A Pathway-Based Genomic Approach to Identify Medications: Application to Alcohol Use Disorder


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Human Gene Expression Datasets
2.2. Drug Gene Expression Datasets
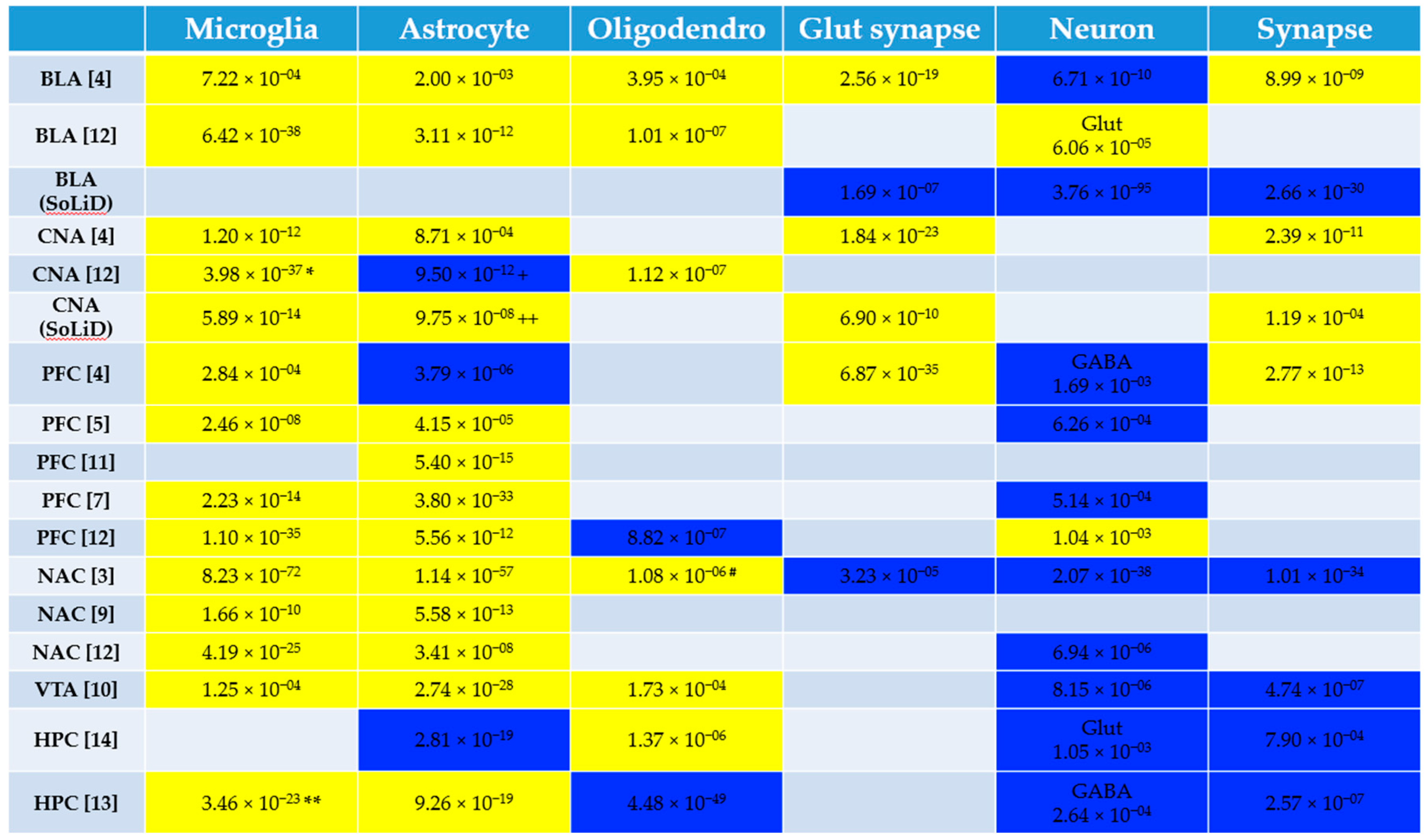
2.3. Pathway and Cell Type Enrichment Analysis
2.4. R Shiny Interactive Webpage
3. Results
3.1. Pathway and Cell Type Enrichment Analysis of Human Alcohol Gene Expression Datasets
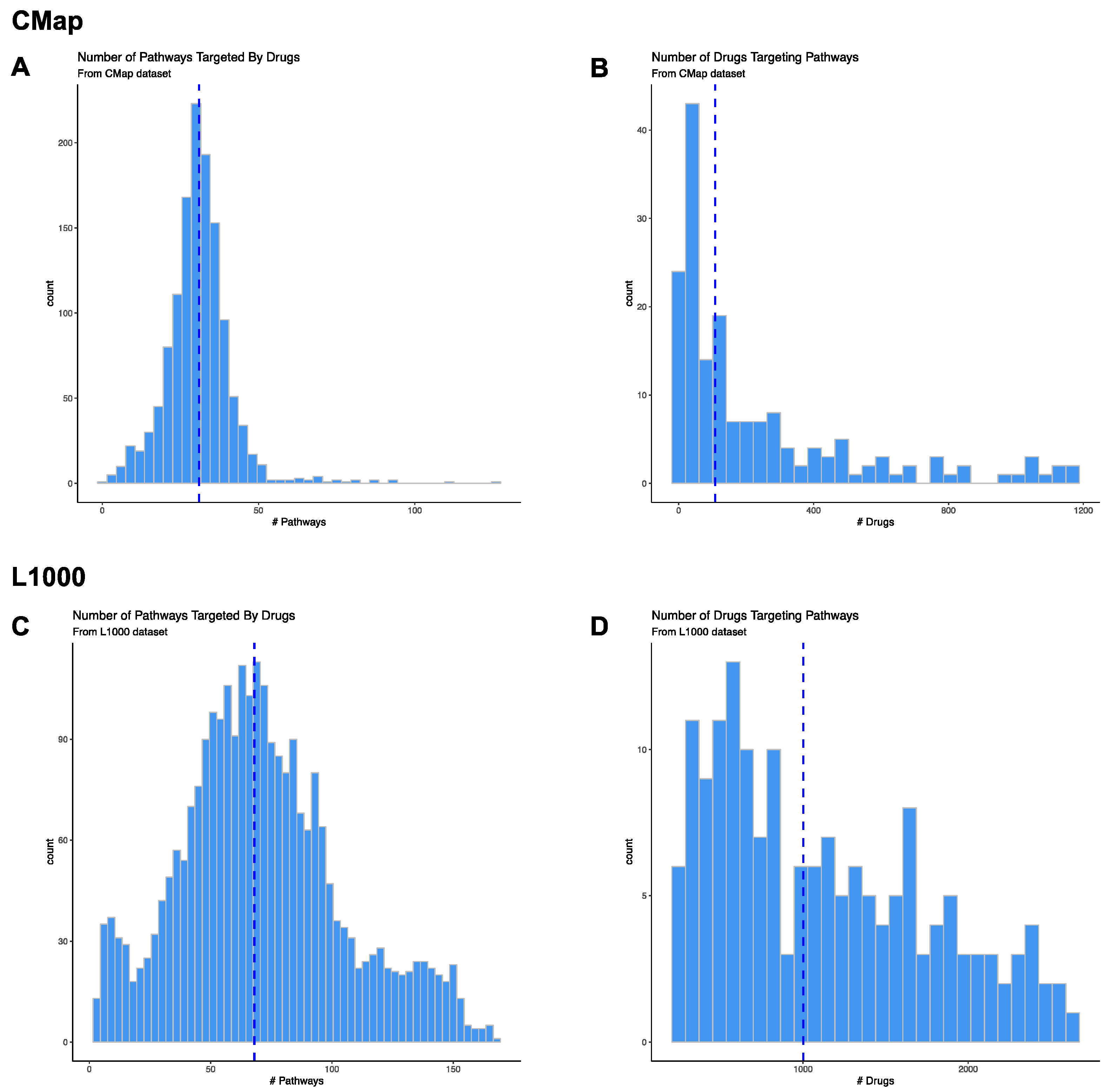
3.2. Pathway Level Analysis of Drug Gene Expression Datasets
3.2.1. CMap Pathways
3.2.2. CMap Drugs
3.2.3. L1000 Pathways
3.2.4. L1000 Drugs
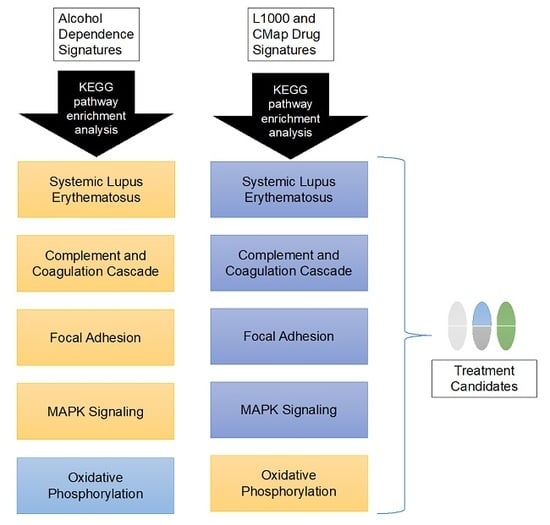
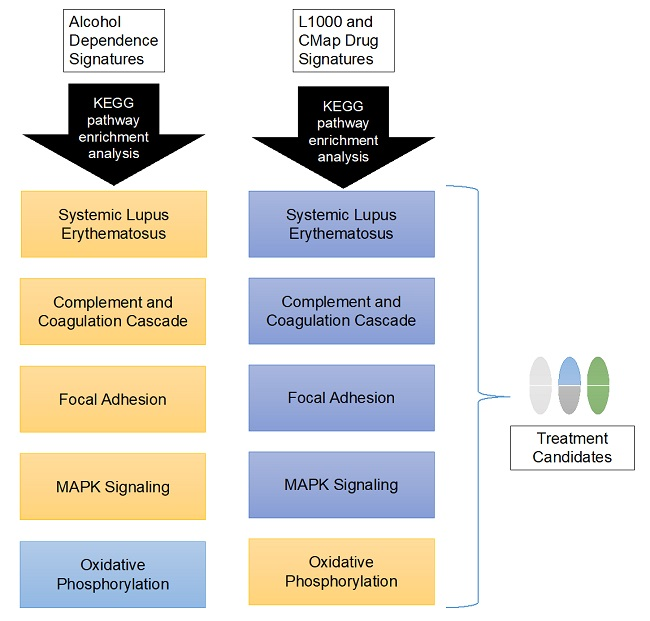
3.3. Candidate Treatment Selection
3.4. Interactive Website to Identify Compounds Predicted to Target Biological Pathways of Interest
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. DSM-5 Task Force. In Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013; p. 947. [Google Scholar]
- Grant, B.F.; Goldstein, R.B.; Saha, T.D.; Chou, S.P.; Jung, J.; Zhang, H.; Pickering, R.P.; Ruan, W.J.; Smith, S.M.; Huang, B.; et al. Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamdani, M.; Williamson, V.; McMichael, G.O.; Blevins, T.; Aliev, F.; Adkins, A.; Hack, L.; Bigdeli, T.; van der Vaart, A.D.; Web, B.T.; et al. Integrating mRNA and miRNA Weighted Gene Co-Expression Networks with eQTLs in the Nucleus Accumbens of Subjects with Alcohol Dependence. PLoS ONE 2015, 10, e0137671. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, I.; Wang, S.; Zhang, L.; Harris, R.A.; Mayfield, R.D. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 2012, 32, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Farris, S.P.; Arasappan, D.; Hunicke-Smith, S.; Harris, R.A.; Mayfield, R.D. Transcriptome organization for chronic alcohol abuse in human brain. Mol. Psychiatry 2015, 20, 1438–1447. [Google Scholar] [CrossRef] [Green Version]
