
Sulfate Aerosols from Non-Explosive Volcanoes: Chemical-Radiative Effects in the Troposphere and Lower Stratosphere

 ,
,
Abstract
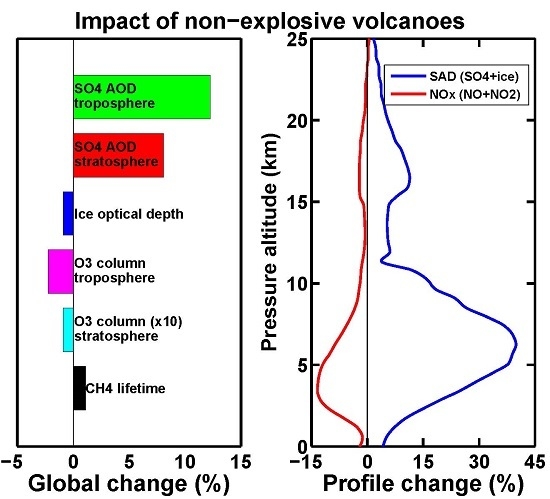
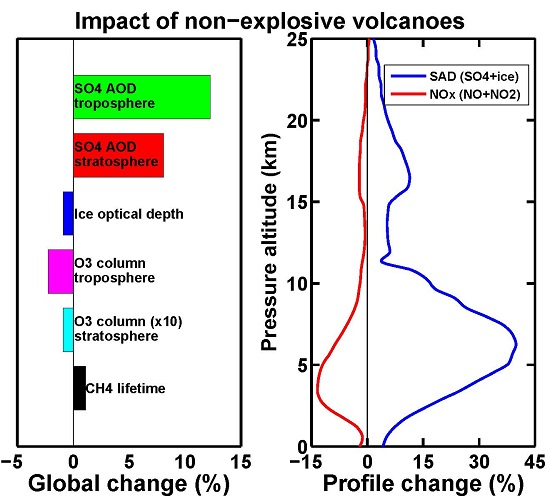
:
1. Introduction
2. Experimental Section
2.1. ULAQ-CCM
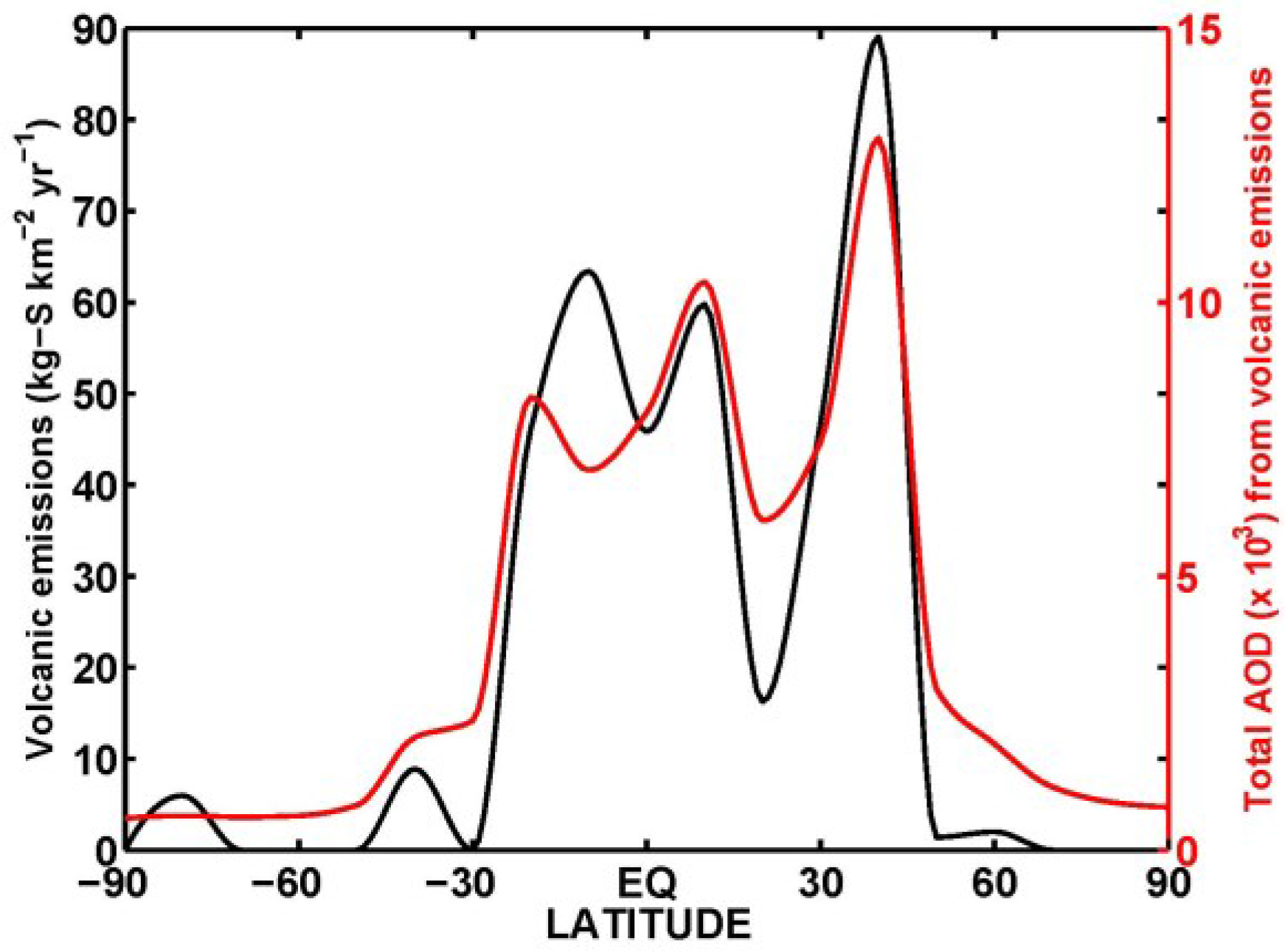
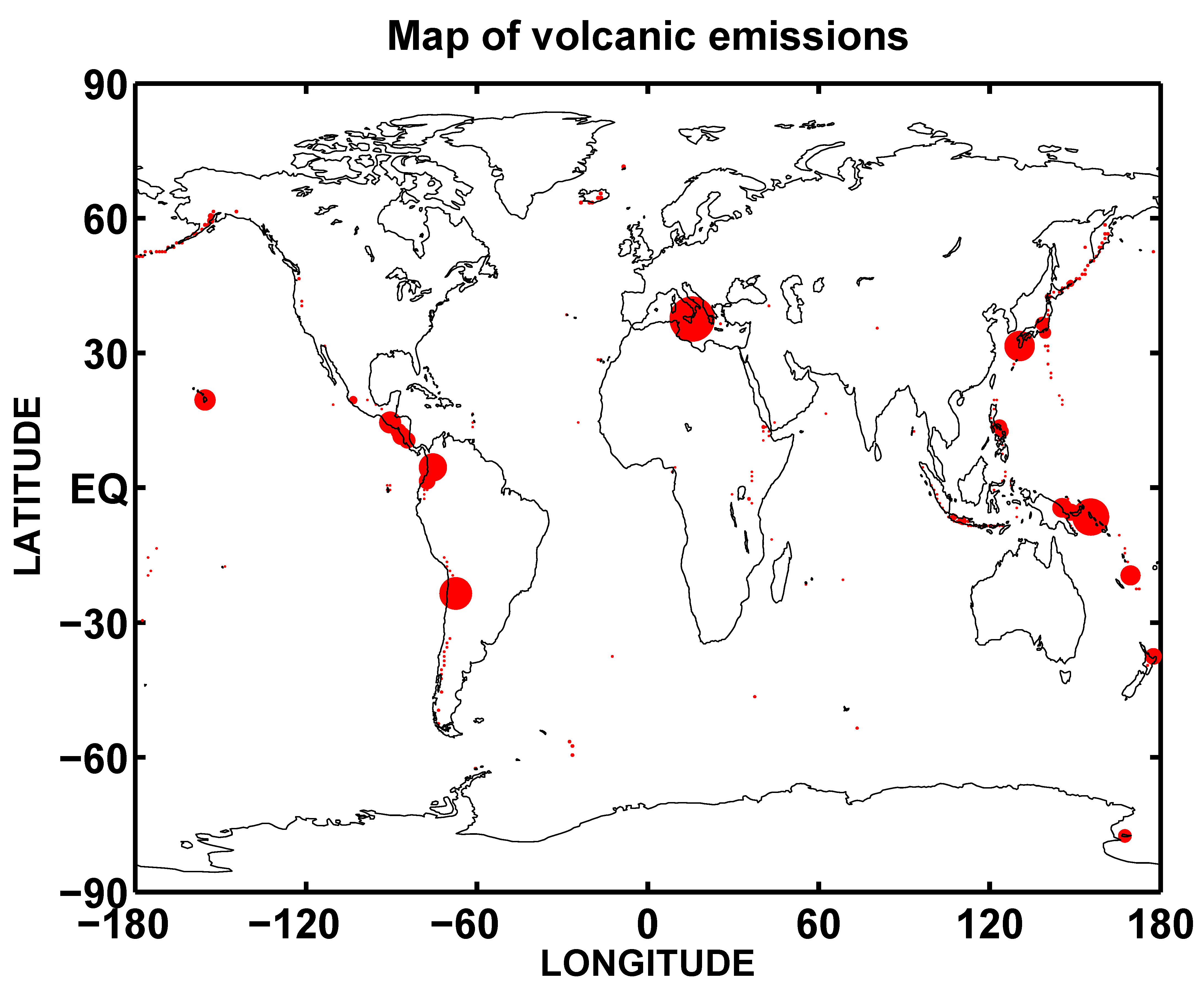
2.2. Numerical Experiment Setup and Sulfur Budget
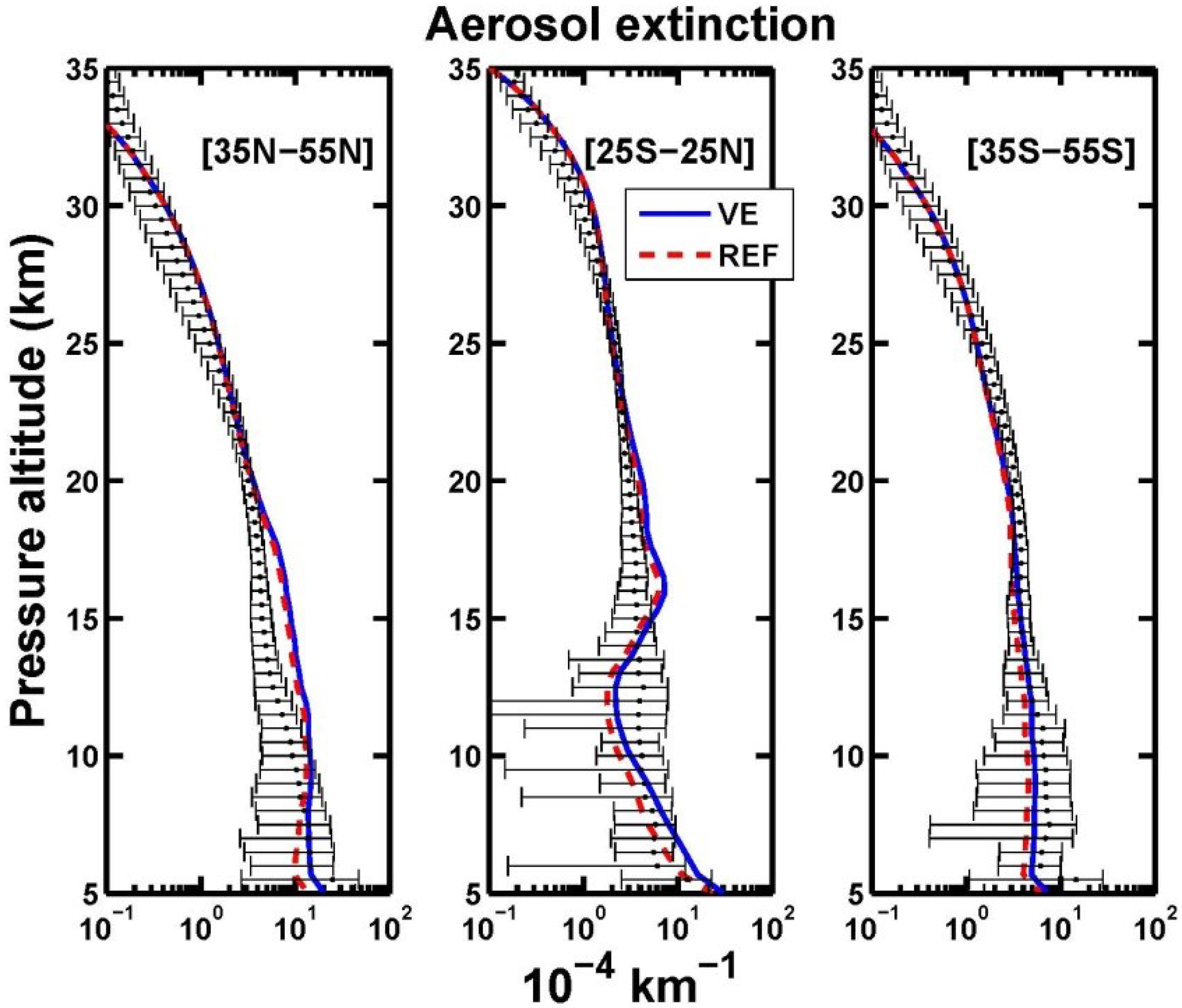
3. Results and Discussion
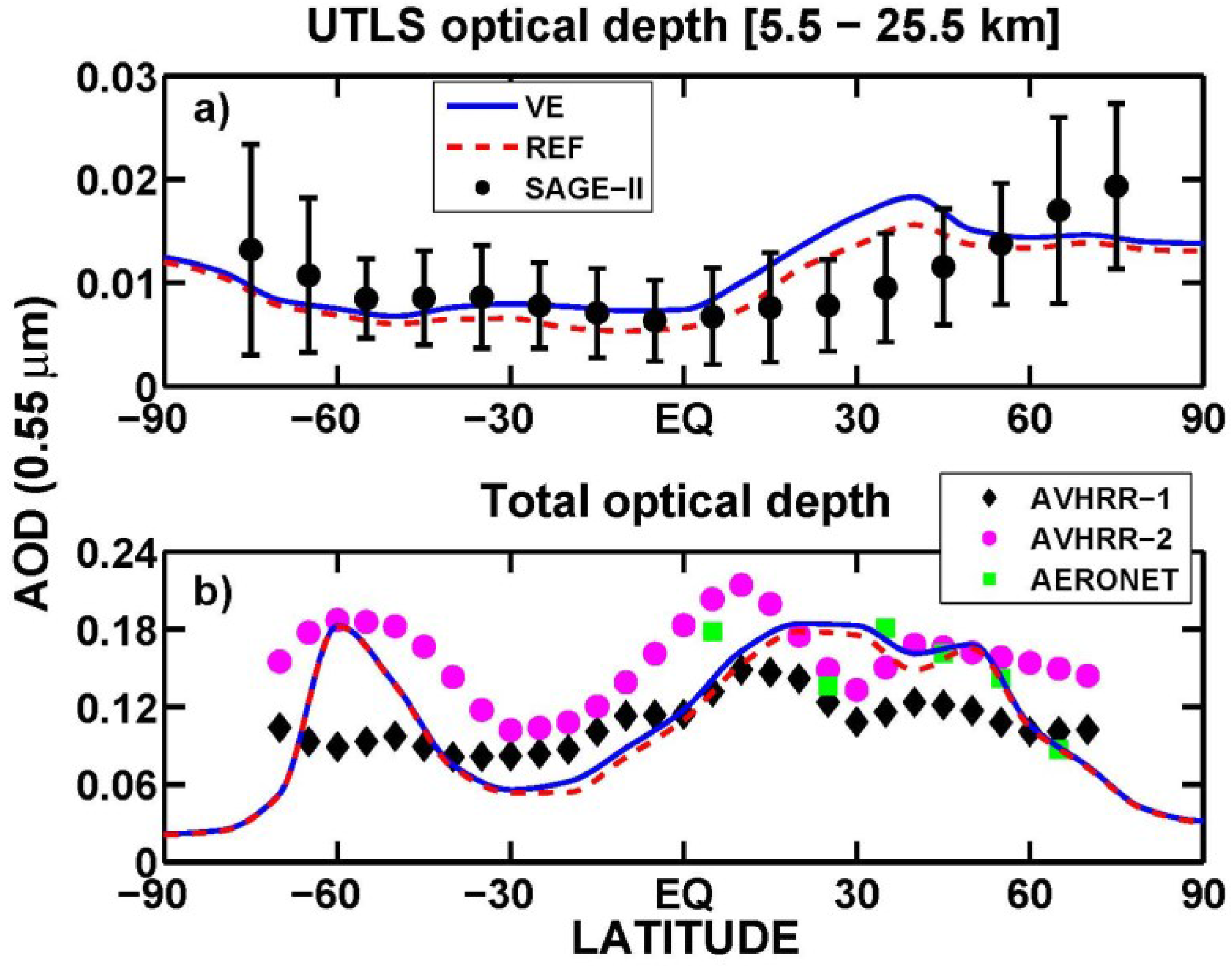
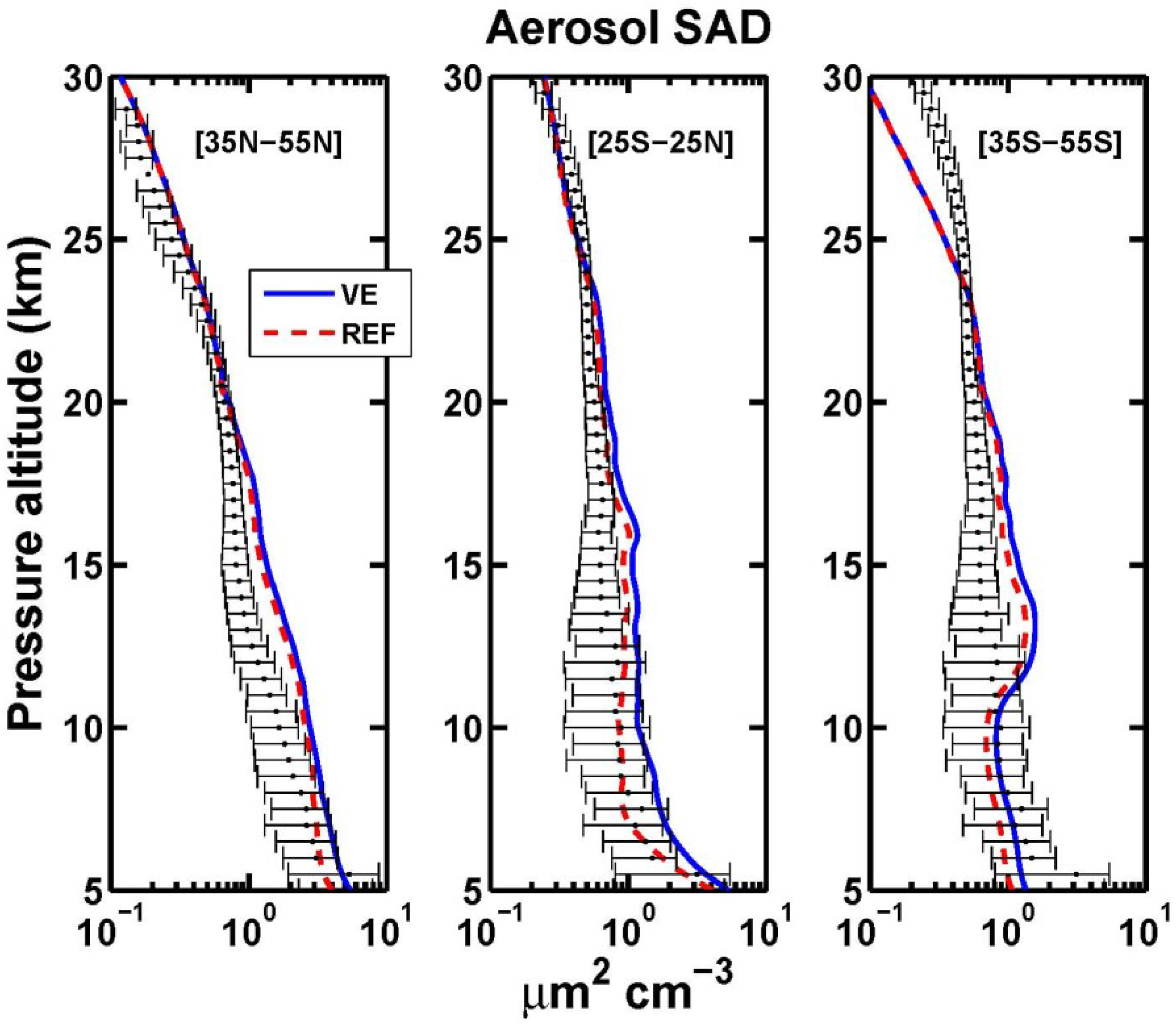
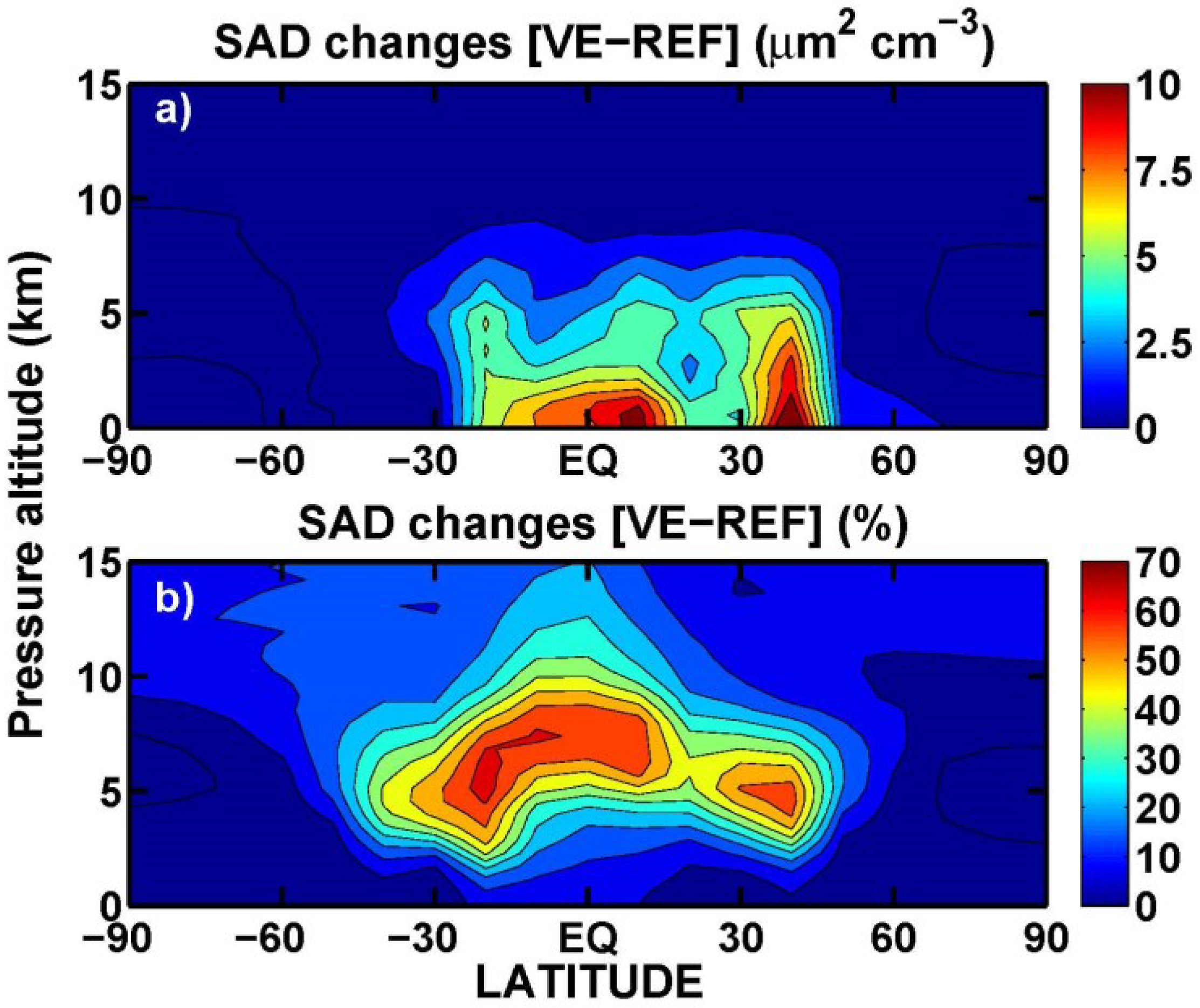
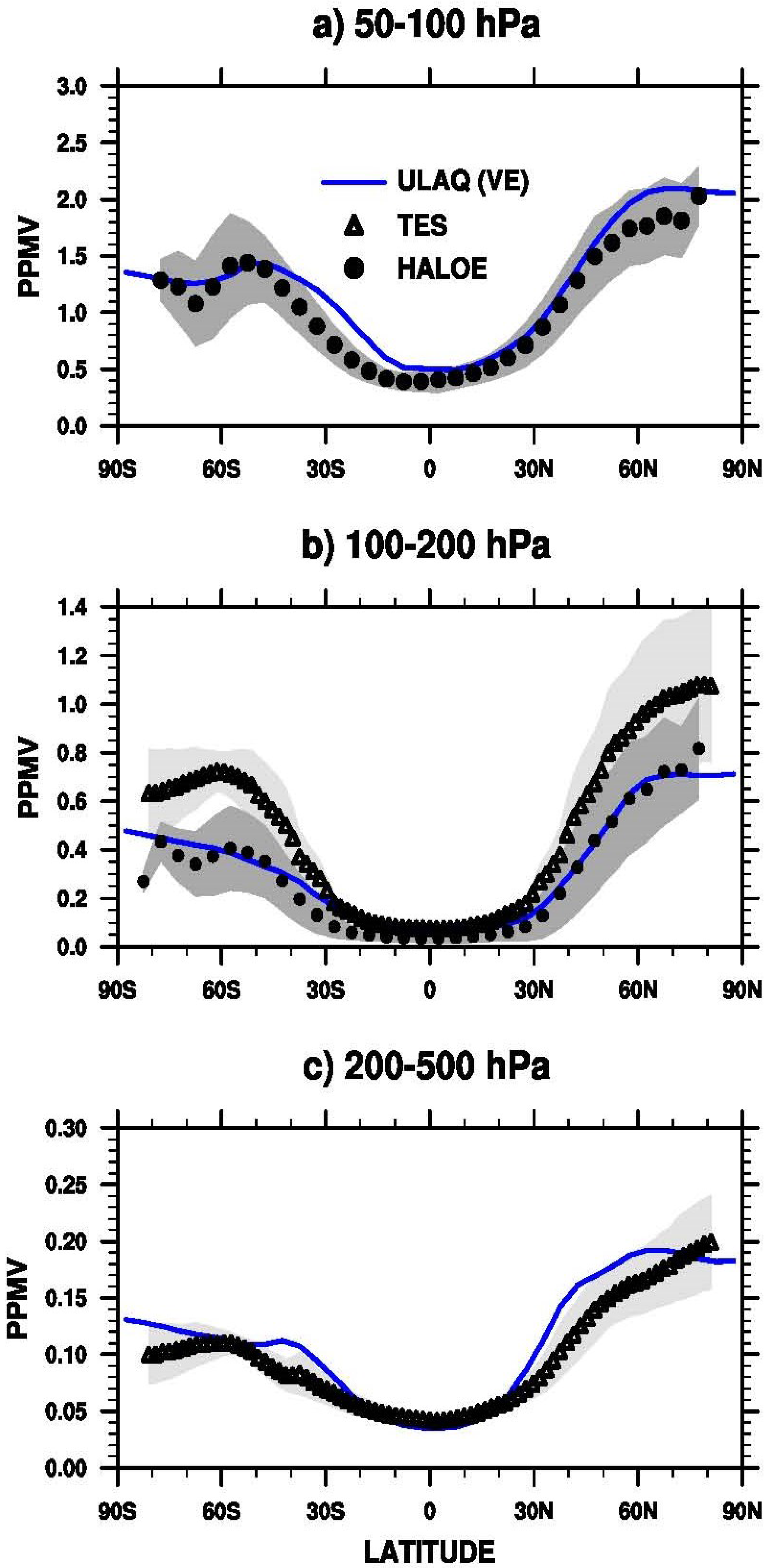
3.1. Aerosol Products
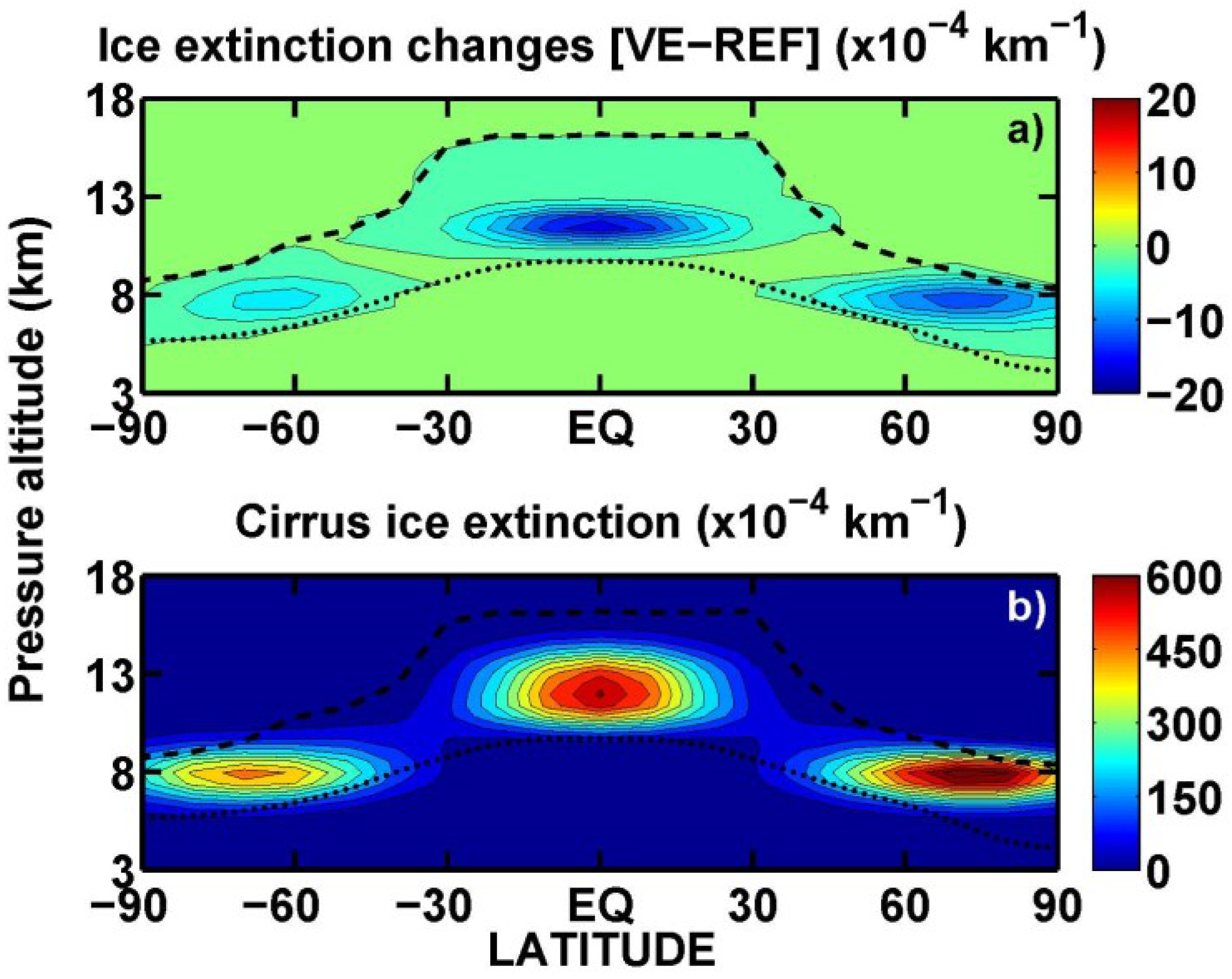
3.2. Upper Tropospheric Cirrus Ice Perturbation
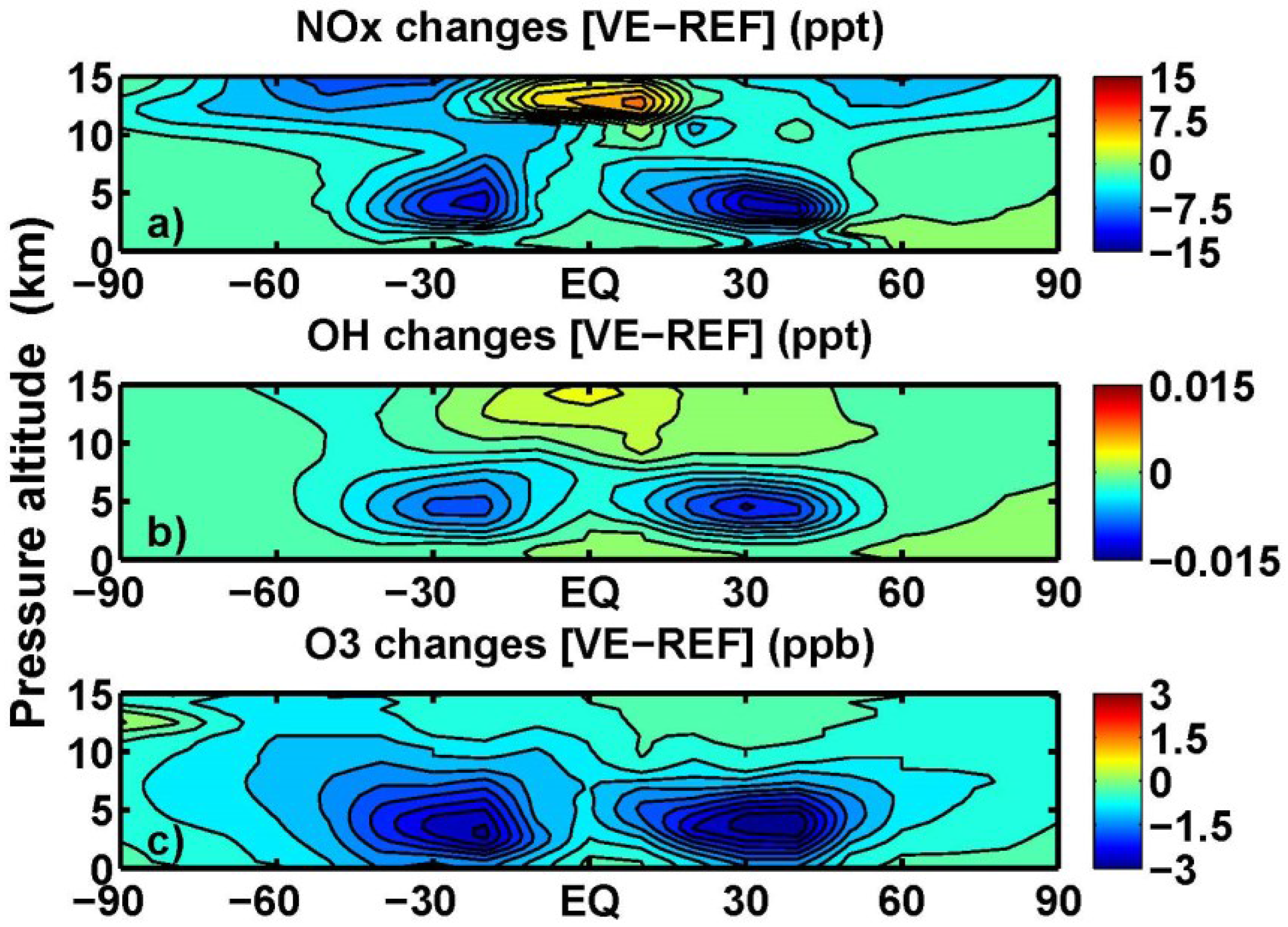
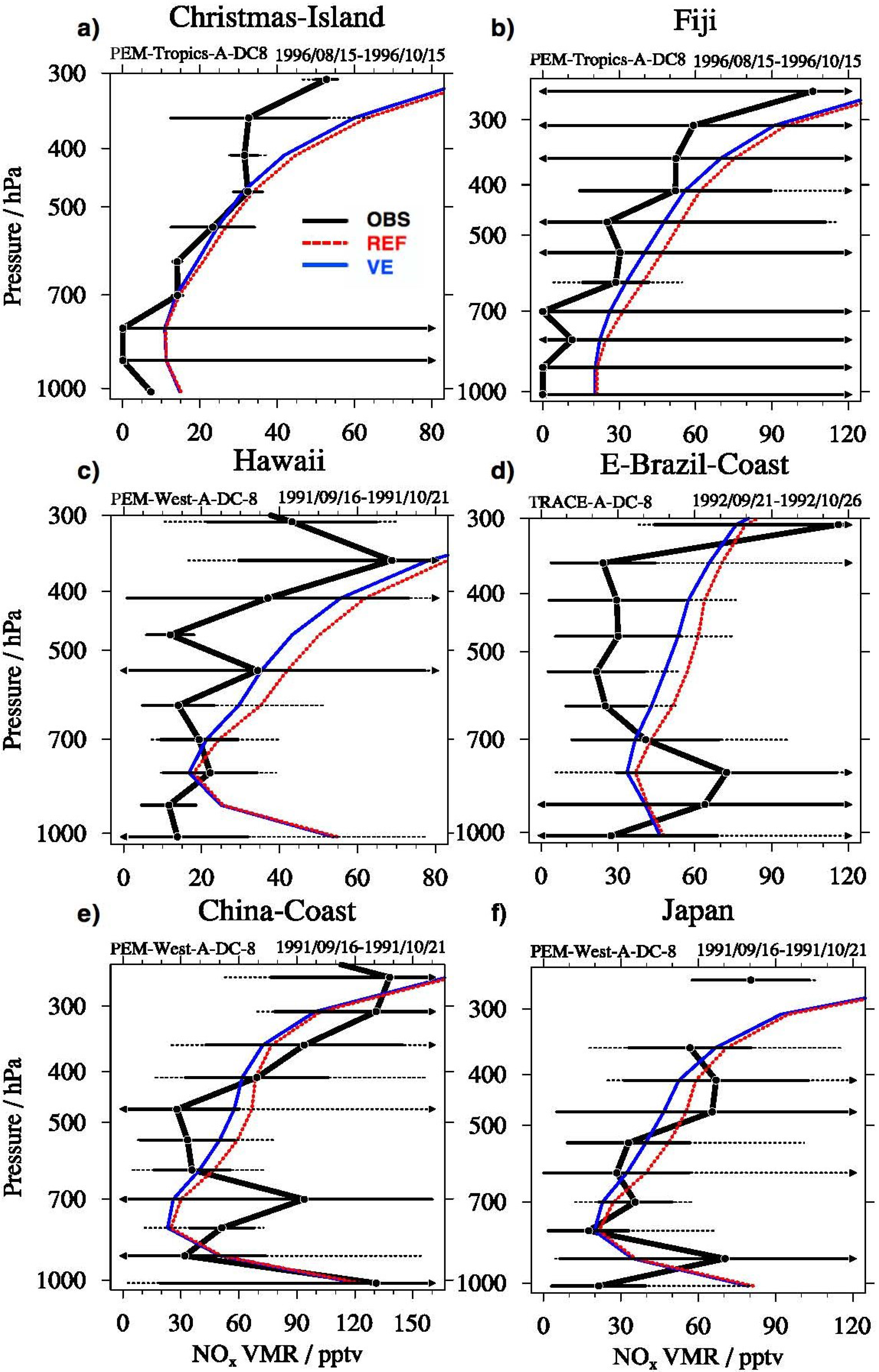
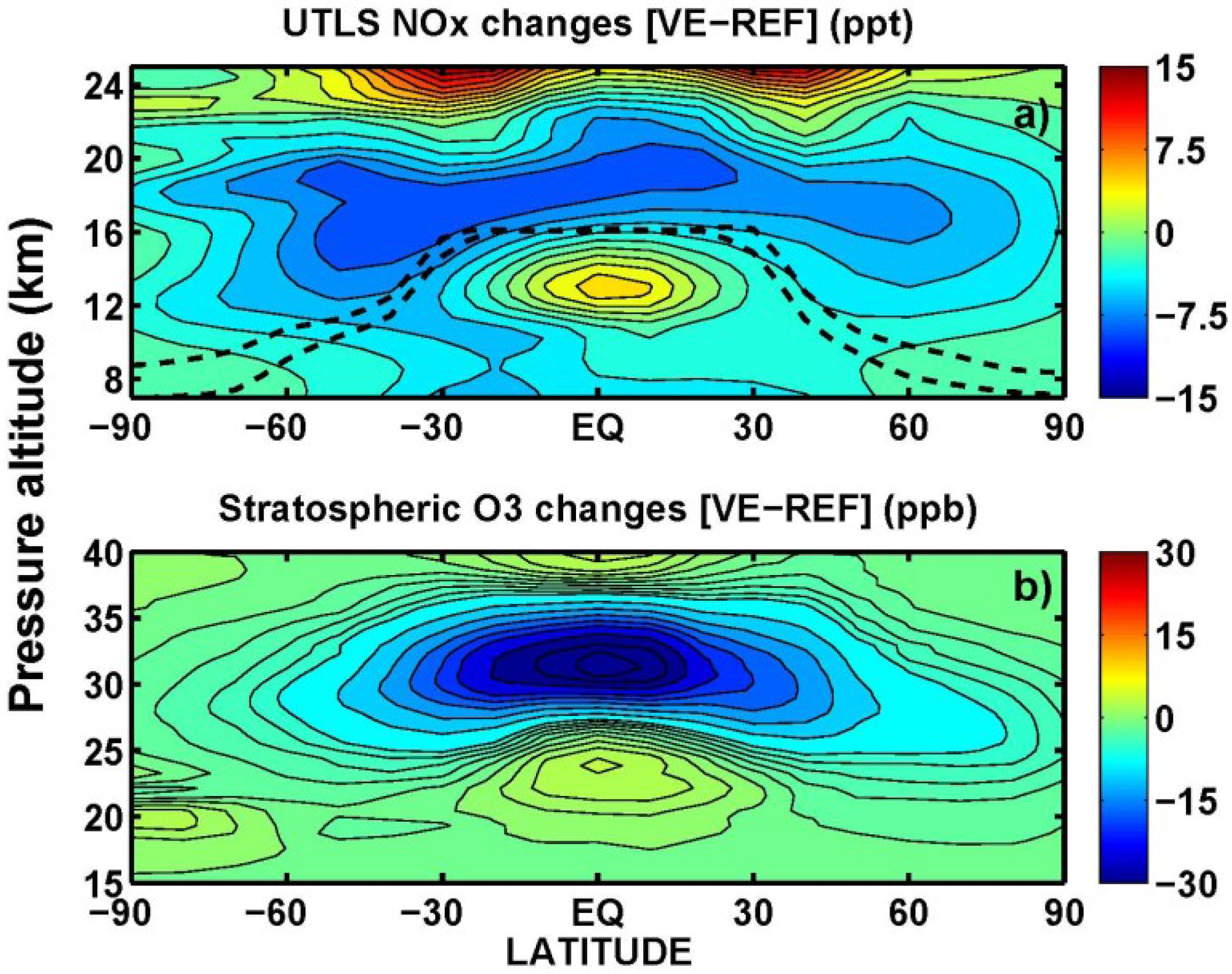
3.3. Tropospheric Chemistry Perturbation
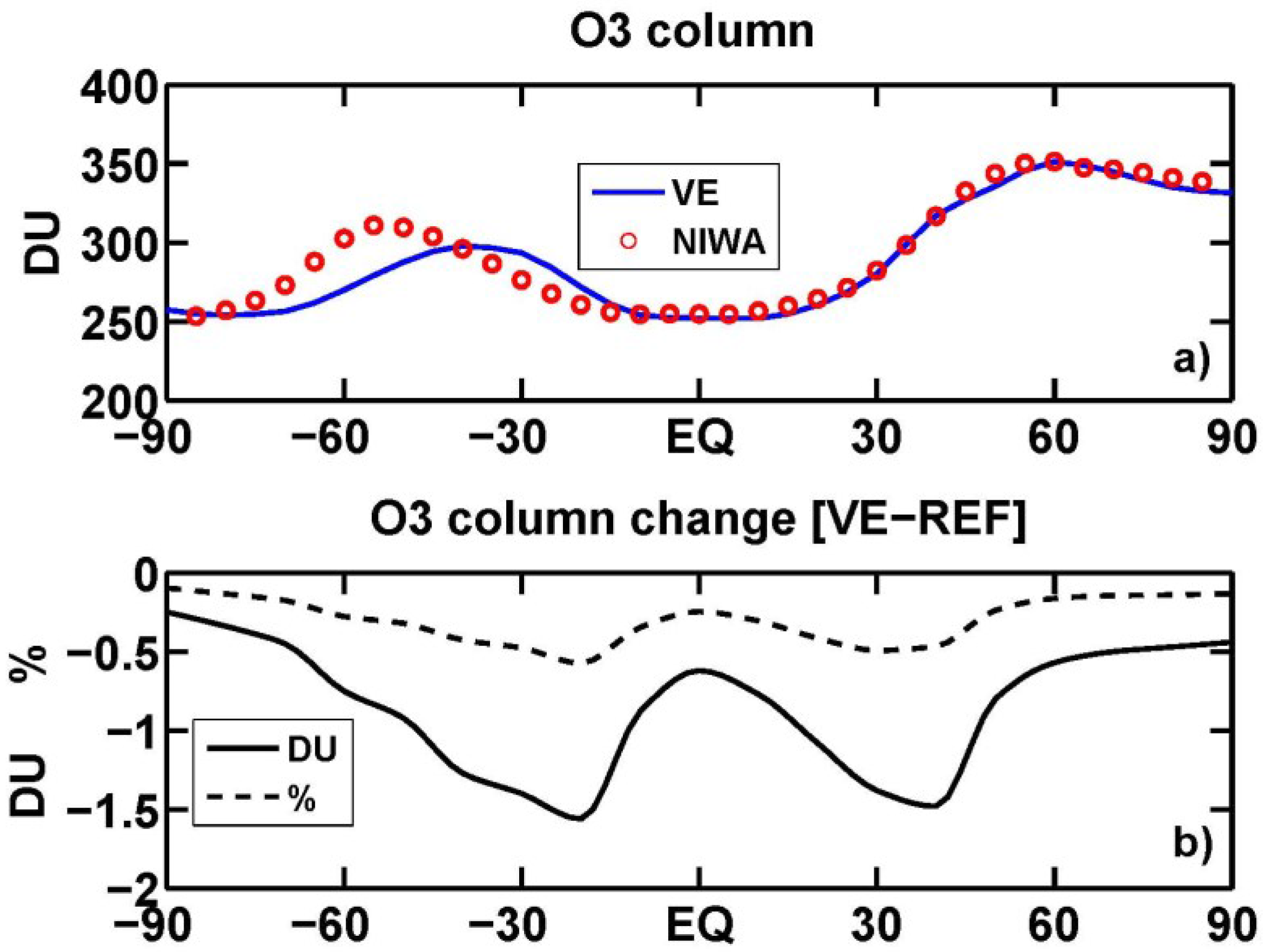
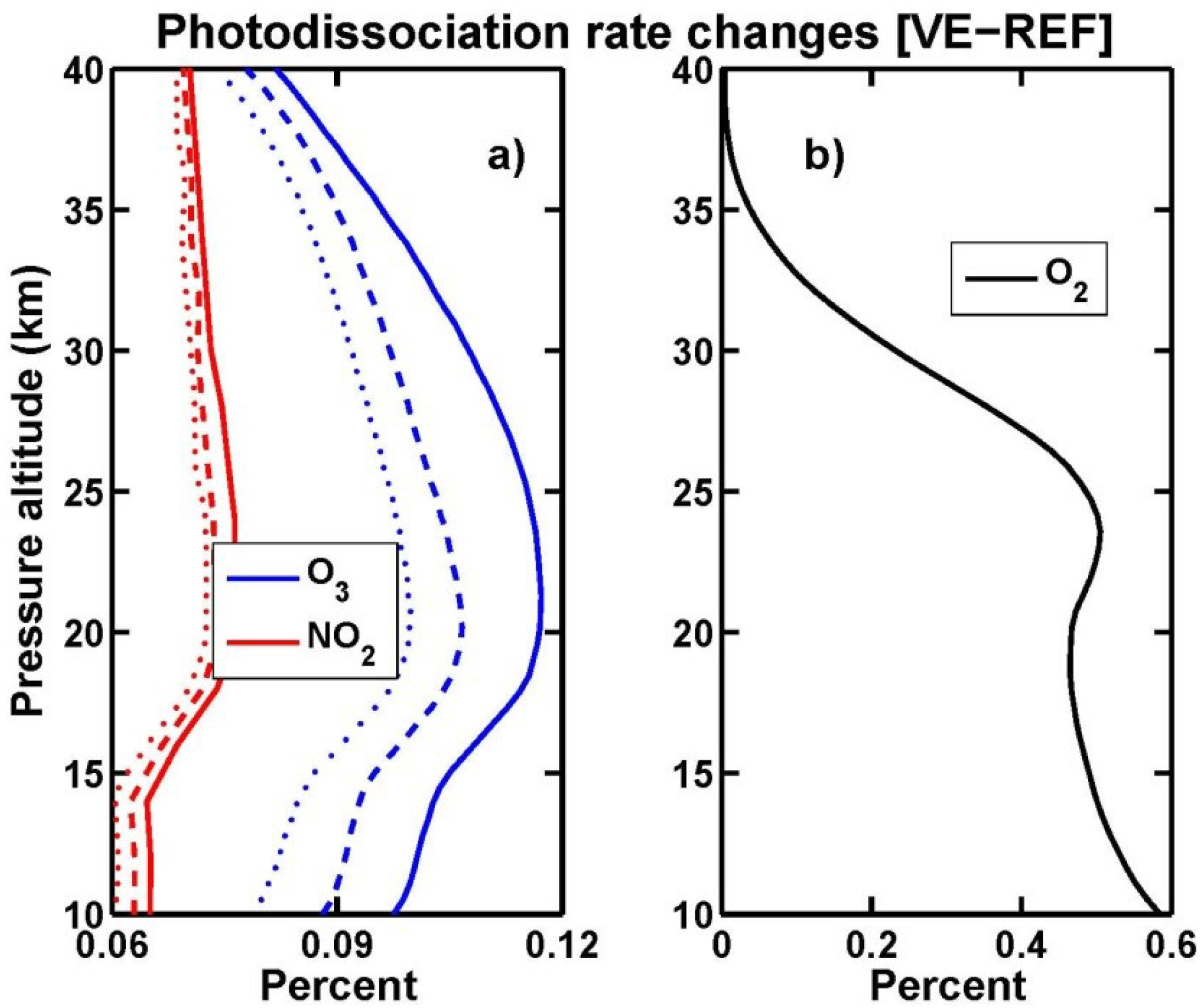
3.4. Stratospheric Chemistry Perturbation
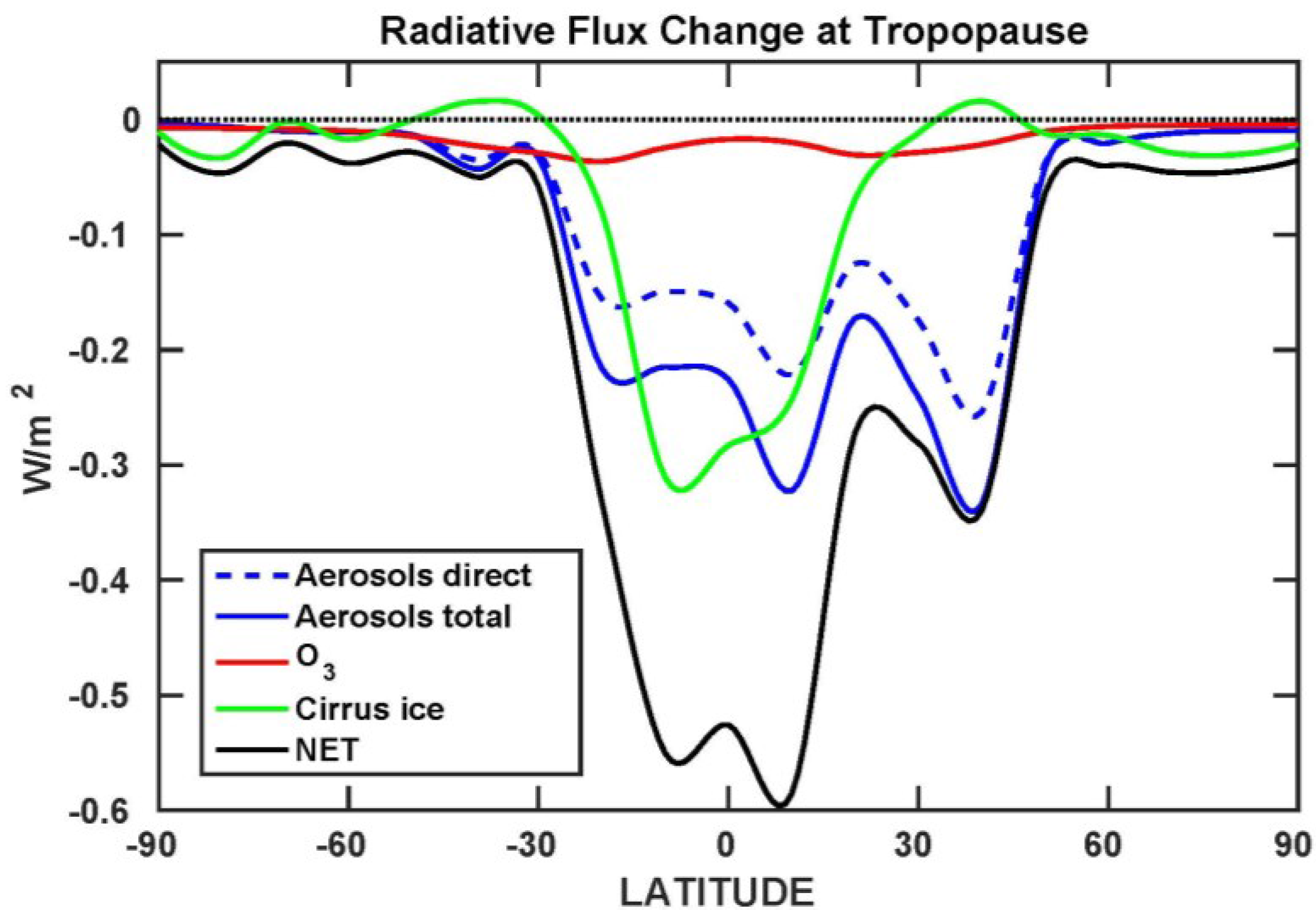
3.5. Tropopause Radiative Flux Changes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
Appendix 1. Dynamics
Appendix 2. Transport


Appendix 3. Radiation
Appendix 4. Sulfate Aerosols
Appendix 5. Cirrus Ice (via Homogeneous Freezing)
References
- Andreae, M.O. Climatic effects of changing atmospheric aerosol levels. In World Survey of Climatology, Volume 16: Future Climates of the World; Henderson-Sellers, A., Ed.; Elsevier: Amsterdam, The Netherlands, 1995; pp. 341–392. [Google Scholar]
