The Role of Reactive Oxygen Species in Arsenic Toxicity
by
 ,
,
Yuxin Hu
1 ,
,
Jin Li
2,
Bin Lou
2,
Ruirui Wu
2,
Gang Wang
1,
Chunwei Lu
1,
Huihui Wang
2,
Jingbo Pi
2 and
Yuanyuan Xu
2,* 1
Experimental Teaching Center, School of Public Health, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, China
2
Program of Environmental Toxicology, School of Public Health, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, China
*
Author to whom correspondence should be addressed.
Biomolecules 2020, 10(2), 240; https://doi.org/10.3390/biom10020240
Submission received: 13 January 2020
/
Revised: 28 January 2020
/
Accepted: 29 January 2020
/
Published: 5 February 2020
(This article belongs to the Special Issue Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches)
Abstract
:Arsenic poisoning is a global health problem. Chronic exposure to arsenic has been associated with the development of a wide range of diseases and health problems in humans. Arsenic exposure induces the generation of intracellular reactive oxygen species (ROS), which mediate multiple changes to cell behavior by altering signaling pathways and epigenetic modifications, or cause direct oxidative damage to molecules. Antioxidants with the potential to reduce ROS levels have been shown to ameliorate arsenic-induced lesions. However, emerging evidence suggests that constructive activation of antioxidative pathways and decreased ROS levels contribute to chronic arsenic toxicity in some cases. This review details the pathways involved in arsenic-induced redox imbalance, as well as current studies on prophylaxis and treatment strategies using antioxidants.
1. Introduction
Arsenic is the 33rd element in the periodic table of elements. It displays features of both a metal and a non-metal, and thus called metalloid. However, it is referred as a heavy metal from a toxicological point of view [1]. Arsenic can be found in soil, water, and air from natural and anthropogenic sources. Over time, arsenic has accumulated a variety of uses, for example in cosmetics, wood preservatives, cotton desiccants, pesticides, and even in the treatment of acute promyelocytic leukemia (APL) [2,3,4]. However, evidence of arsenic poisoning has occurred from oral ingestion of arsenic, when found as a contaminant in food or potable water and when used as a therapeutic. The major route of exposure is via drinking water due to natural contamination of groundwater by inorganic arsenic in the earth’s crust, which threatens the health of more than 140 million people worldwide [5,6]. Chronic exposure to arsenic has been associated with the development of a wide range of human cancers (e.g., lung, skin, liver, bladder, and kidney) [7], as well as other nonmalignant disorders (e.g., respiratory illnesses, cardiovascular diseases, diabetes, neurotoxicity, and renal diseases) [6,8,9,10].
Arsenic exists in nature as both an inorganic trivalent (arsenite: iAs III) and inorganic pentavalent forms (arsenate: iAs V), and is metabolized via biomethylation involving a two-electron reduction of pentavalent arsenicals followed by oxidative methylation to form organic pentavalent arsenicals [11,12]. Inorganic arsenic can be methylated to organics by S-adenosyl-L-methionine (SAM), including monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) in humans with trivalent (MMA III and DMA III, respectively) and pentavalent forms (MMA III and DMA III, respectively) [8]. Inorganic arsenicals are considered more toxic than organics [13]. This is substantiated by the observation that acute toxicity of inorganic arsenic is greater than organics, and methylated arsenicals are easier to excrete [14,15]. Therefore, arsenic methylation was once considered as a detoxification pathway. However, trivalent methylated arsenicals are found to be at least as cytotoxic as their parent compounds if not more so in several human cells [16,17]. MMA III and DMA III show a higher capacity to inhibit activities of many enzymes and are more genotoxic than iAs III [18,19]. Moreover, MMA III and DMA III are more potent inhibitors of glucose-stimulated insulin secretion (GSIS) in isolated islets than iAs III [20]. These studies obscure the role of arsenic methylation in its toxicity, and again suggest that variance in arsenic methylation contributes to inter-individual susceptibility to arsenic-induced adverse health effects [21,22,23].
Oxidative stress mediated by reactive oxygen species (ROS) is a common denominator in arsenic toxicity. Here, we summarized ROS-related pathways in arsenic toxicity, as well as prophylaxis and therapeutic potential of antioxidants to combat arsenic toxicity.
2. ROS and Arsenic Toxicity
The toxic mechanisms of arsenic are complex and not fully understood. At a biochemical level, iAs V can replace phosphate in several reactions. Arsenite (iAs III) and trivalent organic (methylated) arsenicals react with thiols (-SH) in proteins and inhibit their activity. Other mechanisms include epigenetic alteration, oxidative stress, inflammation, and autophagic defects [24,25,26]. ROS are formed in biological systems during the reduction of molecular oxygen and include the superoxide radical anion (O2−•), hydrogen peroxide (H2O2), hydroxyl radical (•OH), hydroperoxyl radical (HOO•), singlet oxygen (1O2), and peroxyl radical (ROO•) [27]. Arsenic induces formation of 1O2, O2−•, H2O2, •OH, and ROO•. Formation of O2−• and H2O2 in response to arsenic exposure in different cell lines are summarized by Shi et al. [28]. Mechanisms responsible for generation of ROS induced by arsenic are proposed as follows (Figure 1). (i) Mitochondria: Mitochondrial complexes I and III in electron transport chain are responsible for the production of O2−•. Arsenic shows mitochondrial toxicity by inhibiting succinic dehydrogenase activity and uncoupling oxidative phosphorylation with production of O2−•, which gives rise to other forms of ROS [29]. (ii) Nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidase (Nox): Nox is a membrane-associated enzyme involved in ROS generation in response to arsenic. The evidence mainly comes from endothelial cells and is reviewed by Ellinsworth [30]. (iii) Generation of ROS during formation of intermediate arsine species [31,32]. For example, dimethylarsenic peroxyl radical is formed during the metabolic processing of DMA [33]. (iv) Redox-active iron released from ferritin caused by methylated arsenic species [34]. (v) Under physiological conditions, the formation of ROS by arsenic lay on the oxidation of arsenite to arsenate [35]. (vi) Endoplasmic reticulum (ER): ER is suggested to be a source of ROS caused by DMA III [36]. (vii) Interference with cellular antioxidants, for example, superoxide dismutase (SOD), catalase (CAT) [37], glutathione (GSH), and GSH-related enzymes [38,39], which indirectly result in increased ROS levels.
Despite of ROS overproduction reported in earlier studies applying high-dose arsenic in acute exposure, emerging evidence suggests that ROS do not increase when cells are exposed to an environmentally relevant dose of arsenic [40,41], especially after chronic exposure [42]. Treatment with 1 μM of arsenite for 4 h did not alter the amount of ROS in human bronchial epithelial cells (Beas-2B) [43]. Similarly, acute exposure to arsenite (2 or 4 h) below 10 μM of arsenite did not alter the intracellular amount of ROS in various cell lines, even with a more sensitive ROS detection method (electron paramagnetic resonance spectroscopy) [40,41]. Reduced intracellular ROS levels were observed in arsenic-transformed Beas-2B cells, and moreover were indicated as contributing to the acquisition of malignant phenotypes [42]. Our recent study found that arsenite-transformed human keratinocytes showed dysregulated autophagy with enhanced p62-NRF2 (nuclear factor (erythroid-derived 2)-like 2) feedback loop and decreased intracellular ROS levels [44]. In this scenario, constitutive Nrf2-mediated antioxidant response is frequently observed (see below). Thus, it is debated that adaptive antioxidant response is involved in toxicity caused by low-dose arsenic and especially during chronic exposure. Further understanding of the role of ROS in arsenic toxicity is needed.
3. Involvement of ROS-Mediated Pathways in Arsenic Toxicity
Signal transduction pathways transmit extracellular signals from series intracellular signaling molecules into genes [45]. Arsenic can alter signal transduction via ROS alteration or reversible oxidation of -SH group in proteins, which leads to activation or inhibition of transcription factors and regulates gene transcription [46]. The major ROS-affected pathways in response to arsenic include Nrf2-antioxidant response element (ARE) signaling pathway, microRNAs (miRNAs), mitophagy pathway, tyrosine phosphorylation system, mitogen-activated protein kinases (MAPKs), nuclear factor κB (NF-κB), and activator protein-1 (AP-1) [47,48,49,50] (Table 1).
3.1. Nrf2-ARE Pathway
Nrf2 is a master transcription factor in antioxidant system. Under physiological conditions, the protein level of Nrf2 is low because it binds to its negative regulatory factor kelch-like epichlorohydrin-associated protein 1 (Keap1), which forms the E3 ubiquitin ligase complex and facilitates ubiquitination and subsequent degradation of Nrf2 by the 26S proteasome [51]. When excessive ROS are generated, certain cysteine residues (C273, C288, and C151) in Keap1 sense the stress and are S-alkylated [52,53], leading to impairment of Keap1-mediated Nrf2 degradation [54]. Nrf2 then accumulates in the cytoplasm, translocates in the nucleus, dimerizes with small musculo-aponeurotic fibrosarcoma (Maf) proteins, and binds to the ARE motif in the promotor region of target genes, including various antioxidant enzymes and detoxification enzymes [55,56,57]. Therefore, the role of Nrf2-ARE pathway attracts much more attention in studies on arsenic toxicity.
Arsenic is an activator of Nrf2-Keap1 pathway [58,59,60,61,62]. Arsenite binds to the Ring finger domain of Ring-box 1 (Rbx1), which leads to the suppression of Cullin 3(Cul3)-Rbx1 E3 ubiquitin ligase activity, thereby activating the Nrf2-induced antioxidant signaling pathways [62]. Meanwhile, many research groups report that arsenic induces Nrf2 activation via the noncanonical mechanism, specifically by p62 accumulation due to dysregulated autophagy flux [43,63,64,65,66]. Accumulation of p62 results in sequestration of Keap1 in the autophagosomes and impairs Nrf2 degradation [67]. On the other hand, p62 is a downstream gene of Nrf2, forming a positive feedback loop [63]. The loop may act as a critical molecular alteration in arsenic carcinogenesis [68]. In addition, arsenite is found to induce acetylation of Nrf2 by p300/CREB (cAMP response element binding protein) binding protein (CBP), which enhances Nrf2 binding capacity to promoter-specific DNA [69] (Figure 2). However, a significant down-regulation was found in cardiac Nrf2 and peroxisome proliferator-activated receptor-γ (PPAR-γ) mRNA expression in arsenic-treated Sprague-Dawley (SD) rats compared with control rats [70]. Different response of Nrf2 to arsenic exposure is related to the strains and age of murines, as well as exposure time and dose of arsenic.
Nrf2 is considered as a protective factor against arsenic toxicity by reducing oxidative stress. Our previous study found that stable knockdown (KD) of NRF2 in human keratinocytes (HaCaT) significantly increased the sensitivity to acute cytotoxicity of inorganic arsenite, whereas KEAP1-KD cells showed a significant resistance to arsenite toxicity [94]. When mouse macrophage cells (RAW 264.7) were exposed to arsenite, a marked increase in ROS occurred in Nrf2-KD cells compared to scramble cells [76]. However, abnormal activation of Nrf2 is suggested to be cancer-promoting [95,96]. On one hand, NRF2-dependent antioxidant and detoxification enzymes promote the detoxification and elimination of ROS to attenuate arsenic carcinogenesis. On the other hand, NRF2 activation may provide cell proliferation or survival advantage by mediating metabolic reprogramming [56,97] and contributing to apoptotic resistance [98,99,100,101], which are important events in the process of arsenic-induced malignant transformation. Long-term exposure to an environmentally relevant dose of arsenic may induce constitutive activation of Nrf2, leading to adaptive antioxidant response, and subsequently contributing to malignant transformation [60,74,102,103]. In chronic arsenic-exposed Chang human hepatocytes, protein levels of nuclear Nrf2 peaked at 8 weeks and significantly elevated afterwards, whereas cytosol Nrf2 did not show significant change, which suggests that chronic arsenic exposure may constitutively activate NRF2 by post-transcriptional mechanism [60]. Furthermore, downstream genes of NRF2; NAD(P)H dehydrogenase [quinone] 1 (NQO1); aldo-keto reductase family 1, member C2 (AKR1C2); and aldo-keto reductase family 1, member C3 (AKR1C3) were overexpressed in chronic arsenic-exposed HaCaT cells [102]. Our recent studies found that silencing NRF2 in HaCaT cells abolished arsenic-induced acquisition of invasion capacity [44]. These data provide direct proof for the oncogenic role of Nrf2 in arsenic carcinogenesis. Taken together, Nrf2 pathway may exert dual roles in arsenic toxicity depending on the dose, exposure time, and cell types. Thus, concerns about strategy of using natural compounds, such as daphnetin (Daph) as an Nrf2 activator, for arsenic detoxification have been raised [104].
3.2. microRNAs
Epigenetic modifications contribute to toxic effects by arsenic exposure [105]. Alteration in miRNAs is one of these modifications and is closely related with intracellular ROS levels. He et al. found that chronic arsenic exposure lead to an overproduction of ROS, which induced activation of the miR-199a-5p/hypoxia inducible factor-1α (HIF-1α)/cyclooxygenase-2 (COX-2) pathway [77]. ROS inhibited miR-199a expression through increasing the promoter methylation of the miR-199a gene by DNA methyltransferase 1 [106]. miR-214 expression was transcriptionally repressed by Nrf2 through ARE within its promoter region in response to arsenic exposure in erythroid cells, and this repression was ROS dependent [78]. Not all alterations in miRNAs in arsenic-induced malignant transformation are related with ROS. Chen et al. found that Nrf2 was modulated by miR-155 in the process of malignant transformation induced by arsenite in human bronchial epithelial cells (16-HBE). However, there was no significant alteration in ROS production as determined by dichlorodihydrofluorescein diacetate (DCFH-DA) probe in the arsenic-transformed cells [107].
The levels of ROS can be regulated by miRNAs, as found in diseases such as cerebral ischemia, ischemia/reperfusion injury, and spinal cord injury. Overexpression of miRNA-20b increased the levels of adenosine 5’ triphosphate (ATP) and ROS in the cerebral ischemia of SD rats, whereas suppression of miRNA-20b decreased the levels of ROS [108]. Overexpression of miR-451 decreased apoptosis rate, ROS levels, and cleaved caspase-3 expression in the oxygen and glucose deprivation/reoxygenation cells [109]. Lycium barbarum polysaccharides (LBPs) reduced levels of ROS and nitric oxide (NO) induced by H2O2 through down-regulating miR-194 in PC-12 cells [110].
Understanding the molecular mechanisms of arsenic-induced toxicity, such as dynamics of ROS generation, miRNA expression, and the relationship between ROS and miRNAs, will certainly shed new light for future strategies against arsenic toxicity. However, so far, there are only a few studies exploring the molecular mechanism of arsenic toxicity in this regard. More studies are preferably needed to understand the potential relationship between ROS alteration and miRNA expression in response to arsenic exposure, of which research on the role of ROS in epigenetic dysregulation would be top priority.
3.3. Mitophagy
The mitochondrion is the major site for ROS production and leakage [111,112]. Moderate ROS levels are essential for cell proliferation and survival by mitophagy [113,114]. Excessive levels of ROS induce apoptotic signaling pathways. Furthermore, unceasing generated ROS in mitochondria lead to autophagy, apoptosis, or necrosis. On one hand, arsenic has mitochondrial toxicity, resulting in mitochondrial swelling and crista fragmentation, disturbing respiratory complex, and giving rise to ROS [115]. On the other hand, excessive ROS generation causes mitochondrial dysfunction [47,49].
