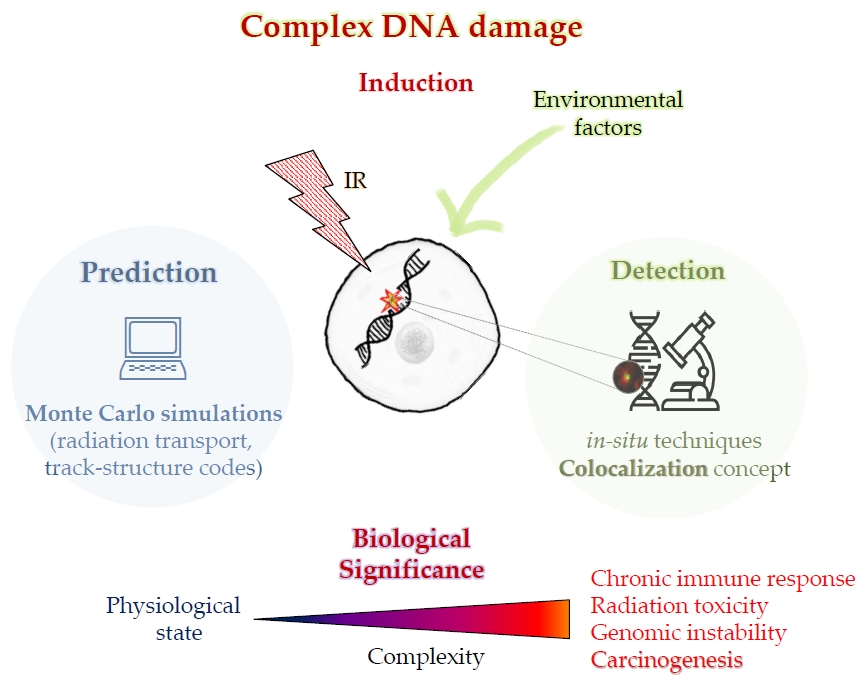
Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance

 ,
, 

Abstract
:
1. Introduction
2. Radiobiological Modeling and Simulations: A Useful Tool for Prediction and Experimental Data Interpretation
2.1. Monte Carlo Simulations for DNA Damage
2.2. Modeling of Radiation Effects in Biological Material
2.3. Radiation Transport and Track-Structure Codes for Radiation-Induced Damage
2.4. Key Conclusions
- The majority of damage events are of simple type containing an SSB. Strand breaks of greater complexity appear at high frequencies.
- The complexity of damage increases with LET.
- The yield of both single- and double-strand breaks per gray and per Dalton is nearly constant over a wide range of LETs.
- For low-LET radiations, nearly 20% of the DSBs are of complex type. The proportion of this clustered damage increases with LET, reaching ~70% or higher for high-LET radiations.
- When base damage is also taken into account, the proportion of complex DSBs increases for all radiations, reaching more than 90% for the higher LETs.
- Experiments show a slower rate of repair of DSBs produced by high-LET radiations.
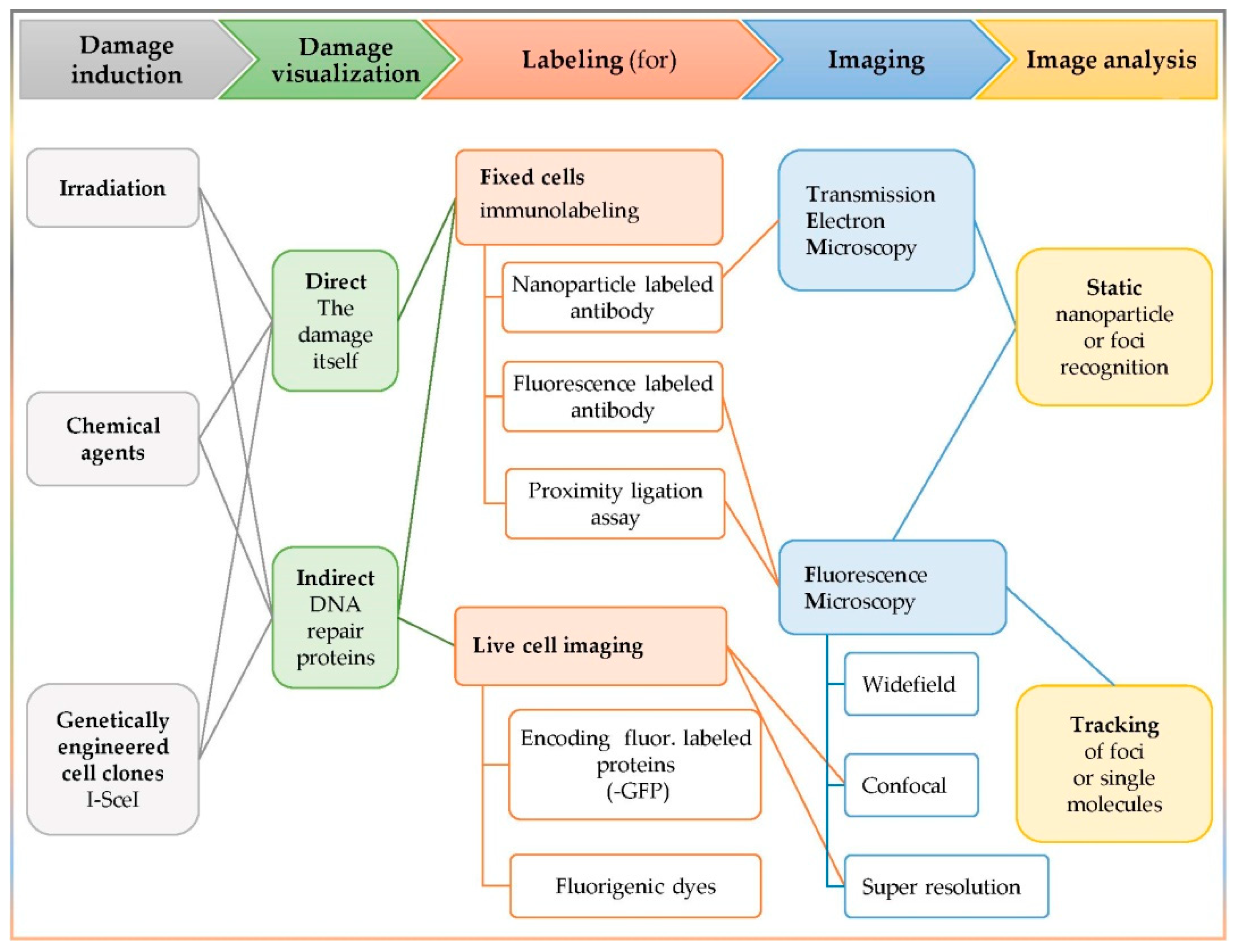
3. Detection of Complex DNA Damage
3.1. The In Situ Detection of Complex DNA Damage and the Colocalization Concept
- The damaging factor (irradiation, chemical reagent, or genetically engineered clones);
- The visualization target (the damage itself or a participating repair enzyme);
- The labeling method (immuno-labeling or fluorescent labeling);
- The imaging apparatus (static or live cell imaging);
- The image analysis (static or tracking).
3.2. Damage Induction
3.3. Damage Visualization: Direct or Indirect
3.4. Labeling Techniques
3.4.1. Immunolabeling—Fixed Cells
In Situ Immunofluorescence-Fluorophore Conjugated Antibodies
Transmission Electron Microscopy (TEM) Analysis—Nanoparticle Conjugated Antibodies
Proximity Ligation Assay―In Situ PLA
3.4.2. Live Cell Imaging
Encoding Fluorescence Labeled Proteins
Fluorogenic Dyes
3.5. Imaging: Microscopy and Image Analysis
3.5.1. Microscopy: TEM and Fluorescence Microscopy
Conventional (Widefield) Fluorescence Microscopy
Confocal Microscopy: Scanning Laser or Spinning Disk
Super Resolution Microscopy (SRM): Beyond the Diffraction Limit
3.5.2. Image Analysis: Co-Localization Coefficients, Parameters, and Methods
Defining an Extra Type of Foci
3.6. DNA Sequencing for Genome-Wide Nucleotide-Resolution DNA Damage Identification
- NGR detects and labels the nick-gaps, induced during DNA replication [118].
