
Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
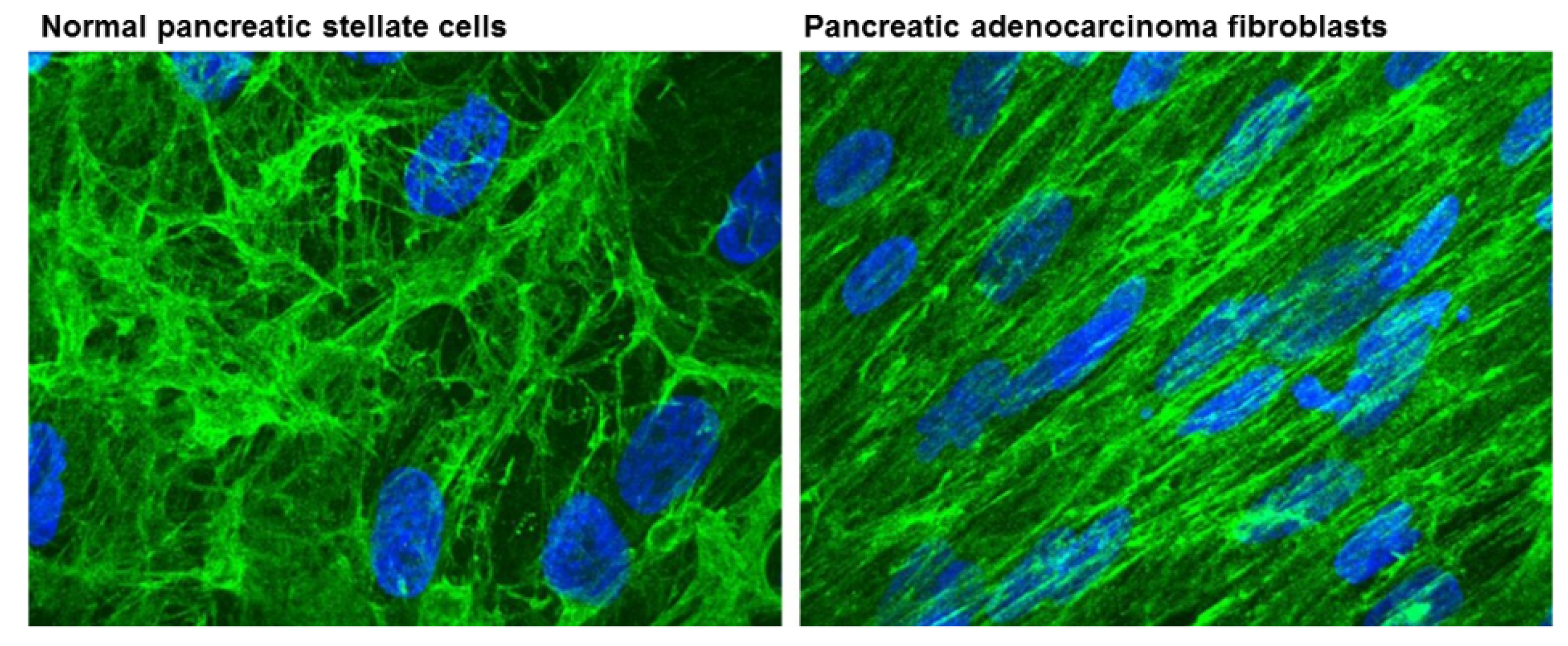
2. Cancer Associated Fibroblasts (CAFs)
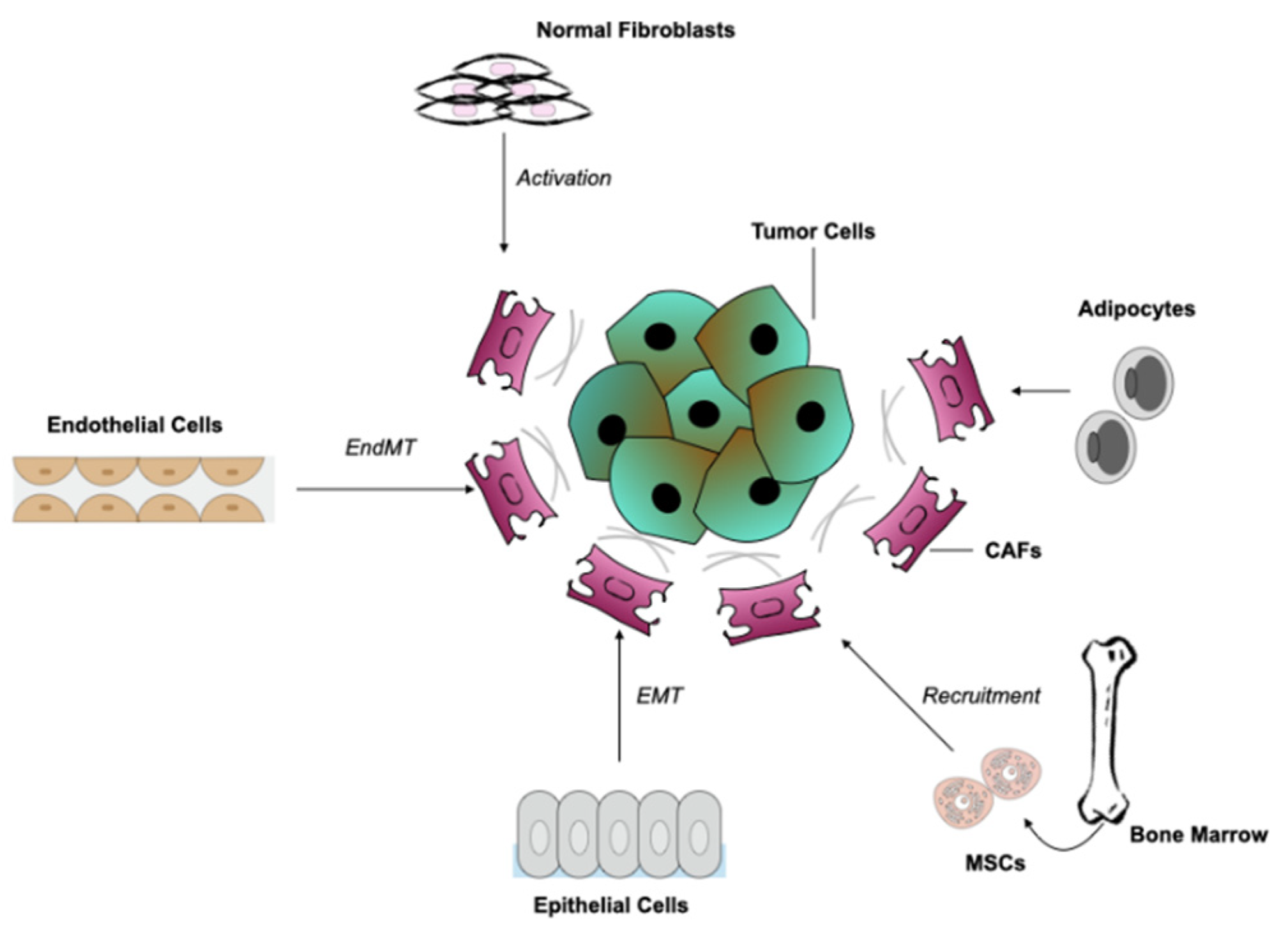
3. Origins of CAFs
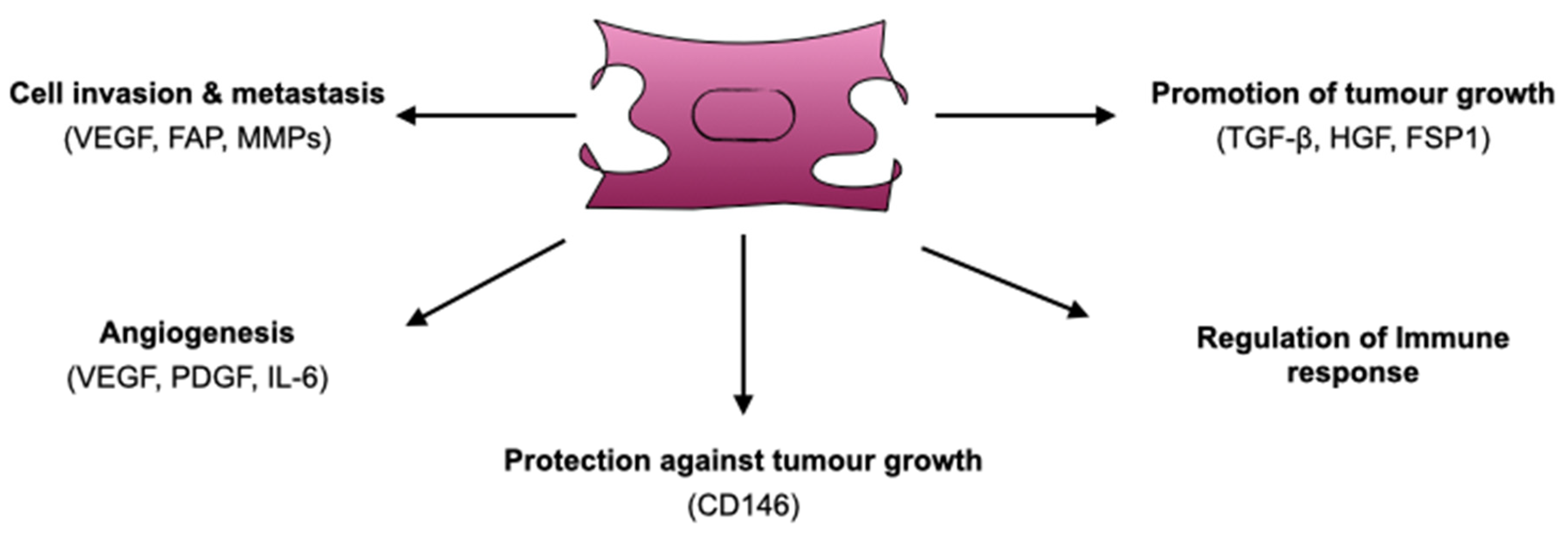
4. Functions of CAFs
4.1. Promotion of Tumour Growth
4.2. Protection Against Tumour Growth
4.3. Cell Invasion and Metastasis
4.4. Angiogenesis
4.5. CAFs and Immune Response
4.6. The Role of miRNAs in CAFs
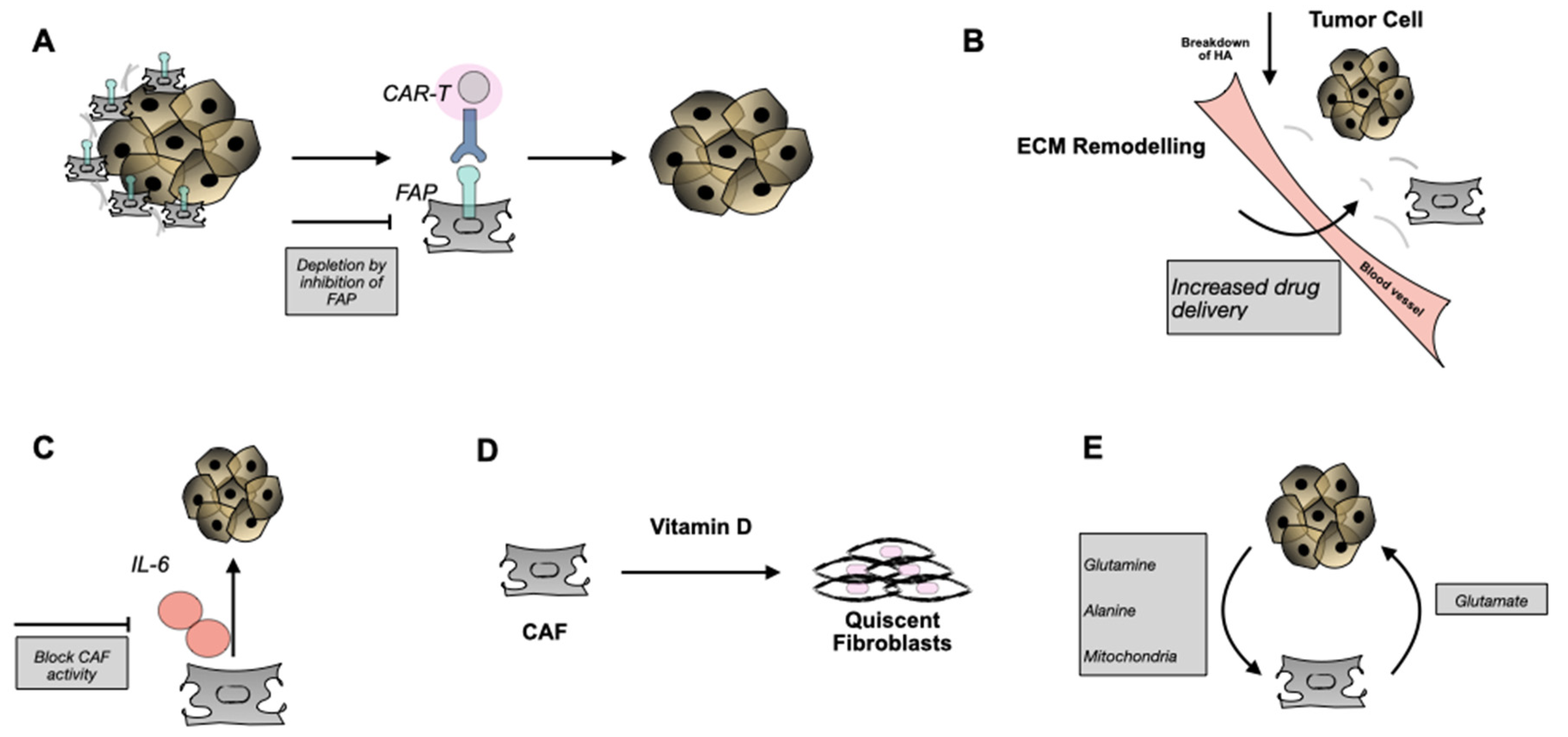
5. Targeting CAFs as an Approach to Anti-Cancer Therapy
5.1. Use of Antibodies, Small Molecules, or CAR-T Cells
5.2. Blocking CAF Function with IL-6
5.3. Hijacking the Metabolic Needs
5.4. Conversion of CAFs to a Quiescent Phenotype