- Penner, J.; Hegg, D.; Andreae, M.; Leaitch, D.; Pitari, G.; Annegarn, H.; Murphy, D.; Nganga, J.; Barrie, L.; Feichter, H. IPCC, Climate Change 2001: Aerosols and Indirect Cloud Effects; IPCC Third Assessment Report; Cambridge University Press: Cambridge, UK, 2001; pp. 289–348. [Google Scholar]
- Kristiansen, N.I.; Stohl, A.; Olivie, D.J.L.; Croft, B.; Sovde, O.A.; Klein, H.; Christoudias, T.; Kunkel, D.; Leadbetter, S.J.; Lee, Y.H.; et al. Evaluation of observed and modelled aerosol lifetimes using radioactive tracers of opportunity and an ensemble of 19 global models. Atmos. Chem. Phys. 2016, 16, 3525–3561. [Google Scholar] [CrossRef]
- Stoiber, R.E.; Williams, S.N.; Huebert, B. Annual contribution of sulphur dioxide to the atmosphere by volcanoes. J. Volcanol. Geotherm. Res. 1987, 33, 1–8. [Google Scholar] [CrossRef]
- Graf, H.-F.; Langmann, B.; Feichter, J. The contribution of Earth degassing to the atmospheric sulphur budget. Chem. Geol. 1998, 147, 131–145. [Google Scholar] [CrossRef]
- Hobbs, P.V.; Radket, L.F.; Eltgrot, W.; Hegg, D.A. Airborne studies of the emissions from the volcanic eruptions of Mt. St. Helens. Science 1981, 211, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Feichter, J.; Kjellström, E.; Rodhe, H.; Dentener, F.; Lelieveld, J.; Roelofs, G.-J. Simulation of the global sulfur cycle in a global climate model. Atmos. Environ. 1996, 30, 1693–1707. [Google Scholar] [CrossRef]
