Current Perspectives in Cancer Immunotherapy
by

Theodoulakis Christofi
1, 
Stavroula Baritaki
2,
Luca Falzone
3,4, 
Massimo Libra
3,5 and
Apostolos Zaravinos
1,*
1
Department of Life Sciences, School of Sciences, European University Cyprus, 1516 Nicosia, Cyprus
2
Division of Surgery, School of Medicine, University of Crete, P.O. Box 2208, Voutes, 71003 Heraklion, Crete, Greece
3
Department of Biomedical and Biotechnological Sciences, Oncologic, Clinic and General Pathology Section, University of Catania, 95123 Catania, Italy
4
Epidemiology Unit, IRCCS Istituto Nazionale Tumori “Fondazione G. Pascale”, 80131 Napoli, Italy
5
Research Center for Prevention, Diagnosis and Treatment of Cancer, University of Catania, 95123 Catania, Italy
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(10), 1472; https://doi.org/10.3390/cancers11101472
Submission received: 16 August 2019
/
Revised: 20 September 2019
/
Accepted: 26 September 2019
/
Published: 30 September 2019
(This article belongs to the Special Issue New Insights into Cancer Vaccines and Immunotherapy)
Abstract
:Different immunotherapeutic approaches have proved to be of significant clinical value to many patients with different types of advanced cancer. However, we need more precise immunotherapies and predictive biomarkers to increase the successful response rates. The advent of next generation sequencing technologies and their applications in immuno-oncology has helped us tremendously towards this aim. We are now moving towards the realization of personalized medicine, thus, significantly increasing our expectations for a more successful management of the disease. Here, we discuss the current immunotherapeutic approaches against cancer, including immune checkpoint blockade with an emphasis on anti-PD-L1 and anti-CTLA-4 monoclonal antibodies. We also analyze a growing list of other co-inhibitory and co-stimulatory markers and emphasize the mechanism of action of the principal pathway for each of these, as well as on drugs that either have been FDA-approved or are under clinical investigation. We further discuss recent advances in other immunotherapies, including cytokine therapy, adoptive cell transfer therapy and therapeutic vaccines. We finally discuss the modulation of gut microbiota composition and response to immunotherapy, as well as how tumor-intrinsic factors and immunological processes influence the mutational and epigenetic landscape of progressing tumors and response to immunotherapy but also how immunotherapeutic intervention influences the landscape of cancer neoepitopes and tumor immunoediting.
1. Introduction
During the last decade, immunotherapy has provided remarkable clinical responses to cancer patients. This turning point has significantly increased our expectations for successfully treating the disease [1]. Successful immunotherapies against melanoma, non–small cell lung cancer (NSCLC) and renal cell cancer have led to active clinical trials [2,3,4,5,6,7,8,9,10,11,12]. However, the percentage of responders is still rather low, underscoring the need to identify those patients who respond best to different immunotherapeutic approaches. Therefore, a suitable selection of cancer patients, based on their genetic profile along with other non-genetic determinants, seems to be critical in order to achieve the most successful therapeutic responses. The advent of next-generation sequencing, along with continuous technological advances in the field of sequencing, has provided us the ability to evaluate the required genomic information that we need in order to predict a patient’s response to immunotherapies. Here, we discuss the current immunotherapeutic strategies against cancer highlighting the pros and cons of each one. We also discuss the value of tumor neoantigens in the patients’ response to immunotherapy.
2. Immunotherapeutic Approaches against Cancer
Overall, there are four different immunotherapeutic strategies. These include immune checkpoint blockade, cytokine therapy, cellular therapy and therapeutic vaccines. To understand how immune checkpoint blockade works, it is critical to comprehend the constant interaction between tumor and immune cells in the continuous process of cancer development. Tumor cells exploit various immune-regulatory mechanisms to achieve immune escape, thus suppressing immune responses against them within the tumor microenvironment. This is more obvious as the disease progresses. Several immune-related cells work towards the establishment of an immunosuppressive microenvironment, including regulatory T cells (Tregs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs) and regulatory B cells. Therefore, within the tumor microenvironment, cancer cells along with immune cells excrete inhibitory cytokines and express checkpoint inhibitors that dampen the anti-tumor activity of specific T cells. During the last decade, a rapid understanding of the mechanisms that most cancer cells use to hide from the immune system has led to the improvement of new, immunotherapeutic approaches against cancer. Especially, the use of anti-PD-1 or anti-PD-L1 monoclonal antibodies (mab) has yielded top-notch medical responses in several cancers. The most recent advances in cancer immunotherapy are summarized in Table 1 and discussed below.
2.1. Cancer Immunotherapy with Immune Checkpoint Blockade
2.1.1. Monoclonal Antibodies against CTLA-4 and the PD-1/PD-L1 Axis
The idea of immune checkpoint blockade involves the inhibition of the immune system’s intrinsic regulatory mechanisms, thus, driving the activation of a better anti-cancer immunological response. Since the recent encouraging clinical responses observed in patients who had been treated with mabs against the immune checkpoint proteins cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed cell death (PD-1), the interest of the scientific community shifted in using such immune checkpoint blockade for treating cancer patients [63]. CTLA-4 is a membrane receptor on effector T cells that binds CD80/CD86 (B7.1/2) on antigen-presenting cells (APC) inducing the arrest of T-cells [64]. Likewise, PD-1 is another cell membrane receptor of activated T cells that interacts with PD-L1/2, inducing T-cell inhibition [65,66].
CTLA-4 blockade stimulates T-cells against cancer. Ipilimumab is an FDA approved anti–CTLA-4 mab against metastatic melanoma, that launched a new era in immunotherapy. We have witnessed a great progress since then, with the launch of novel immune checkpoint inhibitors, including two anti–PD-1 mabs, Pembrolizumab and Nivolumab. Although both mabs were initially approved against metastatic melanoma [67], NSCLC, renal cell carcinoma (RCC) [68,69], head and neck squamous cell carcinoma (HNSCC) after platinum-based chemotherapy [70,71,72] and refractory classic Hodgkin’s lymphoma (cHL), later on, they were approved for any unresectable or metastatic solid tumor characterized by mismatch repair deficiency (dMMR) or microsatellite instability (MSI+) [17]. This was the first time the FDA approved a cancer drug based totally on tumor genetics, instead of the tissue type or the location of the tumor. Such immune checkpoints could induce a durable clinical benefit, including a long-lasting regression of the tumor and an extended stabilization in patients with advanced NSCLC, melanoma and RCC [18,73,74].
Anti-PD-L1/2 targeting, has also attracted much interest in the clinical setting. We now know that PD-L1/2 expression on cancer cells or APCs suppresses CD8+ T cells (CTLs) and induces the function of T–regulatory (Treg) cells [66]. More than a few clinical trials have examined anti-PD-L1/2 alone or in combination with anti–PD-1 mabs [4,5,74,75].
Pidilizumab is another anti-PD-1 mab, tested in B cell lymphoma after autologous stem cell transfer and in relapsed follicular lymphoma, with 34% and 52% response rates, respectively [76,77].
In addition, the new anti-PD-L1 mab, Atezolizumab, showed low response rates (18%) in stage III–IV NCSLC and locally advanced and metastatic urothelial cancer (23%) [14,78,79]. In a recent comparison among these anti-PD-1 and anti-PD-L1 drugs, Nivolumab and Pembrolizumab were found to be related to increased objective response rates, compared to Atezolizumab and Nivolumab, which are correlated to fewer adverse events versus other drugs [80].
On the other hand, although Tremelimumab (anti-CTLA-4 mab) exhibited a good safety profile, it could not extend the overall survival of malignant mesothelioma suffering patients, versus the placebo [81,82]. Furthermore, blockade both PD-1 and CTLA-4 could reverse T cell suppression at both priming and effector cells [83].
At present, Pembrolizumab is the most successful immunotherapeutic approach for melanoma and other cancer types [84]. A phase III clinical trial compared anti-CTLA-4 and anti-PD-1 mabs and showed that Nivolumab-treated patients exhibited better response and survival rates compared to those treated with Ipilimumab. Patients treated with both checkpoint blockers, had better responses and survival [85]. In a similar study, combination therapy of Nivolumab and Ipilimumab on stage III/IV melanoma patients, yielded 57.6 objective response rates and 11.5% complete response rates, versus single therapies [86].
2.1.2. Further Co-Inhibitory and Co-Stimulatory Markers
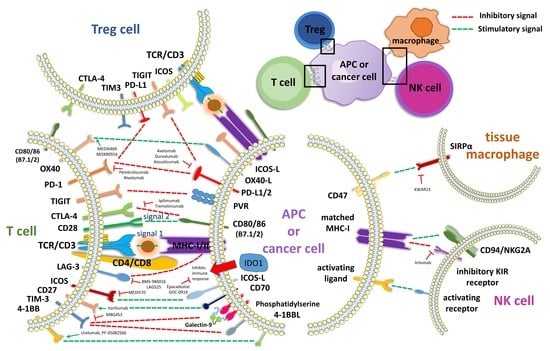
Although the above-mentioned mabs gave encouraging results in metastatic patients, only a fraction of them are good responders. Lately, new approaches targeting further immune-regulating mechanisms are becoming available. These, comprise co-inhibitory and co-stimulatory immune receptors (markers) (Table 2). Such markers embrace T lymphocyte markers, macrophage markers and natural killer (NK) cell markers. Co-inhibitory signaling stem from the signals between TIGIT and PD-L1/2, PD-L1 and PD-1, TIGIT and PVR, CTLA-4 and CD80/86 (B7.1/2), LAG3 and MHCI/II, TIM-3 and Phosphatidylserine, TIM-3 and 4-1BBL or MHC-I and inhibitory KIR receptors. On the other hand, co-stimulatory signaling results from the interaction between OX40 and OX40L, CD28 and CD80/86(B7.1/2), ICOS and ICOS-L, CD27 and CD70, 4-1BB and 4-1BB-L, CD47 and SIRPa or MHC-1 and CD94/NKG2A, among other (Figure 1).
T lymphocyte markers include T-cell Ig and mucin-domain containing-3 (TIM-3) (previously recognized as hepatitis A virus cellular receptor 2, HAVCR2) [126], lymphocyte-activation gene 3 (LAG-3) [110], B7-H3 (CD276), B7-H4 (V-set domain-containing T-cell activation inhibitor 1, VTCN1), V-domain containing Ig suppressor of T cell activation (VISTA), cell surface transmembrane glycoprotein CD200 receptor 1 (CD200R), Inducible Costimulator (ICOS/ICOS-L), T cell immunoglobulin and ITIM domain (TIGIT), CD27/CD70 and glucocorticoid-induced TNF receptor (GITR). On the other hand, macrophage markers include CD47/signal-regulatory protein alpha (SIRPα) and indoleamine-2,3-dioxygenase (IDO1 or INDO); and NK cell markers include CD94/NKG2A and the killer immunoglobulin-like receptor (KIR) family (Figure 1).
Both TIM-3 and LAG-3 receptors are co-expressed on PD-1-expressing T cells, rendering them good candidates for combinatorial targeting with anti–PD-1 agents in PD-1 expressing tumors. TIM-3 is expressed on CD4+ Th1, Th17 and CD8+ CTLs and it is a T cell inhibitory receptor. TIM-3 is composed of a transmembrane domain, an extracellular glycosylated mucin domain and a variable immunoglobulin domain (IgV). TIM-3 has four relevant ligands, including Gal-9, HMGB1, Ceacam-1 and PtdSer. Gal-9 binding to TIM-3 induces an influx of Ca2+ to the intracellular region of Th1 cells, inducing T cell apoptosis. Binding of Gal-9, PtdSer and HMGB1 to TIM-3 functions as a positive regulator of dendritic cells (DCs), while that between Ceacam-1 and TIM-3 leads to a negative regulation of T cell responses [109,110]. Cells co-expressing TIM3 and PD-1, present more defects in cellular division and cytokine production compared to those expressing only PD-1 and blockade of both checkpoints can reverse T cell exhaustion [83]. TIM-3 blockade showed antitumor effects in many mouse tumor models and these effects were even greater once it was combined with CTLA-4 blockade [127].
LAG-3 is another co-inhibitory immune checkpoint [128] found on activated T, B, NK and plasmacytoid dendritic cells (pDCs) [124,129,130], where it predominantly binds to the antigen-MHC-II complex [131], negatively regulating T-cells [132] and activating Treg cells [126,128,133]. LAG-3 have four IgG domains with high structural homology with CD4 molecules while amino acid is <20% homologous to CD4. LAG-3 contains a unique “extraloop” in the membrane distal D1 domain, through which it binds to MHC-II molecules [134]. Combined PD-1 and LAG-3 blockade could reverse T cell function via different inhibitory pathways [83,126]. LAG-3-expressing Treg cells can effectively suppress cytotoxic T cells [135].
B7-H3 is a receptor found on the membrane of APCs, cancer cells and host cells, where it mainly suppresses T cell function. The anti-B7-H3 mab Enoblituzumab (MGA271) mediates potent ab-dependent cytotoxicity against various cancer types [136].
B7-H4 is a costimulatory membrane receptor expressed on APCs and cancer cells, interacting with the CD28 ligand and receptors on T lymphocytes causing an inhibitory effect. It has been associated with gastric carcinomas [137], breast cancer [138,139], non-metastatic clear cell renal cell carcinoma [140] and glioblastoma [141], among other tumor types. Its loss was also recently associated with the development of autoimmune diabetes [142].
VISTA is a checkpoint belonging to the B7 family. It is expressed on APCs and T cells in tissues with a significant number of infiltrating leukocytes [143] and it controls peripheral tolerance and anti-tumor immunity [120]. VISTA also suppresses T cell activation in a T-cell autonomous manner when expressed on CD4+ T cells. VISTA expression was found to be higher after Ipilimumab therapy in prostate cancer patients [144]. In NSCLC VISTA expression is associated with high number of tumor infiltrating lymphocytes (TIL load), PD-1, specific genomic alterations and patient clinical outcome [145].
ICOS is another transmembrane receptor expressed primarily on Treg cells but also in tonsillar T cells [91,146]. Its structure and function is similar to CD28 [91] and interacts with ICOS-L enhancing Treg cell function, T-cell proliferation, secretion of cytokines such as IL-10, IL-4, IL-5, IFNg and IL-17. The ICOS-ICOS-L interaction can also induce the excretion of antibodies by B cells. It also promotes the collaboration between T and B cells via the CD40/CD40L pathway [147]. ICOS expression is induced rapidly after T-cell activation [148]. A phase I clinical trial (NCT02520791) evaluated the anti-ICOS mAb (MEDI-570) in patients with relapsed/refractory peripheral T-cell follicular lymphoma and angioimmunoblastic T-cell lymphoma [149].
CD200R is another inhibitory glycoprotein expressed on the membrane of myeloid and lymphoid cells [150] and CD4+ T cells [151,152]. The CD200-CD200R pathway signals also stimulates the proliferation and activity of Treg cells and upregulates IDO activity [153,154]. The absence of the CD200R signaling was shown to inhibit the development of an endogenous tumor irrespective of the expression of CD200 [155]. A clinical trial using the humanized anti-CD200 mab, Samalizumab is on the way to treating metastatic bladder cancer [32].
