
Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches



 , ,
, ,  , and
, and 
Abstract
:Simple Summary
Abstract
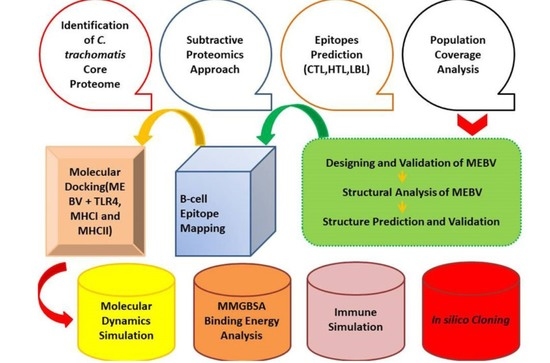
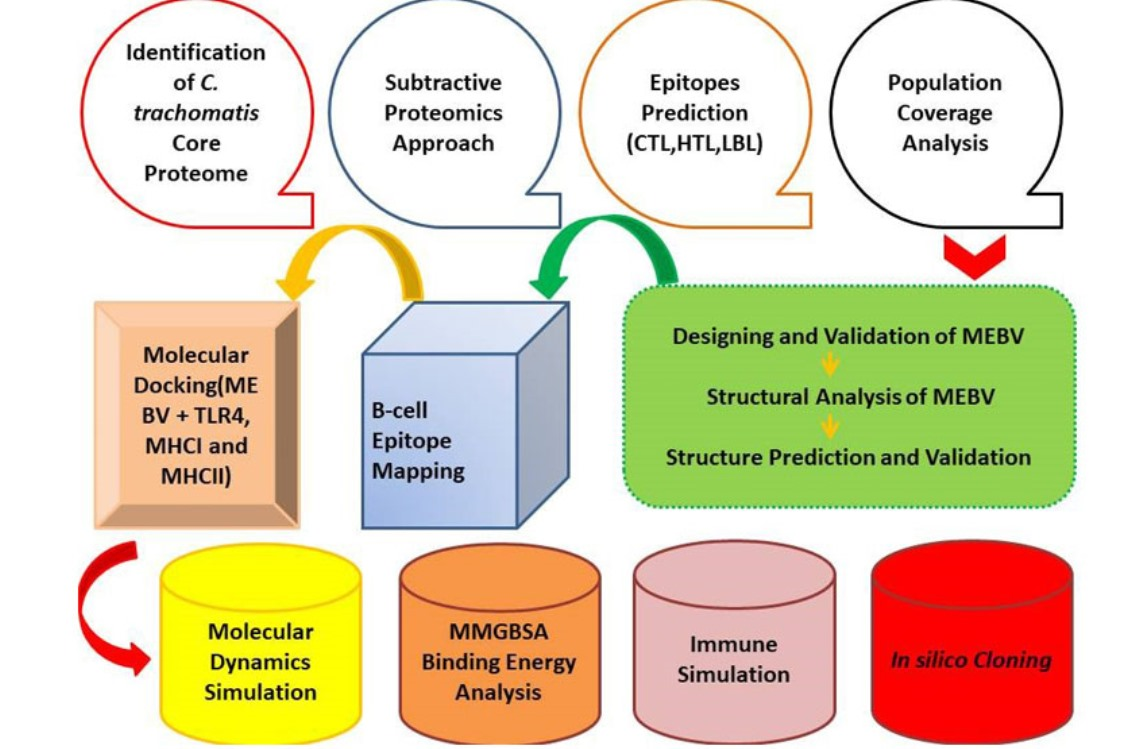
1. Introduction
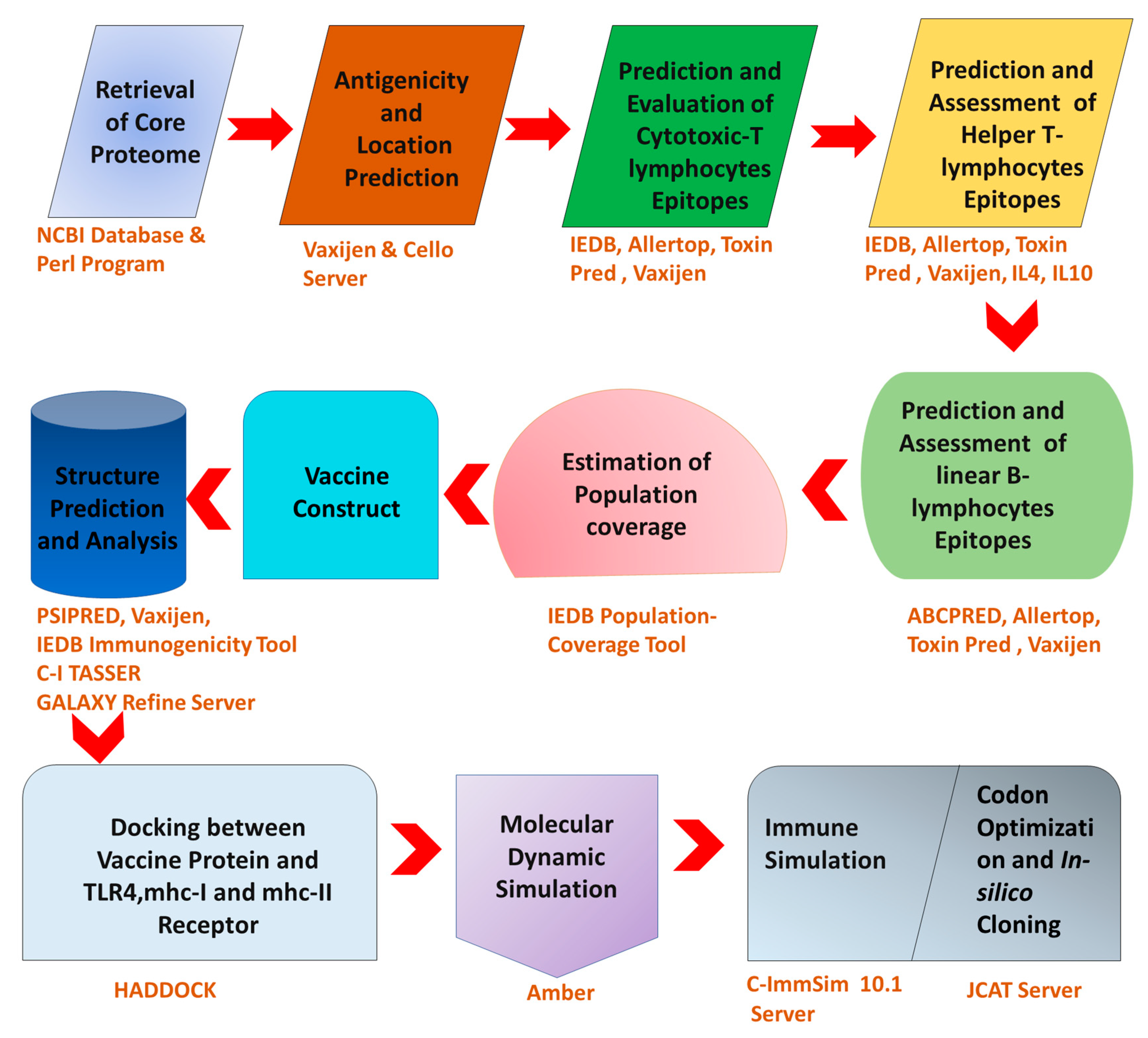
2. Materials and Methods
2.1. Identification of C. trachomatis Core Proteome
2.2. Subtractive Proteomics Approach
2.3. Epitopes Prediction
2.3.1. HTL Epitopes
2.3.2. CTL Epitopes
2.3.3. LBL Epitopes
2.4. World Population Coverage Analysis