6. Targeting CAFs for Imaging and Therapy
7. Future Opportunities for FAP-Mediated Imaging and Treatment
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cancer [Internet]. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 19 November 2020).
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, L.; Yao, H.H.; Zhu, Z.Q.; Ning, Z.L.; Huang, Q. Stromal myofibroblasts are associated with poor prognosis in solid cancers: A meta-analysis of published studies. PLoS ONE 2016, 11, e0159947. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, M.; Farah, F.; Zouari, B.; Jaafoura, M.H.; Kharrat, M. An immunoscore system based on cd3+ and cd8+ infiltrating lymphocytes densities to predict the outcome of patients with colorectal adenocarcinoma. Onco. Targets. Ther. 2019, 12, 8663–8673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 6, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Underhill, L.H.; Dvorak, H.F. Tumors: Wounds That Do Not Heal. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [Green Version]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar] [PubMed]
- Bu, L.; Baba, H.; Yoshida, N.; Miyake, K.; Yasuda, T.; Uchihara, T.; Tan, P.; Ishimoto, T. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 2019, 38, 4887–4901. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast—Like differentiation of human MSC. Cancer 2009, 68, 4331–4339. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.C.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Tomiyama, K.; Murase, N.; Stolz, D.B.; Toyokawa, H.; O’Donnell, D.R.; Smith, D.M.; Dudas, J.R.; Rubin, J.P.; Marra, K.G. Characterization of transplanted GFP+ bone marrow cells into adipose tissue. Stem Cells 2008, 26, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Barth, P.J.; Ebrahimsade, S.; Ramaswamy, A.; Moll, R. CD34+ fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows Arch. 2002, 440, 298–303. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. DMM Dis. Model. Mech. 2018, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Hanahan, D.; Weinberg, R.a. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. 6 Moses. 2004, p. 432. Available online: www.nature.com/nature (accessed on 25 February 2021).