Mitophagy is a type of autophagy physiologically responsible for mitochondrial quality control and mitochondrial ROS balance by removing damaged mitochondria [116,117,118,119]. Phosphate and tension homology deleted on chromsome ten (PTEN)-induced putative kinase 1 (PINK1)/Parkin is considered as one of the classical pathways required for mitophagy induction [119,120]. PINK1/Parkin-mediated mitophagy plays a protective role in some diseases [113,121]. For example, PINK1/Parkin-mediated mitophagy prevented renal tubular epithelial cells (RTEC) from apoptosis and tissue damage in contrast-induced acute kidney injury (CI-AKI) through reducing mitochondrial ROS and subsequent nucleotide-binding oligomerization domain-like receptors (NLR) protein 3 containing pyrin domain (NLRP3) inflammasome activation [113]. Increased mitochondrial fission promoted the survival of hepatocellular carcinoma (HCC) cells by facilitating autophagy and inhibiting mitochondria-dependent apoptosis, which was mediated via elevated ROS production [121].
Arsenic inhibits complex I in the mitochondrial electron transport chain, which leads to excessive generation of ROS, giving rise to lipid peroxidation and protein damage and the subsequent formation of mitochondrial permeability transition (MPT) [122]. Arsenite induces mitophagy via mitochondrial ROS and MPT [80]. PINK1/Parkin is activated upon mitochondrial membrane depolarization, a signal of mitochondrial dysfunction that results from multiple causes including hypoxia and impaired electron transport [123]. In arsenic toxicity, mitophagy exerts a dual role, facilitating cell survival either by eliminating damaged mitochondria or causing cell death. Arsenic-induced apoptosis in the pancreas of rats and insulinoma β-cell (INS-1) through impairment of mitophagy mediated by the ROS/Pparγ/PINK1/Parkin pathway [79]. Mitochondrial B cell lymphoma/leukemia-2 (Bcl-2)/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), a pro-apoptotic Bcl-2 homology 3 (BH3)-only protein, was activated as an upstream signal to increase the expression of caspase 3 and sequestosome 1 (SQSTM1), and contributed to increased cell death caused by arsenic [124]. BNIP3 also interacted with LC3 to target the damaged mitochondria and initiate mitophagy [125].
3.4. Alternative Pathways
3.4.1. Tyrosine Phosphorylation
Tyrosine phosphorylation is an important posttranslational modification that is known to regulate receptor kinase (RK)-mediated signaling in mammals [126,127]. Auto-phosphorylation of specific tyrosine residues increases catalytic efficiency of the RK itself, whereas phosphorylation of additional tyrosine residues creates docking sites for downstream signaling molecules [128,129]. The tyrosine phosphorylation system is mediated by two important classes, receptor tyrosine kinases (RTKs) and nonreceptor tyrosine kinases (NTKs). The former includes growth receptors such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial grow factor (VEGF). The latter includes Src-family protein kinases. Activation of EGFR leads to activation of MAPK pathways. Arsenic exposure increases tyrosine phosphorylation in numerous proteins, with phosphorylation of EGFR as a central target [81,130,131]. The proposed mechanisms involve interaction of -SH groups on EGFR with arsenic and structural changes or dimerization of EGFR caused by ROS [81,130,131,132]. In addition, ROS may inactivate negative regulators of EGFR (namely protein tyrosine phosphatases, PTPs) via oxidation of cysteine residues in the active sites of these enzymes [133,134]. Following EGFR activation, Shc is recruited and phosphorylated at its tyrosine residues, which leads to enhanced Shc-Grb2 interaction and downstream signaling transduction including MAPKs [81,133]. In vivo and in vitro studies also found that arsenic induced Src activation in various cell lines [83,84,135], which is proposed to be mediated by direct interactions between arsenic and vicinal -SH groups of Src [135]. ROS may participate in arsenic induced Src activation, but the evidence remains unclear. In addition, Src acts as the upstream of EGFR and MAPK signaling in response to arsenic exposure [135].
3.4.2. MAPK Pathway
MAPKs regulate proliferation, gene expression, differentiation, mitosis, survival, and apoptosis in the cell, and can be activated by arsenic in a time- and dose-dependent manner when the concentrations range from 0.1 to 500 μM [136,137]. The mRNA levels of p38, extracellular signal-regulated kinase (ERK)1, ERK2, and Jun N-terminal kinase (JNK) and the protein levels of phosphorylated-JNK (p-JNK)/JNK were significantly elevated in cardiomyocytes by arsenic [137]. Arsenic damaged intestinal epithelial cells (HT-29) by increasing p38 phosphorylation [138]. It has been suggested that arsenic activates MAPK via EGFR-dependent signaling transduction, such as EGFR/MEK, EGFR/Ras/MEK, and Src/EGFR cascades [29,83,134,139,140,141,142,143], or EGF-independent signaling pathways, such as Shc [135]. Arsenic induced the expression of COX-2, VEGF, HIF-1, and interleukin- 6 (IL-6), which were regulated by ERK, JNK, and p38 MAPK in simian virus-40 (SV-40) immortalized human uroepithelial cells and mouse lymphatic endothelial cells, mediated by ROS [75,86,88]. In addition, arsenic differentially activated MAPK pathways to exert opposing toxic effects, which were time-, dose-, arsenic species-, and cell-type-dependent. For example, acute arsenic exposure activated ERK1/2 and JNK, whereas chronic arsenic exposure induced continuous p38 activation in human hepatocytes [60]. JNKs were activated and prevented from arsenic-induced malignant transformation through apoptosis induction in mouse epidermal JB6 cells [144]. On the contrary, JNKs were essential in regulating expression of mineral dust-induced gene (Mdig), and thereby mediated arsenic-induced malignant transformation of BEAS-2B cells [145].
3.4.3. NF-κB Pathway
NF-κB is a transcription factor regulating cell-to-cell interaction, intracellular communication, cell recruitment, and transmigration. NF-κB activation induced by arsenic is dependent on the duration of exposure, dose of arsenic, and cell types. Arsenic ranging from 1 μM to 10 μM activated NF-κB [136,146], which is suggested to involve generation of ROS [147], and not depended on inhibitory protein κB (IκB) phosphorylation and degradation [148,149]; however, arsenic above 10 μM inhibited NF-κB [136,146] via Iκκ [150,151,152,153] or interference with DNA binding of NF-κB [146]. In addition, there is a dynamic interaction between Nrf2 and NF-κB pathways, which is complex and under elucidation [154]. Nrf2 is proposed to suppress production of cytokines driven by NF-κB [155]. Nrf2-deficient mouse astrocytes exhibited increased DNA-binding activity and overproduction of pro-inflammatory cytokines [156]. The overexpression of Nrf2 decreased the level of p65 in inflammation reaction in human adenocarcinoma alveolar basal epithelial cells (A549 cell) [157]. p65 has a dual role in Nrf2 activation. In the cell where Nrf2 and NF-κB were simultaneously activated, p65 inhibited Nrf2-ARE-driven transcription activity by competing CREB binding protein (CBP) from Nrf2, and promoted recruitment of histone deacetylase 3 to ARE, leading to local histone hypoacetylation [158,159]. On the other hand, in acute myeloid leukemia cells, NF-κB subunits p50 and p65 induced transcription of Nrf2 by binding to κB sites in Nrf2 proximal promoter [160]. In human embryonic kidney (HEK) 293T cells, Ras-related C3 botulinum toxin substrate 1 (RAC1) induced NRF2 activation through NF-κB [161]. NF-κB signaling pathway disrupted PPARα/δ-mediated lipid homeostasis, which was NRF2-independent in arsenic-exposed BEAS-2B cells [90].
3.4.4. AP-1 Pathway
AP-1 is a complex composed of homodimers or heterodimers of Jun and Fos proteins. It has a vital role in cell growth and apoptosis [162]. All MAPK cascades can induce AP-1 activation in response to arsenic [84,163,164,165]. Effects of arsenic on AP-1 are closely related to arsenic species, as well as time and concentration of exposure [163,166]. Trivalent methylated arsenicals were more potent than trivalent inorganic arsenic as inducers of c-Jun phosphorylation and AP-1 activation [163]. In cells transiently transfected with an AP-1-dependent promoter-reporter construct, methylated trivalent arsenicals methylarsine oxide (MAs III O) was more effective than iAs III in inducing the AP-1-dependent gene transcription [163]. Acute arsenic exposure increased AP-1 binding to DNA via c-Jun and c-Fos, whereas chronic exposure attenuated DNA-binding capacity of AP-1 [163,166,167]. c-Jun/AP-1 pathway-mediated cyclin D1 was indicated as one of the key events in cell malignant transformation caused by low-dose arsenic exposure [168].
3.4.5. p53 Pathway
p53 is a well-known tumor suppressor and plays an importance role in DNA repair, cell cycle, and apoptosis. Reports on the role of arsenic exposure on p53 are controversial, varying with arsenic species, exposure time, and cell types. In a human lyphoblastoid cell line, arsenic induced p53 expression by an ataxia telangiectasia mutated (ATM) (a member of PI3-kinase-related protein kinase)-dependent pathway, which phosphorylated p53 at serine 15 [169,170]. In HaCaT cells, chronic arsenic exposure inactivated p53 via poly(ADP-ribosyl)ation [171]. However, HaCaT cells are immortalized with SV40, which are known to interfere with p53 expression. In human telomerase reverse transcriptase (hTERT)-immortalized human keratinocytes, exposure to low-dose arsenite inhibited p53 expression by transcriptionally upregulating murine double minute 2 (MDM2) or ERK2-mediated overexpression of MDM2 [172]. In arsenic-exposed MCF-7 cells, an S-phase cell cycle arrest was found to depend on activation of p53 downstream cellular defense enzymes (i.e., sestrin 1 (SESN1) and activating transcription factor 3 (ATF3)) that was triggered by ROS generation in the early stage [93]. Recently, it has been shown that MDM2 is a downstream gene of Nrf2 and serves a link between Nrf2 and p53 in pancreas cancer [173]. Yet, involvement of Nrf2-MDM2-p53 pathway in arsenic-induced cancer is not clarified.
3.4.6. Stress Granules (SGs) Pathway
The overproduction of ROS induced by arsenic exposure causes tremendously harmful outcomes to cells, organs, and organisms [174]. To protect against arsenic toxicity, cells quickly activate antioxidant systems. Stress granules (SGs), the non-membranous cytosolic structures consisting of mRNAs and proteins, are formed and have antioxidant activity during arsenic exposure [175,176,177]. After the formation of SGs, the elevation of ROS was suppressed and ROS-dependent apoptosis was inhibited [175]. Beyond their function in defencing arsenic toxicity, SGs have been proposed to alter multiple signaling pathways, such as the JNK, Wnt, and mammalian target of rapamycin (mTOR) pathways, by intercepting and sequestering signaling components [178]. Accordingly, SG formation is a marker of chemoresistance and is upregulated by the production of a prostaglandin (15d-PGJ2), which is controlled by NRF2, in mutant v-Ki-ras 2 Kirsten rat sarcoma viral oncogene homolog (KRAS) cells [179]. The two antioxidant systems mentioned above, NRF2 and SGs, may intertwine in response to environmental stress, although the underling mechanism is not fully understood. Further understanding of the role of ROS along with NRF2 and SGs in arsenic toxicity is needed.
3.4.7. Metabolism Pathway
Metabolic reprograming is a feature of cancer cells, which usually show a strong dependence on aerobic glycolysis (the Warburg effect), increase in glutaminolysis, enhancement of macromolecule production and mitochondrial biogenesis, activation of the pentose phosphate pathway, and upregulation of amino acid and lipid metabolism [180]. Although metabolic reprograming has been implicated in carcinogenesis [181], few studies have been performed to investigate the role of ROS in arsenic-relevant cancer metabolism. The feature of metabolic reprograming in response to arsenic challenge is still not fully understood, albeit there is some evidence suggesting that arsenic induces overproduction of ROS and aerobic glycolysis. Chronic arsenite (75 ppb) exposure was shown to induce aerobic glycolysis while inhibiting mitochondrial oxidative phosphorylation in primary human cells and multiple cell lines (BEAS-2B, human prostate epithelial cell line (RWPE-1), human pulmonary epithelial carcinoma cell line (A549), primary human urothelial cells (HUC), and human dermal fibroblasts (HDF)) [182]. A similar phenomenon was also observed in Caenorhabditis elegans following 48 h arsenite exposure (50 to 500 μM) [183]. When human hepatocyte cells (HL-7702) were treated with different concentrations of arsenite (1 to 5 μM, 12 h), overproduction of ROS resulted from activated nicotine adenine disphosphonucleotide (NADPH) oxidase-mitochondria axis inactivated prolyl hydroxylases (PHDs), which led to protein accumulation of HIF-1α [184]. The latter is recognized as an inducer of aerobic glycolysis [182,185]. Reciprocal crosstalk between ROS and metabolism is vital to function and fate of cancer cells [186,187,188]. Thus, the mitochondria, a major source of ROS production and ATP synthesis, represent a potentially target for cancer therapy [188]. Some antioxidants (e.g., NADPH and GSH) and redox cofactors (e.g., Nicotinamide adenine diuncleotide hydrogen (NADH) and reduced Flavin adenine dinucleotide (FADH)) act as a bridge in redox regulation and metabolic reprograming [189,190,191]. ROS can consume reducing agents (NADPH and GSH) key for cell metabolism [192,193], and meanwhile activate Nrf2, AMPK, and HIF-1, which regulate metabolism and in turn fine tune ROS levels [194,195,196]. Therefore, the overproduction of ROS induced by arsenic is linked to metabolic reprogramming by direct or indirect ways. However, the exact order for the evolution of ROS and cell metabolism and their specific roles in arsenic carcinogenesis remain to be further investigated.
4. Potential Application of Antioxidants to Rescue Arsenic Toxicity
The first line of defense against acute arsenic toxicity is to reduce the amount of arsenic in the body [197]. The ideal arsenic-removal drug can interfere with the interactions of arsenic and molecules in the tissue. 2,3-dimercaptopropane-1-sulfonic acid (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) are hydrophilic and belong to the mercapto family, which have vicinal dithiol moiety for the binding of metals [198,199]. These drugs or their analogs offer therapeutic benefit in acute arsenic poisoning when administered promptly [200]. Due to the limited ability of crossing the blood–brain barrier, loss of essential metals in the body, low cellular membrane penetration, and potential side effects in the kidney and liver, the use of metal chelators is limited [197,201].
Because it is well accepted that excessive generation of ROS plays an important role in the molecular mechanism of arsenic-induced toxicity and related diseases, application of antioxidants, especially extracts from plants, has been widely studied as therapeutics to counteract arsenic-induced toxicity. Some antioxidants involved in the methylation-mediated arsenic detoxification-excretion process, for example, GSH, can mitigate toxicity [202]. Some antioxidants decrease intracellular ROS levels via inhibiting mitochondrial respiratory chain complex I (e.g., metformin) [203]. Others present a protective role against arsenic-induced toxicity by regulating apoptosis-related molecular changes (e.g., diallyl trisulfide) [204,205]. Different types of antioxidants used for rescuing arsenic toxicity and their possible mechanisms are listed in Table 2. These antioxidants are classified as ROS scavengers, oxidative enzyme inhibitors, metal chelators, and antioxidant enzyme cofactors. The protective mechanisms of antioxidants extracted from the natural plants against arsenic-induced toxicity are shown in Figure 3. Besides reducing ROS, these antioxidants are involved in regulating signaling pathways such as Nrf2, NF-κB, MAPKs, transforming growth factor beta/Smad (TGF-β/Smad), and mammalian target of rapamycin/Akt (mTOR/Akt) [104,203,206,207,208,209,210,211].
