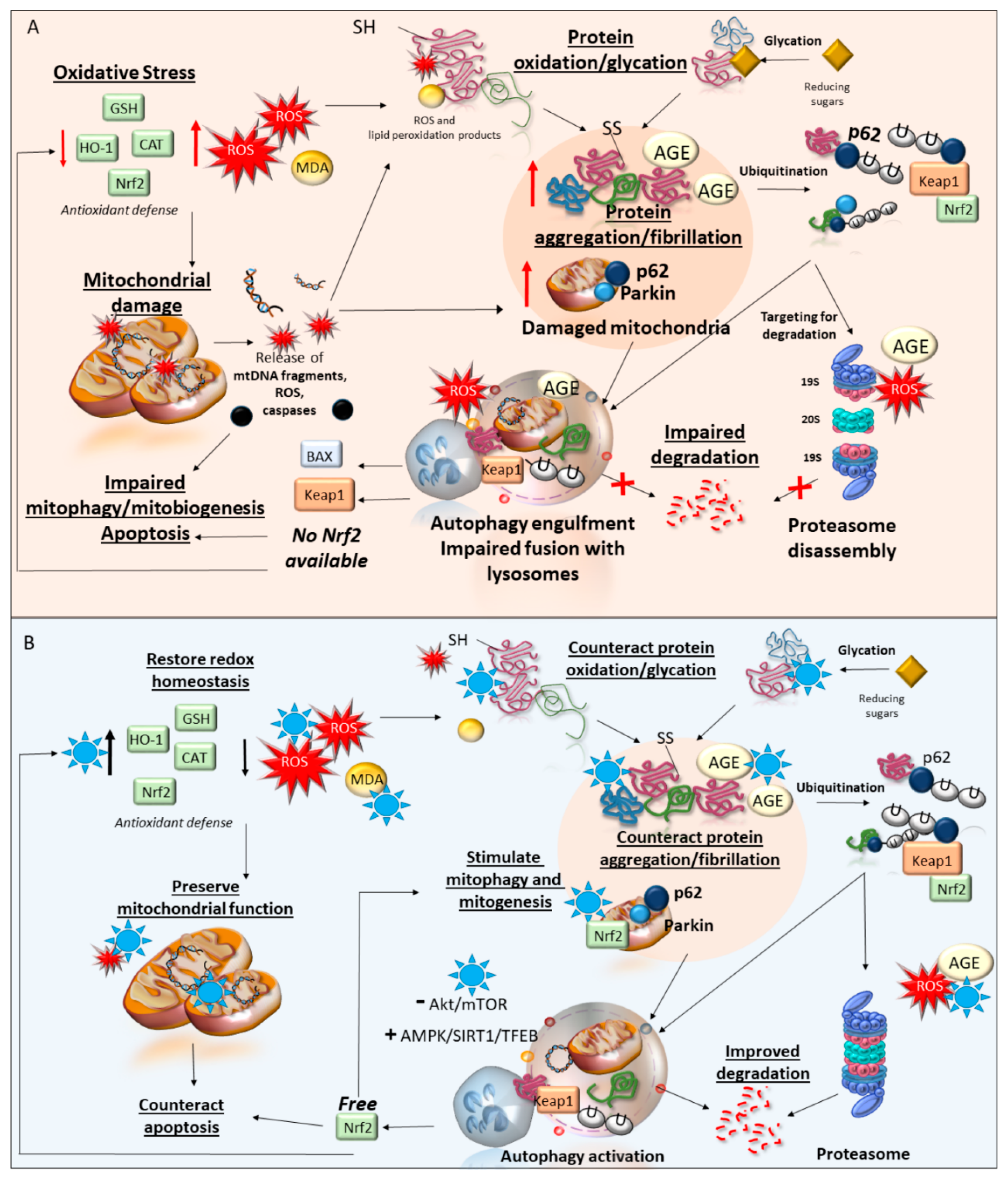
3. Phytochemicals and Oxidative Stress
3.1. Autophagy-Related Antioxidant Effects of Phytochemicals
3.1.1. An Overview of the Autophagy Pathway
3.1.2. Phytochemicals Bridging Autophagy Induction and Prevention of Oxidative Stress
4. Phytochemicals and Mitochondrial Damage
Autophagy-Related Mitochondrial Protection by Phytochemicals
5. Phytochemicals and Proteostasis
5.1. Alterations of Proteostasis in Neurodegeneration
5.2. Anti-Aggregation Mechanisms of Phytochemicals
5.3. Autophagy-Related Anti-Aggregation Effects of Phytochemicals
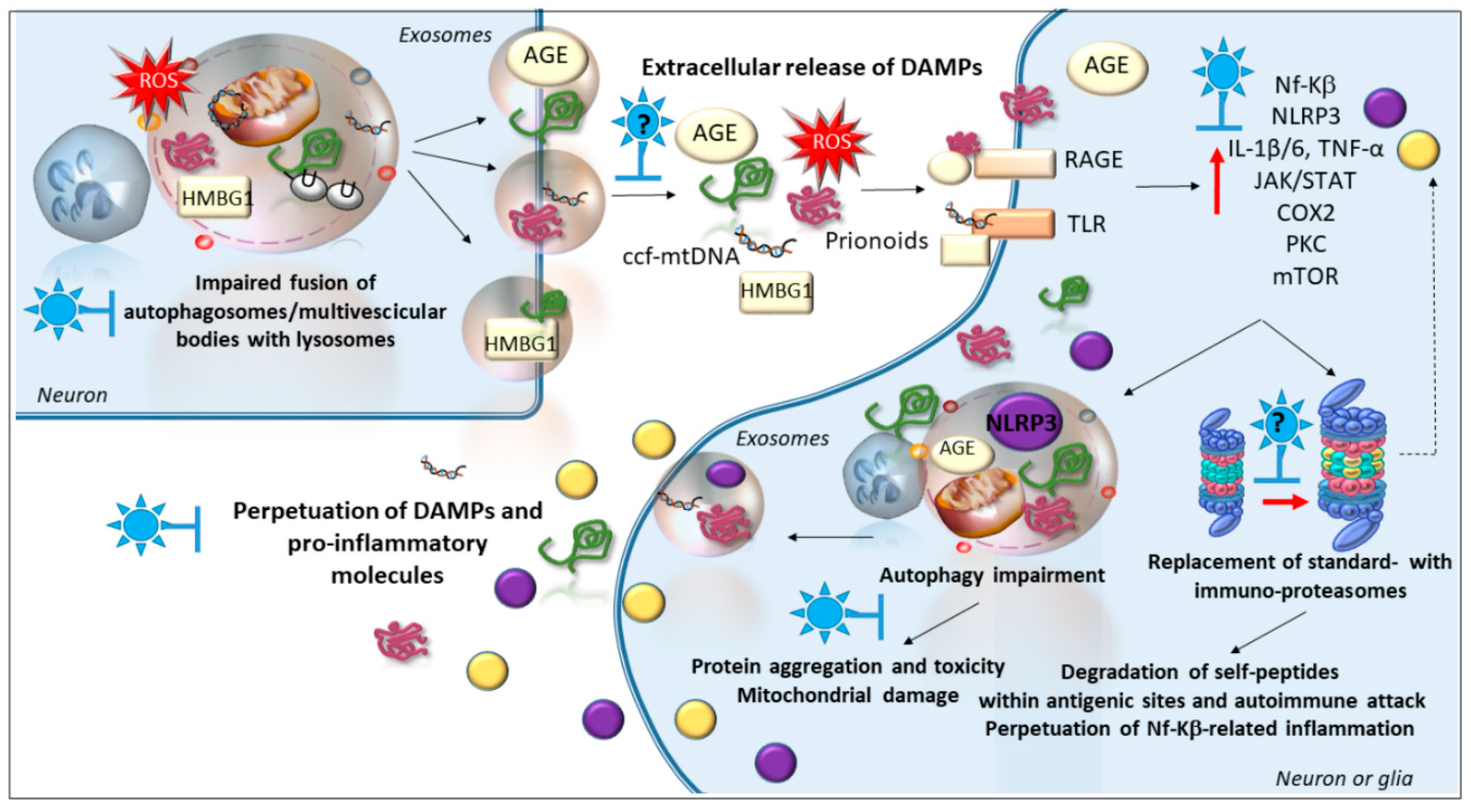
6. Phytochemicals and Inflammatory Pathways
Is There a Role of Autophagy and Immunoproteasome in the Anti-Inflammatory Effects of Phytochemicals?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D.A.; Docea, A.O.; Mahomoodally, M.F.; Lobine, D.; Chazot, P.L.; Kurt, B.; Tumer, T.B.; Moreira, A.C.; et al. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Polzella, M.; Frati, A.; Fornai, F. Phytochemicals Bridging Autophagy Induction and Alpha-Synuclein Degradation in Parkinsonism. Int. J. Mol. Sci. 2019, 20, 3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordoni, L.; Gabbianelli, R. Mitochondrial DNA and Neurodegeneration: Any Role for Dietary Antioxidants? Antioxidants 2020, 9, 764. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, J.; Perumal, M.K.; Vallikannan, B. A critical review on anti-angiogenic property of phytochemicals. J. Nutr. Biochem. 2019, 71, 1–15. [Google Scholar] [CrossRef]
- Abdullah, A.; Mohd Murshid, N.; Makpol, S. Antioxidant Modulation of mTOR and Sirtuin Pathways in Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Pinelli, R.; Biagioni, F.; Limanaqi, F.; Bertelli, M.; Scaffidi, E.; Polzella, M.; Busceti, C.L.; Fornai, F. A Re-Appraisal of Pathogenic Mechanisms Bridging Wet and Dry Age-Related Macular Degeneration Leads to Reconsider a Role for Phytochemicals. Int. J. Mol. Sci. 2020, 21, 5563. [Google Scholar] [CrossRef]
- Rebas, E.; Rzajew, J.; Radzik, T.; Zylinska, L. Neuroprotective Polyphenols: A Modulatory Action on Neurotransmitter Pathways. Curr. Neuropharmacol. 2020, 18, 431–445. [Google Scholar] [CrossRef]
- Momtaz, S.; Memariani, Z.; El-Senduny, F.F.; Sanadgol, N.; Golab, F.; Katebi, M.; Abdolghaffari, A.H.; Farzaei, M.H.; Abdollah, M. Targeting Ubiquitin-Proteasome Pathway by Natural Products: Novel Therapeutic Strategy for Treatment of Neurodegenerative Diseases. Front. Physiol. 2020, 11, 361. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Ryskalin, L.; Busceti, C.L.; Fornai, F. Molecular Mechanisms Linking ALS/FTD and Psychiatric Disorders, the Potential Effects of Lithium. Front. Cell. Neurosci. 2019, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Busceti, C.L.; Limanaqi, F.; Biagioni, F.; Gambardella, S.; Fornai, F. A Focus on the Beneficial Effects of Alpha Synuclein and a Re-Appraisal of Synucleinopathies. Curr. Protein Pept. Sci. 2018, 19, 598–611. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2013, 2, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Khan, H.; Shahab, U.; Rehman, S.; Rafi, Z.; Khan, M.Y.; Ansari, A.; Siddiqui, Z.; Ashraf, J.M.; Abdullah, S.M.; et al. Protein oxidation: An overview of metabolism of sulphur containing amino acid, cysteine. Front. Biosci. (Schol. Ed.) 2017, 9, 71–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aman, Y.; Ryan, B.; Torsetnes, S.B.; Knapskog, A.B.; Watne, L.O.; McEwan, W.A.; Fang, E.F. Enhancing mitophagy as a therapeutic approach for neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 155, 169–202. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Limanaqi, F.; Biagioni, F.; Gaglione, A.; Busceti, C.L.; Fornai, F. A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef] [Green Version]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Fornai, F. The effects of proteasome on baseline and methamphetamine-dependent dopamine transmission. Neurosci. Biobehav. Rev. 2019, 102, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Ryskalin, L.; Fornai, F. Interdependency between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front. Mol. Neurosci. 2018, 11, 299. [Google Scholar] [CrossRef] [Green Version]
- Limanaqi, F.; Busceti, C.L.; Biagioni, F.; Cantini, F.; Lenzi, P.; Fornai, F. Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA. Int. J. Mol. Sci. 2020, 21, 4021. [Google Scholar] [CrossRef]
- Song, J.X.; Lu, J.H.; Liu, L.F.; Chen, L.L.; Durairajan, S.S.; Yue, Z.; Zhang, H.Q.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Lai, M.; Yao, H.; Shah, S.Z.A.; Wu, W.; Wang, D.; Zhao, Y.; Wang, L.; Zhou, X.; Zhao, D.; Yang, L. The NLRP3-Caspase 1 Inflammasome Negatively Regulates Autophagy via TLR4-TRIF in Prion Peptide-Infected Microglia. Front. Aging Neurosci. 2018, 10, 116. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, C.; Zhang, J.; Wu, J.; Zhao, J. Inhibition of GSK-3β alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int. Immunopharmacol. 2019, 68, 234–241. [Google Scholar] [CrossRef]
- Lázaro, D.F.; Bellucci, A.; Brundin, P.; Outeiro, T.F. Editorial: Protein Misfolding and Spreading Pathology in Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 12, 312. [Google Scholar] [CrossRef] [Green Version]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.J.; Miranda, H.C.; Frankowski, H.; Batlevi, Y.; Young, J.E.; Le, A.; Ivanov, N.; Sopher, B.L.; Carromeu, C.; Muotri, A.R.; et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 2014, 17, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262, [published correction appears in Front. Mol. Neurosci. 2020, 12, 311]. [Google Scholar] [CrossRef] [Green Version]
- Natale, G.; Pompili, E.; Biagioni, F.; Paparelli, S.; Lenzi, P.; Fornai, F. Histochemical approaches to assess cell-to-cell transmission of misfolded proteins in neurodegenerative diseases. Eur. J. Histochem. 2013, 57, e5. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef]
- Zhang, M.; Schekman, R. Cell biology. Unconventional secretion, unconventional solutions. Science 2013, 340, 559–561. [Google Scholar] [CrossRef]
- Grimm, S.; Ernst, L.; Grotzinger, N.; Hohn, A.; Breusing, N.; Reinheckel, T.; Grune, T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic. Res. 2010, 44, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ott, C.; Hörlacher, M.; Weber, D.; Höhn, A.; Grune, T. Advanced-glycation-end-product-induced formation of immunoproteasomes: Involvement of RAGE and Jak2/STAT1. Biochem. J. 2012, 448, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla, A.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death Dis. 2014, 5, e1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hu, L.; Jiang, J.; Li, H.; Wu, Q.; Ooi, K.; Wang, J.; Feng, Y.; Zhu, D.; Xia, C. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J. Neuroinflamm. 2020, 17, 15. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Pietrocola, F.; Mariño, G.; Lissa, D.; Vacchelli, E.; Malik, S.A.; Niso-Santano, M.; Zamzami, N.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Pro-autophagic polyphenols reduce the acetylation of cytoplasmic proteins. Cell Cycle 2012, 11, 3851–3860. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Ma, K.; Wu, H.Y.; Wu, Y.P.; Li, B.X. Isoflavones Induce BEX2-Dependent Autophagy to Prevent ATR-Induced Neurotoxicity in SH-SY5Y Cells. Cell Physiol. Biochem. 2017, 43, 1866–1879. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, H.; Yin, T.; Gong, Y.; Yuan, G.; Chen, L.; Liu, J. Quercetin-modified gold-palladium nanoparticles as a potential autophagy inducer for the treatment of Alzheimer’s disease. J. Colloid. Interface Sci. 2019, 552, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sun, S.; Sun, Y.; Song, Q.; Zhu, J.; Song, N.; Chen, M.; Sun, T.; Xia, M.; Ding, J.; et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 2019, 15, 1860–1881. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Z.; Ardah, M.; Haikal, C.; Svanbergsson, A.; Diepenbroek, M.; Vaikath, N.N.; Li, W.; Wang, Z.Y.; Outeiro, T.F.; El-Agnaf, O.M.; et al. Dihydromyricetin and Salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl. Neurodegener. 2019, 8, 18. [Google Scholar] [CrossRef]
