
The Role of Epigenetics in the Progression of Clear Cell Renal Cell Carcinoma and the Basis for Future Epigenetic Treatments

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Current Management of Renal Cell Carcinoma
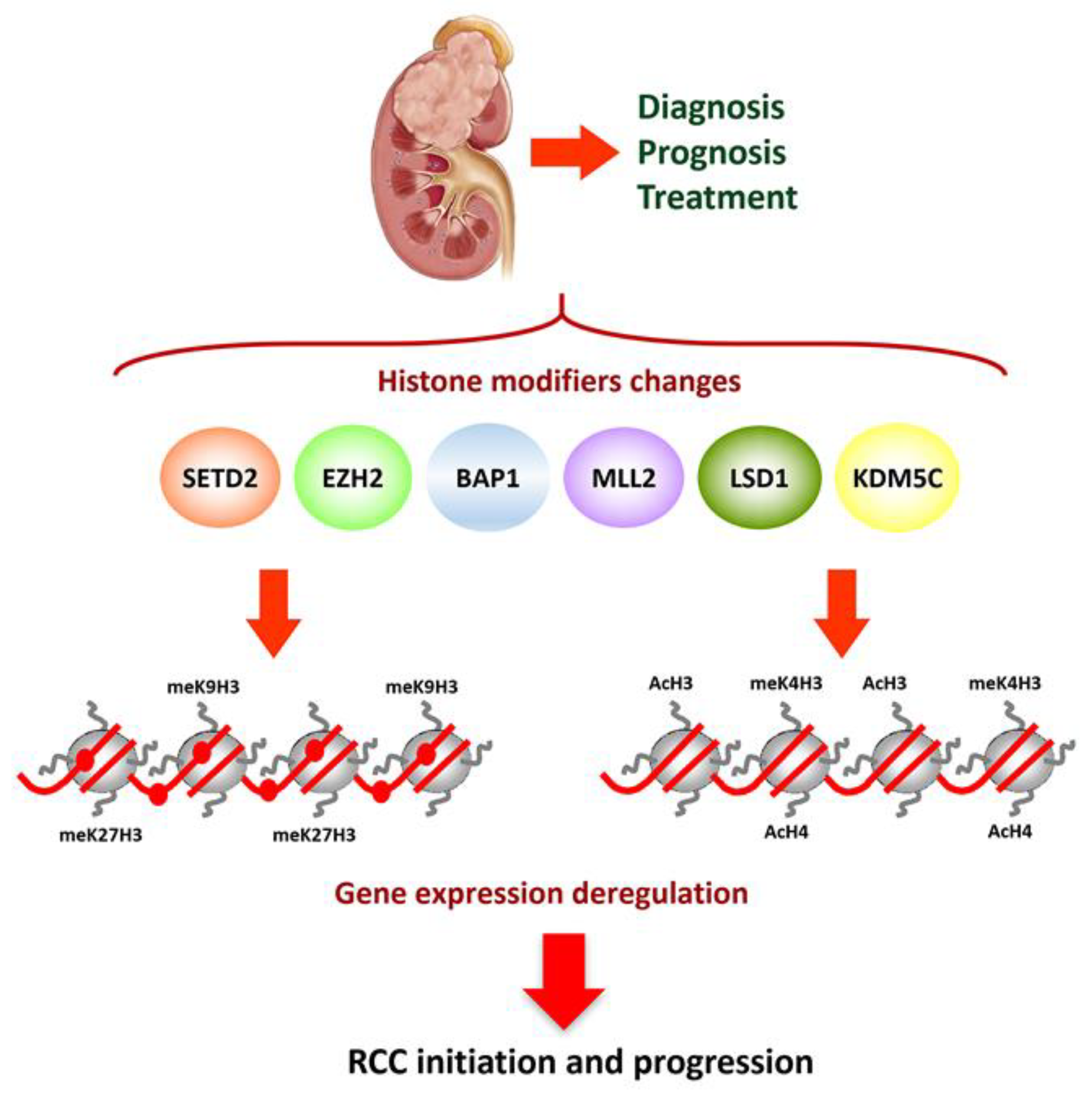
2. Epigenetics of Clear Cell Renal Cell Carcinoma
2.1. Abnormal DNA Methylation
2.1.1. DNA Methylation as Marker of RCC Diagnoses
2.1.2. DNA Methylation as Marker of RCC Subtyping
2.1.3. DNA Methylation as Marker of RCC Prognosis
2.2. Methyl-Binding Proteins
2.3. Post-Translational Histone Modifications
2.4. miRNAs
2.5. lncRNAs
2.6. RNA Methylation
3. Epigenetic-Based Therapeutic Opportunities in ccRCC
3.1. DNMT Inhibition Alone or in Combination with Other Therapies
3.1.1. Azacytidine (5-Azacytidine)
3.1.2. Decitabine (5-Aza-2′-Deoxycytidine)
3.1.3. MG98
3.2. HDAC Inhibition Alone or in Combination with Other Therapies
3.2.1. Vorinostat
3.2.2. Panobinostat
3.2.3. Entinostat (MS-275)
3.2.4. Other HDACi
3.3. Other Epigenetic Therapies
3.4. Caveats and Limitations of Epigenetic Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of Renal Cell Carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef]
- Bot, F.J.; Godschalk, J.C.J.; Krishnadath, K.K.; Van Der Kwast, T.H.; Bosman, F.T.; But, F.J.; Der Van Kwast, T.H.M. Prognostic factors in renal-cell carcinoma: Immunohistochemical detection of p53 protein versus clinico-pathological parameters. Int. J. Cancer 1994, 57, 634–637. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhu, W.; Yu, S.; Li, K.; Ding, Y.; Wu, Q.; Tang, Q.; Zhao, Q.; Lu, C.; Guo, C. Development and validation of a nomogram to predict overall survival for patients with metastatic renal cell carcinoma. BMC Cancer 2020, 20, 1066. [Google Scholar] [CrossRef] [PubMed]
- Trpkov, K.; Hes, O.; Williamson, S.R.; Adeniran, A.J.; Agaimy, A.; Alaghehbandan, R.; Amin, M.B.; Argani, P.; Chen, Y.-B.; Cheng, L.; et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021, 1–33. [Google Scholar] [CrossRef]
- Srigley, J.R.; Delahunt, B.; Eble, J.N.; Egevad, L.; Epstein, J.I.; Grignon, D.; Hes, O.; Moch, H.; Montironi, R.; Tickoo, S.K.; et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am. J. Surg. Pathol. 2013, 37, 1469–1489. [Google Scholar] [CrossRef]
- Maher, E.R. Genomics and epigenomics of renal cell carcinoma. Semin. Cancer Biol. 2013, 23, 10–17. [Google Scholar] [CrossRef]
- Moch, H. An overview of renal cell cancer: Pathology and genetics. Semin. Cancer Biol. 2013, 23, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Pinheiro, P.; Montezuma, D.; Henrique, R.; Jerónimo, C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics 2015, 7, 1003–1015. [Google Scholar] [CrossRef]
- Solano-Iturri, J.D.; Errarte, P.; Etxezarraga, M.C.; Echevarria, E.; Angulo, J.; López, J.I.; Larrinaga, G. Altered Tissue and Plasma Levels of Fibroblast Activation Protein-α (FAP) in Renal Tumours. Cancers 2020, 12, 3393. [Google Scholar] [CrossRef]