- Clegg, S.M.; Abbatt, P.J.D. Oxidation of SO2 by H2O2 on ice surfaces at 228 K: A sink for SO2 in ice clouds. Atmos. Chem. Phys. 2001, 1, 73–78. [Google Scholar] [CrossRef]
- Pinto, J.P.; Turco, R.P.; Toon, O.B. Self-limiting physical and chemical effects in volcanic eruption clouds. J. Geophys. Res. 1989, 94, 11165–11174. [Google Scholar] [CrossRef]
- Bekki, S.; Pyle, J.A.; Zhong, W.; Toumi, R.; Haigh, J.D.; Pyle, D.M. The role of microphysical and chemical processes in prolonging the climate forcing of the Toba eruption. Geophys. Res. Lett. 1996, 23, 2669–2672. [Google Scholar] [CrossRef]
- Read, W.G.; Froidevaux, L.; Waters, J.W. Microwave limb sounder measurements of stratospheric SO2 from the Mt. Pinatubo volcano. Geophys. Res. Lett. 1993, 20, 1299–1302. [Google Scholar] [CrossRef]
- Pitari, G.; Di Genova, G.; Mancini, E.; Visioni, D.; Gandolfi, I.; Cionni, I. Stratospheric aerosols from major volcanic eruptions: A composition-climate model study of the aerosol cloud dispersal and e-folding time. Atmosphere 2016, 7, 75. [Google Scholar] [CrossRef]
- Spiro, P.A.; Jacob, D.J.; Logan, J.A. Global inventory of sulphur emissions with 1 × 1 resolution. J. Geophys. Res. 1992, 97, 6023–6036. [Google Scholar] [CrossRef]
- Graf, H.-F.; Feichter, J.; Langmann, B. Volcanic sulphur emissions: Estimates of source strength and its contribution to the global sulphate distribution. J. Geophys. Res. 1997, 102, 10727–10738. [Google Scholar] [CrossRef]