- Kuang, L.; Cao, X.; Lu, Z. Baicalein Protects against Rotenone-Induced Neurotoxicity through Induction of Autophagy. Biol. Pharm. Bull. 2017, 40, 1537–1543. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lin, S.; Xu, C.; Zhang, P.; Mei, X. Triggering of Autophagy by Baicalein in Response to Apoptosis after Spinal Cord Injury: Possible Involvement of the PI3K Activation. Biol. Pharm. Bull. 2018, 41, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yao, S.; Li, H.; Meng, Z.; Sun, X. Curcumin promotes functional recovery and inhibits neuronal apoptosis after spinal cord injury through the modulation of autophagy. J. Spinal Cord Med. 2019, 1–9, Epub ahead of print. [Google Scholar] [CrossRef]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Chuang, Y.C.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef]
- Vidoni, C.; Secomandi, E.; Castiglioni, A.; Melone, M.A.B.; Isidoro, C. Resveratrol protects neuronal-like cells expressing mutant Huntingtin from dopamine toxicity by rescuing ATG4-mediated autophagosome formation. Neurochem. Int. 2018, 117, 174–187. [Google Scholar] [CrossRef]
- Moon, J.H.; Lee, J.H.; Park, J.Y.; Kim, S.W.; Lee, Y.J.; Kang, S.J.; Seol, J.W.; Ahn, D.C.; Park, S.Y. Caffeine prevents human prion protein-mediated neurotoxicity through the induction of autophagy. Int. J. Mol. Med. 2014, 34, 553–558. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, Y.; Yu, H.; Li, M.; Hang, L.; Xu, X. Apigenin Protects Mouse Retina against Oxidative Damage by Regulating the Nrf2 Pathway and Autophagy. Oxid. Med. Cell. Longev. 2020, 2020, 9420704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, Q.; Zhang, L.; Wang, Q.; Yang, Z.; Liu, J.; Feng, L. Caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of Parkinson’s disease. Pharmacol. Res. 2019, 150, 104538. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Jeong, J.K.; Park, S.Y. Sulforaphane-induced autophagy flux prevents prion protein-mediated neurotoxicity through AMPK pathway. Neuroscience 2014, 278, 31–39. [Google Scholar] [CrossRef]
- Wang, N.; He, J.; Pan, C.; Wang, J.; Ma, M.; Shi, X.; Xu, Z. Resveratrol Activates Autophagy via the AKT/mTOR Signaling Pathway to Improve Cognitive Dysfunction in Rats With Chronic Cerebral Hypoperfusion. Front. Neurosci. 2019, 13, 859. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Guo, Y.; Dang, R.; Yang, M.; Liao, D.; Li, H.; Sun, Z.; Feng, Q.; Xu, P. Salvianolic acid B protects against lipopolysaccharide-induced behavioral deficits and neuroinflammatory response: Involvement of autophagy and NLRP3 inflammasome. J. Neuroinflamm. 2017, 14, 239. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Mei, X.; Cao, Y.; Guo, Z.; Yuan, Y.; Zhao, Z.; Song, C.; Guo, Y.; Shen, Z. Berberine attenuated pro-inflammatory factors and protect against neuronal damage via triggering oligodendrocyte autophagy in spinal cord injury. Oncotarget 2017, 8, 98312–98321. [Google Scholar] [CrossRef] [Green Version]
- Sigrist, S.J.; Carmona-Gutierrez, D.; Gupta, V.K.; Bhukel, A.; Mertel, S.; Eisenberg, T.; Madeo, F. Spermidine-triggered autophagy ameliorates memory during aging. Autophagy 2014, 10, 178–179. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xu, S.; Qian, Y.; Xiao, Q. Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav. Immun. 2017, 64, 162–172. [Google Scholar] [CrossRef]
- Zhuang, X.X.; Wang, S.F.; Tan, Y.; Song, J.X.; Zhu, Z.; Wang, Z.Y.; Wu, M.Y.; Cai, C.Z.; Huang, Z.J.; Tan, J.Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Mahapatra, K.K.; Mishra, S.R.; Mallick, S.; Negi, V.D.; Sarangi, I.; Patil, S.; Patra, S.K.; Bhutia, S.K. Bacopa monnieri inhibits apoptosis and senescence through mitophagy in human astrocytes. Food Chem. Toxicol. 2020, 141, 111367. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.H. Potential synergy of phytochemicals in cancer prevention: Mechanism of action. J. Nutr. 2004, 134, 3479S–3485S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos-Vega, R.; Oomah, B.D. Chemistry and classification of phytochemicals. In Handbook of Plant Food Phytochemicals; Tiwari, B., Brunton, N.P., Brennan, C.S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Joseph, J.; Cole, G.; Head, E.; Ingram, D. Nutrition, brain aging, and neurodegeneration. J. Neurosci. 2009, 29, 12795–12801. [Google Scholar] [CrossRef] [Green Version]
- Potì, F.; Santi, D.; Spaggiari, G.; Zimetti, F.; Zanotti, I. Polyphenol Health Effects on Cardiovascular and Neurodegenerative Disorders: A Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed Pharmacother. 2016, 84, 892–908. [Google Scholar] [CrossRef]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxid. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, N.K.; Modi, P.; Sharma, S.; Deep, S. Quercetin and Baicalein Act as Potent Antiamyloidogenic and Fibril Destabilizing Agents for SOD1 Fibrils. ACS Chem. Neurosci. 2020, 11, 1129–1138. [Google Scholar] [CrossRef]
- Ip, P.; Sharda, P.R.; Cunningham, A.; Chakrabartty, S.; Pande, V.; Chakrabartty, A. Quercitrin and quercetin 3-β-d-glucoside as chemical chaperones for the A4V SOD1 ALS-causing mutant. Protein Eng. Des. Sel. 2017, 30, 431–440. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, Y.; Shaw, A.M.; Goldfine, H.; Tian, J.; Cai, J. Enhancing TFEB-Mediated Cellular Degradation Pathways by the mTORC1 Inhibitor Quercetin. Oxid. Med. Cell. Longev. 2018, 2018, 5073420. [Google Scholar] [CrossRef] [Green Version]
- El-Horany, H.E.; El-Latif, R.N.; ElBatsh, M.M.; Emam, M.N. Ameliorative Effect of Quercetin on Neurochemical and Behavioral Deficits in Rotenone Rat Model of Parkinson’s Disease: Modulating Autophagy (Quercetin on Experimental Parkinson’s Disease). J. Biochem. Mol. Toxicol. 2016, 30, 360–369. [Google Scholar] [CrossRef]
- Pakrashi, S.; Chakraborty, J.; Bandyopadhyay, J. Neuroprotective Role of Quercetin on Rotenone-Induced Toxicity in SH-SY5Y Cell Line through Modulation of Apoptotic and Autophagic Pathways. Neurochem. Res. 2020, 45, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.R.; Wani, W.Y.; Sunkaria, A.; Kandimalla, R.J.; Sharma, R.K.; Verma, D.; Bal, A.; Gill, K.D. Quercetin attenuates neuronal death against aluminum-induced neurodegeneration in the rat hippocampus. Neuroscience 2016, 324, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Inden, M.; Shirai, K.; Sekine, S.I.; Masaki, Y.; Kurita, H.; Ichihara, K.; Inuzuka, T.; Hozumi, I. The effects of Brazilian green propolis that contains flavonols against mutant copper-zinc superoxide dismutase-mediated toxicity. Sci. Rep. 2017, 7, 2882. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.Y.; Lin, L.; Liu, B.L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef]