- Kuperwasser, C.; Chavarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. PNAS 2004, 101, 4966–4971. [Google Scholar] [CrossRef] [Green Version]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Satyamoorthy, K.; Meier, F.; Berking, C.; Bogenrieder, T.; Herlyn, M. Function and regulation of melanoma-stromal fibroblast interactions: When seeds meet soil. Oncogene 2003, 22, 3162–3171. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.W. Myofibroblasts: Paracrine cells important in health and disease. Trans. Am. Clin. Climatol. Assoc. 2000, 111, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xiao, W.; Song, X.; Wang, W.; Dong, X. Recent Advances in Tumor Microenvironment Hydrogen Peroxide-Responsive Materials for Cancer Photodynamic Therapy. Nano-Micro Lett. 2020, 12, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to retrain rather than support pdac. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, M.E.; Werb, Z. ECM signalling: Orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998, 8, 437–441. [Google Scholar] [CrossRef]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–24. [Google Scholar] [CrossRef]
- Li, J.; Jia, Z.; Kong, J.; Zhang, F.; Fang, S.; Li, X.; Li, W.; Yang, X.; Luo, Y.; Lin, B.; et al. Carcinoma-associated fibroblasts lead the invasion of salivary gland adenoid cystic carcinoma cells by creating an invasive track. PLoS ONE 2016, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, H.; Liu, L.; Yu, J.; Ren, X. Fibroblast activation protein: A potential therapeutic target in cancer. Cancer Biol. Ther. 2012, 13, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Yu, D.M.T.; McCaughan, G.W.; Gorrell, M.D. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology 2005, 42, 935–945. [Google Scholar] [CrossRef]
- Henry, L.R.; Lee, H.O.; Lee, J.S.; Klein-Szanto, A.; Watts, P.; Ross, E.A.; Chen, W.T.; Cheng, J.D. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin. Cancer Res. 2007, 13, 1736–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef]
- Ju, M.J.; Qiu, S.J.; Fan, J.; Xiao, Y.S.; Gao, Q.; Zhou, J.; Li, Y.W.; Tang, Z.Y. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. Am. J. Clin. Pathol. 2009, 131, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, L.; Tsionou, C.; Hahnfeldt, P.; Coleman, C.N. Mammary Fibroblasts May Influence Breast Tumor Angiogenesis Via Hypoxia-Induced Vascular Endothelial Growth Factor Up-Regulation And Protein Expression. Cancer Res. 1994, 54, 6083–6086. [Google Scholar]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Gomes, F.G.; Nedel, F.; Alves, A.M.; Nör, J.E.; Tarquinio, S.B.C. Tumor angiogenesis and lymphangiogenesis: Tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2014, 92, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M.; Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2011, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaki, T.; Hara, M.; Shiga, K.; Takeyama, H. Relationship between inflammation and cancer progression: Recent advances in interleukin-6 signaling and its blockage in cancer therapy. Recept. Clin. Investig. 2014, 1–7. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.; Sainson, R.C.A. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef]
- An, Y.; Liu, F.; Chen, Y.; Yang, Q. Crosstalk between cancer-associated fibroblasts and immune cells in cancer. J. Cell. Mol. Med. 2020, 24, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Herrera, M.; Herrera, A.; Domínguez, G.; Silva, J.; García, V.; García, J.M.; Gómez, I.; Soldevilla, B.; Muñoz, C.; Provencio, M.; et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013, 104, 437–444. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, A.; Hendrix, A.; Maynard, D.; Van Bockstal, M.; Daniëls, A.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 2013, 13, 379–388. [Google Scholar] [CrossRef]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Bone 2011, 23, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Pena, C.; Cespedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edqvist, P.H.; Hagglof, C.; Birgisson, H.; Bojmar, L.; Jirstrom, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013, 73, 1287–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Huang, L.; Abdelrahim, M.; Cai, Q.; Truong, A.; Bick, R.; Poindexter, B.; Sheikh-Hamad, D. Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial UCP2. J. Leukoc. Biol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Brooks, H.; Zhang, P.; Smith, W.; McReynolds, M.R.; Hoying, J.B.; Bick, R.; Truong, L.; Poindexter, B.; Lan, H.; et al. Stanniocalcin-1 regulates endothelial gene expression and modulates transendothelial migration of leukocytes. Am. J. Physiol. Ren. Physiol. 2007, 292, 895–904. [Google Scholar] [CrossRef]