- Andres, R.J.; Kasgnoc, A.D. A time-averaged inventory of sub-aerial volcanic sulphur emissions. J. Geophys. Res. Atmos. 1998, 103, 25251–25261. [Google Scholar] [CrossRef]
- Newhall, G.G.; Self, S. The volcanic explosivity index (VEI): An estimate of explosive magnitude of historic eruptions. J. Geophys. Res 1982, 87, 1231–1238. [Google Scholar] [CrossRef]
- Eyring, V.; Lamarque, J.-F.; Hess, P.; Arfeuille, F.; Bowman, K.; Chipperfield, M.P.; Duncan, B.; Fiore, A.; Gettelman, A.; Giorgetta, M.A.; et al. Overview of IGAC/SPARC Chemistry-Climate Model. Initiative (CCMI) Community Simulations in Support of Upcoming Ozone and Climate Assessments; SPARC Newsletters: Zurich, Switzerland, 2013; Volume 40, pp. 48–66. [Google Scholar]
- Lamarque, J.F.; Bond, T.C.; Eyring, V.; Granier, C.; Heil, A.; Klimont, Z.; Lee, D.; Liousse, C.; Mieville, A.; Owen, B.; et al. Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: Methodology and application. Atmos. Chem. Phys. 2010, 10, 7017–7039. [Google Scholar] [CrossRef] [Green Version]
- Benkovitz, C.M.; Berkowitz, C.M.; Easter, R.C.; Nemesure, S.; Wagner, R.; Schwartz, S.E. Sulphate over the North Atlantic and adjacent regions: Evaluation for October and November 1986 using a three-dimensional model driven by observation-derived meteorology. J. Geophys. Res. 1994, 99, 20725–20756. [Google Scholar] [CrossRef]