2.5. Designing and Validation of MEBV
2.6. Structural Analysis of MEBV Construct
2.7. Tertiary Structure Prediction and Validation
2.8. B-Cell Epitope Mapping
2.9. Molecular Docking (MEBV + TLR4, MHCI and MHCII)
2.10. Molecular Dynamics Simulation
2.11. MMGBSA Binding Energy Analysis
2.12. Immune Simulation
2.13. In Silico Cloning
3. Results
3.1. Core Proteome Analysis
3.2. Identification of Target Proteins
3.3. Epitopes Prediction
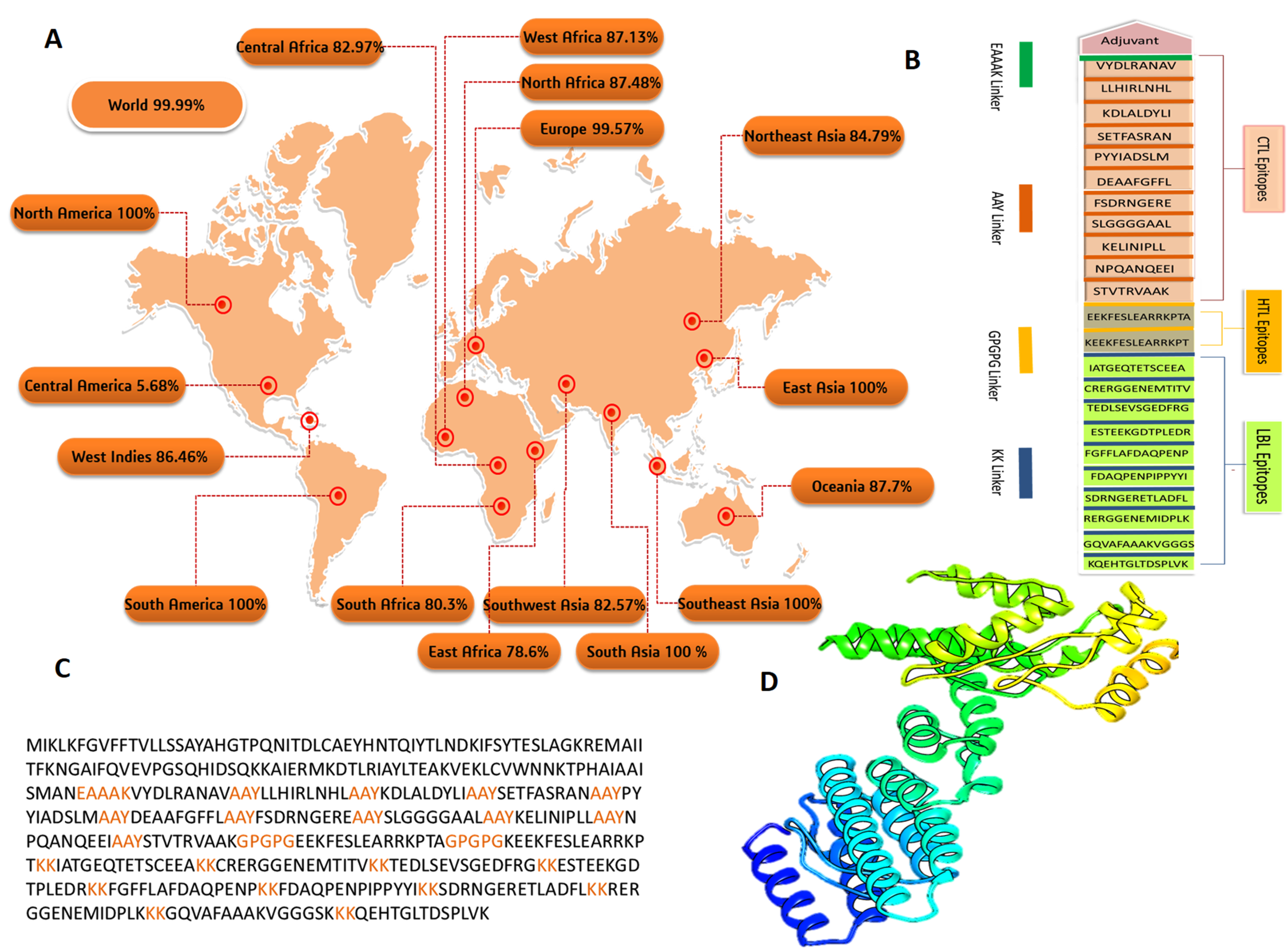