- Barnas, J.L.; Simpson-Abelson, M.R.; Yokota, S.J.; Kelleher, R.J.; Bankert, R.B. T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron. 2010, 3, 29–47. [Google Scholar] [CrossRef] [Green Version]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [Green Version]
- Corver, W.E.; Haar, N.T.; Fleuren, G.J.; Oosting, J. Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations. Cell. Oncol. 2011, 34, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, N.; Shah, V.H.; Urrutia, R. Membrane-to-Nucleus Signals and Epigenetic Mechanisms for Myofibroblastic Activation and Desmoplastic Stroma: Potential Therapeutic Targets for Liver Metastasis? Physiol. Behav. 2015, 13, 604–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, J.; Mishra, R.; Li, X.; Ii, R.S.J.; Sharma, A.; Bhowmick, N.A. A reciprocal role of prostate cancer on stromal DNA damage. Oncogene 2013, 33, 4924–4931. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [Green Version]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of activated fibroblasts for imaging and therapy. EJNMMI Radiopharm. Chem. 2019, 4, 1–15. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; Brien, S.O.; Evans, R.A.; Bajor, D.J.; Durham, A.C.; et al. Tumor-promoting desmoplasia is disrupted by depleting FAP- expressing stromal cells. Cancer Res. 2016, 75, 2800–2810. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Fassnacht, M.; Nair, S.; Boczkowski, D.; Gilboa, E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. 2005, 65, 11156–11163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Phillips, M.; Alice, A.; Bambina, S.; Zebertavage, L.; Friedman, D.; Cottam, B.; Newell, P.; et al. Blockade of fibroblast activation protein in combination with radiation treatment in murine models of pancreatic adenocarcinoma. PLoS ONE 2019, 14, e0211117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.-T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I Dose-Escalation Study of Sibrotuzumab in Patients with Advanced or Metastatic Fibroblast Activation Protein-Positive Cancer 1. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Pargellis, C.A.; Campbell, S.J.; Pav, S.; Graham, E.T.; Pitner, T.P. Inhibition of dipeptidyl peptidase IV (CD26) by peptide boronic acid dipeptides. J. Enzyme Inhib. 1997, 11, 151–169. [Google Scholar] [CrossRef] [Green Version]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef] [Green Version]
- Jansen, K.; De Winter, H.; Heirbaut, L.; Cheng, J.D.; Joossens, J.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; Van Der Veken, P. Selective inhibitors of fibroblast activation protein (FAP) with a xanthine scaffold. Medchemcomm 2014, 5, 1700–1707. [Google Scholar] [CrossRef]
- Ryabtsova, O.; Jansen, K.; Van Goethem, S.; Joossens, J.; Cheng, J.D.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; Van Der Veken, P. Acylated Gly-(2-cyano)pyrrolidines as inhibitors of fibroblast activation protein (FAP) and the issue of FAP/prolyl oligopeptidase (PREP)-selectivity. Bioorganic Med. Chem. Lett. 2012, 22, 3412–3417. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; Heirbaut, L.; Verkerk, R.; Cheng, J.D.; Joossens, J.; Cos, P.; Maes, L.; Lambeir, A.M.; De Meester, I.; Augustyns, K.; et al. Extended structure-activity relationship and pharmacokinetic investigation of (4-quinolinoyl)glycyl-2-cyanopyrrolidine inhibitors of fibroblast activation protein (FAP). J. Med. Chem. 2014, 57, 3053–3074. [Google Scholar] [CrossRef]
- Šimková, A.; Bušek, P.; Šedo, A.; Konvalinka, J. Molecular recognition of fibroblast activation protein for diagnostic and therapeutic applications. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140409. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK Signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [Green Version]
- Jesnowski, R.; Fürst, D.; Ringel, J.; Chen, Y.; Schrödel, A.; Kleeff, J.; Kolb, A.; Schareck, W.D.; Löhr, M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: Deactivation is induced by matrigel and N-acetylcysteine. Lab. Investig. 2005, 85, 1276–1291. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Melissari, M.T.; Chalkidi, N.; Sarris, M.E.; Koliaraki, V. Fibroblast Reprogramming in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2020, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; Tseng, T.W.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2015, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernández Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef] [PubMed]
- Calais, J. FAP: The next billion dollar nuclear theranostics target? J. Nucl. Med. 2020, 61, 163–165. [Google Scholar] [CrossRef]
- Fischer, E.; Chaitanya, K.; Wuëst, T.; Wadle, A.; Scott, A.M.; Van Den Broek, M.; Schibli, R.; Bauer, S.; Renner, C. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin. Cancer Res. 2012, 18, 6208–6218. [Google Scholar] [CrossRef] [Green Version]
