
Novel Insights into the Immunotherapy of Soft Tissue Sarcomas: Do We Need a Change of Perspective?

 ,
, 
Abstract
:1. Introduction
2. Methods
3. T Cell Infiltration
4. T Cell Immunotherapies in Soft Tissue Sarcomas
5. Regulatory T Cell (Treg) Infiltration
6. Targeting Tregs in Soft Tissue Sarcoma
7. Natural Killer (NK) Cell Infiltration
8. NK Cell-Based Immunotherapies in Soft Tissue Sarcomas
9. Macrophages
10. Therapies Targeting Macrophages
11. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Ren, Z.; Du, X.; Hao, M.; Zhou, W. The role of mesenchymal stem/progenitor cells in sarcoma: Update and dispute. Stem Cell Investig. 2014, 1, 18. [Google Scholar] [CrossRef]
- Merry, E.; Thway, K.; Jones, R.L.; Huang, P.H. Predictive and prognostic transcriptomic biomarkers in soft tissue sarcomas. npj Precis. Oncol. 2021, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.D.; Manning, M.A.; Al-Refaie, W.B.; Miettinen, M.M. Soft-Tissue Sarcomas of the Abdomen and Pelvis: Radiologic-Pathologic Features, Part 1-Common Sarcomas: From the Radiologic Pathology Archives. Radiographics 2017, 37, 462–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamboa, A.C.; Gronchi, A.; Cardona, K. Soft-tissue sarcoma in adults: An update on the current state of histiotype-specific management in an era of personalized medicine. CA Cancer J. Clin. 2020, 70, 200–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penel, N.; Grosjean, J.; Robin, Y.M.; Vanseymortier, L.; Clisant, S.; Adenis, A. Frequency of certain established risk factors in soft tissue sarcomas in adults: A prospective descriptive study of 658 cases. Sarcoma 2008, 2008, 459386. [Google Scholar] [CrossRef] [PubMed]
- Popovich, J.R.; Kashyap, S.; Cassaro, S. Sarcoma. In StatPearls; StatPearls Publishing Copyright© 2021; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Cates, J.M.M. The AJCC 8th Edition Staging System for Soft Tissue Sarcoma of the Extremities or Trunk: A Cohort Study of the SEER Database. J. Natl. Compr. Cancer Netw. JNCCN 2018, 16, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Sekimizu, M.; Ogura, K.; Yasunaga, H.; Matsui, H.; Tanaka, S.; Inagaki, K.; Kawai, A. Development of nomograms for prognostication of patients with primary soft tissue sarcomas of the trunk and extremity: Report from the Bone and Soft Tissue Tumor Registry in Japan. BMC Cancer 2019, 19, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.S. The New American Joint Commission on Cancer Staging System for Soft Tissue Sarcomas: Splitting versus Lumping. Ann. Surg. Oncol. 2018, 25, 1101–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronchi, A.; Ferrari, S.; Quagliuolo, V.; Broto, J.M.; Pousa, A.L.; Grignani, G.; Basso, U.; Blay, J.Y.; Tendero, O.; Beveridge, R.D.; et al. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): An international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet. Oncol. 2017, 18, 812–822. [Google Scholar] [CrossRef]
- Komdeur, R.; Hoekstra, H.J.; van den Berg, E.; Molenaar, W.M.; Pras, E.; de Vries, E.G.E.; van der Graaf, W.T.A. Metastasis in Soft Tissue Sarcomas: Prognostic Criteria and Treatment Perspectives. Cancer Metastasis Rev. 2002, 21, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Crettenand, F.; Martin, D.; Cherix, S.; Demartines, N.; Matter, M. Occurrence and prognosis of lymph node metastases in patients selected for isolated limb perfusion with soft tissue sarcoma. J. Cancer 2018, 9, 3311–3315. [Google Scholar] [CrossRef] [PubMed]