Interestingly, different natural compounds used as antioxidants exert varied roles in Nrf2 activation in reducing arsenic toxicity. For example, tetramethylpyrazine (TMP) (50 μM or 100 μM) protected against arsenic-induced nephron toxicity by inhibiting Nrf2 activation, and accordingly reducing Heme oxygenase-1 (HO-1) expression [209]. Pomegranate fruit extract (PFE) (0.2 mL of 0.2% of extract) reduced ROS generation in hepatocytes, thereby reducing arsenic-induced Nrf2 activation [207]. However, most antioxidants showed an effect on promoting Nrf2 activation in response to arsenic [26,70,104,211]. Dietary supplementation with SF (80 mg/kg BW) protected against arsenic-induced nephrotoxicity via the Phosphoinositide 3-kinase (PI3K)/Akt-mediated Nrf2 signaling pathway in the rat kidney [212]. Grape seed proanthocyanidin extract (GSPE) (10, 25, and 50 mg/L) activated Nrf2 signaling pathway to antagonize arsenic-induced oxidative damage, promoted arsenic methylation metabolism, and relieved arsenic-induced hepatotoxicity [26]. Alleviated arsenic toxicity in the liver and reproductive system by eriodictyol and lutein was via activating Nrf2 signaling pathway [213,214]. Co-treatment of antioxidant vitamins L-ascorbic acid and α-tocopherol attenuated toxicity induced by arsenic trioxide in H9c2 cardiomyocytes through activation of Nrf2 and Bcl2 transcription factors [215]. In contrast, PFE (0.2 mL of 0.2% of extract) reversed arsenic-induced hepatotoxicity with reduction in arsenic-induced Nrf2 activation [207], suggesting that other Nrf2-independent mechanisms are involved in attenuation of arsenic toxicity by this antioxidant.
Arsenic activates pro-inflammatory signal pathway and induces ROS generation, thereby resulting the activation of NF-κB [208]. TMP and PFE inhibited arsenic-induced NF-κB-inflammatory pathway [207,209]. In renal tubular epithelial cells, arsenic exposure leads to dynamic alterations of COX-2 expression, which was regulated by NF-κB. TMP and NAC pretreatment reversed COX-2 dynamic changes via suppression of NF-κB activation and β-catenin protein expression [208].
Some antioxidants attenuated arsenic-induced toxicity via MAPK activation. TMP inhibited arsenic-induced activation of MAPK family, and further reduced the expression of arsenic response protein 2 (ARS2), which contributed to its nephron protective effects [209]. As a prospective remedial agent, all-trans retinoic acid (ATRA) (0.5 mg/kg) reversed arsenic-induced oxidative stress and apoptosis by inhibiting the MAPK signaling pathways and repressing p53-dependent apoptosis in the rat uterus [206]. When hepatic cells were exposed to arsenic, p38 and JNK signaling pathways were activated. Carnosic acid (CA), which was commonly found in Rosmarinus officinalis and Salvia officinalis, significantly attenuated arsenic-induced phosphorylation of p38 and JNK [235].
TGF-β1 is a potent profibrogenic cytokine and can be initiated by oxidative stress [236]. Arsenic-induced rat renal injury was correlated with TGF-β1 induced fibrosis, as demonstrated by increased levels of TGF-β1 and pSmad2/3 in renal tissue. Grape seed extract (GSE) (100 mg/kg for male SD rats; 100 mg/L for rat hepatic stellate cells) reduced arsenic-stimulated oxidative stress, and thereby specifically suppressed TGF-β/Smad signaling [228]. (-)-Epigallocatechin-3-gallate (EGCG), eriodictyol, lutein, folic acid, zinc, and selenium were all able to elevate the expression of SOD, GSH, and CAT, and reduce the level of malondialdehyde (MDA) caused by arsenic, resulting in amelioration of arsenic-induced toxicity [214,220,232,233].
Generally speaking, accumulating evidence suggests that antioxidants have the potential to alleviate arsenic toxicity through reducing ROS generation, enhancing antioxidant capacity, regulating ROS-related signaling pathways, and keeping the balance of inflammation and immunomodulation. Natural antioxidants extracted from plants are more promising due to their rich sources, diversity, and few side effects. However, many studies on the role of plant extracts have not been systematically conducted. In vitro assessment results do not provide exact therapeutic implications because effectiveness plant extracts may be influenced by several physiopharmacological processes, such as absorption, distribution, metabolism, storage, and excretion, as well as bioavailability, and presence of co-antioxidants and ions [237]. Hence, a commonly suggested strategy, in which in vitro mechanic analysis goes after in vivo effect evaluation [238], is proposed to assess therapeutic potential of plant extracts in arsenic toxicity. In addition, there is a need to first identify the major target of arsenic-induced pathophysiological alterations. Many plant extracts have multiple functions in addition to severing as an antioxidant. Finally, plant extracts should be screened for low molecular antioxidants that are able to cross blood–brain barriers and reach other target organs. In addition to the above, more and more physiologically-based pharmacokinetic (PBPK) models have been used to predict the pharmacokinetic behavior of drugs in humans on the basis of preclinical data [239,240]. PBPK modeling, a good tool for evaluating and optimizing clinical trial design, provides an approach that enables the plasma concentration-time profiles to be predicted from in vitro and in vivo data [239,241,242], which can be used for choosing the dose of antioxidants.
5. Conclusions
An effectively preventive and therapeutic strategy for arsenic poisoning is still a challenge worldwide due to the incomplete understanding of underlying mechanisms. ROS alteration is a widely accepted key event in arsenic toxicity. Earlier studies with acute exposure and relatively high dose arsenic suggest that excessive levels of ROS disturb cellular signaling pathways, as well as damage macromolecules. The effects of antioxidants on arsenic toxicity are also assessed in a scenario of relatively high dose of arsenic. However, the mechanism and function of ROS may be different in the realistic environment, in which a relatively low-dose and long-term exposure to arsenic affects millions of people. In addition, in vitro assay concentrations of arsenic may misrepresent potential in vivo toxic effects and do not provide dose-response data that can be used for a risk assessment [243,244]; hence, the in vitro-to-in vivo extrapolation (IVIVE) by physiologically based toxicokinetic (PBTK) modeling was used to serve in vitro-in silico-based risk assessment [245]. It is of great importance to apply physiologically relevant doses during toxicological research. The role of ROS in arsenic toxicity should be fully clarified before the application of antioxidants. Of all the antioxidants investigated in arsenic toxicity, natural antioxidants extracted from plants are promising due to their rich sources, diversity, and few side effects, especially those with low molecular weight. More effective therapeutic value from plant extracts is expected on the basis of arsenic-induced pathophysiology targeting combined with in vitro and in vivo assessment.
Author Contributions
Y.H., J.L., and B.L. designed the outline of the article and wrote it. R.W., G.W., and C.L. designed the outline of the article initial draft, and revised and expanded the manuscript. H.W., J.P., and Y.X. helped with intellectual contributions. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (81573187); LiaoNing Revitalization Talents Program (XLYC1807225); Liaoning Pandeng Scholar Program (to Y.X.) from the Education Department of Liaoning Province, China; Key R&D Plan Guidance Project (to Y.X.) from Department of Science and Technology of Liaoning Province, China.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Mandal, B.K.; Suzuki, K.T. Arsenic round the world: A review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad Med. J. 2003, 79, 391–396. [Google Scholar] [CrossRef]
- Ravandi, F.; Koumenis, I.; Johri, A.; Tallman, M.; Roboz, G.J.; Strickland, S.; Garcia-Manero, G.; Borthakur, G.; Naqvi, K.; Meyer, M.; et al. Oral arsenic trioxide ORH-2014 pharmacokinetic and safety profile in patients with advanced hematologic disorders. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Hu, J.; Lo-Coco, F.; Jin, J. The simpler, the better: Oral arsenic for acute promyelocytic leukemia. Blood 2019, 134, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Garty, M.; Deka-Starosta, A.; Chang, P.; Kopin, I.J.; Goldstein, D.S. Effects of clonidine on renal sympathetic nerve activity and norepinephrine spillover. J. Pharmacol. Exp. Ther. 1990, 254, 1068–1075. [Google Scholar]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef] [Green Version]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Leon, France, 2012; Volume 100C, pp. 41–48. [Google Scholar]
- Abernathy, C.O.; Liu, Y.P.; Longfellow, D.; Aposhian, H.V.; Beck, B.; Fowler, B.; Goyer, R.; Menzer, R.; Rossman, T.; Thompson, C.; et al. Arsenic: Health effects, mechanisms of actions, and research issues. Environ. Health Perspect. 1999, 107, 593–597. [Google Scholar] [CrossRef]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Parvez, F.; Chen, Y.; Brandt-Rauf, P.W.; Bernard, A.; Dumont, X.; Slavkovich, V.; Argos, M.; D’Armiento, J.; Foronjy, R.; Hasan, M.R.; et al. Nonmalignant respiratory effects of chronic arsenic exposure from drinking water among never-smokers in Bangladesh. Environ. Health Perspect. 2008, 116, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Aposhian, H.V.; Zakharyan, R.A.; Avram, M.D.; Sampayo-Reyes, A.; Wollenberg, M.L. A review of the enzymology of arsenic metabolism and a new potential role of hydrogen peroxide in the detoxication of the trivalent arsenic species. Toxicol. Appl. Pharmacol. 2004, 198, 327–335. [Google Scholar] [CrossRef]
- Hayakawa, T.; Kobayashi, Y.; Cui, X.; Hirano, S. A new metabolic pathway of arsenite: Arsenic-glutathione complexes are substrates for human arsenic methyltransferase Cyt19. Arch. Toxicol. 2005, 79, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Gebel, T.W. Arsenic methylation is a process of detoxification through accelerated excretion. Int. J. Hyg. Environ. Health 2002, 205, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Davis, T.P.; Wilson, P. Organic Arsenicals as Functional Motifs in Polymer and Biomaterials Science. Macromol. Rapid Commun. 2018, 39, e1800205. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.S.; Ayala-Fierro, F.; Cullen, W.R.; Carter, D.E.; Vasken Aposhian, H. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol. Appl. Pharmacol. 2000, 163, 203–207. [Google Scholar] [CrossRef]
- Styblo, M.; Del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef]
- Dopp, E.; von Recklinghausen, U.; Diaz-Bone, R.; Hirner, A.V.; Rettenmeier, A.W. Cellular uptake, subcellular distribution and toxicity of arsenic compounds in methylating and non-methylating cells. Environ. Res. 2010, 110, 435–442. [Google Scholar] [CrossRef]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Douillet, C.; Stýblo, M. Arsenite and its trivalent methylated metabolites inhibit glucose-stimulated calcium influx and insulin secretion in murine pancreatic islets. Arch. Toxicol. 2019, 93, 2525–2533. [Google Scholar] [CrossRef]
- Chen, C.J.; Hsu, L.I.; Wang, C.H.; Shih, W.L.; Hsu, Y.H.; Tseng, M.P.; Lin, Y.C.; Chou, W.L.; Chen, C.Y.; Lee, C.Y.; et al. Biomarkers of exposure, effect, and susceptibility of arsenic-induced health hazards in Taiwan. Toxicol. Appl. Pharmacol. 2005, 206, 198–206. [Google Scholar] [CrossRef]
- Lindberg, A.L.; Rahman, M.; Persson, L.A.; Vahter, M. The risk of arsenic induced skin lesions in Bangladeshi men and women is affected by arsenic metabolism and the age at first exposure. Toxicol. Appl. Pharmacol. 2008, 230, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Arsenic methylation, urinary arsenic metabolites and human diseases: Current perspective. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2007, 25, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Tian, M.; Wang, X.; Zhang, J.; Huang, Q.; Liu, L.; Shen, H. Cortex and hippocampus DNA epigenetic response to a long-term arsenic exposure via drinking water. Environ. Pollut. 2018, 234, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Goel, R.K.; Kaur, T. Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 2011, 18, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Niu, Q.; Hu, Y.; Feng, G.; Wang, H.; Li, S. Proanthocyanidins Antagonize Arsenic-Induced Oxidative Damage and Promote Arsenic Methylation through Activation of the Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 8549035. [Google Scholar] [CrossRef] [Green Version]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [CrossRef]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef]
- Corsini, E.; Asti, L.; Viviani, B.; Marinovich, M.; Galli, C.L. Sodium arsenate induces overproduction of interleukin-1alpha in murine keratinocytes: Role of mitochondria. J. Investig. Dermatol. 1999, 113, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Ellinsworth, D.C. Arsenic, reactive oxygen, and endothelial dysfunction. J. Pharmacol. Exp. Ther. 2015, 353, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Hayashi, H.; Tachikawa, M.; Kato, K.; Hasegawa, A.; Oku, N.; Okada, S. Metabolic methylation is a possible genotoxicity-enhancing process of inorganic arsenics. Mutat. Res. 1997, 394, 95–101. [Google Scholar] [CrossRef]
- Yamanaka, K.; Okada, S. Induction of lung-specific DNA damage by metabolically methylated arsenics via the production of free radicals. Environ. Health Perspect. 1994, 102 (Suppl. 3), 37–40. [Google Scholar] [PubMed]
- Kato, K.; Hayashi, H.; Hasegawa, A.; Yamanaka, K.; Okada, S. DNA damage induced in cultured human alveolar (L-132) cells by exposure to dimethylarsinic acid. Environ. Health Perspect. 1994, 102 (Suppl. 3), 285–288. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Kitchin, K.T.; Cullen, W.R. Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch. Biochem. Biophys. 2000, 382, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Del Razo, L.M.; Quintanilla-Vega, B.; Brambila-Colombres, E.; Calderon-Aranda, E.S.; Manno, M.; Albores, A. Stress proteins induced by arsenic. Toxicol. Appl. Pharmacol. 2001, 177, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Naranmandura, H.; Xu, S.; Koike, S.; Pan, L.Q.; Chen, B.; Wang, Y.W.; Rehman, K.; Wu, B.; Chen, Z.; Suzuki, N. The endoplasmic reticulum is a target organelle for trivalent dimethylarsinic acid (DMAIII)-induced cytotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Nordenson, I.; Beckman, L. Is the genotoxic effect of arsenic mediated by oxygen free radicals? Hum. Hered. 1991, 41, 71–73. [Google Scholar] [CrossRef]