- Jang, J.H.; Lee, S.H.; Jung, K.; Yoo, H.; Park, G. Inhibitory Effects of Myricetin on Lipopolysaccharide-Induced Neuroinflammation. Brain Sci. 2020, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M. Baicalein attenuates α-synuclein aggregation, inflammasome activation and autophagy in the MPP+-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef]
- Baluchnejadmojarad, T.; Zeinali, H.; Roghani, M. Scutellarin alleviates lipopolysaccharide-induced cognitive deficits in the rat: Insights into underlying mechanisms. Int. Immunopharmacol. 2018, 54, 311–319. [Google Scholar] [CrossRef]
- Shin, J.W.; Kweon, K.J.; Kim, D.K.; Kim, P.; Jeon, T.D.; Maeng, S.; Sohn, N.W. Scutellarin Ameliorates Learning and Memory Deficit via Suppressing β-Amyloid Formation and Microglial Activation in Rats with Chronic Cerebral Hypoperfusion. Am. J. Chin. Med. 2018, 46, 1203–1223. [Google Scholar] [CrossRef]
- Che, D.N.; Cho, B.O.; Kim, J.S.; Shin, J.Y.; Kang, H.J.; Jang, S.I. Luteolin and Apigenin Attenuate LPS-Induced Astrocyte Activation and Cytokine Production by Targeting MAPK, STAT3, and NF-κB Signaling Pathways. Inflammation 2020, 43, 1716–1728. [Google Scholar] [CrossRef]
- Chesser, A.S.; Ganeshan, V.; Yang, J.; Johnson, G.V. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr. Neurosci. 2016, 19, 21–31. [Google Scholar] [CrossRef]
- Gu, H.F.; Nie, Y.X.; Tong, Q.Z.; Tang, Y.L.; Zeng, Y.; Jing, K.Q.; Zheng, X.L.; Liao, D.F. Epigallocatechin-3-gallate attenuates impairment of learning and memory in chronic unpredictable mild stress-treated rats by restoring hippocampal autophagic flux. PLoS ONE 2014, 9, e112683, Erratum in PLoS ONE 2015, 10, e0117649. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, J.; Wei, H.; Gu, T.; Wang, J.; Wu, Z.; Yang, Q. Genistein Attenuates Acute Cerebral Ischemic Damage by Inhibiting the NLRP3 Inflammasome in Reproductively Senescent Mice. Front. Aging Neurosci. 2020, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Cao, L.; Guan, T.; Chen, L.; Xin, H.; Li, Y.; Zheng, R.; Yu, D. Protection by genistein on cortical neurons against oxidative stress injury via inhibition of NF-kappaB, JNK and ERK signaling pathway. Pharm. Biol. 2015, 53, 1124–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddadi, R.; Nayebi, A.M.; Eyvari Brooshghalan, S. Silymarin prevents apoptosis through inhibiting the Bax/caspase-3 expression and suppresses toll like receptor-4 pathway in the SNc of 6-OHDA intoxicated rats. Biomed Pharmacother. 2018, 104, 127–136. [Google Scholar] [CrossRef]
- Haddadi, R.; Eyvari-Brooshghalan, S.; Nayebi, A.M.; Sabahi, M.; Ahmadi, S.A. Neuronal degeneration and oxidative stress in the SNc of 6-OHDA intoxicated rats; improving role of silymarin long-term treatment. Naunyn Schmiedebergs Arch. Pharmacol. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Lu, X.; Dong, J.; Zheng, D.; Li, X.; Ding, D.; Xu, H. Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia-reperfusion injury in rats. Nanomedicine 2020, 28, 102208. [Google Scholar] [CrossRef]
- Wu, Y.L.; Chang, J.C.; Lin, W.Y.; Li, C.C.; Hsieh, M.; Chen, H.W.; Wang, T.S.; Wu, W.T.; Liu, C.S.; Liu, K.L. Caffeic acid and resveratrol ameliorate cellular damage in cell and Drosophila models of spinocerebellar ataxia type 3 through upregulation of Nrf2 pathway. Free Radic. Biol. Med. 2018, 115, 309–317. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef]
- Guo, D.; Xie, J.; Zhao, J.; Huang, T.; Guo, X.; Song, J. Resveratrol protects early brain injury after subarachnoid hemorrhage by activating autophagy and inhibiting apoptosis mediated by the Akt/mTOR pathway. Neuroreport 2018, 29, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Le, K.; Chibaatar Daliv, E.; Wu, S.; Qian, F.; Ali, A.I.; Yu, D.; Guo, Y. SIRT1-regulated HMGB1 release is partially involved in TLR4 signal transduction: A possible anti-neuroinflammatory mechanism of resveratrol in neonatal hypoxic-ischemic brain injury. Int. Immunopharmacol. 2019, 75, 105779. [Google Scholar] [CrossRef]
- Yu, W.; Tao, M.; Zhao, Y.; Hu, X.; Wang, M. 4′-Methoxyresveratrol Alleviated AGE-Induced Inflammation via RAGE-Mediated NF-κB and NLRP3 Inflammasome Pathway. Molecules 2018, 23, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, A.; Muñoz-Arenas, G.; Caporal-Hernandez, K.; Vázquez-Roque, R.; Lopez-Lopez, G.; Kozina, A.; Espinosa, B.; Flores, G.; Treviño, S.; Guevara, J. Gallic acid improves recognition memory and decreases oxidative-inflammatory damage in the rat hippocampus with metabolic syndrome. Synapse 2020, e22186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Hsu, C.C.; Huang, H.J.; Chang, C.J.; Sun, S.H.; Lin, A.M. Gallic Acid Attenuated LPS-Induced Neuroinflammation: Protein Aggregation and Necroptosis. Mol. Neurobiol. 2020, 57, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Ogut, E.; Sekerci, R.; Akcay, G.; Yildirim, F.B.; Derin, N.; Aslan, M.; Sati, L. Protective effects of syringic acid on neurobehavioral deficits and hippocampal tissue damages induced by sub-chronic deltamethrin exposure. Neurotoxicol. Teratol. 2019, 76, 106839. [Google Scholar] [CrossRef] [PubMed]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.; Haylett, W.L.; Johnson, G.; Carr, J.A.; Bardien, S. Antioxidant effects of curcumin in models of neurodegeneration, aging, oxidative and nitrosative stress: A review. Neuroscience 2019, 406, 1–21. [Google Scholar] [CrossRef]
- Bohlmann, J.; Keeling, C.I. Terpenoid biomaterials. Plant J. 2008, 54, 656–669. [Google Scholar] [CrossRef]
- Aguiar, S.; Borowski, T. Neuropharmacological review of the nootropic herb Bacopa monnieri. Rejuvenation Res. 2013, 16, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Srivastav, S.; Fatima, M.; Mondal, A.C. Bacopa monnieri alleviates paraquat induced toxicity in Drosophila by inhibiting jnk mediated apoptosis through improved mitochondrial function and redox stabilization. Neurochem Int. 2018, 121, 98–107. [Google Scholar] [CrossRef]
- Wadhwa, R.; Konar, A.; Kaul, S.C. Nootropic potential of Ashwagandha leaves: Beyond traditional root extracts. Neurochem. Int. 2016, 95, 109–118. [Google Scholar] [CrossRef]
- Dutta, K.; Patel, P.; Julien, J.P. Protective effects of Withania somnifera extract in SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2018, 309, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, M.J.; Muralidhara. Standardized extract of Withania somnifera (Ashwagandha) markedly offsets rotenone-induced locomotor deficits, oxidative impairments and neurotoxicity in Drosophila melanogaster. J. Food Sci. Technol. 2015, 52, 1971–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent Advances in Studies on the Therapeutic Potential of Dietary Carotenoids in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yan, W.; Beight, C. Lutein and Zeaxanthin Isomers Protect against Light-Induced Retinopathy via Decreasing Oxidative and Endoplasmic Reticulum Stress in BALB/cJ Mice. Nutrients 2018, 10, 842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, G.; Rasul, A.; Anwar, H.; Aziz, N.; Razzaq, A.; Wei, W.; Ali, M.; Li, J.; Li, X. Role of Plant Derived Alkaloids and Their Mechanism in Neurodegenerative Disorders. Int. J. Biol. Sci. 2018, 14, 341–357. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Tang, H.; Li, W.; Gong, Y.; Li, S.; Huang, J.; Fang, Y.; Yuan, W.; Liu, Y.; Wang, S.; et al. AMPK and its activator Berberine in the Treatment of Neurodegenerative Diseases. Curr. Pharm. Des. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Liang, Y.; Chen, H.; Ji, X.; Huang, M. Berberine mitigates cognitive decline in an Alzheimer’s Disease Mouse Model by targeting both tau hyperphosphorylation and autophagic clearance. Biomed. Pharmacother. 2020, 121, 109670. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef]