- Emori, M.; Tsuchie, H.; Nagasawa, H.; Sonoda, T.; Tsukamoto, A.; Shimizu, J.; Murahashi, Y.; Mizushima, E.; Takada, K.; Murase, K.; et al. Early Lymph Node Metastasis May Predict Poor Prognosis in Soft Tissue Sarcoma. Int. J. Surg. Oncol. 2019, 2019, 6708474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamenovic, D.; Hohenberger, P.; Roessner, E. Pulmonary metastasectomy in soft tissue sarcomas: A systematic review. J. Thorac. Dis. 2021, 13, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Matsuoka, M.; Soma, T.; Arai, R.; Kato, H.; Harabayashi, T.; Adachi, H.; Shinohara, T.; Sagawa, T.; Nishiyama, N.; et al. Metastases of soft tissue sarcoma to the liver: A Historical Cohort Study from a Hospital-based Cancer Registry. Cancer Med. 2020, 9, 6159–6165. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.M.; Lindsay, A.D.; Spiguel, A.R.; Scarborough, M.T.; Gibbs, C.P. Brain metastases from Truncal and extremity bone and soft tissue sarcoma: Single institution study of oncologic outcomes. Rare Tumors 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Younis, M.H.; Summers, S.; Pretell-Mazzini, J. Bone metastasis in extremity soft tissue sarcomas: Risk factors and survival analysis using the SEER registry. Musculoskelet. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- de Juan Ferré, A.; Álvarez Álvarez, R.; Casado Herráez, A.; Cruz Jurado, J.; Estival González, A.; Martín-Broto, J.; Martínez Marín, V.; Moreno Vega, A.; Sebio García, A.; Valverde Morales, C. SEOM Clinical Guideline of management of soft-tissue sarcoma (2020). Clin. Transl. Oncol. 2021, 23, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Sambri, A.; Caldari, E.; Fiore, M.; Zucchini, R.; Giannini, C.; Pirini, M.G.; Spinnato, P.; Cappelli, A.; Donati, D.M.; De Paolis, M. Margin Assessment in Soft Tissue Sarcomas: Review of the Literature. Cancers 2021, 13, 1687. [Google Scholar] [CrossRef] [PubMed]
- Spolverato, G.; Callegaro, D.; Gronchi, A. Defining Which Patients Are at High Risk for Recurrence of Soft Tissue Sarcoma. Curr. Treat. Options Oncol. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Wiltink, L.M.; Haas, R.L.M.; Gelderblom, H.; van de Sande, M.A.J. Treatment Strategies for Metastatic Soft Tissue Sarcomas. Cancers 2021, 13, 1722. [Google Scholar] [CrossRef]
- Rehders, A.; Stoecklein, N.H.; Poremba, C.; Alexander, A.; Knoefel, W.T.; Peiper, M. Reexcision of soft tissue sarcoma: Sufficient local control but increased rate of metastasis. World J. Surg. 2009, 33, 2599–2605. [Google Scholar] [CrossRef] [PubMed]
- Bartelstein, M.K.; Yerramilli, D.; Christ, A.B.; Kenan, S.; Ogura, K.; Fujiwara, T.; Fabbri, N.; Healey, J.H. Postradiation Fractures after Combined Modality Treatment in Extremity Soft Tissue Sarcomas. Sarcoma 2021, 2021, 8877567. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.; Verma, V.; Takiar, R.; Vajapey, R.; Amarnath, S.; Murphy, E.; Mesko, N.W.; Lietman, S.; Joyce, M.; Anderson, P.; et al. Radiation Therapy in the Management of Soft Tissue Sarcoma: A Clinician’s Guide to Timing, Techniques, and Targets. Am. J. Clin. Oncol. 2016, 39, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Oh, R.J.; Miura, H.; Masai, N.; Shiomi, H.; Inoue, T. Outcomes and toxicity of radiotherapy for refractory bone and soft tissue sarcomas. Mol. Clin. Oncol. 2016, 4, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, A.; Gupta, V.G.; Bakhshi, S. Newer medical therapies for metastatic soft tissue sarcoma. Expert Rev. Anticancer Ther. 2017, 17, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.; Patel, S.R. Chemotherapy for soft tissue sarcoma. Cancer 2016, 122, 2952–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronchi, A. Surgery in soft tissue sarcoma: The thin line between a surgical or more conservative approach. Future Oncol. 2021, 17, 3–6. [Google Scholar] [CrossRef]