3.4. Population Coverage Analysis
3.5. Construction of Multi-Epitope Based Vaccine (MEBV)
3.6. Structural Analysis of the Vaccine Construct
3.7. Prediction of B Cell Epitopes of MEBV
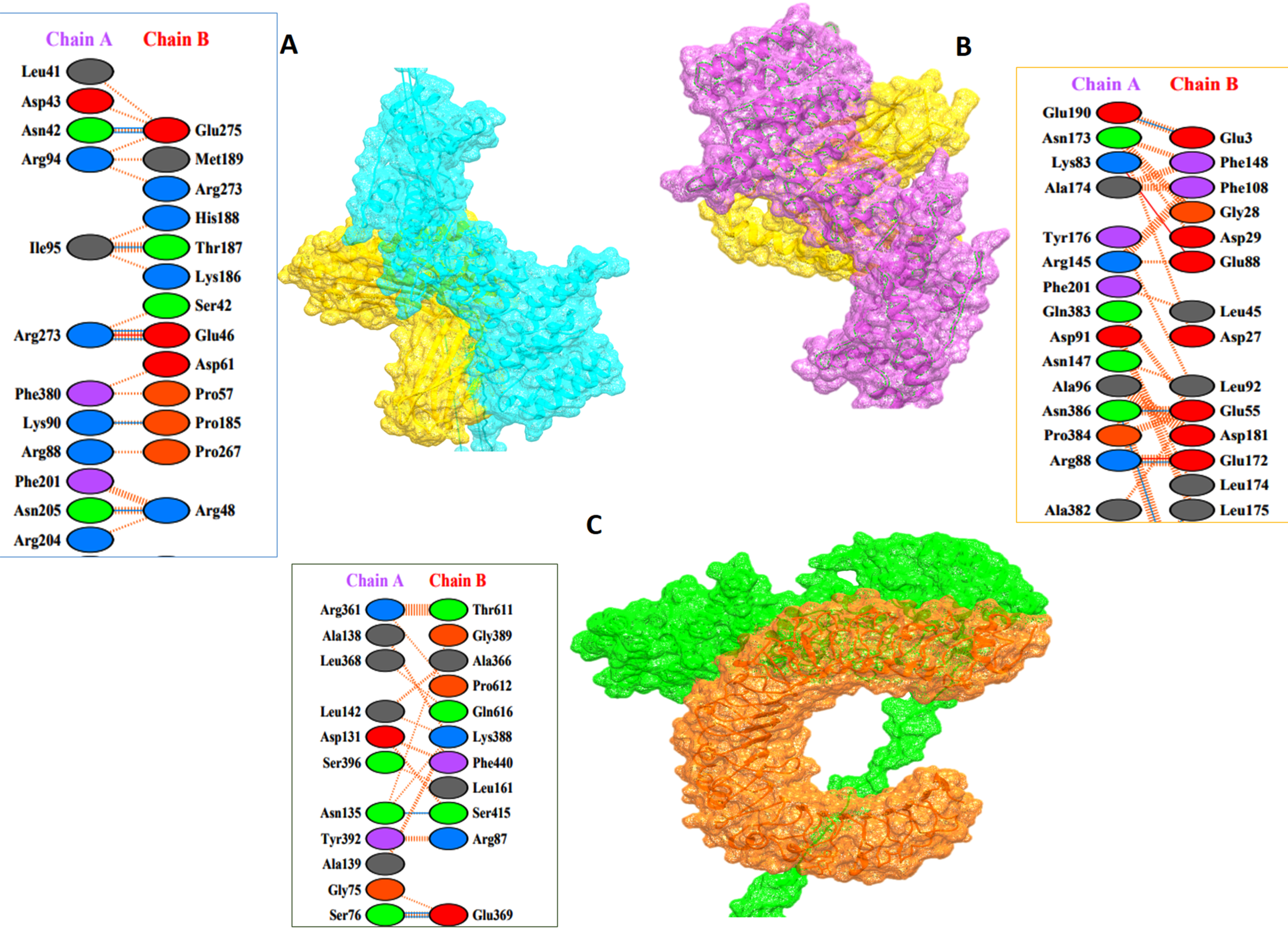
3.8. Protein–Protein Docking
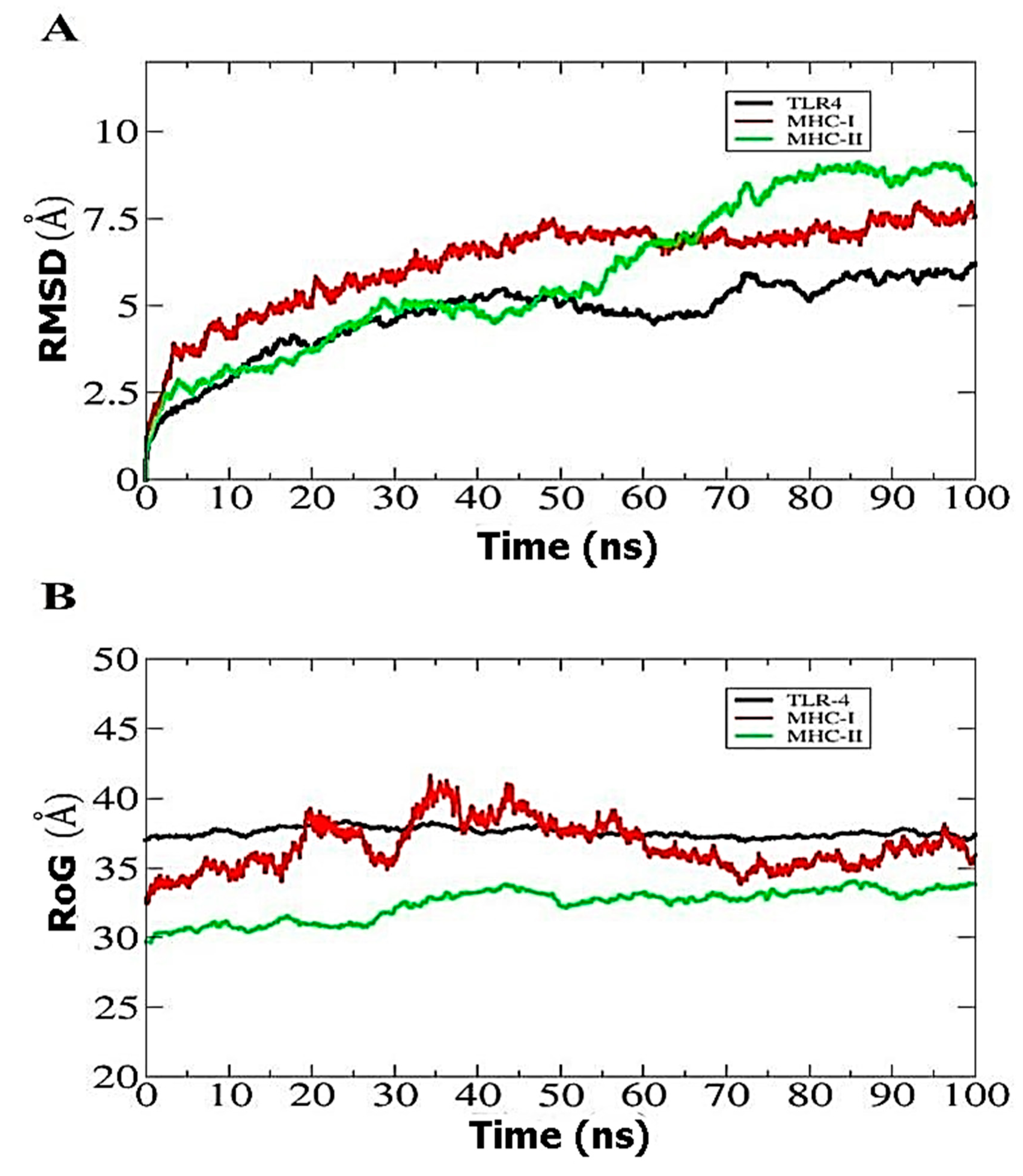
3.9. Molecular Dynamics Simulation
3.10. Binding Free Energies
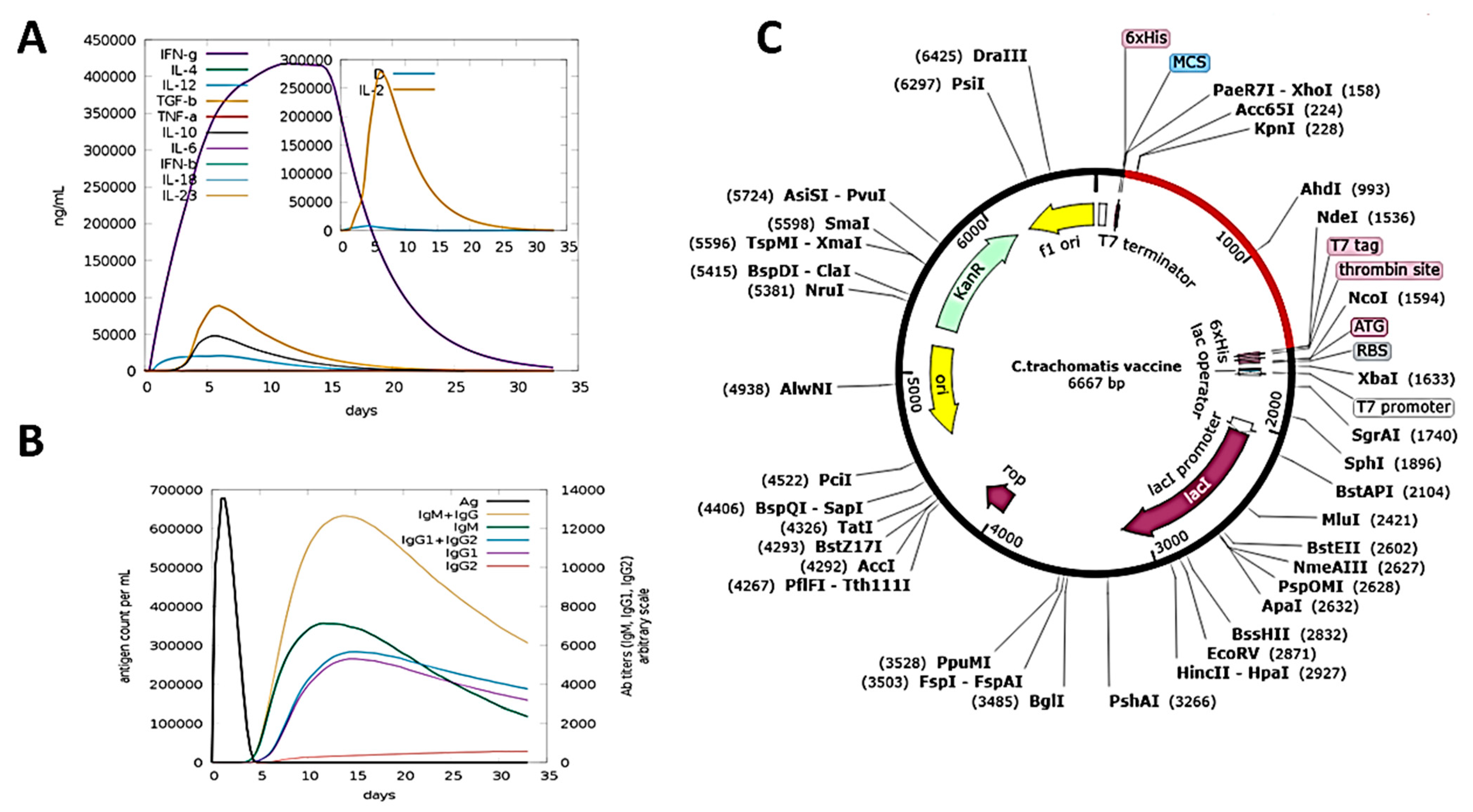
3.11. Immune Simulation
3.12. In Silico Cloning and Codon Optimization
4. Discussion
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- Nunes, A.; Gomes, J.P. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 2014, 23, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Norashikin, S.; Gangaram, H.; Hussein, S.H. Prevalence of Chlamydia trachomatis in Genito-urinary Medicine Clinic, Hospital Kuala Lumpur: A 5-year Retrospective Analysis. Available online: http://dermatology.org.my/pdf/journal%202007D.pdf (accessed on 26 July 2021).
