Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects?

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Endothelial Function
1.2. Endothelial Dysfunction
2. Endothelial Dysfunction in Diabetes
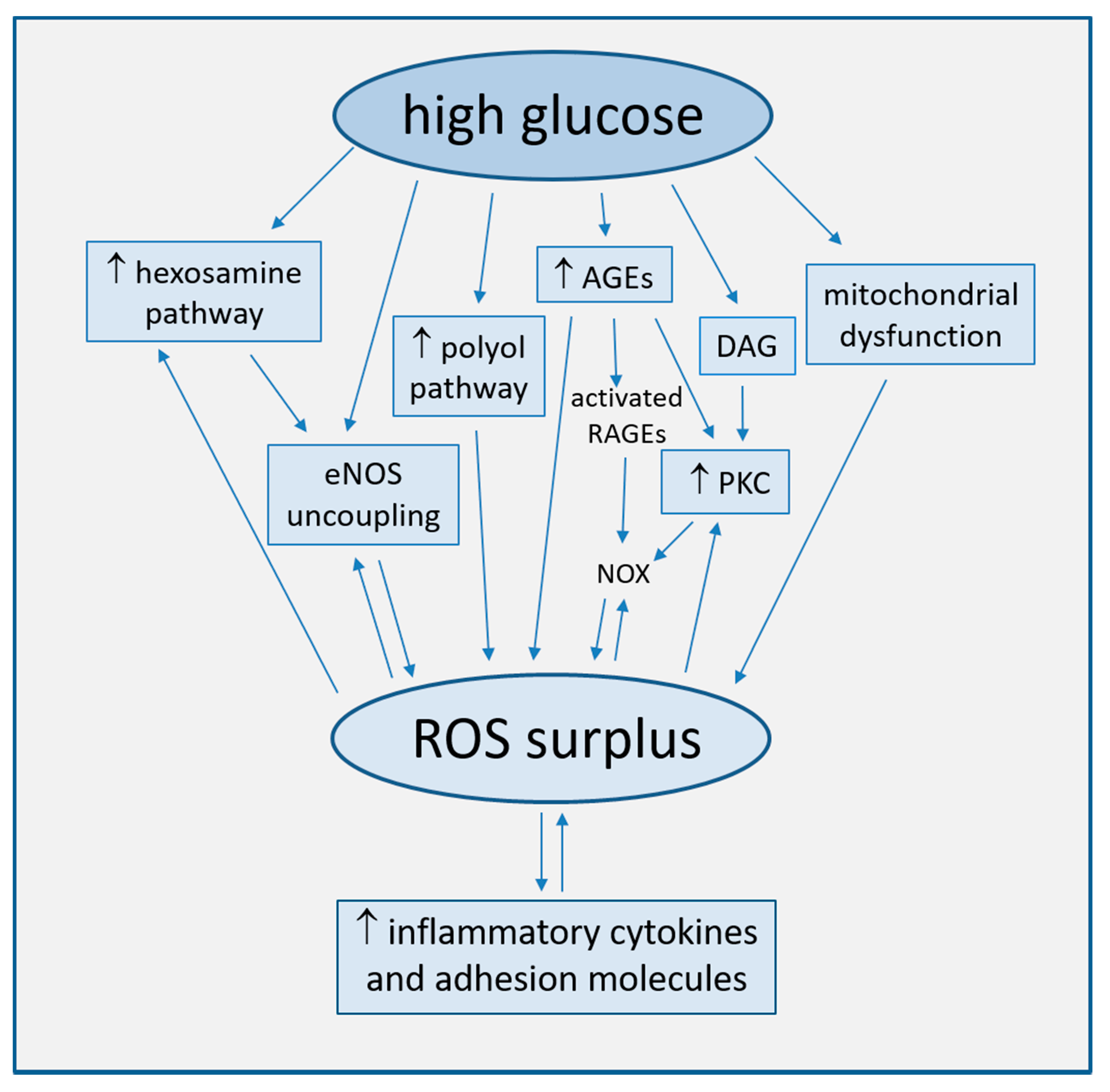
2.1. Increased ROS Production
2.1.1. Uncoupling of eNOS
2.1.2. Mitochondrial Dysfunction
2.1.3. Activation of the Polyol Pathway
2.1.4. Generation of Advanced Glycation End-Products (AGEs)
2.1.5. Activation of Protein Kinase C (PKC)
2.2. Endothelial Apoptosis and Senescence
2.3. Other Pathogenetic Mechanisms of Vascular Dysfunction
3. Metformin Promotes Cardiovascular Health
3.1. Overview on Metformin
3.1.1. Historical Notes
3.1.2. Pharmacological Effects on Glucose Metabolism
3.1.3. Activation of AMPK
3.2. Metformin Reduces Cardiovascular Mortality in Diabetes
4. Protective Properties of Metformin on Endothelium
4.1. Metformin Improves Endothelium-Dependent Vascular Response
4.1.1. Role of Insulin Resistance Correction
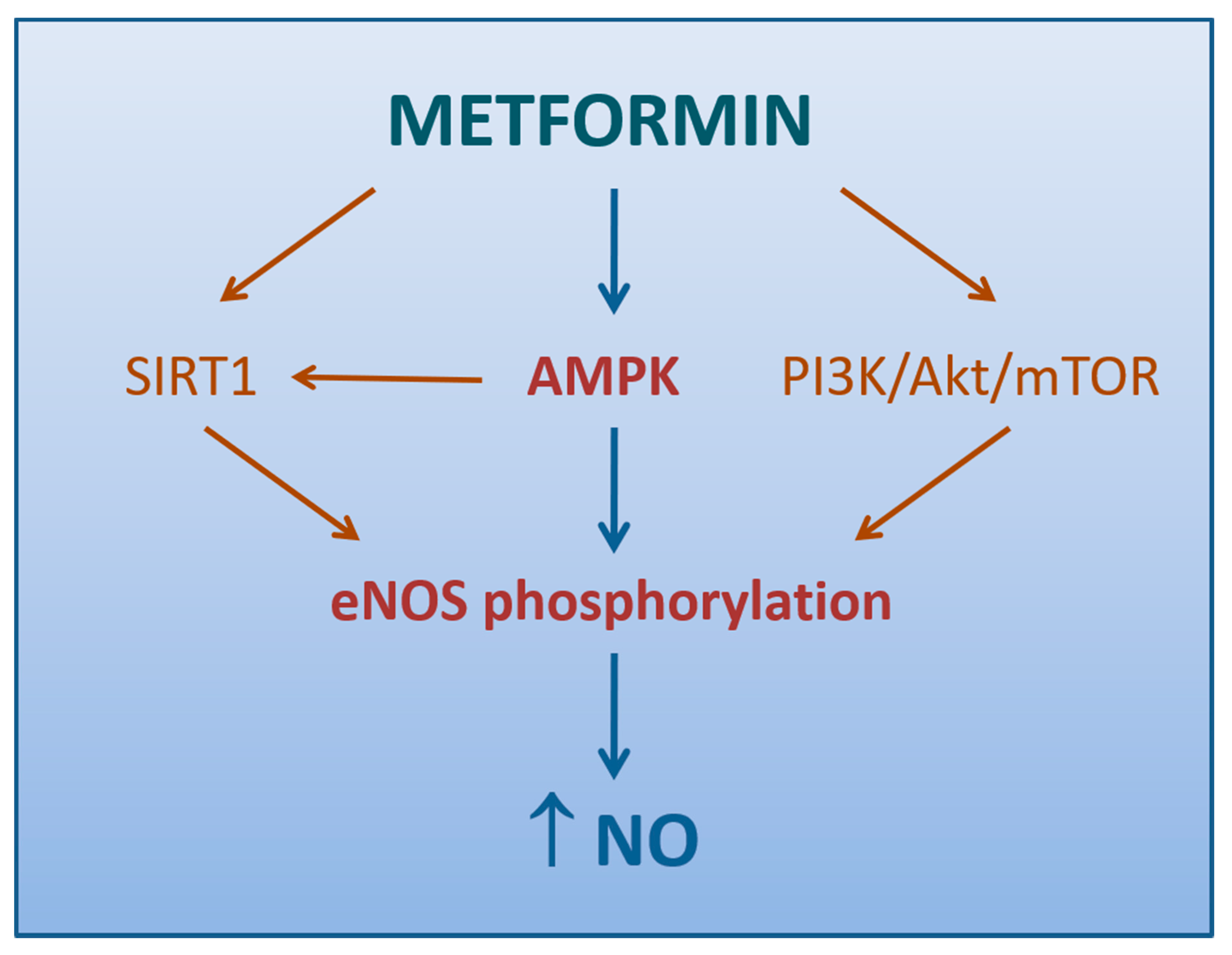
4.1.2. Role of AMPK Activation
4.1.3. Other Mechanisms
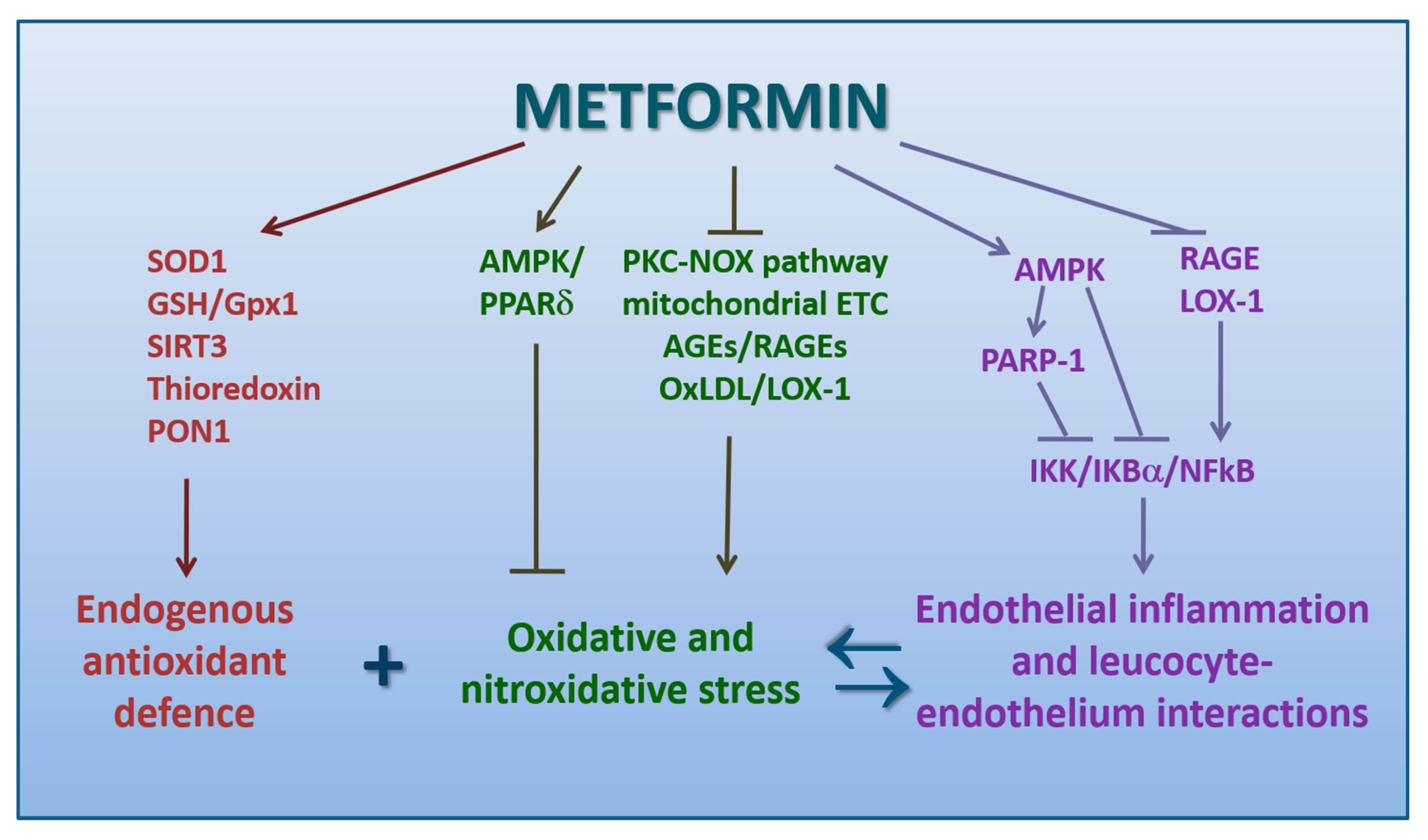
4.2. Metformin Promotes Antioxidation
4.3. Metformin Counteracts the Pro-Atherogenic Role of oxLDL and LOX-1
4.4. Metformin Inhibits Endothelial Inflammation and Leukocyte-Endothelium Interactions
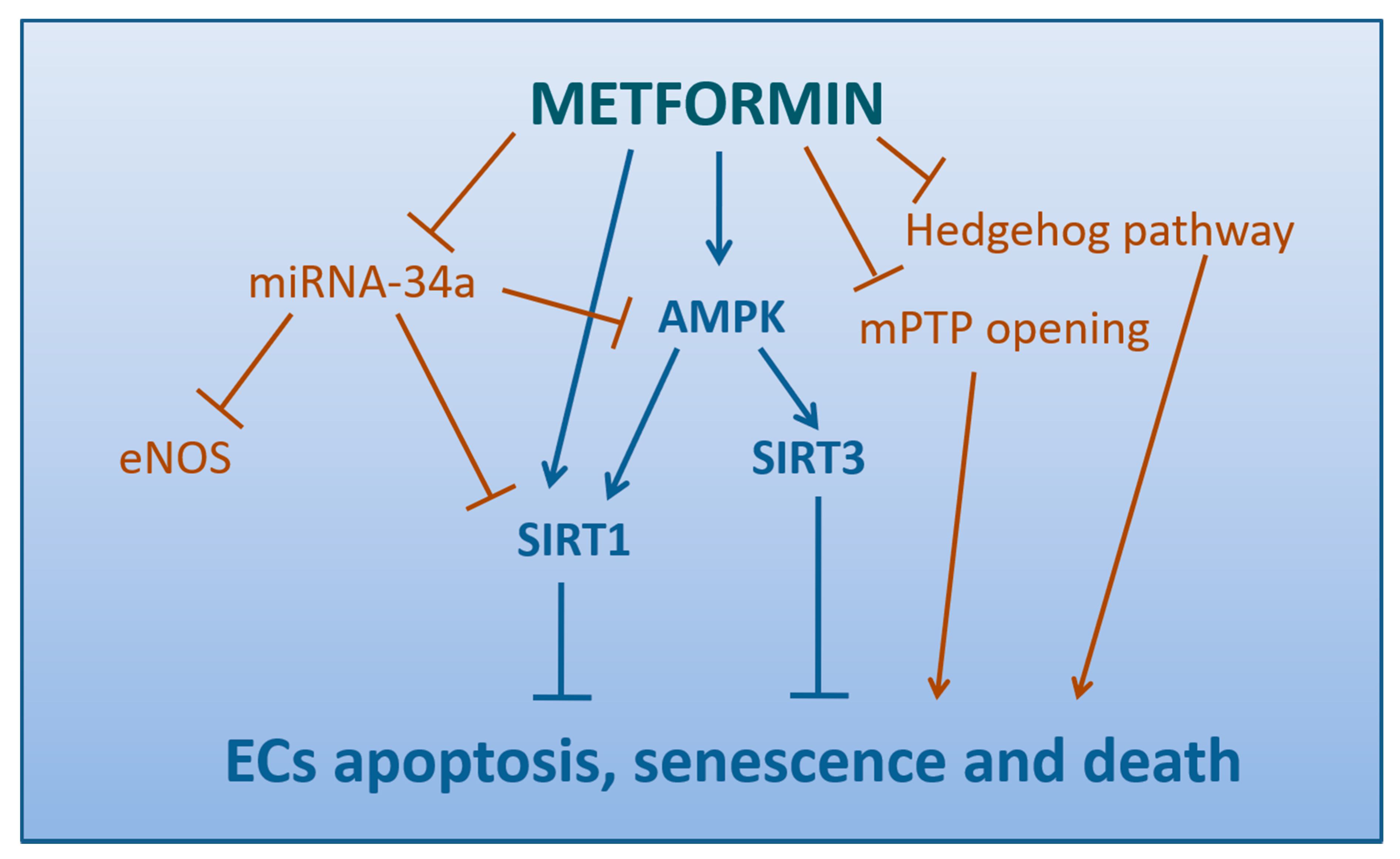
4.5. Metformin Attenuates the Apoptosis, Senescence, and Death of Endothelial Cells
4.6. Metformin Inhibits Mitochondrial Fission
4.7. Other Protective Vascular Actions by Metformin
5. Conclusions
Funding
Conflicts of Interest
References
- Haffner, S.M.; Lehto, S.; Rönnemaa, T.; Pyörälä, K.; Laakso, M. Mortality from Coronary Heart Disease in Subjects with Type 2 Diabetes and in Nondiabetic Subjects with and without Prior Myocardial Infarction. New Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42 (Suppl. 1), S90–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Das, A.; Chen, J.; Wu, P.; Li, X.; Fang, Z. Metformin in patients with and without diabetes: A paradigm shift in cardiovascular disease management. Cardiovasc. Diabetol. 2019, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Revin, V.V.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular Endothelium: Functioning in Norm, Changes in Atherosclerosis and Current Dietary Approaches to Improve Endothelial Function. Mini-Rev. Med. Chem. 2015, 15, 338–350. [Google Scholar] [CrossRef]
- Nishida, K.; Harrison, D.G.; Navas, J.P.; Fisher, A.A.; Dockery, S.P.; Uematsu, M.; Nerem, R.M.; Alexander, R.W.; Murphy, T.J. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Investig. 1992, 90, 2092–2096. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, R.M.; Murad, F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ. Res. 1983, 52, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease —A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Nafisa, A.; Gray, S.G.; Cao, Y.; Wang, T.; Xu, S.; Wattoo, F.H.; Barras, M.; Cohen, N.D.; Kamato, D.; Little, P.J. Endothelial function and dysfunction: Impact of metformin. Pharmacol. Ther. 2018, 192, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, H.; Yasutake, H.; Fujii, K.; Owada, M.K.; Nakaike, R.; Fukumoto, Y.; Takayanagi, T.; Nagao, T.; Egashira, K.; Fujishima, M.; et al. The Importance of the Hyperpolarizing Mechanism Increases as the Vessel Size Decreases in Endothelium-Dependent Relaxations in Rat Mesenteric Circulation. J. Cardiovasc. Pharmacol. 1996, 28, 703–711. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Sasso, F.C.; Cozzolino, D.; Saccomanno, F.; Assaloni, R.; Ceriello, A.; Giugliano, D. Effect of a single high-fat meal on endothelial function in patients with the metabolic syndrome: Role of tumor necrosis factor-α. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marfella, R.; Ferraraccio, F.; Rizzo, M.R.; Portoghese, M.; Barbieri, M.; Basilio, C.; Nersita, R.; Siniscalchi, L.I.; Sasso, F.C.; Ambrosino, I.; et al. Innate Immune Activity in Plaque of Patients with Untreated andl-Thyroxine-Treated Subclinical Hypothyroidism. J. Clin. Endocrinol. Metab. 2011, 96, 1015–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial Dysfunction, Oxidative Stress, and Risk of Cardiovascular Events in Patients with Coronary Artery Disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.J.; Phillips, S.A. Assessment and Prognosis of Peripheral Artery Measures of Vascular Function. Prog. Cardiovasc. Dis. 2015, 57, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; DeFelice, A.F.; Hanig, J.P.; Colatsky, T. Biomarkers of Endothelial Cell Activation Serve as Potential Surrogate Markers for Drug-induced Vascular Injury. Toxicol. Pathol. 2010, 38, 856–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Williams, S.B.; Goldfine, A.B.; Timimi, F.K.; Ting, H.H.; Roddy, M.-A.; Simonson, D.C.; Creager, M.A. Acute Hyperglycemia Attenuates Endothelium-Dependent Vasodilation in Humans In Vivo. Circulation 1998, 97, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Kawano, H.; Motoyama, T.; Hirashima, O.; Hirai, N.; Miyao, Y.; Sakamoto, T.; Kugiyama, K.; Ogawa, H.; Yasue, H. Hyperglycemia rapidly suppresses flow-mediated endothelium- dependent vasodilation of brachial artery. J. Am. Coll. Cardiol. 