- Morgan, S.S.; Cranmer, L.D. Systematic therapy for unresectable or metastatic soft-tissue sarcomas: Past, present, and future. Curr. Oncol. Rep. 2011, 13, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Nixon, N.A.; Blais, N.; Ernst, S.; Kollmannsberger, C.; Bebb, G.; Butler, M.; Smylie, M.; Verma, S. Current landscape of immunotherapy in the treatment of solid tumours, with future opportunities and challenges. Curr. Oncol. 2018, 25, e373–e384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovelli, R.; Ciccarese, C.; Schutz, F.A.; Tortora, G.; de Velasco, G. Complete response to immune checkpoint inhibitors-based therapy in advanced renal cell carcinoma patients. A meta-analysis of randomized clinical trials. Urol. Oncol. 2020, 38, 798.e717–798.e724. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Zhang, L.; Tong, G.; Ou, Z.; Wang, Z.; Zhang, H.; Qiao, G. Pathologic complete response to preoperative immunotherapy in a lung adenocarcinoma patient with bone metastasis: A case report. Thorac. Cancer 2020, 11, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutkin, P.M.; Hiniker, S.M.; Swetter, S.M.; Reddy, S.A.; Knox, S.J. Complete Response of Metastatic Melanoma to Local Radiation and Immunotherapy: 6.5 Year Follow-Up. Cureus 2018, 10, e3723. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.-M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumarianou, A.; Duran-Moreno, J. The Sarcoma Immune Landscape: Emerging Challenges, Prognostic Significance and Prospective Impact for Immunotherapy Approaches. Cancers 2021, 13, 363. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Mowery, Y.M.; Riedel, R.F.; Kirsch, D.G. Rationale and emerging strategies for immune checkpoint blockade in soft tissue sarcoma. Cancer 2018, 124, 3819–3829. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zeng, W.; Kong, W.; Shi, Y.; Mou, X. The Study of Sarcoma Microenvironment Heterogeneity Associated With Prognosis Based on an Immunogenomic Landscape Analysis. Front. Bioeng. Biotechnol. 2020, 8, 1003. [Google Scholar] [CrossRef] [PubMed]
- Levine, L.S.; Mahuron, K.M.; Tsai, K.K.; Wu, C.; Mattis, D.M.; Pauli, M.L.; Oglesby, A.; Lee, J.C.; Spitzer, M.H.; Krummel, M.F.; et al. Tumor Immune Profiling-Based Neoadjuvant Immunotherapy for Locally Advanced Melanoma. Ann. Surg. Oncol. 2020, 27, 4122–4130. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.F.; Omar, H.A. Immunotherapy, an evolving approach for the management of triple negative breast cancer: Converting non-responders to responders. Crit. Rev. Oncol. Hematol. 2018, 122, 202–207. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. CMLS 2018, 75, 689–713. [Google Scholar] [CrossRef] [Green Version]
- Klaver, Y.; Rijnders, M.; Oostvogels, A.; Wijers, R.; Smid, M.; Grünhagen, D.; Verhoef, C.; Sleijfer, S.; Lamers, C.; Debets, R. Differential quantities of immune checkpoint-expressing CD8 T cells in soft tissue sarcoma subtypes. J. Immunother. Cancer 2020, 8, e000271. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef] [Green Version]