- Shiragannavar, S.; Madagi, S.; Hosakeri, J.; Barot, V. In silico vaccine design against Chlamydia trachomatis infection. Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- De Schryver, A.; Meheus, A. Epidemiology of sexually transmitted diseases: The global picture. Bull. WHO 1990, 68, 639. [Google Scholar] [PubMed]
- Paavonen, J.; Eggert-Kruse, W. Chlamydia trachomatis: Impact on human reproduction. Hum. Reprod. Update 1999, 5, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: An update. Indian J. Med. Res. 2013, 138, 303. [Google Scholar]
- Malik, A.; Jain, S.; Hakim, S.; Shukla, I.; Rizvi, M. Chlamydia trachomatis infection & female infertility. Indian J. Med. Res. 2006, 123, 770. [Google Scholar]
- Fan, H.; Zhong, G. Chlamydia trachomatis. In Molecular Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1449–1469. [Google Scholar]
- Suchland, R.J.; Jeffrey, B.M.; Xia, M.; Bhatia, A.; Chu, H.G.; Rockey, D.D.; Stamm, W.E. Identification of concomitant infection with Chlamydia trachomatis IncA-negative mutant and wild-type strains by genomic, transcriptional, and biological characterizations. Infect. Immun. 2008, 76, 5438–5446. [Google Scholar] [CrossRef] [Green Version]
- Suchland, R.J.; Rockey, D.D.; Bannantine, J.P.; Stamm, W.E. Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane. Infect. Immun. 2000, 68, 360–367. [Google Scholar] [CrossRef] [Green Version]
- de la Maza, L.M.; Zhong, G.; Brunham, R.C. Update on Chlamydia trachomatis vaccinology. Clin. Vaccine Immunol. 2017, 24, e00543-16. [Google Scholar] [CrossRef] [Green Version]
- Mishori, R.; McClaskey, E.L.; WinklerPrins, V. Chlamydia trachomatis infections: Screening, diagnosis, and management. Am. Fam. Physician 2012, 86, 1127–1132. [Google Scholar]
- Wolle, M.A.; West, S.K. Ocular Chlamydia trachomatis infection: Elimination with mass drug administration. Expert Rev. Anti-Infect. Ther. 2019, 17, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Contini, C.; Rotondo, J.C.; Magagnoli, F.; Maritati, M.; Seraceni, S.; Graziano, A.; Poggi, A.; Capucci, R.; Vesce, F.; Tognon, M. Investigation on silent bacterial infections in specimens from pregnant women affected by spontaneous miscarriage. J. Cell. Physiol. 2019, 234, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schautteet, K.; De Clercq, E.; Vanrompay, D. Chlamydia trachomatis vaccine research through the years. Infect. Dis. Obstet. Gynecol. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, S.M.; McKay, P.F. Chlamydia trachomatis: Cell biology, immunology and vaccination. Vaccine 2021, 39, 2965–2975. [Google Scholar] [CrossRef] [PubMed]
- Hafner, L.M.; Wilson, D.P.; Timms, P. Development status and future prospects for a vaccine against Chlamydia trachomatis infection. Vaccine 2014, 32, 1563–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somani, J.; Bhullar, V.B.; Workowski, K.A.; Farshy, C.E.; Black, C.M. Multiple drug-resistant Chlamydia trachomatis associated with clinical treatment failure. J. Infect. Dis. 2000, 181, 1421–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, F.; Noor, A.; Ishaq, A.R.; Farzeen, I.; Saleem, M.H.; Ghaffar, K.; Aslam, M.F.; Aslam, S.; Chen, J.-T. Recent Advances in Diagnostic and Therapeutic Approaches for Breast Cancer: A Comprehensive Review. Curr. Pharm. Des. 2021, 27, 2344–2365. [Google Scholar] [CrossRef]
- Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahbar, M.R.; Ghasemi, Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol. Biosyst. 2017, 13, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef]
- Mamede, L.D.; de Paula, K.G.; de Oliveira, B.; Dos Santos, J.S.C.; Cunha, L.M.; Junior, M.C.; Jung, L.R.C.; Taranto, A.G.; de Oliveira Lopes, D.; Leclercq, S.Y. Reverse and structural vaccinology approach to design a highly immunogenic multi-epitope subunit vaccine against Streptococcus pneumoniae infection. Infect. Genet. Evol. 2020, 85, 104473. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.; Javaid, A.; Shahid, F.; Ashfaq, U.A. Rational design of multimeric based subunit vaccine against Mycoplasma pneumonia: Subtractive proteomics with immunoinformatics framework. Infect. Genet. Evol. 2021, 91, 104795. [Google Scholar] [CrossRef]
- Rehman, A.; Ashfaq, U.A.; Shahid, F.; Noor, F.; Aslam, S. The Screening of phytochemicals against NS5 Polymerase to treat Zika Virus infection: Integrated computational based approach. Comb. Chem. High Throughput Screen. 2021, 24, 1–14. [Google Scholar] [CrossRef]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef]
- ul Ain, Q.; Ahmad, S.; Azam, S.S. Subtractive proteomics and immunoinformatics revealed novel B-cell derived T-cell epitopes against Yersinia enterocolitica: An etiological agent of Yersiniosis. Microb. Pathog. 2018, 125, 336–348. [Google Scholar] [CrossRef]
- Gul, H.; Ali, S.S.; Saleem, S.; Khan, S.; Khan, J.; Wadood, A.; Rehman, A.U.; Ullah, Z.; Ali, S.; Khan, H. Subtractive proteomics and immunoinformatics approaches to explore Bartonella bacilliformis proteome (virulence factors) to design B and T cell multi-epitope subunit vaccine. Infect. Genet. Evol. 2020, 85, 104551. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Saba Ismail, S.A.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 12. [Google Scholar]