1999, 34, 146–154. [Google Scholar] [CrossRef] [Green Version]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Sasso, F.C.; Siniscalchi, M.; Paolisso, P.; Rizzo, M.R.; Ferraro, F.; Stabile, E.; Sorropago, G.; Calabrò, P.; Carbonara, O.; et al. Peri-Procedural Tight Glycemic Control during Early Percutaneous Coronary Intervention Is Associated with a Lower Rate of In-Stent Restenosis in Patients with Acute ST-Elevation Myocardial Infarction. J. Clin. Endocrinol. Metab. 2012, 97, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmey, H.A.L.; Ye, X.; Ding, H.; Triggle, C.R.; Garland, C.J.; Dora, K.A. Hyperglycaemia disrupts conducted vasodilation in the resistance vasculature of db/db mice. Vasc. Pharmacol. 2018, 29–35. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nat. Cell Biol. 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Vascular Adhesion Molecules in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Minutolo, R.; Sasso, F.C.; Chiodini, P.; Cianciaruso, B.; Carbonara, O.; Zamboli, P.; Tirino, G.; Pota, A.; Torella, R.; Conte, G.; et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: A comparative analysis in nephrology, diabetology and primary care settings. J. Hypertens. 2006, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, R.; Gabbai, F.B.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Garofalo, C.; Sasso, F.C.; Santoro, D.; Bellizzi, V.; Conte, G.; et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018, 33, 1942–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriello, A.; Russo, P.D.; Amstad, P.; Cerutti, P. High Glucose Induces Antioxidant Enzymes in Human Endothelial Cells in Culture: Evidence Linking Hyperglycemia and Oxidative Stress. Diabetes 1996, 45, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Förstermann, U.; Munzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Aljofan, M.; Ding, H. High glucose increases expression of cyclooxygenase-2, increases oxidative stress and decreases the generation of nitric oxide in mouse microvessel endothelial cells. J. Cell. Physiol. 2009, 222, 669–675. [Google Scholar] [CrossRef]
- Pannirselvam, M.; Verma, S.; Anderson, T.J.; Triggle, C.R. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db−/−) mice: Role of decreased tetrahydrobiopterin bioavailability. Br. J. Pharmacol. 2002, 136, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Moens, A.L.; Kass, D.A. Tetrahydrobiopterin and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2006, 26, 2439–2444. [Google Scholar] [CrossRef]
- Wever, R.M.; Van Dam, T.; Van Rijn, H.J.; De Groot, F.; Rabelink, T.J. Tetrahydrobiopterin Regulates Superoxide and Nitric Oxide Generation by Recombinant Endothelial Nitric Oxide Synthase. Biochem. Biophys. Res. Commun. 1997, 237, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Tatham, A.L.; Hale, A.B.; Alp, N.J.; Channon, K. Critical Role for Tetrahydrobiopterin Recycling by Dihydrofolate Reductase in Regulation of Endothelial Nitric-oxide Synthase Coupling: Relative importance of the de novo biopterin synthesis versus salvage pathways. J. Biol. Chem. 2009, 284, 28128–28136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channon, K.M. Tetrahydrobiopterin and Nitric Oxide Synthase Recouplers. Bone Regul. Osteoporos. Ther. 2020, 1–14. [Google Scholar] [CrossRef]
- Ihlemann, N.; Rask-Madsen, C.; Perner, A.; Dominguez, H.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am. J. Physiol. Circ. Physiol. 2003, 285, H875–H882. [Google Scholar] [CrossRef] [Green Version]
- Pannirselvam, M.; Simon, V.; Verma, S.; Anderson, T.; Triggle, C.R. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br. J. Pharmacol. 2003, 140, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Franscini, N.; Bächli, E.; Blau, N.; Fischler, M.; Walter, R.B.; Schaffner, A.; Schoedon, G. Functional Tetrahydrobiopterin Synthesis in Human Platelets. Circulation 2004, 110, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wu, Y.; Song, P.; Zhang, M.; Wang, S.; Zou, M.-H. Proteasome-Dependent Degradation of Guanosine 5′-Triphosphate Cyclohydrolase I Causes Tetrahydrobiopterin Deficiency in Diabetes Mellitus. Circulation 2007, 116, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.-H. In Vivo Activation of AMP-Activated Protein Kinase Attenuates Diabetes-Enhanced Degradation of GTP Cyclohydrolase I. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hatley, M.E.; Bolick, D.T.; Palmer, L.A.; Edelstein, D.; Brownlee, M.; Hedrick, C.C. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 2004, 47, 1727–1734. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Shen, H.; Liu, H.-N.; Wang, Y.; Bai, Y.; Wu, N. Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovasc. Diabetol. 2016, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hart, P.C.; Germain, D.; Bonini, M.G. SOD2 and the Mitochondrial UPR: Partners Regulating Cellular Phenotypic Transitions. Trends Biochem. Sci. 2016, 41, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Tsai, Y.-S. Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 2011, 32, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabe-Nishimura, C. Aldose reductase in glucose toxicity: A potential target for the prevention of diabetic complications. Pharmacol. Rev. 1998, 50, 21–34. [Google Scholar]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose Reductase, Oxidative Stress, and Diabetic Mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.B.M.; Ramana, K.V.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Aldose Reductase Regulates High Glucose-Induced Ectodomain Shedding of Tumor Necrosis Factor (TNF)-α via Protein Kinase C-δ and TNF-α Converting Enzyme in Vascular Smooth Muscle Cells. Endocrinology 2008, 150, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Yadav, U.C.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibition suppresses airway inflammation. Chem. Interact. 2011, 191, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Li, Q.; Zhang, Y.C.; Ma, G.; Feng, Y.; Zhu, Q.; Dai, Q.; Chen, Z.; Yao, Y.; Chen, L.; et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed. Pharmacother. 2010, 64, 35–43. [Google Scholar] [CrossRef]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar] [PubMed]