- Chang, C.F.; Lee, Y.C.; Lee, K.H.; Lin, H.C.; Chen, C.L.; Shen, C.J.; Huang, C.C. Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 2016, 23, 72. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Cristofani, R.; Tedesco, B.; Ferrari, V.; Messi, E.; Piccolella, M.; Casarotto, E.; Chierichetti, M.; Cicardi, M.E.; Galbiati, M.; et al. Enhanced Clearance of Neurotoxic Misfolded Proteins by the Natural Compound Berberine and Its Derivatives. Int. J. Mol. Sci. 2020, 21, 3443. [Google Scholar] [CrossRef]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Wang, N.N.; Qi, H.; Qiu, Y.Y.; Zhang, C.H.; Brown, E.; Kong, H.; Kong, L. Up-Regulation Thioredoxin Inhibits Advanced Glycation End Products-Induced Neurodegeneration. Cell. Physiol. Biochem. 2018, 50, 1673–1686. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, S.; Zhang, Y.; Lin, X.; Song, Y.; Xue, Z.; Qian, H.; Wang, S.; Wan, G.; Zheng, X.; et al. Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. 2017, 8, e2738. [Google Scholar] [CrossRef]
- Vijayan, B.; Raj, V.; Nandakumar, S.; Kishore, A.; Thekkuveettil, A. Spermine protects alpha-synuclein expressing dopaminergic neurons from manganese-induced degeneration. Cell. Biol. Toxicol. 2019, 35, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Sankhe, R.; Mudgal, J.; Arora, D.; Nampoothiri, M. Spermidine, an autophagy inducer, as a therapeutic strategy in neurological disorders. Neuropeptides 2020, 102083. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Gambardella, S.; Biagioni, F.; Busceti, C.L.; Fornai, F. Epigenetic Effects Induced by Methamphetamine and Methamphetamine-Dependent Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 4982453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Said Ahmed, M.; Hung, W.Y.; Zu, J.S.; Hockberger, P.; Siddique, T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J. Neurol. Sci. 2000, 176, 88–94. [Google Scholar] [CrossRef]
- Arredondo, F.; Echeverry, C.; Abin-Carriquiry, J.A.; Blasina, F.; Antúnez, K.; Jones, D.P.; Go, Y.M.; Liang, Y.L.; Dajas, F. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic. Biol. Med. 2010, 49, 738–747. [Google Scholar] [CrossRef]
- Wang, J.; Mao, J.; Wang, R.; Li, S.; Wu, B.; Yuan, Y. Kaempferol Protects Against Cerebral Ischemia Reperfusion Injury Through Intervening Oxidative and Inflammatory Stress Induced Apoptosis. Front. Pharmacol. 2020, 11, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, R.M.; Mohamed, W.R.; Omar, H.A. A neuroprotective role of kaempferol against chlorpyrifos-induced oxidative stress and memory deficits in rats via GSK3β-Nrf2 signaling pathway. Pestic. Biochem. Physiol. 2018, 152, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Dhanraj, V.; Karuppaiah, J.; Balakrishnan, R.; Elangovan, N. Myricetin attenuates neurodegeneration and cognitive impairment in Parkinsonism. Front. Biosci (Elite Ed.) 2018, 10, 481–494. [Google Scholar]
- Wu, S.; Yue, Y.; Peng, A.; Zhang, L.; Xiang, J.; Cao, X.; Ding, H.; Yin, S. Myricetin ameliorates brain injury and neurological deficits via Nrf2 activation after experimental stroke in middle-aged rats. Food Funct. 2016, 7, 2624–2634. [Google Scholar] [CrossRef]
- Choi, S.M.; Kim, B.C.; Cho, Y.H.; Choi, K.H.; Chang, J.; Park, M.S.; Kim, M.K.; Cho, K.H.; Kim, J.K. Effects of Flavonoid Compounds on β-amyloid-peptide-induced Neuronal Death in Cultured Mouse Cortical Neurons. Chonnam. Med. J. 2014, 50, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Liu, G.Q. Protection against hydrogen peroxide-induced cytotoxicity in PC12 cells by scutellarin. Life Sci. 2004, 74, 2959–2973. [Google Scholar] [CrossRef]
- Chao, X.J.; Chen, Z.W.; Liu, A.M.; He, X.X.; Wang, S.G.; Wang, Y.T.; Liu, P.Q.; Ramassamy, C.; Mak, S.H.; Cui, W.; et al. Effect of tacrine-3-caffeic acid, a novel multifunctional anti-Alzheimer’s dimer, against oxidative-stress-induced cell death in HT22 hippocampal neurons: Involvement of Nrf2/HO-1 pathway. CNS Neurosci. Ther. 2014, 20, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective Effect of Caffeic Acid Phenethyl Ester in A Mouse Model of Alzheimer’s Disease Involves Nrf2/HO-1 Pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.; Kang, I.J.; Kim, B.; Sim, H.J.; Kim, D.W.; Ahn, J.H.; Lee, J.C.; Ryoo, S.; Shin, M.C.; Cho, J.H.; et al. Experimental Pretreatment with Chlorogenic Acid Prevents Transient Ischemia-Induced Cognitive Decline and Neuronal Damage in the Hippocampus through Anti-Oxidative and Anti-Inflammatory Effects. Molecules 2020, 25, 3578. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Q.; Lu, Y.; Wan, J.; Dai, H.; Zhou, X.; Lv, S.; Chen, X.; Zhang, X.; Hang, C.; et al. Cerebroprotection by salvianolic acid B after experimental subarachnoid hemorrhage occurs via Nrf2- and SIRT1-dependent pathways. Free Radic. Biol. Med. 2018, 124, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.J.; Lian, Y.G.; Zhao, H.Y.; Xu, Q.L. Curcumin protects from oxidative stress and inhibits α-synuclein aggregation in MPTP induced parkinsonian mice. Int. J. Clin. Exp. Med. 2016, 9, 2654–2665. [Google Scholar]
- Cui, Q.; Li, X.; Zhu, H. Curcumin ameliorates dopaminergic neuronal oxidative damage via activation of the Akt/Nrf2 pathway. Mol. Med. Rep. 2016, 13, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palle, S.; Neerati, P. Improved neuroprotective effect of resveratrol nanoparticles as evinced by abrogation of rotenone-induced behavioral deficits and oxidative and mitochondrial dysfunctions in rat model of Parkinson’s disease. Naunyn. Schmiedebergs Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, P.; Jain, C.K.; Mathur, A. Comparative evaluation of four triterpenoid glycoside saponins of bacoside A in alleviating sub-cellular oxidative stress of N2a neuroblastoma cells. J. Pharm. Pharmacol. 2018, 70, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Parihar, P.; Shetty, R.; Ghafourifar, P.; Parihar, M.S. Increase in oxidative stress and mitochondrial impairment in hypothalamus of streptozotocin treated diabetic rat: Antioxidative effect of Withania somnifera. Cell. Mol. Biol. (Noisy-le-grand). 2016, 62, 73–83. [Google Scholar]
- Ademowo, O.S.; Dias, I.H.K.; Diaz-Sanchez, L.; Sanchez-Aranguren, L.; Stahl, W.; Griffiths, H.R. Partial Mitigation of Oxidized Phospholipid-Mediated Mitochondrial Dysfunction in Neuronal Cells by Oxocarotenoids. J. Alzheimers Dis. 2020, 74, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.; Huang, C.; Yang, M.; Fan, C.; Li, Q.; Zhao, J.; Gan, D.; Li, A.; Zhu, L.; Lu, D. The Secretion from Bone Marrow Mesenchymal Stem Cells Pretreated with Berberine Rescues Neurons with Oxidative Damage Through Activation of the Keap1-Nrf2-HO-1 Signaling Pathway. Neurotox. Res. 2020, 38, 59–73. [Google Scholar] [CrossRef]