- Chouchane, S.; Snow, E.T. In vitro effect of arsenical compounds on glutathione-related enzymes. Chem. Res. Toxicol. 2001, 14, 517–522. [Google Scholar] [CrossRef]
- Styblo, M.; Serves, S.V.; Cullen, W.R.; Thomas, D.J. Comparative inhibition of yeast glutathione reductase by arsenicals and arsenothiols. Chem. Res. Toxicol. 1997, 10, 27–33. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Harder, B.; Castro-Portuguez, R.; Rodrigues, S.D.; Wong, P.K.; Chapman, E.; Zhang, D.D. Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol. Appl. Pharmacol. 2018, 341, 106–113. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Pan, J.; Wang, X.; Zhang, Z.; Chen, F.; Shi, X. Reduced reactive oxygen species-generating capacity contributes to the enhanced cell growth of arsenic-transformed epithelial cells. Cancer Res. 2010, 70, 5127–5135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Sun, R.; Wang, H.; Yang, B.; Wang, F.; Xu, H.; Chen, S.; Zhao, R.; Pi, J.; Xu, Y. Enhanced p62-NRF2 Feedback Loop due to Impaired Autophagic Flux Contributes to Arsenic-Induced Malignant Transformation of Human Keratinocytes. Oxid. Med. Cell. Longev. 2019, 2019, 1038932. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Xi, S. A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes. Regul. Toxicol. Pharmacol. 2018, 99, 78–88. [Google Scholar] [CrossRef]
- Platanias, L.C. Biological responses to arsenic compounds. J. Biol. Chem. 2009, 284, 18583–18587. [Google Scholar] [CrossRef] [Green Version]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Flora, S.J. Arsenic-induced oxidative stress and its reversibility. Free Radic. Biol. Med. 2011, 51, 257–281. [Google Scholar] [CrossRef]
- Parodi-Rullán, R.M.; Soto-Prado, J.; Vega-Lugo, J.; Chapa-Dubocq, X.; Díaz-Cordero, S.I.; Javadov, S. Divergent Effects of Cyclophilin-D Inhibition on the Female Rat Heart: Acute Versus Chronic Post-Myocardial Infarction. Cell. Physiol. Biochem. 2018, 50, 288–303. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Chen, F.; Fu, J.; Xu, Y.; Hou, Y.; Kou, H.H.; Zhai, C.; Nelson, M.B.; Zhang, Q.; et al. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic. Biol. Med. 2016, 99, 544–556. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [Green Version]
- Aono, J.; Yanagawa, T.; Itoh, K.; Li, B.; Yoshida, H.; Kumagai, Y.; Yamamoto, M.; Ishii, T. Activation of Nrf2 and accumulation of ubiquitinated A170 by arsenic in osteoblasts. Biochem. Biophys. Res. Commun. 2003, 305, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Gao, S.; Li, J.; Wu, L.; Zhang, Y.; Li, W.; Zhao, L.; Chen, J.; Yang, S.; Sun, G.; et al. Acute arsenic exposure induces inflammatory responses and CD4(+) T cell subpopulations differentiation in spleen and thymus with the involvement of MAPK, NF-kB, and Nrf2. Mol. Immunol. 2017, 81, 160–172. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; Wang, H.; Xu, Y. Induction of glutathione synthesis in human hepatocytes by acute and chronic arsenic exposure: Differential roles of mitogen-activated protein kinases. Toxicology 2014, 325, 96–106. [Google Scholar] [CrossRef]
- Janasik, B.; Reszka, E.; Stanislawska, M.; Jablonska, E.; Kuras, R.; Wieczorek, E.; Malachowska, B.; Fendler, W.; Wasowicz, W. Effect of Arsenic Exposure on NRF2-KEAP1 Pathway and Epigenetic Modification. Biol. Trace Elem. Res. 2017. [Google Scholar] [CrossRef]
- Jiang, J.; Tam, L.M.; Wang, P.; Wang, Y. Arsenite Targets the RING Finger Domain of Rbx1 E3 Ubiquitin Ligase to Inhibit Proteasome-Mediated Degradation of Nrf2. Chem. Res. Toxicol. 2018, 31, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ling, M.; Chen, C.; Luo, F.; Yang, P.; Wang, D.; Chen, X.; Xu, H.; Xue, J.; Yang, Q.; et al. Impaired autophagic flux and p62-mediated EMT are involved in arsenite-induced transformation of L-02 cells. Toxicol. Appl. Pharmacol. 2017, 334, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Ng, A.C.; Hara, T.; Hipp, M.S.; Lage, K.; Xavier, R.J.; Ryu, K.Y.; Taguchi, K.; et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: A potential role for protein aggregation in autophagic substrate selection. J. Cell Biol. 2010, 191, 537–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Trinh, E.; Qiang, L.; Xie, L.; Hu, W.Y.; Prins, G.S.; Pi, J.; He, Y.Y. Arsenic Induces p62 Expression to Form a Positive Feedback Loop with Nrf2 in Human Epidermal Keratinocytes: Implications for Preventing Arsenic-Induced Skin Cancer. Molecules 2017, 22, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, X.; Shao, F.; Lv, G.; Lv, H.; Lee, J.H.; Qian, X.; Wang, Z.; Xia, Y.; Du, L.; et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci. Adv. 2019, 5, eaav4570. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.A.; Kandhare, A.D.; Bodhankar, S.L. Elucidation of protective efficacy of Pentahydroxy flavone isolated from Madhuca indica against arsenite-induced cardiomyopathy: Role of Nrf-2, PPAR-γ, c-fos and c-jun. Environ. Toxicol. Pharmacol. 2017, 56, 172–185. [Google Scholar] [CrossRef]
- Zhong, L.; Hao, H.; Chen, D.; Hou, Q.; Zhu, Z.; He, W.; Sun, S.; Sun, M.; Li, M.; Fu, X. Arsenic trioxide inhibits the differentiation of fibroblasts to myofibroblasts through nuclear factor erythroid 2-like 2 (NFE2L2) protein and the Smad2/3 pathway. J. Cell. Physiol. 2019, 234, 2606–2617. [Google Scholar] [CrossRef]
- Massrieh, W.; Derjuga, A.; Blank, V. Induction of endogenous Nrf2/small maf heterodimers by arsenic-mediated stress in placental choriocarcinoma cells. Antioxid. Redox Signal. 2006, 8, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Chen, W.; Li, Y.; Villeneuve, N.F.; Zhang, D.D. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: Enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharmacol. 2008, 230, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, J.; Diwan, B.A.; Sun, Y.; Liu, J.; Qu, W.; He, Y.; Styblo, M.; Waalkes, M.P. Arsenic-induced malignant transformation of human keratinocytes: Involvement of Nrf2. Free Radic. Biol. Med. 2008, 45, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kou, M.C.; Weng, C.Y.; Hu, L.W.; Wang, Y.J.; Wu, M.J. Arsenic modulates heme oxygenase-1, interleukin-6, and vascular endothelial growth factor expression in endothelial cells: Roles of ROS, NF-κB, and MAPK pathways. Arch. Toxicol. 2012, 86, 879–896. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, Y.; Li, L.; Yang, Y.; Jia, J.; Hong, Z.; Li, T.; Xu, Y.; Fu, J.; Sun, Y.; et al. Nrf2 deficiency aggravates the increase in osteoclastogenesis and bone loss induced by inorganic arsenic. Toxicol. Appl. Pharmacol. 2019, 367, 62–70. [Google Scholar] [CrossRef]
- He, J.; Wang, M.; Jiang, Y.; Chen, Q.; Xu, S.; Xu, Q.; Jiang, B.H.; Liu, L.Z. Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1α/COX-2 pathway. Environ. Health Perspect. 2014, 122, 255–261. [Google Scholar] [CrossRef]
- Gao, M.; Liu, Y.; Chen, Y.; Yin, C.; Chen, J.J.; Liu, S. miR-214 protects erythroid cells against oxidative stress by targeting ATF4 and EZH2. Free Radic. Biol. Med. 2016, 92, 39–49. [Google Scholar] [CrossRef]
- Zhang, Q.; Bai, J.; Yao, X.; Jiang, L.; Wu, W.; Yang, L.; Gao, N.; Qiu, T.; Yang, G.; Habtemariam Hidru, T.; et al. Taurine rescues the arsenic-induced injury in the pancreas of rat offsprings and in the INS-1 cells. Biomed. Pharmacother. 2019, 109, 815–822. [Google Scholar] [CrossRef]
- Guidarelli, A.; Fiorani, M.; Cerioni, L.; Scotti, M.; Cantoni, O. Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition. Biofactors 2017, 43, 673–684. [Google Scholar] [CrossRef]
- Chen, W.; Martindale, J.L.; Holbrook, N.J.; Liu, Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol. Cell. Biol. 1998, 18, 5178–5188. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.Y.; Liu, Z.M.; Huang, H.S. NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes. Arch. Toxicol. 2012, 86, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Barchowsky, A.; Roussel, R.R.; Klei, L.R.; James, P.E.; Ganju, N.; Smith, K.R.; Dudek, E.J. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol. Appl. Pharmacol. 1999, 159, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Luster, M.I.; Simeonova, P.P. Arsenic and urinary bladder cell proliferation. Toxicol. Appl. Pharmacol. 2004, 198, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.H.; Tseng, T.J.; Su, C.C.; Tang, F.C.; Yen, C.C.; Liu, Y.Y.; Yang, C.Y.; Wu, C.C.; Chen, K.L.; Hung, D.Z.; et al. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-regulated pathways. Toxicol. Lett. 2014, 224, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, S.; Xi, S.; Yan, L.; Wang, H.; Song, Y.; Sun, G. Arsenic induces the expressions of angiogenesis-related factors through PI3K and MAPK pathways in SV-HUC-1 human uroepithelial cells. Toxicol. Lett. 2013, 222, 303–311. [Google Scholar] [CrossRef]
- Liu, S.; Wang, F.; Yan, L.; Zhang, L.; Song, Y.; Xi, S.; Jia, J.; Sun, G. Oxidative stress and MAPK involved into ATF2 expression in immortalized human urothelial cells treated by arsenic. Arch. Toxicol. 2013, 87, 981–989. [Google Scholar] [CrossRef]
- Wang, H.; Xi, S.; Xu, Y.; Wang, F.; Zheng, Y.; Li, B.; Li, X.; Zheng, Q.; Sun, G. Sodium arsenite induces cyclooxygenase-2 expression in human uroepithelial cells through MAPK pathway activation and reactive oxygen species induction. Toxicol. In Vitro 2013, 27, 1043–1048. [Google Scholar] [CrossRef]
- Saha, S.; Sadhukhan, P.; Mahalanobish, S.; Dutta, S.; Sil, P.C. Ameliorative role of genistein against age-dependent chronic arsenic toxicity in murine brains via the regulation of oxidative stress and inflammatory signaling cascades. J. Nutr. Biochem. 2018, 55, 26–40. [Google Scholar] [CrossRef]
- Stueckle, T.A.; Lu, Y.; Davis, M.E.; Wang, L.; Jiang, B.H.; Holaskova, I.; Schafer, R.; Barnett, J.B.; Rojanasakul, Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol. 2012, 261, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Chatterjee, R.; Giri, A.K.; Mandal, C.; Chaudhuri, K. Arsenic-induced cell proliferation is associated with enhanced ROS generation, Erk signaling and CyclinA expression. Toxicol. Lett. 2010, 198, 263–271. [Google Scholar] [CrossRef]
- Felix, K.; Manna, S.K.; Wise, K.; Barr, J.; Ramesh, G.T. Low levels of arsenite activates nuclear factor-kappaB and activator protein-1 in immortalized mesencephalic cells. J. Biochem. Mol. Toxicol. 2005, 19, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Pozo-Molina, G.; Ponciano-Gómez, A.; Rivera-González, G.C.; Hernández-Zavala, A.; Garrido, E. Arsenic-induced S phase cell cycle lengthening is associated with ROS generation, p53 signaling and CDC25A expression. Chem. Biol. Interact. 2015, 238, 170–179. [Google Scholar] [CrossRef]
- Zhao, R.; Hou, Y.; Zhang, Q.; Woods, C.G.; Xue, P.; Fu, J.; Yarborough, K.; Guan, D.; Andersen, M.E.; Pi, J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ. Health Perspect. 2012, 120, 583–589. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, F.; Huber, M.; Kuehne, A.; Kramer, S.; Haertel, E.; Muzumdar, S.; Wagner, J.; Tanner, Y.; Bohm, F.; Smola, S.; et al. Nrf2 Activation Promotes Keratinocyte Survival during Early Skin Carcinogenesis via Metabolic Alterations. Cancer Res. 2015, 75, 4817–4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Elsby, R.; Kitteringham, N.R.; Goldring, C.E.; Lovatt, C.A.; Chamberlain, M.; Henderson, C.J.; Wolf, C.R.; Park, B.K. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J. Biol. Chem. 2003, 278, 22243–22249. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Hu, L.; Chen, Y. Rutaecarpine may improve neuronal injury, inhibits apoptosis, inflammation and oxidative stress by regulating the expression of ERK1/2 and Nrf2/HO-1 pathway in rats with cerebral ischemia-reperfusion injury. Drug Des. Devel. Ther. 2019, 13, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lin, J.; Ge, J.; Wang, L.L.; Li, N.; Sun, X.T.; Cao, H.B.; Li, J.L. Selenium triggers Nrf2-mediated protection against cadmium-induced chicken hepatocyte autophagy and apoptosis. Toxicol In Vitro 2017, 44, 349–356. [Google Scholar] [CrossRef]
- Mir, S.A.; Pinto, S.M.; Paul, S.; Raja, R.; Nanjappa, V.; Syed, N.; Advani, J.; Renuse, S.; Sahasrabuddhe, N.A.; Prasad, T.S.; et al. SILAC-based quantitative proteomic analysis reveals widespread molecular alterations in human skin keratinocytes upon chronic arsenic exposure. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Qu, W.; Reece, J.M.; Kumagai, Y.; Waalkes, M.P. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell Res. 2003, 290, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Li, Y.; Xiao, Q.; Li, D. Daphnetin activates the Nrf2-dependent antioxidant response to prevent arsenic-induced oxidative insult in human lung epithelial cells. Chem. Biol. Interact. 2019, 302, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Momtaz, S.; Niaz, K.; Hassan, F.I.; Abdollahi, M. Epigenetic mechanisms underlying the toxic effects associated with arsenic exposure and the development of diabetes. Food Chem. Toxicol. 2017, 107, 406–417. [Google Scholar] [CrossRef]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Jiang, X.; Gu, S.; Zhang, Z. MicroRNA-155 regulates arsenite-induced malignant transformation by targeting Nrf2-mediated oxidative damage in human bronchial epithelial cells. Toxicol. Lett. 2017, 278, 38–47. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Dong, L.; Sun, S.; Li, L. miRNA-20b inhibits cerebral ischemia-induced inflammation through targeting NLRP3. Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Y.; Zhang, M.; Yan, Y.; Yu, H.; Ge, L. microRNA-451 protects neurons against ischemia/reperfusion injury-induced cell death by targeting CELF2. Neuropsychiatr. Dis. Treat. 2018, 14, 2773–2782. [Google Scholar] [CrossRef] [Green Version]
- Niu, T.; Jin, L.; Niu, S.; Gong, C.; Wang, H. Lycium Barbarum Polysaccharides Alleviates Oxidative Damage Induced by H2O2 Through Down-Regulating MicroRNA-194 in PC-12 and SH-SY5Y Cells. Cell. Physiol. Biochem. 2018, 50, 460–472. [Google Scholar] [CrossRef]