- Italiano, A.; Bellera, C.; D’Angelo, S. PD1/PD-L1 targeting in advanced soft-tissue sarcomas: A pooled analysis of phase II trials. J. Hematol. Oncol. 2020, 13, 55. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.-X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Akiyama, R.; Huang, R.R.; Shintaku, I.P.; Wang, X.; Tumeh, P.C.; Singh, A.; Chmielowski, B.; Denny, C.; Federman, N.; et al. Infiltration of CD8 T Cells and Expression of PD-1 and PD-L1 in Synovial Sarcoma. Cancer Immunol. Res. 2017, 5, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 3360. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, H.; Wu, N.; Cong, L.; Cong, X. Prognostic Significance of Tumor-Infiltrating Lymphocyte Grade in Melanoma: A Meta-Analysis. Dermatology 2020, 236, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Lequerica-Fernández, P.; Suárez-Canto, J.; Rodriguez-Santamarta, T.; Rodrigo, J.P.; Suárez-Sánchez, F.J.; Blanco-Lorenzo, V.; Domínguez-Iglesias, F.; García-Pedrero, J.M.; de Vicente, J.C. Prognostic Relevance of CD4+, CD8+ and FOXP3+ TILs in Oral Squamous Cell Carcinoma and Correlations with PD-L1 and Cancer Stem Cell Markers. Biomedicines 2021, 9, 653. [Google Scholar] [CrossRef]
- Rodrigo, J.P.; Sánchez-Canteli, M.; López, F.; Wolf, G.T.; Hernández-Prera, J.C.; Williams, M.D.; Willems, S.M.; Franchi, A.; Coca-Pelaz, A.; Ferlito, A. Tumor-Infiltrating Lymphocytes in the Tumor Microenvironment of Laryngeal Squamous Cell Carcinoma: Systematic Review and Meta-Analysis. Biomedicines 2021, 9, 486. [Google Scholar] [CrossRef] [PubMed]
- Strizova, Z.; Bartunkova, J.; Smrz, D. The challenges of adoptive cell transfer in the treatment of human renal cell carcinoma. Cancer Immunol. Immunother. CII 2019, 68, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Wustrack, R.L.; Shao, E.; Sheridan, J.; Zimel, M.; Cho, S.-J.; Horvai, A.E.; Luong, D.; Kwek, S.S.; Fong, L.; Okimoto, R.A. Tumor morphology and location associate with immune cell composition in pleomorphic sarcoma. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; von Mehren, M.; Van Tine, B.A. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234–12247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Q.; Liu, Y.; Yuan, T.; Wang, H.; Li, B.; Jiang, Y.; Mo, X.; Lei, Y.; Xiao, Y.; Dong, S.; et al. Predicted CD4+ T cell infiltration levels could indicate better overall survival in sarcoma patients. J. Int. Med Res. 2021, 49, 0300060520981539. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, M.; Bolognesi, M.M.; Antoranz, A.; Mancari, R.; Carinelli, S.; Faretta, M.; Bosisio, F.M.; Cattoretti, G. The Adaptive and Innate Immune Cell Landscape of Uterine Leiomyosarcomas. Sci. Rep. 2020, 10, 702. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.; Kim, Y.-M.; Kim, S.-B.; Kim, C.-S.; Kwon, S.-W.; Kim, Y.; Kim, H.; Lee, H. Therapeutic DC vaccination with IL-2 as a consolidation therapy for ovarian cancer patients: A phase I/II trial. Cell. Mol. Immunol. 2015, 12, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, A.; López, M.; Serrano, A.; Ramirez, M.; Pérez, C.; Aguirre, A.; González, R.; Alfaro, J.; Larrondo, M.; Fodor, M.; et al. Dendritic cell immunizations alone or combined with low doses of interleukin-2 induce specific immune responses in melanoma patients. Clin. Exp. Immunol. 2005, 142, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Desbois, M.; Béal, C.; Charrier, M.; Besse, B.; Meurice, G.; Cagnard, N.; Jacques, Y.; Béchard, D.; Cassard, L.; Chaput, N. IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: Implications for antimetastatic treatment. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, S.H.; Heo, Y.S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, R.; Sugiura, D.; Shimizu, K.; Maruhashi, T.; Watada, M.; Okazaki, I.