- Zhang, C.; Li, C.; Chen, S.; Li, Z.; Jia, X.; Wang, K.; Bao, J.; Liang, Y.; Wang, X.; Chen, M.; et al. Berberine protects against 6-OHDA-induced neurotoxicity in PC12 cells and zebrafish through hormetic mechanisms involving PI3K/AKT/Bcl-2 and Nrf2/HO-1 pathways. Redox Biol. 2017, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kumar, P.; Deshmukh, R. Neuroprotective potential of spermidine against rotenone induced Parkinson’s disease in rats. Neurochem. Int. 2018, 116, 104–111. [Google Scholar] [CrossRef]
- Costa, L.G.; de Laat, R.; Dao, K.; Pellacani, C.; Cole, T.B.; Furlong, C.E. Paraoxonase-2 (PON2) in brain and its potential role in neuroprotection. Neurotoxicology 2014, 43, 3–9. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef] [PubMed]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation - at the Center of Autophagy Regulation. Front. Cell. Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008, 4, 527–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tan, S.H.; Nicolas, V.; Bauvy, C.; Yang, N.D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The Autophagoproteasome a Novel Cell Clearing Organelle in Baseline and Stimulated Conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.Y.; Wang, Y.N.; Zhang, G.J. The influence of oxidation of membrane thiol groups on lysosomal proton permeability. Biochem. J. 2001, 360, 355–362. [Google Scholar] [CrossRef]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef] [Green Version]
- Janda, E.; Lascala, A.; Carresi, C.; Parafati, M.; Aprigliano, S.; Russo, V.; Savoia, C.; Ziviani, E.; Musolino, V.; Morani, F.; et al. Parkinsonian toxin-induced oxidative stress inhibits basal autophagy in astrocytes via NQO2/quinone oxidoreductase 2: Implications for neuroprotection. Autophagy 2015, 11, 1063–1080. [Google Scholar] [CrossRef]
- He, X.; Yuan, W.; Li, Z.; Hou, Y.; Liu, F.; Feng, J. 6-Hydroxydopamine induces autophagic flux dysfunction by impairing transcription factor EB activation and lysosomal function in dopaminergic neurons and SH-SY5Y cells. Toxicol. Lett. 2018, 283, 58–68. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, A.; Bell, B.A.; Peachey, N.S.; Daniele, L.L.; Reyes-Reveles, J.; Sharp, R.C.; Jun, B.; Bazan, N.G.; Sparrow, J.R.; Kim, H.J.; et al. Microtubule-Associated Protein 1 Light Chain 3B, (LC3B) Is Necessary to Maintain Lipid-Mediated Homeostasis in the Retinal Pigment Epithelium. Front. Cell. Neurosci. 2018, 12, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.D.; Zhao, J.J. TFEB Participates in the Aβ-Induced Pathogenesis of Alzheimer’s Disease by Regulating the Autophagy-Lysosome Pathway. DNA Cell Biol. 2015, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Mangano, G.; Durando, L.; Ragni, L.; Martini, C. The Nootropic Drug A-Glyceryl-Phosphoryl-Ethanolamine Exerts Neuroprotective Effects in Human Hippocampal Cells. Int. J. Mol. Sci. 2020, 21, 941. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Karsli-Uzunbas, G.; Poillet-Perez, L.; Sawant, A.; Hu, Z.S.; Zhao, Y.; Moore, D.; Hu, W.; White, E. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 2020, 34, 688–700. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell. 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, R.M.C.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Pérez, C.; Martinez-Gonzalez, L.; Paglione, M.; Di Schiavi, E.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- He, Y.; Jia, K.; Li, L.; Wang, Q.; Zhang, S.; Du, J.; Du, H. Salvianolic acid B attenuates mitochondrial stress against Aβ toxicity in primary cultured mouse neurons. Biochem. Biophys. Res. Commun. 2018, 498, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Ruffoli, R.; Bartalucci, A.; Frati, A.; Fornai, F. Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Nichols, M.; Zhang, J.; Polster, B.M.; Elustondo, P.A.; Thirumaran, A.; Pavlov, E.V.; Robertson, G.S. Synergistic neuroprotection by epicatechin and quercetin: Activation of convergent mitochondrial signaling pathways. Neuroscience 2015, 308, 75–94. [Google Scholar] [CrossRef]
- Cakir, Z.; Funk, K.; Lauterwasser, J.; Todt, F.; Zerbes, R.M.; Oelgeklaus, A.; Tanaka, A.; van der Laan, M.; Edlich, F. Parkin promotes proteasomal degradation of misregulated BAX. J. Cell Sci. 2017, 130, 2903–2913. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, X.; Xu, L.; Sun, R.; Ma, Z.; Deng, X.; Liu, B.; Fu, Q.; Qu, R.; Ma, S. Baicalin attenuates in vivo and in vitro hyperglycemia-exacerbated ischemia/reperfusion injury by regulating mitochondrial function in a manner dependent on AMPK. Eur. J. Pharmacol. 2017, 815, 118–126. [Google Scholar] [CrossRef]
- Wang, W.; Xu, J. Curcumin Attenuates Cerebral Ischemia-reperfusion Injury through Regulating Mitophagy and Preserving Mitochondrial Function. Curr. Neurovasc. Res. 2020, 17, 113–122. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Sun, X.; Shi, X.; Wang, L.; Huang, L.; Zhou, W. Evaluation of the neuroprotective effect of EGCG: A potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018, 9, 6349–6359. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, M.; Dai, Y.; Sun, Y.; Aman, Y.; Xu, Y.; Yu, P.; Zheng, Y.; Yang, J.; Zhu, X. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging (Albany NY) 2020, 12, 16852–16866. [Google Scholar] [CrossRef]
- Hang, W.; He, B.; Chen, J.; Xia, L.; Wen, B.; Liang, T.; Wang, X.; Zhang, Q.; Wu, Y.; Chen, Q.; et al. Berberine Ameliorates High Glucose-Induced Cardiomyocyte Injury via AMPK Signaling Activation to Stimulate Mitochondrial Biogenesis and Restore Autophagic Flux. Front. Pharmacol. 2018, 9, 1121. [Google Scholar] [CrossRef] [PubMed]
- Toti, L.; Bartalucci, A.; Ferrucci, M.; Fulceri, F.; Lazzeri, G.; Lenzi, P.; Soldani, P.; Gobbi, P.; La Torre, A.; Gesi, M. High-intensity exercise training induces morphological and biochemical changes in skeletal muscles. Biol. Sport. 2013, 30, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as Caloric Restriction Mimetics Regulating Mitochondrial Biogenesis and Mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Interaction between pathogenic proteins in neurodegenerative disorders. J. Cell. Mol. Med. 2012, 16, 1166–1183. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimón, J.; Lleó, A. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Charles, V.; Mezey, E.; Reddy, P.H.; Dehejia, A.; Young, T.A.; Polymeropoulos, M.H.; Brownstein, M.J.; Tagle, D.A. α-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci. Lett. 2000, 289, 29–32. [Google Scholar] [CrossRef]
- Takei, Y.; Oguchi, K.; Koshihara, H.; Hineno, A.; Nakamura, A.; Ohara, S. α-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum. Pathol. 2013, 44, 1171–1176. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol 2017, 134, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Ryskalin, L.; Biagioni, F.; Gambardella, S.; Busceti, C.L.; Falleni, A.; Lazzeri, G.; Fornai, F. Methamphetamine increases Prion Protein and induces dopamine-dependent expression of protease resistant PrPsc. Arch. Ital. Biol. 2017, 155, 81–97. [Google Scholar] [CrossRef] [PubMed]
