
Expression Profile and Prognostic Value of Wnt Signaling Pathway Molecules in Colorectal Cancer

 ,
, 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culture Method, and Reagents
2.2. Cell Transfections
2.3. Cell Viability Assay
2.4. Focal Formation Assays
2.5. Quantitative Reverse-Transcription Polymerase Chain Reaction
2.6. CellMiner Data Mining and Analysis
2.7. UALCAN Analysis
2.8. Oncomine Database Analysis
2.9. Survival Analysis
2.10. Gene Ontology, Functional, and Signaling Pathway Analyses
2.11. cBioPortal Analyses
2.12. Gene Expression Profiling Interactive Analysis
2.13. Analysis of CTNNB1 and Its Corresponding Target Genes in Multiple CRC Clinical Datasets
2.14. Statistical Analysis
3. Results
3.1. mRNA Expression of Top 10 Upregulated CRC-Associated Genes in Various Human Cancer Cell Lines and Tissues
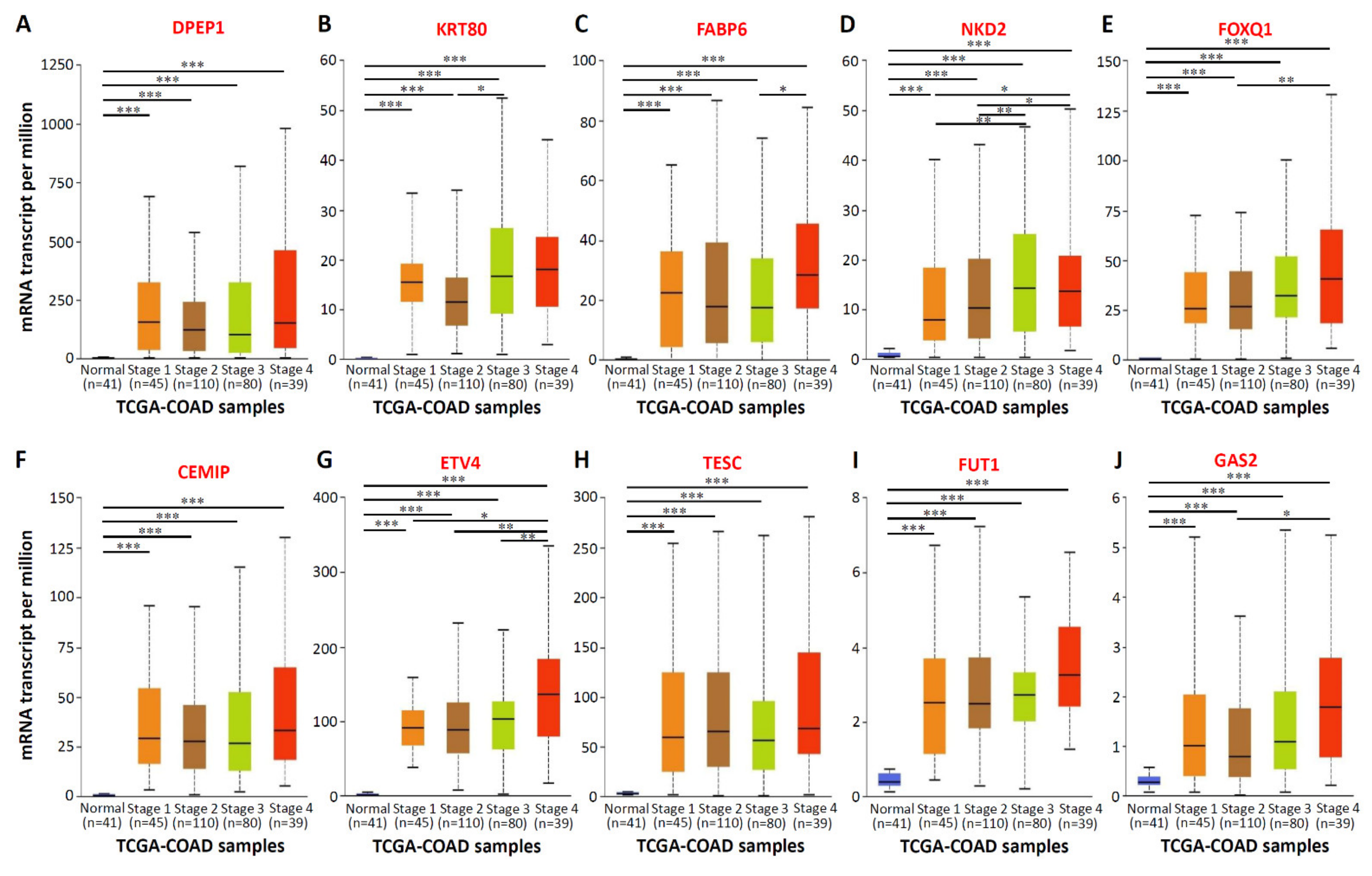
3.2. mRNA Expression and Clinical Relevance of the Top 10 Upregulated CRC-Associated Genes in CRC Tissues and Patients
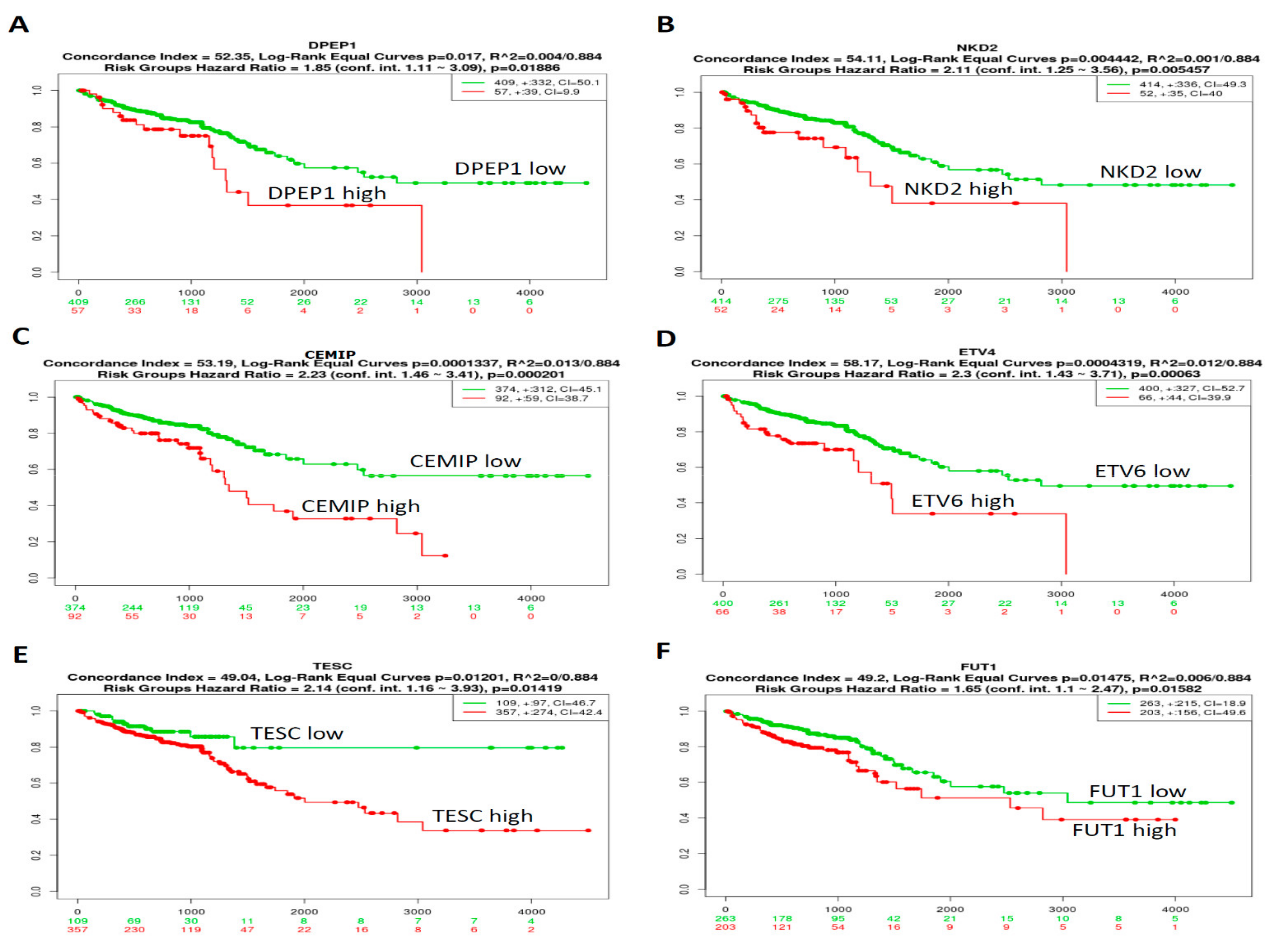
3.3. Prognosis of the Top 10 Upregulated CRC-Associated Genes in CRC Samples Was Analyzed Using Kaplan–Meier Analyses
3.4. Molecular Network and Functional Enrichment Analyses of the Top 10 Upregulated CRC-Associated Genes
3.5. Top 10 Upregulated CRC-Associated Genes in Genetic Alterations and Their Correlation with CTNNB1 in CRC
3.6. CTNNB1 Plays the Central Role to Regulate NKD2, FOXQ1, and CEMIP Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. Ca-Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppede, F. Genetics, Cytogenetics, and Epigenetics of Colorectal Cancer. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarkavelis, G.; Boussios, S.; Papadaki, A.; Katsanos, K.H.; Christodoulou, D.K.; Pentheroudakis, G. Current and future biomarkers in colorectal cancer. Ann. Gastroenterol. 2017, 30, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xuan, Y.; Gao, B.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Jiao, W. Identification of an eight-gene prognostic signature for lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Ferreira, C.G. Colorectal Cancer: Epidemiology, Disease Mechanisms and Interventions to Reduce Onset and Mortality. Clin. Colorectal. Cancer 2016, 15, 195–203. [Google Scholar] [CrossRef]