- BLISS can detect DSBs induced by endonucleases, using their corresponding unique molecular identifiers (UMIs) [119].
- BLESS is able to detect DSBs at nucleotide resolution. It is independent of proteins that bind to DNA or single-stranded DNA, which are both sources of bias. BLESS’ innovation is that it uses an amplification step for the fragments created by two DSBs [120].
- i-BLESS is a BLESS adaptation that is suitable for very small and fragile cell genomes (yeast), while it achieves the incredible detection accuracy of one DSB per 105 cells [121].
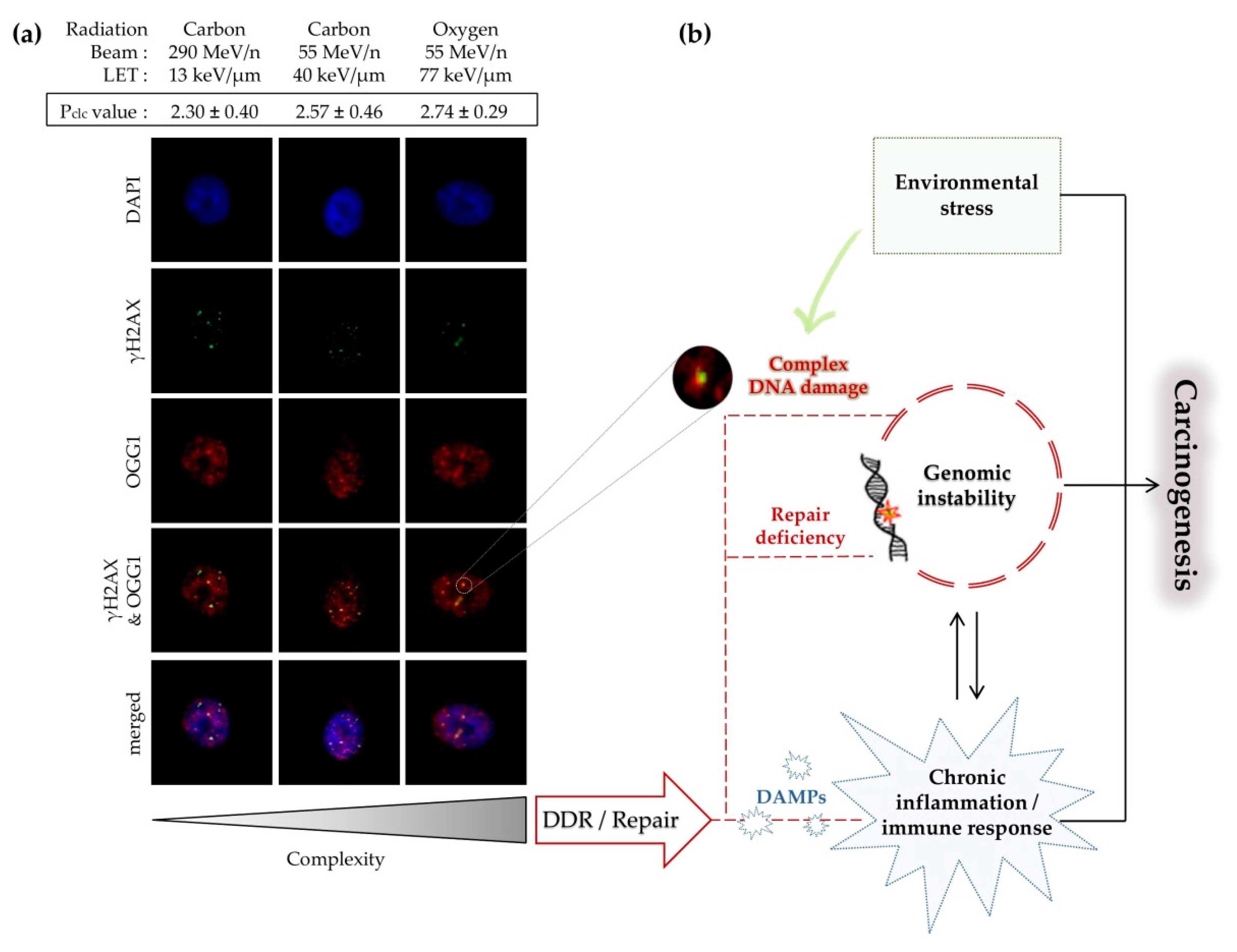
4. Biological Response to Clustered DNA Damage and Its Significance
4.1. The Role of Delayed Repair
4.2. Double-Strand Break Clustering
4.3. Carcinogenesis Associated with Clustered DNA Damage
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paganetti, H. Proton Relative Biological Effectiveness–Uncertainties and Opportunities. Int. J. Part. Ther. 2018, 5, 2–14. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Matsuura, T.; Wada, M.; Egashira, Y.; Nishio, T.; Furusawa, Y. Enhanced radiobiological effects at the distal end of a clinical proton beam: In vitro study. J. Radiat. Res. 2014, 55, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Carante, M.P.; Aimè, C.; Cajiao, J.J.T.; Ballarini, F. BIANCA, a biophysical model of cell survival and chromosome damage by protons, C-ions and He-ions at energies and doses used in hadrontherapy. Phys. Med. Boil. 2018, 63, 075007. [Google Scholar] [CrossRef] [PubMed]
- Ballarini, F.; Altieri, S.; Bortolussi, S.; Giroletti, E.; Protti, N. A Model of Radiation-Induced Cell Killing: Insights into Mechanisms and Applications for Hadron Therapy. Radiat. Res. 2013, 180, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Paganetti, H.; Van Luijk, P. Biological Considerations When Comparing Proton Therapy with Photon Therapy. Semin. Radiat. Oncol. 2013, 23, 77–87. [Google Scholar] [CrossRef]
- Tommasino, F.; Durante, M. Proton radiobiology. Cancers 2015, 7, 353–381. [Google Scholar] [CrossRef]
- Durante, M.; Loeffler, J.S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2009, 7, 37–43. [Google Scholar] [CrossRef]
- Sato, T.; Sihver, L.; Iwase, H.; Nakashima, H.; Niita, K. Simulations of an accelerator-based shielding experiment using the particle and heavy-ion transport code system PHITS. Adv. Space Res. 2005, 35, 208–213. [Google Scholar] [CrossRef]
- Sirunyan, A.; Tumasyan, A.; Adam, W.; Asilar, E.; Bergauer, T.; Brandstetter, J.; Brondolin, E.; Dragicevic, M.; Erö, J.; Flechl, M.; et al. Particle-flow reconstruction and global event description with the CMS detector. J. Instrum. 2017, 12, P10003. [Google Scholar] [CrossRef]
- Nikjoo, H.; Emfietzoglou, D.; Liamsuwan, T.; Taleei, R.; Liljequist, D.; Uehara, S. Radiation track, DNA damage and response—A review. Rep. Prog. Phys. 2016, 79, 116601. [Google Scholar] [CrossRef]
- Ward, J. The Complexity of DNA Damage: Relevance to Biological Consequences. Int. J. Radiat. Boil. 1994, 66, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. The Initial Physical Damage Produced by Ionizing Radiations. Int. J. Radiat. Boil. 1989, 56, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Charlton, D.; Goodhead, D. Monte Carlo track structure studies of energy deposition and calculation of initial DSB and RBE. Adv. Space Res. 1994, 14, 161–180. [Google Scholar] [CrossRef]
- Holley, W.R.; Chatterjee, A. Clusters of DNA induced by ionizing radiation: Formation of short DNA fragments. I. Theoretical modeling. Radiat. Res. 1996, 145, 188–199. [Google Scholar] [CrossRef]
- Pinto, M.; Prise, K.M.; Michael, B.D. A Monte Carlo model of DNA double-strand break clustering and rejoining kinetics for the analysis of pulsed-field gel electrophoresis data. Radiat. Res. 2004, 162, 453–463. [Google Scholar] [CrossRef]
- Stewart, R.D.; Yu, V.K.; Georgakilas, A.G.; Koumenis, C.; Park, J.H.; Carlson, D.J. Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat. Res. 2011, 176, 587–602. [Google Scholar] [CrossRef]
- Friedland, W.; Dingfelder, M.; Kundrát, P.; Jacob, P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 28–40. [Google Scholar] [CrossRef]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of Radiation-Induced Clustered Non-DSB Damage – A Monte Carlo Track Structure Modeling and Calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef]
- Rydberg, B. Clusters of DNA Damage Induced by Ionizing Radiation: Formation of Short DNA Fragments. II. Experimental Detection. Radiat. Res. 1996, 145, 200–209. [Google Scholar] [CrossRef]
- Prise, K.M.; Pullar, C.H.; Michael, B.D. A study of endonuclease III-sensitive sites in irradiated DNA: Detection of alpha-particle-induced oxidative damage. Carcinogenesis 1999, 20, 905–909. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered damages and total lesions induced in DNA by ionizing radiation: Oxidized bases and strand breaks. Biochemistry 2000, 39, 8026–8031. [Google Scholar] [CrossRef] [PubMed]
- Fakir, H.; Sachs, R.K.; Stenerlöw, B.; Hofmann, W. Clusters of DNA Double-Strand Breaks Induced by Different Doses of Nitrogen Ions for Various LETs: Experimental Measurements and Theoretical Analyses. Radiat. Res. 2006, 166, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Asaithamby, A.; Chen, D.J. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat. Res. 2011, 711, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy–The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Mladenov, E.; Mangelis, A.; Laskaratou, D.A.; Fragkoulis, G.I.; Hellweg, C.E.; Martin, O.A.; Emfietzoglou, D.; et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free. Radic. Res. 2016, 50 (Suppl. S1), S64–S78. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-LET irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Sage, E.; Harrison, L. Clustered DNA lesion repair in eukaryotes: Relevance to mutagenesis and cell survival. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 123–133. [Google Scholar] [CrossRef]
- Magnander, K.; Elmroth, K. Biological consequences of formation and repair of complex DNA damage. Cancer Lett. 2012, 327, 90–96. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free. Radic. Boil. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Georgakilas, A.G.; Ravanat, J.-L. Targeted and Off-Target (Bystander and Abscopal) Effects of Radiation Therapy: Redox Mechanisms and Risk/Benefit Analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J. The linear-quadratic model is an appropriate methodology for determining isoeffective doses at large doses per fraction. Semin. Radiat. Oncol. 2008, 18, 234–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, D.J.; Hlatky, L.R.; Hahnfeldt, P.J.; Huang, Y.; Sachs, R.K. The linear-quadratic model and most other common radiobiological models result in similar predictions of time-dose relationships. Radiat. Res. 1998, 150, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Elsässer, T.; Kramer, M.; Scholz, M. Accuracy of the Local Effect Model for the Prediction of Biologic Effects of Carbon Ion Beams In Vitro and In Vivo. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 866–872. [Google Scholar] [CrossRef]