- Giesel, F.L.; Kratochwil, C.; Lindner, T.; Marschalek, M.M.; Loktev, A.; Lehnert, W.; Debus, J.; Jäger, D.; Flechsig, P.; Altmann, A.; et al. 68 Ga-FAPI PET/CT: Biodistribution and preliminary dosimetry estimate of 2 DOTA-containing FAP-targeting agents in patients with various cancers. J. Nucl. Med. 2019, 60, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Koerber, S.A.; Staudinger, F.; Kratochwil, C.; Adeberg, S.; Haefner, M.F.; Ungerechts, G.; Rathke, H.; Winter, E.; Lindner, T.; Syed, M.; et al. The Role of 68Ga-FAPI PET/CT for Patients with Malignancies of the Lower Gastrointestinal Tract: First Clinical Experience. J. Nucl. Med. 2020, 61, 1331–1336. [Google Scholar] [CrossRef]
- Syed, M.; Flechsig, P.; Liermann, J.; Windisch, P.; Staudinger, F.; Akbaba, S.; Koerber, S.A.; Freudlsperger, C.; Plinkert, P.K.; Debus, J.; et al. Fibroblast activation protein inhibitor (FAPI) PET for diagnostics and advanced targeted radiotherapy in head and neck cancers. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2836–2845. [Google Scholar] [CrossRef]
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics Targeting Fibroblast Activation Protein in the Tumor Stroma: 64Cu- and 225Ac-Labeled FAPI-04 in Pancreatic Cancer Xenograft Mouse Models. J. Nucl. Med. 2020, 61, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Toms, J.; Kogler, J.; Maschauer, S.; Daniel, C.; Schmidkonz, C.; Kuwert, T.; Prante, O. Targeting Fibroblast Activation Protein: Radiosynthesis and Preclinical Evaluation of an 18 F-labeled FAP Inhibitor. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer uptake in 28 different kinds of cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, T.; Altmann, A.; Krämer, S.; Kleist, C.; Loktev, A.; Kratochwil, C.; Giesel, F.; Mier, W.; Marme, F.; Debus, J.; et al. Design and Development of 99mTc-Labeled FAPI Tracers for SPECT Imaging and 188Re Therapy. J. Nucl. Med. 2020, 61, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loktev, A.; Lindner, T.; Burger, E.M.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Marmé, F.; Jäger, D.; Mier, W.; et al. Development of fibroblast activation protein-targeted radiotracers with improved tumor retention. J. Nucl. Med. 2019, 60, 1421–1429. [Google Scholar] [CrossRef]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A tumor-imaging method targeting cancer-associated fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Dahlbom, M.; Lindner, T.; Vauclin, S.; Mona, C.; Slavik, R.; Czernin, J.; Haberkorn, U.; Calais, J. Radiation Dosimetry and Biodistribution of 68Ga-FAPI-46 PET Imaging in Cancer Patients. J. Nucl. Med. 2020, 61, 1171–1177. [Google Scholar] [CrossRef]
- Röhrich, M.; Loktev, A.; Wefers, A.K.; Altmann, A.; Paech, D.; Adeberg, S.; Windisch, P.; Hielscher, T.; Flechsig, P.; Floca, R.; et al. IDH-wildtype glioblastomas and grade III/IV IDH-mutant gliomas show elevated tracer uptake in fibroblast activation protein–specific PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2569–2580. [Google Scholar] [CrossRef]
- Shi, X.; Xing, H.; Yang, X.; Li, F.; Yao, S.; Zhang, H.; Zhao, H.; Hacker, M.; Huo, L.; Li, X. Fibroblast imaging of hepatic carcinoma with 68Ga-FAPI-04 PET/CT: A pilot study in patients with suspected hepatic nodules. Eur. J. Nucl. Med. Mol. Imaging 2020. [Google Scholar] [CrossRef]
- Poeppel, T.D.; Binse, I.; Petersenn, S.; Lahner, H.; Schott, M.; Antoch, G.; Brandau, W.; Bockisch, A.; Boy, C. 68Ga-DOTATOC versus 68Ga-DOTATATE PET/CT in functional imaging of neuroendocrine tumors. J. Nucl. Med. 2011, 52, 1864–1870. [Google Scholar] [CrossRef] [Green Version]
- Kurash, M.M.; Gill, R.; Khairulin, M.; Harbosh, H.; Keidar, Z. 68Ga-labeled PSMA-11 (68Ga-isoPROtrace-11) synthesized with ready to use kit: Normal biodistribution and uptake characteristics of tumour lesions. Sci. Rep. 2020, 10, 3109. [Google Scholar] [CrossRef] [Green Version]
- Giesel, F.; Adeberg, S.; Syed, M.; Lindner, T.; Jimenez, L.D.; Mavriopoulou, E.; Staudinger, F.; Tonndorf-Martini, E.; Regnery, S.; Rieken, S.; et al. FAPI-74 PET/CT Using Either 18 F-AlF or Cold-kit 68 Ga-labeling: Biodistribution, Radiation Dosimetry and Tumor Delineation in Lung Cancer Patients. J. Nucl. Med. 2020. [Google Scholar] [CrossRef]
- Chen, H.; Pang, Y.; Wu, J.; Zhao, L.; Hao, B.; Wu, J.; Wei, J.; Wu, S.; Zhao, L.; Luo, Z.; et al. Comparison of [68Ga]Ga-DOTA-FAPI-04 and [18F] FDG PET/CT for the diagnosis of primary and metastatic lesions in patients with various types of cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1820–1832. [Google Scholar] [CrossRef]
- Imaging of Solid Tumors Using 68Ga-FAP-2286—Full Text View—ClinicalTrials.gov [Internet]. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04621435?term=FAP-2286&draw=2&rank=1 (accessed on 18 February 2021).