- López, J.I. Intratumor heterogeneity in clear cell renal cell carcinoma: A review for the practicing pathologist. APMIS 2016, 124, 153–159. [Google Scholar] [CrossRef]
- Rydzanicz, M.; Wrzesiński, T.; Bluyssen, H.A.; Wesoły, J. Genomics and epigenomics of clear cell renal cell carcinoma: Recent developments and potential applications. Cancer Lett. 2013, 341, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Onay, H.; Pehlivan, S.; Koyuncuoglu, M.; Kirkali, Z.; Özkinay, F. Multigene Methylation Analysis of Conventional Renal Cell Carcinoma. Urol. Int. 2009, 83, 107–112. [Google Scholar] [CrossRef]
- Lasseigne, B.N.; Burwell, T.C.; Patil, M.A.; Absher, D.M.; Brooks, J.D.; Myers, R.M. DNA methylation profiling reveals novel diagnostic biomarkers in renal cell carcinoma. BMC Med. 2014, 12, 235. [Google Scholar] [CrossRef]
- Pires-Luís, A.S.; Costa-Pinheiro, P.; Ferreira, M.J.; Antunes, L.; Lobo, F.; Oliveira, J.; Henrique, R.; Jerónimo, C. Identification of clear cell renal cell carcinoma and oncocytoma using a three-gene promoter methylation panel. J. Transl. Med. 2017, 15, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasseigne, B.N.; Brooks, J.D. The Role of DNA Methylation in Renal Cell Carcinoma. Mol. Diagn. Ther. 2018, 22, 431–442. [Google Scholar] [CrossRef]
- Ellinger, J.; Müller, S.C.; Dietrich, D. Epigenetic biomarkers in the blood of patients with urological malignancies. Expert Rev. Mol. Diagn. 2015, 15, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-H.; Haddad, A.; Wu, K.-J.; Zhao, H.-W.; Kapur, P.; Zhang, Z.-L.; Zhao, L.-Y.; Chen, Z.-H.; Zhou, Y.-Y.; Zhou, J.-C.; et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat. Commun. 2015, 6, 8699. [Google Scholar] [CrossRef] [Green Version]
- Malouf, G.G.; Su, X.; Zhang, J.; Creighton, C.J.; Ho, T.H.; Lu, Y.; Raynal, N.J.-M.; Karam, J.A.; Tamboli, P.; Allanick, F.; et al. DNA Methylation Signature Reveals Cell Ontogeny of Renal Cell Carcinomas. Clin. Cancer Res. 2016, 22, 6236–6246. [Google Scholar] [CrossRef] [Green Version]
- Angulo, J.C.; López, J.I.; Ropero, S. DNA Methylation and Urological Cancer, a Step Towards Personalized Medicine: Current and Future Prospects. Mol. Diagn. Ther. 2016, 20, 531–549. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, L.Y.; Fu, S.; Hlady, R.A.; Wagner, R.T.; Wang, L.; Eckel-Passow, J.E.; Castle, E.P.; Stanton, M.L.; Thompson, R.H.; Parker, A.S.; et al. Identification of DNA methylation signatures associated with poor outcome in lower-risk Stage, Size, Grade and Necrosis (SSIGN) score clear cell renal cell cancer. Clin. Epigenetics 2021, 13, 1–16. [Google Scholar] [CrossRef]
- Mir, M.C.; Derweesh, I.; Porpiglia, F.; Zargar, H.; Mottrie, A.; Autorino, R. Partial Nephrectomy Versus Radical Nephrectomy for Clinical T1b and T2 Renal Tumors: A Systematic Review and Meta-analysis of Comparative Studies. Eur. Urol. 2017, 71, 606–617. [Google Scholar] [CrossRef]