- Dahle, T.J.; Magro, G.; Ytre-Hauge, K.S.; Stokkevåg, C.H.; Choi, K.; Mairani, A. Sensitivity study of the microdosimetric kinetic model parameters for carbon ion radiotherapy. Phys. Med. Boil. 2018, 63, 225016. [Google Scholar] [CrossRef]
- Tello Cajiao, J.J.; Carante, M.P.; Bernal Rodriguez, M.A.; Ballarini, F. Proximity effects in chromosome aberration induction: Dependence on radiation quality, cell type and dose. DNA Repair (Amst) 2018, 64, 45–52. [Google Scholar] [CrossRef]
- Nikjoo, H.; Uehara, S.; Emfietzoglou, D. Interaction of Radiation with Matter; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Friedland, W.; Kundrat, P. Modeling of Radiation Effects in Cells and Tissues. In Comprehensive Biomedical Physics; Brahme, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 105–142. [Google Scholar]
- Nikjoo, H.; Uehara, S.; Emfietzoglou, D.; Cucinotta, F. Track-structure codes in radiation research. Radiat. Meas. 2006, 41, 1052–1074. [Google Scholar] [CrossRef]
- Kyriakou, I.; Ivanchenko, V.; Sakata, D.; Bordage, M.; Guatelli, S.; Incerti, S.; Emfietzoglou, D. Influence of track structure and condensed history physics models of Geant4 to nanoscale electron transport in liquid water. Phys. Medica Eur. J. Med. Phys. 2019, 58, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Nikjoo, H.; Uehara, S.; E Wilson, W.; Hoshi, M.; Goodhead, D.T. Track structure in radiation biology: Theory and applications. Int. J. Radiat. Boil. 1998, 73, 355–364. [Google Scholar] [CrossRef]
- Zaider, M.; Brenner, D.J.; Wilson, W.E. The Applications of Track Calculations to Radiobiology I. Monte Carlo Simulation of Proton Tracks. Radiat. Res. 1983, 95, 231–247. [Google Scholar] [CrossRef]
- Uehara, S.; Nikjoo, H. Monte Carlo simulation of water radiolysis for low-energy charged particles. J. Radiat. Res. 2006, 47, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikjoo, H.; Uehara, S. Track Structure Studies of Biological Systems, in Charged Particle and Photon Interactions with Matter: Chemical, Physicochemical, and Biological Consequences with Applications; Mozumder, A., Hatano, Y., Eds.; CRC Press: Boca Raton, FL, USA, 2003; pp. 491–531. [Google Scholar]
- Dingfelder, M. Track-structure simulations for charged particles. Health Phys. 2012, 103, 590–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, W.R.; Hirayama, H.; Rogers, D.W.O. The EGS4 Code System. In SLAC Report 265; Stanford Linear Accelerator Center, Stanford University: Stanford, CA, USA, 1985. [Google Scholar]
- Battistoni, G.; Cerutti, F.; Fassò, A.; Ferrari, A.; Muraro, S.; Ranft, J.; Roesler, S.; Sala, P.R. The FLUKA code: description and benchmarking. AIP Conf. Proc. 2007, 896, 31–49. [Google Scholar]
- Ferrari, A.; Sala, R.P.; Fasso, A.; Ranft, J. FLUKA: A Multi-Particle Transport Code; Stanford Linear Accelerator Center: Stanford, CA, USA, 2005. [Google Scholar]
- Agostinelli, S.; Allisonas, J.; Amakoe, K.; Apostolakisa, J.; Araujoaj, H.; Arcelmxa, P.; Asaigai, M.; Axenit, D.; Banerjeebil, S.; Barrand, G.; et al. GEANT4―A simulation toolkit. Nucl. Instrum. Methods 2003, 506, 250–303. [Google Scholar] [CrossRef] [Green Version]
- Incerti, S.; Kyriakou, I.; Bernal, M.A.; Bordage, M.C.; Francis, Z.; Guatelli, S.; Ivanchenko, V.; Karamitros, M.; Lampe, N.; Lee, S.B. Geant4-DNA example applications for track structure simulations in liquid water: A report from the Geant4-DNA Project. Med. Phys. 2018, 45, e722–e739. [Google Scholar] [CrossRef] [Green Version]
- Bernal, M.A.; Bordage, M.C.; Brown, J.M.C.; Davídková, M.; Delage, E.; El Bitar, Z.; Enger, S.A.; Francis, Z.; Guatelli, S.; Ivanchenko, V.N.; et al. Track structure modeling in liquid water: A review of the Geant4-DNA very low energy extension of the Geant4 Monte Carlo simulation toolkit. Phys. Med. 2015, 31, 861–874. [Google Scholar] [CrossRef]
- Liamsuwan, T.; Emfietzoglou, D.; Uehara, S.; Nikjoo, H. Microdosimetry of low-energy electrons. Int. J. Radiat. Biol. 2012, 88, 899–907. [Google Scholar] [CrossRef]
- Uehara, S.; Nikjoo, H.; Goodhead, D.T. Cross sections for water vapour for Monte Carlo track structure code from 10 eV to 10 MeV region. Phys. Med. Biol. 1993, 38, 1841–1858. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Papamichael, G.; Kostarelos, K.; Moscovitch, M. A Monte Carlo track structure code for electrons (~10 eV-10 keV) and protons (~0.3-10 MeV) in water: partitioning of energy and collision events. Phys. Med. Biol. 2000, 45, 3171–3194. [Google Scholar] [CrossRef]
- Emfietzoglou, D.; Karava, K.; Papamichael, G.; Moscovitch, M. Monte Carlo simulation of the energy loss of low-energy electrons in liquid water. Phys. Med. Biol. 2003, 48, 2355–2371. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D.; Papamichael, G.; Nikjoo, H. Monte Carlo Electron Track Structure Calculations in Liquid Water Using a New Model Dielectric Response Function. Radiat. Res. 2017, 188, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Emfietzoglou, D.; Papamichael, G.; Karava, K.; Androulidakis, I.; Pathak, A.; Phillips, G.W.; Moscovitch, M.; Kostarelos, K.A. Monte-Carlo code for the detailed simulation of electron and light-ion tracks in condensed matter. Radiat. Prot. Dosimetry 2006, 119, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Semenenko, V.A.; Stewart, R.D. A fast Monte Carlo algorithm to simulate the spectrum of DNA damages formed by ionizing radiation. Radiat. Res. 2004, 161, 451–457. [Google Scholar] [CrossRef]
- Forster, R.A.; Godfrey, T.N.K. MCNP―A General Monte Carlo Code For Neutron And Photon Transport. In Monte-Carlo Methods and Applications in Neutronics, Photonics and Statistical Physics. Lecture Notes in Physics; Alcouffe, R., Dautray, R., Forster, A., Ledanois, G., Mercier, B., Eds.; Springer: Berlin\Heidelberg, Germany, 1985. [Google Scholar]
- Dingfelder, M.; Travia, A.; McLawhorn, R.A.; Shinpaugh, J.L.; Toburen, L.H. Electron emission from foils and biological materials after proton impact. Radiat. Phys. Chem. 2008, 77, 1213–1217. [Google Scholar] [CrossRef] [Green Version]
- Salvat, F.; Fernández-Varea, J.M.; Sempau, J. PENELOPE: A Code System for Monte Carlo Simulation of Electron and Photon Transport. 2001. Available online: https://moreira.tamu.edu/BAEN625/TOC_files/penelope.pdf (accessed on 14 October 2019).
- Siantar, C.L.H.; Moses, E.I. The PEREGRINE programmer: using physics and computer simulation to improve radiation therapy for cancer. Eur. J. Phys. 1998, 19, 513–552. [Google Scholar] [CrossRef]