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell. Physiol. 2007, 212, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Taye, A.; Saad, A.H.; Kumar, A.H.; Morawietz, H. Effect of apocynin on NADPH oxidase-mediated oxidative stress-LOX-1-eNOS pathway in human endothelial cells exposed to high glucose. Eur. J. Pharmacol. 2010, 627, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent High Glucose Enhances Apoptosis Related to Oxidative Stress in Human Umbilical Vein Endothelial Cells: The Role of Protein Kinase C and NAD(P)H-Oxidase Activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoguchi, T.; Sonta, T.; Tsubouchi, H.; Etoh, T.; Kakimoto, M.; Sonoda, N.; Sato, N.; Sekiguchi, N.; Kobayashi, K.; Sumimoto, H.; et al. Protein Kinase C-Dependent Increase in Reactive Oxygen Species (ROS) Production in Vascular Tissues of Diabetes: Role of Vascular NAD(P)H Oxidase. J. Am. Soc. Nephrol. 2003, 14, 227S–232S. [Google Scholar] [CrossRef] [Green Version]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Inoguchi, T.; Kern, T.S.; Engerman, R.L.; Oates, P.J.; King, G.L. Characterization of the Mechanism for the Chronic Activation of Diacylglycerol-Protein Kinase C Pathway in Diabetes and Hypergalactosemia. Diabetes 1994, 43, 1122–1129. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef] [Green Version]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E-/- mice. Sci. Rep. 2015, 4, 5519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, T.; Fukuo, K.; Yasuda, O.; Hotta, M.; Miyazaki, J.; Takemura, Y.; Kawamoto, H.; Ichijo, H.; Ogihara, T. Apoptosis Signal-Regulating Kinase 1 Mediates Cellular Senescence Induced by High Glucose in Endothelial Cells. Diabetes 2006, 55, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated Collagen I Induces Premature Senescence-Like Phenotypic Changes in Endothelial Cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Sheu, S.-S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Zou, G.; Gu, J.; Zhang, J. L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res. Clin. Pr. 2010, 89, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Matsui-Hirai, H.; Hayashi, T.; Yamamoto, S.; Ina, K.; Maeda, M.; Kotani, H.; Iguchi, A.; Ignarro, L.J.; Hattori, Y. Dose-Dependent Modulatory Effects of Insulin on Glucose-Induced Endothelial Senescence In Vitro and In Vivo: A Relationship between Telomeres and Nitric Oxide. J. Pharmacol. Exp. Ther. 2011, 337, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-H.; Mo, X.; Gong, Q.; Pan, Q.; Yang, X.; Cai, W.; Li, C.; Ma, J.-X.; He, Y.; Gao, G. Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis 2008, 13, 1331–1343. [Google Scholar] [CrossRef]
- Ho, F.M.; Lin, W.-W.; Chen, B.C.; Chao, C.M.; Yang, C.-R.; Lin, L.Y.; Lai, C.C.; Liu, S.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-κB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell. Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic Anhydrase Activation Is Associated with Worsened Pathological Remodeling in Human Ischemic Diabetic Cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Jiang, S.; Yang, Y.; Liu, S.; Ponnusamy, M.; Xin, H.; Yu, T. Noncoding RNAs as therapeutic targets in atherosclerosis with diabetes mellitus. Cardiovasc. Ther. 2018, 36, e12436. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Tarallo, R.; Marino, F.; Giurato, G.; Veneziano, C.; Aquila, I.; Scalise, M.; Mancuso, T.; Cianflone, E.; et al. miRNA Regulation of the Hyperproliferative Phenotype of Vascular Smooth Muscle Cells in Diabetes. Diabetes 2018, 67, 2554–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinlay, S.; Behrendt, D.; Fang, J.C.; Delagrange, D.; Morrow, J.; Witztum, J.L.; Rifai, N.; Selwyn, A.P.; Creager, M.A.; Ganz, P. long-term effect of combined vitamins e and c on coronary and peripheral endothelial function. J. Am. Coll. Cardiol. 2004, 43, 629–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvin, E.; Bolen, S.; Yeh, H.-C.; Wiley, C.; Wilson, L.M.; Marinopoulos, S.S.; Feldman, L.; Vassy, J.L.; Wilson, R.; Bass, E.B.; et al. Cardiovascular Outcomes in Trials of Oral Diabetes Medications: A systematic review. Arch. Intern. Med. 2008, 168, 2070–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yandrapalli, S.; Jolly, G.; Horblitt, A.; Sanaani, A.; Aronow, W. Cardiovascular benefits and safety of non-insulin medications used in the treatment of type 2 diabetes mellitus. Postgrad. Med. 2017, 129, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Roumie, C.L.; Hung, A.M.; Greevy, R.A.; Grijalva, C.G.; Liu, X.; Murff, H.J.; Elasy, T.A.; Griffin, M.R. Comparative Effectiveness of Sulfonylurea and Metformin Monotherapy on Cardiovascular Events in Type 2 Diabetes Mellitus: A cohort study. Ann. Intern. Med. 2012, 157, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, R.; Travert, F.; Pasquet, B.; Wilson, P.W.; Smith, S.C.; Goto, S.; Ravaud, P.; Marre, M.; Porath, A.; Bhatt, D.L.; et al. Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch. Intern. Med. 2010, 170, 1892–1899. [Google Scholar] [CrossRef] [Green Version]