- Passardi, A.; Scarpi, E.; Ulivi, P. Molecular and Translational Research on Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 4105. [Google Scholar] [CrossRef]
- Grzadkowski, M.R.; Sendorek, D.H.; P’ng, C.; Huang, V.; Boutros, P.C. A comparative study of survival models for breast cancer prognostication revisited: The benefits of multi-gene models. BMC Bioinform. 2018, 19, 400. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Tang, W.C.; Cheng, Y.W.; Sia, P.; Huang, C.C.; Lee, Y.C.; Jiang, H.Y.; Wu, M.H.; Lai, I.L.; Lee, J.W.; et al. Targeting of multiple oncogenic signaling pathways by Hsp90 inhibitor alone or in combination with berberine for treatment of colorectal cancer. Biochim. Biophys. Acta 2015, 1853, 2261–2272. [Google Scholar] [CrossRef] [Green Version]
- Hung, K.C.; Huang, T.C.; Cheng, C.H.; Cheng, Y.W.; Lin, D.Y.; Fan, J.J.; Lee, K.H. The Expression Profile and Prognostic Significance of Metallothionein Genes in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Yang, J. Identification of Genes Related to Clinicopathological Characteristics and Prognosis of Patients with Colorectal Cancer. DNA Cell Biol. 2020, 39, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]
- Ambler, G.; Seaman, S.; Omar, R.Z. An evaluation of penalised survival methods for developing prognostic models with rare events. Stat Med. 2012, 31, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.N.; Wang, Z.C.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.F.; Li, C.W.; Kang, B.X.; Gao, G.; Li, C.; Zhang, Z.M. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Cortazar, A.R.; Torrano, V.; Martin-Martin, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martin, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Jorissen, R.N.; Gibbs, P.; Christie, M.; Prakash, S.; Lipton, L.; Desai, J.; Kerr, D.; Aaltonen, L.A.; Arango, D.; Kruhoffer, M.; et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin. Cancer Res. 2009, 15, 7642–7651. [Google Scholar] [CrossRef] [Green Version]