- Plante, I.; Cucinotta, F.A. Multiple CPU Computing: The Example of the Code RITRACKS. In Computational Intelligence Methods for Bioinformatics and Biostatistics. CIBB 2012. Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Schuemann, J.; McNamara, A.L.; Ramos-Méndez, J.; Perl, J.; Held, K.D.; Paganetti, H.; Incerti, S.; Faddegon, B. TOPAS-nBio: An Extension to the TOPAS Simulation Toolkit for Cellular and Sub-cellular Radiobiology. Radiat. Res. 2019, 191, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Wälzlein, C.; Scifoni, E.; Krämer, M.; Durante, M. Simulations of dose enhancement for heavy atom nanoparticles irradiated by protons. Phys. Med. Biol. 2014, 59, 1441–1458. [Google Scholar] [CrossRef]
- Solov’yov, I.A.; Korol, A.V.; Solov’yov, A.V. Multiscale Modeling of Complex Molecular Structure and Dynamics with MBN Explorer; Springer: Berlin, Germany, 2017. [Google Scholar]
- Surdutovich, E.; Yakubovich, A.V.; Solov’Yov, A.V. Multiscale approach to radiation damage induced by ion beams: Complex DNA damage and effects of thermal spikes. Eur. Phys. J. D 2010, 60, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Torfeh, E.; Simon, M.; Muggiolu, G.; Devès, G.; Vianna, F.; Bourret, S.; Incerti, S.; Barberet, P.; Seznec, H. Monte-Carlo dosimetry and real-time imaging of targeted irradiation consequences in 2-cell stage Caenorhabditis elegans embryo. Sci. Rep. 2019, 9, 10568. [Google Scholar] [CrossRef]
- Ward, J. DNA Damage Produced by Ionizing Radiation in Mammalian Cells: Identities, Mechanisms of Formation, and Reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar] [PubMed]
- Ward, J.F. Radiation Mutagenesis: The Initial DNA Lesions Responsible. Radiat. Res. 1995, 142, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Regulus, P.; Duroux, B.; Bayle, P.-A.; Favier, A.; Cadet, J.; Ravanat, J.-L. Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion. Proc. Natl. Acad. Sci. USA 2007, 104, 14032–14037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikjoo, H.; O’Neill, P.; Wilson, W.E.; Goodhead, D.T. Computational Approach for Determining the Spectrum of DNA Damage Induced by Ionizing Radiation. Radiat. Res. 2001, 156, 577–583. [Google Scholar] [CrossRef]
- Semenenko, V.A.; Stewart, R.D. Fast Monte Carlo simulation of DNA damage formed by electrons and light ions. Phys. Med. Boil. 2006, 51, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M. Radiation Track Structure: How the Spatial Distribution of Energy Deposition Drives Biological Response. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Hill, M.A. Track to the future: Historical perspective on the importance of radiation track structure and DNA as a radiobiological target. Int. J. Radiat. Biol. 2018, 94, 759–768. [Google Scholar] [CrossRef]
- Schuemann, J.; McNamara, A.L.; Warmenhoven, J.W.; Henthorn, N.T.; Kirkby, K.J.; Merchant, M.J.; Ingram, S.; Paganetti, H.; Held, K.D.; Ramos-Mendez, J.; et al. A New Standard DNA Damage (SDD) Data Format. Radiat. Res. 2019, 191, 76–92. [Google Scholar] [CrossRef]
- Nikjoo, H. Radiation track and DNA damage. Iran. J. Radiat. Res. 2003, 1, 3–16. [Google Scholar]
- Nikjoo, H.; Emfietzoglou, D.; Watanabe, R.; Uehara, S. Can Monte Carlo track structure codes reveal reaction mechanism in DNA damage and improve radiation therapy? Radiat. Phys. Chem. 2008, 77, 1270–1279. [Google Scholar] [CrossRef]
- Nikjoo, H.; Taleei, R.; Liamsuwan, T.; Liljequist, D.; Emfietzoglou, D. Perspectives in radiation biophysics: From radiation track structure simulation to mechanistic models of DNA damage and repair. Radiat. Phs. Chem. 2016, 128, 3–10. [Google Scholar] [CrossRef]
- Pechatnikov, V.A.; Afanasyev, V.N.; A Korol, B.; Korneev, V.N.; Yua, R.; Umansky, S.R. Flow cytometry analysis of DNA degradation in thymocytes of gamma-irradiated or hydrocortisone treated rats. Gen. Physiol. Biophys. 1986, 5, 273–284. [Google Scholar] [PubMed]
- Hong, Z.; Jiang, J.; Hashiguchi, K.; Hoshi, M.; Lan, L.; Yasui, A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J. Cell Sci. 2008, 121 Pt 19, 3146–3154. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046.e5–1061.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauptner, A.; Krücken, R.; Greubel, C.; Hable, V.; Dollinger, G.; Drexler, G.A.; Deutsch, M.; Löwe, R.; Friedl, A.A.; Dietzel, S.; et al. DNA-repair protein distribution along the tracks of energetic ions. Radiat. Prot. Dosim. 2006, 122, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Friedland, W.; Kundrát, P.; Schmitt, E.; Becker, J.; Ilicic, K.; Greubel, C.; Reindl, J.; Siebenwirth, C.; Schmid, T.E.; Dollinger, G. Modeling Studies on Dicentrics Induction after Sub-Micrometer Focused Ion Beam Grid Irradiation. Radiat. Prot. Dosim. 2019, 183, 40–44. [Google Scholar] [CrossRef]
- Jain, S.; Coulter, J.A.; Hounsell, A.R.; Butterworth, K.T.; McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Dickson, G.R.; Prise, K.M.; Currell, F.J.; et al. Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.S.; Patel, K.; Ahmad, J.; Holloway, K.; Evans, M.D.; Lunec, J. Monoclonal Antibody to Single-Stranded DNA: A Potential Tool for DNA Repair Studies. Biochem. Biophys. Res. Commun. 2001, 284, 232–238. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Petrini, J.H.J. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003, 1, 207–218. [Google Scholar]
- Jurvansuu, J.; Raj, K.; Stasiak, A.; Beard, P. Viral Transport of DNA Damage That Mimics a Stalled Replication Fork. J. Virol. 2005, 79, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015, 11, 1486–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorat, Y.; Schanz, S.; Ruebe, C.E. Ultrastructural insights into the biological significance of persisting DNA damage foci after low doses of ionizing radiation. Clin. Cancer Res. 2016, 22, 5300–5311. [Google Scholar] [CrossRef]
- Gustafsdottir, S.M.; Schallmeiner, E.; Fredriksson, S.; Gullberg, M.; Söderberg, O.; Jarvius, M.; Jarvius, J.; Howell, M.; Landegren, U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005, 345, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Mladenova, V.; Sharif, M.; Chaudhary, S.; Mavragani, I.V.; Soni, A.; Saha, J.; Schipler, A.; Mladenov, E. Defined Biological Models of High-Let Radiation Lesions. Radiat. Prot. Dosim. 2018, 183, 1–9. [Google Scholar] [CrossRef]
- Galbiati, A.; Di Fagagna, F.D. DNA Damage In Situ Ligation Followed by Proximity Ligation Assay (DI-PLA). Methods Mol. Biol. 2019, 1896, 11–20. [Google Scholar]
- Edwards, S.K.; Ono, T.; Wang, S.; Jiang, W.; Franzini, R.M.; Jung, J.W.; Chan, K.M.; Kool, E.T. In Vitro Fluorogenic Real-Time Assay of the Repair of Oxidative DNA Damage. ChemBioChem 2015, 16, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Beharry, A.A.; Nagel, Z.D.; Samson, L.D.; Kool, E.T. Fluorogenic Real-Time Reporters of DNA Repair by MGMT, a Clinical Predictor of Antitumor Drug Response. PLoS ONE 2016, 11, 0152684. [Google Scholar] [CrossRef] [Green Version]