- ul Qamar, M.T.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse vaccinology assisted designing of multiepitope-based subunit vaccine against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 132. [Google Scholar] [CrossRef]
- Ahmad, S.; Shahid, F.; Tahir ul Qamar, M.; Abbasi, S.W.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A.; Ullah Saeed, H.F. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Noor, F.; Saleem, M.H.; Chen, J.-T.; Javed, M.R.; Al-Megrin, W.A.; Aslam, S. Integrative bioinformatics approaches to map key biological markers and therapeutic drugs in Extramammary Paget’s disease of the scrotum. PLoS ONE 2021, 16, e0254678. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Ajmal, A.; Ali, F.; Rastrelli, L. Core proteome mediated therapeutic target mining and multi-epitope vaccine design for Helicobacter pylori. Genomics 2020, 112, 3473–3483. [Google Scholar] [CrossRef]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Madden, T. The BLAST sequence analysis tool. In The NCBI Handbook [Internet], 2nd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. [Google Scholar]
- Shenoy, P.J.; Vin, H.M. Cello: A disk scheduling framework for next generation operating systems. ACM Sigmetrics Perform. Eval. Rev. 1998, 26, 44–55. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, M.; Guyard-Nicodème, M.; Hirchaud, E.; Parra, A.; Chemaly, M.; Dory, D. Identification of novel vaccine candidates against Campylobacter through reverse vaccinology. J. Immunol. Res. 2016, 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.K.; Avashthi, H.; Tiwari, A.; Jain, P.A.; Ramkete, P.W.; Kayastha, A.M.; Singh, V.K. MFPPI–multi FASTA ProtParam interface. Bioinformation 2016, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Modeling 2014, 20, 2278. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood J. Am. Soc. Hematol. 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Gupta, S.; Vir, P.; Raghava, G. Prediction of IL4 inducing peptides. Clin. Dev. Immunol. 2013, 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, G.; Usmani, S.S.; Dhanda, S.K.; Kaur, H.; Singh, S.; Sharma, M.; Raghava, G.P. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci. Rep. 2017, 7, 42851. [Google Scholar] [CrossRef] [Green Version]
- Halary, F.; Peyrat, M.A.; Champagne, E.; Lopez-Botet, M.; Moretta, A.; Moretta, L.; Vié, H.; Fournié, J.J.; Bonneville, M. Control of self-reactive cytotoxic T lymphocytes expressing γ δ T cell receptors by natural killer inhibitory receptors. Eur. J. Immunol. 1997, 27, 2812–2821. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.; He, Y.; Yang, Z. Epitope promiscuity and population coverage of Mycobacterium tuberculosis protein antigens in current subunit vaccines under development. Infect. Genet. Evol. 2020, 80, 104186. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Deléage, G. Alignsec: Viewing protein secondary structure predictions within large multiple sequence alignments. Bioinformatics 2017, 33, 3991–3992. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.H.; Bushweller, J.H.; Sodano, P.; Billeter, M.; Björnberg, O.; Holmgren, A.; Wüthrich, K. NMR structure of oxidized Escherichia coli glutaredoxin: Comparison with reduced E. coli glutaredoxin and functionally related proteins. Protein Sci. 1992, 1, 310–321. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B., III; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Li, C.; Tan, T.; Zhang, H.; Feng, W. Analysis of the conformational stability and activity of Candida antarctica lipase B in organic solvents: Insight from molecular dynamics and quantum mechanics/simulations. J. Biol. Chem. 2010, 285, 28434–28441. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef] [PubMed]
- McLennan, A.; Bates, A.; Turner, P.; White, M. Bios Instant Notes in Molecular Biology; Taylor & Francis: Oxfordshire, UK, 2012. [Google Scholar]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. PCCP 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson—Boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Sakharkar, K.R.; Sakharkar, M.K.; Chow, V.T. A novel genomics approach for the identification of drug targets in pathogens, with special reference to Pseudomonas aeruginosa. Silico Biol. 2004, 4, 355–360. [Google Scholar]
- Hafner, L.M.; Timms, P. Development of a vaccine for Chlamydia trachomatis: Challenges and current progress. Vaccine Dev. Ther. 2015, 5, 45–58. [Google Scholar]
- Kelly, K.A. Cellular immunity and Chlamydia genital infection: Induction, recruitment, and effector mechanisms. Int. Rev. Immunol. 2003, 22, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Refeidi, A.; El Mekki, A. Etiology and clinical features of acute epididymo-orchitis. Ann. Saudi Med. 1996, 16, 171–174. [Google Scholar] [PubMed]
- Ohshige, K.; Morio, S.; Mizushima, S.; Kitamura, K.; Tajima, K.; Suyama, A.; Usuku, S.; Tia, P.; Hor, L.; Heng, S. Behavioural and serological human immunodeficiency virus risk factors among female commercial sex workers in Cambodia. Int. J. Epidemiol. 2000, 29, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Anttila, T.; Saikku, P.; Koskela, P.; Bloigu, A.; Dillner, J.; Ikäheimo, I.; Jellum, E.; Lehtinen, M.; Lenner, P.; Hakulinen, T. Serotypes of Chlamydia trachomatis and risk for development of cervical squamous cell carcinoma. JAMA 2001, 285, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Prevalence and Incidence of Selected Curable Sexually Transmitted Infections: Overview and Estimates; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- Miller, W.C.; Ford, C.A.; Morris, M.; Handcock, M.S.; Schmitz, J.L.; Hobbs, M.M.; Cohen, M.S.; Harris, K.M.; Udry, J.R. Prevalence of chlamydial and gonococcal infections among young adults in the United States. JAMA 2004, 291, 2229–2236. [Google Scholar] [CrossRef] [Green Version]