- Sassen, K.; Starr, D.O’C.; Mace, G.G.; Poellot, M.R.; Melfi, S.H.; Eberhard, W.L.; Spinhirne, J.D.; Eloranta, E.W.; Hagen, D.E.; Hallett, J. The 5–6 December 1991 FIRE IFO II jet stream cirrus case study: Possible influences of volcanic aerosols. J. Atmos. Sci. 1995, 52, 97–123. [Google Scholar] [CrossRef]
- Song, N.; Starr, D.O’C.; Wuebbles, D.J.; Williams, A.; Larson, S.M. Volcanic aerosols and inter-annual variation of high clouds. Geophys. Res. Lett. 1996, 23, 2657–2660. [Google Scholar] [CrossRef]
- Kärcher, B.; Lohmann, U. A parameterization of cirrus cloud formation: Homogeneous freezing of supercooled aerosols. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Kuebbeler, M.; Lohmann, U.; Feichter, J. Effects of stratospheric sulfate aerosol geo-engineering on cirrus clouds. Geophys. Res. Lett. 2012, 39, L23803. [Google Scholar] [CrossRef]
- Pitari, G.; Aquila, V.; Kravitz, B.; Robock, A.; Watanabe, S.; Cionni, I.; De Luca, N.; Di Genova, G.; Mancini, E.; Tilmes, S. Stratospheric ozone response to sulfate geoengineering: Results from the Geoengineering Model Intercomparison Project (GeoMIP). J. Geophys. Res. 2014, 119, 2629–2653. [Google Scholar] [CrossRef]
- Sander, S.P.; Abbatt, J.; Barker, J.R.; Burkholder, J.B.; Friedl, R.R.; Golden, D.M.; Huie, R.E.; Kolb, C.E.; Kurylo, M.J.; Moortgat, G.K.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies; Evaluation No. 17; Jet Propulsion Laboratory: Pasadena, CA, USA, 2011. [Google Scholar]