TIGIT is found both on T cells and NK cells and it regulates T-cell mediated immunity via the CD226/TIGIT-PVR (poliovirus receptor or CD155) pathway [114]. TIGIT is mainly expressed on lymphocytes, including TILs and Tregs that infiltrate different types of tumors [156]. It has been shown to attenuate the immune response via direct signaling, inducing ligand signaling and competition with and disruption of signaling by the costimulatory receptor CD226 (also known as DNAM-1). PVR is also broadly expressed in tumors, suggesting that the TIGIT-PVR axis provides a major immune escape mechanism for cancer cells. Notably, TIGIT expression is tightly correlated with that of PD-1 and both are co-expressed on TILs. Combined blockade of both checkpoints could further increase cell proliferation, cytokine production and degranulation of both tumor antigen-specific CD8⁺ T cells and TILs in melanoma [157]. TIGIT could also synergize with TIM-3 to suppress an anticancer immune response [158].
Another co-stimulatory immune checkpoint of great interest is the TNF receptor family CD27. This binds to CD70 [159] regulating the activation of B-cells [160,161], mainly through the NF-κB and MAPK8/JNK pathways. Prolonged CD27-CD70 interactions have been show to exhaust T cells and induce lethal immunodeficiency [90]. Varlilumab is a new agonist CD27 mab that has been shown to activate T-cells in tumor models [33,34].
GITR is another co-stimulatory immune checkpoint molecule, encoded by the TNF receptor superfamily gene TNFRSF18 in humans. GITR expression is elevated upon T-cell activation and is critical in dominant immunological self-tolerance maintained by CD25+/CD4+ Tregs. GITR inhibits Tregs, extending the survival of effector T cells. High GITR levels have been reported on the surface of Tregs in mouse models but also on any activated T cells in humans, challenging its usefulness as a Treg marker [162]. Recently, PD-1 blockade and activation of GITR could synergistically induce the activation of T cells [163]. In addition, agonistic targeting of GITR was shown to augment TIL functionality in hepatocellular carcinoma [164].
The macrophage inhibitory marker SIRPα interacts with CD47, a broadly expressed transmembrane receptor, negatively controlling effector function of innate immune cells. The interaction of SIRPα with CD47 inhibits macrophage-mediated phagocytosis [165] similar to the self-signals provided by MHC-I molecules to NK cells via Ig-like or Ly49 receptors [166,167]. Cancer cells expressing high levels of CD47 could activate SIRPα and inhibit macrophage-mediated destruction. Engineered high-affinity variants of SIRPα antagonized CD47 on tumor cells and enhanced phagocytosis [168]. Furthermore, anti-SIRPα antibodies (e.g., KWAR23) were also found to help macrophages to diminish cancer progression and metastasis [35,36].
IDO1 along with IDO2 and tryptophan-2,3-dioxygenase (TDO), metabolize tryptophan to kynurenine, which is then metabolized to kynurenic acid [169,170]. Recent indication shows that IDO is upregulated in cancer progression, aiding tumor cells through pathogenic inflammatory processes to escape elimination by immune cells by inducing immune tolerance against tumor antigens [171,172]. IDO is expressed by some alternatively activated macrophages and other immune-regulatory cells [173], suppressing T cells and NK cells [174] and activating Tregs and MDSCs and promotes angiogenesis [171]. Contrarily, the pro-angiogenic vascular endothelial growth factor (VEGF) has been shown to increase the expression and activity of IDO in dendritic cells (DCs) thereby suppressing antigen-specific and mitogen-stimulated lymphocyte proliferation [175]. Moreover, the WNT signaling pathway and particularly the metabolic reprogramming of fatty acid oxidation (FAO) in DCs increases IDO expression while suppresses IL-6 and IL-12 cytokines; thus, aiding the tolerization of DCs within the tumor microenvironment and further contributes to anti-PD-1 immunotherapy resistance [176]. Targeting the WNT pathway with the specific tankyrase (TNKS) inhibitor, XAV-939, can enhance the immune response against pancreatic ductal adenocarcinoma (PDAC) cells with lymph node-positive metastasis [177]. IDO has been found to be upregulated in prostate cancer, colorectal cancer, pancreatic, cervical, gastric, ovarian, head, lung and other cancer types [178]. Epacadostat (INCB024360) combined with Pembrolizumab was recently announced to target IDO1 and provide encouraging antitumor activity in NSCLC patients and other tumors (ECHO-202/KEYNOTE-037) [31]. Navoximod (GDC-0919), a novel IDO1-specific checkpoint inhibitor, could also transiently decrease plasma kynurenine in patients with recurrent tumors. In this trial (NCT02048709), a response rate of 9% was reported [29]. The same checkpoint inhibitor, when administered with Atezolizumab in progressive solid cancers, exhibited activity but without a clear evidence for benefit of dual treatment [30]. Indoximod (previously termed as D-1MT) is an immunometabolic adjuvant recently shown to enhance T cell activity in cancer [179].
Nonetheless, a tight correlation exists between immune-infiltrate, angiogenesis and cancer progression and dissemination to distant sites and to nodal compartment. Indeed, CD8+ T cells and immune cells come and go across the permeable capillaries. Because of these intimate interactions, it happens that local dendritic cells (DCs) within the tumor microenvironment become tolerant to immunotherapeutic strategies and thus, promote immune evasion and immunotherapy resistance. One such mechanism was described in melanoma by Zhao F et al. [176], where blockade of the Wnt5a-β-catenin-peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling pathway augmented anti-melanoma immunity, enhanced the activity of anti-PD-1 antibody immunotherapy and suppressed disease progression in a transgenic melanoma model. Increased WNT activation and a peculiar immune microenvironment were also shown to increase the likelihood of lymphatic dissemination in pancreatic ductal adenocarcinoma (PDAC) [177]. In this study, regional lymph node positive PDACs were enriched in M2 macrophages and activated DCs [177]. Furthermore, endothelial cells (ECs) also interfere with the efficiency of DC maturation. VEGF was found to increase the expression and activity of IDO in DCs, having a suppressive effect on Ag-specific and mitogen-stimulated lymphocyte proliferation [175].
CD94/NKG2 is a dimer between a C-type lectin receptor (NKG2) and the CD94 molecule, primarily expressed on NK and a few CD8+ T cells [180,181]. Activating CD94/NKG2 receptors stimulate cytotoxic activity of NK cells, whereas inhibitory receptors impede it [182]. The CD94/NKG2 dimer recognizes non-classical MHC-I glycoproteins [183]. NKG2A and NKG2B receptors transmit inhibitory signals through their Immunoreceptor Tyrosine-based Inhibitory Motif (ITIM) tails, while NKG2C, NKG2D, NKG2E and NKG2H are activating receptors.
KIR receptors are membrane glycoproteins found on NK cells and T cells [184,185], suppressing their cytotoxic activity via inhibitory signals that they produce [186]. Only a few KIR receptors are activating, showing that their recognition of MHC molecules triggers the cytotoxic activity of their cell [187]. Recently prolonged KIR blockade using the anti-KIR2D mAb, lirilumab, was announced to be safe and well tolerated [188]. Nevertheless, the EffiKIR clinical trial (NCT 01687387) failed to show clinical effects on AML patients [189].
2.1.3. Adverse Effects Associated with Checkpoint Blockade
Inhibition of immune-checkpoint receptors using the above-mentioned therapeutic mabs for treating cancer patients is linked to many side effects of various rates, which resemble autoimmune reactions [190]. Furthermore, despite many cancer patients respond well to immune checkpoint blockade, a significant proportion of these tumors will eventually develop tumor resistance and will progress. Therefore, finding the proper balance between immune-therapies and/or finding new therapeutic tools to overcome such a resistance will significantly improve the clinical outcome [191].
In particular, the rate of incidence and the types of adverse events related to immune-checkpoint inhibitors depend on the specific features of patients and on the type of agent used [192]. Several studies demonstrated that the checkpoint inhibitors are more tolerated compared to classical chemotherapeutic agents; in particular it was demonstrated that PD-1/PD-L1 inhibitors have less toxicity compared to the anti-CTLA-4 antibody Ipilimumab [193]. Overall, the most common immune-checkpoint inhibitors-related toxicity are systemic, endocrine, dermatologic and gastrointestinal side effects [192,193]. Regarding the systemic adverse events, these encompass fatigue and fever, asthenia, hypotension, dyspnea and other symptoms due to infusion reactions whose incidence is about 25% of patients treated with immunotherapy [16,194]. Other studies paid the attention to the endocrine, dermatological and gastrointestinal adverse events showing that treatments with Ipilimumab, Nivolumab and Pembrolizumab were able to induce skin toxicities due to allergic reaction to the compounds [195]. Ntali and colleagues (2017) have made a careful review of the literature regarding the endocrine sequelae of cancer immunotherapy, showing that these treatments may lead to hypophysitis, adrenalitis and both hypothyroidism and hyperthyroidism depending on specific characteristics of patients [196]. Finally, as observed for several chemotherapeutic agents, also the administration of immune checkpoint inhibitors is correlated to several gastrointestinal side effects mainly including diarrhea, colitis, nausea, vomiting and abdominal pains [197]. Furthermore, adverse effects including rash or pruritus, thyroiditis, hypothyroidism or hypophysitis and asymptomatic pancreatitis were reported in dMMR cancer patients treated with PD-1 blockade [198].
2.2. Cancer Immunotherapy with Cytokine Therapy
The tumor microenvironment (TME) consists of cancer cells and stroma including non-malignal endothelial, fat, blood and immune cells of the surrounding niche [199]. Tumor progression is depended on the interaction of cancer cells with their microenvironment, influencing cancer cell survival and growth, invasion and metastasis, through an orchestrated signaling crosstalk that drives tumor evolution [200]. There, tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs), innate lymphoid cells (ILCs), MDSCs, mast cells, T cells and NK cells produce various factors such as enzymes, chemokines and cytokines which can increase angiogenesis but also modulate the local immunity and establish immunosuppression [201,202,203].
Cytokines are molecules that promote inter-communication between the immune cells and were initially used as an immunotherapeutic approach. Proinflammatory cytokines stimulate crucial immune effectors, including T cells and NK cells. In the tumor’s microenvironment, cytokines mediate cancer progression in solid and hematologic tumors. For example, in multiple myeloma several cell types within the bone marrow, including cells of the immune system, mesenchymal stem cells and bone marrow stromal cells, can contribute to the development of myeloma bone disease [204]. The cytokine- and cell-adhesion-dependent bone marrow niche and stromal microenvironment support the formation of new vessels and the proliferation of multiple myeloma, irrespective of immune-surveillance [205,206,207,208]. Leone et al. provided evidence that the intimate interaction between endothelial cells, tumor cells and CD8+ T cells creates a permissive immune-microenvironment within the bone marrow that allows undisturbed cancer proliferation [209]. They demonstrated that endothelial cells act as APCs, stimulating a central memory CD8+ T cell population, which negatively regulates the effector memory CD8+ T cells with anti-tumor activity. Remarkably, a defective immunosurveillance allows for the persistence and proliferation of multiple myeloma cells—an immune-microenvironment disease evolution characterized by exhausted CD8+ cells, overexpressing check point molecules such as LAG3 and PD1, in preclinical models offers suitable targets for increased survival in in vivo models [205,206,210]. In a clinical setting, a patient with a larger CD8 cytokine profile, along with competent CD8 T cells and dendritic cells had an increased overall survival and time to progression [206,211]. Therefore, it is likely that angiogenesis within the bone marrow, a recognized hallmark of multiple myeloma progression, parallels multiple myeloma evasion from T cell immune surveillance [208,209,212].
Importantly, cancer-associated fibroblasts (CAFs) are also implicated in mediating tumor-promoting inflammation by secreting cytokines and chemokines that mediate the recruitment and activation of immune cells and by their reciprocal interactions with immune cells in the tumor microenvironment [213,214]. CAFs also contribute to immune escape by upregulating immunosuppressive cytokines and immune checkpoint ligands, as well as via the exclusion of anti-tumor CD8+ T cells from cancer cells and by affecting the functional differentiation of tumor infiltrating inflammatory cells [215]. Furthermore, the intimate interaction between endothelial cells, tumor cells and CD8+ T cells creates a permissive immune microenvironment that allows undisturbed cancer proliferation [209].
Thus far, two cytokines are FDA approved as anti-cancer therapeutic agents—IL-2 against metastatic melanoma and kidney cancer and IFN-α as adjuvant therapy against stage III melanoma. IFN-α is an all and more preferable treatment for diseases like Philadelphia-negative myeloproliferative neoplasms (MPNs), essential thrombocytosis, polycythemia vera (PV) and primary myelofibrosis (PMF) [216,217]. Nevertheless, IFNα treatment has been linked with side effects, even at low-doses and of its pegylated form [42,217,218,219,220]. Among the major pathways by which IFN-α2 acts, is the JAK/STAT signaling pathway [221,222,223,224]. IFNα2 has anti-proliferative, pro-apoptotic, antiangiogenic and immunomodulatory mechanisms of action [221,222,224,225] and it can also downregulate telomerase reverse transcriptase and telomerase activity [226].
Modern approaches can also interfere with cytokine-related immune effect. For example, cytokine production was shown to be induced by CAR-T cells, such as HER2-specific designed ankyrin repeat protein (DARPin28z) CAR T-cells, upon antigen binding [227]. Furthermore, the multi-specific DARPin® molecule, MP0250, was shown to strongly neutralize VEGF and HGF causing a significant reduction in the number of vessels and was defined as a novel combination drug for treatment of multiple myeloma patients [228]. Profiling for angiogenesis related cytokines of endothelial cells isolated from bone marrow of multiple myeloma patients treated with MP0250, confirmed that neutralization of VEGF and HGF results in a change of the cytokine profile towards in favor of antiangiogenic acting cytokines [228].
2.3. Cancer Immunotherapy with T-Cells Redirected against Tumor Antigens
Many hopes have been put on the adoptive cell transfer (ACT) technology for treating B cell malignancies among many other cancer types. In ACT, autologous TILs are initially isolated from an existing tumor mass, co-cultured with IL2 to grow ex vivo [229] and subpopulations of these proliferating cells are tested in vitro against the patient’s original tumor. Following, the number of cells that are active against the tumor is increased [18,73]. Then, the high numbers of TILs are reinfused to the patient to provoke in vivo immune response. In fact, ACT has produced major clinical response in metastatic cancer patients [230,231,232]. Although this technology could be used against different types of cancer, efficacy has been shown only against a few types, including melanoma, possibly related to its immunogenicity and its higher affinity for immune clearance. Therefore, the interest has now shifted into the development of ACT strategies using modified patient-specific T cells that carry genetically engineered antigen receptors in the form of either native or recombinant TCRs or chimeric molecules (CARs) [233].
2.3.1. TCR Engineered-T Cell Immunotherapy
In addition to TIL-based immunotherapy where patient’s TILs are activated and expanded ex vivo, following re-infusion into the patient’s blood circulation, patient’s T lymphocytes can also be genetically engineered to express TCRs obtained from tumor-reactive T cell clones, thereby creating in vivo large quantities of tumor reactive T-cell populations [234]. Given that naturally existing TCRs recognize processed peptides presented on MHC, the TCR engineered-T cells may target a large number of both surface and intracellular tumor-associated antigens. However, TCRs investigated thus far for ACT, have been limited to mostly MHC-I-restricted candidates [235]. Efforts to better engineer high-affinity TCRs have yielded recombinant TCR-carrying T cells that are better effectors than endogenous tumor reactive T cells [235]. The list of ongoing clinical trials using TCR-engineered T cells is progressively expanding, with most of them directed against well characterized tumor-expressed antigens such as gp100 and tyrosinase for melanoma, WT1 for AML and NSCLC, HPV-6 for HPV-associated cancers and others [235,236].