-M.; Okazaki, T. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front. Immunol. 2019, 10, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet. Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet. Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37 (Suppl. 1), 95–100. [Google Scholar] [CrossRef] [Green Version]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolle, M.A.; Herbsthofer, L.; Granegger, B.; Goda, M.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; Pichler, M.; Gerger, A.; et al. T-regulatory cells predict clinical outcome in soft tissue sarcoma patients: A clinico-pathological study. Br. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Tsai, J.-W.; Ali, A.M.; Cormier, J.N.; Bishop, A.J.; Guadagnolo, B.A.; Torres, K.E.; Somaiah, N.; Hunt, K.K.; Wargo, J.A.; et al. Analysis of the immune infiltrate in undifferentiated pleomorphic sarcoma of the extremity and trunk in response to radiotherapy: Rationale for combination neoadjuvant immune checkpoint inhibition and radiotherapy. OncoImmunology 2018, 7, e1385689. [Google Scholar] [CrossRef]
- Que, Y.; Xiao, W.; Guan, Y.X.; Liang, Y.; Yan, S.M.; Chen, H.Y.; Li, Q.Q.; Xu, B.S.; Zhou, Z.W.; Zhang, X. PD-L1 Expression Is Associated with FOXP3+ Regulatory T-Cell Infiltration of Soft Tissue Sarcoma and Poor Patient Prognosis. J. Cancer 2017, 8, 2018–2025. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Doberstein, S.K. Bempegaldesleukin (NKTR-214): A CD-122-biased IL-2 receptor agonist for cancer immunotherapy. Expert Opin. Biol. Ther. 2019, 19, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Chua, H.L.; Serov, Y.; Brahmi, Z. Regulation of FasL expression in natural killer cells. Hum. Immunol. 2004, 65, 317–327. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Smyth, M.J.; Trapani, J.A. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 2006, 6, 940–952. [Google Scholar] [CrossRef]
- Gaggero, S.; Witt, K.; Carlsten, M.; Mitra, S. Cytokines Orchestrating the Natural Killer-Myeloid Cell Crosstalk in the Tumor Microenvironment: Implications for Natural Killer Cell-Based Cancer Immunotherapy. Front. Immunol. 2020, 11, 621225. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Carlsten, M.; Björklund, A.; Sohlberg, E.; Bryceson, Y.T.; Ljunggren, H.G. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 2017, 31, 20–29. [Google Scholar] [CrossRef]
- Sorbye, S.W.; Kilvaer, T.K.; Valkov, A.; Donnem, T.; Smeland, E.; Al-Shibli, K.; Bremnes, R.M.; Busund, L.-T. Prognostic impact of CD57, CD68, M-CSF, CSF-1R, Ki67 and TGF-beta in soft tissue sarcomas. BMC Clin. Pathol. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bücklein, V.; Adunka, T.; Mendler, A.N.; Issels, R.; Subklewe, M.; Schmollinger, J.C.; Noessner, E. Progressive natural killer cell dysfunction associated with alterations in subset proportions and receptor expression in soft-tissue sarcoma patients. OncoImmunology 2016, 5, e1178421. [Google Scholar] [CrossRef] [Green Version]