- Farris, S.P.; Harris, R.A.; Ponomarev, I. Epigenetic modulation of brain gene networks for cocaine and alcohol abuse. Front. Neurosci. 2015, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Wang, J.C.; Farris, S.P.; Liu, Y.; McClintick, J.; Gupta, I.; Meyers, J.L.; Bertelsen, S.; Chao, M.; Nurnberger, J.; et al. Analysis of whole genome-transcriptomic organization in brain to identify genes associated with alcoholism. Transl. Psychiatry 2019, 9, 89. [Google Scholar] [CrossRef]
- Rao, X.; Thapa, K.S.; Chen, A.B.; Lin, H.; Gao, H.; Reiter, J.L.; Hargreaves, K.A.; Ipe, J.; Lai, D.; Xuei, X.; et al. Allele-Specific Expression and High-Throughput Reporter Assay Reveal Functional Variants in Human Brains with Alcohol Use Disorders. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Flatscher-Bader, T.; Harrison, E.; Matsumoto, I.; Wilce, P.A. Genes associated with alcohol abuse and tobacco smoking in the human nucleus accumbens and ventral tegmental area. Alcohol. Clin. Exp. Res. 2010, 34, 1291–1302. [Google Scholar] [CrossRef]
- Flatscher-Bader, T.; Zuvela, N.; Landis, N.; Wilce, P.A. Smoking and alcoholism target genes associated with plasticity and glutamate transmission in the human ventral tegmental area. Hum. Mol. Genet. 2008, 17, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, F.; Xu, H.; Liu, Y.; Liu, J.; Zhao, H.; Gelernter, J. Differentially co-expressed genes in postmortem prefrontal cortex of individuals with alcohol use disorders: Influence on alcohol metabolism-related pathways. Hum. Genet. 2014, 133, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Thapa, K.S.; Chen, A.B.; Lin, H.; Gao, H.; Reiter, J.L.; Hargreaves, K.A.; Ipe, J.; Lai, D.; Xuei, X.; et al. Allele-specific expression and high-throughput reporter assay reveal functional genetic variants associated with alcohol use disorders. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintick, J.N.; Xuei, X.; Tischfield, J.A.; Goate, A.; Foroud, T.; Wetherill, L.; Ehringer, M.A.; Edenberg, H.J. Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol 2013, 47, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Yuan, Q.; Mash, D.C.; Goldman, D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl. Acad. Sci. USA 2011, 108, 6626–6631. [Google Scholar] [CrossRef] [Green Version]