- da Luz, M.H.; Peres, I.T.; Santos, T.G.; Martins, V.R.; Icimoto, M.Y.; Lee, K.S. Dopamine induces the accumulation of insoluble prion protein and affects autophagic flux. Front. Cell. Neurosci. 2015, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, D.; English, A.M. SOD1 oxidation and formation of soluble aggregates in yeast: Relevance to sporadic ALS development. Redox Biol. 2014, 2, 632–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamoto-Nagai, M.; Hisaka, S.; Naoi, M.; Maruyama, W. Modification of α-synuclein by lipid peroxidation products derived from polyunsaturated fatty acids promotes toxic oligomerization: Its relevance to Parkinson disease. J. Clin. Biochem. Nutr. 2018, 62, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Vicente, M.H.; Outeiro, T.F. The sour side of neurodegenerative disorders: The effects of protein glycation. J. Pathol. 2010, 221, 13–25. [Google Scholar]
- Adrover, M.; Mariño, L.; Sanchis, P.; Pauwels, K.; Kraan, Y.; Lebrun, P.; Vilanova, B.; Muñoz, F.; Broersen, K.; Donoso, J. Mechanistic insights in glycation-induced protein aggregation. Biomacromolecules 2014, 15, 3449–3462. [Google Scholar] [CrossRef]
- Zhu, M.; Han, S.; Fink, A.L. Oxidized quercetin inhibits α-synuclein fibrillization. Biochim. Biophys. Acta 2013, 1830, 2872–2881. [Google Scholar] [CrossRef]
- Medvedeva, M.; Barinova, K.; Melnikova, A.; Semenyuk, P.; Kolmogorov, V.; Gorelkin, P.; Erofeev, A.; Muronetz, V. Naturally occurring cinnamic acid derivatives prevent amyloid transformation of alpha-synuclein. Biochimie 2020, 170, 128–139. [Google Scholar] [CrossRef]
- Taebnia, N.; Morshedi, D.; Yaghmaei, S.; Aliakbari, F.; Rahimi, F.; Arpanaei, A. Curcumin-loaded amine-functionalized mesoporous silica nanoparticles inhibit α-synuclein fibrillation and reduce its cytotoxicity-associated effects. Langmuir 2016, 32, 13394–13402. [Google Scholar] [CrossRef] [PubMed]
- Šneideris, T.; Baranauskiene, L.; Cannon, J.G.; Rutkiene, R.; Meškys, R.; Smirnovas, V. Looking for a generic inhibitor of amyloid-like fibril formation among flavone derivatives. Peer J. 2015, 3, e1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic acid interacts with α-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta 2014, 1844, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughey, B.; Raymond, L.D.; Raymond, G.J.; Maxson, L.; Silveira, J.; Baron, G.S. Inhibition of protease-resistant prion protein accumulation in vitro by curcumin. J. Virol. 2003, 77, 5499–5502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner-Bratkovic, I.; Gaspersic, J.; Smid, L.M.; Bresjanac, M.; Jerala, R. Curcumin binds to the alpha-helical intermediate and to the amyloid form of prion protein—A new mechanism for the inhibition of PrPSc accumulation. J. Neurochem. 2008, 104, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Yu, K.H.; Jheng, C.P.; Chung, R.; Lee, C.I. Curcumin reduces amyloid fibrillation of prion protein and decreases reactive oxidative stress. Pathogens 2013, 2, 506–519. [Google Scholar] [CrossRef]
- Ladner-Keay, C.L.; Ross, L.; Perez-Pineiro, R.; Zhang, L.; Bjorndahl, T.C.; Cashman, N.; Wishart, D.S. A simple in vitro assay for assessing the efficacy, mechanisms and kinetics of anti-prion fibril compounds. Prion 2018, 12, 280–300. [Google Scholar] [CrossRef]
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed beta-amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2005, 336, 444–449. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-beta aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladiwala, A.R.A.; Lin, J.C.; Bale, S.S.; Marcelino-Cruz, A.M.; Bhattacharya, M.; Dordick, J.S.; Tessier, P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid a beta into off-pathway conformers. J. Biol. Chem. 2010, 285, 24228–24237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.T.; Yang, H.Y.; Wang, W.; Wu, Q.Q.; Tian, Y.R.; Jia, J.P. Sulforaphane Inhibits the Generation of Amyloid-β Oligomer and Promotes Spatial Learning and Memory in Alzheimer’s Disease (PS1V97L) Transgenic Mice. J. Alzheimers. Dis. 2018, 62, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.T.; Samarat, K.; Takamura, Y.; Azo-Oussou, A.F.; Nakazono, Y.; Vestergaard, M.C. Polyphenols Modulate Alzheimer’s Amyloid Beta Aggregation in a Structure-Dependent Manner. Nutrients 2019, 11, 756. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S. Molecular Pharmacology of Rosmarinic and Salvianolic Acids: Potential Seeds for Alzheimer’s and Vascular Dementia Drugs. Int. J. Mol. Sci. 2018, 19, 458. [Google Scholar] [CrossRef] [Green Version]
- Dariya, B.; Nagaraju, G.P. Advanced glycation end products in diabetes, cancer and phytochemical therapy. Drug Discov. Today 2020, 25, 1614–1623. [Google Scholar] [CrossRef]
- Rebollo-Hernanz, M.; Fernández-Gómez, B.; Herrero, M.; Aguilera, Y.; Martín-Cabrejas, M.A.; Uribarri, J.; Del Castillo, M.D. Inhibition of the Maillard Reaction by Phytochemicals Composing an Aqueous Coffee Silverskin Extract via a Mixed Mechanism of Action. Foods 2019, 8, 438. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.F.; Zhang, Y.J.; Zhou, H.Y.; Wang, H.M.; Tian, L.P.; Liu, J.; Ding, J.Q.; Chen, S.D. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Buratta, S.; Chiaradia, E.; Tognoloni, A.; Gambelunghe, A.; Meschini, C.; Palmieri, L.; Muzi, G.; Urbanelli, L.; Emiliani, C.; Tancini, B. Effect of Curcumin on Protein Damage Induced by Rotenone in Dopaminergic PC12 Cells. Int. J. Mol. Sci. 2020, 21, 2761. [Google Scholar] [CrossRef]
- Regitz, C.; Dußling, L.M.; Wenzel, U. Amyloid-beta (Aβ₁₋₄₂)-induced paralysis in Caenorhabditis elegans is inhibited by the polyphenol quercetin through activation of protein degradation pathways. Mol. Nutr. Food Res. 2014, 58, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Rossignol, J.; Dunbar, G.L. Curcumin Modulates Molecular Chaperones and Autophagy-Lysosomal Pathways In Vitro after Exposure to Aβ42. J. Alzheimers Dis. Parkinsonism. 2017, 7, 299. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits α-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wei, W.; Gaertig, M.A.; Li, S.; Li, X.-J. Therapeutic Effect of Berberine on Huntington’s Disease Transgenic Mouse Model. PLoS ONE 2015, 10, e0134142. [Google Scholar] [CrossRef]
- Deng, J.; Koutras, C.; Donnelier, J.; Alshehri, M.; Fotouhi, M.; Girard, M.; Casha, S.; McPherson, P.S.; Robbins, S.M.; Braun, J.E.A. Neurons Export Extracellular Vesicles Enriched in Cysteine String Protein and Misfolded Protein Cargo. Sci. Rep. 2017, 7, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ugras, S.; Daniels, M.J.; Fazelinia, H.; Gould, N.S.; Yocum, A.K.; Luk, K.C.; Luna, E.; Ding, H.; McKennan, C.; Seeholzer, S.; et al. Induction of the immunoproteasome subunit Lmp7 links proteostasis and immunity in α-synuclein aggregation disorders. EBioMedicine 2018, 31, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Wagner, L.K.; Gilling, K.E.; Schormann, E.; Kloetzel, P.M.; Heppner, F.L.; Krüger, E.; Prokop, S. Immunoproteasome deficiency alters microglial cytokine response and improves cognitive deficits in Alzheimer’s disease-like APPPS1 mice. Acta Neuropathol. Commun. 2017, 5, 52. [Google Scholar] [CrossRef]