- Judge, S.J.; Darrow, M.A.; Thorpe, S.W.; Gingrich, A.A.; O’Donnell, E.F.; Bellini, A.R.; Sturgill, I.R.; Vick, L.V.; Dunai, C.; Stoffel, K.M.; et al. Analysis of tumor-infiltrating NK and T cells highlights IL-15 stimulation and TIGIT blockade as a combination immunotherapy strategy for soft tissue sarcomas. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wen, B.; Anton, O.M.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V.; et al. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E10915–E10924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal. Transduct. Target. Ther. 2020, 29, 250. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanuza, P.M.; Pesini, C.; Arias, M.A.; Calvo, C.; Ramirez-Labrada, A.; Pardo, J. Recalling the Biological Significance of Immune Checkpoints on NK Cells: A Chance to Overcome LAG3, PD1, and CTLA4 Inhibitory Pathways by Adoptive NK Cell Transfer? Front. Immunol. 2020, 10, 3010. [Google Scholar] [CrossRef] [Green Version]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Pinette, A.; McMichael, E.; Courtney, N.B.; Duggan, M.; Benner, B.N.; Choueiry, F.; Yu, L.; Abood, D.; Mace, T.A.; Carson, W.E., 3rd. An IL-15-based superagonist ALT-803 enhances the NK cell response to cetuximab-treated squamous cell carcinoma of the head and neck. Cancer Immunol. Immunother. CII 2019, 68, 1379–1389. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hatano, H.; Ariizumi, T.; Sasaki, T.; Yamagishi, T.; Umezu, H.; Endo, N. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci. 2018, 109, 3043–3054. [Google Scholar] [CrossRef] [Green Version]
- Tsagozis, P.; Augsten, M.; Zhang, Y.; Li, T.; Hesla, A.; Bergh, J.; Haglund, F.; Tobin, N.P.; Ehnman, M. An immunosuppressive macrophage profile attenuates the prognostic impact of CD20-positive B cells in human soft tissue sarcoma. Cancer Immunol. Immunother. CII 2019, 68, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Raj, S.K.; Kooshki, M.; Winters, M.; Russell, G.B.; Miller, L.D.; Laurini, J.A.; Pierre, T.; Savage, P.D. Prognostic implications of tumor associated macrophages (TAMs) in soft tissue sarcoma. J. Clin. Oncol. 2019, 37, e22548. [Google Scholar] [CrossRef]
- Lee, C.H.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef] [Green Version]
- Ganjoo, K.N.; Witten, D.; Patel, M.; Espinosa, I.; La, T.; Tibshirani, R.; van de Rijn, M.; Jacobs, C.; West, R.B. The prognostic value of tumor-associated macrophages in leiomyosarcoma: A single institution study. Am. J. Clin. Oncol. 2011, 34, 82–86. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef] [Green Version]
- Nabeshima, A.; Matsumoto, Y.; Fukushi, J.; Iura, K.; Matsunobu, T.; Endo, M.; Fujiwara, T.; Iida, K.; Fujiwara, Y.; Hatano, M.; et al. Tumour-associated macrophages correlate with poor prognosis in myxoid liposarcoma and promote cell motility and invasion via the HB-EGF-EGFR-PI3K/Akt pathways. Br. J. Cancer 2015, 112, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for Soft Tissue Sarcoma: Current Status and Future Perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scurr, M. Histology-driven chemotherapy in soft tissue sarcomas. Curr. Treat. Options Oncol. 2011, 12, 32–45. [Google Scholar] [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumor-Associated Macrophages in Sarcomas. Cancers 2021, 13, 1086. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.; Palmerini, E.; Pollack, S.M. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncol. Annu. Meet. 2018, 38, 925–938. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Bui, N.; Bolleddu, S.; Lohman, M.; Becker, H.C.; Ganjoo, K. Nivolumab plus ipilimumab for soft tissue sarcoma: A single institution retrospective review. Immunotherapy 2020, 12, 1303–1312. [Google Scholar] [CrossRef]
- Ayodele, O.; Razak, A.R.A. Immunotherapy in soft-tissue sarcoma. Curr. Oncol. 2020, 27, 17–23. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]



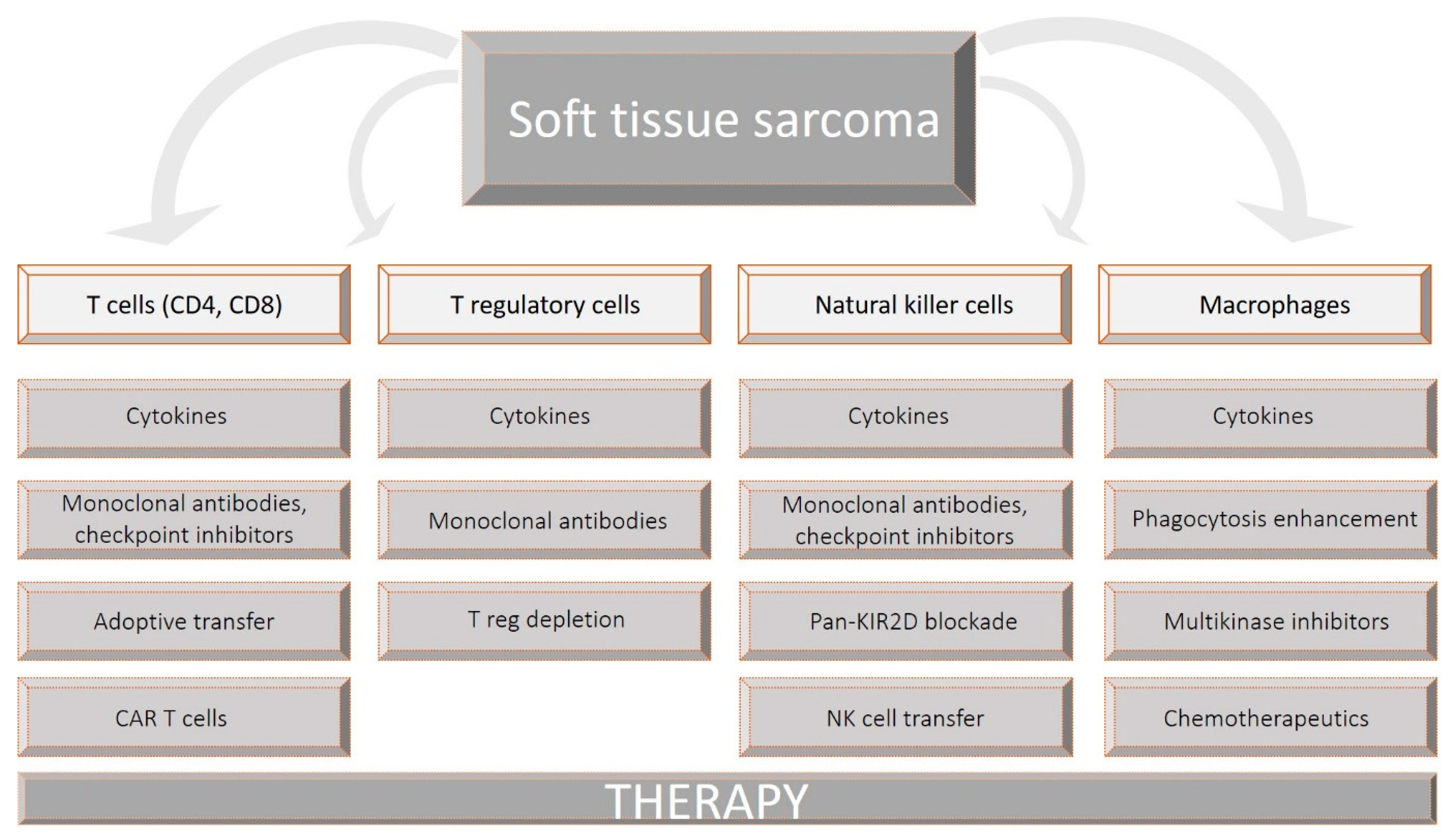{kind=link}