- Scheen, A.J.; Paquot, N. Metformin revisited: A critical review of the benefit-risk balance in at-risk patients with type 2 diabetes. Diabetes Metab. 2013, 39, 179–190. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Ríos, J.L.; Francini, F.; Schinella, G.R. Natural Products for the Treatment of Type 2 Diabetes Mellitus. Planta Med. 2015, 81, 975–994. [Google Scholar] [CrossRef] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Eva, A.-R.; Ranal-Muino, E.; Fernandez-Fernandez, C.; Pazos-Garcia, C.; Vila-Altesor, M. Metabolic Effects of Metformin in Humans. Curr. Diabetes Rev. 2019, 15, 328–339. [Google Scholar] [CrossRef]
- Buse, J.B.; DeFronzo, R.A.; Rosenstock, J.; Kim, T.; Burns, C.; Skare, S.; Baron, A.; Fineman, M. The Primary Glucose-Lowering Effect of Metformin Resides in the Gut, Not the Circulation. Results From Short-term Pharmacokinetic and 12-Week Dose-Ranging Studies. Diabetes Care 2016, 39, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiernsperger, N.F. Metformin as a cellular protector; a synoptic view of modern evidences. J. Nephropharmacol. 2015, 4, 31–36. [Google Scholar] [PubMed]
- Nath, M.; Bhattacharjee, K.; Choudhury, Y. Pleiotropic effects of anti-diabetic drugs: A comprehensive review. Eur. J. Pharmacol. 2020, 884, 173349. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J.; Welsh, P.; Petrie, J.R. Metformin, lipids and atherosclerosis prevention. Curr. Opin. Lipidol. 2018, 29, 346–353. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase—An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; De Montellano, P.R.O.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Lee, I.K.; Kim, H.S.; Kim, Y.M.; Koh, E.H.; Won, J.C.; Han, S.M.; Kim, M.-S.; Jo, I.; Oh, G.T.; et al. α-Lipoic Acid Prevents Endothelial Dysfunction in Obese Rats via Activation of AMP-Activated Protein Kinase. Arter. Thromb. Vasc. Biol. 2005, 25, 2488–2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.-H.; Wu, Y. Amp-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin. Exp. Pharmacol. Physiol. 2007, 35, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; Depinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin Activates AMP-Activated Protein Kinase in Endothelial Cells via a Pathway Involving Ca2+/Calmodulin-Dependent Protein Kinase Kinase β. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-Carboxamide Ribonucleoside. A Specific Method for Activating AMP-Activated Protein Kinase in Intact Cells? JBIC J. Biol. Inorg. Chem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2011, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin Activates AMP Kinase through Inhibition of AMP Deaminase. J. Biol. Chem. 2010, 286, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Ye, K.; Triggle, C.R. Impact of currently used anti-diabetic drugs on myoendothelial communication. Curr. Opin. Pharmacol. 2019, 45, 1–7. [Google Scholar] [CrossRef]
- Logie, L.; Harthill, J.; Patel, K.; Bacon, S.; Hamilton, D.L.; Macrae, K.; McDougall, G.; Wang, H.-H.; Xue, L.; Jiang, H.; et al. Cellular Responses to the Metal-Binding Properties of Metformin. Diabetes 2012, 61, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; Macdonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nat. Cell Biol. 2014, 510, 542–546. [Google Scholar] [CrossRef] [Green Version]
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An Old Drug for the Treatment of Diabetes but a New Drug for the Protection of the Endothelium. Med Princ. Pr. 2015, 24, 401–415. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 854–865. [CrossRef]
- Saenz, A.; Fernandez-Esteban, I.; Mataix, A.; Segura, M.A.; Roque, M.; Moher, D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2005, 20, CD002966. [Google Scholar] [CrossRef] [Green Version]
- Eurich, D.T.; McAlister, F.A.; Blackburn, D.F.; Majumdar, S.R.; Tsuyuki, R.T.; Varney, J.; Johnson, J.A. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: Systematic review. BMJ 2007, 335, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, C.; Monami, M.; Marchionni, N.; Mannucci, E. Effect of metformin on cardiovascular events and mortality: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2011, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Leaver, J.K.; Irving, G.J. Impact of metformin on cardiovascular disease: A meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, C.H.; Gislason, G.; Andersson, C.; Ahlehoff, O.; Charlot, M.; Schramm, T.K.; Vaag, A.; Abildstrøm, S.Z.; Torp-Pedersen, C.; Hansen, T.W. Effects of oral glucose-lowering drugs on long term outcomes in patients with diabetes mellitus following myocardial infarction not treated with emergent percutaneous coronary intervention—A retrospective nationwide cohort study. Cardiovasc. Diabetol. 2010, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151. [Google Scholar] [CrossRef]
- De Jager, J.; Kooy, A.; Schalkwijk, C.; Van Der Kolk, J.; Lehert, P.; Bets, D.; Wulffelé, M.G.; Donker, A.J.; Stehouwer, C.D.A. Long-term effects of metformin on endothelial function in type 2 diabetes: A randomized controlled trial. J. Intern. Med. 2014, 275, 59–70. [Google Scholar] [CrossRef]
- Chait, A.; Bornfeldt, K.E. Diabetes and atherosclerosis: Is there a role for hyperglycemia? J. Lipid Res. 2008, 50, S335–S339. [Google Scholar] [CrossRef] [Green Version]