- Laibe, S.; Lagarde, A.; Ferrari, A.; Monges, G.; Birnbaum, D.; Olschwang, S.; Project, C.O.L. A seven-gene signature aggregates a subgroup of stage II colon cancers with stage III. OMICS 2012, 16, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Reinhold, W.C.; Sunshine, M.; Liu, H.; Varma, S.; Kohn, K.W.; Morris, J.; Doroshow, J.; Pommier, Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012, 72, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koni, M.; Pinnaro, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef]
- Sveen, A.; Kopetz, S.; Lothe, R.A. Biomarker-guided therapy for colorectal cancer: Strength in complexity. Nat. Rev. Clin. Oncol. 2020, 17, 11–32. [Google Scholar] [CrossRef]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204. [Google Scholar] [CrossRef] [Green Version]
- Van Es, J.H.; Barker, N.; Clevers, H. You Wnt some, you lose some: Oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 2003, 13, 28–33. [Google Scholar] [CrossRef]
- Chen, I.C.; Lee, K.H.; Hsu, Y.H.; Wang, W.R.; Chen, C.M.; Cheng, Y.W. Expression Pattern and Clinicopathological Relevance of the Indoleamine 2,3-Dioxygenase 1/Tryptophan 2,3-Dioxygenase Protein in Colorectal Cancer. Dis. Markers 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Yi, G.; Neelakandan, A.K.; Gontarek, B.C.; Vollbrecht, E.; Becraft, P.W. The naked endosperm Genes Encode Duplicate INDETERMINATE Domain Transcription Factors Required for Maize Endosperm Cell Patterning and Differentiation. Plant. Physiol. 2015, 167, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Obsil, T.; Obsilova, V. Structure/function relationships underlying regulation of FOXO transcription factors. Oncogene 2008, 27, 2263–2275. [Google Scholar] [CrossRef] [Green Version]
- Birkenkamp-Demtroder, K.; Maghnouj, A.; Mansilla, F.; Thorsen, K.; Andersen, C.L.; Oster, B.; Hahn, S.; Orntoft, T.F. Repression of KIAA1199 attenuates Wnt-signalling and decreases the proliferation of colon cancer cells. Brit. J. Cancer 2011, 105, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wang, P.L.; Huang, L.; Qiao, J.X.; Li, J.H. Bioinformatic analysis reveals an exosomal miRNA-mRNA network in colorectal cancer. BMC Med. Genom. 