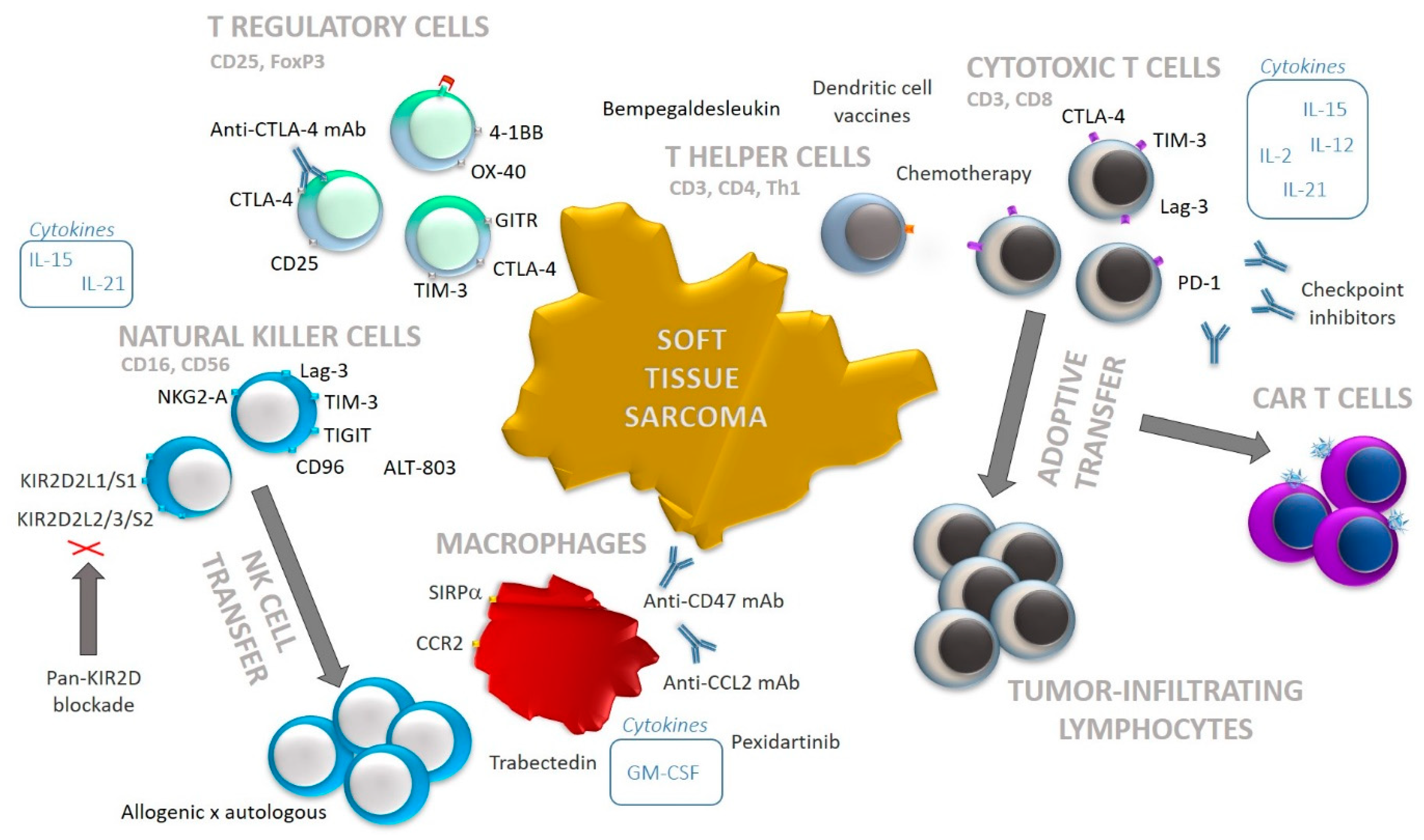{kind=link}
| Phase II + III Clinical Trials | ||||||
|---|---|---|---|---|---|---|
| Drug Name | Diagnosis | Trial Design | Setting | CPI Dosage Regimen | Estimated Study Completion | Identifier |
| Nivolumab | Soft tissue sarcoma | Non-randomized | Combination therapy: Ipilimumab, Cryoablation | 3 mg/kg every 3 weeks x 4 doses. | October 2025 | NCT04118166 |
| Nivolumab | Soft tissue sarcoma | Randomized | Combination therapy: Relatlimab | 240 mg every 2 weeks | September 2024 | NCT04095208 |
| Nivolumab | Soft tissue sarcoma | Non-randomized | Combination therapy: Trabectedin | 240 mg every 3 weeks | October 2022 | NCT03590210 |
| Nivolumab | Soft tissue sarcoma | Randomized | Combination: Cabozantinib, Ipilimumab | 3 mg/kg every 3 weeks x 4 doses, followed by 480 mg every 4 weeks | January 2027 | NCT04551430 |
| Nivolumab | Sarcoma, Desmoid, Chondroma | Non-randomized | Combination: Trabectedin, Talimogene Laherparepvec | 240 mg every 2 weeks | December 2022 | NCT03886311 |
| Nivolumab | Advanced/metastatic sarcoma | Non-randomized | Combination: NKTR-214 | 360 mg every 3 weeks | September 2023 | NCT03282344 |
| Nivolumab | Advanced/metastatic sarcoma | Non-randomized | Combination: Trabectedin, Ipilimumab | 3 mg/kg every 2 weeks up to 26 doses | March 2022 | NCT03138161 |
| Nivolumab | Advanced/metastatic sarcoma | Non-randomized | Combination: Gemcitabine, Doxorubicin, Docetaxel | 240 mg IV on Day 1 of each cycle | December 2025 | NCT04535713 |
| Nivolumab | Soft tissue sarcoma | Non-randomized | Combination: Sunitinib | 240 mg every 2 weeks | September 2022 | NCT03277924 |
| Nivolumab | Recurrent/refractory sarcoma | Non-randomized | Monotherapy | 240 mg every 2 weeks | March 2029 | NCT03465592 |
| Nivolumab | Resectable or recurrent dedifferentiated/undifferentiated pleomorfic sarcoma | Randomized | A: monotherapy; B combination with Ipilimumab; C combination with RT; D combination with Ipilimumab and RT | IV on days 1, 15 and 29 in A, B; IV over 1 h on days 1, 15, 29 and 43 in C, D | October 2021 | NCT03307616 |
| Nivolumab | Angiosarcoma | Randomized | A: nivolumab, paclitaxel; B paclitaxel; C: nivolumab, cabozantinib S-malate | I.V. on Day 1 of each cycle, cycles repeat every 4 weeks | September 2023 | NCT04339738 |
| Nivolumab | Soft tissue sarcoma | Non-randomized | Combination: AL3818 | 240 mg every 2 weeks | December 2022 | NCT04165330 |
| Nivolumab | Epitheloid sarcoma | Non-randomized | Combination: Ipilimumab | Nivo and Ipi at predetermined dosage day 1 of a 21-day cycle for 4 cycles. | October 2025 | NCT04416568 |
| Nivolumab | Uterine sarcomas | Non-randomized | Monotherapy | 480 mg IV once every 4 weeks | August 2022 | NCT03241745 |
| Nivolumab | Soft tissue sarcoma | Non-randomized | Combination: BA3011 | Unspecified | January 2022 | NCT03425279 |
| Nivolumab | Sarcoma 2nd-line and relapsed/refractory | Non-randomized | Combination: NKTR-262, bempegaldesleukin | 360 mg every 3 weeks | December 2021 | NCT03435640 |
| Nivolumab | Leiomyosarcoma | Non-randomized | Combination: Rucaparib | 480 mg i.v. on day 1 of every four-week cycle | November 2022 | NCT04624178 |
| Nivolumab | Angiosarcoma, endometrial carcinosarcoma | Non-randomized | Monotherapy, Combination: Ipilimumab | Unspecified | August 2021 | NCT02834013 |
| Nivolumab | Advanced/metastatic sarcoma | Randomized | Combination: Ipilimumab | Nivolumab: 3 mg/kg i.v. every 2 weeks for 4 cycles; Ipilimumab 1 mg/kg IV over 60 min every 6 weeks for 4 cycles | August 2025 |  NCT04741438 |