- De Aguiar, L.G.K.; Kraemer-Aguiar, L.G.; Bahia, L.R.; Villela, N.; Laflor, C.; Sicuro, F.; Wiernsperger, N.F.; Bottino, D.; Bouskela, E. Metformin Improves Endothelial Vascular Reactivity in First-Degree Relatives of Type 2 Diabetic Patients With Metabolic Syndrome and Normal Glucose Tolerance. Diabetes Care 2006, 29, 1083–1089. [Google Scholar] [CrossRef]
- Marfella, R.; Acampora, R.; Verrazzo, G.; Ziccardi, P.; De Rosa, N.; Giunta, R.; Giugliano, D. Metformin Improves Hemodynamic and Rheological Responses to L-Arginine in NIDDM Patients. Diabetes Care 1996, 19, 934–939. [Google Scholar] [CrossRef]
- Kapur, N. Nitric oxide: An emerging role in cardioprotection? Heart 2001, 86, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Mather, K.J.; Verma, S.; Anderson, T.J. Improved endothelial function with metformin in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2001, 37, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, H.O.; Brechtel, G.; Johnson, A.; Fineberg, N.; Baron, A.D. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J. Clin. Investig. 1994, 94, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Investig. 1996, 98, 894–898. [Google Scholar] [CrossRef] [Green Version]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Chaker, H.; Leaming, R.; Johnson, A.; Brechtel, G.; Baron, A.D. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J. Clin. Investig. 1996, 97, 2601–2610. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Investig. 1997, 100, 1230–1239. [Google Scholar] [CrossRef]
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefèbvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Balletshofer, B.; Rittig, K.; Maerker, E.; Häring, H.; Volk, A.; Jacob, S.; Rett, K. Impaired Non-Esterified Fatty Acid Suppression is Associated with Endothelial Dysfunction in Insulin Resistant Subjects. Horm. Metab. Res. 2001, 33, 428–431. [Google Scholar] [CrossRef]
- Michell, B.J.; Chen, Z.-P.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated Control of Endothelial Nitric-oxide Synthase Phosphorylation by Protein Kinase C and the cAMP-dependent Protein Kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Cacicedo, J.M.; Itani, S.; Yagihashi, N.; Saha, A.K.; Ye, J.M.; Chen, K.; Zou, M.; Carling, D.; Boden, G.; et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): Possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem. Soc. Trans. 2003, 31, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Katakam, P.V.G.; Ujhelyi, M.R.; Hoenig, M.; Miller, A.W. Metformin Improves Vascular Function in Insulin-Resistant Rats. Hypertension 2000, 35, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, C.; Matafome, P.; Louro, T.; Nunes, E.; Fernandes, R.; Seiça, R.M. Metformin restores endothelial function in aorta of diabetic rats. Br. J. Pharmacol. 2011, 163, 424–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, C.; Mercuro, G.; Cornoldi, A.; Fini, M.; Volterrani, M.; Rosano, G. Metformin improves endothelial function in patients with metabolic syndrome. J. Intern. Med. 2005, 258, 250–256. [Google Scholar] [CrossRef]
- Tack, C.J.J.; Ong, M.K.E.; Lutterman, J.A.; Smits, P. Insulin-induced vasodilatation and endothelial function in obesity/insulin resistance. Effects of troglitazone. Diabetologia 1998, 41, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.K.; Papathanassiou, K.; Bechlioulis, A.; Pappas, K.; Kazakos, N.; Kanioglou, C.; Kostoula, A.; Vezyraki, P.; Makriyiannis, D.; Tsatsoulis, A.; et al. Effects of pioglitazone and metformin on vascular endothelial function in patients with type 2 diabetes treated with sulfonylureas. Diabetes Vasc. Dis. Res. 2011, 9, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Pitocco, D.; Zaccardi, F.; Tarzia, P.; Milo, M.; Scavone, G.; Rizzo, P.; Pagliaccia, F.; Nerla, R.; Di Franco, A.; Manto, A.; et al. Metformin improves endothelial function in type 1 diabetic subjects: A pilot, placebo-controlled randomized study. Diabetes Obes. Metab. 2013, 15, 427–431. [Google Scholar] [CrossRef]
- Salt, I.P.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.J.; Xie, Z.; Viollet, B.; Zou, M.-H. Activation of the AMP-Activated Kinase by Antidiabetes Drug Metformin Stimulates Nitric Oxide Synthesis In Vivo by Promoting the Association of Heat Shock Protein 90 and Endothelial Nitric Oxide Synthase. Diabetes 2006, 55, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Bosselaar, M.; Boon, H.; Van Loon, L.J.C.; Broek, P.H.H.V.D.; Smits, P.; Tack, C.J. Intra-arterial AICA-riboside administration induces NO-dependent vasodilation in vivo in human skeletal muscle. Am. J. Physiol. Metab. 2009, 297, E759–E766. [Google Scholar] [CrossRef]