Although the TCR-engineered-based ACT strategies remain an exciting and rapidly evolving field, limitations on the treatment efficacy and the number of patients that can be treated still exist. The MHC restriction especially this of HLA class I molecules which are often downregulated in cancer cells, the dependence on co-receptors such as CD8 and the competition between the transgene receptor and the endogenous TCR for interaction with the available CD3 complex components, usually minimize the therapeutic effectiveness of the approach [236,237]. In addition, the presence of endogenous TCR may allow for chain mispairing between endogenous and introduced TCR a and β chains, leading to reduced receptor expression and often to serious adverse effects propagated by the unpredictable generation of self-reactive TCRs [238,239]. Currently, such “off target, off tumor” effects as the result of TCR chain mispairing or high affinity TCRs recognizing “off target” epitopes, are alleviated by several strategies including among others, codon optimization of the TCR a and β genes and the use of single chain recombinant TCRs [240,241].
2.3.2. CAR Engineered T-Cell Immunotherapy versus T-Cell Activating Polyspecific Antibodies
Unlike the engineered TCRs, chimeric antigen receptors (CARs) are “synthetic molecules” and they do not exist naturally [242]. CAR technology was initially introduced 25 years ago via T cells transduced, by a disarmed virus, to express a chimeric antigen receptor that consists of an extracellular mAb-derived single-chain variable fragment (scFv) region and various intracellular signaling domains. A CD8 or IgG4-derived hinge-transmembrane fragment is used to connect the intracellular and extracellular domains of CAR [243]. Depending on the number of the co-stimulatory domains included in the cytoplasmic fraction of the receptor, together with the CD3ζ TCR constant signaling region, the CARs can be classified into 1st, 2nd or 3rd generation receptors [235] (Figure 2).
Encouraging early clinical results with second-generation anti-CD19 CARs were observed in patients with lymphoma [244,245]. However, the high affinity for target cells conferred by the Ig component of CARs, combined with amplified non-physiologic T-cell signaling in 2nd and 3rd -generation constructs, has been associated with serious adverse events [246]. Reducing on-target toxicities while maintaining antitumor efficacy is an important goal of current investigations [247].
The incorporation of co-stimulatory domains optimizes not only the production of the cytokines necessary for the proliferation of the CAR T cells but also contributes to the more efficient and long-lasting signal transmission by the receptor, thus overcoming most of the immune checkpoint-derived restrictions [248]. 4-IBB, CD28, DAP10, ICOS or OX-40 are within the list of the most commonly utilized co-stimulatory molecules for the construction of the 2nd and 3rd generation CARs. Both generations have shown in vitro and in vivo good efficacies on T cell function following antigen stimulation in multiple malignancies [249,250,251,252].
As CAR technology is moving forward, a 4th generation of chimeric antigen receptors has been recently constructed on the base of the 2nd generation CARs with the inclusion of a cytokine producing gene cassette driven by an NFAT sensitive promoter (Figure 2). These chimeric receptors, widely known as T-cell redirected for universal cytokine mediated killing (TRUCKs) are able to better facilitate and sustain T cell activation compared to alternative and potentially less toxic but effective approaches, namely Bispecific T cell engagers (BiTEs), Tri-specific T cell engagers (TiTEs) and Dual-affinity Re-targeting Antibody (DART) molecules. TRUCKs manage to do so, while attracting innate immune cell populations in order to increase receptor specificity by eliminating antigen-negative cancer cells in the targeted microenvironment [253].
Ideally, CAR T cell therapy could redirect T cell killing to cells that express the antibody’s cognate tumor associated surface antigen, in a MHC-independent manner [254]. Since 2017, CARs have been in the lead against hematologic malignancies, including chronic (CLL) and acute (ALL) leukemia [255,256,257], as well as relapsed/refractory multiple myeloma (MM) [251]. Most of the FDA approved CARs or currently in clinical trials, are directed against B-cell restricted antigens, including CD19, CD138, kappa-light chain and B cell maturation antigen (BCMA) [251,258,259]. To date, the administration of CAR T-cell immunotherapy in hematologic cancer patients has yielded encouraging results with the clinical outcomes to depend mostly on the nature of the targeted antigen, the CAR construct, the dose used and the disease stage [251,260]. On the other hand, CARs designed to treat solid malignancies are still in an infant phase and only recently have yielded some promising results [258,261,262]. Currently, in clinical trials are 2nd and 3rd generation CARs designed to target a wide variety of antigens expressed on solid tumors, including among others the HER2 and EGFR-positive malignancies, carcinoembryonic antigen (CEA) for metastatic tumors of various origin, PCMA antigen for prostate cancer, EGFRvIII for glioblastoma, GD2 and Li-CAM for neuroblastoma, VEGFR-II and cMet for melanoma, Muc-1 for various malignancies and GPC3 for hepatocellular carcinoma [235,262,263]. Ongoing clinical trials using 4th generation CARs (TRUCKs) are directed against PSMA and GD2 in bladder and neuroblastoma cancers, respectively [263]. Currently the CAR technology is moving forward and multi-specific designed CAR T cells can address heterogeneous tumors [263].
Although CARs provide MHC- independent antigen recognition and lack of restrictions related to endogenous co-stimulation of the receptor, the high affinity for target cells conferred by the Ig component of CARs, combined with amplified non-physiologic T-cell signaling in 2nd and 3rd-generation constructs, have been associated with serious adverse events [246,264]. The so called “on target, off tumor” phenomenon which is translated in effective cytotoxic action of CAR-T cells against non-tumor cells that express the targeted antigen, has been noted in most CAR applications resulting in significant limitations of their tumor-specific effectiveness [246,247,264]. In addition, CAR recognition is only limited to surface antigens, while targeted antigen loss, anti-transgene immunogenicity especially against murine-derived scFv or activation of immune-checkpoint pathways such as PD-1/PD-L1 by MDSCs can further decrease the efficiency of CAR-T cell based ACT trials [235,265]. Other serious adverse effects include but are not limited to, cytokine storm [266] and tumor lysis syndrome [264], suggesting that continuing efforts are required to decrease the toxicities related to CAR-T cell immunotherapeutic applications and improve the clinical outcomes.
New approaches by bispecific antibodies and antibody-associated immune modulation can now offer intriguing opportunities, as discussed in Reference [267]. On this axis, intriguing opportunities for redirecting T cells against cancer cells have been offered by the development of alternative antibody-associated immune-modulating approaches, including polyspecific antibodies such as BiTEs, TiTEs and DART molecules [267,268]. Polyspecific antibodies can be directed against multiple tumor antigens to eradicate tumor cells more precisely and effectively [269].
Bispecific antibodies with ability to engage two different antigens are the most commonly used polyspecific antibodies [270]. BiTEs, also known as T-cell activating bispecific antibodies (TABs), are usually composed of two tandem single chain variable fragments (scFvs), each with a unique antigen specificity [268,270,271,272]. Most BiTEs, including Blinatumomab, the only bispecific antibody approved for the treatment of ALL, simultaneously recognize CD3 and a tumor-associated antigen (TAA), thus enabling engagement of effector T cells with cancer cells [268,269,273]. This crosslinking leads to T cell activation and direct cytotoxicity regardless of TCR specificity and MHC restrictions, while induction of cytokine responses may further trigger a bystander effect on adjacent cells [268,274]. DART proteins, are even smaller than BiTEs and have a diabody format where one VL chain is followed by the VH chain of the second binder and the two polypeptide chains align in a head-to-tail fashion [268].
Although BiTEs may redirect the large reservoir of resident T cells to tumors, while other T-cell activating approaches, such as CARs or TRUCKS rely on significant in vivo expansion to exert antitumor activity, it is still unclear whether BiTEs may potentially have a comparative advantage over CARs, with respect to their toxicity, specificity and binding activity on the target epitopes [275]. Among the major limitations in the activity and effectiveness of BiTEs appear to be T cell anergy and exhaustion triggered mainly by PD1/PD-L1 activation [268]. Efforts to bypass the aforementioned limitations by combinations of BiTEs with antibodies against immune checkpoint inhibitors (checkpoint blockage) or other molecules, have already achieved promising preclinical results; however further research is needed to validate the efficacy of the method and the durability of the results for most BiTEs [268]. Moreover, and contrary to the considerable activities of most BiTEs on hematologic malignancies—such as ALL—the therapeutic benefit of newly developed BiTEs on solid tumors is still under investigation [269]. Mechanisms associated with TAB’s failure to kill solid tumors mainly include, antibody inability to access the target tissue, dose-limiting toxicities and low half-lives, on target but off-tumor toxicities, fatal cytokine release syndrome and other adverse effects similar to those observed with CAR-T cell approaches, as described above [268]. DARTs also face the problem of low in vivo half-life, which can be partially solved by fusion of an Fc domain that prolongs antibody stability in the serum [268,276]. However, the two linkers on the diabody format may reduce the mobility of the antigen binding sites, thus restricting antigen recognition [277].
Complimentary to T-cell activating polyspecific antibodies, innovative techniques have also introduced BiKEs and TriKEs, in an effort to enhance the therapeutic benefit of NK cell-based immunotherapy. BiKEs and TriKEs directly trigger NK activation and cytotoxicity against various TAAs on tumor cells after engagement to CD16 [269,278,279]. Although in vivo safety is still uncertain for those molecules, they keep high promise for use in triggering cytokine production [280].
As discussed by Rancie et al., there is no doubt that new generation polyspecific T and NK-cell engagers have successfully joined CARs on the cancer immunotherapy highway [280]; however, major differences between the two strategies determine not only their performance but also their suitability for use depending on the type of cancer and the targeted TAA [269]. Although CAR T cell production is significantly costly, as it is patient specific, however it appears that new CAR T cell constructs have the comparative advantages of being capable to (1) bind TAAs even at low density causing direct lysis, (2) naturally extravasate and travel between endothelial barriers, thus allowing better distribution and (3) expand in vivo and establishing a memory cell pool that facilitates prolonged response [269] Overall, efforts to enhance the therapeutic benefit of CAR-T based approaches by developing strategies to reduce the adverse effects and especially on-target toxicities, while maintaining antitumor efficacy, are important subjects of intense research on CAR-T cell immunotherapy era [247].
3. Therapeutic Vaccines
Vaccines against tumor initiation-involved viruses can provide a precautionary measure against tumor development. The human papilloma virus (HPV) is linked to cervical and oropharyngeal head and HNSCC and vaccination with a recombinant vaccine against certain subtypes of HPV can now decrease the occurrence of these cancers [56,57,71,281]. Likewise, vaccination against hepatitis B virus (HBV) can help prevent hepatocellular carcinoma [55].
Apart from preventative vaccines, therapeutic vaccines against tumor antigens can also yield encouraging immunotherapeutic outcomes. Engineered vaccines against both tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) can provoke immunological response against cancer cells, which persists in the future with the help of memory T and B cells. So far, the only FDA approved therapeutic cancer vaccine, is Sipuleucel-T, administered in prostate cancer patients. Autologous lymphocytes are isolated from the patient and exposed in vitro to a conjugate of prostatic acid phosphatase with granulocyte-macrophage colony-stimulating factor (PAP-GM-CSF), before being reinfused to the patient. These immune cells have mainly differentiated into DCs and present the PAP antigen, which is found in the majority of the prostrate cells, to T cells provoking an immune response [59]. Sipuleucel-T increased prostate cancer patients’ overall survival by 4 months and provided better survival rates versus the controls [59]. A trial using a vaccine against gp100 combined with immune-modulators (e.g., IL2) is under way for melanoma patients [60].
In addition, stimulator of interferon genes (STING) is a transmembrane protein that induces type I interferon production in cells infected with intracellular pathogens. Combination therapy of STING with anti–PD-1 could promote antitumor innate immunity and improved response [61].
GVAX is another therapeutic vaccine against pancreatic cancer. It is composed of whole tumor cells that have been genetically engineered to express GM-CSF. Initial trials with GVAX did not improve the overall survival of patients with metastatic pancreatic cancer but when combined with the mesothelin-secreting vaccine, CRS-207, it synergistically produced longer overall survival [62]. Nevertheless, the incapability of most vaccines to produce immune response, when administered alone, shows the ability that tumor cells have to evade the immune system and the necessity for successful combinatorial therapies. As most of the therapeutic vaccines have yielded modest clinical benefit to patients with advanced cancers, their value is now underscored.
Additionally, the introduction of personalized mRNA/DNA-based vaccines against TSAs of the patient, provides the prospect for better anticancer vaccines.
A novel approach suggests that the immune system can neutralize various immune-suppressive signals, through self-reactive, pro-inflammatory T cells which can target inhibitory Treg cells and were therefore called anti-regulatory T cells (anti-Tregs) [282]. The activation of these anti-Treg cells was suggested to offer a new path to block immune inhibitory pathways within the tumor microenvironment [283]. Thus, if successfully targeted, a therapeutic vaccination aiming to activate anti-Tregs could enhance anti-tumor immunity by relieving immune suppression and potentiating effective anti-tumor T cell responses [284]. The potential use of an anti-Treg-activating vaccine was recently suggested that it could attract T cells into the tumor microenvironment, eventually producing more susceptible targets to immune checkpoint inhibition [284].
4. Mutational and Epigenetic Landscape in Tumor Progression and Response to Therapy
The genetic code of cancer cells includes loss- and gain-of-function mutations, derived from DNA replication errors or caused by genotoxic agents [285,286]. These mutations can be inherited or acquired (germline or somatic) in pre-malignant cells and provide the stage for further genetic alterations, driving cancer pathogenesis [287,288]. Several theories try to explain the mutational landscape evolution of tumors [289,290,291]. Been widely adopted, the clonal evolution concept suggests that driver mutations are positively selected based on their ability to provide a fitness advantage to tumor cells, such as promoting survival and growth, where they clonally expand in expense to less fit tumor cells [289,292]. On average, tumors carry 4 coding substitutions in driver genes (ranging from 1 to 10 depending on the tumor type) half of which are not in known cancer genes [293]. Negative selection is mostly absent in cancer genomes with less than 1 coding substitution in a given gene, as the majority of these mutations lead to cell death and are eventually lost [293]. Moreover, neutral or passenger mutations dominate cancer genomes but are predominantly synonymous with no selective advantage. However, some synonymous mutations, such as the T125T hotspot mutation in TP53, can affect gene splicing but also protein folding and act as driver mutations [293,294,295,296].
Besides exogenous mutagens, cell intrinsic processes and especially DNA replication errors is the primary source of genomic instability and ultimately malignancy [297]. DNA polymerases and mismatch repair (MMR) proteins are responsible for correcting DNA replication errors thereby maintaining genomic stability [298]. Mutations in these proteins significantly impair DNA replicative fidelity, frequently resulting in microsatellite instability (MSI) [299,300,301,302,303].
The mutational landscape of tumors can shape cancer immunity and response to immunotherapy and other chemotherapeutic strategies [304] (Figure 3). Tumor neoantigens (TNAs) are generated by nonsynonymous mutations in malignant cells, providing a novel target for immune responses [305,306,307,308]. Some neoantigens can be immunogenic and induce host immune responses against tumors. On the other hand, a high mutational burden and the number of TNAs can exhibit adverse effects and favor immunoevasion of cancer cells from host immune responses [309,310]. Moreover, these hypermutated tumors can also induce immunosuppression by expressing immunoinhibitory molecules like PD-L1 and CTLA-4, thus developing adaptive resistance [310,311,312]. The tumor microenvironment, TNA immunogenicity, intratumoral heterogeneity (ITH) and other factors seem to play a role on how tumors respond to immunotherapy [313,314,315]. The immunophenotype is also a key determinant that drives clinical decision in cancer and is now considered an additional challenge for immunotherapy. For example, Atezolizumab (anti-PD-L1) has shown encouraging activity against metastatic colorectal cancer when combined with chemotherapy and/or targeted therapies, especially with MEK inhibitor Cobimetinib [316]. Similarly, combination therapies of immunotherapy with existing chemotherapy, radiation or other immunotherapy with different mechanisms of action has been suggested to be evaluated to achieve excellent outcomes in patients with esophageal cancer [317].