- Albiges, L.; Powles, T.; Staehler, M.; Bensalah, K.; Giles, R.H.; Hora, M.; Kuczyk, M.A.; Lam, T.B.; Ljungberg, B.; Marconi, L.; et al. Updated European Association of Urology Guidelines on Renal Cell Carcinoma: Immune Checkpoint Inhibition Is the New Backbone in First-line Treatment of Metastatic Clear-cell Renal Cell Carcinoma. Eur. Urol. 2019, 76, 151–156. [Google Scholar] [CrossRef]
- Jonasch, E. NCCN Guidelines Updates: Management of Metastatic Kidney Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 587–589. [Google Scholar]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Angulo, J.C.; Shapiro, O. The Changing Therapeutic Landscape of Metastatic Renal Cancer. Cancers 2019, 11, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khagi, Y.; Kurzrock, R.; Patel, S.P. Next generation predictive biomarkers for immune checkpoint inhibition. Cancer Metastasis Rev. 2017, 36, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaggio, A.; Epaillard, N.; Pobel, C.; Moreira, M.; Oudard, S.; Vano, Y.-A. Tumor Microenvironment Features as Predictive Biomarkers of Response to Immune Checkpoint Inhibitors (ICI) in Metastatic Clear Cell Renal Cell Carcinoma (mccRCC). Cancers 2021, 13, 231. [Google Scholar] [CrossRef]
- Larrinaga, G.; Solano-Iturri, J.D.; Errarte, P.; Unda, M.; Loizaga-Iriarte, A.; Pérez-Fernández, A.; Echevarría, E.; Asumendi, A.; Manini, C.; Angulo, J.C.; et al. Soluble PD-L1 Is an Independent Prognostic Factor in Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 667. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Selby, P.J.; Banks, R.E. Renal cancer biomarkers: The promise of personalized care. BMC Med. 2012, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.J.; Chen, D.; Wang, P.I.; Marker, M.; Redzematovic, A.; Chen, Y.-B.; Selcuklu, S.D.; Weinhold, N.; Bouvier, N.; Huberman, K.H.; et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur. Urol. 2017, 71, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Taby, R.; Issa, J.-P.J. Cancer Epigenetics. CA Cancer J. Clin. 2010, 60, 376–392. [Google Scholar] [CrossRef]
- Valdés-Mora, F.; Clark, S.J. Prostate cancer epigenetic biomarkers: Next-generation technologies. Oncogene 2015, 34, 1609–1618. [Google Scholar] [CrossRef]
- Xing, T.; He, H. Epigenomics of clear cell renal cell carcinoma: Mechanisms and potential use in molecular pathology. Chin. J. Cancer Res. 2016, 28, 80–91. [Google Scholar] [PubMed]
- Joosten, S.C.; Deckers, I.A.; Aarts, M.J.; Hoeben, A.; Van Roermund, J.G.; Smits, K.M.; Melotte, V.; Van Engeland, M.; Tjan-Heijnen, V.C. Prognostic DNA methylation markers for renal cell carcinoma: A systematic review. Epigenomics 2017, 9, 1243–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lommen, K.; Vaes, N.; Aarts, M.J.; van Roermund, J.G.; Schouten, L.J.; Oosterwijk, E.; Melotte, V.; Tjan-Heijnen, V.C.; van Engeland, M.; Smits, K.M. Diagnostic DNA Methylation Biomarkers for Renal Cell Carcinoma: A Systematic Review. Eur. Urol. Oncol. 2021, 4, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.A.; Sørensen, K.D. Epigenetics of Urological Cancers. Int. J. Mol. Sci. 2019, 20, 4775. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, L.; Zhang, D.; Fan, Y.; Jia, Z.; Qin, P.; Yu, J.; Zheng, S.; Yang, F. Identification of potential serum biomarkers for Wilms tumor after excluding confounding effects of common systemic inflammatory factors. Mol. Biol. Rep. 2012, 39, 5095–5104. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, J.H.; Jang, H.J.; Han, B.; Zang, D.Y. Clinicopathologic Significance of VHL Gene Alteration in Clear-Cell Renal Cell Carcinoma: An Updated Meta-Analysis and Review. Int. J. Mol. Sci. 2018, 19, 2529. [Google Scholar] [CrossRef]
- Huang, J.; Yao, X.; Zhang, J.; Dong, B.; Chen, Q.; Xue, W.; Liu, D.; Huang, Y. Hypoxia-induced downregulation of miR-30c promotes epithelial-mesenchymal transition in human renal cell carcinoma. Cancer Sci. 2013, 104, 1609–1617. [Google Scholar] [CrossRef]
- Zhai, W.; Sun, Y.; Jiang, M.; Wang, M.; Gasiewicz, T.A.; Zheng, J. Differential regulation of LncRNA-SARCC suppresses VHL-mutant RCC cell proliferation yet promotes VHL-normal RCC cell proliferation via modulating androgen receptor/HIF-2α/C-MYC axis under hypoxia. Oncogene 2016, 35, 4866–4880. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [Green Version]
- Thévenin, A.; Ein-Dor, L.; Ozery-Flato, M.; Shamir, R. Functional gene groups are concentrated within chromosomes, among chromosomes and in the nuclear space of the human genome. Nucleic Acids Res. 2014, 42, 9854–9861. [Google Scholar] [CrossRef] [Green Version]
- Mehdi, A.; Riazalhosseini, Y. Epigenome Aberrations: Emerging Driving Factors of the Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 1774. [Google Scholar] [CrossRef] [Green Version]
- De Cubas, A.A.; Rathmell, W.K. Epigenetic modifiers: Activities in renal cell carcinoma. Nat. Rev. Urol. 2018, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Nalejska, E.; Mączyńska, E.; Lewandowska, M.A. Prognostic and Predictive Biomarkers: Tools in Personalized Oncology. Mol. Diagn. Ther. 2014, 18, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrypkina, I.; Tsyba, L.; Onyshchenko, K.; Morderer, D.; Kashparova, O.; Nikolaienko, O.; Panasenko, G.; Vozianov, S.; Romanenko, A.; Rynditch, A. Concentration and Methylation of Cell-Free DNA from Blood Plasma as Diagnostic Markers of Renal Cancer. Dis. Markers 2016, 2016, 3693096. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, P.V.; Berchuck, J.E.; Korthauer, K.; Spisak, S.; Nassar, A.H.; Alaiwi, S.A.; Chakravarthy, A.; Shen, S.Y.; Bakouny, Z.; Boccardo, F.; et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat. Med. 2020, 26, 1041–1043. [Google Scholar] [CrossRef]