- Warden, A.S.; Mayfield, R.D. Gene expression profiling in the human alcoholic brain. Neuropharmacology 2017, 122, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Butte, A.J. Leveraging big data to transform target selection and drug discovery. Clin. Pharmacol. Ther. 2016, 99, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Ma, L.; Paik, H.; Sirota, M.; Wei, W.; Chua, M.S.; So, S.; Butte, A.J. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.B.; Harris, R.A.; Mayfield, R.D. From gene networks to drugs: Systems pharmacology approaches for AUD. Psychopharmacology 2018. [Google Scholar] [CrossRef]
- Ferguson, L.B.; Ozburn, A.R.; Ponomarev, I.; Metten, P.; Reilly, M.; Crabbe, J.C.; Harris, R.A.; Mayfield, R.D. Genome-Wide Expression Profiles Drive Discovery of Novel Compounds that Reduce Binge Drinking in Mice. Neuropsychopharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genom. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.H.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergushichev, A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Sugino, K.; Hempel, C.M.; Miller, M.N.; Hattox, A.M.; Shapiro, P.; Wu, C.; Huang, Z.J.; Nelson, S.B. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006, 9, 99–107. [Google Scholar] [CrossRef]
- Ait-Ghezala, G.; Mathura, V.S.; Laporte, V.; Quadros, A.; Paris, D.; Patel, N.; Volmar, C.H.; Kolippakkam, D.; Crawford, F.; Mullan, M. Genomic regulation after CD40 stimulation in microglia: Relevance to Alzheimer’s disease. Brain Res. Mol. Brain Res. 2005, 140, 73–85. [Google Scholar] [CrossRef]