- Hirmas, N.; Jadaan, R.; Al-Ibraheem, A. Peptide Receptor Radionuclide Therapy and the Treatment of Gastroentero-pancreatic Neuroendocrine Tumors: Current Findings and Future Perspectives. Nucl. Med. Mol. Imaging 2018, 52, 190–199. [Google Scholar] [CrossRef]
- Wang, Y.; Weng, J.; Wen, X.; Hu, Y.; Ye, D. Recent advances in stimuli-responsive in situ self-assembly of small molecule probes for in vivo imaging of enzymatic activity. Biomater. Sci. 2021. [Google Scholar] [CrossRef]
- De Decker, A.; Vliegen, G.; Van Rompaey, D.; Peeraer, A.; Bracke, A.; Verckist, L.; Jansen, K.; Geiss-Friedlander, R.; Augustyns, K.; De Winter, H.; et al. Novel Small Molecule-Derived, Highly Selective Substrates for Fibroblast Activation Protein (FAP). ACS Med. Chem. Lett. 2019, 10, 1173–1179. [Google Scholar] [CrossRef]
- Wang, Q.; Guan, J.; Wan, J.; Li, Z. Disulfide based prodrugs for cancer therapy. RSC Adv. 2020, 10, 24397–24409. [Google Scholar] [CrossRef]
- Giang, I.; Boland, E.L.; Poon, G.M.K. Prodrug applications for targeted cancer therapy. AAPS J. 2014, 16, 899–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Bhuniya, S.; Lee, H.; Kim, H.M.; Cheong, C.; Maiti, S.; Hong, K.S.; Kim, J.S. An activatable prodrug for the treatment of metastatic tumors. J. Am. Chem. Soc. 2014, 136, 13888–13894. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tracer | Chemical Structure | Radionuclides | Application | References |
|---|---|---|---|---|
| FAPI-2 |  | PET: 68Ga Therapy: 177Lu | Various Cancers | [98] |
| FAPI-4 |  | PET: 68Ga, 64Cu Therapy: 90Y, 177Lu, 225Ac | Breast Cancer | [105] |
| FAPI-21 |  | PET: 68Ga Therapy: 177Lu | Ovarian and Colorectal Cancer | [106] |
| FAPI-46 |  | PET: 68Ga Therapy: 90Y, 177Lu | Colorectal, Mammary, Oropharyngeal Cancer | [106] |
| FAPI-34 |  | SPECT: 99mTc Therapy: 188Re | Pancreatic, Ovarian Cancer | [104] |
| FAPI-74 |  | PET:18F, 68Ga | Lung Cancer | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koustoulidou, S.; Hoorens, M.W.H.; Dalm, S.U.; Mahajan, S.; Debets, R.; Seimbille, Y.; de Jong, M. Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy. Cancers 2021, 13, 1100. https://doi.org/10.3390/cancers13051100
Koustoulidou S, Hoorens MWH, Dalm SU, Mahajan S, Debets R, Seimbille Y, de Jong M. Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy. Cancers. 2021; 13(5):1100. https://doi.org/10.3390/cancers13051100
Chicago/Turabian StyleKoustoulidou, Sofia, Mark W. H. Hoorens, Simone U. Dalm, Shweta Mahajan, Reno Debets, Yann Seimbille, and Marion de Jong. 2021. "Cancer-Associated Fibroblasts as Players in Cancer Development and Progression and Their Role in Targeted Radionuclide Imaging and Therapy" Cancers 13, no. 5: 1100. https://doi.org/10.3390/cancers13051100