- Xie, X.; Bi, H.L.; Lai, S.; Zhang, Y.L.; Li, N.; Cao, H.J.; Han, L.; Wang, H.X.; Li, H.H. The immunoproteasome catalytic β5i subunit regulates cardiac hypertrophy by targeting the autophagy protein ATG5 for degradation. Sci. Adv. 2019, 5, eaau0495. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zou, L.X.; Lin, Q.Y.; Yan, X.; Bi, H.L.; Xie, X.; Wang, S.; Wang, Q.S.; Zhang, Y.L.; Li, H.H. Resveratrol as a new inhibitor of immunoproteasome prevents PTEN degradation and attenuates cardiac hypertrophy after pressure overload. Redox Biol. 2019, 20, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Z.; Zhang, Z.; Liu, H.; Li, W.; Guo, X.; Zhang, Z.; Liu, Y.; Jia, L.; Li, Y.; Ren, Y.; et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ. 2019, 26, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Jamwal, S.; Kumar, P. Neuroprotective potential of Quercetin in combination with piperine against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. Neural Regen. Res. 2017, 12, 1137–1144. [Google Scholar] [CrossRef]
- Cordaro, M.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Peritore, A.F.; D’Amico, R.; Gugliandolo, E.; di Paola, R.; Cuzzocrea, S. 2-Pentadecyl-2-Oxazoline Reduces Neuroinflammatory Environment in the MPTP Model of Parkinson Disease. Mol. Neurobiol. 2018, 55, 9251–9266. [Google Scholar] [CrossRef]
- Du. Y.: Luo. M.: Du. Y.: Xu, M.; Yao, Q.; Wang, K.; He, G. Liquiritigenin Decreases Aβ Levels and Ameliorates Cognitive Decline by Regulating Microglia M1/M2 Transformation in AD Mice. Neurotox. Res. 2020. Epub ahead of print. [Google Scholar] [CrossRef]
- Chen, L.; Pan, H.; Bai, Y.; Li, H.; Yang, W.; Lin, Z.X.; Cui, W.; Xian, Y.F. Gelsemine, a natural alkaloid extracted from Gelsemium elegans Benth. alleviates neuroinflammation and cognitive impairments in Aβ oligomer-treated mice. Psychopharmacology (Berl). 2020, 237, 2111–2124. [Google Scholar] [CrossRef]
- Yang, E.J.; Mahmood, U.; Kim, H.; Choi, M.; Choi, Y.; Lee, J.P.; Cho, J.Y.; Hyun, J.W.; Kim, Y.S.; Chang, M.J.; et al. Phloroglucinol ameliorates cognitive impairments by reducing the amyloid β peptide burden and pro-inflammatory cytokines in the hippocampus of 5XFAD mice. Free Radic. Biol. Med. 2018, 126, 221–234. [Google Scholar] [CrossRef]
- Mattioli, R.; Francioso, A.; d’Erme, M.; Trovato, M.; Mancini, P.; Piacentini, L.; Casale, A.M.; Wessjohann, L.; Gazzino, R.; Costantino, P.; et al. Anti-Inflammatory Activity of A Polyphenolic Extract from Arabidopsis thaliana in In Vitro and In Vivo Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 708. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a New Therapeutic Strategy to Control Neuroinflammation: Neuroprotective Effects in Experimental Models of Spinal Cord and Brain Injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223. [Google Scholar] [CrossRef] [PubMed]
- Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caltagirone, C.; Cisari, C.; Schievano, C.; Di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S.; Stroke Study Group. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: From Rodent to Man. Transl. Stroke Res. 2016, 7, 54–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozkurt, A.A.; Mustafa, G.; Tarık, A.; Adile, O.; Murat, S.H.; Mesut, K.; Yıldıray, K.; Coskun, S.; Murat, C. Syringaldehyde exerts neuroprotective effect on cerebral ischemia injury in rats through anti-oxidative and anti-apoptotic properties. Neural Regen. Res. 2014, 9, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective Effect of Quercetin Against the Detrimental Effects of LPS in the Adult Mouse Brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Koza, L.A.; Winter, A.N.; Holsopple, J.; Baybayon-Grandgeorge, A.N.; Pena, C.; Olson, J.R.; Mazzarino, R.C.; Patterson, D.; Linseman, D.A. Protocatechuic Acid Extends Survival, Improves Motor Function, Diminishes Gliosis, and Sustains Neuromuscular Junctions in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Nutrients 2020, 12, 1824. [Google Scholar] [CrossRef]
- Winter, A.N.; Ross, E.K.; Wilkins, H.M.; Stankiewicz, T.R.; Wallace, T.; Miller, K.; Linseman, D.A. An anthocyanin-enriched extract from strawberries delays disease onset and extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Nutr. Neurosci. 2018, 21, 414–426. [Google Scholar] [CrossRef]
- Huang, Q.; Ye, X.; Wang, L.; Pan, J. Salvianolic acid B abolished chronic mild stress-induced depression through suppressing oxidative stress and neuro-inflammation via regulating NLRP3 inflammasome activation. J. Food Biochem. 2019, 43, e12742. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, F.; Guan, X. Baicalin reverse depressive-like behaviors through regulation SIRT1-NF-kB signalling pathway in olfactory bulbectomized rats. Phytother. Res. 2019, 33, 1480–1489. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Polzella, M.; Fabrizi, C.; Fornai, F. Potential Antidepressant Effects of Scutellaria baicalensis, Hericium erinaceus and Rhodiola rosea. Antioxidants 2020, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, G.; Gamba, P.; Badilli, U.; Gargiulo, S.; Maina, M.; Guina, T.; Calfapietra, S.; Biasi, F.; Cavalli, R.; Poli, G.; et al. Loading into nanoparticles improves quercetin’s efficacy in preventing neuroinflammation induced by oxysterols. PLoS ONE 2014, 9, e96795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silswal, N.; Reddy, N.S.; Qureshi, A.A.; Qureshi, N. Resveratrol Downregulates Biomarkers of Sepsis Via Inhibition of Proteasome’s Proteases. Shock 2018, 5, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Xie, S.P.; Saw, W.T.; Ho, P.G.H.; Wang, H.; Lei, Z.; Yi, Z.; Tan, E.K. The Therapeutic Implications of Tea Polyphenols Against Dopamine (DA) Neuron Degeneration in Parkinson’s Disease (PD). Cells 2019, 8, 911. [Google Scholar] [CrossRef] [Green Version]
- Farkhondeh, T.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Folgado, S.L.; Rajabpour-Sanati, A.; Khazdair, M.R.; Samarghandian, S. Green tea catechins inhibit microglial activation which prevents the development of neurological disorders. Neural Regen. Res. 2020, 15, 1792–1798. [Google Scholar]
- Bai, D.; Jin, G.; Zhang, D.; Zhao, L.; Wang, M.; Zhu, Q.; Zhu, L.; Sun, Y.; Liu, X.; Chen, X.; et al. Natural silibinin modulates amyloid precursor protein processing and amyloid-β protein clearance in APP/PS1 mice. J. Physiol. Sci. 2019, 69, 643–652. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Models of Neuroinflammation | Anti-Inflammatory Phytochemicals |
|---|---|
| Parkinson’s disease (PD) SNCA- and LPS-induced neurodegeneration in mice [53] Cells transfected with C-terminal modified α-syn and BAC-α-syn-GFP transgenic mice [54] 6-OHDA-treated rats [95] MPTP-treated rats [235] and mice [236] | Kaempferol [53] Dihydromyricetin and Salvianolic acid [54] Silymarin [95] Quercetin [235] 2-pentadecyl-2-oxazoline [236] |
| Alzheimer’s disease (AD) APP/PS1 transgenic mice [237] Aβ oligomer-treated mice [238] 5XFAD mice [239] Aβ25⁻35 peptide-treated BV2 murine microglia cells and human Aβ1⁻42-expressing flies [240] | Liquiritigenin [237] Gelsemine [238] Phloroglucinol [239] Arabidopsis extract (caffeic acid, kaempferol, quercetin, synapic acid, luteolin) [240] |
| Traumatic Brain Injury (TBI) | 2-pentadecyl-2-oxazoline [241] Artemisin [242] |
| Spinal Cord Injury (SCI) | Berberine [67] Curcumin [57] 2-pentadecyl-2-oxazoline [241] |
| Brain hemorrhage Prechiasmatic cistern blood injection [243] and internal carotid rupture [101] and/or Brain ischemia/hypoperfusion Middle Cerebral Artery Occlusion (MCAO) [93,244,245,246] | Curcumin [243] Genistein [93] Resveratrol [101] 2-pentadecyl-2-oxazoline [244] Palmitoylethanolamide-luteolin [245] Syringaldehyde [246] |
| Lipopolysaccharide (LPS) | Salvianolic acid [66] Resveratrol [69] Quercetin [247] |