- Slater, A.A.; Alokail, M.; Gentle, D.; Yao, M.; Kovacs, G.; Maher, E.R.; Latif, F. DNA methylation profiling distinguishes histological subtypes of renal cell carcinoma. Epigenetics 2013, 8, 252–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Shenoy, N.; Vallumsetla, N.; Zou, Y.; Galeas, J.N.; Shrivastava, M.; Hu, C.; Susztak, K.; Verma, A. Role of DNA methylation in renal cell carcinoma. J. Hematol. Oncol. 2015, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, A.M.; Cairns, P. Epigenetics of kidney cancer and bladder cancer. Epigenomics 2011, 3, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Joosten, S.C.; Feng, Z.; De Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Karami, S.; Andreotti, G.; Liao, L.M.; Pfeiffer, R.M.; Weinstein, S.J.; Purdue, M.P.; Hofmann, J.N.; Albanes, D.; Männistö, S.; Moore, L.E. LINE1 methylation levels in pre-diagnostic leukocyte DNA and future renal cell carcinoma risk. Epigenetics 2015, 10, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [Green Version]
- Battagli, C.; Uzzo, R.G.; Dulaimi, E.; De Caceres, I.I.; Krassenstein, R.; Al-Saleem, T.; E Greenberg, R.; Cairns, P. Promoter hypermethylation of tumor suppressor genes in urine from kidney cancer patients. Cancer Res. 2003, 63, 8695–8699. [Google Scholar]
- Hoque, M.O.; Begum, S.; Topaloglu, O.; Jeronimo, C.; Mambo, E.; Westra, W.H.; Califano, J.A.; Sidransky, D. Quantitative Detection of Promoter Hypermethylation of Multiple Genes in the Tumor, Urine, and Serum DNA of Patients with Renal Cancer. Cancer Res. 2004, 64, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- Ibanez de Caceres, I.; Dulaimi, E.; Hoffman, A.M.; Al-Saleem, T.; Uzzo, R.G.; Cairns, P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006, 66, 5021–5028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, V.L.; Henrique, R.; Ribeiro, F.R.; Pinto, M.; Oliveira, J.; Lobo, F.; Teixeira, M.R.; Jerónimo, C. Quantitative promoter methylation analysis of multiple cancer-related genes in renal cell tumors. BMC Cancer 2007, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Peters, I.; Rehmet, K.; Wilke, N.; A Kuczyk, M.; Hennenlotter, J.; Eilers, T.; Machtens, S.; Jonas, U.; Serth, J. RASSF1A promoter methylation and expression analysis in normal and neoplastic kidney indicates a role in early tumorigenesis. Mol. Cancer 2007, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.R.; Ricketts, C.J.; Gentle, D.; McRonald, F.; Carli, N.; Khalili, H.; Brown, M.; Kishida, T.; Yao, M.; Banks, R.E.; et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene 2011, 30, 1390–1401. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.; Zahalka, T.; Fechner, G.; Müller, S.C.; Ellinger, J. Serum DNA hypermethylation in patients with kidney cancer: Results of a prospective study. Anticancer. Res. 2013, 33, 4651–4656. [Google Scholar]
- Urakami, S.; Shiina, H.; Enokida, H.; Hirata, H.; Kawamoto, K.; Kawakami, T.; Kikuno, N.; Tanaka, Y.; Majid, S.; Nakagawa, M.; et al. Wnt Antagonist Family Genes as Biomarkers for Diagnosis, Staging, and Prognosis of Renal Cell Carcinoma Using Tumor and Serum DNA. Clin. Cancer Res. 2006, 12, 6989–6997. [Google Scholar] [CrossRef] [Green Version]