- Azadmanesh, J.; Lutz, W.E.; Weiss, K.L.; Coates, L.; Borgstahl, G.E.O. Redox manipulation of the manganese metal in human manganese superoxide dismutase for neutron diffraction. Acta Crystallogr. F Struct. Biol. Commun. 2018, 74, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhao, H.; Wang, Y.; Shao, Y.; Zong, H.; Zeng, X.; Xing, M. Arsenic trioxide and/or copper sulfate induced apoptosis and autophagy associated with oxidative stress and perturbation of mitochondrial dynamics in the thymus of Gallus gallus. Chemosphere 2019, 219, 227–235. [Google Scholar] [CrossRef]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Xu, H.; Wang, Z.; Mao, Y.; Yuan, L.; Luo, W.; Cui, Z.; Cui, T.; Wang, X.L.; Shen, Y.H. PINK1-Parkin-Mediated Mitophagy Protects Mitochondrial Integrity and Prevents Metabolic Stress-Induced Endothelial Injury. PLoS ONE 2015, 10, e0132499. [Google Scholar] [CrossRef]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, S.H.; Kwak, H.B. Role of exercise in age-related sarcopenia. J. Exerc. Rehabil. 2018, 14, 551–558. [Google Scholar] [CrossRef]
- Williams, J.A.; Ding, W.X. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Nutt, L.K.; Gogvadze, V.; Uthaisang, W.; Mirnikjoo, B.; McConkey, D.J.; Orrenius, S. Indirect effects of Bax and Bak initiate the mitochondrial alterations that lead to cytochrome c release during arsenic trioxide-induced apoptosis. Cancer Biol. Ther. 2005, 4, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Zeinvand-Lorestani, M.; Kalantari, H.; Khodayar, M.J.; Teimoori, A.; Saki, N.; Ahangarpour, A.; Rahim, F.; Khorsandi, L. Dysregulation of Sqstm1, mitophagy, and apoptotic genes in chronic exposure to arsenic and high-fat diet (HFD). Environ. Sci. Pollut. Res. Int. 2018, 25, 34351–34359. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 2014, 6, a020644. [Google Scholar] [CrossRef] [Green Version]
- Macho, A.P.; Lozano-Durán, R.; Zipfel, C. Importance of tyrosine phosphorylation in receptor kinase complexes. Trends Plant Sci. 2015, 20, 269–272. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; Pawson, T. Phosphotyrosine signaling: Evolving a new cellular communication system. Cell 2010, 142, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Suc, I.; Meilhac, O.; Lajoie-Mazenc, I.; Vandaele, J.; Jurgens, G.; Salvayre, R.; Negre-Salvayre, A. Activation of EGF receptor by oxidized LDL. FASEB J. 1998, 12, 665–671. [Google Scholar] [CrossRef]
- Wu, W.; Graves, L.M.; Jaspers, I.; Devlin, R.B.; Reed, W.; Samet, J.M. Activation of the EGF receptor signaling pathway in human airway epithelial cells exposed to metals. Am. J. Physiol. 1999, 277, L924–L931. [Google Scholar] [CrossRef]
- Wu, W.; Jaspers, I.; Zhang, W.; Graves, L.M.; Samet, J.M. Role of Ras in metal-induced EGF receptor signaling and NF-kappaB activation in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1040–L1048. [Google Scholar] [CrossRef] [PubMed]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Sumi, D. Arsenic: Signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, P.P.; Luster, M.I. Arsenic carcinogenicity: Relevance of c-Src activation. Mol. Cell. Biochem. 2002, 234–235, 277–282. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The paradox of arsenic: Molecular mechanisms of cell transformation and chemotherapeutic effects. Crit. Rev. Oncol. Hematol. 2002, 42, 5–24. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Liu, J.; Guo, M.; Fei, D.; Yu, H.; Xing, M. The cardiotoxicity of the common carp (Cyprinus carpio) exposed to environmentally relevant concentrations of arsenic and subsequently relieved by zinc supplementation. Environ. Pollut. 2019, 253, 741–748. [Google Scholar] [CrossRef]
- Jeong, C.H.; Seok, J.S.; Petriello, M.C.; Han, S.G. Arsenic downregulates tight junction claudin proteins through p38 and NF-κB in intestinal epithelial cell line, HT-29. Toxicology 2017, 379, 31–39. [Google Scholar] [CrossRef]
- Barchowsky, A.; Klei, L.R.; Dudek, E.J.; Swartz, H.M.; James, P.E. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic. Biol. Med. 1999, 27, 1405–1412. [Google Scholar] [CrossRef]
- Lynn, S.; Gurr, J.R.; Lai, H.T.; Jan, K.Y. NADH oxidase activation is involved in arsenite-induced oxidative DNA damage in human vascular smooth muscle cells. Circ. Res. 2000, 86, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Hoshino, M.; Okamoto, M.; Sawamura, R.; Hasegawa, A.; Okada, S. Induction of DNA damage by dimethylarsine, a metabolite of inorganic arsenics, is for the major part likely due to its peroxyl radical. Biochem. Biophys. Res. Commun. 1990, 168, 58–64. [Google Scholar] [CrossRef]
- Yamanaka, K.; Ohba, H.; Hasegawa, A.; Sawamura, R.; Okada, S. Mutagenicity of dimethylated metabolites of inorganic arsenics. Chem. Pharm. Bull. 1989, 37, 2753–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.H.; Shen, Y.L.; Jing, Y.K.; Cai, X.; Jia, P.M.; Huang, Y.; Tang, W.; Shi, G.Y.; Sun, Y.P.; Dai, J.; et al. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J. Natl. Cancer Inst. 1999, 91, 772–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Ma, W.Y.; Li, J.; Dong, Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res. 1999, 59, 3053–3058. [Google Scholar]
- Sun, J.; Yu, M.; Lu, Y.; Thakur, C.; Chen, B.; Qiu, P.; Zhao, H.; Chen, F. Carcinogenic metalloid arsenic induces expression of mdig oncogene through JNK and STAT3 activation. Cancer Lett. 2014, 346, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Shi, X. Signaling from toxic metals to NF-kappaB and beyond: Not just a matter of reactive oxygen species. Environ. Health Perspect. 2002, 110 (Suppl. 5), 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchowsky, A.; Dudek, E.J.; Treadwell, M.D.; Wetterhahn, K.E. Arsenic induces oxidant stress and NF-kappa B activation in cultured aortic endothelial cells. Free Radic. Biol. Med. 1996, 21, 783–790. [Google Scholar] [CrossRef]
- Huang, C.; Li, J.; Ding, M.; Wang, L.; Shi, X.; Castranova, V.; Vallyathan, V.; Ju, G.; Costa, M. Arsenic-induced NFkappaB transactivation through Erks- and JNKs-dependent pathways in mouse epidermal JB6 cells. Mol. Cell. Biochem. 2001, 222, 29–34. [Google Scholar] [CrossRef]
- Li, M.; Cai, J.F.; Chiu, J.F. Arsenic induces oxidative stress and activates stress gene expressions in cultured lung epithelial cells. J. Cell. Biochem. 2002, 87, 29–38. [Google Scholar] [CrossRef]
- Kapahi, P.; Takahashi, T.; Natoli, G.; Adams, S.R.; Chen, Y.; Tsien, R.Y.; Karin, M. Inhibition of NF-kappa B activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J. Biol. Chem. 2000, 275, 36062–36066. [Google Scholar] [CrossRef] [Green Version]
- Lemarie, A.; Morzadec, C.; Merino, D.; Micheau, O.; Fardel, O.; Vernhet, L. Arsenic trioxide induces apoptosis of human monocytes during macrophagic differentiation through nuclear factor-kappaB-related survival pathway down-regulation. J. Pharmacol. Exp. Ther. 2006, 316, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Mahieux, R.; Pise-Masison, C.; Gessain, A.; Brady, J.N.; Olivier, R.; Perret, E.; Misteli, T.; Nicot, C. Arsenic trioxide induces apoptosis in human T-cell leukemia virus type 1- and type 2-infected cells by a caspase-3-dependent mechanism involving Bcl-2 cleavage. Blood 2001, 98, 3762–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, R.R.; Barchowsky, A. Arsenic inhibits NF-kappaB-mediated gene transcription by blocking IkappaB kinase activity and IkappaBalpha phosphorylation and degradation. Arch. Biochem. Biophys. 2000, 377, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediat. Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef]
- Dai, P.; Shen, D.; Shen, J.; Tang, Q.; Xi, M.; Li, Y.; Li, C. The roles of Nrf2 and autophagy in modulating inflammation mediated by TLR4-NFκB in A549cell exposed to layer house particulate matter 2.5 (PM). Chemosphere 2019, 235, 1134–1145. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. (Berl) 1996, 74, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Drobna, Z.; Jaspers, I.; Thomas, D.J.; Styblo, M. Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals. FASEB J. 2003, 17, 67–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.G.; Shi, C.; Bode, A.M.; Dong, Z. p38alpha MAPK is required for arsenic-induced cell transformation. Mol. Carcinog. 2016, 55, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkus, R.; Weiner, L.M.; Daniel, V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappaB, and glutathione S-transferase gene expression. J. Biol. Chem. 1996, 271, 13422–13429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Jin, X.; Snow, E.T. Effect of arsenic on transcription factor AP-1 and NF-kappaB DNA binding activity and related gene expression. Toxicol. Lett. 2002, 133, 33–45. [Google Scholar] [CrossRef]
- Simeonova, P.P.; Wang, S.; Kashon, M.L.; Kommineni, C.; Crecelius, E.; Luster, M.I. Quantitative relationship between arsenic exposure and AP-1 activity in mouse urinary bladder epithelium. Toxicol. Sci. 2001, 60, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, J.; Gao, J.; Huang, C. c-Jun/AP-1 pathway-mediated cyclin D1 expression participates in low dose arsenite-induced transformation in mouse epidermal JB6 Cl41 cells. Toxicol. Appl. Pharmacol. 2009, 235, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Menendez, D.; Mora, G.; Salazar, A.M.; Ostrosky-Wegman, P. ATM status confers sensitivity to arsenic cytotoxic effects. Mutagenesis 2001, 16, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Yih, L.H.; Lee, T.C. Arsenite induces p53 accumulation through an ATM-dependent pathway in human fibroblasts. Cancer Res. 2000, 60, 6346–6352. [Google Scholar]
- Komissarova, E.V.; Rossman, T.G. Arsenite induced poly(ADP-ribosyl)ation of tumor suppressor P53 in human skin keratinocytes as a possible mechanism for carcinogenesis associated with arsenic exposure. Toxicol. Appl. Pharmacol. 2010, 243, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zeng, W.; Qi, M.; Duan, Y.; Su, J.; Zhao, S.; Zhong, W.; Gao, M.; Li, F.; He, Y.; et al. Low dose arsenite confers resistance to UV induced apoptosis via p53-MDM2 pathway in ketatinocytes. Oncogenesis 2017, 6, e370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Higuchi, M.; Matsuki, H.; Yoshita, M.; Ohsawa, T.; Oie, M.; Fujii, M. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 2013, 33, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabocka, E.; Bar-Sagi, D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016, 167, 1803–1813.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, M.; Courtney, S.C.; Brinton, M.A. Arsenite-induced stress granule formation is inhibited by elevated levels of reduced glutathione in West Nile virus-infected cells. PLoS Pathog. 2017, 13, e1006240. [Google Scholar] [CrossRef]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Lee, C.H.; Yu, H.S. Role of mitochondria, ROS, and DNA damage in arsenic induced carcinogenesis. Front. Biosci. (Schol Ed.) 2016, 8, 312–320. [Google Scholar] [CrossRef]
- Zhao, F.; Severson, P.; Pacheco, S.; Futscher, B.W.; Klimecki, W.T. Arsenic exposure induces the Warburg effect in cultured human cells. Toxicol. Appl. Pharmacol. 2013, 271, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luz, A.L.; Godebo, T.R.; Bhatt, D.P.; Ilkayeva, O.R.; Maurer, L.L.; Hirschey, M.D.; Meyer, J.N. From the Cover: Arsenite Uncouples Mitochondrial Respiration and Induces a Warburg-like Effect in Caenorhabditis elegans. Toxicol. Sci. 2016, 152, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.N.; Xi, M.M.; Guo, Y.; Hai, C.X.; Yang, W.L.; Qin, X.J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014, 224, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef]
- Purohit, V.; Simeone, D.M.; Lyssiotis, C.A. Metabolic Regulation of Redox Balance in Cancer. Cancers 2019, 11, 955. [Google Scholar] [CrossRef] [Green Version]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour. Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Pal, S.; Mohammed, A.; Farooqui, M.; Doescher, M.P.; Asch, A.S.; Yamada, H.Y. Biological effects and epidemiological consequences of arsenic exposure, and reagents that can ameliorate arsenic damage in vivo. Oncotarget 2017, 8, 57605–57621. [Google Scholar] [CrossRef] [Green Version]
- Abu El-Saad, A.M.; Al-Kahtani, M.A.; Abdel-Moneim, A.M. N-acetylcysteine and meso-2,3-dimercaptosuccinic acid alleviate oxidative stress and hepatic dysfunction induced by sodium arsenite in male rats. Drug Des. Devel. Ther. 2016, 10, 3425–3434. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Shi, N.; Geng, Z.; Li, X.; Hu, X.; Wang, Z. Inhibitory mechanism of dimercaptopropanesulfonic acid (DMPS) in the cellular biomethylation of arsenic. Biochimie 2014, 106, 167–174. [Google Scholar] [CrossRef]
- Kosnett, M.J. The role of chelation in the treatment of arsenic and mercury poisoning. J. Med. Toxicol. 2013, 9, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Mutter, J.; Aaseth, J. Metal chelators and neurotoxicity: Lead, mercury, and arsenic. Arch. Toxicol. 2017, 91, 3787–3797. [Google Scholar] [CrossRef]
- Wang, D.; Lin, L.; Li, X.; Sun, G.F. Effects of glutathione on the in vivo metabolism and oxidative stress of arsenic in mice. J. Toxicol. Sci. 2015, 40, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Xie, H.; Yang, F.; Shan, Q.; Dai, H.; Zhuo, J.; Wei, X.; Song, P.; Zhou, L.; Xu, X.; et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: Roles of p38 MAPK, ERK3, and mTORC1. J. Hematol. Oncol. 2017, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Miltonprabu, S.; Sumedha, N.C. Arsenic-induced hepatic mitochondrial toxicity in rats and its amelioration by diallyl trisulfide. Toxicol. Mech. Methods 2014, 24, 124–135. [Google Scholar] [CrossRef]
- Sumedha, N.C.; Miltonprabu, S. Diallyl trisulfide ameliorates arsenic-induced hepatotoxicity by abrogation of oxidative stress, inflammation, and apoptosis in rats. Hum. Exp. Toxicol. 2015, 34, 506–525. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chatterji, U. All-Trans Retinoic Acid Ameliorates Arsenic-Induced Oxidative Stress and Apoptosis in the Rat Uterus by Modulating MAPK Signaling Proteins. J. Cell. Biochem. 2017, 118, 3796–3809. [Google Scholar] [CrossRef]