- Albright, A.V.; Gonzalez-Scarano, F. Microarray analysis of activated mixed glial (microglia) and monocyte-derived macrophage gene expression. J. Neuroimmunol. 2004, 157, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Ye, S.; Chu, A.; Anton, K.; Yi, S.; Vincent, V.A.; von Schack, D.; Chin, D.; Murray, J.; Lohr, S.; et al. Identification of cathepsin B as a mediator of neuronal death induced by Abeta-activated microglial cells using a functional genomics approach. J. Biol. Chem. 2004, 279, 5565–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.M.; Francescutti-Verbeem, D.M.; Kuhn, D.M. Gene expression profile of activated microglia under conditions associated with dopamine neuronal damage. FASEB J. 2006, 20, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Horvath, S.; Geschwind, D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 12698–12703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Bachoo, R.M.; Kim, R.S.; Ligon, K.L.; Maher, E.A.; Brennan, C.; Billings, N.; Chan, S.; Li, C.; Rowitch, D.H.; Wong, W.H.; et al. Molecular diversity of astrocytes with implications for neurological disorders. Proc. Natl. Acad. Sci. USA 2004, 101, 8384–8389. [Google Scholar] [CrossRef] [Green Version]
- Bayes, A.; van de Lagemaat, L.N.; Collins, M.O.; Croning, M.D.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.C.; Cheng, J.; Allaire, J.J.; Xie, Y.; McPherson, J. Shiny: Web Application Framework for R. R Package Version 1.3.2. 2019. Available online: https://CRAN.R-project.org/package=shiny (accessed on 14 December 2019).
- Wickham, H. Tidyverse: Easily Install and Load the ‘Tidyverse’. R Package Version 1.2.1. 2017. Available online: https://CRAN.R-project.org/package=tidyverse (accessed on 14 December 2019).
- Dowle, M.S.A. Data.Table: Extension of Data.Frame. R Package Version 1.12.2. 2019. Available online: https://CRAN.R-project.org/package=data.table (accessed on 14 December 2019).
- Urbanek, S. Png: Read and Write PNG Images. R Package Version 0.1-7. 2013. Available online: https://CRAN.R-project.org/package=png (accessed on 14 December 2019).