- Beharry, A.A.; Lacoste, S.; O’Connor, T.R.; Kool, E.T. Fluorescence Monitoring of the Oxidative Repair of DNA Alkylation Damage by ALKBH3, a Prostate Cancer Marker. J. Am. Chem. Soc. 2016, 138, 3647–3650. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.L.; Beharry, A.A.; Srivastava, A.; O’Connor, T.R.; Kool, E.T. Fluorescence Probes for ALKBH2 Allow the Measurement of DNA Alkylation Repair and Drug Resistance Responses. Angew. Chem. Int. Ed. Engl. 2018, 57, 12896–12900. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.M.; Lacoste, J.; Swift, J.L.; Brown, C.M. Live-cell microscopy - tips and tools. J. Cell Sci. 2009, 122, 753–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Boil. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imrichova, T.; Hubackova, S.; Kucerova, A.; Kosla, J.; Bartek, J.; Hodny, Z.; Vasicova, P. Dynamic PML protein nucleolar associations with persistent DNA damage lesions in response to nucleolar stress and senescence-inducing stimuli. Aging 2019, 11, 7206–7235. [Google Scholar] [CrossRef] [PubMed]
- Varga, D.; Majoros, H.; Ujfaludi, Z.; Erdélyi, M.; Pankotai, T. Quantification of DNA damage induced repair focus formation via super-resolution dSTORM localization microscopy. Nanoscale 2019, 11, 14226–14236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumhardt, P.; Stein, J.; Mücksch, J.; Stehr, F.; Bauer, J.; Jungmann, R.; Schwille, P. Photo-Induced Depletion of Binding Sites in DNA-PAINT Microscopy. Molecules 2018, 23, 3165. [Google Scholar] [CrossRef] [Green Version]
- Chow, T.T.; Shi, X.; Wei, J.-H.; Guan, J.; Stadler, G.; Huang, B.; Blackburn, E.H. Local enrichment of HP1alpha at telomeres alters their structure and regulation of telomere protection. Nat. Commun. 2018, 9, 3583. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, S.M.; Zhao, T.; Rozario, A.M.; Rootes, C.L.; McMillan, P.J.; Purcell, A.W.; Woon, A.; Marsh, G.A.; Lieu, K.G.; Wang, L.-F.; et al. Viral regulation of host cell biology by hijacking of the nucleolar DNA-damage response. Nat. Commun. 2018, 9, 3057. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, M.; Wagner, E.; Lee, J.-H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuß, F.; Port, M.; Bestvater, F.; Scherthan, H.; et al. Super-resolution localization microscopy of radiation-induced histone H2AX-phosphorylation in relation to H3K9-trimethylation in HeLa cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef]
- Eryilmaz, M.; Schmitt, E.; Krufczik, M.; Theda, F.; Lee, J.-H.; Cremer, C.; Bestvater, F.; Schaufler, W.; Hausmann, M.; Hildenbrand, G. Localization Microscopy Analyses of MRE11 Clusters in 3D-Conserved Cell Nuclei of Different Cell Lines. Cancers 2018, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; Van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103 Pt 3, 857–862. [Google Scholar]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, O.A.; Ivashkevich, A.; Choo, S.; Woodbine, L.; Jeggo, P.A.; Martin, R.F.; Lobachevsky, P. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: Cell cycle effect and implications for prediction of radiosensitivity. DNA Repair 2013, 12, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Plante, I.; Emfietzoglou, D.; Iliakis, G.; Georgakilas, A.G. Non-DSB clustered DNA lesions. Does theory colocalize with the experiment? Radiat. Phys. Chem. 2016, 128, 26–35. [Google Scholar] [CrossRef]
- Ivashkevich, A.N.; Martin, O.A.; Smith, A.J.; Redon, C.E.; Bonner, W.M.; Martin, R.F.; Lobachevsky, P.N. Gamma H2AX foci as a measure of DNA damage: A computational approach to automatic analysis. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, M.-C.; Massonneau, J.; LeDuc, F.; Arguin, M.; Brazeau, M.-A.; Boissonneault, G. Quantification and genome-wide mapping of DNA double-strand breaks. DNA Repair 2016, 48, 63–68. [Google Scholar] [CrossRef]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [Green Version]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biernacka, A.; Zhu, Y.; Skrzypczak, M.; Forey, R.; Pardo, B.; Grzelak, M.; Nde, J.; Mitra, A.; Kudlicki, A.; Crosetto, N.; et al. i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks. Commun. Boil. 2018, 1, 181. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Biernacka, A.; Pardo, B.; Dojer, N.; Forey, R.; Skrzypczak, M.; Fongang, B.; Nde, J.; Yousefi, R.; Pasero, P.; et al. qDSB-Seq is a general method for genome-wide quantification of DNA double-strand breaks using sequencing. Nat. Commun. 2019, 10, 2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwman, B.A.M.; Crosetto, N. Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadet, J.; Carell, T.; Cellai, L.; Chatgilialoglu, C.; Gimisis, T.; Miranda, M.A.; O’Neill, P.; Ravanat, J.-L.; Robert, M. DNA Damage and Radical Reactions: Mechanistic Aspects, Formation in Cells and Repair Studies. Chim. Int. J. Chem. 2008, 62, 742–749. [Google Scholar] [CrossRef]
- Vitti, T.E.; Parsons, L.J. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers 2019, 11, 946. [Google Scholar] [CrossRef] [Green Version]
- Friedland, W.; Schmitt, E.; Kundrát, P.; Dingfelder, M.; Baiocco, G.; Barbieri, S.; Ottolenghi, A. Comprehensive track-structure based evaluation of DNA damage by light ions from radiotherapy-relevant energies down to stopping. Sci. Rep. 2017, 7, 45161. [Google Scholar] [CrossRef]
- Ushigome, T.; Shikazono, N.; Fujii, K.; Watanabe, R.; Suzuki, M.; Tsuruoka, C.; Tauchi, H.; Yokoya, A. Yield of single- and double-strand breaks and nucleobase lesions in fully hydrated plasmid DNA films irradiated with high-LET charged particles. Radiat. Res. 2011, 177, 614–627. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both Complexity and Location of DNA Damage Contribute to Cellular Senescence Induced by Ionizing Radiation. PLoS ONE 2016, 11, 0155725. [Google Scholar] [CrossRef]
- Goodhead, D. Initial Events in the Cellular Effects of Ionizing Radiations: Clustered Damage in DNA. Int. J. Radiat. Boil. 1994, 65, 7–17. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA Damage Induced by High Linear Energy Transfer Alpha-Particles and Protons Triggers a Specific Cellular DNA Damage Response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Brzozowska, B.; Sollazzo, A.; Lundholm, L.; Lisowska, H.; Haghdoost, S.; Wojcik, A. Simultaneous induction of dispersed and clustered DNA lesions compromises DNA damage response in human peripheral blood lymphocytes. PLoS ONE 2018, 13, e0204068. [Google Scholar] [CrossRef] [PubMed]
- Boreyko, A.V.; Bugay, A.N.; Bulanova, T.S.; Dushanov, E.B.; Jezkova, L.; Kulikova, E.A.; Smirnova, E.V.; Zadneprianetc, M.G.; Krasavin, E.A. Clustered DNA Double-Strand Breaks and Neuroradiobiological Effects of Accelerated Charged Particles. Phys. Part. Nucl. Lett. 2018, 15, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Averbeck, N.B.; Ringel, O.; Herrlitz, M.; Jakob, B.; Durante, M.; Taucher-Scholz, G. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle 2014, 13, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Narang, H.; Sarma, A.; Krishna, M. DNA damage response signaling in lung adenocarcinoma A549 cells following gamma and carbon beam irradiation. Mutat. Res. Mol. Mech. Mutagen. 2011, 716, 10–19. [Google Scholar] [CrossRef]