- Matsumoto, T.; Noguchi, E.; Ishida, K.; Kobayashi, T.; Yamada, N.; Kamata, K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2008, 295, H1165–H1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enkhjargal, B.; Godo, S.; Sawada, A.; Suvd, N.; Saito, H.; Noda, K.; Satoh, K.; Shimokawa, H. Endothelial AMP-Activated Protein Kinase Regulates Blood Pressure and Coronary Flow Responses Through Hyperpolarization Mechanism in Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Vanhoutte, P.M.; Leung, S.W.S. Vascular adenosine monophosphate-activated protein kinase: Enhancer, brake or both? Basic Clin. Pharmacol. Toxicol. 2019, 127, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunachalam, G.; Samuel, S.M.; Marei, I.; Ding, H.; Triggle, C.R. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br. J. Pharmacol. 2013, 171, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lakshmanan, A.P.; Hwang, M.J.; Kubba, H.; Mushannen, A.; Triggle, C.R.; Ding, H. Metformin improves endothelial function in aortic tissue and microvascular endothelial cells subjected to diabetic hyperglycaemic conditions. Biochem. Pharmacol. 2015, 98, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.H.C.; Vanhoutte, P.M. Endothelial dysfunction: A strategic target in the treatment of hypertension? Pflügers Archiv. 2010, 459, 995–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.-H.; Kirkpatrick, S.S.; Davis, B.J.; Nelson, J.S.; Wiles, W.G.; Schlattner, U.; Neumann, D.; Brownlee, M.; Freeman, M.B.; Goldman, M.H. Withdrawal: Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo: Role of mitochondrial reactive nitrogen species. J. Biol. Chem. 2019, 294, 13525. [Google Scholar] [CrossRef] [Green Version]
- Bellin, C.; De Wiza, D.H.; Wiernsperger, N.F.; Rosen, P. Generation of Reactive Oxygen Species by Endothelial and Smooth Muscle Cells: Influence of Hyperglycemia and Metformin. Horm. Metab. Res. 2006, 38, 732–739. [Google Scholar] [CrossRef]
- Faure, P.; Rossini, E.; Wiernsperger, N.; Richard, M.J.; Favier, A.; Halimi, S. An insulin sensitizer improves the free radical defense system potential and insulin sensitivity in high fructose-fed rats. Diabetes 1999, 48, 353–357. [Google Scholar] [CrossRef]
- Gargiulo, P.; Caccese, D.; Pignatelli, P.; Brufani, C.; De Vito, F.; Marino, R.; Lauro, R.; Violi, F.; Di Mario, U.; Sanguigni, V. Metformin decreases platelet superoxide anion production in diabetic patients. Diabetes/Metab. Res. Rev. 2002, 18, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Ouslimani, N.; Peynet, J.; Bonnefont-Rousselot, D.; Thérond, P.; Legrand, A.; Beaudeux, J.-L. Metformin decreases intracellular production of reactive oxygen species in aortic endothelial cells. Metabolism 2005, 54, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.-Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS Production Induced by a Reverse-Electron Flux at Respiratory-Chain Complex 1 is Hampered by Metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.S.; Singh, A.K.; Akhtar, F.; Chaudhary, A.; Rizvi, S.I. Metformin protects red blood cells against rotenone induced oxidative stress and cytotoxicity. Arch. Physiol. Biochem. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosen, P.; Wiernsperger, N.F. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto–Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes/Metab. Res. Rev. 2006, 22, 323–330. [Google Scholar] [CrossRef]
- Sultuybek, G.K.; Ozdas, S.B.; Curgunlu, A.; Tezcan, V.; Onaran, I. Does metformin prevent short term oxidant-induced DNA damage? In vitro study on lymphocytes from aged subjects. J. Basic Clin. Physiol. Pharmacol. 2007, 18, 129–140. [Google Scholar] [CrossRef]
- Sambe, T.; Mason, R.P.; Dawoud, H.; Sherratt, S.C.; Malinski, T. Metformin treatment decreases nitroxidative stress, restores nitric oxide bioavailability and endothelial function beyond glucose control. Biomed. Pharmacother. 2018, 98, 149–156. [Google Scholar] [CrossRef]
- Khouri, H.; Collin, F.; Bonnefont-Rousselot, D.; Legrand, A.; Jore, D.; Gardes-Albert, M. Radical-induced oxidation of metformin. JBIC J. Biol. Inorg. Chem. 2004, 271, 4745–4752. [Google Scholar] [CrossRef]
- Mithieux, G.; Guignot, L.; Bordet, J.-C.; Wiernsperger, N. Intrahepatic mechanisms underlying the effect of metformin in decreasing basal glucose production in rats fed a high-fat diet. Diabetes 2002, 51, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Forouzandeh, F.; Salazar, G.; Patrushev, N.; Xiong, S.; Hilenski, L.; Fei, B.; Alexander, R.W. Metformin Beyond Diabetes: Pleiotropic Benefits of Metformin in Attenuation of Atherosclerosis. J. Am. Heart Assoc. 2014, 3, e001202. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-N.; Song, J.; Zhang, L.; Lemaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 Pathway Reduces Fatty Acid-Induced Increase in Intracellular Reactive Oxygen Species by Upregulating Thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Song, J.; Li, X.-N.; Zhang, L.; Wang, X.; Chen, L.; Shen, Y.H. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem. Biophys. Res. Commun. 2010, 396, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Takasaka, N.; Yoshida, M.; Tsubouchi, K.; Minagawa, S.; Araya, J.; Saito, N.; Fujita, Y.; Kurita, Y.; Kobayashi, K.; et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, W.; Wei, R.; Ke, J.; Yang, J.; Liu, Y.; Wang, X.; Wang, G.; Hong, T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J. Diabetes Complicat. 2016, 30, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Bułdak, Ł.; Łabuzek, K.; Bułdak, R.J.; Machnik, G.; Bołdys, A.; Basiak, M.; Bogusław, O. Metformin reduces the expression of NADPH oxidase and increases the expression of antioxidative enzymes in human monocytes/macrophages cultured in vitro. Exp. Ther. Med. 2016, 13, 794. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, M.; Liang, B.; Xu, J.; Xie, Z.; Liu, C.; Viollet, B.; Yan, D.; Zou, M.-H. AMPKα2 Deletion Causes Aberrant Expression and Activation of NAD(P)H Oxidase and Consequent Endothelial Dysfunction In Vivo. Circ. Res. 2010, 106, 1117–1128. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Cooper, M.E.; Thallas, V.; Burns, W.; Thomas, M.C.; Brammar, G.C.; Lee, F.; Grant, S.L.; Burrell, L.M.; Jerums, G.; et al. Reduction of the Accumulation of Advanced Glycation End Products by ACE Inhibition in Experimental Diabetic Nephropathy. Diabetes 2002, 51, 3274–3282. [Google Scholar] [CrossRef]