- Johnson, B.A.; Roache, J.D.; Javors, M.A.; DiClemente, C.C.; Cloninger, C.R.; Prihoda, T.J.; Bordnick, P.S.; Ait-Daoud, N.; Hensler, J. Ondansetron for Reduction of Drinking Among Biologically Predisposed Alcoholic Patients. JAMA 2000, 284, 963. [Google Scholar] [CrossRef]
- Tunstall, B.J.; Lorrai, I.; McConnell, S.A.; Gazo, K.L.; Zallar, L.J.; de Guglielmo, G.; Hoang, I.; Haass-Koffler, C.L.; Repunte-Canonigo, V.; Koob, G.F.; et al. Probenecid Reduces Alcohol Drinking in Rodents. Is Pannexin1 a Novel Therapeutic Target for Alcohol Use Disorder? Alcohol Alcohol. 2019, 54, 497–502. [Google Scholar] [CrossRef]
- Wen, R.T.; Zhang, M.; Qin, W.J.; Liu, Q.; Wang, W.P.; Lawrence, A.J.; Zhang, H.T.; Liang, J.H. The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol. Clin. Exp. Res. 2012, 36, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, D.D.; Alexander, L.; Malone, J.; Federoff, D.; Froehlich, J.C. The alpha2-adrenergic receptor agonist, clonidine, reduces alcohol drinking in alcohol-preferring (P) rats. Alcohol 2014, 48, 543–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggio, G.M.; Camillieri, G.; Platania, C.B.; Castorina, A.; Marrazzo, G.; Torrisi, S.A.; Nona, C.N.; D’Agata, V.; Nobrega, J.; Stark, H.; et al. Dopamine D3 receptor is necessary for ethanol consumption: An approach with buspirone. Neuropsychopharmacology 2014, 39, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, I.; Stelly, C.E.; Morikawa, H.; Blednov, Y.A.; Mayfield, R.D.; Harris, R.A. Mechanistic insights into epigenetic modulation of ethanol consumption. Alcohol 2017, 60, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Maccioni, P.; Pes, D.; Orru, A.; Froestl, W.; Gessa, G.L.; Carai, M.A.; Colombo, G. Reducing effect of the positive allosteric modulator of the GABA(B) receptor, GS39,783, on alcohol self-administration in alcohol-preferring rats. Psychopharmacology 2007, 193, 171–178. [Google Scholar] [CrossRef]
- Wolstenholme, J.T.; Warner, J.A.; Capparuccini, M.I.; Archer, K.J.; Shelton, K.L.; Miles, M.F. Genomic analysis of individual differences in ethanol drinking: Evidence for non-genetic factors in C57BL/6 mice. PLoS ONE 2011, 6, e21100. [Google Scholar] [CrossRef] [Green Version]
- Qiang, M.; Li, J.G.; Denny, A.D.; Yao, J.M.; Lieu, M.; Zhang, K.; Carreon, S. Epigenetic mechanisms are involved in the regulation of ethanol consumption in mice. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef]
- Warnault, V.; Darcq, E.; Levine, A.; Barak, S.; Ron, D. Chromatin remodeling—A novel strategy to control excessive alcohol drinking. Transl. Psychiatry 2013, 3, e231. [Google Scholar] [CrossRef]
- Al Ameri, M.; Al Mansouri, S.; Al Maamari, A.; Bahi, A. The histone deacetylase (HDAC) inhibitor valproic acid reduces ethanol consumption and ethanol-conditioned place preference in rats. Brain Res. 2014, 1583, 122–131. [Google Scholar] [CrossRef]
- Truitt, J.M.; Blednov, Y.A.; Benavidez, J.M.; Black, M.; Ponomareva, O.; Law, J.; Merriman, M.; Horani, S.; Jameson, K.; Lasek, A.W.; et al. Inhibition of IKKbeta Reduces Ethanol Consumption in C57BL/6J Mice. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Black, M.; Benavidez, J.M.; Stamatakis, E.E.; Harris, R.A. PPAR Agonists: I. Role of Receptor Subunits in Alcohol Consumption in Male and Female Mice. Alcohol. Clin. Exp. Res. 2016, 40, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, G.M.; Warden, A.S.; Bridges, C.R.; Blednov, Y.A.; Harris, R.A. Chronic ethanol consumption: Role of TLR3/TRIF-dependent signaling. Addict. Biol. 2018, 23, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besheer, J.; Lindsay, T.G.; O’Buckley, T.K.; Hodge, C.W.; Morrow, A.L. Pregnenolone and ganaxolone reduce operant ethanol self-administration in alcohol-preferring p rats. Alcohol. Clin. Exp. Res. 2010, 34, 2044–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadda, F.; Garau, B.; Colombo, G.; Gessa, G.L. Isradipine and other calcium channel antagonists attenuate ethanol consumption in ethanol-preferring rats. Alcohol. Clin. Exp. Res. 1992, 16, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.W.; Neill, J.C.; Costall, B. An investigation into the effects of 5-HT agonists and receptor antagonists on ethanol self-administration in the rat. Alcohol 1998, 16, 249–270. [Google Scholar] [CrossRef]