Interestingly, the number of TNAs is highly correlated with the mutational load and the infiltration of TILs [318]. Furthermore, predicted immunogenic mutations in cancer patients extracted from The Cancer Genome Atlas (TCGA) database, linked mutational epitopes with higher tumor CTL content and increased patient survival [319]. The positive prognostic value of elevated mutational and TNA burden is also prevalent in a variety of cancers that show active immunological responses and signatures, regardless of treatment that are associated with improved survival of cancer patients [313,320,321,322,323,324]. The recognition of TNAs by host T cells makes them ideal immune targets and gives hope for further immunotherapeutic intervention by targeting them [325,326,327].
In addition, several studies demonstrated that the evaluation of the epigenetic status of patients, as well as induced epigenetics modification, might predict the response rate of patients and boost the efficacy of cancer immunotherapies [328,329]. Several studies focused the attention on the role of methylation events and microRNAs (miRNAs) alterations in cancer development, aggressiveness and response to therapies [330]. In particular, the methylation status of PD-L1 may be associated with the survival rate of acute myeloid leukemia patients, where patients with hypomethylation of PD-L1 promoter have a lower overall survival compared to patients with high levels of methylation [331]. Other studies demonstrated that the treatment with hypomethylating agents before the administration of immune checkpoint inhibitors are able to improve the response rate in several tumors [332,333,334].
Regarding miRNAs, numerous studies have identified specific miRNAs associated with the development, progression and response to therapies in different cancer types [335,336,337,338,339,340,341]. In particular, it was demonstrated that miRNAs may serve for the development of new diagnostic, prognostic and therapeutic strategies in the context of cancer immunotherapy. Boeri M et al. (2019) identify a set of miRNAs whose expression levels were associated with the response rate of NSCLC patients to PD1/PD-L1 inhibitors [342]. Furthermore, in a recent review of literature it was reported that several miRNAs, of which miR-34a and miR-200 represent two of the most representative miRNAs, are able to directly regulate the PD-L1 expression leading to a poor prognosis of mesothelioma patients [343]. Despite the growing body of evidences about the assessment of the mutational and epigenetics landscape of patients to predict the response to immune checkpoint inhibitors, further studies are needed to identify specific biomarkers and clarify the functional roles of genetic and epigenetics modification during cancer immunotherapy.
5. Modulation of Gut Microbiota Composition and Response to Immunotherapy
In the last decade, several studies demonstrated that the human gut microbiota is involved not only in gastrointestinal diseases but play also crucial roles in the development and progression of other pathological conditions, including neurodegenerative diseases and cancer [344]. In particular, it was demonstrated that the composition of human gut microbiota may influence the response rate of cancer patients treated with chemotherapy and immune checkpoint inhibitors enhancing or reducing the overall survival and progression-free survival of patients [345,346,347]. With a particular regard to immunotherapy, it was demonstrated that the modulation of gut microbiota with the administration of specific probiotics strains, including Lactobacillus rhamnosus GG, Bifidobacterium longum, Enterococcus faecium and Collinsella aerofaciens, may influence the patient’s response to anti-CTLA-4 and anti-PD-1/PD-L1 inhibitors [346,348,349].
To further strengthen the important role of gut microbiota homeostasis during immunotherapy, other studies demonstrated that antibiotic treatments before the administration of immune checkpoint inhibitors lead to a lower response rate to immune checkpoint inhibitors [350].
Finally, it was also demonstrated that microbiota modulation through fecal microbial transplantation (FMT) could be a good strategy to enhance the responsiveness of patients treated with immunotherapy [351].
6. Evolution of the Landscape on Cancer Neoepitopes during Immunotherapy
In cancers, approximately 99% of somatic substitutions are well tolerated and accumulate in malignant cells, often leading to hypermutation [352,353]. Prediction models estimate TNA numbers to be associated with mutational load; but experimental validation reveals that only a small fraction of neoepitopes can bind to MHC, recognized by TCR and be immunogenic [354]. The highly immunogenic TNAs generated by nonsynonymous mutations are selectively depleted by the host immune surveillance thereby shaping tumor evolution [355,356]. A model for evolution of Tumor-Immune associations proposes that tumor intrinsic factors like TNAs elicit immune infiltrates which kill immunogenic clones; driving the growth of immune resistant or immune suppressing subclones [356] (Figure 3).
Studies show that the TNA landscape evolves heterogeneously through multiple distinct tumor immune microenvironments, such as in metastatic lesions, over the course of tumor progression and treatment status [357,358,359]. Furthermore, in a case of long term cancer survivors, neoantigen quality rather than quantity is identified as a biomarker of immunogenic tumors, that could be used to better direct immunomodulatory treatments [313]. Moreover, the number of TNAs per missense mutation, referred to as neoantigen frequency but not the number of missense mutations or total TNAs, correlates with clinical outcomes and could act as a prognostic factor and potential biomarker for cancer immunotherapy [360]. Tumor heterogeneity seems to favor TNA diversity; in addition to high clonal TNA burden, tumors appear to respond better to immune checkpoint blockers and have improved prognosis compared to low clonal TNA bearing tumors [314,361,362].
Despite the significant contribution of immune checkpoint blockers in cancer immunotherapy, during immune checkpoint blockade, the dynamics of mutational landscapes affect tumor neoantigens through genomic changes to truncal and subclonal mutations that eliminate immunogenic TNAs and develop clones with acquired resistance, further complicating cancer treatment [307,363]. In addition, immune checkpoint blockers are found to exert T cell-dependent immunoselective pressure in tumor progression, effectively potentiating cancer immunoediting [308,364].
Tumor and microenvironment changes are observed in response to anti-PD-1 therapy. Responding patients exhibit reduction in mutation and neoantigen burden as well as clonal evolution-directed immunoediting [365]. Furthermore, expansion of the T cell repertoire and production of specific T cell clonotypes target tumor neoantigens during anti-PD-1 treatment, which also appears to upregulate an array of immune checkpoint-related genes [365]. Moreover, immunotherapy with anti-CTLA-4 antibodies seems to enhance T cell priming and induce newly detected T cell responses broadening the TCR repertoire [366,367]. Mobilization and increase of the TCR repertoire is also observed after immunotherapy with anti-CD4 monoclonal antibody or TIL and is associated with increased antitumor immunity and improved treatment response [368,369,370].
Strategies implementing longitudinal and multiregional sampling of tumors throughout cancer progression and treatment of individual patients provide the best information of tumor neoantigen and microenvironment evolution [326]. Interestingly, despite the huge challenges, researchers were able to investigate tumor response to immune checkpoint blockers over time and identified potential mechanisms of therapeutic resistance as well as adaptive immune signatures on early treated biopsies that predict response to immune checkpoint blockers [371,372].
7. Conclusions
In summary, the immunotherapeutic developments during the last years have significantly increased our hopes for successfully treating different cancer types. However, the development of new, more effective anticancer immunotherapeutic agents, urges a thorough understanding of the aspects that allow cancer cells to escape elimination by immune cells. Many cancer patients have shown a better clinical response when treated with a single of the combined immunotherapies. Still, a broader group of patients needs to benefit from these immunotherapies. Furthermore, we need to keep in mind that these immunotherapies do not come without adverse effects and complications. Therefore, the focus has already shifted towards the design of novel immunotherapies, which are tailored to the patient’s genetic profile and the appropriate selection of cancer patients who are more likely to present durable responses.
Author Contributions
Conceptualization, A.Z.; methodology, T.C., A.Z.; validation, T.C., S.B., L.F., M.L. and A.Z.; writing—original draft preparation, T.C., S.B., L.F., M.L. and A.Z.; writing—review and editing, T.C., A.Z.; visualization, T.C., S.B., A.Z.; supervision, A.Z.; project administration, A.Z.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Tykodi, S.S. PD-1 as an emerging therapeutic target in renal cell carcinoma: Current evidence. Onco Targets Ther. 2014, 7, 1349–1359. [Google Scholar] [CrossRef]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Fountain, E.; Bassett, R.L.; Cain, S.; Posada, L.; Gombos, D.S.; Hwu, P.; Bedikian, A.; Patel, S.P. Adjuvant Ipilimumab in High-Risk Uveal Melanoma. Cancers 2019, 11, 152. [Google Scholar] [CrossRef]
- Inozume, T.; Yaguchi, T.; Ariyasu, R.; Togashi, Y.; Ohnuma, T.; Honobe, A.; Nishikawa, H.; Kawakami, Y.K.T. Analysis of the Tumor Reactivity of Tumor-Infiltrating Lymphocytes in a Metastatic Melanoma Lesion that Lost Major Histocompatibility Complex Class I Expression after Anti-PD-1 Therapy. J. Investig. Dermatol. 2019, 139, 1490–1496. [Google Scholar] [CrossRef]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.C.; Jegede, O.; Shukla, S.A.; Braun, D.A.; Horak, C.E.; Wind-Rotolo, M.; Ishii, Y.; Catalano, P.J.; Grosha, J.; Flaifel, A.; et al. irRECIST for the Evaluation of Candidate Biomarkers of Response to Nivolumab in Metastatic Clear Cell Renal Cell Carcinoma: Analysis of a Phase II Prospective Clinical Trial. Clin. Cancer Res. 2019, 25, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, Q.; Yu, X.; Khan, K.; Weng, X.; Guan, M. Complete response associated with immune checkpoint inhibitors in advanced non-small-cell lung cancer: A meta-analysis of nine randomized controlled trials. Cancer Manag. Res. 2019, 11, 1623–1629. [Google Scholar] [CrossRef]
- Ning, Y.; Suzman, D.; Maher, V.E.; Zhang, L.; Tang, S.; Ricks, T.; Palmby, T.; Fu, W.; Liu, Q.; Goldberg, K.B.; et al. FDA Approval Summary: Atezolizumab for the Treatment of Patients with Progressive Advanced Urothelial Carcinoma after Platinum-Containing Chemotherapy. Oncologist 2017, 22, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marté, J.L.; Lepone, L.M.; et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.; Lee, J.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Updated efficacy and tolerability of durvalumab in locally advanced or metastatic urothelial carcinoma. J. Clin. Oncol. 2017, 35, 286. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Iversen, T.Z.; Engell-Noerregaard, L.; Mellemgaard, A.; Andersen, M.H.; Svane, I.M. Durable clinical responses and long-term follow-up of stage III-IV non-small-cell lung cancer (NSCLC) patients treated with IDO peptide vaccine in a phase i study-a brief research report. Front. Immunol. 2018, 9, 2145. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van Den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, W.; Zhang, X.; Ding, Y.; Du, Q.; Hu, R. 1-L-MT, an IDO inhibitor, prevented colitis-associated cancer by inducing CDC20 inhibition-mediated mitotic death of colon cancer cells. Int. J. Cancer 2018, 143, 1516–1529. [Google Scholar] [CrossRef]
- Zakharia, Y.; Rixe, O.; Ward, J.H.; Drabick, J.J.; Shaheen, M.F.; Milhem, M.M.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; Link, C.J.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 9512. [Google Scholar] [CrossRef]
- Jha, G.G.; Gupta, S.; Tagawa, S.T.; Koopmeiners, J.S.; Vivek, S.; Dudek, A.Z.; Cooley, S.A.; Blazar, B.R.; Miller, J.S. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35 (Suppl. S15), 3066. [Google Scholar] [CrossRef]
- Bravo-Alegria, J.; Maegawa, S.; Callegari, K.; Kennis, B.; Ma, W.; Brugmann, W.; Rao, A.; Lee, D.A.; Khatua, S.; Zaky, W.; et al. IMMU-22. Natural Killer Cell Immunotherapy For Diffuse Intrinsic Pontine Glioma. Neuro Oncol. 2018, 20, 103. [Google Scholar] [CrossRef]
- Emadi, A.; Holtzman, N.G.; Imran, M.; El Chaer, F.; Koka, M.; Singh, Z.; Shahlaee, A.; Sausville, E.A.; Law, J.; Lee, S.T.; et al. Indoximod in combination with idarubicin and cytarabine for upfront treatment of patients with newly diagnosed acute myeloid leukemia (AML): Phase 1 report. Haematologica 2017, 102, 375. [Google Scholar]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolušić, E.; Frédérick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Nayak-Kapoor, A.; Hao, Z.; Sadek, R.; Dobbins, R.; Marshall, L.; Vahanian, N.N.; Jay Ramsey, W.; Kennedy, E.; Mautino, M.R.; Link, C.J.; et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J. Immunother. Cancer 2018, 6, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.H.; LoRusso, P.M.; Burris, H.A.; Gordon, M.S.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223. [Google Scholar] [CrossRef] [PubMed]
- Rexin, P.; Tauchert, A.; Hänze, J.; Heers, H.; Schmidt, A.; Hofmann, R.; Hegele, A. The Immune Checkpoint Molecule CD200 Is Associated with Tumor Grading and Metastasis in Bladder Cancer. Anticancer Res. 2018, 38, 2749–2754. [Google Scholar] [PubMed]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, W.E.; et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, V.; Sundarapandiyan, K.; Zhao, B.; Bylesjo, M.; Marsh, H.C.; Keler, T. Characterization of the human T cell response to in vitro CD27 costimulation with varlilumab. J. Immunother. Cancer 2015, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Yanagita, T.; Murata, Y.; Tanaka, D.; Motegi, S.; Arai, E.; Daniwijaya, E.W.; Hazama, D.; Washio, K.; Saito, Y.; Kotani, T.; et al. Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2017, 2, e89140. [Google Scholar] [CrossRef] [PubMed]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Perez-Gracia, J.L.; Martin-Algarra, S.; et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2-/-IL2Rγnull immunodeficient mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef]
- Faden, D.L.; Concha-Benavente, F.; Chakka, A.B.; McMichael, E.L.; Chandran, U.; Ferris, R.L. Immunogenomic correlates of response to cetuximab monotherapy in head and neck squamous cell carcinoma. Head Neck 2019, 41, 2591–2601. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Yalniz, F.F.; Daver, N.; Rezvani, K.; Kornblau, S.; Ohanian, M.; Borthakur, G.; DiNardo, C.D.; Konopleva, M.; Burger, J.; Gasior, Y.; et al. A Pilot Trial of Lirilumab With or Without Azacitidine for Patients With Myelodysplastic Syndrome. Clin. Lymphoma Myeloma Leuk. 2018, 18, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Masarova, L.; Patel, K.P.; Newberry, K.J.; Cortes, J.; Borthakur, G.; Konopleva, M.; Estrov, Z.; Kantarjian, H.; Verstovsek, S. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: A post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017, 4, e165–e175. [Google Scholar] [CrossRef]