- Chipperfield, M.; Liang, Q.; Abraham, L.; Bekki, S.; Braesicke, P.; Dhomse, S.; Di Genova, G.; Fleming, E.L.; Hardiman, S.; Iachetti, D.; et al. Multi-model estimates of atmospheric lifetimes of long-lived ozone-depleting substances: Present and future. J. Geophys. Res 2014, 119, 2555–2573. [Google Scholar]
- Randles, C.A.; Kinne, S.; Myhre, G.; Schulz, M.; Stier, P.; Fischer, J.; Doppler, L.; Highwood, E.; Ryder, C.; Harris, B.; et al. Intercomparison of shortwave radiative transfer schemes in global aerosol modeling: Results from the AeroCom Radiative Transfer Code Experiment. Atmos. Chem. Phys 2013, 13, 2347–2379. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, O.; Giorgetta, M.A.; Shibata, K.; Eyring, V.; Waugh, D.W.; Shepherd, T.G.; Akiyoshi, H.; Austin, J.; Baumgärtner, A.; Bekki, S.; et al. A review of CCMVal-2 models and simulations. J. Geophys. Res. 2010, 115, D00M02. [Google Scholar]
- Butchart, N.; Cionni, I.; Eyring, V.; Waugh, D.W.; Akiyoshi, H.; Austin, J.; Brühl, C.; Chipperfield, M.P.; Cordero, E.; Dameris, M.; et al. Chemistry-climate model simulations of 21st century stratospheric climate and circulations changes. J. Clim. 2010, 23, 5349–5374. [Google Scholar] [CrossRef]
- Pitari, G.; Di Genova, G.; De Luca, N. A modelling study of the impact of on-road diesel emissions on Arctic black carbon and solar radiation transfer. Atmosphere 2015, 6, 318–340. [Google Scholar] [CrossRef]
- Chou, M.-D.; Suarez, M.J.; Liang, X.-Z.; Yan, M.M.-H. A Thermal Infrared Radiation Parameterization for Atmospheric Studies; NASA/TM-2001–104606; Goddard Space Flight Center: Greenbelt, MD, USA, 2001.