- Singleton, B.K.; Griffin, C.S.; Thacker, J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. Cancer Res. 2002, 62, 6263. [Google Scholar]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. BioSyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Goodarzi, A.A.; Jeggo, P.; Löbrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Bennett, P.V.; Wilson, D.M.; Sutherland, B.M. Processing of bistranded abasic DNA clusters in γ-irradiated human hematopoietic cells. Nucleic Acids Res. 2004, 32, 5609–5620. [Google Scholar] [CrossRef]
- Dianov, G.L.; O’Neill, P.; Goodhead, D.T. Securing genome stability by orchestrating DNA repair: Removal of radiation-induced clustered lesions in DNA. BioEssays 2001, 23, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and Repair of Clustered DNA Lesions: What Do We Know So Far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Jin, C. Histone variants in environmental-stress-induced DNA damage repair. Mutat. Res. 2019, 780, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Goodhead, D.T. Mechanisms for the biological effectiveness of high-LET radiations. Radiat. Res. 1999, 40, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prise, K.M.; Pinto, M.; Newman, H.C.; Michael, B.D. A Review of Studies of Ionizing Radiation-Induced Double-Strand Break Clustering. Radiat. Res. 2001, 156, 572–576. [Google Scholar] [CrossRef]
- Ostashevsky, J.Y. A Model Relating Cell Survival to DNA Fragment Loss and Unrepaired Double-Strand Breaks. Radiat. Res. 1989, 118, 437–466. [Google Scholar] [CrossRef] [PubMed]
- Yatagai, F.; Suzuki, M.; Ishioka, N.; Ohmori, H.; Honma, M. Repair of I-SceI Induced DSB at a specific site of chromosome in human cells: Influence of low-dose, low-dose-rate gamma-rays. Radiat. Environ. Biophys. 2008, 47, 439–444. [Google Scholar] [CrossRef]
- Honma, M.; Sakuraba, M.; Koizumi, T.; Takashima, Y.; Sakamoto, H.; Hayashi, M. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair 2007, 6, 781–788. [Google Scholar] [CrossRef]
- Yoshihiko, H.; Takahiro, O.; Atsuko, N.; Motohiro, Y.; Hiro, S.; Siripan, L.; Kathryn, D.H.; Takashi, N.; Atsushi, S. Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J. Radiat. Res. 2018, 60, 69–79. [Google Scholar]
- Mladenova, V.; Mladenov, E.; Iliakis, G. Novel Biological Approaches for Testing the Contributions of Single DSBs and DSB Clusters to the Biological Effects of High LET Radiation. Front. Oncol. 2016, 6, 163. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Saha, J.; Iliakis, G. Processing-Challenges Generated by Clusters of DNA Double-Strand Breaks Underpin Increased Effectiveness of High-LET Radiation and Chromothripsis. In Chromosome Translocation; Zhang, Y., Ed.; Springer: Singapore, 2018; pp. 149–168. [Google Scholar]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Jeggo, P. DNA Double-strand Break Repair in a Cellular Context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keszenman, D.J.; Kolodiuk, L.; Baulch, J.E. DNA damage in cells exhibiting radiation-induced genomic instability. Mutagenesis 2015, 30, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Carante, M.P.; Altieri, S.; Bortolussi, S.; Postuma, I.; Protti, N.; Ballarini, F. Modeling radiation-induced cell death: Role of different levels of DNA damage clustering. Radiat. Environ. Biophys. 2015, 54, 305–316. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res./Rev. Mol. Mech. Mutagen. 2010, 704, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Okayasu, R. Repair of DNA damage induced by accelerated heavy ions—A mini review. Int. J. Cancer 2012, 130, 991–1000. [Google Scholar] [CrossRef]
- Hada, M.; Cucinotta, F.A.; Gonda, S.R.; Wu, H. mBAND Analysis of Chromosomal Aberrations in Human Epithelial Cells Exposed to Low- and High-LET Radiation. Radiat. Res. 2007, 168, 98–105. [Google Scholar] [CrossRef]
- Ritter, S.; Durante, M. Heavy-ion induced chromosomal aberrations: A review. Mutat. Res. Toxicol. Environ. Mutagen. 2010, 701, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Cucinotta, F.A. Heavy ion carcinogenesis and human space exploration. Nat. Rev. Cancer 2008, 8, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Villanueva, M.; Wu, H. Radiation and microgravity–Associated stress factors and carcinogensis. REACH 2019, 13, 100027. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Boil. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Adams, C.; Balmain, A.; Costes, S.V.; DeMaria, S.; Illa-Bochaca, I.; Mao, J.H.; Ouyang, H.; Sebastiano, C.; Tang, J. Systems biology perspectives on the carcinogenic potential of radiation. J. Radiat. Res. 2014, 55 (Suppl. S1), i145–i154. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Balázs, K.; Kis, E.; Badie, C.; Bogdándi, E.N.; Candéias, S.; Garcia, L.C.; Dominczyk, I.; Frey, B.; Gaipl, U.; Jurányi, Z.; et al. Radiotherapy-Induced Changes in the Systemic Immune and Inflammation Parameters of Head and Neck Cancer Patients. Cancers 2019, 11, 1324. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, L.; Sahoo, N.; Aliru, M.; Krishnan, S. Dependence of Immunogenic Modulation of Tumor Cells by Proton Radiation on the Linear Energy Transfer. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, E607. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Oike, T.; Okonogi, N.; Suzuki, Y.; Ando, K.; Sato, H.; Noda, S.-E.; Isono, M.; Mimura, K.; Kono, K.; et al. Carbon-ion beams induce production of an immune mediator protein, high mobility group box 1, at levels comparable with X-ray irradiation. J. Radiat. Res. 2015, 56, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; He, M.; Tu, W.; Konishi, T.; Liu, W.; Xie, Y.; Dang, B.; Li, W.; Uchihori, Y.; Hei, T.K.; et al. The differential role of human macrophage in triggering secondary bystander effects after either gamma-ray or carbon beam irradiation. Cancer Lett. 2015, 363, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, A.; Ueda, Y.; Yamada, S.; Harada, Y.; Shimada, H.; Hasegawa, M.; Tsujii, H.; Ochiai, T.; Yonemitsu, Y. Carbon-ion beam treatment induces systemic antitumor immunity against murine squamous cell carcinoma. Cancer 2010, 116, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, Y.; Iwakawa, M.; Seino, K.-I.; Nakawatari, M.; Wada, H.; Kamijuku, H.; Nakamura, E.; Nakano, T.; Imai, T. Combining Carbon Ion Radiotherapy and Local Injection of α-Galactosylceramide–Pulsed Dendritic Cells Inhibits Lung Metastases in an In Vivo Murine Model. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Imadome, K.; Iwakawa, M.; Nojiri, K.; Tamaki, T.; Sakai, M.; Nakawatari, M.; Moritake, T.; Yanagisawa, M.; Nakamura, E.; Tsujii, H.; et al. Upregulation of stress-response genes with cell cycle arrest induced by carbon ion irradiation in multiple murine tumors models. Cancer Biol. Ther. 2008, 7, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Burns, F.J.; Chen, H.; Chen, S.; Wu, F. Alterations in Gene Expression in Rat Skin Exposed to56Fe Ions and Dietary Vitamin A Acetate. Radiat. Res. 2006, 165, 570–581. [Google Scholar] [CrossRef]