| Pembrolizumab | Advanced sarcoma | Non-randomized | Combination: Lenvatinib | 200 mg as a 30-min IV infusion, Q3W +/−3 days | March 2024 | NCT04784247 |
| Pembrolizumab | Advanced sarcoma | Non-randomized | Combination: Metronomic Cyclophosphamide | 200 mg every 3 weeks on day 8 for 3 weeks | August 2021 | NCT02406781 |
| Pembrolizumab | Soft tissue sarcoma of the Extremity | Randomized | Combination: Radiotherapy | 200 mg i.v. every 3 weeks | July 2025 | NCT03092323 |
| Pembrolizumab | Soft tissue sarcoma | Non-randomized | Combination: Axitinib | 200 mg i.v. infusion every 21 weeks, max up to 2 years | December 2022 | NCT02636725 |
| Pembrolizumab | Advanced/metastatic sarcoma | Non-randomized | Combination: Epacadostat | 200 mg/dose Day 1, Q 3 weeks | January 2022 | NCT03414229 |
| Pembrolizumab | Soft tissue sarcoma | Non-randomized | Combination: Eribulin | Pembrolizumab every 3 weeks | August 2024 | NCT03899805 |
| Pembrolizumab | Advanced/metastatic soft tissue sarcoma | Non-randomized | Combination: Doxorubicin | 200 mg intravenously every 3 weeks | February 2025 | NCT03056001 |
| Pembrolizumab | Soft tissue sarcoma | Non-randomized | Combination: Radiotherapy | i.v. every 3 weeks for 3 months | June 2023 | NCT03338959 |
| Pembrolizumab | Advanced/metastatic sarcoma | Non-randomized | Combination: Alimogene Laherparepvec (T-VEC) | Every 3 weeks | March 2022 | NCT03069378 |
| Pembrolizumab | Sarcoma of extremities | Non-randomized | Combination: Isolated Limb infusion (ILI) | 200 mg i.v. every 3 weeks | April 2023 | NCT04332874 |
| Pembrolizumab | Leiomyosarcoma and Undifferentiated Pleomorphic Sarcoma | Non-randomized | Combination: Gemcitabine | 200 mg every 3 weeks | December 2020 | NCT03123276 |
| Pembrolizumab | Advanced/metastatic Synovial Sarcoma | Non-randomized | Combination: Interferon gamma-1b | 200 mg i.v. every 3 weeks | April 2022 | NCT03063632 |
| Pembrolizumab | Soft tissue sarcoma | Non-randomized | Combination: Intra-tumoral BT-001 | 200 mg intravenously every 3 weeks | November 2024 | NCT04725331 |
| Pembrolizumab | Sarcoma | Non-randomized | Monotherapy | 200 mg i.v. every 3 weeks | December 2023 | NCT03012620 |
| Ipilimumab | Undifferentiated Pleomorphic Sarcoma Or Myxofibrosarcoma | Randomized | Combination: Envafolimab | 1 mg/kg every 3 weeks for a total of 4 doses | July 2022 | NCT04480502 |
| Ipilimumab | Soft tissue sarcoma | Non-randomized | Combination: Aldesleukin, nivolumab, fludarabine, cyclophosphamide | Unspecified | June 2024 | NCT03449108 |
| Ipilimumab | Sarcoma | Non-randomized | Combination: INT230-6 | Day 1 every 3 weeks for four treatments | July 2022 | NCT03058289 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozaniak, A.; Vachtenheim, J., Jr.; Lischke, R.; Bartunkova, J.; Strizova, Z. Novel Insights into the Immunotherapy of Soft Tissue Sarcomas: Do We Need a Change of Perspective? Biomedicines 2021, 9, 935. https://doi.org/10.3390/biomedicines9080935
Ozaniak A, Vachtenheim J Jr., Lischke R, Bartunkova J, Strizova Z. Novel Insights into the Immunotherapy of Soft Tissue Sarcomas: Do We Need a Change of Perspective? Biomedicines. 2021; 9(8):935. https://doi.org/10.3390/biomedicines9080935
Chicago/Turabian StyleOzaniak, Andrej, Jiri Vachtenheim, Jr., Robert Lischke, Jirina Bartunkova, and Zuzana Strizova. 2021. "Novel Insights into the Immunotherapy of Soft Tissue Sarcomas: Do We Need a Change of Perspective?" Biomedicines 9, no. 8: 935. https://doi.org/10.3390/biomedicines9080935