| Amyotrophic Lateral Sclerosis (ALS) SOD1G93A mice [112,248,249] | Ashwagandha [112] Protocatechuic acid [248] Anthocyanin-enriched strawberries extract [249] |
| Anxiety and depression Chronic mild stress, and olfactory bulbectomized rat model of depression [250,251] | Salvianolic acid [250] Baicalin [251,252] |
| Phytochemical Experimental Model Polyphenols | Anti-Oxidant Effects | Mitochondrial Protection | Anti-Apoptotic Effects | Proteostasis | Anti-Inflammatory Effects | Autophagy-Related Effects | |
|---|---|---|---|---|---|---|---|
| Quercetin | PD [50,81,82,235]; AD [51,222]; Metal neurotoxicity [83]; LPS [247] | ↓LDH ↓ROS ↓MDA ↑GSH [51,81,82,235] | ↑MMP ↓ultrastructural alterations [82,83] | ↓caspase-3 ↓BAX/Bcl-2 ↓CHOP ↓DNA fragmentation [51,158,159,160,161,247] | Aβ [51,211,222] α-syn [200] | ↓Reactive gliosis (GFAP) ↓TLR-4 ↓COX-2 ↓IL-1β, IL-6, TNF-α [235,247] | ↑LC3II ↑Beclin-1 ↓P62 ↑LAMP-2 ↑flux [50,51,81,82,222] |
| Kaempferol | PD [52,53,84]; Ischemia [85,132] | ↓ROS ↓SDH ↓MDA ↑NRF2 ↑SOD ↑GSH [52,84,85,132] | ↑MMP ↑ATP ↑mitophagy ↓cyt-c [52,85] | ↓caspase-3/9 ↓JNK/p38MAPK [52] | SOD-1 [84] Aβ [211] | ↓GFAP ↓NLRP3 ↓TNF-α, IL-1β [53,132] | ↑LC3II ↓P62 ↑flux ↑AMPK [52,53,84,85] |
| (Dihydro)-Myricetin | PD [54,134]; LPS [86]; Ischemia [135] | ↓ROS ↓MDA, LPO ↑NRF2 ↑SOD ↑GSH ↑CAT [134,135] | ↑MMP ↑ATP ↓cyt-c [134,135] | ↓caspase-3/9 ↓JNK/p38MAPK ↓BAX/Bcl-2 [86,134] | α-syn [54] PrP [209] Aβ [211] | ↓GFAP ↓iNOS, COX-2, ↓PGE2 ↓IL-1β, TNF-α [54,86] | ↑LC3II ↑LAMP-1/2A [54] |
| Scutellarin | LPS [88]; Chronic hypoperfusion [89]; H2O2 neurotoxicity [137] | ↓ROS ↓MDA ↓LDH ↑NRF2 ↑SOD ↑GSH [88,137] | ↑MMP [137] | ↓sub-G1 peak in flow cytometry [137] | Aβ [89,203] α-syn [203] | ↓NF-κB ↓ IL-1β, TNF-α ↓IBA-1 microgliosis [88,89] | ↑LC3II ↑Beclin-1 ↓P62 ↓mTOR [88] |
| Baicalein | PD [55,87]; SCI [56]; Ischemia [180] | ↓ROS [180] | ↑mitophagy ↑MMP ↓fission [55,180] | ↓caspase-3/9/12 ↓BAX/Bcl-2 [55,56,87,180] | α-syn [87] | ↓Microglial inflammasome ↓IL-1β [87] | ↑PI3K ↑LC3II ↑Beclin-1 ↓P62 ↑AMPK [55,56,180] |
| Apigenin | AMD [61]; LPS [90]; | ↑NRF2 ↑HO-1, NQO-1 ↑SOD, GSH-Px ↓ROS, MDA [61] | - | ↓ ERK, STAT3 [90] | - | ↓Astrocyte activation ↓NF-κB ↓IL-31 and IL-33 [90] | ↑LC3II ↓P62 [61] |
| Catechins | PD [50,255]; Prion [63]; AD [91]; Chronic stress [92]; Brain hemorrhage [182] | ↓ROS ↑NRF2 ↑PGC-1α [91,182,255] | ↑mitophagy ↓fragmentation ↓mtDNA copy number ↓cyt-c [63,182] | ↓BAX/Bcl-2 ↓TUNEL [63,92] | α-syn [203,205,255] p-tau [91] Aβ [92,203,205,211,216] | ↑M2 microglia polarization ↓NLRP3 ↓NF-κB ↓IL-1β, TNF-α ↓iNOS ↓COX-2 [256] | ↑LC3II ↓P62 ↑SIRT-1 ↓mTOR ↑flux [50,63,92,182] |
| Genistein | Ischemia [93]; H2O2 neurotoxicity [94] | ↓LDH, ROS [93,94] | - | ↓BAX/Bcl-2 ↓caspase-1/3/9 ↓JNK, ERK [93,94] | - | ↓NLRP3 in microglia and neurons ↓NF-κB ↓IL-1β, TNF-α [93,94] | - |
| Silymarin/ Silibinin | PD [95,96]; AD [257] | ↑SOD, GSH-Px, CAT ↓MDA [95,96,257] | - | ↓BAX/Bcl-2 ↓caspase-3/9 ↓TLR4 [95] | APP and Aβ [257] | - | - |
| Resveratrol | PD [50,58,144]; Chronic hypoperfusion [65]; LPS [69]; Ischemia [97,101]; AD [99,214]; Brain hemorrhage [100]; AGEs-induced neuroinflammation [102] | ↓ROS ↓MDA ↑SOD, GSH, CAT [58,59,65,97,99,144] | ↓fragmentation ↑MMP ↑mitophagy ↑Complex-I activity [58,99,144] | ↓TUNEL ↓BAX/Bcl-2 ↓caspase-3/9 ↓ p38 and JNK [65,97,99,100] | polyQ-Htt [59] PrP [209] Aβ [213,214] | ↑M2 microglia polarization ↓STAT3/6 ↑PGC-1α ↓TLR-4/RAGE— NF-κB ↓HMGB1, NLRP3 ↓iNOS, COX-2 [69,101,102] | ↑ATG4 ↑LC3II ↓P62 ↑flux ↓Akt/mTOR ↑SIRT1 [50,58,59,65,99,100,101] |
| Syringic/gallic acid | Metabolic syndrome [103]; LPS [104]; Deltamethrin neurotoxicity [105] | ↓ROS ↑SOD, CAT [103,105] | - | ↓caspase-3 [105] | α-syn [104,204] | ↓Reactive gliosis (↓GFAP, ED-1) ↓iNOS, IL-1β, TNF-α [103] | - |
| Caffeic and chlorogenic acids | PD [62]; Spinocerebellar ataxia [98]; Glutamate excitotoxicity [138]; AD [139]; Ischemia [140] | ↓ROS ↑NRF2 ↑SOD [98,138,139,140] | ↑MMP [138] | ↓caspase-9 [139] | α-syn [62] polyQ-ataxin3 [98] | ↓Reactive gliosis (↓GFAP, ED-1) ↓IL-2, TNF-α [139,140] | ↑JNK/Bcl-2 ↑LC3II ↓P62 [62,98] |
| Rosmarinic and Salvianolic acid | PD [54]; LPS [66]; Brain hemorrhage [141]; AD [172] | ↓ROS ↓MDA ↑SOD, GSH-Px, ↑GSH, CAT ↑NRF2 ↑HO-1, NQO-1 [141,172] | ↓fragmentation ↑MMP ↑ATP [172] | - | α-syn [54] Aβ [212] | ↓GFAP and IBA-1 ↓NLRP3 ↓IL-1β, IL6, and TNF-α [54,66] | ↑LC3II ↑Beclin-1 ↑LAMP-1/2A [54,66] |
| Curcumin | PD [50,70,106,142,143,220,224]; SCI [57]; Ischemia [181]; AD [223]; Brain hemorrhage [243] | ↓ROS ↓MDA ↑GSH ↑NRF2 [70,106,142,143,224] | ↑MMP ↑ATP ↑mitophagy [70,181] | ↓BAX/Bcl-2 Caspase-3/9 [106,224] | APP and Aβ [106,223] α-syn [142,202,220,224] PrP [206,207,208,209] | ↓Gliosis ↓iNOS ↓TNF-α, IL-1β, IL-1α ↓TLR4 ↑M2 microglia polarization [57,224,243] | ↑LC3II ↓P62 ↓Akt/mTOR ↑TFEB [50,57,70,106,181,220,223] |
| Terpenes | |||||||
| Bacosides | Benzo[a]pyrene [71]; PD [110]; H2O2 neurotoxicity [145] | ↓ROS ↑SOD, CAT ↑NRF2 [71,110,145] | ↓ cyt-c ↑mitophagy ↑ATP ↑MMP [71,110,145] | ↓TUNEL, Annexin-V ↓JNK ↓caspase-3 [71,110,145] | - | - | ↑LC3II, Beclin-1, ATG5, ULK1 [71] |
| Withanolides | ALS [111]; PD [112]; Diabetes [146] | ↓ROS, LPO ↑GSH, SOD [112,146] | ↑complex I–III and complex II–III activity ↓mitochondrial permeabilization [112,146] | - | SOD-1 [111] | ↓Gliosis (↓GFAP and IBA-1) ↓NF-κB ↓COX-2 [111] | ↑LC3II ↓ P62 [111] |
| Carotenoids | AMD [115]; AD [147] | ↓ROS ↑GSH ↑NRF2 [115,147] | ↓mitochondrial uncoupling [147] | ↓JNK and ↓ER stress [115] | - | - | - |
| Alkaloids | |||||||
| Berberine | SCI [67]; AD [118,119]; ALS/FTD [120]; SBMA [121]; tert-butyl hydroperoxide neurotoxicity [148]; PD [149]; HD [225] | ↓ROS ↑NRF2—HO-1 [148,149] | ↑MMP [148] | ↓caspase-3 ↓BAX/Bcl-2 [67,148,149] | p-tau [118] Aβ, APP [119] TDP-43 [120] ARpolyQ [121] polyQ-Htt [225] | ↓IL-1β, TNF-α [67] | ↑LC3B, ATG16L, and ATG7 ↓P62 ↓GSK3β ↓mTOR ↑ⅢPI3K/Beclin-1 [67,118,119] |
| Caffeine | Prion [60] | - | - | ↓JNK ↓DNA strand breakage [60] | PrP [60] | - | ↑LC3II ↑flux [60] |
| Other | |||||||
| Sulforaphane | Prion [64]; PD [123]; Diabetes [124]; AD [215] | ↓ROS, LDH, MDA ↑NRF-2, GSH, NQO1, THx [64,123,124] | - | ↓TUNEL, Annexin-V ↓caspase-3 [64,123] | PrP [64] AGE in retina [124] Aβ [215] | - | ↑LC3II ↓P62 ↑AMPK ↓mTOR [64,123] |
| Spermine/ Spermidine | Ischemia [125]; PD [126,150,183]; AD [183] | ↓MDA ↑GSH [150] | ↑mitophagy ↓ cyt-c [125,183] | ↓caspase-3 [125] | α-syn [126] | ↓TNF-α, IL-1β, IL-6 [150] | ↑LC3II ↑Beclin-1 ↑flux [125,126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limanaqi, F.; Biagioni, F.; Mastroiacovo, F.; Polzella, M.; Lazzeri, G.; Fornai, F. Merging the Multi-Target Effects of Phytochemicals in Neurodegeneration: From Oxidative Stress to Protein Aggregation and Inflammation. Antioxidants 2020, 9, 1022. https://doi.org/10.3390/antiox9101022
Limanaqi F, Biagioni F, Mastroiacovo F, Polzella M, Lazzeri G, Fornai F. Merging the Multi-Target Effects of Phytochemicals in Neurodegeneration: From Oxidative Stress to Protein Aggregation and Inflammation. Antioxidants. 2020; 9(10):1022. https://doi.org/10.3390/antiox9101022
Chicago/Turabian StyleLimanaqi, Fiona, Francesca Biagioni, Federica Mastroiacovo, Maico Polzella, Gloria Lazzeri, and Francesco Fornai. 2020. "Merging the Multi-Target Effects of Phytochemicals in Neurodegeneration: From Oxidative Stress to Protein Aggregation and Inflammation" Antioxidants 9, no. 10: 1022. https://doi.org/10.3390/antiox9101022