The Novel Phosphatidylinositol-3-Kinase (PI3K) Inhibitor Alpelisib Effectively Inhibits Growth of PTEN-Haploinsufficient Lipoma Cells

 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Effect of Alpelisib on Proliferation of Lipoma Cells
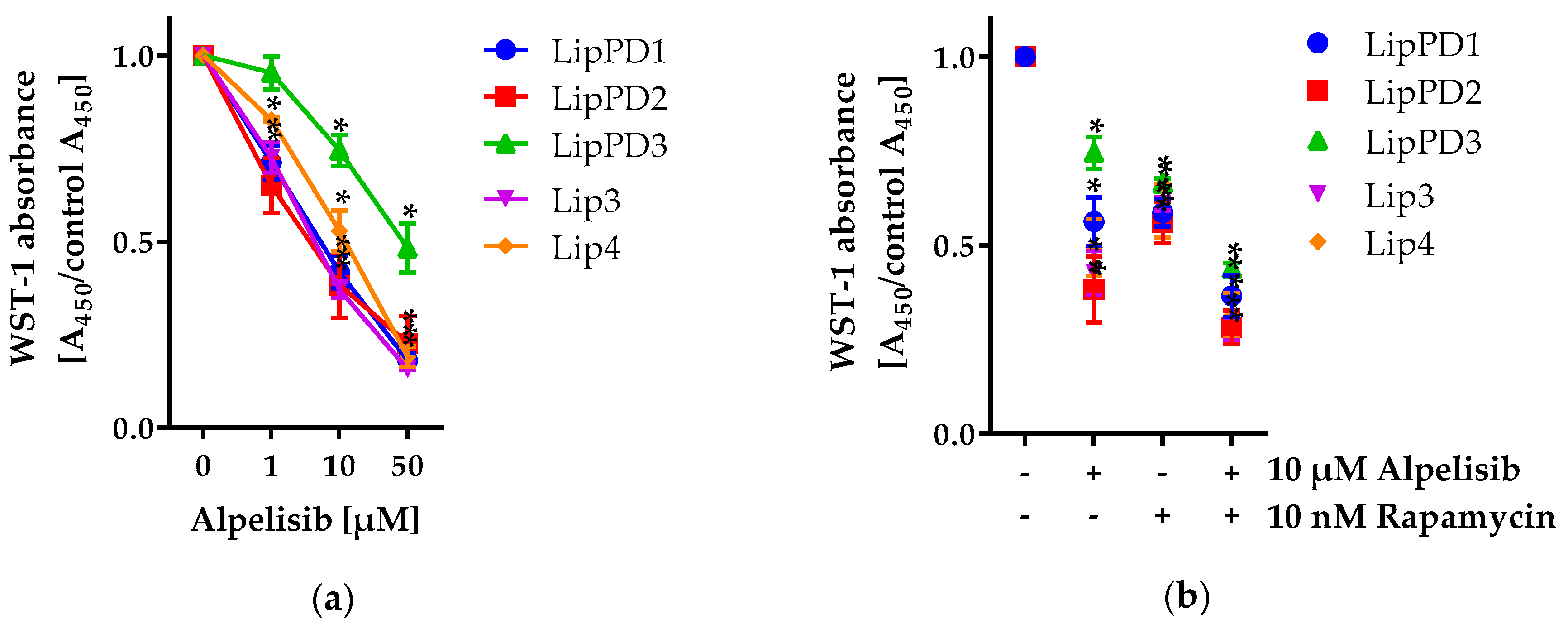
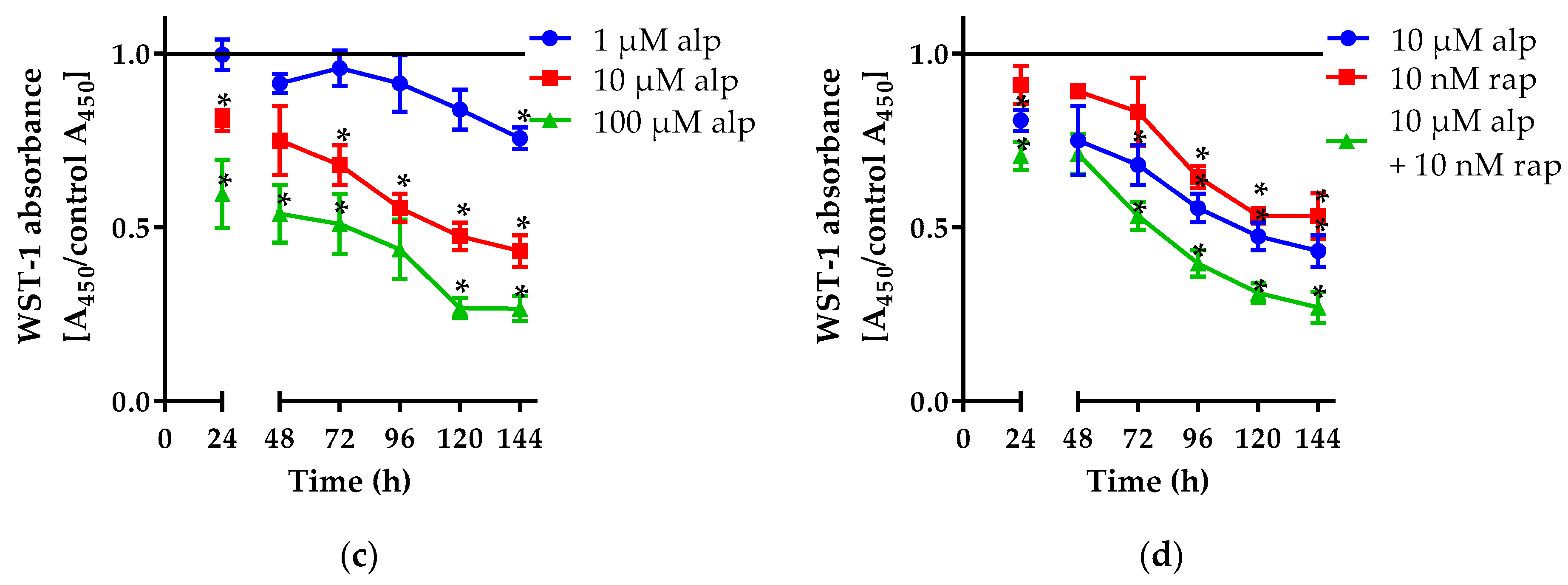
2.1.1. Alpelisib Reduced Cell Viability in a Dose- and Time-Dependent Manner
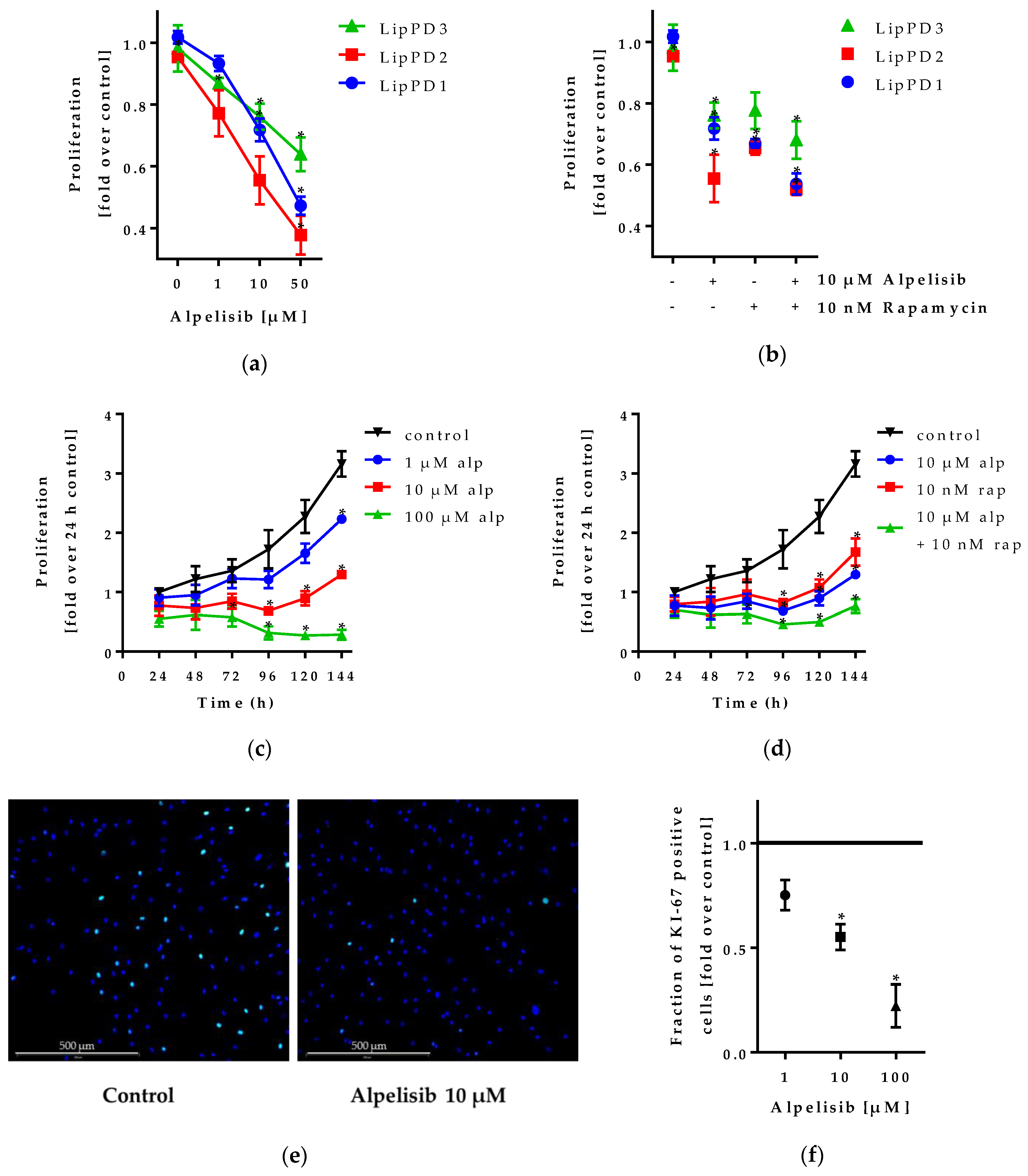
2.1.2. Alpelisib Inhibited Lipoma Cell Proliferation
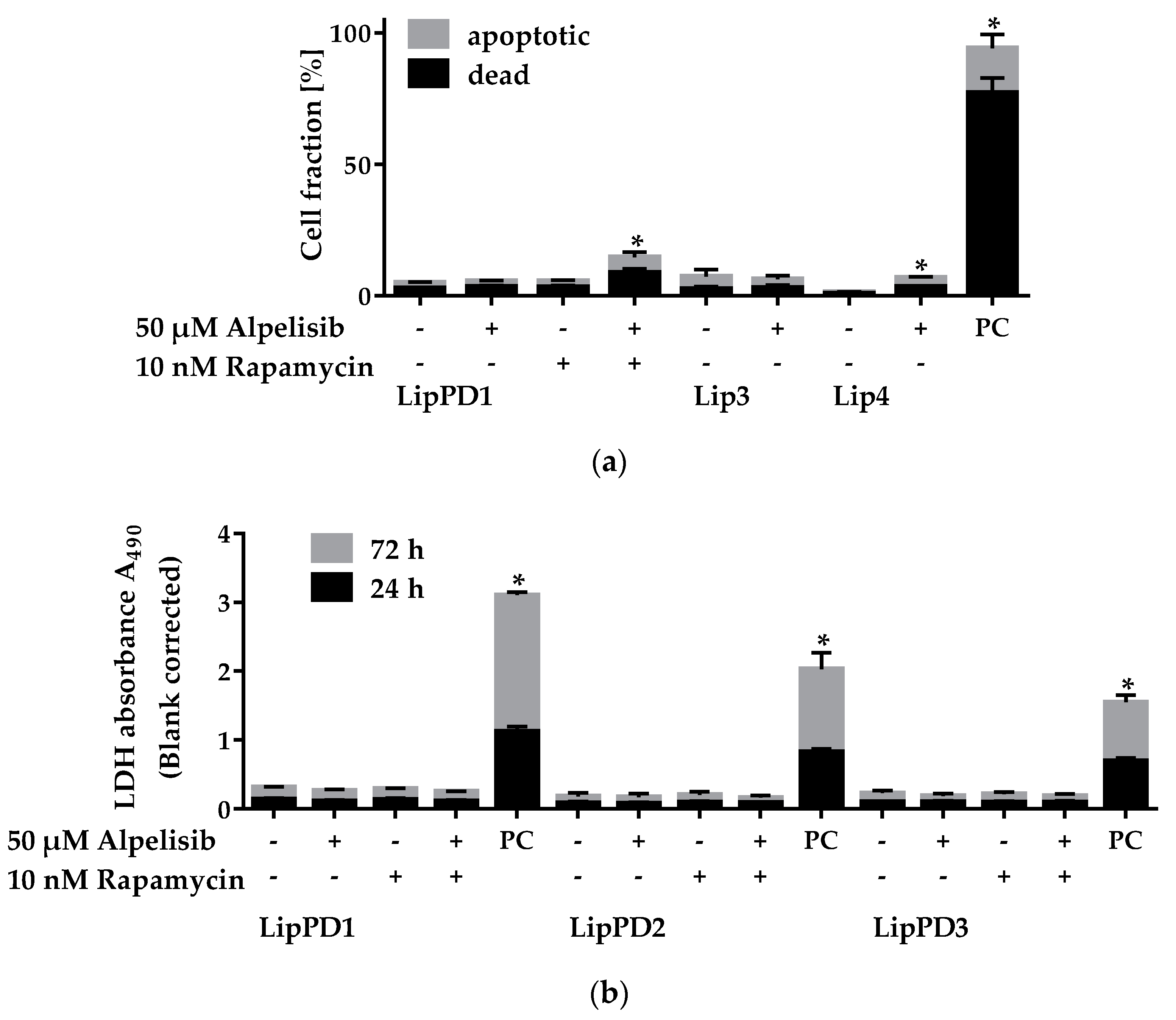
2.2. Cytotoxicity of Alpelisib in Lipoma Cells
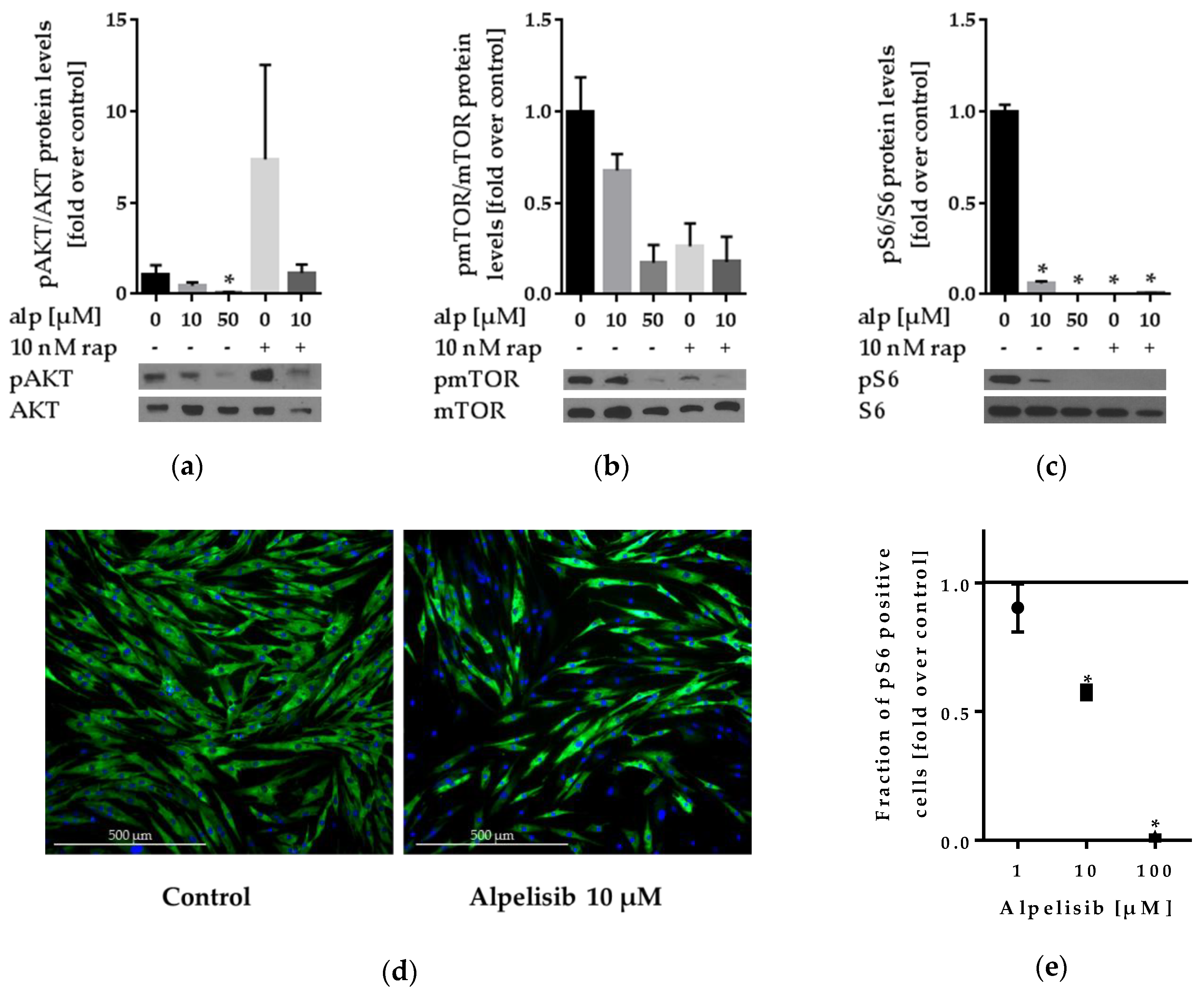
2.3. Effect of Alpelisib on PI3K Signaling
2.3.1. Alpelisib Reduced PI3K/AKT/mTOR Pathway Activation in Lipoma Cells
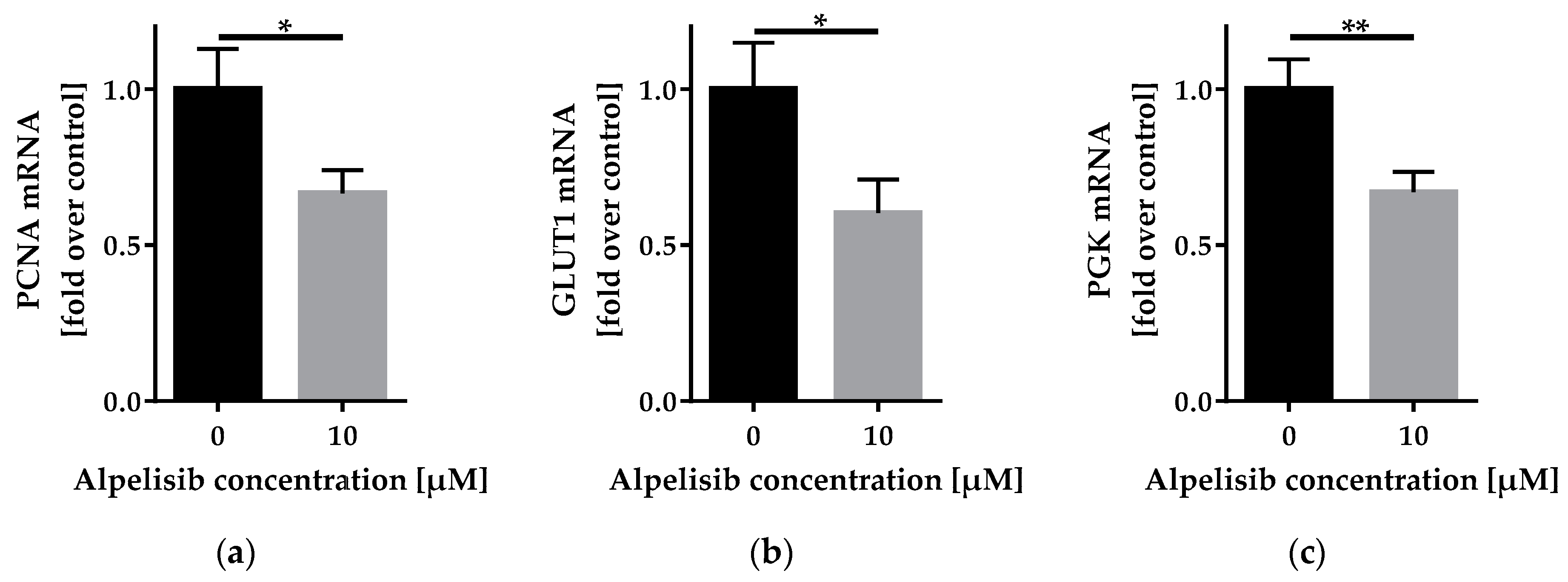
2.3.2. Alpelisib Reduced PCNA, GLUT1, and PGK mRNA Expression
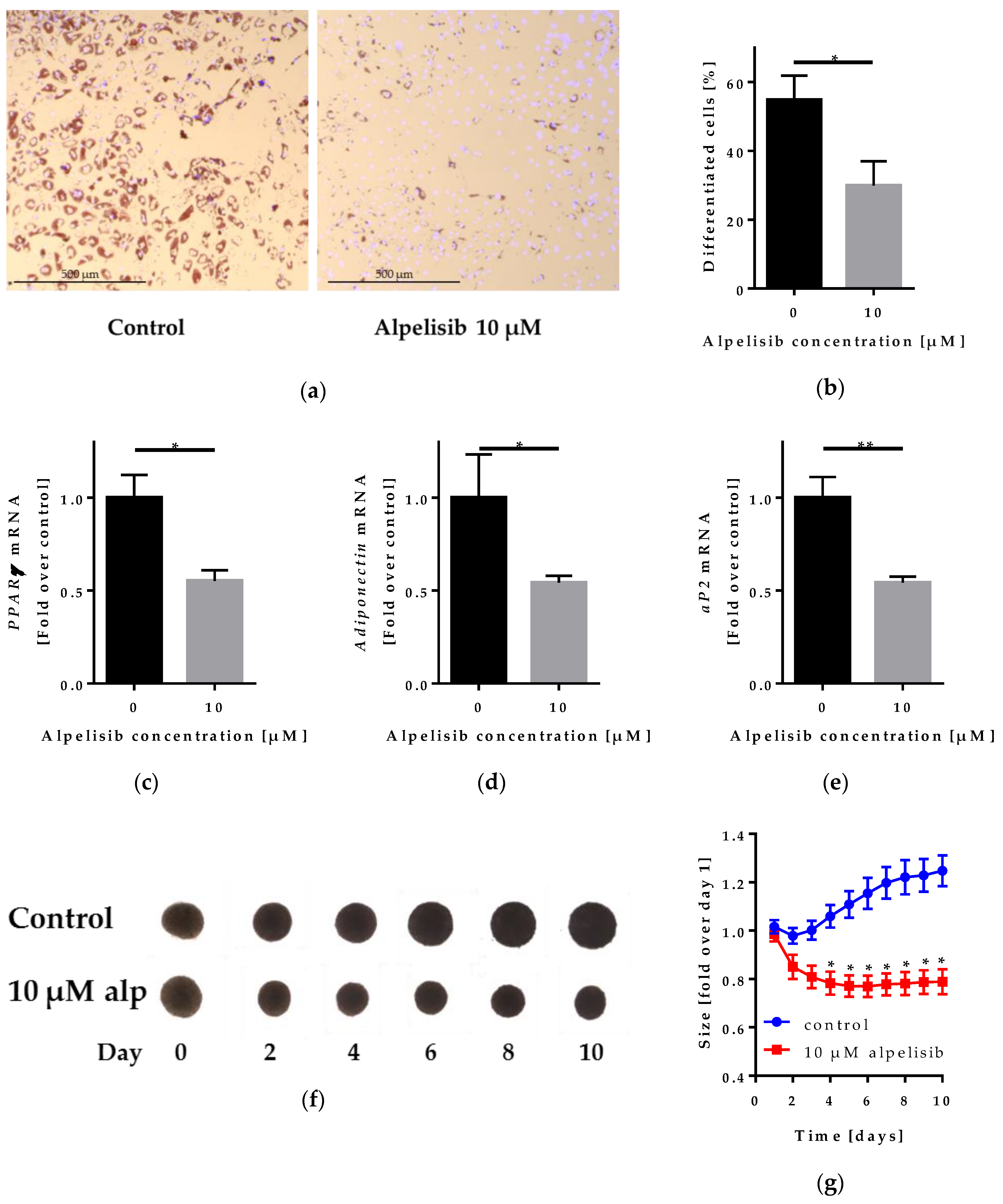
2.4. Effect of Alpelisib on Adipocyte Differentiation in 2D and 3D Models
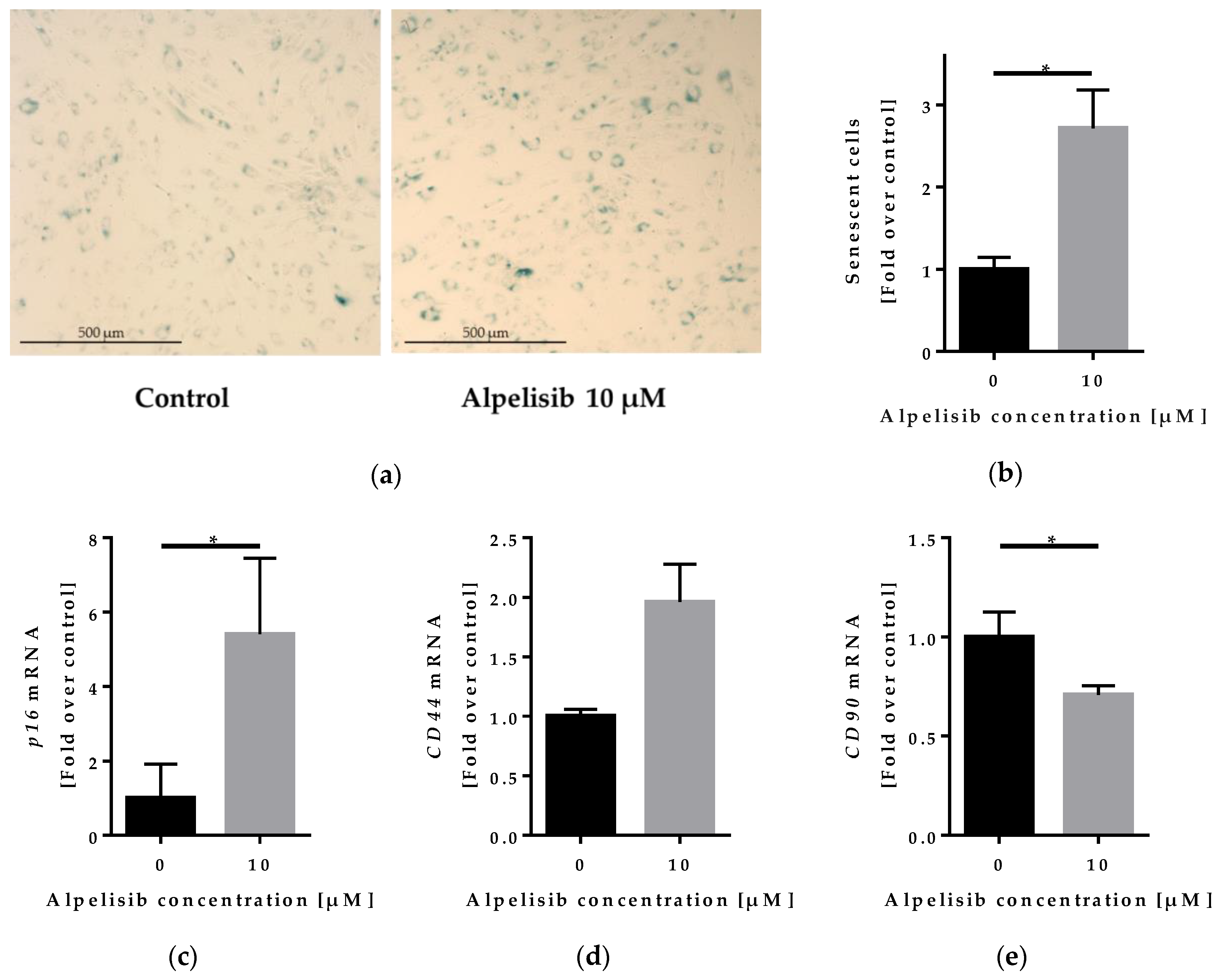
2.5. Effect of Alpelisib on Senescence of Lipoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Adipocyte Differentiation
4.2. Cell Viability/Proliferation
4.3. Apotosis Assay
4.4. LDH Cytotoxcicity Assay
4.5. Western Blot Analysis
4.6. Immunofluorescence Staining