2021, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Scola, L.; Zanghi, A.; Biondi, A.; Di Cataldo, A.; Libra, M.; Candido, S. Integrated analysis of colorectal cancer microRNA datasets: Identification of microRNAs associated with tumor development. Aging 2018, 10, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.Y.; Chen, X.L.; Mo, S.Y.; Li, J.W.; Huang, Z.Z.; Huang, S.F.; Xu, J.Y.; He, B.Y.; Zou, Y.; Chen, J.F.; et al. Meta-signature LncRNAs serve as novel biomarkers for colorectal cancer: Integrated bioinformatics analysis, experimental validation and diagnostic evaluation. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, X.; Lu, Y.; Wu, J.Z.; Feng, J.F. DNA-methylated gene markers for colorectal cancer in TCGA database. Exp. Med. 2020, 19, 3042–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnov, G.S.; Kudryavtseva, A.V.; Snezhkina, A.V.; Lakunina, V.A.; Beniaminov, A.D.; Melnikova, N.V.; Dmitriev, A.A. Pan-Cancer Analysis of TCGA Data Revealed Promising Reference Genes for qPCR Normalization. Front. Genet. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ledesma, E.; Verhaak, R.G.W.; Trevino, V. Identification of a multi-cancer gene expression biomarker for cancer clinical outcomes using a network-based algorithm. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-F.; Wang, C.-Y.; Tang, W.-C.; Lee, Y.-C.; Ta, H.D.K.; Lin, L.-C.; Pan, S.-R.; Ni, Y.-C.; Anuraga, G.; Lee, K.-H. Expression Profile and Prognostic Value of Wnt Signaling Pathway Molecules in Colorectal Cancer. Biomedicines 2021, 9, 1331. https://doi.org/10.3390/biomedicines9101331
Wu Y-F, Wang C-Y, Tang W-C, Lee Y-C, Ta HDK, Lin L-C, Pan S-R, Ni Y-C, Anuraga G, Lee K-H. Expression Profile and Prognostic Value of Wnt Signaling Pathway Molecules in Colorectal Cancer. Biomedicines. 2021; 9(10):1331. https://doi.org/10.3390/biomedicines9101331
Chicago/Turabian StyleWu, Yung-Fu, Chih-Yang Wang, Wan-Chun Tang, Yu-Cheng Lee, Hoang Dang Khoa Ta, Li-Chia Lin, Syu-Ruei Pan, Yi-Chun Ni, Gangga Anuraga, and Kuen-Haur Lee. 2021. "Expression Profile and Prognostic Value of Wnt Signaling Pathway Molecules in Colorectal Cancer" Biomedicines 9, no. 10: 1331. https://doi.org/10.3390/biomedicines9101331