- Pitari, G.; Di Genova, G.; Coppari, E.; De Luca, N.; Di Carlo, P.; Iarlori, M.; Rizi, V. Desert dust transported over Europe: Lidar observations and model evaluation of the radiative impact. J. Geophys. Res. 2015, 120, 2881–2898. [Google Scholar] [CrossRef]
- Rayner, N.A.; Parker, D.E.; Horton, E.B.; Folland, C.K.; Alexander, L.V.; Rowell, D.P.; Kent, E.C.; Kaplan, A. Global analyses of sea surface temperature, sea ice, and night marine air temperature since the late nineteenth century. J. Geophys. Res. 2003, 108, 4407. [Google Scholar] [CrossRef]
- Lacis, A.; Hansen, J.E.; Sato, M. Climate forcing by stratospheric aerosols. Geophys. Res. Lett. 1992, 19, 1607–1610. [Google Scholar] [CrossRef]
- Lee, C.; Martin, R.V.; Van Donkelaar, A.; Lee, A.; Dickerson, R.R.; Hains, J.C.; Krotkov, N.; Richter, A.; Vinnikov, K.; Schwab, J.J. SO2 emissions and lifetimes: Estimate from inverse modeling using in situ and global, space-based (SCIAMACHY and OMI) observations. J. Geophys. Res. 2011, 116. [Google Scholar] [CrossRef]
- Langner, J.; Rodhe, H. A global three-dimensional model of the tropospheric sulphur cycle. J. Atmos. Chem. 1991, 13, 225–263. [Google Scholar] [CrossRef]
- Ganzeveld, L.; Lelieveld, J.; Roelofs, G.-J. A dry deposition parameterization for sulfur oxides in a chemistry and general circulation model. J. Geophys. Res. 1998, 103, 5679–5694. [Google Scholar] [CrossRef]
- Feichter, J.; Lohmann, U.; Schult, I. The atmospheric sulphur cycle in ECHAM-4 and its impact on the shortwave radiation. Clim. Dyn. 1997, 13, 235–246. [Google Scholar] [CrossRef]
- Stratosphere-troposphere Processes and Their Role in Climate (SPARC). Assessment of Stratospheric Aerosol Properties (ASAP); Thomason, L., Peter, T., Eds.; WCRP-124, WMO/TD-1295, SPARC Report #4; SPARC: Zurich, Switzerland, 2006. [Google Scholar]
- Bergman, J.W.; Jensen, E.J.; Pfister, L.; Yang, Q. Seasonal differences of vertical-transport efficiency in the tropical tropopause layer: On the interplay between tropical deep convection, large-scale vertical ascent, and horizontal circulations. J. Geophys. Res. 2012, 117, D05302. [Google Scholar] [CrossRef]
- Höpfner, M.; Glatthor, N.; Grabowski, U.; Kellmann, S.; Kiefer, M.; Linden, A.; Orphal, J.; Stiller, G.; Von Clarmann, T.; Funke, B. Sulfur dioxide (SO2) as observed by MIPAS/Envisat: Temporal development and spatial distribution at 15–45 km altitude. Atmos. Chem. Phys. 2013, 13, 10405–10423. [Google Scholar] [CrossRef]
- Zhao, X.-P.; Chan, P.K. NOAA CDR Program NOAA Climate Data Record (CDR) of AVHRR Daily and Monthly Aerosol Optical Thickness over Global Oceans, Version 2.0; NOAA National Centers for Environmental Information: Asheville, NC, USA, 2014.
- Mishchenko, M.K.; Geogdzhayev, I.V.; Cairns, B.; Rossow, W.B.; Lacis, A.A. Aerosol retrievals over the ocean using channel 1 and 2 AVHRR data: A sensitivity analysis and preliminary results. Appl. Opt. 1999, 38, 7325–7341. [Google Scholar] [CrossRef] [PubMed]
- Holben, B.N.; Tanre, D.; Smirnov, A.; Eck, T.F.; Slutsker, I.; Abuhassan, N.; Newcomb, W.W.; Schafer, J.S.; Chatenet, B.; Lavenuet, F.; et al. An emerging ground-based aerosol climatology: Aerosol optical depth from AERONET. J. Geophys. Res. 2001, 106, 12067–12097. [Google Scholar] [CrossRef]
- English, J.M.; Toon, O.B.; Mills, M.J.; Yu, F. Microphysical simulations of new particle formation in the upper troposphere and lower stratosphere. Atmos. Chem. Phys. 2011, 11, 9303–9322. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, J.; Kärcher, B.; Lohmann, U. Effects of ice nuclei on cirrus clouds in a global climate model. J. Geophys. Res. 2011, 116, D18206. [Google Scholar] [CrossRef]
- Pitari, G.; Mancini, E.; Rizi, V.; Shindell, D.T. Impact of future climate and emission changes on stratospheric aerosols and ozone. J. Atmos. Sci. 2002, 59, 414–440. [Google Scholar] [CrossRef]
- Koop, T.; Luo, B.; Peter, T. Water activity as the determinant for homogeneous ice nucleation in aqueous solutions. Nature 2000, 406, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, U.; Kärcher, B. First interactive simulations of cirrus clouds formed by homogeneous freezing in the ECHAM GCM. J. Geophys. Res. 2002, 107, 4141. [Google Scholar] [CrossRef]
- Ström, J.; Strauss, B.; Anderson, T.; Schroder, F.; Heintzenberg, J.; Wiedding, R. In situ observations of the microphysical properties of young cirrus clouds. J. Atmos. Sci. 1997, 54, 2542–2553. [Google Scholar] [CrossRef]
- Fahey, D.W.; Kawa, S.R.; Woodbridge, E.L.; Tin, P.; Wilson, J.C.; Jonsson, H.H.; Dye, J.E.; Baumgardner, D.; Borrmann, S.; Toohey, D.W.; et al. In-situ measurements constraining the role of sulphate aerosols in mid-latitude ozone depletion. Nature 1993, 363, 509–514. [Google Scholar] [CrossRef]
- Naik, V.; Voulgarakis, A.; Fiore, A.M.; Horowitz, L.W.; Lamarque, J.-F.; Lin, M.; Prather, M.J.; Young, P.J.; Bergmann, D.; Cameron-Smith, P.J.; et al. Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 5277–5298. [Google Scholar] [CrossRef] [Green Version]
- Voulgarakis, A.; Naik, V.; Lamarque, J.-F.; Shindell, D.T.; Young, P.J.; Prather, M.J.; Wild, O.; Field, R.D.; Bergmann, D.; Cameron-Smith, P.; et al. Analysis of present day and future OH and methane lifetime in the ACCMIP simulations. Atmos. Chem. Phys. 2013, 13, 2563–2587. [Google Scholar] [CrossRef] [Green Version]
- Emmons, L.K.; Hauglustaine, D.A.; Müller, J.-F.; Carroll, M.A.; Brasseur, G.P.; Brunner, D.; Staehelin, J.; Thouret, V.; Marenco, A. Data composites of airborne observations of tropospheric ozone and its precursors. J. Geophys. Res. 2000, 105, 20497–20538. [Google Scholar] [CrossRef]
- Bodeker, G.E.; Shiona, H.; Eskes, H. Indicators of Antarctic ozone depletion. Atmos. Chem. Phys. 2005, 5, 2603–2615. [Google Scholar] [CrossRef]
- Grooss, J.-U.; Russell, J.M., III. A stratospheric climatology for O3, H2O, CH4, NOx, HCl and HF derived from HALOE measurements. Atmos. Chem. Phys. 2005, 5, 2797–2807. [Google Scholar] [CrossRef]
- TES/Aura L3 Ozone (O3) Monthly (TL3O3M). Available online: https://eosweb.larc.nasa.gov/project/tes/tes_tl3o3m_table (accessed on 23 June 2016).