4.7. Reverse Transcription Quantitative PCR (RT-qPCR)
4.8. Senescence β-Galactosidase Staining
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blumenthal, G.M.; Dennis, P.A. PTEN hamartoma tumor syndromes. Eur. J. Hum. Genet. 2008, 16, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlin, R.J.; Cohen, M.M.; Condon, L.M.; Burke, B.A. Bannayan-Riley-Ruvalcaba syndrome. Am. J. Med. Genet. 1992, 44, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Marsh, D.J.; Trahair, T.N.; Martin, J.L.; Chee, W.Y.; Walker, J.; Kirk, E.P.; Baxter, R.C.; Marshall, G.M. Rapamycin treatment for a child with germline PTEN mutation. Nat. Rev. Clin. Oncol. 2008, 5, 357. [Google Scholar] [CrossRef] [PubMed]
- Schmid, G.L.; Kässner, F.; Uhlig, H.H.; Körner, A.; Kratzsch, J.; Händel, N.; Zepp, F.-P.; Kowalzik, F.; Laner, A.; Starke, S.; et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatr. Res. 2014, 75, 527–534. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell. Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.-P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 558, 540. [Google Scholar] [CrossRef]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Jain, S.; Santa-Maria, C.A.; Rademaker, A.; Giles, F.J.; Cristofanilli, M.; Gradishar, W.J. Phase I study of alpelisib (BYL-719) and T-DM1 in HER2-positive metastatic breast cancer after trastuzumab and taxane therapy. JCO 2017, 35, 1026. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.M.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. LBA3_PRAlpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): Results of the phase III SOLAR-1 trial. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Wang, G.; Cao, X.; Lai, S.; Luo, X.; Feng, Y.; Xia, X.; Yen, P.M.; Gong, J.; Hu, J. PI3K stimulates DNA synthesis and cell-cycle progression via its p55PIK regulatory subunit interaction with PCNA. Mol. Cancer Ther. 2013, 12, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, C.; Thorburn, A. Transcriptional Activation of the Glucose Transporter GLUT1 in Ventricular Cardiac Myocytes by Hypertrophic Agonists. J. Biol. Chem. 1999, 274, 9006–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J. Biol. Chem. 2014, 289, 22785–22797. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Gourronc, F.A.; Chaly, A.; Wadkins, D.A.; Burand, A.J.; Markan, K.R.; Idiga, S.O.; Wu, M.; Potthoff, M.J.; Ankrum, J.A. Scaffold-free generation of uniform adipose spheroids for metabolism research and drug discovery. Sci. Rep. 2018, 8, 523. [Google Scholar] [CrossRef]
- Kässner, F.; Sauer, T.; Penke, M.; Richter, S.; Landgraf, K.; Körner, A.; Kiess, W.; Händel, N.; Garten, A. Simvastatin induces apoptosis in PTEN-haploinsufficient lipoma cells. Int. J. Mol. Med. 2018, 41, 3691–3698. [Google Scholar] [CrossRef]
- Tang, H.; Xue, G. Major Physiological Signaling Pathways in the Regulation of Cell Proliferation and Survival. In Mechanisms of Drug Resistance in Cancer Therapy; Mandalà, M., Romano, E., Eds.; Springer: Cham, Switzerland, 2018; pp. 13–30. ISBN 978-3-030-10507-5. [Google Scholar]
- Maiuri, T.; Ho, J.; Stambolic, V. Regulation of adipocyte differentiation by distinct subcellular pools of protein kinase B (PKB/Akt). J. Biol. Chem. 2010, 285, 15038–15047. [Google Scholar] [CrossRef]
- Keam, B.; Kim, S.; Ahn, Y.-O.; Kim, T.M.; Lee, S.-H.; Kim, D.-W.; Heo, D.S. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. Anticancer Res. 2015, 35, 175–182. [Google Scholar]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation, and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Stallock, J.P.; Ng, J.C.; Reinhard, C.; Neufeld, T.P. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000, 14, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.-L.; Manning, B.D. Insulin Stimulates Adipogenesis through the Akt-TSC2-mTORC1 Pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef]