- Shaw, K.; Coleman, D.; O’Sullivan, M.; Stephens, N. Public health policies and management strategies for genital Chlamydia trachomatis infection. Risk Manag. Healthc. Policy 2011, 4, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huston, W.M.; Harvie, M.; Mittal, A.; Timms, P.; Beagley, K.W. Vaccination to protect against infection of the female reproductive tract. Expert Rev. Clin. Immunol. 2012, 8, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igietseme, J.U.; Eko, F.O.; Black, C.M. Chlamydia vaccines: Recent developments and the role of adjuvants in future formulations. Expert Rev. Vaccines 2011, 10, 1585–1596. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ul Qamar, M.T.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [Green Version]
- María, R.; Arturo, C.; Alicia, J.A.; Paulina, M.; Gerardo, A.O. The impact of bioinformatics on vaccine design and development. In Vaccines; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Seib, K.L.; Zhao, X.; Rappuoli, R. Developing vaccines in the era of genomics: A decade of reverse vaccinology. Clin. Microbiol. Infect. 2012, 18, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Bulir, D.C.; Liang, S.; Lee, A.; Chong, S.; Simms, E.; Stone, C.; Kaushic, C.; Ashkar, A.; Mahony, J.B. Immunization with chlamydial type III secretion antigens reduces vaginal shedding and prevents fallopian tube pathology following live C. muridarum challenge. Vaccine 2016, 34, 3979–3985. [Google Scholar] [CrossRef]
- Chellas-Géry, B.; Linton, C.N.; Fields, K.A. Human GCIP interacts with CT847, a novel Chlamydia trachomatis type III secretion substrate, and is degraded in a tissue-culture infection model. Cell. Microbiol. 2007, 9, 2417–2430. [Google Scholar] [CrossRef] [PubMed]
- Fields, K.; Hackstadt, T. Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol. Microbiol. 2000, 38, 1048–1060. [Google Scholar] [CrossRef]
- Fields, K.A.; Fischer, E.R.; Mead, D.J.; Hackstadt, T. Analysis of putative Chlamydia trachomatis chaperones Scc2 and Scc3 and their use in the identification of type III secretion substrates. J. Bacteriol. 2005, 187, 6466–6478. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, K.P.; Yu, H.; Jiang, X.; Chan, Q.; Moon, K.-M.; Foster, L.J.; Brunham, R.C. Outer membrane proteins preferentially load MHC class II peptides: Implications for a Chlamydia trachomatis T cell vaccine. Vaccine 2015, 33, 2159–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infect. Genet. Evol. 2017, 49, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Liu, Y.; Hsi, J.; Wang, H.; Tao, R.; Shao, Y. Cholera toxin B subunit acts as a potent systemic adjuvant for HIV-1 DNA vaccination intramuscularly in mice. Hum. Vaccines Immunother. 2014, 10, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Nguyen, M.T. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A. Computational screening of medicinal plant phytochemicals to discover potent pan-serotype inhibitors against dengue virus. Sci. Rep. 2019, 9, 1433. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; ul Qamar, M.T.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the molecular mechanism of staphylococcal DNA gyrase inhibitors: A combined ligand-based and structure-based resources pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Monath, T.P. Flaviviruses; Army Medical Research Inst of Infectious Diseases Fort Detrick Md: Frederick, MD, USA, 1990. [Google Scholar]
- Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, C.; Sharma, A.; Bhattacharya, M.; Sharma, G.; Agoramoorthy, G.; Lee, S. Diabetes and COVID-19: A major challenge in pandemic period? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 11409–11420. [Google Scholar] [PubMed]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef]
- Pourhajibagher, M.; Bahador, A. Designing and in silico analysis of PorB protein from Chlamydia trachomatis for developing a vaccine candidate. Drug Res. 2016, 66, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Gomes, J.P.; Karunakaran, K.P.; Brunham, R.C. Bioinformatic analysis of Chlamydia trachomatis polymorphic membrane proteins PmpE, PmpF, PmpG and PmpH as potential vaccine antigens. PLoS ONE 2015, 10, e0131695. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Accession No | Location | Antigenicity | Transmembrane Helices | Molecular Weight |
|---|---|---|---|---|---|
| Type III secretion system translocon subunit CopD2 | WP_009873465 | Outer Membrane | 0.5273 | 0 | 55.01 kDa |
| SctW family type III secretion system gatekeeper subunit CopN | WP_009873547 | Extracellular | 0.6041 | 0 | 45.2 kDa |
| SycD/LcrH family type III secretion system chaperone Scc2 | WP_009873911 | Outer Membrane | 0.6154 | 0 | 27.09 kDa |
| Hypothetical protein CTDEC_0668 | ADI51345 | Periplasmic | 0.5663 | 0 | 24.45 kDa |
| CT847 family type III secretion system effector | WP_009873454 | Extracellular | 0.6101 | 0 | 19.97 kDa |