- Gonzalgo, M.L.; Yegnasubramanian, S.; Yan, G.; Rogers, C.G.; Nicol, T.L.; Nelson, W.G.; Pavlovich, C.P. Molecular Profiling and Classification of Sporadic Renal Cell Carcinoma by Quantitative Methylation Analysis. Clin. Cancer Res. 2004, 10, 7276–7283. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.R.; Gentle, D.; Abdulrahman, M.; Maina, E.N.; Gupta, K.; Banks, R.E.; Wiesener, M.S.; Kishida, T.; Yao, M.; Teh, B.; et al. Tumor Suppressor Activity and Epigenetic Inactivation of Hepatocyte Growth Factor Activator Inhibitor Type 2/SPINT2 in Papillary and Clear Cell Renal Cell Carcinoma. Cancer Res. 2005, 65, 4598–4606. [Google Scholar] [CrossRef] [Green Version]
- McRonald, F.E.; Morris, M.R.; Gentle, D.; Winchester, L.; Baban, D.; Ragoussis, J.; Clarke, N.W.; Brown, M.D.; Kishida, T.; Yao, M.; et al. CpG methylation profiling in VHL related and VHL unrelated renal cell carcinoma. Mol. Cancer 2009, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Brennan, K.; Metzner, T.J.; Kao, C.-S.; Massie, C.E.; Stewart, G.D.; Haile, R.W.; Brooks, J.D.; Hitchins, M.P.; Leppert, J.T.; Gevaert, O. Development of a DNA Methylation–Based Diagnostic Signature to Distinguish Benign Oncocytoma From Renal Cell Carcinoma. JCO Precis. Oncol. 2020, 4, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhuang, J.; Wang, P.P.; Jiang, J.; Lin, C.; Zeng, P.; Liang, Y.; Zhang, X.; Dai, Y.; Diao, H. DNA methylation-based classification and identification of renal cell carcinoma prognosis-subgroups. Cancer Cell Int. 2019, 19, 185. [Google Scholar] [CrossRef]
- Evelönn, E.A.; Landfors, M.; Haider, Z.; Köhn, L.; Ljungberg, B.; Roos, G.; Degerman, S. DNA methylation associates with survival in non-metastatic clear cell renal cell carcinoma. BMC Cancer 2019, 19, 65. [Google Scholar] [CrossRef] [Green Version]
- Van Vlodrop, I.J.H.; Joosten, S.C.; De Meyer, T.; Smits, K.M.; Van Neste, L.; Melotte, V.; Baldewijns, M.M.L.L.; Schouten, L.J.; van den Brandt, P.A.; Jeschke, J.; et al. A Four-Gene Promoter Methylation Marker Panel Consisting of GREM1, NEURL, LAD1, and NEFH Predicts Survival of Clear Cell Renal Cell Cancer Patients. Clin. Cancer Res. 2017, 23, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef]
- Peters, I.; Dubrowinskaja, N.; Hennenlotter, J.; Antonopoulos, W.I.; Von Klot, C.A.; Tezval, H.; Stenzl, A.; Kuczyk, M.A.; Serth, J. DNA methylation of neural EGFL like 1 (NELL1) is associated with advanced disease and the metastatic state of renal cell cancer patients. Oncol. Rep. 2018, 40, 3861–3868. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Zeng, W.; Liu, X. A Gene Signature of Survival Prediction for Kidney Renal Cell Carcinoma by Multi-Omic Data Analysis. Int. J. Mol. Sci. 2019, 20, 5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Zhou, Y.; Jin, L.; Cao, C.; Gao, C.; Zhou, J.; Yang, D.; Zhu, J. Development and validation of an integrative methylation signature and nomogram for predicting survival in clear cell renal cell carcinoma. Transl. Androl. Urol. 2020, 9, 1082–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, L.; Ju, L.; Qian, K.; Wang, X.; Xiao, Y.; Wang, G. Epigenetic signature predicts overall survival clear cell renal cell carcinoma. Cancer Cell Int. 2020, 20, 564. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Q.; Zhu, Q.; Liu, C.; Nan, X.; Wang, F.; Fang, L.; Liu, J.; Xie, C.; Fu, S.; et al. Identification of methylation-driven genes related to prognosis in clear-cell renal cell carcinoma. J. Cell. Physiol. 2019, 235, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W.; Park, H.; Seo, S.P.; Byun, Y.J.; Piao, X.-M.; Kim, S.M.; Kim, W.T.; Yun, S.-J.; Jang, W.; Shon, H.S.; et al. Methylation Signature for Prediction of Progression Free Survival in Surgically Treated Clear Cell Renal Cell Carcinoma. J. Korean Med. Sci. 2019, 34, e144. [Google Scholar] [CrossRef]
- Shi, S.; Ye, S.; Wu, X.; Xu, M.; Zhuo, R.; Liao, Q.; Xi, Y. A Two-DNA Methylation Signature to Improve Prognosis Prediction of Clear Cell Renal Cell Carcinoma. Yonsei Med. J. 2019, 60, 1013–1020. [Google Scholar] [CrossRef]
- Kim, Y.; Jang, W.; Piao, X.; Yoon, H.; Byun, Y.J.; Kim, J.S.; Kim, S.M.; Lee, S.K.; Seo, S.P.; Kang, H.W.; et al. ZNF492 and GPR149 methylation patterns as prognostic markers for clear cell renal cell carcinoma: Array-based DNA methylation profiling. Oncol. Rep. 2019, 42, 453–460. [Google Scholar] [CrossRef]
- Breault, J.E.; Shiina, H.; Igawa, M.; Ribeiro-Filho, L.A.; Deguchi, M.; Enokida, H.; Urakami, S.; Terashima, M.; Nakagawa, M.; Kane, C.J.; et al. Methylation of the gamma-catenin gene is associated with poor prognosis of renal cell carcinoma. Clin. Cancer Res. 2005, 11, 557–564. [Google Scholar] [PubMed]
- Peters, I.; Eggers, H.; Atschekzei, F.; Hennenlotter, J.; Waalkes, S.; Tränkenschuh, W.; Großhennig, A.; Merseburger, A.S.; Stenzl, A.; Kuczyk, M.A.; et al. GATA5CpG island methylation in renal cell cancer: A potential biomarker for metastasis and disease progression. BJU Int. 2012, 110, E144–E152. [Google Scholar] [CrossRef] [PubMed]
- Peters, I.; Gebauer, K.; Dubrowinskaja, N.; Atschekzei, F.; Kramer, M.W.; Hennenlotter, J.; Tezval, H.; Abbas, M.; Scherer, R.; Merseburger, A.S.; et al. GATA5 CpG island hypermethylation is an independent predictor for poor clinical outcome in renal cell carcinoma. Oncol. Rep. 2014, 31, 1523–1530. [Google Scholar] [CrossRef] [Green Version]
- Van Vlodrop, I.J.H.; Baldewijns, M.M.L.; Smits, K.M.; Schouten, L.J.; van Neste, L.; Van Criekinge, W.; van Poppel, H.; Lerut, E.; Schuebel, K.E.; Ahuja, N.; et al. Prognostic significance of Gremlin1 (GREM1) promoter CpG island hypermethylation in clear cell renal cell carcinoma. Am. J. Pathol. 2010, 176, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, H.; Steffens, S.; Grosshennig, A.; Becker, J.U.; Hennenlotter, J.; Stenzl, A.; Merseburger, A.S.; Serth, J. Prognostic and diagnostic relevance of hypermethylated in cancer 1 (HIC1) CpG island methylation in renal cell carcinoma. Int. J. Oncol. 2012, 40, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-L.; Wang, Y.-L.; Fu, X.-L.; Ma, J.-G. Aberrant Methylation of PCDH8 is a Potential Prognostic Biomarker for Patients with Clear Cell Renal Cell Carcinoma. Med. Sci. Monit. 2014, 20, 2380–2385. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.L.; Seligson, D.; Liu, X.; Janzen, N.; Bui, M.H.; Yu, H.; Shi, T.; Belldegrun, A.S.; Horvath, S.; Figlin, R.A. Using tumor markers to predict the survival of patients with metastatic renal cell carcinoma. J. Urol. 2005, 173, 1496–1501. [Google Scholar] [CrossRef]