- Dutton, J.W., 3rd; Chen, H.; You, C.; Brodie, M.S.; Lasek, A.W. Anaplastic lymphoma kinase regulates binge-like drinking and dopamine receptor sensitivity in the ventral tegmental area. Addict. Biol. 2017, 22, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Chung, F.H.; Jin, Z.H.; Hsu, T.T.; Hsu, C.L.; Liu, H.C.; Lee, H.C. Gene-Set Local Hierarchical Clustering (GSLHC)—A Gene Set-Based Approach for Characterizing Bioactive Compounds in Terms of Biological Functional Groups. PLoS ONE 2015, 10, e0139889. [Google Scholar] [CrossRef]
- Siavelis, J.C.; Bourdakou, M.M.; Athanasiadis, E.I.; Spyrou, G.M.; Nikita, K.S. Bioinformatics methods in drug repurposing for Alzheimer’s disease. Brief. Bioinform. 2016, 17, 322–335. [Google Scholar] [CrossRef]
- Mayfield, J.; Ferguson, L.; Harris, R.A. Neuroimmune signaling: A key component of alcohol abuse. Curr. Opin. Neurobiol. 2013, 23, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.; Most, D.; Ferguson, L.B.; Mayfield, J.; Harris, R.A.; Blednov, Y.A. Neuroimmune pathways in alcohol consumption: Evidence from behavioral and genetic studies in rodents and humans. Int. Rev. Neurobiol. 2014, 118, 13–39. [Google Scholar]
- Erickson, E.K.; Grantham, E.K.; Warden, A.S.; Harris, R.A. Neuroimmune signaling in alcohol use disorder. Pharmacol. Biochem. Behav. 2019, 177, 34–60. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Erickson, E.; Robinson, G.; Harris, R.A.; Mayfield, R.D. The neuroimmune transcriptome and alcohol dependence: Potential for targeted therapies. Pharmacogenomics 2016, 17, 2081–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, F.T.; Lawrimore, C.J.; Walter, T.J.; Coleman, L.G., Jr. The role of neuroimmune signaling in alcoholism. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Vetreno, R.P. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 2016, 233, 1543–1557. [Google Scholar] [CrossRef] [Green Version]
- Lasek, A.W. Effects of Ethanol on Brain Extracellular Matrix: Implications for Alcohol Use Disorder. Alcohol. Clin. Exp. Res. 2016, 40, 2030–2042. [Google Scholar] [CrossRef] [Green Version]
- Lasek, A.W.; Chen, H.; Chen, W.Y. Releasing Addiction Memories Trapped in Perineuronal Nets. Trends Genet. 2018, 34, 197–208. [Google Scholar] [CrossRef]
- Chen, H.; He, D.; Lasek, A.W. Repeated Binge Drinking Increases Perineuronal Nets in the Insular Cortex. Alcohol. Clin. Exp. Res. 2015, 39, 1930–1938. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lasek, A.W. Perineuronal nets in the insula regulate aversion-resistant alcohol drinking. Addict. Biol. 2019. [Google Scholar] [CrossRef]
- Xue, Y.X.; Xue, L.F.; Liu, J.F.; He, J.; Deng, J.H.; Sun, S.C.; Han, H.B.; Luo, Y.X.; Xu, L.Z.; Wu, P.; et al. Depletion of perineuronal nets in the amygdala to enhance the erasure of drug memories. J. Neurosci. 2014, 34, 6647–6658. [Google Scholar] [CrossRef] [Green Version]
- Stefaniuk, M.; Beroun, A.; Lebitko, T.; Markina, O.; Leski, S.; Meyza, K.; Grzywacz, A.; Samochowiec, J.; Samochowiec, A.; Radwanska, K.; et al. Matrix Metalloproteinase-9 and Synaptic Plasticity in the Central Amygdala in Control of Alcohol-Seeking Behavior. Biol. Psychiatry 2017, 81, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Adkins, A.E.; Hack, L.M.; Bigdeli, T.B.; Williamson, V.S.; McMichael, G.O.; Mamdani, M.; Edwards, A.C.; Aliev, F.; Chan, R.F.; Bhandari, P.; et al. Genomewide Association Study of Alcohol Dependence Identifies Risk Loci Altering Ethanol-Response Behaviors in Model Organisms. Alcohol. Clin. Exp. Res. 2017, 41, 911–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edenberg, H.J.; Koller, D.L.; Xuei, X.; Wetherill, L.; McClintick, J.N.; Almasy, L.; Bierut, L.J.; Bucholz, K.K.; Goate, A.; Aliev, F.; et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol. Clin. Exp. Res. 2010, 34, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Joslyn, G.; Ravindranathan, A.; Brush, G.; Schuckit, M.; White, R.L. Human variation in alcohol response is influenced by variation in neuronal signaling genes. Alcohol. Clin. Exp. Res. 2010, 34, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Howland, R.H. Methylene Blue: The Long and Winding Road From Stain to Brain: Part 2. J. Psychosoc. Nurs. Ment. Health Serv. 2016, 54, 21–26. [Google Scholar] [PubMed]