- Cho, H.J.; Park, J.; Lee, H.W.; Lee, Y.S.; Kim, J.B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem. Biophys. Res. Commun. 2004, 321, 942–948. [Google Scholar] [CrossRef]
- Martini, H.; Iacovoni, J.S.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Itier, R.; Dambrin, C.; Pizzinat, N.; Mialet-Perez, J.; et al. Aging induces cardiac mesenchymal stromal cell senescence and promotes endothelial cell fate of the CD90 + subset. Aging Cell 2019, e13015. [Google Scholar] [CrossRef]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Muntión, S.; Preciado, S.; Puig, N.; López-Ruano, G.; Hernández-Hernández, Á.; Redondo, A.; Ortega, R.; Rodríguez, C.; et al. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal. 2016, 14. [Google Scholar] [CrossRef]
- Mun, G.I.; Boo, Y.C. Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2102–H2111. [Google Scholar] [CrossRef] [Green Version]
- Fischer-Posovszky, P.; Newell, F.S.; Wabitsch, M.; Tornqvist, H.E. Human SGBS Cells—A Unique Tool for Studies of Human Fat Cell Biology. Obes. Facts 2008, 1, 184–189. [Google Scholar] [CrossRef]
- Rockstroh, D.; Löffler, D.; Kiess, W.; Landgraf, K.; Körner, A. Regulation of human adipogenesis by miR125b-5p. Adipocyte 2016, 5, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-A.; Chen, Y.-J.; Chen, J.J. Testing for differentially expressed genes with microarray data. Nucleic Acids Res. 2003, 31, e52. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sex | Age at Resection | Mutation |
|---|---|---|---|
| LipPD1 | male | 3 | Heterozygous deletion of PTEN exons 2-9 of 9 |
| LipPD2 | male | 3 | PTEN heterozygous point mutation (c.404T>A, p.I135K) |
| LipPD3 | female | 4 | PTEN heterozygous point mutation (exon 1, c.76A>C, p.T26P) |
| Lip3 | female | 14 | Mosaic PI3KCA mutation (10%) (exon 10, c.1624G>A, p.(Glu542Lys) |
| Lip4 | male | 13 | Mosaic PI3KCA mutation (30-40 %) (exon 8, c.1340_1366del, p.(Pro447_Leu455del) |
| Primary Antibody | Dilution | Supplier | Cat. no |
|---|---|---|---|
| mTOR (7C10) Rabbit mAb | 1:1000 TBS-T 5%BSA (Wb) | CST | #2983 |
| Phospho-mTOR (Ser2448) Rabbit mAb (D9C2) | 1:1000 TBS-T 5%BSA (Wb) | CST | #5536 |
| AKT antibody Rabbit polyclonal Ab | 1:1000 TBS-T 5%BSA (Wb) | CST | #9272 |
| Phospho-AKT (Thr308) (224F9) Rabbit mAb | 1:500 TBS-T 5%BSA (Wb) | CST | #4056 |
| S6 ribosomal protein (5G10) Rabbit mAb | 1:1000 TBS-T 5%BSA (Wb) | CST | #2217 |
| Phospho-S6 Ribosomal protein (Ser235/236) (D57.2.2E) XP® Rabbit mAb | 1:1000 TBS-T 5%BSA (Wb) 1:500 IF-buffer (IF) | CST | #4858 |