- Atkins, M.B.; Kunkel, L.; Sznol, M.; Rosenberg, S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J. Sci. Am. 2000, 6 (Suppl. S1), S11–S14. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 Consecutive Patients With Metastatic Melanoma or Renal Cell Cancer Using High-Dose Bolus Interleukin 2. JAMA J. Am. Med. Assoc. 1994, 271, 907–913. [Google Scholar] [CrossRef]
- Mocellin, S.; Pasquali, S.; Rossi, C.R.; Nitti, D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2010, 102, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory DLBCL. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453. [Google Scholar] [CrossRef]
- Kurokawa, T.; Oelke, M.; Mackensen, A. Induction and clonal expansion of tumor-specific cytotoxic T lymphocytes from renal cell carcinoma patients after stimulation with autologous dendritic cells loaded with tumor cells. Int. J. Cancer 2001, 91, 749–756. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Parsonage, G.; Zhuang, X.; Machado, L.R.; James, C.H.; Salman, A.; Searle, P.F.; Hui, E.P.; Chan, A.T.; Lee, S.P. Human leukocyte antigen (HLA) A∗1101-restricted epstein-barr virus-specific t-cell receptorgenetransfer to target nasopharyngeal carcinoma. Cancer Immunol. Res. 2015, 3, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Chemin, I. Evaluation of a hepatitis B vaccination program in Taiwan: Impact on hepatocellular carcinoma development. Future Oncol. 2010, 6, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Mammas, I.N.; Sourvinos, G.; Zaravinos, A.; Spandidos, D.A. Vaccination against human papilloma virus (HPV): Epidemiological evidence of HPV in non-genital cancers. Pathol. Oncol. Res. 2011, 17, 103–119. [Google Scholar] [CrossRef]
- Kreimer, A.R. Prospects for prevention of HPV-driven oropharynx cancer. Oral Oncol. 2014, 50, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Paavonen, J.; Naud, P.; Salmerón, J.; Wheeler, C.M.; Chow, S.N.; Apter, D.; Kitchener, H.; Castellsague, X.; Teixeira, J.C.; Skinner, S.R.; et al. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): Final analysis of a double-blind, randomised study in young women. Lancet 2009, 374, 301–314. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Gibney, G.T.; Atkins, M.B. Advances in immunotherapy for melanoma. BMC Med. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Donskov, F.; McDermott, D.F.; Lee, J.L.; Szczylik, C.; Malik, J.; Alekseev, B.Y.; Larkin, J.M.G.; Matveev, V.B.; Airatovich Gafanov, R.; Tomczak, P.; et al. 871PKEYNOTE-427 cohort A: Pembrolizumab monotherapy as first-line therapy in advanced clear cell renal cell carcinoma (ccRCC). Ann. Oncol. 2018, 29 (Suppl. S8), viii303–viii331. [Google Scholar] [CrossRef]
- Economopoulou, P.; Agelaki, S.; Perisanidis, C.; Giotakis, E.I.; Psyrri, A. The promise of immunotherapy in head and neck squamous cell carcinoma. Ann. Oncol. 2016, 27, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget 2014, 5, 3956–3969. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Sunwoo, J.B. Immunotherapy for Head and Neck Squamous Cell Carcinoma. Hematol. Oncol. Clin. N. Am. 2015, 29, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar]
- Armand, P.; Nagler, A.; Weller, E.A.; Devine, S.M.; Avigan, D.E.; Chen, Y.B.; Kaminski, M.S.; Holland, H.K.; Winter, J.N.; Mason, J.R.; et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large b-cell lymphoma: Results of an international phase II trial. J. Clin. Oncol. 2013, 31, 4199–4206. [Google Scholar] [CrossRef]
- Bryan, L.J.; Gordon, L.I. Pidilizumab in the treatment of diffuse large B-cell lymphoma. Expert Opin. Biol. Ther. 2014, 14, 1361–1368. [Google Scholar] [CrossRef]
- Hamilou, Z.; Lavaud, P.; Loriot, Y. Atezolizumab in urothelial bladder carcinoma. Future Oncol. 2018, 14, 331–341. [Google Scholar] [CrossRef]
- Gkolfinopoulos, S.; Mountzios, G. Recent clinical trials of immunotherapy in non-small-cell lung cancer. Immunotherapy 2019, 11, 461–466. [Google Scholar] [CrossRef]
- Passiglia, F.; Galvano, A.; Rizzo, S.; Incorvaia, L.; Listì, A.; Bazan, V.; Russo, A. Looking for the best immune-checkpoint inhibitor in pre-treated NSCLC patients: An indirect comparison between nivolumab, pembrolizumab and atezolizumab. Int. J. Cancer 2018, 142, 1277–1284. [Google Scholar] [CrossRef]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Thomas, A.; Hassan, R. CTLA-4 blockade in mesothelioma: Ineffective or reason for optimism? Lancet Oncol. 2017, 18, 1150–1151. [Google Scholar] [CrossRef]
- Li, J.; Ni, L.; Dong, C. Immune checkpoint receptors in cancer: Redundant by design? Curr. Opin. Immunol. 2017, 45, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 19, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, M.E. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270. [Google Scholar] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4 and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- DeBenedette, M.A.; Randall Chu, N.; Pollok, K.E.; Hurtado, J.; Wade, W.F.; Kwon, B.S.; Watts, T.H. Role of 4-1BB ligand in costimulation of T lymphocyte growth and its upregulation on M12 B lymphomas by camp. J. Exp. Med. 1995, 181, 985–992. [Google Scholar] [CrossRef]
- Gramaglia, I.; Weinberg, A.D.; Lemon, M.; Croft, M. Ox-40 ligand: A potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998, 161, 6510–6517. [Google Scholar]
- Tesselaar, K.; Arens, R.; Van Schijndel, G.M.W.; Baars, P.A.; Van Der Valk, M.A.; Borst, J.; Van Oers, M.H.J.; Van Lier, R.A.W. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat. Immunol. 2003, 4, 49–54. [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Nocentini, G.; Giunchi, L.; Ronchetti, S.; Krausz, L.T.; Bartoli, A.; Moraca, R.; Migliorati, G.; Riccardi, C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6216–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janakiram, M.; Chinai, J.M.; Zhao, A.; Sparano, J.A.; Zang, X. HHLA2 and TMIGD2: New immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology 2015, 4, e1026534. [Google Scholar] [CrossRef] [PubMed]
- Tamada, K.; Shimozaki, K.; Chapoval, A.I.; Zhai, Y.; Su, J.; Chen, S.F.; Hsieh, S.L.; Nagata, S.; Ni, J.; Chen, L. LIGHT, a TNF-Like Molecule, Costimulates T Cell Proliferation and Is Required for Dendritic Cell-Mediated Allogeneic T Cell Response. J. Immunol. 2000, 164, 4105–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenberger, S.P.; Toes, R.E.M.; Van Dervoort, E.I.H.; Offringa, R.; Melief, C.J.M. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD4OL interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Tahara-Hanaoka, S.; Miyamoto, A.; Hara, A.; Honda, S.I.; Shibuya, K.; Shibuya, A. Identification and characterization of murine DNAM-1 (CD226) and its poliovirus receptor family ligands. Biochem. Biophys. Res. Commun. 2005, 329, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-M.; Bhawan, S.; Majima, T.; Wei, H.; Nishimura, M.I.; Yagita, H.; Kumar, V. Cutting Edge: The NK Cell Receptor 2B4 Augments Antigen-Specific T Cell Cytotoxicity Through CD48 Ligation on Neighboring T Cells. J. Immunol. 2003, 170, 4881–4885. [Google Scholar] [CrossRef]
- Seiffert, M.; Brossart, P.; Cant, C.; Cella, M.; Colonna, M.; Brugger, W.; Ullrich, A.; Bühring, H.J. Signal-regulatory protein α (SIRPα) but not SIRPβ is involved in T-cell activation, binds to CD47 with high affinity and is expressed on immature CD34+CD38- hematopoietic cells. Blood 2001, 97, 2741–2749. [Google Scholar] [CrossRef]
- Fernandez, N.C.; Treiner, E.; Vance, R.E.; Jamieson, A.M.; Lemieux, S.; Raulet, D.H. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood 2005, 105, 4416–4423. [Google Scholar] [CrossRef] [Green Version]
- Hodge, J.W.; Greiner, J.W.; Tsang, K.Y.; Sabzevari, H.; Kudo-Saito, C.; Grosenbach, D.W.; Gulley, J.L.; Arlen, P.M.; Marshall, J.L.; Panicali, D.; et al. Costimulatory molecules as adjuvants for immunotherapy. Front. Biosci. 2006, 11, 788–803. [Google Scholar] [CrossRef]
- Van Seventer, G.A.; Shimizu, Y.; Horgan, K.J.; Shaw, S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J. Immunol. 1990, 144, 4579–4586. [Google Scholar]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.H.G.; Wolkers, M.C.; Van Den Boom, M.D.; Van Den Valk, M.A.; Schumacher, T.N.M. Immunotherapy through TCR gene transfer. Nat. Immunol. 2001, 2, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, M.L.; Lucas, C.L.; Buhler, L.; Rayat, G.; Rodriguez-Barbosa, J.I. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J. Leukoc. Biol. 2010, 87, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2011, 208, 1331. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 Interactions Promote Cytokine and NK Cell-Mediated Suppression of Poorly Immunogenic Melanoma Metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretz-Rommel, A.; Qin, F.; Dakappagari, N.; Ravey, E.P.; McWhirter, J.; Oltean, D.; Frederickson, S.; Maruyama, T.; Wild, M.A.; Nolan, M.J.; et al. CD200 Expression on Tumor Cells Suppresses Antitumor Immunity: New Approaches to Cancer Immunotherapy. J. Immunol. 2007, 178, 5595–5605. [Google Scholar] [CrossRef] [Green Version]
- Suh, W.K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A.; et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Husain, B.; Ramani, S.R.; Chiang, E.; Lehoux, I.; Paduchuri, S.; Arena, T.A.; Patel, A.; Wilson, B.; Chan, P.; Franke, Y.; et al. A platform for extracellular interactome discovery identifies novel functional binding partners for the immune receptors B7-H3/CD276 and PVR/CD155. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef]
- Sica, G.L.; Choi, I.H.; Zhu, G.; Tamada, K.; Wang, S.D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-H4, a Molecule of the B7 Family, Negatively Regulates T Cell Immunity. Immunity 2003, 18, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Lines, J.L.; Sempere, L.F.; Broughton, T.; Wang, L.; Noelle, R. VISTA Is a Novel Broad-Spectrum Negative Checkpoint Regulator for Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 510–517. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Chinai, J.M.; Buhl, S.; Scandiuzzi, L.; Ray, A.; Jeon, H.; Ohaegbulam, K.C.; Ghosh, K.; Zhao, A.; Scharff, M.D.; et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc. Natl. Acad. Sci. USA 2013, 110, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell aloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A.A. LAG-3 Regulates Plasmacytoid Dendritic Cell Homeostasis. J. Immunol. 2009, 182, 1885–1891. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.A.; Pardoll, D.M.; Drake, C.G. Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T Cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, M.V.; Drake, C.G. LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 2010, 344, 269–278. [Google Scholar]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4 + CD25highFoxp3+ Regulatory T Cells That Are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.; Alderson, R.F.; Chen, F.Z.; Huang, L.; Zhang, W.; Gorlatov, S.; Burke, S.; Ciccarone, V.; Li, H.; Yang, Y.; et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin. Cancer Res. 2012, 18, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, Z.; Zhang, Y.; Quan, Q.; Huang, L.; Xu, Y.; Cao, L.; Zhang, X. Association of increased B7 protein expression by infiltrating immune cells with progression of gastric carcinogenesis. Medicine (Baltimore) 2019, 98, e14663. [Google Scholar] [CrossRef]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-cell-engaging B7-H4/CD3 bispecific Fab-scFv antibody targets human breast cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef]
- Altan, M.; Kidwell, K.M.; Pelekanou, V.; Carvajal-Hausdorf, D.E.; Schalper, K.A.; Toki, M.I.; Thomas, D.G.; Sabel, M.S.; Hayes, D.F.; Rimm, D.L. Association of B7-H4, PD-L1 and tumor infiltrating lymphocytes with outcomes in breast cancer. NPJ Breast Cancer 2018, 4, 40. [Google Scholar] [CrossRef]
- Azuma, T.; Sato, Y.; Ohno, T.; Azuma, M.; Kume, H. Serum soluble B7-H4 is a prognostic marker for patients with non-metastatic clear cell renal cell carcinoma. PLoS ONE 2018, 13, e0199719. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, F.; Tang, C.; Chen, D.; Qin, Z.; Hua, W.; Xu, M.; Zhong, P.; Yu, S.; Chen, D.; et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: An exploratory randomized phase II clinical trial. Cancer Immunol. Immunother. 2018, 67, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Radichev, I.A.; Maneva-Radicheva, L.V.; Amatya, C.; Salehi, M.; Parker, C.; Ellefson, J.; Burn, P.; Savinov, A.Y. Loss of Peripheral Protection in Pancreatic Islets by Proteolysis-Driven Impairment of VTCN1 (B7-H4) Presentation Is Associated with the Development of Autoimmune Diabetes. J. Immunol. 2016, 196, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroel-Espindola, F.; Yu, X.; Datar, I.; Mani, N.; Sanmamed, M.; Velcheti, V.; Syrigos, K.; Toki, M.; Zhao, H.; Chen, L.; et al. Spatially resolved and quantitative analysis of vista/pd-1h as a novel immunotherapy target in human non–small cell lung cancer. Clin. Cancer Res. 2018, 24, 1562–1573. [Google Scholar] [CrossRef]
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef]
- McAdam, A.J.; Greenwald, R.J.; Levin, M.A.; Chernova, T.; Malenkovich, N.; Ling, V.; Freeman, G.J.; Sharpe, A.H. Icos is critical for CD40-mediated antibody class switching. Nature 2001, 409, 102–105. [Google Scholar] [CrossRef]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Amatore, F.; Gorvel, L.; Olive, D. Inducible co-stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Targets 2018, 22, 343–351. [Google Scholar] [CrossRef]
- Wright, G.J.; Puklavec, M.J.; Willis, A.C.; Hoek, R.M.; Sedgwick, J.D.; Brown, M.H.; Barclay, A.N. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 2000, 13, 233–242. [Google Scholar] [CrossRef]
- Caserta, S.; Nausch, N.; Sawtell, A.; Drummond, R.; Barr, T.; MacDonald, A.S.; Mutapi, F.; Zamoyska, R. Chronic infection drives expression of the inhibitory receptor CD200R and its ligand CD200, by mouse and human CD4 T cells. PLoS ONE 2012, 7, e35466. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Pan, Y.; Zhang, Q.; Sun, W. Role of CD200/CD200R Signaling Pathway in Regulation of CD4+T Cell Subsets During Thermal Ablation of Hepatocellular Carcinoma. Med. Sci. Monit. 2019, 25, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, E.S.K.; de Ruiter, T.; Baridi, A.; Veninga, H.; Hoek, R.M.; Meyaard, L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol. Immunol. 2008, 45, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, K.W.; Kwon, Y.R.; Kim, B.M.; Kim, Y.J. Forced expression of CD200 improves the differentiation capability and immunoregulatory functions of mesenchymal stromal cells. Biotechnol. Lett. 2018, 40, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, T.P.; Karnam, G.; Goverse, G.; Van Der Marel, A.P.J.; Greuter, M.J.; Van Schaarenburg, R.A.; Visser, W.F.; Brenkman, A.B.; Molenaar, R.; Hoek, R.M.; et al. CD200-CD200R signaling suppresses anti-tumor responses independently of CD200 expression on the tumor. Oncogene 2012, 31, 2979–2988. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8 + T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8 + T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.F.; Joller, N.; Tan, D.J.; Teng, M.W.L.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [Green Version]