- Howland, R.H. Methylene Blue: The Long and Winding Road from Stain to Brain: Part 1. J. Psychosoc. Nurs. Ment. Health Serv. 2016, 54, 21–24. [Google Scholar] [PubMed]
- Scobie, K.N.; Damez-Werno, D.; Sun, H.; Shao, N.; Gancarz, A.; Panganiban, C.H.; Dias, C.; Koo, J.; Caiafa, P.; Kaufman, L.; et al. Essential role of poly(ADP-ribosyl)ation in cocaine action. Proc. Natl. Acad. Sci. USA 2014, 111, 2005–2010. [Google Scholar] [CrossRef] [Green Version]
- Vengeliene, V.; Rossmanith, M.; Takahashi, T.T.; Alberati, D.; Behl, B.; Bespalov, A.; Spanagel, R. Targeting Glycine Reuptake in Alcohol Seeking and Relapse. Pharmacol. Exp. Ther. 2018, 365, 202–211. [Google Scholar] [CrossRef]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov. 2013, 12, 866–885. [Google Scholar] [CrossRef]
- Townsend, K.P.; Pratico, D. Novel therapeutic opportunities for Alzheimer’s disease: Focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005, 19, 1592–1601. [Google Scholar] [CrossRef]
- Mercer, L.D.; Higgins, G.C.; Lau, C.L.; Lawrence, A.J.; Beart, P.M. MDMA-induced neurotoxicity of serotonin neurons involves autophagy and rilmenidine is protective against its pathobiology. Neurochem. Int. 2017, 105, 80–90. [Google Scholar] [CrossRef]
- Rose, C.; Menzies, F.M.; Renna, M.; Acevedo-Arozena, A.; Corrochano, S.; Sadiq, O.; Brown, S.D.; Rubinsztein, D.C. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 2144–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, B.R.; Green-Thompson, Z.W.; Pugh, P.J.; Lazic, S.E.; Mason, S.L.; Griffin, J.; Jones, P.S.; Rowe, J.B.; Rubinsztein, D.C.; Barker, R.A. An open-label study to assess the feasibility and tolerability of rilmenidine for the treatment of Huntington’s disease. J. Neurol. 2017, 264, 2457–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magiatis, P.; Polychronopoulos, P.; Skaltsounis, A.L.; Lozach, O.; Meijer, L.; Miller, D.B.; O’Callaghan, J.P. Indirubins deplete striatal monoamines in the Intact and MPTP-treated mouse brain and block kainate-induced striatal astrogliosis. Neurotoxicol. Teratol. 2010, 32, 212–219. [Google Scholar] [CrossRef]
- Suzuki, H.; Kaneko, T.; Mizokami, Y.; Narasaka, T.; Endo, S.; Matsui, H.; Yanaka, A.; Hirayama, A.; Hyodo, I. Therapeutic efficacy of the Qing Dai in patients with intractable ulcerative colitis. World J. Gastroenterol. 2013, 19, 2718–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhao, G.; Fang, J.S.; Yuan, T.Y.; Liu, A.L.; Du, G.H. Discovery of the neuroprotective effects of alvespimycin by computational prioritization of potential anti-Parkinson agents. FEBS J. 2014, 281, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pennings, J.L.; Massingham, L.J.; Wick, H.C.; Siegel, A.E.; Tantravahi, U.; Bianchi, D.W. An Integrated Human/Murine Transcriptome and Pathway Approach To Identify Prenatal Treatments For Down Syndrome. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Qiu, C.; Cui, Q. Drug-Path: A database for drug-induced pathways. Database 2015, 2015. [Google Scholar] [CrossRef]
- Napolitano, F.; Carrella, D.; Mandriani, B.; Pisonero-Vaquero, S.; Sirci, F.; Medina, D.L.; Brunetti-Pierri, N.; di Bernardo, D. Gene2drug: A computational tool for pathway-based rational drug repositioning. Bioinformatics 2018, 34, 1498–1505. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Study | Brain Area | GEO Accession | Platform | Case/Controls |
|---|---|---|---|---|
| Mamdani 2015 [3] | Nucleus accumbens | GSE62699 | Microarray | 18 cases, 18 controls (1 female per group) |
| Ponomarev 2012 [4] | Central nucleus of the amygdala Basolateral amygdala Prefrontal cortex | NA | Microarray | 17 cases, 15 controls (1 female per group) |
| Farris 2015 [5] | Prefrontal cortex | NA | RNAseq | 16 cases, 15 controls (all males) |