| GAPDH (6C5) Mouse mAb | 1:50,000 TBS-T 5% milk (Wb) | Merck | MAB374 |
| KI-67 (MIB-1) Mouse mAb | 1:200 IF-buffer (IF) | Dako | P0447 |
| Secondary antibody | Dilution | Supplier | Cat. no |
| Polyclonal goat anti-rabbit immunoglobulin/HRP | 1:2000 TBS-T 5% milk (Wb) | Dako | P0447 |
| Polyclonal goat anti-mouse immunoglobulin/HRP | 1:2000 TBS-T 5% milk (Wb) | Dako | P0448 |
| Alexa Fluor 488 goat anti-mouse IgG H+L | 1:1000 IF-buffer (IF) | Invitrogen | A11001 |
| Alexa Fluor 488 goat anti-rabbit IgG H+L | 1:1000 IF-buffer (IF) | Invitrogen | A11034 |
| Gene | Forward | Reverse | Probe |
|---|---|---|---|
| TBP | TTGTAAACTTGACCTAAAGACCATTGC | TTCGTGGCTCTCTTATCCTCATG | AACGCCGAATATAATCCCAAGCGGTTTG |
| β-actin | CGA GCG CGG CTA CAG CTT | TGC TTG TGT TGG GTG GAT ATT G | TGG CCA CCG ACT CCT ACA AGG TTA CTC AC |
| HPRT | GGC AGT ATA ATC CAA AGA TGG TCA A | GTC TGG CTT ATA TCC AAC ACT TCG T | CAA GCT TGC TGG TGA AAA GGA CCC C |
| GLUT1 | TGGCATCAACGCTGTCTTCT | AGCCAATGGTGGCATACACA | |
| PGK | GAATGGGAAGCTTTTGCCCG | GCAGTGTCTCCACCACCTATG | |
| PCNA | CTAAAATGCGCCGGCAATGA | TCTCCTGGTTTGGTGCTTCA | |
| Adiponectin | GGC CGT GAT GGC AGA GAT | CCT TCA GCC CCG GGT ACT | CGATGTCTCCCTTAGGACCAATAAGACCTGG |
| aP2 | GCTTTTGTAGGTACCTGGAAACTTG | ACA CTG ATG ATC ATG TTA GGT TTG G | CCTGGTGGCAAAGCCCACTCCTCAT |