- Choudhury, S.; Ghosh, S.; Mukherjee, S.; Gupta, P.; Bhattacharya, S.; Adhikary, A.; Chattopadhyay, S. Pomegranate protects against arsenic-induced p53-dependent ROS-mediated inflammation and apoptosis in liver cells. J. Nutr. Biochem. 2016, 38, 25–40. [Google Scholar] [CrossRef]
- Gong, X.; Ivanov, V.N.; Davidson, M.M.; Hei, T.K. Tetramethylpyrazine (TMP) protects against sodium arsenite-induced nephrotoxicity by suppressing ROS production, mitochondrial dysfunction, pro-inflammatory signaling pathways and programed cell death. Arch. Toxicol. 2015, 89, 1057–1070. [Google Scholar] [CrossRef]
- Gong, X.; Ivanov, V.N.; Hei, T.K. 2,3,5,6-Tetramethylpyrazine (TMP) down-regulated arsenic-induced heme oxygenase-1 and ARS2 expression by inhibiting Nrf2, NF-κB, AP-1 and MAPK pathways in human proximal tubular cells. Arch. Toxicol. 2016, 90, 2187–2200. [Google Scholar] [CrossRef] [Green Version]
- Han, X.D.; Zhang, Y.Y.; Wang, K.L.; Huang, Y.P.; Yang, Z.B.; Liu, Z. The involvement of Nrf2 in the protective effects of (-)-Epigallocatechin-3-gallate (EGCG) on NaAsO2-induced hepatotoxicity. Oncotarget 2017, 8, 65302–65312. [Google Scholar] [CrossRef] [Green Version]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Mangiferin alleviates arsenic induced oxidative lung injury via upregulation of the Nrf2-HO1 axis. Food Chem. Toxicol. 2019, 126, 41–55. [Google Scholar] [CrossRef]
- Thangapandiyan, S.; Ramesh, M.; Miltonprabu, S.; Hema, T.; Jothi, G.B.; Nandhini, V. Sulforaphane potentially attenuates arsenic-induced nephrotoxicity via the PI3K/Akt/Nrf2 pathway in albino Wistar rats. Environ. Sci. Pollut. Res. Int. 2019, 26, 12247–12263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.G.; Xu, S.Z.; Niu, Q.; Ding, Y.S.; Pang, L.J.; Ma, R.L.; Jing, M.X.; Wang, K.; Ma, X.M.; Feng, G.L.; et al. Lutein alleviates arsenic-induced reproductive toxicity in male mice via Nrf2 signaling. Hum. Exp. Toxicol. 2016, 35, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Meng, X.; Wang, F.; Bao, Y.; Huo, J. Eriodictyol attenuates arsenic trioxide-induced liver injury by activation of Nrf2. Oncotarget 2017, 8, 68668–68674. [Google Scholar] [CrossRef] [Green Version]
- Vineetha, R.C.; Binu, P.; Arathi, P.; Nair, R.H. L-ascorbic acid and α-tocopherol attenuate arsenic trioxide-induced toxicity in H9c2 cardiomyocytes by the activation of Nrf2 and Bcl2 transcription factors. Toxicol. Mech. Methods 2018, 28, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Maity, M.; Perveen, H.; Dash, M.; Jana, S.; Khatun, S.; Dey, A.; Mandal, A.K.; Chattopadhyay, S. Arjunolic Acid Improves the Serum Level of Vitamin B. Biol. Trace Elem. Res. 2018, 182, 78–90. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Houshmand, G.; Goudarzi, M.; Sezavar, S.H.; Mehrzadi, S.; Mansouri, E.; Kalantar, M. Ameliorative effect of gallic acid on sodium arsenite-induced spleno-, cardio- and hemato-toxicity in rats. Life Sci. 2019, 217, 91–100. [Google Scholar] [CrossRef]
- Miltonprabu, S.; Sumedha, N.C.; Senthilraja, P. Diallyl trisulfide, a garlic polysulfide protects against As-induced renal oxidative nephrotoxicity, apoptosis and inflammation in rats by activating the Nrf2/ARE signaling pathway. Int. Immunopharmacol. 2017, 50, 107–120. [Google Scholar] [CrossRef]
- Yu, N.H.; Pei, H.; Huang, Y.P.; Li, Y.F. (-)-Epigallocatechin-3-Gallate Inhibits Arsenic-Induced Inflammation and Apoptosis through Suppression of Oxidative Stress in Mice. Cell. Physiol. Biochem. 2017, 41, 1788–1800. [Google Scholar] [CrossRef] [Green Version]
- Guvvala, P.R.; Ravindra, J.P.; Rajani, C.V.; Sivaram, M.; Selvaraju, S. Protective role of epigallocatechin-3-gallate on arsenic induced testicular toxicity in Swiss albino mice. Biomed. Pharmacother. 2017, 96, 685–694. [Google Scholar] [CrossRef]
- Rizwan, S.; Naqshbandi, A.; Farooqui, Z.; Khan, A.A.; Khan, F. Protective effect of dietary flaxseed oil on arsenic-induced nephrotoxicity and oxidative damage in rat kidney. Food Chem. Toxicol. 2014, 68, 99–107. [Google Scholar] [CrossRef]
- Kumar, M.; Thakur, R. Seed Extract Ameliorates Arsenic-Induced Blood Cell Genotoxicity and Hepatotoxicity in Wistar Albino Rats. Rep. Biochem. Mol. Biol. 2018, 7, 110–118. [Google Scholar]
- Kadirvel, R.; Sundaram, K.; Mani, S.; Samuel, S.; Elango, N.; Panneerselvam, C. Supplementation of ascorbic acid and alpha-tocopherol prevents arsenic-induced protein oxidation and DNA damage induced by arsenic in rats. Hum. Exp. Toxicol. 2007, 26, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Das, D.; Mukherjee, M.; Das, A.S.; Mitra, C. Synergistic effect of folic acid and vitamin B12 in ameliorating arsenic-induced oxidative damage in pancreatic tissue of rat. J. Nutr. Biochem. 2006, 17, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, Z.; Liu, W.; Wang, J.; He, X.; Huang, H.; Zhang, J.; Yang, Z. Melatonin protects against arsenic trioxide-induced liver injury by the upregulation of Nrf2 expression through the activation of PI3K/AKT pathway. Oncotarget 2017, 8, 3773–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Wang, Z.; Shu, Z.; Li, Z.; Ning, Y.; Yun, K.; Bai, H.; Liu, R.; Liu, W. Effect and mechanism of Sorbus pohuashanensis (Hante) Hedl. flavonoids protect against arsenic trioxide-induced cardiotoxicity. Biomed. Pharmacother. 2017, 88, 1–10. [Google Scholar] [CrossRef]
- Zhou, J.; Ci, X.; Ma, X.; Yu, Q.; Cui, Y.; Zhen, Y.; Li, S. Pterostilbene Activates the Nrf2-Dependent Antioxidant Response to Ameliorate Arsenic-Induced Intracellular Damage and Apoptosis in Human Keratinocytes. Front. Pharmacol. 2019, 10, 497. [Google Scholar] [CrossRef]
- Pan, X.; Dai, Y.; Li, X.; Niu, N.; Li, W.; Liu, F.; Zhao, Y.; Yu, Z. Inhibition of arsenic-induced rat liver injury by grape seed exact through suppression of NADPH oxidase and TGF-β/Smad activation. Toxicol. Appl. Pharmacol. 2011, 254, 323–331. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, X.; Li, N.; Li, X.; Wang, Y.; Liu, X.; Yin, X.; Yu, Z. Grape seed extract attenuates arsenic-induced nephrotoxicity in rats. Exp. Ther. Med. 2014, 7, 260–266. [Google Scholar] [CrossRef]
- Ling, S.; Shan, Q.; Liu, P.; Feng, T.; Zhang, X.; Xiang, P.; Chen, K.; Xie, H.; Song, P.; Zhou, L.; et al. Metformin ameliorates arsenic trioxide hepatotoxicity via inhibiting mitochondrial complex I. Cell Death Dis. 2017, 8, e3159. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Rashid, K.; Sadhukhan, P.; Agarwal, N.; Sil, P.C. Attenuative role of mangiferin in oxidative stress-mediated liver dysfunction in arsenic-intoxicated murines. Biofactors 2016, 42, 515–532. [Google Scholar] [CrossRef]
- Kumar, A.; Malhotra, A.; Nair, P.; Garg, M.; Dhawan, D.K. Protective role of zinc in ameliorating arsenic-induced oxidative stress and histological changes in rat liver. J. Environ. Pathol. Toxicol. Oncol. 2010, 29, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Z.; Li, J.J.; Chen, C.; Zhang, P.C.; Dong, L.; Chen, J.H.; Chen, Q.; Zhang, X.T.; Wang, Z.L. Protective effects of selenium on oxidative damage and oxidative stress related gene expression in rat liver under chronic poisoning of arsenic. Food Chem. Toxicol. 2013, 58, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Uson-Lopez, R.A.; Sikder, M.T.; Tan, G.; Hosokawa, T.; Saito, T.; Kurasaki, M. Ameliorative effects of selenium on arsenic-induced cytotoxicity in PC12cells via modulating autophagy/apoptosis. Chemosphere 2017, 196, 453–466. [Google Scholar] [CrossRef]
- Das, S.; Joardar, S.; Manna, P.; Dua, T.K.; Bhattacharjee, N.; Khanra, R.; Bhowmick, S.; Kalita, J.; Saha, A.; Ray, S.; et al. Carnosic Acid, a Natural Diterpene, Attenuates Arsenic-Induced Hepatotoxicity via Reducing Oxidative Stress, MAPK Activation, and Apoptotic Cell Death Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 1421438. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, S.P.; Higgins, P.J. TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 2012, 347, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Kasote, D.M.; Katyare, S.S.; Hegde, M.V.; Bae, H. Significance of antioxidant potential of plants and its relevance to therapeutic applications. Int. J. Biol. Sci. 2015, 11, 982–991. [Google Scholar] [CrossRef] [Green Version]
- Holst, B.; Williamson, G. Nutrients and phytochemicals: From bioavailability to bioefficacy beyond antioxidants. Curr. Opin. Biotechnol. 2008, 19, 73–82. [Google Scholar] [CrossRef]
- Zhuang, X.; Lu, C. PBPK modeling and simulation in drug research and development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.P.; Schuhmacher, M.; Kumar, V. The development of a pregnancy PBPK Model for Bisphenol A and its evaluation with the available biomonitoring data. Sci. Total Environ. 2018, 624, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef]
- Verheijen, M.; Schrooders, Y.; Gmuender, H.; Nudischer, R.; Clayton, O.; Hynes, J.; Niederer, S.; Cordes, H.; Kuepfer, L.; Kleinjans, J.; et al. Bringing in vitro analysis closer to in vivo: Studying doxorubicin toxicity and associated mechanisms in 3D human microtissues with PBPK-based dose modelling. Toxicol. Lett. 2018, 294, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Fabian, E.; Gomes, C.; Birk, B.; Williford, T.; Hernandez, T.R.; Haase, C.; Zbranek, R.; van Ravenzwaay, B.; Landsiedel, R. In Vitro-to-in vivo extrapolation (IVIVE) by PBTK modeling for animal-free risk assessment approaches of potential endocrine-disrupting compounds. Arch. Toxicol. 2019, 93, 401–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, M.P.; Liao, C.M.; Tsai, J.W.; Chen, B.C. A PBTK/TD modeling-based approach can assess arsenic bioaccumulation in farmed tilapia (Oreochromis mossambicus) and human health risks. Integr. Environ. Assess. Manag. 2005, 1, 40–54. [Google Scholar] [CrossRef] [Green Version]
- Rowland, M.A.; Perkins, E.J.; Mayo, M.L. Physiological fidelity or model parsimony? The relative performance of reverse-toxicokinetic modeling approaches. BMC Syst. Biol. 2017, 11, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mechanisms of generation of reactive oxygen species (ROS) induced by arsenic. Arsenic (As) induces significant ROS generation mainly through the mitochondrial (Mit) electron transport chain. Activation of nicotine adenine disphosphonucleotide (NADPH) oxidase (Nox) also contributes to the generation of superoxide anion (O2−•). Additionally, arsenic metabolism leads to the generation of ROS in cells. 1O2, singlet oxygen; H2O2, hydrogen peroxide; •OH, hydroxyl radicals; ER, endoplasmic reticulum; SOD, superoxide dismutase.
Figure 1.
Mechanisms of generation of reactive oxygen species (ROS) induced by arsenic. Arsenic (As) induces significant ROS generation mainly through the mitochondrial (Mit) electron transport chain. Activation of nicotine adenine disphosphonucleotide (NADPH) oxidase (Nox) also contributes to the generation of superoxide anion (O2−•). Additionally, arsenic metabolism leads to the generation of ROS in cells. 1O2, singlet oxygen; H2O2, hydrogen peroxide; •OH, hydroxyl radicals; ER, endoplasmic reticulum; SOD, superoxide dismutase.

Figure 2.
Regulatory models of the Nrf2-ARE pathway induced by arsenic. Under basal condition, Nrf2 is associated with Keap1 and degraded by proteasomes. Under arsenic-exposed condition, Nrf2 is activated via the canonical and noncanonical mechanisms. Arsenic binds to the Ring finger domain of RING-box 1 (Rbx1), which leads to the suppression of Cul3-Rbx1 E3 ubiquitin ligase activity, thereby activating the Nrf2-induced antioxidant signaling pathway via the canonical mechanism. Arsenic induces Nrf2 activation via the noncanonical mechanism by p62 accumulation due to dysregulated autophagy flux. p62 is a downstream gene of Nrf2, forming a positive feedback loop with Nrf2.
Figure 2.
Regulatory models of the Nrf2-ARE pathway induced by arsenic. Under basal condition, Nrf2 is associated with Keap1 and degraded by proteasomes. Under arsenic-exposed condition, Nrf2 is activated via the canonical and noncanonical mechanisms. Arsenic binds to the Ring finger domain of RING-box 1 (Rbx1), which leads to the suppression of Cul3-Rbx1 E3 ubiquitin ligase activity, thereby activating the Nrf2-induced antioxidant signaling pathway via the canonical mechanism. Arsenic induces Nrf2 activation via the noncanonical mechanism by p62 accumulation due to dysregulated autophagy flux. p62 is a downstream gene of Nrf2, forming a positive feedback loop with Nrf2.

Figure 3.
Mechanism of protective effects against arsenic-induced toxicity by antioxidants extracted from plants. The representative natural antioxidants extracted from plants show various mechanisms for protection against arsenic-induced toxicity. Most of them (SFN, Daph, SPF, GSPE, DATS, mangiferin, Pts, EGCG, and eriodictyol) promote the dissociation of Nrf2 with Keap1, promoting the expression of downstream genes of Nrf2, and finally increasing the antioxidant capacity. QTN is suggested to act though Nrf2 without clear evidence. Additionally, GSE protects against arsenic-induced oxidative damage through suppression of NOX-mediated ROS generation. SFN, sulforaphane; Daph, daphnetin; SPF, Sorbus pohuashanensis flavonoids; GSPE, grape seed proanthocyanidin extract; DATS, diallyl trisulfide; Pts, pterostilbene; EGCG, (-)-Epigallocatechin-3-gallate; GSE, grape seed extract; TGFβ, transforming growth factor-β; NOX, Nicotinamide adenine dinucleotide phosphate oxidase; SMADs, drosophila mothers against decapentaplegic protein.
Figure 3.
Mechanism of protective effects against arsenic-induced toxicity by antioxidants extracted from plants. The representative natural antioxidants extracted from plants show various mechanisms for protection against arsenic-induced toxicity. Most of them (SFN, Daph, SPF, GSPE, DATS, mangiferin, Pts, EGCG, and eriodictyol) promote the dissociation of Nrf2 with Keap1, promoting the expression of downstream genes of Nrf2, and finally increasing the antioxidant capacity. QTN is suggested to act though Nrf2 without clear evidence. Additionally, GSE protects against arsenic-induced oxidative damage through suppression of NOX-mediated ROS generation. SFN, sulforaphane; Daph, daphnetin; SPF, Sorbus pohuashanensis flavonoids; GSPE, grape seed proanthocyanidin extract; DATS, diallyl trisulfide; Pts, pterostilbene; EGCG, (-)-Epigallocatechin-3-gallate; GSE, grape seed extract; TGFβ, transforming growth factor-β; NOX, Nicotinamide adenine dinucleotide phosphate oxidase; SMADs, drosophila mothers against decapentaplegic protein.