- Ogata, T.; Teshima, T.; Kagawa, K.; Hishikawa, Y.; Takahashi, Y.; Kawaguchi, A.; Suzumoto, Y.; Nojima, K.; Furusawa, Y.; Matsuura, N. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005, 65, 113. [Google Scholar]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.-L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, I.V.; Nikitaki, Z.; Hada, M.; Georgakilas, A.G. Invited talk: Uniting ionizing radiation, DNA damage and immune response through an integrative experimental and in silico analysis. In Proceedings of the 43rd Annual Meeting of the European Radiation Research Society, Essen, Germany, 17–21 September 2017. [Google Scholar]
- Mavragani, I.V.; Laskaratou, D.A.; Frey, B.; Candéias, S.M.; Gaipl, U.S.; Lumniczky, K.; Georgakilas, A.G. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review. Toxicol. Res. 2016, 5, 12–33. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Code | Particles | Pros | Cons | Ref. |
|---|---|---|---|---|
| EGS4 | photons & e- | Developed specifically for dose calculations in RT applications | Not applicable at the nanoscale due to limitations of the physics models | [47] |
| FLUKA | Large set of particles | Multipurpose code covering medical, space, nuclear, and high-energy applications | Closed-source code. Not applicable at the nanoscale due to limitations of the physics models | [48,49] |
| Geant4 | large set of particles | Open-source toolkit covering medical, space, nuclear, and high-energy applications. Contains powerful visualization packages. Includes low-energy physics models for sub-keV transport. Maintained by a large international collaboration | Complex toolkit. Computationally intensive. Requires users with advanced programming skills. | [41,50] |
| Geant4-DNA | Large set of particles | Performs event-by-event simulation for track-structure applications in liquid water. Includes a variety of low-energy electron models. Includes chemistry (water radiolysis) and biology (DNA damage and repair models) Maintained by a large international collaboration and continuously upgraded | Complex toolkit. Computationally intensive. Requires users with advanced programming skills. | [51,52] |
| KURBUC | Photons, neutrons, electrons, protons, alpha, carbon ions | Performs event-by-event simulations down to very low energies for track-structure applications. Includes physics models for both gaseous and liquid water medium. Includes chemistry (water radiolysis) and biology (DNA damage and repair models) | Proprietary code. Specific to water medium. | [53,54] |
| MC4 | e-, protons, α-particles | Performs event-by-event simulations down to low-energies for track-structure applications. Includes physics models for both gaseous and liquid water medium. Known for its upgraded models for the liquid phase. | Proprietary code. Specific to water. Does not extend to relativistic energies. | [55,56,57,58] |
| MCDS | e-, p, alpha particles & ions | Simulates in a very fast way (ranging from seconds to minutes) the induction and clustering of DNA lesions. | Lacks accuracy. Impossible to generate damage configurations for e- with energy lower than 80 eV. | [59] |
| MCNP | Large set of particles | Multipurpose code covering nuclear and medical applications. Known for its accurate neutron models. | Not a free access code. | [60] |
| PARTRAC | e- & ions | Performs event-by-event simulation for track-structure applications. Uses physics models specifically developed for liquid water. Includes chemistry (water radiolysis) and biology (DNA damage and repair models) | Proprietary code. Specific to water. Does not include relativistic energies. The parameterization of the DNA model does not consider the DNA atomic composition. | [17,61] |
| PENELOPE | Photons, e- & e+ | Developed for dose calculations in radiotherapy applications. Known for its electron models that extend to low-energies. Event-by-event simulations possible for application to microdosimetry | Limited track-structure applications due the incomplete simulation of electron track ends. Requires users with advanced programming skills to develop their own applications. | [62] |
| PEREGRINE | Large set of particles | Developed for radiotherapy treatment planning. | Gives results through a computer cluster. Not applicable at the nanoscale due to limitations of the physics models | [63] |
| RITRACKS | e- & ions | Performs event-by-event simulation up to relativistic energies. Simulation of radiation tracks without the need of extensive knowledge of computer programming. Simple in use. | Distributed only to authorized users. Specific to water. Uses atomic cross sections that are not reliable for nanoscale transport in liquid water. | [64] |
| Topas-nBio | large set of particles | Uses Geant4-DNA as its transport engine. Simply to use software specifically developed for radiobiological applications at the (sub) cellular level. | Not (yet) open-sourced. Specific to water medium. | [65] |
| TRAX | e- & ions | Performs event-by-event simulation for track-structure applications in various media. | Uses atomic cross sections that are not reliable for nanoscale transport in condensed-phase media. | [66] |
| Cell Type | Radiation Type | Biological Response | Ref. |
|---|---|---|---|
| HeLa and oropharyngeal squamous cell carcinoma (UMSCC74A and UMSCC6) cells | High-LET α-particles (121 keV/μm) or protons (12 keV/μm), versus low-LET protons (1 keV/μm) and X-rays | Τhe signaling and repair of complex DNA damage, particularly induced by high-LET IR is coordinated through the specific induction of H2Bub catalyzed by MSL2 and RNF20/40, a mechanism that contributes to decreased cell survival after irradiation. | [125,130] |
| Human peripheral blood lymphocytes | Mixed beam of alpha-particles (241Am source, 0.223 Gy/min, LET: 90.9 keV/μm) and X-rays (190 kV, 4.0 mA, 0-2 Gy) | Induced DNA damage was above the level predicted by assuming additivity. The activation levels of DDR proteins and mRNA levels of the studied genes were highest in cells exposed to mixed beams. The repair of damage occurs with a delay. | [131] |
| Human dermal fibroblasts | High-LET IR with carbon ions (9.5 MeV/n; LET 190 keV/μm; calculated mean dose: 1.52 Gy) or calcium ions (7.7 MeV/n; LET 1800 keV/μm; calculated mean dose: 14.4 Gy), versus low-LET IR with 6-MV photons (10 Gy) | High-LET-IR induced clustered DNA damage and triggered profound changes in chromatin structure along particle trajectories. DSBs exhibited delayed repair despite cooperative activity of 53BP1, pATM, pKap-1. | [26] |
| Normal human skin fibroblasts | 60Co γ-rays (LET=0.3 keV/μm), accelerated 11B (E = 8.1 MeV/nucleon, LET=138 keV/μm) ions | It has been found that heavy charged particles induce clustered DNA damage in the genome of cells that can lead not only to gene mutations, but also to large deletions. | [132] |