| PPARγ | GATCCAGTGGTTGCAGATTACAA | GAGGGAGTTGGAAGGCTCTTC | TGACCTGAAACTTCAAGAGTACCAAAGTGCAA |
| FASN | GGCAAATTCGACCTTTCTCAGA | GGACCCCGTGGAATGTCA | CACCCGCTCGGCATGGCTATCTT |
| p16 | CTTCGGCTGACTGGCTGG | TCATCATGACCTGGATCGGC | |
| CD44 | Predesigned assay (ID: Hs01075864_m1) | ||
| CD90 | Predesigned assay (ID: Hs00264235_s1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirstein, A.S.; Augustin, A.; Penke, M.; Cea, M.; Körner, A.; Kiess, W.; Garten, A. The Novel Phosphatidylinositol-3-Kinase (PI3K) Inhibitor Alpelisib Effectively Inhibits Growth of PTEN-Haploinsufficient Lipoma Cells. Cancers 2019, 11, 1586. https://doi.org/10.3390/cancers11101586
Kirstein AS, Augustin A, Penke M, Cea M, Körner A, Kiess W, Garten A. The Novel Phosphatidylinositol-3-Kinase (PI3K) Inhibitor Alpelisib Effectively Inhibits Growth of PTEN-Haploinsufficient Lipoma Cells. Cancers. 2019; 11(10):1586. https://doi.org/10.3390/cancers11101586
Chicago/Turabian StyleKirstein, Anna S., Adrien Augustin, Melanie Penke, Michele Cea, Antje Körner, Wieland Kiess, and Antje Garten. 2019. "The Novel Phosphatidylinositol-3-Kinase (PI3K) Inhibitor Alpelisib Effectively Inhibits Growth of PTEN-Haploinsufficient Lipoma Cells" Cancers 11, no. 10: 1586. https://doi.org/10.3390/cancers11101586