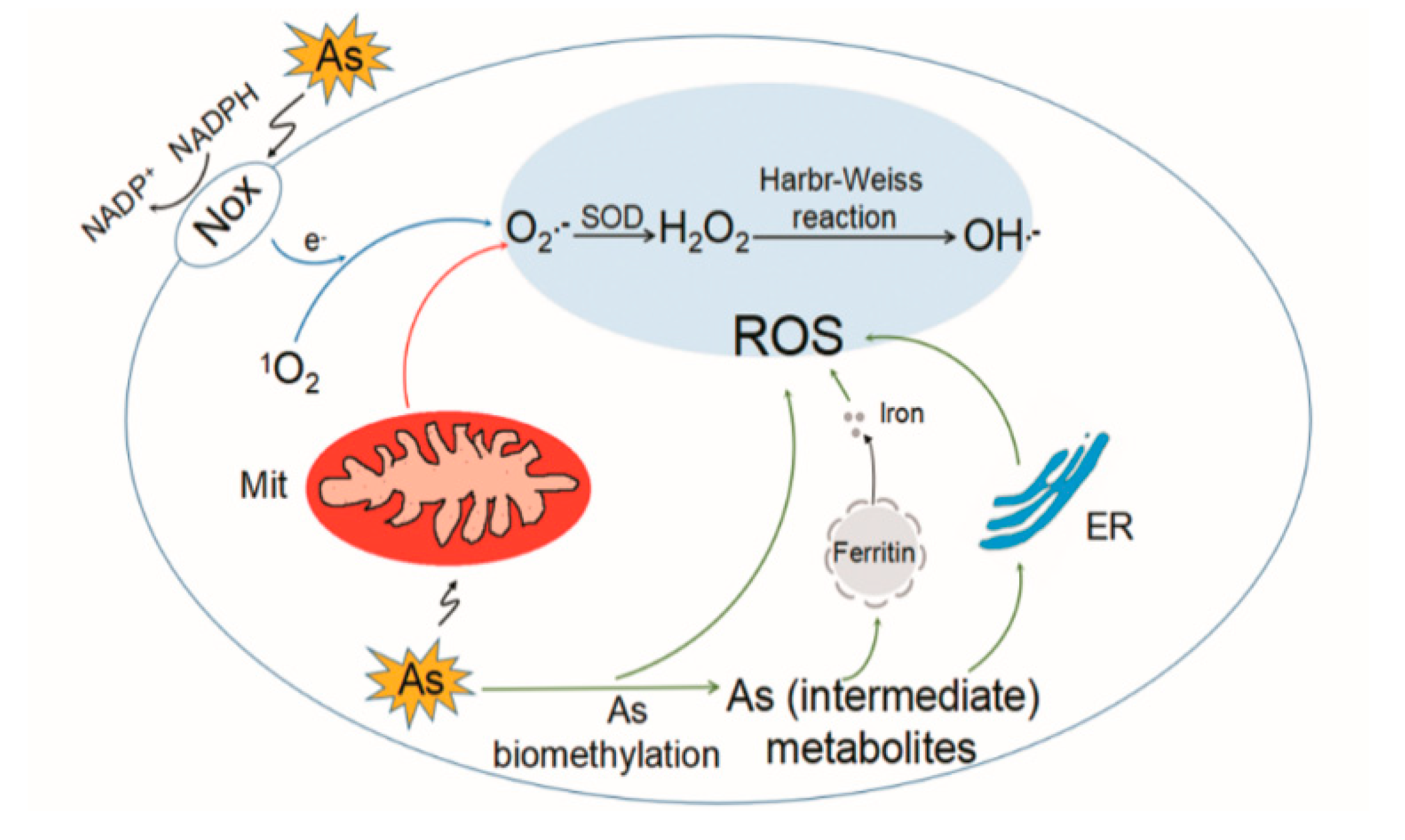{kind=link}
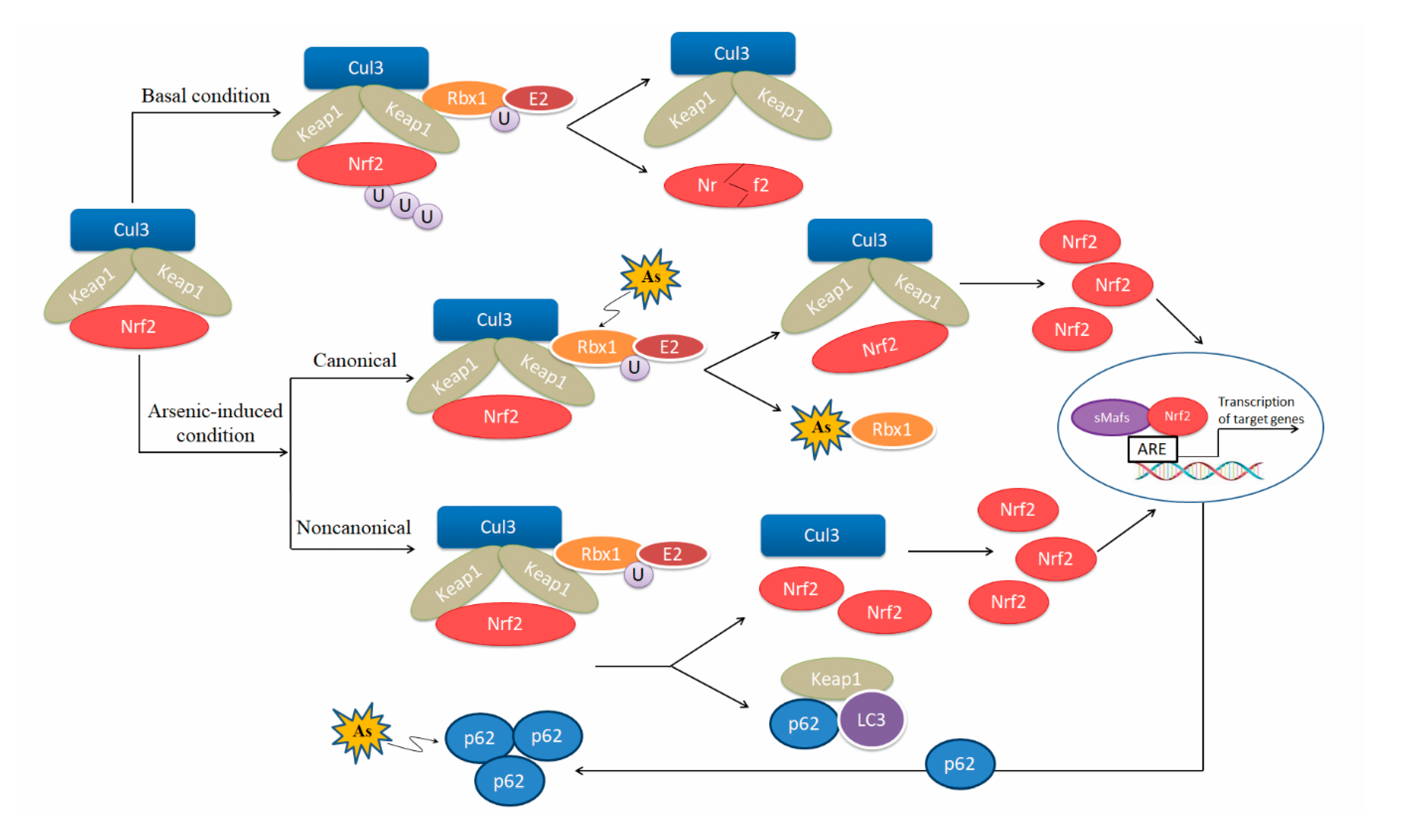{kind=link}
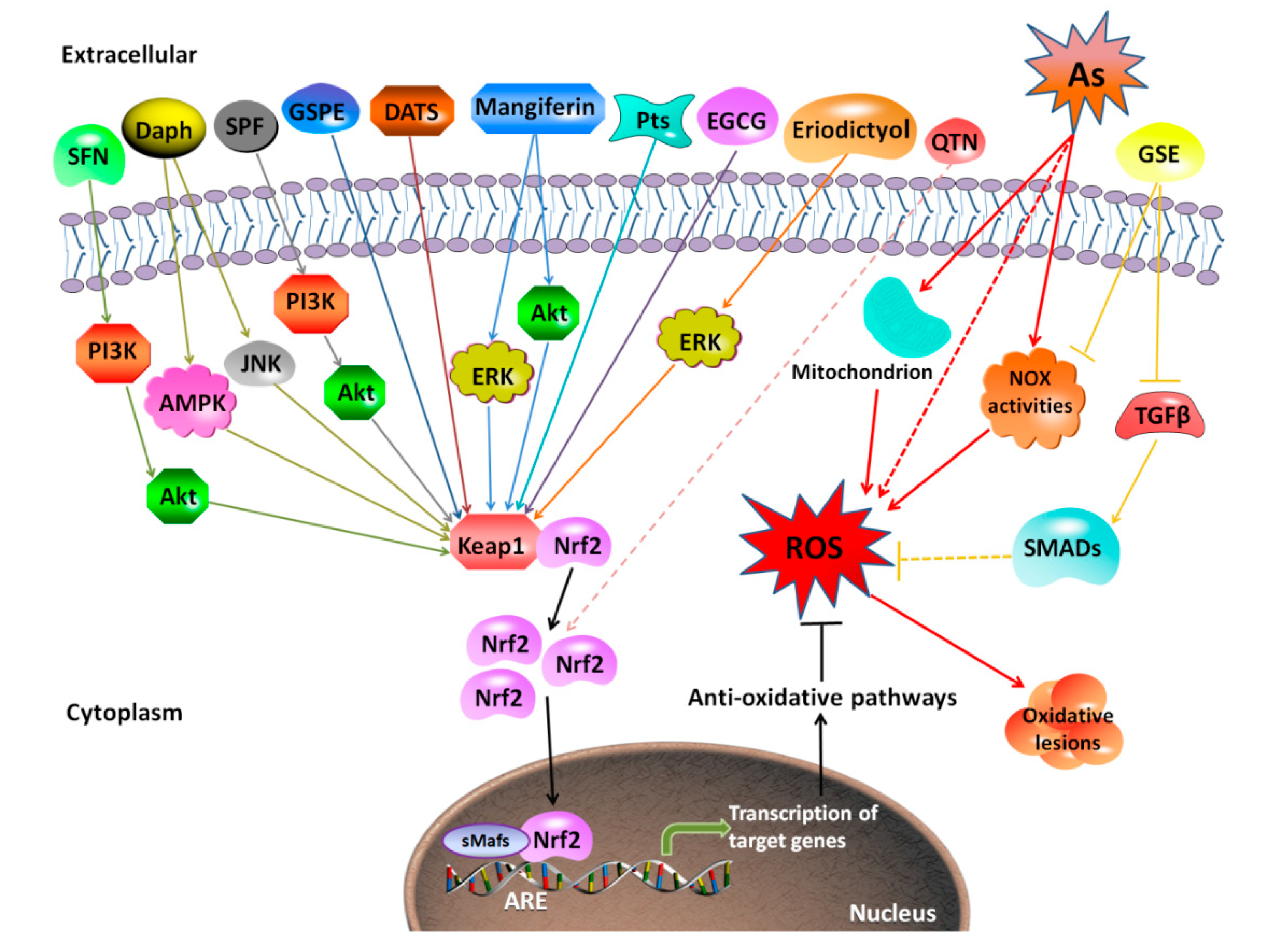{kind=link}
Table 1.
Pathways related with ROS in arsenic toxicity.
| Classification | Pathway | Model | Treatment | Key Findings | References |
|---|---|---|---|---|---|
| Nrf2-ARE (nuclear factor (erythroid-derived 2)-like 2- arsenic include Nrf2-antioxidant response element) | Nrf2/Heme oxygenase 1 (HO-1)/ROS | Human skin fibroblasts | 0, 2.5, 5, or 10 μM As2O3 for 24 h | Arsenic exposure leads to upregulated expression of Nrf2 and its downstream target gene HO-1 with reduced levels of ROS. | [71] |
| Nrf2/HO-1, A170, Prx I | MC3T3-E1 cells | 200-800 μM NaAsO3 or 50-100 μM NaAsO2 for 0, 4, 8, 12, 16, or 24 h | Arsenic induces Nrf2 activation, resulting in the transcriptional activation of A170, HO-1, and Prx I and accumulating high molecular mass forms of A170. | [58] | |
| H2O2/Nrf2/HO-1 | JAR cells | 5 µM As2O3 for 2 to 24 h; 0-10 µM As2O3 for 6 h | Arsenic exposure causes H2O2 over-production, and leads to increase of Nrf2/small Maf DNA binding activity and HO-1 expression. | [72] | |
| Nrf2/HO-1, NAD(P)H dehydrogenase [quinone] 1 (NQO1) | MDA-MB-231 cells | 0-50 µM NaAsO2 for 16 h; 0-2.5 µM monomethylarsonic acid (MMA) (III) for 16 h | Arsenic inhibits the activity of the kelch-like epichlorohydrin-associated protein 1 (Keap1)-Cul3 E3 ubiquitin ligase and then induces the Nrf2-dependent response. | [73] | |
| Nrf2/HO-1, NQO1, Glutamate-cysteine ligase catalytic subunit (GCLC) | HaCaT cells | 100 µM NaAsO2 for 28 weeks | Arsenic-transformed cells show elevated expression of Nrf2 and its target genes, including NQO1, HO-1, and GCLC. | [74] | |
| ROS/Nrf2/HO-1 | Mouse lymphatic endothelial cells | 0, 5, or 7.5 μM As2O3 for 6 h | Arsenic causes ROS over-production and induces Nrf2 activation and HO-1 expressions. | [75] | |
| Nrf2/NQO1, HO-1, GCLC/ROS/p38/ nuclear factor of activated T-cells c1 (NFATc1) | RAW 264.7 cells; mouse bone marrow-derived macrophages | 5 ppm iAs in drinking water for 16 weeks; 0, 0.25, or 0.5 μM iAs for 7 days | Lack of Nrf2 increases arsenic-induced ROS levels and phosphorylation of p38, which aggravates the increase in osteoclastogenesis. | [76] | |
| microRNAs | ROS/miR-199a-5p/HIF-1α/COX-2 | BEAS-2B cells | 1 μM NaAsO2 for 26 weeks, 0, 0.5, 1, or 2 μM NaAsO2 for 24 h | Arsenic-induced ROS inhibit miR-199a expression, and induce the expression of HIF-1α and COX-2. | [77] |
| ROS/Nrf2/miR-214/AFT4 | MEL cells | 10 μM arsenic for 3 h | miR-214 expression is transcriptionally repressed by Nrf2 in a ROS-dependent pathway, which leads to increased ATF4 protein content. | [78] | |
| Mitophagy | ROS/Pparγ/phosphate and tension homology deleted on chromsome ten (PTEN)-induced putative kinase 1 (PINK1)/Parkin | INS-1 cells | 0, 2, 4, or 8 mg/kg As2O3 (from gestation day 6 until pup postnatal day 42) | Arsenic induces ROS, which inhibits the expression of PPARγ and PINK1, and upregulates the expression of Bax. | [79] |
| Mito O2−•, H2O2/ mitochondrial permeability transition (MPT) | U937, MCF-7, HT22, and NCTC-2544 cells | 2.5 µM NaAsO2 for 16 or 48 h | Arsenite induces DNA damage via mitochondrial ROS and induction of mitochondrial permeability transition. | [80] | |
| RTKs (receptor tyrosine kinases)/NTKs (nonreceptor tyrosine kinases) | ROS/epidermal growth factor receptor (EGFR), Shc/Ras/ERK | PC12 cells | 400 μM NaAsO2 for 0, 10, 20, 30, 60 min | EGFR and Shc mediate in the activation of Ras/ERK signaling cascade by arsenite. | [81,82] |
| ROS/c-Src/NF-κB | Porcine aortic endothelial cells | 0 to 100 μM NaAsO2 for 24 h | H2O2 is sufficient for arsenite-induced stimulation of tyrosine kinases and activation of NF-κB. | [83] | |
| ROS/EGFR/MAP/ERK/AP-1 | Mouse urinary bladder | 0.002% or 0.01% NaAsO2 for 16 weeks | Arsenic-induced cell proliferation is correlated with the activation of MAP kinase pathway, leading to activation of ERK kinase and AP-1. | [82,84] | |
| MAPKs (mitogen-activated protein kinases) | ROS/JNK, ERK/ gastrin-releasing peptide 78 (GRP 78), CHOP | Neuro-2a cells | 0, 1, 3, 5, 7, or 10 μM As2O3 for 24 h | Arsenic induces ROS generation, causing cell death via both JNK/ERK-mediated mitochondria-dependent and GRP 78/CHOP-triggered apoptotic pathways. | [85] |
| ROS/MAPK, PI3K/AKT/HIF-1a/COX-2, vascular endothelial grow factor (VEGF) | SV-HUC-1 cells | 0, 1, 2, 4, 8, or 10 μM NaAsO2 for 24 h | Arsenic-induced COX-2, VEGF, and HIF-1 expression, mediated by ROS, is regulated by ERK, JNK, p38 MAPK, and PI3K/AKT. | [86] | |
| ROS/JNK, p38/ATF2 | SV-HUC-1 cells | 0, 1, 2, 4, 8, or 10 μM NaAsO2 for 24 h | Arsenic-induced ROS are involved in activation of JNK and p38 signaling pathways, which are responsible for ATF2 overexpression. | [87] | |
| ROS/ERK, JNK, and p38/COX-2 | SV-HUC-1 cells | 0, 1, 2, 4, 8, or 10 μM NaAsO2 for 24 h | Arsenic induces ROS, which result in an induction of COX-2 expression through activation of the ERK, JNK, and p38 MAPK pathways. | [88] | |
| ROS/ERK, JNK, and p38/IL-6, VEGF | SVEC4-10 cells | 0, 5, or 7.5 μM As2O3 for 6 h | Arsenic causes ROS over-production and induces activation of ERK, JNK, and p38 MAPK, as well as expression of IL-6 and VEGF. | [75] | |
| ROS/ERK1/2/Beclin1, PINK1, Parkin 1, LCIIIB | Male Wistar rats | NaAsO2 (10 mg/kg) orally for 3 months | PKCδ is activated in the arsenic-intoxicated aged brains, which increases the expression of ERK1/2. ERK1/2 activates its downstream autophagic molecules Beclin1, PINK1, Parkin 1, and LCIIIB. | [89] | |
| NF-κB (nuclear factor κB) | ROS/NF-κB/PPARα/δ | BEAS-2B cells | 0, 2.5, 5, 10 or 25 μM As2O3 for 24 h; 2.5 μM As2O3 for 6 months | Arsenic induces ROS generation, enhancing NF-κB signaling and suppressing PPARα/δ signaling. | [90] |
| ROS/c-Src/NF-κB | Porcine aortic endothelial cells | 0 to 100 μM NaAsO2 for 24 h | H2O2 is sufficient for arsenite-induced stimulation of tyrosine kinases and activation of NF-κB. | [83] | |
| AP-1 (activator protein-1) | ROS/ERK/AP-1/cyclin A | HaCaT and Int407 cells | 0, 2 or 20 μM NaAsO2 for 24 h | Arsenic-induced cell proliferation is associated with enhanced ROS generation, ERK signaling, and cyclin A expression. | [91] |
| ROS/AP-1 | 1RB3AN27 cells | 0 to 10 μM NaAsO2 for 2 h or 72 h | Arsenic, in a dose-dependent manner, induces generation of ROS and activation of AP-1. | [92] | |
| p53 | p53/ROS/SESN1/ Cell division cycle 25A (CDC25A) | (MCF-7 (p53+/+)) or H1299 cells | 5 μM NaAsO2 for 6, 12, 24, or 36 h | Arsenic activates p53-dependent transcription of ROS detoxification genes, which could be responsible for the S-phase cell cycle arrest. | [93] |
Table 2.