- Lens, S.M.A.; Tesselaar, K.; Van Oers, M.H.J.; Van Lier, R.A.W. Control of lymphocyte function through CD27-CD70 interactions. Semin. Immunol. 1998, 10, 491–499. [Google Scholar] [CrossRef]
- Kobata, T.; Jacquot, S.; Kozlowski, S.; Agematsu, K.; Schlossman, S.F.; Morimoto, C. CD27-CD70 interactions regulate B-cell activation by T cells. Proc. Natl. Acad. Sci. USA 2006, 92, 11249–11253. [Google Scholar] [CrossRef]
- Borst, J.; Hendriks, J.; Xiao, Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005, 17, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25 + CD4 + regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Maruhashi, T.; Sugiura, D.; Shimizu, K.; Watada, M.; Okazaki, I.; Okazaki, T. PD-1 efficiently inhibits T cell activation even in the presence of co-stimulation through CD27 and GITR. Biochem. Biophys. Res. Commun. 2019, 511, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, A.A.; Zhou, G.; Doukas, M.; Boor, P.P.C.; Noordam, L.; Mancham, S.; Campos Carrascosa, L.; Van Der Heide-Mulder, M.; Polak, W.G.; Ijzermans, J.N.M.; et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int. J. Cancer 2019, 145, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell. Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Barclay, A.N. Signal regulatory protein alpha (SIRPα)/CD47 interaction and function. Curr. Opin. Immunol. 2009, 21, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Stefanidakis, M.; Newton, G.; Lee, W.Y.; Parkos, C.A.; Luscinskas, F.W. Endothelial CD47 interaction with SIRP{gamma} is required for human T-cell transendothelial migration under shear flow conditions in vitro. Blood 2008, 112, 1280–1289. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; Van De Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Ganong, A.H.; Cotman, C.W. Kynurenic Acid and Quinolinic Acid Act at N-Methyl-D-Aspartate Receptors in the Rat Hippocampus. J. Pharmacol. Exp. Ther. 1986, 236, 293–299. [Google Scholar]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Marti, L.C.; Pavon, L.; Severino, P.; Sibov, T.; Guilhen, D.; Moreira-Filho, C.A. Vascular endothelial growth factor-A enhances indoleamine 2,3-dioxygenase expression by dendritic cells and subsequently impacts lymphocyte proliferation. Mem. Inst. Oswaldo Cruz 2014, 109, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.C.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene expression comparison between the lymph node-positive and -negative reveals a peculiar immune microenvironment signature and a theranostic role for WNT targeting in pancreatic ductal adenocarcinoma: A pilot study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Y.; Yin, Z.; Feng, S.; Sun, L.; Li, Z. Research progress of indoleamine 2,3-dioxygenase inhibitors. Future Med. Chem. 2015, 7, 185–201. [Google Scholar] [CrossRef]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8, 370. [Google Scholar] [CrossRef] [Green Version]
- Borrego, F.; Masilamani, M.; Marusina, A.I.; Tang, X.; Coligan, J.E. The CD94/NKG2 family of receptors: From molecules and cells to clinical relevance. Immunol. Res. 2006, 35, 263–277. [Google Scholar] [CrossRef]
- Colonna, M.; Moretta, A.; Vély, F.; Vivier, E. A high-resolution view of NK-cell receptors: Structure and function. Immunol. Today 2000, 21, 428–431. [Google Scholar] [CrossRef]
- Yokoyama, W.M.; Plougastel, B.F.M. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 2003, 3, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Yawata, M.; Yawata, N.; Abi-Rached, L.; Parham, P. Variation Within the Human Killer Cell Immunoglobulin-Like Receptor (KIR) Gene Family. Crit. Rev. Immunol. 2012, 22, 20. [Google Scholar] [CrossRef]
- Bashirova, A.A.; Martin, M.P.; McVicar, D.W.; Carrington, M. The Killer Immunoglobulin-Like Receptor Gene Cluster: Tuning the Genome for Defense. Annu. Rev. Genom. Hum. Genet. 2006, 7, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Vance, R.E.; McMahon, C.W. Regulation of the natural killer cell receptor repertoire. Annu. Rev. Immunol. 2001, 19, 291–330. [Google Scholar] [CrossRef] [PubMed]
- Vilches, C.; Parham, P. KIR: Diverse, Rapidly Evolving Receptors of Innate and Adaptive Immunity. Annu. Rev. Immunol. 2002, 20, 217–251. [Google Scholar] [CrossRef]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [Green Version]
- Vey, N.; Dumas, P.; Recher, C.; Gastaud, L.; Lioure, B.; Bulabois, C.; Pautas, C.; Marolleau, J.; Leprêtre, S.; Raffoux, E.; et al. Randomized Phase 2 Trial of Lirilumab (anti-KIR monoclonal antibody, mAb) As Maintenance Treatment in Elderly Patients (pts) with Acute Myeloid Leukemia (AML): Results of the Effikir Trial. Blood 2017, 130 (Suppl. S1), 889. [Google Scholar]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Draghi, A.; Chamberlain, C.A.; Furness, A.; Donia, M. Acquired resistance to cancer immunotherapy. Semin. Immunopathol. 2019, 41, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Myers, G. Immune-related adverse events of immune checkpoint inhibitors: A brief review. Curr. Oncol. 2018, 25, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.J.; Carlino, M.S. Immune Checkpoint Inhibitor Toxicity. Curr. Oncol. Rep. 2018, 20, 72. [Google Scholar] [CrossRef] [PubMed]
- Kroschinsky, F.; Stölzel, F.; von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. New drugs, new toxicities: Severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Baldo, B.A. Adverse events to monoclonal antibodies used for cancer therapy focus on hypersensitivity responses. Oncoimmunology 2013, 2, e26333. [Google Scholar] [CrossRef]
- Ntali, G.; Kassi, E.; Alevizaki, M. Endocrine sequelae of immune checkpoint inhibitors. Hormones 2017, 16, 341–350. [Google Scholar] [PubMed]
- Mearns, E.S.; Bell, J.A.; Galaznik, A.; Puglielli, S.M.; Cichewicz, A.B.; Boulanger, T.; Garcia-Ribas, I. Gastrointestinal adverse events with combination of checkpoint inhibitors in advanced melanoma: A systematic review. Melanoma Manag. 2018, 5, MMT01. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the tumor microenvironment and tumor-stroma interaction by non-invasive preclinical imaging. Front. Oncol. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.A.; Edwards, C.M.; Croucher, P.I. Tumor-host cell interactions in the bone disease of myeloma. Bone 2011, 48, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kolstad, P. Follow-up study of 232 patients with stage Ia1 and 411 patients with stage Ia2 squamous cell carcinoma of the cervix (microinvasive carcinoma). Gynecol. Oncol. 1989, 33, 265–272. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado, T.J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Kimlinger, T.K.; Greipp, P.R.; Rajkumar, S.V. Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: Evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood 2004, 104, 1159–1165. [Google Scholar] [CrossRef]
- Hose, D.; Moreaux, J.; Meissner, T.; Seckinger, A.; Goldschmidt, H.; Benner, A.; Mahtouk, K.; Hillengass, J.; Rème, T.; De Vos, J.; et al. Induction of angiogenesis by normal and malignant plasma cells. Blood 2009, 114, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone marrow endothelial cells sustain a tumor-specific CD8 + T cell subset with suppressive function in myeloma patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef] [PubMed]
- Croucher, D.C.; Chesi, M.; Li, Z.; Garbitt, V.M.; Sharik, M.E.; Waller, D.; Bergsagel, P.L.; Pugh, T.J.; Trudel, S. A single-cell transcriptional analysis of tumour cells and the immune microenvironment during disease evolution in a transgenic mouse model of myeloma. Blood 2018, 132 (Suppl. S1), 56. [Google Scholar]
- Seymour, F.; Cavenagh, J.; Gribben, J.G. Characterising the Immunological Microenvironment in Newly Diagnosed Multiple Myeloma Bone Marrow By Time of Flight Cytometry Reveals Abnormalities in Antigen Presenting and Effector Lymphocyte Populations with Prognostic Significance. Blood 2018, 132 (Suppl. S1), 58. [Google Scholar]
- Ribatti, D.; Nico, B.; Vacca, A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int. Rev. Cell. Mol. Biol. 2015, 314, 259–282. [Google Scholar] [PubMed]
- Servais, C.; Erez, N. From sentinel cells to inflammatory culprits: Cancer-associated fibroblasts in tumour-related inflammation. J. Pathol. 2013, 229, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences and Perspectives. Mediat. Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.L.; Massonnet, G.; Dutel, J.L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon α-2a. Blood 2006, 108, 2037–2040. [Google Scholar] [CrossRef]
- Masarova, L.; Yin, C.C.; Cortes, J.E.; Konopleva, M.; Borthakur, G.; Newberry, K.J.; Kantarjian, H.M.; Bueso-Ramos, C.E.; Verstovsek, S. Histomorphological responses after therapy with pegylated interferon α-2a in patients with essential thrombocythemia (ET) and polycythemia vera (PV). Exp. Hematol. Oncol. 2017, 6, 30. [Google Scholar]
- Crisà, E.; Cerrano, M.; Beggiato, E.; Benevolo, G.; Lanzarone, G.; Manzini, P.M.; Borchiellini, A.; Riera, L.; Boccadoro, M.; Ferrero, D. Can pegylated interferon improve the outcome of polycythemia vera patients? J. Hematol. Oncol. 2017, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The interferons: 50 Years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A. Interferon: The pathways of discovery I. Molecular and cellular aspects. Cytokine Growth Factor Rev. 2006, 17, 381–409. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Proietti, E.; Belardelli, F. IFN-α and novel strategies of combination therapy for cancer. Ann. N. Y. Acad. Sci. 2007, 1112, 256–268. [Google Scholar] [CrossRef]
- Xu, D.; Erickson, S.; Szeps, M.; Gruber, A.; Sangfelt, O.; Einhorn, S.; Pisa, P.; Grandér, D. Interferon alpha down-regulates telomerase reverse transcriptase and telomerase activity in human malignant and nonmalignant hematopoietic cells. Blood 2000, 96, 4313–4318. [Google Scholar] [PubMed]
- Hammill, J.A.; VanSeggelen, H.; Helsen, C.W.; Denisova, G.F.; Evelegh, C.; Tantalo, D.G.M.; Bassett, J.D.; Bramson, J.L. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J. Immunother. Cancer 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Rao, L.; De Veirman, K.; Giannico, D.; Saltarella, I.; Desantis, V.; Frassanito, M.A.; Solimando, A.G.; Ribatti, D.; Prete, M.; Harstrick, A.; et al. Targeting angiogenesis in multiple myeloma by the VEGF and HGF blocking DARPin® protein MP0250: A preclinical study. Oncotarget 2018, 9, 13366–13381. [Google Scholar] [CrossRef]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef]
- Rosenberg, S.A. Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 2012, 4, 127. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef] [PubMed]
- Sukari, A.; Abdallah, N.; Nagasaka, M. Unleash the power of the mighty T cells-basis of adoptive cellular therapy. Crit. Rev. Oncol. Hematol. 2019, 136, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Debets, R.; Donnadieu, E.; Chouaib, S.; Coukos, G. TCR-engineered T cells to treat tumors: Seeing but not touching? Semin. Immunol. 2016, 28, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Spear, T.T.; Nagato, K.; Nishimura, M.I. Strategies to genetically engineer T cells for cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 153303381983106. [Google Scholar] [CrossRef]
- Leung, W.; Heslop, H.E. Adoptive Immunotherapy with Antigen-Specific T Cells Expressing a Native TCR. Cancer Immunol. Res. 2019, 7, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.; Zimmermann, S.; Arber, C.; Irving, M.; Trueb, L.; Coukos, G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf. 2019, 42, 315–334. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, M.E. T-cell Immunotherapies and the Role of Nonclinical Assessment: The Balance between Efficacy and Pathology. Toxicol. Pathol. 2018, 46, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Chuntova, P.; Downey, K.M.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front. Immunol. 2019, 9, 3062. [Google Scholar] [CrossRef] [Green Version]
- Tendeiro Rego, R.; Morris, E.C.; Lowdell, M.W. T-cell receptor gene-modified cells: Past promises, present methodologies and future challenges. Cytotherapy 2019, 21, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.-L.; Wang, X.-C.; Lu, Y.-J.; Lu, X.-J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.; Tauber, P.A.; Pickl, W.F. The making and function of CAR cells. Immunol. Lett. 2019, 212, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Dotti, G.; Brentjens, R.; Savoldo, B.; Jensen, M.; Cooper, L.J.N.; June, C.H.; Rosenberg, S.; Sadelain, M.; Heslop, H.E. CARs on track in the clinic. Mol. Ther. 2011, 19, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Büning, H.; Uckert, W.; Cichutek, K.; Hawkins, R.E.; Abken, H. Do CARs need a driver’s license? Adoptive cell therapy with chimeric antigen receptor-redirected T cells has caused serious adverse events. Hum. Gene. Ther. 2010, 21, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef]
- Xu, D.; Jin, G.; Chai, D.; Zhou, X.; Gu, W.; Chong, Y.; Song, J.; Zheng, J. The development of CAR design for tumor CAR-T cell therapy. Oncotarget 2018, 9, 13991–14004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Van Schandevyl, S.; Kerre, T. Chimeric antigen receptor T-cell therapy: Design improvements and therapeutic strategies in cancer treatment. Acta Clin. Belg. 2018, 1–7. [Google Scholar] [CrossRef]
- Ormhøj, M.; Bedoya, F.; Frigault, M.J.; Maus, M.V. CARs in the Lead Against Multiple Myeloma. Curr. Hematol. Malig. Rep. 2017, 12, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Han, W.-D. Chimeric antigen receptor modified T-cells for cancer treatment. Chronic Dis. Transl. Med. 2018, 4, 225–243. [Google Scholar] [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-L.; Hua, Z.-C. Chimeric Antigen Receptor T-cell (CAR T) Therapy for Hematologic and Solid Malignancies: Efficacy and Safety—A Systematic Review with Meta-Analysis. Cancers 2019, 11, 47. [Google Scholar] [CrossRef]
- Holzinger, A.; Barden, M.; Abken, H. The growing world of CAR T cell trials: A systematic review. Cancer Immunol. Immunother. 2016, 65, 1433–1450. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Shi, J.; Liu, H. Chimeric antigen receptor T cell targeting B cell maturation antigen immunotherapy is promising for multiple myeloma. Ann. Hematol. 2019, 98, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Heyman, B.; Yang, Y. Chimeric Antigen Receptor T Cell Therapy for Solid Tumors: Current Status, Obstacles and Future Strategies. Cancers 2019, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakoub-Agha, I.; Moreau, A.-S.; Ahmad, I.; Borel, C.; Hadhoum, N.; Masouridi-Levrat, S.; Naudin, J.; Nicolas-Virelizier, E.; Ouachée-Chardin, M.; Platon, L.; et al. Prise en charge pratique du syndrome de relargage des cytokines (CRS) post-CAR-T cells chez l’adulte et l’enfant: Recommandation de la Société francophone de greffe de moelle et de thérapie cellulaire (SFGM-TC). Bull. Cancer 2019, 106, S102–S109. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Ribatti, D.; Vacca, A.; Einsele, H. Targeting B-cell non Hodgkin lymphoma: New and old tricks. Leuk. Res. 2016, 42, 93–104. [Google Scholar] [CrossRef]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies With Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Stieglmaier, J.; Benjamin, J.; Nagorsen, D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin. Biol. Ther. 2015, 15, 1093–1099. [Google Scholar] [CrossRef]