| McClintick 2013 [13] | Hippocampus | GSE44456 | Microarray | 20 cases, 19 controls (6 females per group) |
| Flatscher-Bader 2008 [10] | Ventral tegmental area | GSE9058 | Microarray | 6 cases, 6 controls (all males) |
| Flatscher-Bader 2010 [9] | Nucleus accumbens | GSE20568 | Microarray | 10 cases, 10 controls (1 female per group) |
| Zhou 2011 & Farris 2015 [6,14] | Hippocampus | NA | RNAseq | 8 cases, 8 controls (all males) |
| Zhang 2014 [11] | Prefrontal cortex | GSE49376 | Microarray | 23 cases, 23 controls (7 females per group) |
| Kapoor 2019 [7] | Prefrontal cortex | NA | RNAseq | 65 cases, 73 controls (all males) |
| Rao 2019 [12] | Nucleus accumbens Prefrontal cortex Basolateral amygdala Central nucleus of the amygdala | NA | RNAseq | 30 cases, 30 controls (7 females per group) |
| Mayfield | Basolateral amygdala Central nucleus of the amygdala | NA | RNAseq | 17 cases (2 female) and 16 controls (1 female) |
| Up-Regulated Pathways | Down-Regulated Pathways | ||
|---|---|---|---|
| KEGG Pathway | Number of Datasets (out of 17) | KEGG Pathway | Number of Datasets (out of 17) |
| Complement_and_coagulation_cascades | 8 | Oxidative_phosphorylation | 6 |
| Focal_adhesion | 8 | Parkinsons_disease | 4 |
| Mapk_signaling_pathway | 8 | Proteasome | 4 |
| Systemic_lupus_erythematosus | 8 | Alzheimers_disease | 3 |
| Cytokine_cytokine_receptor_interaction | 7 | Cardiac_muscle_contraction | 2 |
| Ecm_receptor_interaction | 7 | Dna_replication | 2 |
| Cell_adhesion_molecules_cams | 6 | Fructose_and_mannose_metabolism | 2 |
| Leishmania_infection | 6 | Glycolysis_gluconeogenesis | 2 |
| Regulation_of_actin_cytoskeleton | 6 | Huntingtons_disease | 2 |
| Ribosome | 6 | Mismatch_repair | 2 |
| Leukocyte_transendothelial_migration | 5 | Propanoate_metabolism | 2 |
| Natural_killer_cell_mediated_cytotoxicity | 5 | Vibrio_cholerae_infection | 2 |
| Pathways_in_cancer | 5 | ||
| Brain Region | Focal Adhesion | Complement and Coagulation Cascades | Mapk Signaling Pathway | Systemic Lupus Erythematosus |
| BLA (3) | 3 | 2 | 1 | 2 |
| CNA (3) | 1 | 2 | 2 | 1 |
| PFC (5) | 2 | 3 | 3 | 3 |
| NAC (3) | 1 | 1 | ||
| VTA (1) | 1 | |||
| HPC (2) | 1 | 1 | 2 | |
| Brain Region | Oxidative Phosphorylation | Parkinsons Disease | Proteasome | Alzheimers Disease |
| BLA (3) | 1 | |||
| CNA (3) | 1 | 1 | 1 | |
| PFC (5) | 2 | 2 | 1 | |
| NAC (3) | 2 | 1 | 1 | 1 |
| VTA (1) | ||||
| HPC (2) | 1 | 1 | 1 |
| Drug | MOA | Indication | Phase |
|---|---|---|---|
| methylene-blue | guanylyl cyclase inhibitor, nitric oxide production inhibitor | methemoglobinemia | Launched |
| ketotifen | histamine receptor agonist, leukotriene receptor antagonist, phosphodiesterase inhibitor | itching | Launched |
| indirubin | CDK inhibitor, glycogen synthase kinase inhibitor | Phase 2/Phase 3 | |
| diflunisal | prostanoid receptor antagonist | rheumatoid arthritis, osteoarthritis | Launched |
| azelastine | histamine receptor antagonist | conjunctivitis | Launched |
| ALX-5407 | Glycine transporter 1 inhibitor | Phase 1 | |
| rucaparib | PARP inhibitor | Phase 3 | |
| rilmenidine | adrenergic receptor agonist, imidazoline receptor agonist | hypertension | Launched |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferguson, L.B.; Patil, S.; Moskowitz, B.A.; Ponomarev, I.; Harris, R.A.; Mayfield, R.D.; Messing, R.O. A Pathway-Based Genomic Approach to Identify Medications: Application to Alcohol Use Disorder. Brain Sci. 2019, 9, 381. https://doi.org/10.3390/brainsci9120381
Ferguson LB, Patil S, Moskowitz BA, Ponomarev I, Harris RA, Mayfield RD, Messing RO. A Pathway-Based Genomic Approach to Identify Medications: Application to Alcohol Use Disorder. Brain Sciences. 2019; 9(12):381. https://doi.org/10.3390/brainsci9120381
Chicago/Turabian StyleFerguson, Laura B., Shruti Patil, Bailey A. Moskowitz, Igor Ponomarev, Robert A. Harris, Roy D. Mayfield, and Robert O. Messing. 2019. "A Pathway-Based Genomic Approach to Identify Medications: Application to Alcohol Use Disorder" Brain Sciences 9, no. 12: 381. https://doi.org/10.3390/brainsci9120381