| CHLPN 76kDa-like protein | CPR70663.1 | Extracellular | 0.5456 | 0 | 68.91 kDa |
| Protein Name | Allele | Epitopes | Immunogenicity | Antigenicity |
|---|---|---|---|---|
| Type III secretion system translocon subunit CopD2 | HLA−C *14:02, HLA−C *08:02 | VYDLRANAV | 0.10067 | 1.4953 |
| Type III secretion system translocon subunit CopD2 | HLA−B *08:01, HLA−B *14:02 | LLHIRLNHL | 0.19782 | 2.0095 |
| SctW family type III secretion system gatekeeper subunit CopN | HLA−B *40:02, HLA−A *32:01 | KDLALDYLI | 0.03625 | 0.8238 |
| SctW family type III secretion system gatekeeper subunit CopN | HLA−B *44:02, HLA−B *44:03 | SETFASRAN | 0.07931 | 0.892 |
| SycD/LcrH family type III secretion system chaperone Scc2 | HLA−C *14:02, HLA−A *23:01, HLA−C *07:02 | PYYIADSLM | 0.0456 | 0.6918 |
| SycD/LcrH family type III secretion system chaperone Scc2 | HLA−B *18:01, HLA−B *40:01, HLA−B *44:03, HLA−E *01:03, HLA−B *44:02, HLA−E *01:01, HLA−B *38:01, HLA−B *40:02 | DEAAFGFFL | 0.36517 | 0.6844 |
| Hypothetical protein CTDEC_0668 | HLA−C *08:02, HLA−C *05:01 | FSDRNGERE | 0.19962 | 1.889 |
| Hypothetical protein CTDEC_0668 | HLA−B *15:02, HLA−B *07:02 | SLGGGGAAL | 0.16588 | 2.1805 |
| CT847 family type III secretion system effector | HLA−B *40:02, HLA−B *44:03, HLA−B *40:01, HLA−B *48:01, HLA−B *44:02, HLA−C *04:01 | KELINIPLL | 0.23346 | 0.625 |
| CHLPN 76kDa-like protein | HLA−B *51:01, HLA−B *53:01 | NPQANQEEI | 0.02943 | 0.9999 |
| CHLPN 76kDa-like protein | HLA−A *68:01, HLA−A *11:01, HLA−A *03:01, HLA−A *30:01 | STVTRVAAK | 0.1976 | 0.7123 |
| Protein Name | Peptide | Allele | IL4 | IL10 | IFN |
|---|---|---|---|---|---|
| SctW family type III secretion system gatekeeper subunit CopN | EEKFESLEARRKPTA | HLA−DRB5 *01:01, HLA−DRB5 *01:05 | Inducer | Inducer | Positive |
| SctW family type III secretion system gatekeeper subunit CopN | KEEKFESLEARRKPT | HLA−DRB5 *01:01, HLA−DRB5 *01:05 | Inducer | Inducer | Positive |
| Protein Name | Sequence | Position | Score | Immunogenicity | Antigenicity |
|---|---|---|---|---|---|
| Type III secretion system translocon subunit CopD2 | IATGEQTETSCEEA | 25 | 0.77 | 0.13565 | 1.5065 |
| Type III secretion system translocon subunit CopD2 | CRERGGENEMTITV | 1 | 0.67 | 0.33982 | 1.6901 |
| SctW family type III secretion system gatekeeper subunit CopN | RERGGENEMTASGG | 2 | 0.83 | 0.05641 | 2.1554 |
| SctW family type III secretion system gatekeeper subunit CopN | TEDLSEVSGEDFRG | 110 | 0.64 | 0.09196 | 1.3234 |
| SycD/LcrH family type III secretion system chaperone Scc2 | FGFFLAFDAQPENP | 142 | 0.83 | 0.31428 | 1.0071 |
| SycD/LcrH family type III secretion system chaperone Scc2 | FDAQPENPIPPYYI | 148 | 0.71 | 0.06882 | 1.1690 |
| Hypothetical protein CTDEC_0668 | SDRNGERETLADFL | 183 | 0.7 | 0.43964 | 1.2628 |
| Hypothetical protein CTDEC_0668 | FSLGGGGAALDSTV | 48 | 0.54 | 0.09884 | 1.5042 |
| CHLPN 76kDa-like protein | GQVAFAAAKVGGGS | 449 | 0.76 | 0.19157 | 1.0759 |
| CHLPN 76kDa-like protein | KQEHTGLTDSPLVK | 299 | 0.76 | 0.02767 | 1.0426 |
| Parameters | MEBV-TLR4 | MEBV-MHCI | MEBV-MHCII |
|---|---|---|---|
| HADDOCK score | 202.6 ± 13.6 | 221.3 ± 13.2 | 179.4 ±17.3 |
| Cluster size | 6 | 5 | 6 |
| RMSD from the overall lowest-energy structure | 45.4 ± 0.1 | 33.6 ± 0.1 | 9.4 ± 0.5 |
| van der Waals energy | −33.0 ± 2.1 | −48.8 ± 4.6 | −60.1 ± 2.1 |
| Electrostatic energy | −96.3 ± 21.8 | −63.8 ± 10.0 | −261.1 ± 24.8 |
| Desolvation energy | −0.1 ± 2.9 | −1.8 ± 1.3 | −10.7 ± 3.8 |
| Restraint violation energy | 2549.5 ± 170.4 | 2846.2 ± 134.9 | 3024.6 ± 192.4 |
| Buried Surface Area | 1154.6 ± 98.4 | 2101.9 ± 120.2 | 2907.6 ± 63.1 |
| Z-Score | −1.6 | −0.8 | −1.6 |
| Energy Parameter | TLR-4-MEBV Complex | MHC-I-MEBV Complex | MHC-II-MEBV Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −79.80 | −61.96 | −72.10 |
| EEL | −71.77 | −53.07 | −59.00 |
| EGB | 36.45 | 42.58 | 52.13 |
| ESURF | −6.63 | −8.18 | −6.10 |
| Delta G gas | −151.57 | −118.03 | −131.10 |
| Delta G solv | 29.82 | 34.4 | 46.03 |
| Delta Total | −121.75 | −83.63 | −85.07 |
| MM-PBSA | |||
| VDWAALS | −79.80 | −61.96 | −72.10 |
| EEL | −71.77 | −53.07 | −59.00 |
| EPB | 43.58 | 41.55 | 49.65 |
| ENPOLAR | −8.46 | −5.75 | −9.36 |
| Delta G gas | −151.57 | −118.03 | −131.10 |
| Delta G solv | 35.12 | 35.8 | 40.29 |
| Delta Total | −116.45 | −82.23 | −90.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, S.; Ahmad, S.; Noor, F.; Ashfaq, U.A.; Shahid, F.; Rehman, A.; Tahir ul Qamar, M.; Alatawi, E.A.; Alshabrmi, F.M.; Allemailem, K.S. Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches. Biology 2021, 10, 997. https://doi.org/10.3390/biology10100997
Aslam S, Ahmad S, Noor F, Ashfaq UA, Shahid F, Rehman A, Tahir ul Qamar M, Alatawi EA, Alshabrmi FM, Allemailem KS. Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches. Biology. 2021; 10(10):997. https://doi.org/10.3390/biology10100997
Chicago/Turabian StyleAslam, Sidra, Sajjad Ahmad, Fatima Noor, Usman Ali Ashfaq, Farah Shahid, Abdur Rehman, Muhammad Tahir ul Qamar, Eid A. Alatawi, Fahad M. Alshabrmi, and Khaled S. Allemailem. 2021. "Designing a Multi-Epitope Vaccine against Chlamydia trachomatis by Employing Integrated Core Proteomics, Immuno-Informatics and In Silico Approaches" Biology 10, no. 10: 997. https://doi.org/10.3390/biology10100997