- Klein, J.S.; Gnanapragasam, P.N.P.; Galimidi, R.P.; Foglesong, C.P.; West, A.P.; Bjorkman, P.J. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc. Natl. Acad. Sci. USA 2009, 106, 7385–7390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-cell engager (BiTE) antibody construct Blinatumomab for the treatment of Patients with relapsed/refractory non-Hodgkin lymphoma: Final results from a phase I study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Sherman, M.; McElroy, P.L.; Lofgren, J.A.; Moody, G.; Baeuerle, P.A.; Coxon, A.; Arvedson, T. Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS ONE 2017, 12, e0183390. [Google Scholar]
- Velasquez, M.P.; Torres, D.; Iwahori, K.; Kakarla, S.; Arber, C.; Rodriguez-Cruz, T.; Szoor, A.; Bonifant, C.L.; Gerken, C.; Cooper, L.J.; et al. T cells expressing CD19-specific Engager Molecules for the Immunotherapy of CD19-positive Malignancies. Sci. Rep. 2016, 6, 27130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yang, Y.; Fan, D.; Xiong, D. The development of bispecific antibodies and their applications in tumor immune escape. Exp. Hematol. Oncol. 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum. Vaccin. Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Mammas, I.N.; Theodoridou, M.; Kramvis, A.; Thiagarajan, P.; Gardner, S.; Papaioannou, G.; Melidou, A.; Koutsaki, M.; Kostagianni, G.; Achtsidis, V.; et al. Paediatric virology: A rapidly increasing educational challenge (Review). Exp. Ther. Med. 2017, 13, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. Immune Regulation by Self-Recognition: Novel Possibilities for Anticancer Immunotherapy. J. Natl. Cancer Inst. 2015, 107, 154. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. The T-win® technology: Immune-modulating vaccines. Semin. Immunopathol. 2019, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H. Anti-cancer immunotherapy: Breakthroughs and future strategies. Semin. Immunopathol. 2019, 41, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Miñana, B.; Valcárcel, J.; Gabaldón, T.; Lehner, B. Synonymous Mutations Frequently Act as Driver Mutations in Human Cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warneford, S.G.; Witton, L.J.; Townsend, M.L.; Rowe, P.B.; Reddel, R.R.; Dalla-Pozza, L.; Symonds, G. Germ-line splicing mutation of the p53 gene in a cancer-prone family. Cell Growth Differ. 1992, 3, 839–846. [Google Scholar] [PubMed]
- Soudon, J.; Caron de Fromentel, C.; Bernard, O.; Larsen, C.J. Inactivation of the p53 gene expression by a splice donor site mutation in a human T-cell leukemia cell line. Leukemia 1991, 5, 917–920. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA replication fidelity and cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Maple, J.T.; Smyrk, T.C.; Boardman, L.A.; Johnson, R.A.; Thibodeau, S.N.; Chari, S.T.; Real, F.X. Defective DNA mismatch repair in long-term (≥3 years) survivors with pancreatic cancer. Pancreatology 2005, 5, 220–228. [Google Scholar] [CrossRef]
- Plaschke, J.; Engel, C.; Krüger, S.; Holinski-Feder, E.; Pagenstecher, C.; Mangold, E.; Moeslein, G.; Schulmann, K.; Gebert, J.; von Knebel Doeberitz, M.; et al. Lower Incidence of Colorectal Cancer and Later Age of Disease Onset in 27 Families With Pathogenic MSH6 Germline Mutations Compared With Families With MLH1 or MSH2 Mutations: The German Hereditary Nonpolyposis Colorectal Cancer Consortium. J. Clin. Oncol. 2004, 22, 4486–4494. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.F.; Johansen, J.; Bjørnevoll, I.; Sylvander, A.E.; Steinsbekk, K.S.; Sætrom, P.; Sandvik, A.K.; Drabløs, F.; Sjursen, W. A novel POLE mutation associated with cancers of colon, pancreas, ovaries and small intestine. Fam. Cancer 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Siegmund, K.D.; Weisenberger, D.J.; Long, T.I.; Thibodeau, S.N.; Lindor, N.; Young, J.; Jenkins, M.A.; Hopper, J.L.; Baron, J.A.; et al. Molecular Characterization of MSI-H Colorectal Cancer by MLHI Promoter Methylation, Immunohistochemistry and Mismatch Repair Germline Mutation Screening. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3208–3215. [Google Scholar] [CrossRef] [PubMed]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients With BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.-S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; Van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; Van Der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Parcesepe, P.; Giordano, G.; Laudanna, C.; Febbraro, A.; Pancione, M. Cancer-Associated Immune Resistance and Evasion of Immune Surveillance in Colorectal Cancer. Gastroenterol. Res. Pract. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef]
- Bin Lim, S.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Tapia Rico, G.; Price, T.J. Atezolizumab for the treatment of colorectal cancer: The latest evidence and clinical potential. Expert Opin. Biol. Ther. 2018, 18, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakamura, J.; Noshiro, H. Promising immunotherapies for esophageal cancer. Expert Opin. Biol. Ther. 2017, 17, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Finotello, F.; Rieder, D.; Trajanoski, Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front. Immunol. 2017, 8, 1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Boissière-Michot, F.; Lazennec, G.; Frugier, H.; Jarlier, M.; Roca, L.; Duffour, J.; Du Paty, E.; Laune, D.; Blanchard, F.; Le Pessot, F.; et al. Characterization of an adaptive immune response in microsatellite-instable colorectal cancer. Oncoimmunology 2014, 3, e29256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, J.M.; Panda, A.; Zhong, H.; Hirshfield, K.; Damare, S.; Lane, K.; Sokol, L.; Stein, M.N.; Rodriguez-Rodriquez, L.; Kaufman, H.L.; et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Investig. 2016, 126, 2334–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of Polymerase e–Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef]
- Capietto, A.-H.; Jhunjhunwala, S.; Delamarre, L. Characterizing neoantigens for personalized cancer immunotherapy. Curr. Opin. Immunol. 2017, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.-P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef]
- Pan, R.-Y.; Chung, W.-H.; Chu, M.-T.; Chen, S.-J.; Chen, H.-C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef]
- Dunn, J.; Rao, S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging (Albany NY) 2017, 8, 933–944. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Grünen, S.; Dietrich, J.; Kristiansen, G.; Landsberg, J.; Dietrich, D. PD-L1 (CD274) promoter methylation predicts survival in patients with acute myeloid leukemia. Leukemia 2017, 31, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non-Small Cell Lung Cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Lupo, G.; La Rosa, G.R.M.; Crimi, S.; Anfuso, C.D.; Salemi, R.; Rapisarda, E.; Libra, M.; Candido, S. Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 2019, 11, 610. [Google Scholar] [CrossRef]
- Falzone, L.; Romano, G.L.; Salemi, R.; Bucolo, C.; Tomasello, B.; Lupo, G.; Anfuso, C.D.; Spandidos, D.A.; Libra, M.; Candido, S. Prognostic significance of deregulated microRNAs in uveal melanomas. Mol. Med. Rep. 2019, 19, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, R.; Palini, S.; Vento, M.E.; La Ferlita, A.; Lo Faro, M.J.; Caroppo, E.; Borzì, P.; Falzone, L.; Barbagallo, D.; Ragusa, M.; et al. Identification of extracellular vesicles and characterization of miRNA expression profiles in human blastocoel fluid. Sci. Rep. 2019, 9, 84. [Google Scholar] [CrossRef]
- Falzone, L.; Scola, L.; Zanghì, A.; Biondi, A.; Di Cataldo, A.; Libra, M.; Candido, S. Integrated analysis of colorectal cancer microRNA datasets: Identification of microRNAs associated with tumor development. Aging (Albany NY) 2018, 10, 1000–1014. [Google Scholar] [CrossRef]
- Polo, A.; Crispo, A.; Cerino, P.; Falzone, L.; Candido, S.; Giudice, A.; De Petro, G.; Ciliberto, G.; Montella, M.; Budillon, A.; et al. Environment and bladder cancer: Molecular analysis by interaction networks. Oncotarget 2017, 8, 65240–65252. [Google Scholar] [CrossRef]
- Falzone, L.; Candido, S.; Salemi, R.; Basile, M.S.; Scalisi, A.; McCubrey, J.A.; Torino, F.; Signorelli, S.S.; Montella, M.; Libra, M. Computational identification of microRNAs associated to both epithelial to mesenchymal transition and NGAL/MMP-9 pathways in bladder cancer. Oncotarget 2016, 7, 72758–72766. [Google Scholar] [CrossRef] [Green Version]
- Hafsi, S.; Candido, S.; Maestro, R.; Falzone, L.; Soua, Z.; Bonavida, B.; Spandidos, D.A.; Libra, M. Correlation between the overexpression of Yin Yang 1 and the expression levels of miRNAs in Burkitt’s lymphoma: A computational study. Oncol. Lett. 2016, 11, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Boeri, M.; Milione, M.; Proto, C.; Signorelli, D.; Lo Russo, G.; Galeone, C.; Verri, C.; Mensah, M.; Centonze, G.; Martinetti, A.; et al. Circulating miRNAs and PD-L1 Tumor Expression Are Associated with Survival in Advanced NSCLC Patients Treated with Immunotherapy: A Prospective Study. Clin. Cancer Res. 2019, 25, 2166–2173. [Google Scholar] [CrossRef]
- Romano, G.; Kwong, L.N. Diagnostic and therapeutic applications of miRNA-based strategies to cancer immunotherapy. Cancer Metastasis Rev. 2018, 37, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivarelli, S.; Falzone, L.; Basile, M.S.; Nicolosi, D.; Genovese, C.; Libra, M.; Salmeri, M. Benefits of using probiotics as adjuvants in anticancer therapy (Review). World Acad. Sci. J. 2019, 1, 125–135. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Banna, G.L.; Torino, F.; Marletta, F.; Santagati, M.; Salemi, R.; Cannarozzo, E.; Falzone, L.; Ferraù, F.; Libra, M. Lactobacillus rhamnosus GG: An Overview to Explore the Rationale of Its Use in Cancer. Front. Pharmacol. 2017, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; De Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, A.; Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017, 35, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938. [Google Scholar] [CrossRef]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765. [Google Scholar] [CrossRef]
- Matsushita, H.; Hasegawa, K.; Oda, K.; Yamamoto, S.; Nishijima, A.; Imai, Y.; Asada, K.; Ikeda, Y.; Karasaki, T.; Fujiwara, K.; et al. The frequency of neoantigens per somatic mutation rather than overall mutational load or number of predicted neoantigens per se is a prognostic factor in ovarian clear cell carcinoma. Oncoimmunology 2017, 6, e1338996. [Google Scholar] [CrossRef]
- Milo, I.; Bedora-Faure, M.; Garcia, Z.; Thibaut, R.; Périé, L.; Shakhar, G.; Deriano, L.; Bousso, P. The immune system profoundly restricts intratumor genetic heterogeneity. Sci. Immunol. 2018, 3, eaat1435. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Rieder, D.; Klepsch, V.; Charoentong, P.; Finotello, F.; Hackl, H.; Hermann-Kleiter, N.; Löwer, M.; Baier, G.; Krogsdam, A.; et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 2018, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 Blockade Broadens the Peripheral T-Cell Receptor Repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.P.; Van Der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8 + T cell response. Sci. Transl. Med. 2014, 6, 254ra128. [Google Scholar] [CrossRef]
- Kansy, B.A.; Shayan, G.; Jie, H.-B.; Gibson, S.P.; Lei, Y.L.; Brandau, S.; Lang, S.; Schmitt, N.C.; Ding, F.; Lin, Y.; et al. T cell receptor richness in peripheral blood increases after cetuximab therapy and correlates with therapeutic response. Oncoimmunology 2018, 7, e1494112. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Ueha, S.; Shichino, S.; Ogiwara, H.; Hashimoto, S.; Kakimi, K.; Ito, S.; Matsushima, K. TCR Repertoire Analysis Reveals Mobilization of Novel CD8 + T Cell Clones Into the Cancer-Immunity Cycle Following Anti-CD4 Antibody Administration. Front. Immunol. 2019, 9, 3185. [Google Scholar] [CrossRef]
- Kvistborg, P.; Shu, C.J.; Heemskerk, B.; Fankhauser, M.; Thrue, C.A.; Toebes, M.; Van Rooij, N.; Linnemann, C.; Van Buuren, M.M.; Urbanus, J.H.M.; et al. TIL therapy broadens the tumor-reactive CD8 + T cell compartment in melanoma patients. Oncoimmunology 2012, 1, 409–418. [Google Scholar] [CrossRef]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Immune-regulation within the tumor microenvironment is controlled by different checkpoints located on the T cell membrane. These, interact with their ligands found on the surface of antigen presenting cells (APC) or tumor cells, forming axes that provide either stimulatory signals (green) or inhibitory (red) signals between the two cells. Immune-therapeutic drugs belonging to checkpoint inhibitors act by blocking these axes (T).
Figure 1.
Immune-regulation within the tumor microenvironment is controlled by different checkpoints located on the T cell membrane. These, interact with their ligands found on the surface of antigen presenting cells (APC) or tumor cells, forming axes that provide either stimulatory signals (green) or inhibitory (red) signals between the two cells. Immune-therapeutic drugs belonging to checkpoint inhibitors act by blocking these axes (T).

Figure 2.
Motifs of chimeric antigen receptor (CAR) constructs. The basic structure of CARs (1st generation) contains extracellular mAb-derived variable heavy (VH) and light (VL) chains directed against a native tumor-specific antigen (extracellular domain) fused with a TCR CD3ζ chain-containing intracellular signaling domain through a transmembrane linker. Second and third generation CARs provide an improved receptor signaling strength and persistence by incorporation, in the basic CAR structure, of one or two co-stimulatory molecules, respectively. T-cells redirected for universal cytokine mediated killing (TRUCKS) serve as the 4th generation of chimeric antigen receptors constructed on the base of the 2nd generation of CARs with the addition of a NFAT-driven cytokine producing gene cassette, such as IL-12. scFv, single-chain variable fragment.
Figure 2.
Motifs of chimeric antigen receptor (CAR) constructs. The basic structure of CARs (1st generation) contains extracellular mAb-derived variable heavy (VH) and light (VL) chains directed against a native tumor-specific antigen (extracellular domain) fused with a TCR CD3ζ chain-containing intracellular signaling domain through a transmembrane linker. Second and third generation CARs provide an improved receptor signaling strength and persistence by incorporation, in the basic CAR structure, of one or two co-stimulatory molecules, respectively. T-cells redirected for universal cytokine mediated killing (TRUCKS) serve as the 4th generation of chimeric antigen receptors constructed on the base of the 2nd generation of CARs with the addition of a NFAT-driven cytokine producing gene cassette, such as IL-12. scFv, single-chain variable fragment.

Figure 3.
Clonal evolution of tumor progression and tumor neoantigens (TNA) landscape diversity. Cancer pathogenesis is initiated by driver mutations that provide a fitness advantage to cells which evolve into the primary tumor. Additional mutational changes drive tumor evolution and are influenced by intrinsic and extrinsic factors. Regarding immunity, immunotherapeutic intervention and/or host immunosurveillance exert a selective pressure to immunogenic clones leading to the rise of resistant subclones. These tumors can harbor TNAs that are recognized as self and elicit poor immunological responses or induce local tumor microenvironment (TME) immunosuppression. Persistent tumors expand and acquire more mutations leading to intratumoral heterogeneity (ITH) and/or hypermutation, further affecting the TNA repertoire.