| HeLa Kyoto cells | Pulsed UV laser (micro-irradiation, IR) | Recruitment and dissociation of 70 DNA repair proteins to laser-induced complex DNA lesions. | [83] |
| Human uveal melanoma (92–1) cells | Carbon ions (LET: 80 keV/μm) and iron ions (LET: 400 keV/μm) at different doses, versus X-rays (LET: 4 keV/μm) | Heavy ions were more effective at inducing senescence than X-rays. Less-efficient repair was observed when DNA damage was induced by heavy ions compared to X-rays and most of the irreparable damage was complex of SSBs and DSBs, while DNA damage induced by X-rays was mostly repaired in 24 h. Results suggest that DNA damage induced by heavy ion is complex and difficult to repair, thus presenting as persistent DNA damage, and pushes the cell into senescence. | [128] |
| Human dermal fibroblasts, NFFh-TERT foreskin fibroblasts | Low-LET irradiation with 6 MV photons, versus high-LET irradiation with carbon ions (9.5 MeV/n; LET = 190 keV/μm) | High-LET irradiation caused localized energy deposition within the particle tracks and generated highly clustered DNA lesions with multiple DSBs in close proximity. Ηuge DSB clusters predominantly localized in condensed heterochromatin. High-LET irradiation-induced clearly higher DSB yields than low-LET irradiation, and large fractions of these heterochromatic DSBs remained unrepaired. | [24] |
| Human osteosarcoma cell line (U2-OS) | X-rays (250 keV, 16 mA; LET: 2 keV/μm), versus heavy ions: 238U ions (LET: 15,000 keV/μm), 207Pb ions (LET: 13,500 keV/μm), 197Au ions (LET: 13,000 keV/μm), 119Sn ions (LET: 7,880 keV/μm), 59Ni (LET: 3,430 keV/μm), 48Ti (LET: 2,180 keV/μm), 14N ions (LET: 400 keV/μm), and 12C ions (LET: 170 keV/μm) | DSB complexity plays a critical role in the decision for DSB end-resection in G1-cells. CtIP, MRE11, and EXO1 are required for the resection of complex DSBs in G. Repair of complex DSBs relies on resection independent of the cell cycle stage. | [133] |
| Human cells (fibroblasts, HBECs) | 1 Gy of Si (LET: 44 keV/μm) or Fe (LET: 150 keV/μm) ions | Direct visualization of clustered DNA lesions at the single-cell level using 53BP1, XRCC1, and hOGG1 as surrogate markers for DSBs, SSBs, and base damage, respectively, reveals that most complex DNA damage is not repaired in human cells. Unrepaired clustered DNA lesions result in the generation of a spectrum of chromosome aberrations. Checkpoint release before the completion of clustered DNA damage repair is a major cause of chromosomal aberrations. | [23] |
| Human Lung Adenocarcinoma (A5490) cells | 12C ions (62 MeV, LET: 290 keV/μm), versus 60Co γ-rays (1–3 Gy) | Carbon ions were three times more cytotoxic than γ-rays. The observed decrease in number of γ-H2AX foci 4 h after γ-rays irradiation indicates repair of damage and is supported by nearly 100% survival, whilst the decrease in γ-H2AX foci after carbon ion irradiation was not indicative of repair. | [134] |
| HF12 primary male human fibroblast cells | 238Pu α-particles (range, ∼20 μm; peak energy, 3.26 MeV; LET=121.4 keV/μm) | Many α-particle-induced mutations are large deletions. Rejoining at microhomologies characterizes large deletion junctions. Intra- and interchromosomal insertions and inversions occur at the sites of some large deletions. Novel fragments found in complex rearrangements derive from other sites of radiation damage in the same cell. | [135] |
| Biological System/Cell Type | Radiation Type | Immune Response | Ref. |
|---|---|---|---|
| Peripheral blood mononuclear cells (PBMCs) of head and neck (HNSCC) cancer patients | Intensity modulated radiotherapy (IMRT), (51–74 Gy total dose, 1.6–3 Gy dose/fraction) | Expression of the FXDR, SESN1, GADD45, DDB2, and MDM2 radiation-response genes were altered in the PBMCs of patients after RT. All changes were long-lasting, detectable one month after RT. Local tumor irradiation induces systemic changes in the level of immune and inflammation-related plasma proteins. RT induces changes in the immune phenotype of PBMCs of HNSCC patients. | [168] |
| Murine CT26 colorectal cancer cells | 8 Gy proton beams at 1.09 keV/μm (low), 2.58 keV/μm (medium) and 7.7 keV/μm (high) LET. | Increase in percentage expression of immune markers (OX40L, CD40, ICAM-1, and MHC-I) in high-LET irradiated cells. High-LET proton radiation can be used to stimulate better immunogenic phenotype in tumor cells compared to low LET proton radiation. | [169] |
| Human cancer cell lines: TE2, KYSE70, A549, NCI-H460 and WiDr | Carbon ions (290 MeV/n, LET 30 keV/µm) | Carbon-ion beams significantly increased HMGB1 (a damage-associated molecular pattern—DAMP) levels in the culture supernatants. | [170] |
| NCI-H446 (lung tumor cells) | Carbon ions (290 MeV/n, LET 13 keV/µm) | Cyto- and chemokine response release by tumor cells after irradiation. (TNF-α) | [171] |
| Tumor-bearing mice (C3H/He, Balb/c nude mice) | Carbon ion irradiation (290 MeV/n, LET=77 keV/µm) | Increased cytotoxic T-lymphocytes (CTL)-associated lysis of isolated tumor splenocytes after carbon ion irradiation treatment with supplementary intratumoral dendritic cell (DC) injection. | [172] |
| Tumors of mouse squamous cell carcinoma (NR-S1) cells inoculated in the legs of C3H/HeSlc mice | Carbon ions (290 MeV/n, 6-cm spread-out Bragg peak, 6 Gy) | Even when exposed to the same equivalent doses, carbon ion therapy might activate the immune system to a greater extent than conventional RT. | [173] |
| NR-S1 and SCCVII (squamous cell carcinoma), NFSa, #8520 (fibrosarcoma) | Carbon ions (290 MeV/n, LET 50 keV/µm) | Significant C-ion induced upregulation of stress-responsive and cell-communication genes common to different tumor types. | [174] |
| Rat skin | 56Fe ions (1.01 GeV/n) | 56Fe ion radiation significantly induced inflammation-related genes, including many in the categories of ‘immune response’, ‘response to stress’, ‘signal transduction’, and ‘response to biotic stress’, that contribute to carcinogenesis. | [175] |
| Highly aggressive HT1080 human fibrosarcoma and LM8 mouse osteosarcoma cells | Carbon ion beams (290 MeV/n), versus X-rays | When compared with photon irradiation, carbon ion exposure reduced the number of distant lung metastasis in carcinoma models in immunocompetent mice. | [176] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. https://doi.org/10.3390/cancers11111789
Mavragani IV, Nikitaki Z, Kalospyros SA, Georgakilas AG. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers. 2019; 11(11):1789. https://doi.org/10.3390/cancers11111789
Chicago/Turabian StyleMavragani, Ifigeneia V., Zacharenia Nikitaki, Spyridon A. Kalospyros, and Alexandros G. Georgakilas. 2019. "Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance" Cancers 11, no. 11: 1789. https://doi.org/10.3390/cancers11111789