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Lopez-Domenech, S.; Escribano-Lopez, I.; Veses, S.; Jover, A.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M. Does Metformin Protect Diabetic Patients from Oxidative Stress and Leukocyte-Endothelium Interactions? Antioxid. Redox Signal. 2017, 27, 1439–1445. [Google Scholar] [CrossRef]
- Javadipour, M.; Rezaei, M.; Keshtzar, E.; Khodayar, M.J. Metformin in contrast to berberine reversed arsenic-induced oxidative stress in mitochondria from rat pancreas probably via Sirt3-dependent pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22368. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.-W.; Lee, S.S.-T.; Chen, Z.-Y.; Yao, X.; Wang, N.; Huang, Y. Metformin Protects Endothelial Function in Diet-Induced Obese Mice by Inhibition of Endoplasmic Reticulum Stress Through 5′ Adenosine Monophosphate–Activated Protein Kinase–Peroxisome Proliferator–Activated Receptor δ Pathway. Arter. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef]
- Heinloth, A.; Heermeier, K.; Raff, U.; Wanner, C.; Galle, J. Stimulation of NADPH oxidase by oxidized low-density lipoprotein induces proliferation of human vascular endothelial cells. J. Am. Soc. Nephrol. 2000, 11, 1819–1825. [Google Scholar]
- Shiu, S.W.M.; Wong, Y.; Tan, K.C.B. Effect of advanced glycation end products on lectin-like oxidized low density lipoprotein receptor-1 expression in endothelial cells. J. Atheroscler. Thromb. 2012, 19, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Ouslimani, N.; Mahrouf, M.; Peynet, J.; Bonnefont-Rousselot, D.; Cosson, C.; Legrand, A.; Beaudeux, J.-L. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin-like oxidized receptor 1. Metabolism 2007, 56, 308–313. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Choi, H.C.; Lyons, T.J.; Zou, M.-H. Activation of AMP-Activated Protein Kinase Inhibits Oxidized LDL-Triggered Endoplasmic Reticulum Stress In Vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef] [Green Version]
- Valente, A.J.; Irimpen, A.M.; Siebenlist, U.; Chandrasekar, B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: Inhibition by HDL3 and AMPK activators. Free. Radic. Biol. Med. 2014, 70, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-H.; Chan, S.-H.; Chu, P.-M.; Lin, H.-C.; Tsai, K.-L. Metformin regulates oxLDL-facilitated endothelial dysfunction by modulation of SIRT1 through repressing LOX-1-modulated oxidative signaling. Oncotarget 2016, 7, 10773–10787. [Google Scholar] [CrossRef] [Green Version]
- Roxo, D.F.; Arcaro, C.A.; Gutierres, V.O.; Costa, M.C.; Oliveira, J.O.; Lima, T.F.O.; Assis, R.P.; Brunetti, I.L.; Baviera, A.M. Curcumin combined with metformin decreases glycemia and dyslipidemia, and increases paraoxonase activity in diabetic rats. Diabetol. Metab. Syndr. 2019, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirmiranpoor, H.; Mousavizadeh, M.; Noshad, S.; Ghavami, M.; Ebadi, M.; Ghasemiesfe, M.; Nakhjavani, M.; Esteghamati, A. Comparative effects of pioglitazone and metformin on oxidative stress markers in newly diagnosed type 2 diabetes patients: A randomized clinical trial. J. Diabetes its Complicat. 2013, 27, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.P.; Palmer, T.M. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin. Investig. Drugs 2012, 21, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Mamputu, M.; Wiernsperger, N.; Renier, G. Metformin inhibits monocyte adhesion to endothelial cells and foam cell formation. Br. J. Diabetes Vasc. Dis. 2003, 3, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin Inhibits Monocyte-to-Macrophage Differentiation via AMPK-Mediated Inhibition of STAT3 Activation: Potential Role in Atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin Inhibits Cytokine-Induced Nuclear Factor κB Activation Via AMP-Activated Protein Kinase Activation in Vascular Endothelial Cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.; Szabó, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Gongol, B.; Marin, T.; Peng, I.-C.; Woo, B.; Martin, M.; King, S.; Sun, W.; Johnson, D.A.; Chien, S.; Shyy, J.Y.-J. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 2013, 110, 3161–3166. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Zhang, J.; Li, Z.; Zhang, J.; Yin, Y.; Wang, Y.; Marin, T.L.; Gongol, B.; Xiao, H.; Zhang, Y.-Y.; et al. Cardiovascular Protective Effect of Metformin and Telmisartan: Reduction of PARP1 Activity via the AMPK-PARP1 Cascade. PLoS ONE 2016, 11, e0151845. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X. Sirtuin 1-Mediated Cellular Metabolic Memory of High Glucose Via the LKB1/AMPK/ROS Pathway and Therapeutic Effects of Metformin. Diabetes 2011, 61, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, A.-L. Apoptosis and necrosis in health and disease: Role of mitochondria. Adv. Clin. Chem. 2003, 224, 29–55. [Google Scholar] [CrossRef]