Figure 3.
Clonal evolution of tumor progression and tumor neoantigens (TNA) landscape diversity. Cancer pathogenesis is initiated by driver mutations that provide a fitness advantage to cells which evolve into the primary tumor. Additional mutational changes drive tumor evolution and are influenced by intrinsic and extrinsic factors. Regarding immunity, immunotherapeutic intervention and/or host immunosurveillance exert a selective pressure to immunogenic clones leading to the rise of resistant subclones. These tumors can harbor TNAs that are recognized as self and elicit poor immunological responses or induce local tumor microenvironment (TME) immunosuppression. Persistent tumors expand and acquire more mutations leading to intratumoral heterogeneity (ITH) and/or hypermutation, further affecting the TNA repertoire.


{kind=link}
{kind=link}
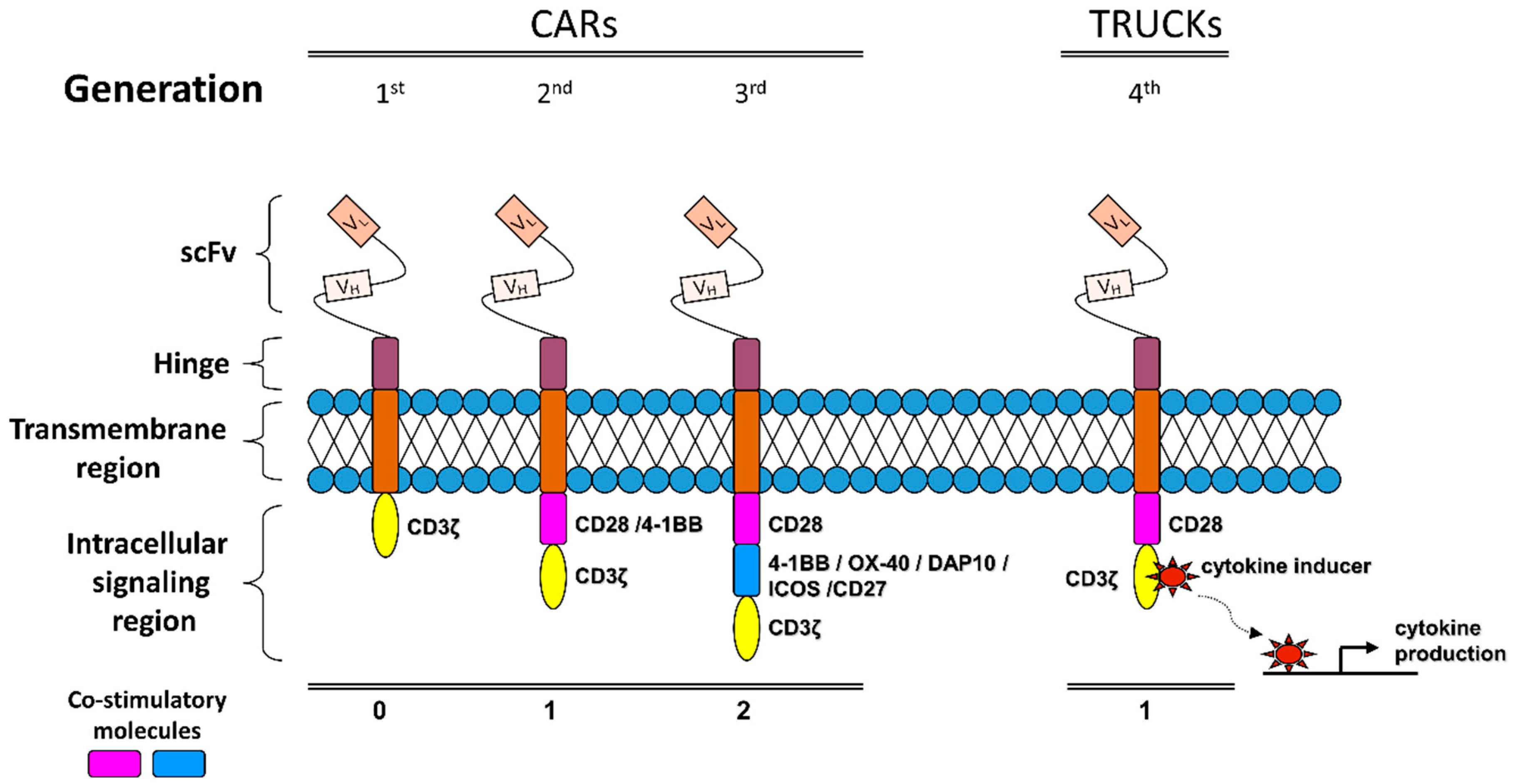{kind=link}
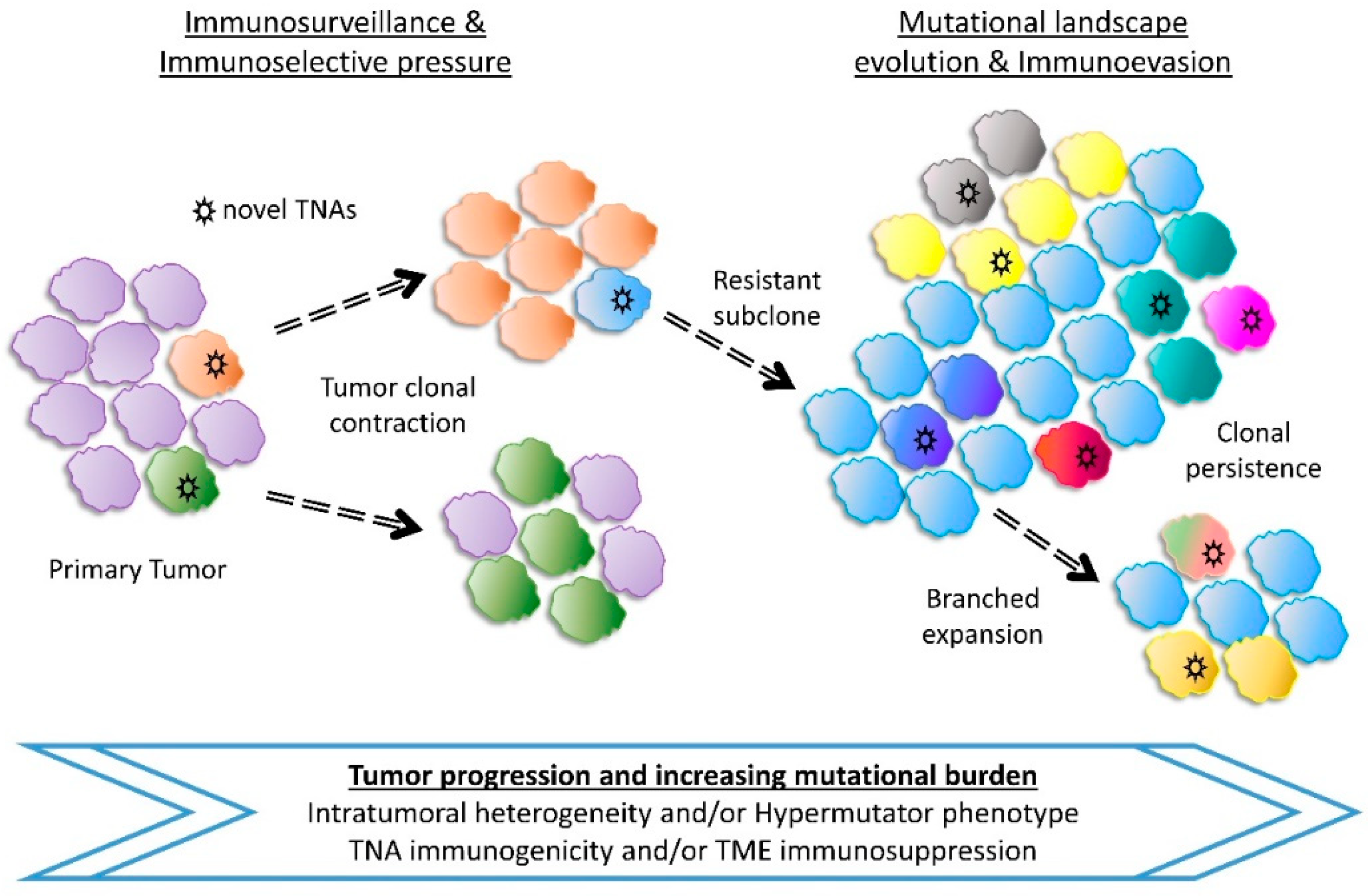{kind=link}
Table 1.
Recent advances in cancer immunotherapy.
| Immune Checkpoint Blockade | Cancer | Organism | References |
|---|---|---|---|
| Ipilimumab (anti–CTLA-4) | Melanoma | Humans | [3] |
| Tremelimumab (anti–CTLA-4) | Hepatocellular carcinoma (HCC) | Humans | [13] |
| Atezolizumab (anti-PD-L1) | Bladder, NSCLC | Humans | [14,15] |
| Avelumab (anti-PD-L1) | Merkel cell carcinoma | Humans | [16] |
| Pembrolizumab (anti-PD-1) | NSCLC, Melanoma, cHL, RCC, HNSCC, dMMR or MSI+ tumors | Humans | [17] |
| Nivolumab (anti-PD-1) | Humans | [18] | |
| Durvalumab (anti-PD-L1) | NSCLC, Urothelial carcinoma | Humans | [19,20] |
| IDO5 (IDO inhibitor) | NSCLC | Humans | [21] |
| 1-L-MT (IDO1 inhibitor) | Mastocytoma, CRC | Mice, cell lines | [22,23] |
| Indoximod (IDO1 inhibitor) | Melanoma, Prostate, Brain, AML | Humans | [24,25,26,27] |
| 680C91 & LM10 (TDO inhibitors) | Various cancer types | Mice, cell lines | [28] |
| Navoximod (IDO1 inhibitor) | Advanced solid tumors | Humans | [29,30] |
| Epacadostat (IDO1 inhibitor) | Multiple advanced solid tumors | Humans | [31] |
| Samalizumab (anti-CD200) | Bladder carcinoma | Humans | [32] |
| Varlilumab (anti-CD27) | Advanced refractory solid tumors | Humans | [33,34] |
| KWAR23 (anti-SIRPa) | Burkitt’s lymphoma, RCC, melanoma | Mice, cell lines | [35,36] |
| Urelumab (anti-CD137) | CRC, Gastric, Lymphoma | Mice, Humans | [37,38] |
| Lirilumab (anti-KIR2D mAb) | HNSCC, Lymphoma, myeloid malignancies | Humans, Mice, cell lines | [39,40,41] |
| Cytokine Therapy | |||
| Interleukin-2 (IL-2) | Melanoma, kidney, polycythemia vera | Humans | [42,43,44] |
| Interferon alpha (IFN-α) | Melanoma | Humans | [45] |
| Cellular Therapy | |||
| Tisagenlecleucel (anti-CD19) | NHL, ALL, DLBCL | Humans | [46,47] |
| Axicabtagene ciloleucel (anti-CD19) | Large B-Cell Lymphoma, NHL | Humans | [48,49] |
| anti-MUC1 CAR-T cells | Seminal vesicle carcinoma | Humans | [50] |
| CD33 knockout hematopoietic stem and progenitor cells (HSPCs) | Acute myeloid leukemia (AML) | Macaques, mice | [51] |
| Tumor antigen-loaded dendritic cells | Renal cell carcinoma (RCC) | Cell lines | [52] |
| IL-12p70-producing DCs | Melanoma | Humans | [53] |
| coTCRcys-transduced T cells | Nasopharyngeal | Cell lines | [54] |
| Therapeutic Vaccines | |||
| Hepatitis B virus (HBV) | HCC | Humans | [55] |
| Human papilloma virus (HPV) | Cervical, HNSCC, Oropharyngeal | Humans | [56,57,58] |
| Sipuleucel-T | Prostate | Humans | [59] |
| anti-gp100 | Melanoma | Humans | [60] |
| STINGVAX & anti–PD-1(G4) | Melanoma, Pancreatic, Colon, Tongue | Mice | [61] |
| GVAX & CRS-207 | Pancreatic | Humans | [62] |
Table 2.
Co-stimulatory and co-inhibitory markers in cancer immunity.
| Immune Receptors | Cancer Cell or APC | T, Treg, M, NK Cells | References |
|---|---|---|---|
| co-stimulatory | CD80/86 (B7.1/2) | CD28 | [87] |
| 4-1BBL | CD137 (4-1BB) | [88] | |
| OX-40L | OX40 | [89] | |
| CD70 | CD27 | [90] | |
| ICOSL (B7RP1) | ICOS | [91] | |
| GITRL | GITR | [92] | |
| B7-H7 (HHLA2) | TMIGD2 (CD28H) | [93] | |
| LIGHT | HVEM (CD270) | [94] | |
| CD40 | CD40L | [95] | |
| PVR (CD155) | DNAM-1 (CD226) | [96] | |
| CD48 | 2B4 (CD244) | [97] | |
| CD47 | SIRPa | [98] | |
| MHC-I | CD94/NKG2 | [99] | |
| LFA3 (CD58) | CD2 | [100] | |
| ICAM | LFA1 | [101] | |
| MHC-I or II | TCR/CD3 | [102,103] | |
| co-inhibitory | CD80/86 (B7.1/2) | CTLA-4 (CD152) | [104] |
| PD-L1/2 | PD-1 | [105] | |
| PD-L1 | B7-1 (CD80) | [106] | |
| IDO1/2, TDO | Tryptophan | [22,107] | |
| HVEM (CD270) | BTLA, CD160 | [108] | |
| GAL9, PtdSer, HMGB1, Ceacam-1 | TIM3 | [109,110] | |
| PVR (CD155) | CD96, TIGIT, DNAM-1 | [111,112] | |
| PVRL2 (CD112) | CD112R, TIGIT | [113,114] | |
| Adenosine | A2aR | [115] | |
| CD200 | CD200R | [116] | |
| B7-H3 (CD276) | ?, IL20RA | [117,118] | |
| B7-H4 (B7S1, VTCN1) | ? | [119] | |
| B7-H5 (VISTA) | ?, VSIG-3 | [120,121] | |
| B7-H7 (HHLA2) | ? | [122] | |
| MHC-I | KIR | [123] | |
| MHC-I or II, FGL1 | LAG-3 | [124,125] |
APC, antigen presenting cell; Treg, T regulatory cell; M, Macrophage; NK cell, natural killer cell.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Christofi, T.; Baritaki, S.; Falzone, L.; Libra, M.; Zaravinos, A. Current Perspectives in Cancer Immunotherapy. Cancers 2019, 11, 1472. https://doi.org/10.3390/cancers11101472
AMA Style
Christofi T, Baritaki S, Falzone L, Libra M, Zaravinos A. Current Perspectives in Cancer Immunotherapy. Cancers. 2019; 11(10):1472. https://doi.org/10.3390/cancers11101472
Chicago/Turabian StyleChristofi, Theodoulakis, Stavroula Baritaki, Luca Falzone, Massimo Libra, and Apostolos Zaravinos. 2019. "Current Perspectives in Cancer Immunotherapy" Cancers 11, no. 10: 1472. https://doi.org/10.3390/cancers11101472
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.