Antioxidants with prophylactic and therapeutic potential to rescue arsenic toxicity.
| Classification | Compound | Model and Tissue | Treatment | Mechanism | References |
|---|---|---|---|---|---|
| ROS scavengers | Arjunolic acid | Female Wistar rats; serum | NaAsO2 (10 mg/kg for two estrous cycles); arjunolic acid (10 mg/kg for two estrous cycles) | malondialdehyde (MDA) ↓ conjugated diene (CD) ↓ ROS ↓ ovarian steroidogenesis ↑ Vitamin B12 ↑ folate↑ | [216] |
| Gallic acid (GA) | Male Wistar rats; heart and spleen | NaAsO2 (10 mg/kg for 21 days) GA (10 or 30 mg/kg for 7 days) | creatine kinase-MB (CK-MB) ↓ nitric oxide (NO) ↓ MDA ↓ glutathione (GSH) ↑ superoxide dismutase (SOD) ↑ glutathione peroxidase (GPx) ↑ catalase (CAT) ↑ | [217] | |
| Grape seed proanthocyanidin extract (GSPE) | Human L-02 cells | NaAsO2 (25 μM for 24 h) GSPE (10, 25, or 50 mg/L for 24 h) | ROS ↓ MDA ↓ Nrf2 ↑ HO-1 ↑ NQO1 ↑ gultathione S transferases (GST) ↑ | [26] | |
| Diallyl trisulfide (DATS) | Male albino rats; serum | Na3AsO4 (5 mg/kg for 28 days) DATS (20, 40, or 80 mg/kg for 28 days) | GSH ↑ SOD ↑ GPx ↑ CAT ↑ GST ↑ alkaline phosphatase (ALP) ↓ alanine aminotransferase (ALT) ↓aminotransferase (AST) ↓ ACP ↓ | [204] | |
| Male albino rats; serum | Na3AsO4 (5 mg/kg for 28 days) DATS (20, 40, or 80 mg/kg for 28 days) | GSH ↑ SOD ↑ GPx ↑ CAT ↑ GST ↑ glutathione reductase (GR) ↑ glucose-6-phosphate dehydrogenase (G6PD) ↑ Total sulfhydryl groups (T-SH) ↑ vitamin C (VC) ↑ vitamin E (VE) ↑ Nrf2 ↑ HO-1 ↑ ROS ↓ NO ↓ | [218] | ||
| (-)-Epigallocatechin-3-gallate (EGCG) | Sprague-Dawley (SD) rats; liver | NaAsO2 (5 mg/kg/day for 30 days) EGCG (50 mg/kg/day for 30 days) | Nrf2 activation ↑ | [210] | |
| Male BALB/c mice; serum, thymus, and spleen | NaAsO2 (10 mg/kg/day for 30 days) EGCG (10 mg/kg/day for 30 days) | ROS ↓ Caspase-3 activation ↓ | [219] | ||
| Swiss albino mice; serum and spermatozoa | Na2HAsO4·7H2O (200 ppm for 40 days) EGCG (20 mg/kg for 40 days) | ROS ↓ GSH ↑ CAT ↑ MDA ↓ | [220] | ||
| Tetramethylpyrazine (TMP) | Human kidney 2 (HK-2) cells | NaAsO2 (10 μM for 24 h) TMP (50 µM or 100 µM for 24 h) | ROS ↓ NF-κB ↓ COX-2 ↓ Mitochondrial dysfunction ↓ GSH ↑ | [208] | |
| Human HK-2 cells | NaAsO2 (10 μM for 6 or 24 h) TMP (50 µM or 100 µM for 6 or 24 h) | Arsenic-induced MAPKs, AP-1, Nrf2, and NF-κB pathways ↓ HO-1 ↓ ARS2 ↓ | [209] | ||
| Flaxseed oil (FXO) | Male Wistar rats; kidney and blood | Na2HAsO4 (20 mg/kg for 4 days) FXO (15% by weight for 18 days) | Free radicals ↓ ROS ↓ Membrane organization and functions ↑ | [221] | |
| Pomegranate fruit extract (PFE) | Male Swiss albino mice; liver | NaAsO2 (0.01, 0.05, or 0.1 mg/L for 30 days) PFE (0.2 mL of 0.2% of extract for 30 days) | ROS ↓ Nrf2 ↓ p53 ↓ miR-34a ↓ Apoptosis ↓ | [207] | |
| Eriodictyol | Male Wistar rats; liver | As2O3 (3 mg/kg at day 1, 4, and 5) Eriodictyol (10, 20, or 40 mg/kg, 1 h before or after As2O3 treatment) | ROS ↓ MDA ↓ SOD ↑ GPx ↑ CAT ↑ Nrf2 ↑ HO-1 ↑ | [214] | |
| Sulforaphane (SFN) | Male albino Wistar rats; renal | NaAsO2 (5 mg/kg for 28 days) SFN (80 mg/kg for 28 days) | B-cell lymphoma 2-associated X protein (Bax) ↓ ROS ↓ SOD ↑ CAT ↑ B-cell lymphoma 2 (Bcl2) ↑ Arsenic-induced nephrotoxicity ↓ PI3K/Akt ↑ Nrf2 ↑ | [212] | |
| Syzygium cumini seed extract (SCE) | Wistar albino rats; blood and liver | NaAsO2 (100 ppm for 60 days) SCE (200, 400 mg/kg for 60 days) | AST ↓ ALP ↓ ALT ↓ SOD ↑ GSH ↑ CAT ↑ | [222] | |
| Daphnetin (Daph) | Human Beas-2B cells | NaAsO2 (25 μM for 20 h) Daph (2.5, 5 or 10 μg/mL for 20 h) | Bax ↓ ROS ↓ Bcl2 ↑ Nrf2 ↑ HO-1 ↑ | [104] | |
| Vitamin C or Vitamin E | Male albino rats; plasma, liver, and kidney | NaAsO2 (100 ppm for 30 days) Ascorbic acid (200 mg/kg for 30 days) α-tocopherol (400 mg/kg for 30 days) | Protein carbonyl content and DNA damage ↓ | [223] | |
| Folic acid | Male albino rats; serum | As2O3 (3 mg/kg/day for 30 days) Folic acid (36 μg/kg/day for 30 days) | NO ↓ OH- ↓ SOD ↑ GSH ↑ CAT ↑ MDA ↓ | [224] | |
| Lutein (LU) | Male Kunming mice; plasma and testis | As2O3 (5 mg/kg/day for 5 weeks) Lutein (40 mg/kg/day for 5 weeks) | Nrf2 ↑ HO-1 ↑ NQO1 ↑ GST ↑ SOD ↑ GSH ↑ MDA ↓ | [213] | |
| All-trans retinoic acid (ATRA) | SD rats Uteri and serum | NaAsO2 (4 ppm for 28 or 56 days) ATRA (0.5 mg/kg for 28 or 56 days) | MAPK signaling components ↓ p53-dependent apoptosis ↓ | [206] | |
| Glutathione | Female albino mice; urine, liver | NaAsO2 (50 mg/L for 10 days) GSH (200, 400, or 800 mg/kg after NaAsO2 treatment) | Arsenic methylation ↑ Arsenic excretion ↑ | [202] | |
| Melatonin | Male Wistar rats; liver | As2O3 (10 mg/mL for 4 days) Melatonin (5, 10, or 20 mg/kg for 8 days) | AST ↓ ALT ↓ MDA ↓ ROS ↓ SOD ↑ GPx ↑ CAT ↑ Nrf2 ↑ HO-1 ↑ p-Akt/Akt ↑ p-PI3K/PI3K ↑ | [225] | |
| Oxidative enzyme inhibitors | Sorbus pohuashanensis flavonoids (SPF) | BALB/c mice; heart H9c2 cells | As2O3 (1 mg/kg for 14 days) SPF (5, 10, or 20 mg/kg for 14 days) | creatine kinase (CK) ↓ CK-MB ↓ glutamic oxaloacetic transaminase (GOT) ↓ lactate dehydrogenase (LDH) ↓ ROS ↓ Caspase-3, -8, -9 ↓ SOD ↑ glutathione peroxidase (GSH-Px) ↑ CAT ↑ Bcl2/Bax ↑ Nrf2 ↑ HO-1 ↑ p-Akt/Akt ↑ | [226] |
| Pterostilbene (Pts) | HaCaT cells and JB6 cells | NaAsO2 (25 mM for 24 h) Pts (3.75, 7.5, 15, or 30 mM for 1 h) | Bax ↓ MDA ↓ ROS ↓ Caspase 3 ↓ Nrf2 ↑ HO-1 ↑ Bcl-2 ↑ SOD ↑ | [227] | |
| Grape seed exact (GSE) | Male SD rats; liver | NaAsO2 (30 ppm for 12 months) GSE (100 mg/kg for 12 months) | p-Smad2/3 ↓ TGF-β ↓ Nox2 ↓ Nox4 ↓ p47phox ↓ | [228] | |
| Rat hepatic stellate cells | NaAsO2 (2 μM for 24 h) GSE (100 mg/L for 20 min) | TGF-β/Smad signaling ↓ NADPH oxidase activities ↓ ROS ↓ | |||
| Male SD rats; renal | NaAsO2 (30 ppm for 12 months) GSE (100 mg/kg for 12 months) | TGF-β/Smad signaling ↓ NADPH oxidase activities ↓ ROS ↓ | [229] | ||
| Metformin | Male Kunming mice; liver | As2O3 (6 μM for 48 h); Metformin (5 mM for 48 h) As2O3 (12 mg/kg/day for 3 days) Metformin (200 mg/kg/day for 3 days) | mitochondrial respiratory chain complex I ↓ NADH/NAD+ ↑ ROS ↓ | [230] | |
| Intrahepatic cholangiocarcinoma cell lines (CCLP-1, RBE, and HCCC-9810) | As2O3 (3 μM for 24 or 72 h); metformin (10 mM for 24 or 72 h) | mTORC1 ↓ p38 MAPK ↓ ERK3 ↑ | [203] | ||
| Metal chelators | Mangiferin | Male Swiss albino mice; lung | NaAsO2 (10 mg/kg for 3 months) Mangiferin (40 mg/kg for 5 weeks) | LDH ↓ MPO ↓ LPO ↓ ROS ↓ TNF-α ↓ Nrf2 ↑ HO-1 ↑ SOD ↑ GST ↑ GR ↑ GPx ↑ GSH ↑ | [211] |
| HepG2 cells | NaAsO2 (10 mg/kg for 28 days) Mangiferin (40 mg/kg for 5 weeks) | ALP ↓ ALT ↓ LDH ↓ ROS ↓ GSSG ↓ MDA ↓ SOD ↑ GST ↑ GR ↑ Catalase ↑ GSH ↑ GSH/GSSG ↑ | [231] | ||
| 3,5,7,3′,4′-Pentahydroxy flavone (QTN) | Male SD rats; serum | NaAsO2 (5 mL/kg for 28 days) QTN (5, 10, or 20 mg/kg for 28 days) | LDH ↓ CK-MB ↓ AST ↓ ALT ↓ ALP ↓LDL-C ↓ VLDL-C ↓ MDA ↓ NO ↓ c-fos ↓ c-jun ↓ HDL-C ↑ SOD ↑ GSH↑ PPAR-γ ↑ Na-K-ATPase ↑ Complex I, II, IV ↑ Nrf2 ↑ | [70] | |
| Antioxidant enzyme cofactors | Zinc | Male Wistar rats; liver | NaAsO2 (100 ppm for 3 months) Zinc (227 mg/L for 3 months) | GSH ↑ SOD ↑ GPx ↑ GR ↑ CAT ↑ LPO ↓ | [232] |
| Selenium | SD rats; serum, liver | NaAsO2 (13 mg/L for 20 weeks) Na2SeO3 (17 mg/L for 20 weeks) | MDA ↓ Heat shock 70 kDa protein (HSP70) ↓ mRNA of SOD1, CAT, GPx, and Thioredoxin Reductase 1 (Txnrd1) ↑ | [233] | |
| PC12 cells | NaAsO2 (5, 10, 20, or 40 μM for 48 h) Na2SeO3 (5, 10, 20, or 40 μM for 48 h) | The cellular accumulation of arsenic ↓ mTOR/Akt autophagy signaling pathway ↑ | [234] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. https://doi.org/10.3390/biom10020240
AMA Style
Hu Y, Li J, Lou B, Wu R, Wang G, Lu C, Wang H, Pi J, Xu Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules. 2020; 10(2):240. https://doi.org/10.3390/biom10020240
Chicago/Turabian StyleHu, Yuxin, Jin Li, Bin Lou, Ruirui Wu, Gang Wang, Chunwei Lu, Huihui Wang, Jingbo Pi, and Yuanyuan Xu. 2020. "The Role of Reactive Oxygen Species in Arsenic Toxicity" Biomolecules 10, no. 2: 240. https://doi.org/10.3390/biom10020240
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.