- Grimm, S.; Brdiczka, D. The permeability transition pore in cell death. Apoptosis 2007, 12, 841–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detaille, D.; Guigas, B.; Chauvin, C.; Batandier, C.; Fontaine, E.; Wiernsperger, N.; Leverve, X. Metformin Prevents High-Glucose-Induced Endothelial Cell Death Through a Mitochondrial Permeability Transition-Dependent Process. Diabetes 2005, 54, 2179–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhamra, G.S.; Hausenloy, D.J.; Davidson, S.M.; Carr, R.D.; Paiva, M.; Wynne, A.M.; Mocanu, M.M.; Yellon, D.M. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res. Cardiol. 2008, 103, 274–284. [Google Scholar] [CrossRef]
- Bai, B.; Liang, Y.; Xu, C.; Lee, M.Y.; Xu, A.; Wu, N.; Vanhoutte, P.M.; Wang, Y. Cyclin-Dependent Kinase 5–Mediated Hyperphosphorylation of Sirtuin-1 Contributes to the Development of Endothelial Senescence and Atherosclerosis. Circulation 2012, 126, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, G.; Lakshmanan, A.P.; Samuel, S.M.; Triggle, C.; Ding, H. Molecular Interplay between microRNA-34a and Sirtuin1 in Hyperglycemia-Mediated Impaired Angiogenesis in Endothelial Cells: Effects of Metformin. J. Pharmacol. Exp. Ther. 2016, 356, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Yang, H.; Xiao, B. Role of microRNAs in endothelial inflammation and senescence. Mol. Biol. Rep. 2011, 39, 4509–4518. [Google Scholar] [CrossRef]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 2010, 398, 735–740. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD + elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef]
- Niu, C.; Chen, Z.; Kim, K.T.; Sun, J.; Xue, M.; Chen, G.; Li, S.; Shen, Y.; Zhu, Z.; Wang, X.; et al. Metformin alleviates hyperglycemia-induced endothelial impairment by downregulating autophagy via the Hedgehog pathway. Autophagy 2019, 15, 843–870. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugus, J.J.; Ngoh, G.A.; Bachschmid, M.M.; Walsh, K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J. Mol. Cell. Cardiol. 2011, 51, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial Dynamics in Diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Escribano-Lopez, I.; De Marañon, A.M.; Lopez-Domenech, S.; Orden, S.; Torres, I.R.; Alvarez, A.; Veses, S.; et al. Are Mitochondrial Fusion and Fission Impaired in Leukocytes of Type 2 Diabetic Patients? Antioxid. Redox Signal. 2016, 25, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and Endothelial Function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Reif, M.M.; Craige, S.M.; Kant, S.; Keaney, J.F. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide 2016, 55, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Meuwese, M.C.; Vink, H.; Hoekstra, J.B.L.; Kastelein, J.J.P.; Stroes, E.S.G. The endothelial glycocalyx: A potential barrier between health and vascular disease. Curr. Opin. Lipidol. 2005, 16, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Van Haare, J.; Kooi, M.; Van Teeffelen, J.W.G.E.; Vink, H.; Slenter, J.; Cobelens, H.; Strijkers, G.J.; Koehn, D.; Post, M.J.; Van Bilsen, M. Metformin and sulodexide restore cardiac microvascular perfusion capacity in diet-induced obese rats. Cardiovasc. Diabetol. 2017, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, B.; Xiao, Y.; Song, F.; Long, X.; Huang, J.; Tian, M.; Deng, S.; Wu, Q. Metformin-induced activation of AMPK inhibits the proliferation and migration of human aortic smooth muscle cells through upregulation of p53 and IFI16. Int. J. Mol. Med. 2017, 41, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Wu, Y.; Li, Z. KLF4/Ch25h axis activated by metformin suppresses EndoMT in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2020, 522, 838–844. [Google Scholar] [CrossRef]
- Kuan, I.H.S.; Savage, R.L.; Duffull, S.B.; Walker, R.J.; Wright, D.F. The Association between Metformin Therapy and Lactic Acidosis. Drug Saf. 2019, 42, 1449–1469. [Google Scholar] [CrossRef]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Caturano, A.; Galiero, R.; Pafundi, P.C. Metformin for Type 2 Diabetes. JAMA 2019, 322, 1312. [Google Scholar] [CrossRef]
- Rhee, M.K.; Herrick, K.A.; Ziemer, D.C.; Vaccarino, V.; Weintraub, W.S.; Narayan, K.V.; Kolm, P.; Twombly, J.G.; Phillips, L.S. Many Americans Have Pre-Diabetes and Should Be Considered for Metformin Therapy. Diabetes Care 2009, 33, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardu, C.; Paolisso, P.; Sacra, C.; Mauro, C.; Minicucci, F.; Portoghese, M.; Rizzo, M.R.; Barbieri, M.; Sasso, F.C.; D’Onofrio, N.; et al. Effects of Metformin Therapy on Coronary Endothelial Dysfunction in Patients With Prediabetes With Stable Angina and Nonobstructive Coronary Artery Stenosis: The CODYCE Multicenter Prospective Study. Diabetes Care 2019, 42, 1946–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2015, 7, 4265–4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Gjeloshi, K.; Masini, F.; Acierno, C.; Di Martino, A.; Albanese, G.; Alfano, M.; Rinaldi, L.; et al. Metformin: A Potential Therapeutic Tool for Rheumatologists. Pharmaceuticals 2020, 13, 234. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2021, 9, 3. https://doi.org/10.3390/biomedicines9010003
Salvatore T, Pafundi PC, Galiero R, Rinaldi L, Caturano A, Vetrano E, Aprea C, Albanese G, Di Martino A, Ricozzi C, et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines. 2021; 9(1):3. https://doi.org/10.3390/biomedicines9010003
Chicago/Turabian StyleSalvatore, Teresa, Pia Clara Pafundi, Raffaele Galiero, Luca Rinaldi, Alfredo Caturano, Erica Vetrano, Concetta Aprea, Gaetana Albanese, Anna Di Martino, Carmen Ricozzi, and et al. 2021. "Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects?" Biomedicines 9, no. 1: 3. https://doi.org/10.3390/biomedicines9010003