- Kremser, S.; Thomason, L.W.; Hobe, M.; Hermann, M.; Deshler, T.; Timmreck, C.; Toohey, M.; Stenke, A.; Schwarz, J.P.; Weigel, R.; et al. Stratospheric aerosol—Observations, processes, and impact on climate. Rev. Geophys. 2016, 54. [Google Scholar] [CrossRef]
- Sheng, J.-X.; Weisenstein, D.K.; Luo, B.-P.; Rozanov, E.; Stenke, A.; Anet, J.; Bingemer, H.; Peter, T. Global atmospheric sulfur budget under volcanically quiescent conditions: Aerosol-chemistry-climate model predictions and validation. J. Geophys. Res. Atmos. 2015, 120, 256–276. [Google Scholar] [CrossRef]
- Mills, M.J.; Toon, O.B.; Turco, R.P.; Kinnison, D.E.; Garcia, R.R. Massive global ozone loss predicted following regional nuclear conflict. PNAS 2008, 105, 5307–5312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tian, W.; Dhomse, S.; Xie, F.; Shu, J.; Austin, J. Stratospheric ozone depletion from future nitrous oxide increases. Atmos. Chem. Phys. 2014, 14, 12967–12982. [Google Scholar] [CrossRef]
- Myhre, G.; Shine, K.; Readel, G.; Gauss, M.; Isaksen, I.; Tang, Q.; Prather, M.; Williams, J.; Van Velthoven, P.; Dessens, O.; et al. Radiative forcing due to changes in ozone and methane caused by the transport sector. Atmos. Environ. 2011, 45, 387–394. [Google Scholar] [CrossRef]
- Prather, M.J.; Ehhalt, D.; Dentener, F.; Derwent, R.; Dlugokencky, E.; Holland, E.; Isaksen, I.; Katima, J.; Kirchhoff, V.; Matson, P.; et al. IPCC, Climate Change 2001: Atmospheric Chemistry and Greenhouse Gases; IPCC Third Assessment Report; Cambridge University Press: Cambridge, UK, 2001; pp. 239–287. [Google Scholar]
- Fuglestvedt, J.; Berntsen, T.; Myhre, G.; Rypdal, K.; Skeie, R.B. Climate forcing from the transport sectors. PNAS 2008, 105, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Chipperfield, M.; Liang, Q.; Abraham, L.; Bekki, S.; Braesicke, P.; Dhomse, S.; Di Genova, G.; Fleming, E.L.; Hardiman, S.; Iachetti, D.; et al. Model estimates of lifetimes. In Lifetimes of Stratospheric Ozone-Depleting Substances, Their Replacements, and Related Species; SPARC: Zurich, Switzerland, 2013. [Google Scholar]
- Stevenson, D.S.; Dentener, F.J.; Schultz, M.; Ellingsen, K.; van Noije, T.; Zeng, G.; Amann, M.; Atherton, C.; Bell, N.; Bergmann, D.; et al. Multi-model ensemble simulations of present-day and near-future tropospheric ozone. J. Geophys. Res. 2006, 111, D08301. [Google Scholar] [CrossRef]
- Pitari, G.; Rizi, V.; Ricciardulli, L.; Visconti, G. High-speed civil transport impact: The role of sulfate, nitric acid trihydrate and ice aerosols studied with a two-dimensional model including aerosol physics. J. Geophys. Res. 1993, 98, 23141. [Google Scholar] [CrossRef]
- Lohmann, U.; McFarlane, N.; Levkov, L.; Abdella, K.; Albers, F. Comparing different cloud schemes of a single column model by using mesoscale forcing and nudging technique. J. Clim. 1999, 12, 438–461. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
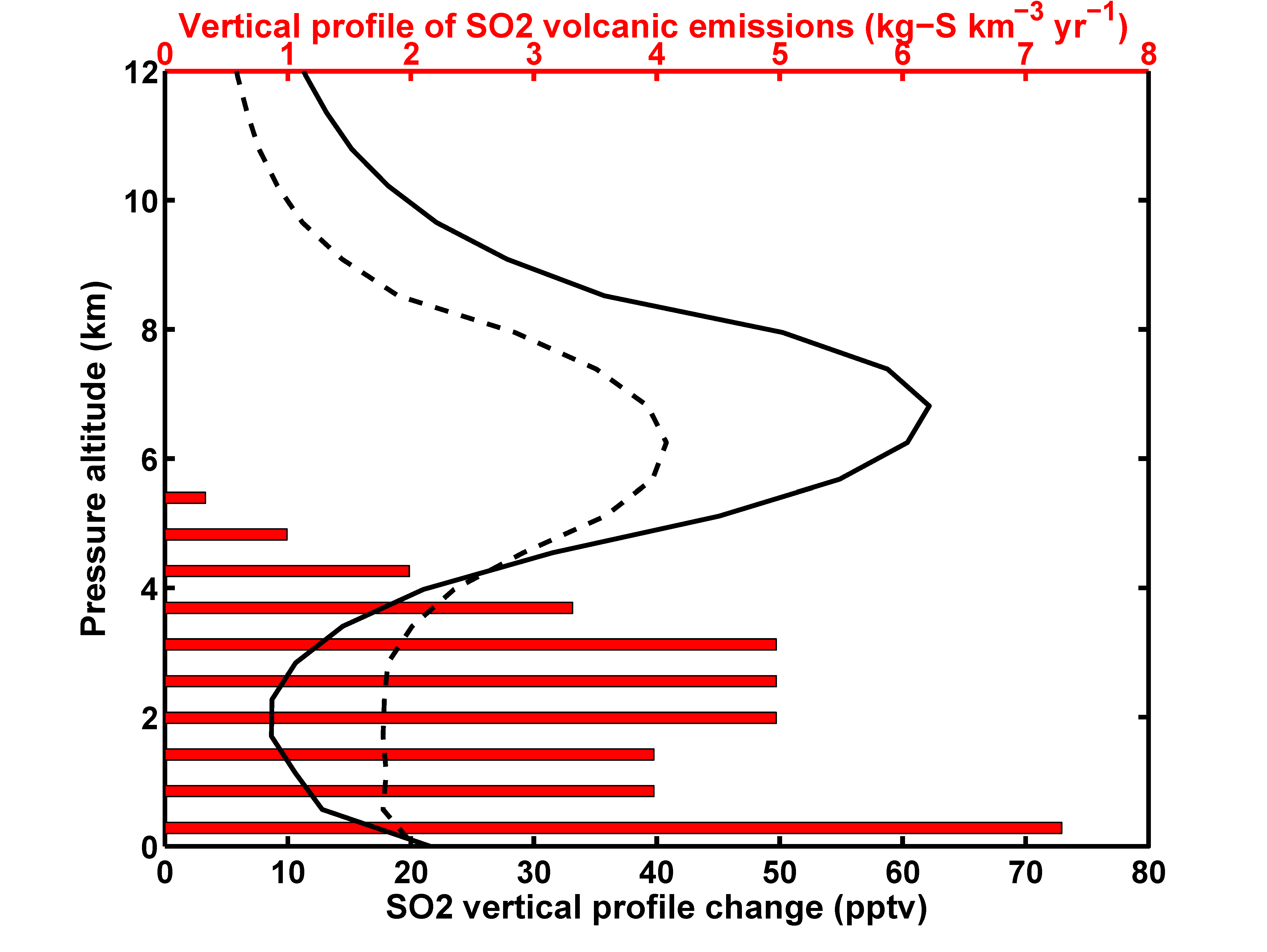{kind=link}
| Sulfur Emission (Tg-S/yr) | SO2 Burden (Tg-S) (%) | SO42− Burden (Tg-S) (%) | Efficiency | |
|---|---|---|---|---|
| Total | 98.4 | 0.56 Tg-S | 0.61 Tg-S | - |
| Anthropogenic Biomass Burning Oceanic (DMS) OCS photolysis | 88.8 | 86% | 84% | 0.93 |
| Non-Explosive Volcanoes | 9.6 | 14% | 16% | 1.64 |
| SO2 Source (Tg-S/yr) | SO2 Burden (Tg-S) (%) | SO42− Source (Tg-S/yr) | SO42− Burden (Tg-S) (%) | |
|---|---|---|---|---|
| Stratospheric | 0.145 | 0.0120 Tg-S | 0.160 | 0.162 Tg-S |
| Anthropogenic Biomass Burning Oceanic (DMS) OCS photolysis | 0.138 | 95% | 0.150 | 94% |
| Non-Explosive Volcanoes | 0.007 | 5.0% | 0.010 | 6.0% |
| Optical Depth (λ = 0.55 µm) | CH4 Lifetime (%) | O3 Column (DU) | RF-SW (mW·m−2) | RF-LW (mW·m−2) | RF-NET (mW·m−2) | |
|---|---|---|---|---|---|---|
| SO42− direct | 5.3 × 10−3 | −105 | 7.0 | −98 | ||
| SO42− indirect (warm clouds) | −44 | - | −44 | |||
| Ice (cirrus clouds) | −1.0 × 10−3 | 9.0 | −90 | −81 | ||
| CH4 | 1.1 | - | 10 | 10 | ||
| O3 | −1.0 | −3.0 | −18 | −21 | ||
| Total RF | −143 | −91 | −234 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitari, G.; Visioni, D.; Mancini, E.; Cionni, I.; Di Genova, G.; Gandolfi, I. Sulfate Aerosols from Non-Explosive Volcanoes: Chemical-Radiative Effects in the Troposphere and Lower Stratosphere. Atmosphere 2016, 7, 85. https://doi.org/10.3390/atmos7070085
Pitari G, Visioni D, Mancini E, Cionni I, Di Genova G, Gandolfi I. Sulfate Aerosols from Non-Explosive Volcanoes: Chemical-Radiative Effects in the Troposphere and Lower Stratosphere. Atmosphere. 2016; 7(7):85. https://doi.org/10.3390/atmos7070085
Chicago/Turabian StylePitari, Giovanni, Daniele Visioni, Eva Mancini, Irene Cionni, Glauco Di Genova, and Ilaria Gandolfi. 2016. "Sulfate Aerosols from Non-Explosive Volcanoes: Chemical-Radiative Effects in the Troposphere and Lower Stratosphere" Atmosphere 7, no. 7: 85. https://doi.org/10.3390/atmos7070085