- Kawai, Y.; Sakano, S.; Suehiro, Y.; Okada, T.; Korenaga, Y.; Hara, T.; Naito, K.; Matsuyama, H.; Hinoda, Y. Methylation level of the RASSF1A promoter is an independent prognostic factor for clear-cell renal cell carcinoma. Ann. Oncol. 2010, 21, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Klacz, J.; Wierzbicki, P.M.; Wronska, A.; Rybarczyk, A.; Stanislawowski, M.; Slebioda, T.; Olejniczak, A.; Matuszewski, M.; Kmiec, Z. Decreased expression of RASSF1A tumor suppressor gene is associated with worse prognosis in clear cell renal cell carcinoma. Int. J. Oncol. 2016, 48, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazdak, M.; Tezval, H.; Callauch, J.C.; Dubrowinskaja, N.; Peters, I.; Bokemeyer, C.; Hennenlotter, J.; Stenzl, A.; Kuczyk, M.A.; Serth, J. DNA methylation of sarcosine dehydrogenase (SARDH) loci as a prognosticator for renal cell carcinoma. Oncol. Rep. 2019, 42, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Atschekzei, F.; Hennenlotter, J.; Jänisch, S.; Großhennig, A.; Tränkenschuh, W.; Waalkes, S.; Peters, I.; Dörk, T.; Merseburger, A.S.; Stenzl, A.; et al. SFRP1CpG island methylation locus is associated with renal cell cancer susceptibility and disease recurrence. Epigenetics 2012, 7, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricketts, C.J.; Hill, V.K.; Linehan, W.M. Tumor-Specific Hypermethylation of Epigenetic Biomarkers, Including SFRP1, Predicts for Poorer Survival in Patients from the TCGA Kidney Renal Clear Cell Carcinoma (KIRC) Project. PLoS ONE 2014, 9, e85621. [Google Scholar] [CrossRef]
- Yang, M.; Hlady, R.A.; Zhou, D.; Ho, T.H.; Robertson, K.D. In silico DNA methylation analysis identifies potential prognostic biomarkers in type 2 papillary renal cell carcinoma. Cancer Med. 2019, 8, 5760–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, L.; Clarke, A.R. The roles of the methyl-CpG binding proteins in cancer. Genes Cancer 2011, 2, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- Li, L.; Li, N.; Liu, N.; Huo, F.; Zheng, J. MBD2 Correlates with a Poor Prognosis and Tumor Progression in Renal Cell Carcinoma. Oncotargets Ther. 2020, 13, 10001–10012. [Google Scholar] [CrossRef]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Goh, X.Y.; Vetter, M.; Pickering, L.; Swanton, C. Epigenetic regulation in RCC: Opportunities for therapeutic intervention? Nat. Rev. Urol. 2012, 9, 147–155. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Ellis, L.; Pili, R. Histone modifications: Implications in renal cell carcinoma. Epigenomics 2013, 5, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Rogenhofer, S.; Kahl, P.; Mertens, C.; Hauser, S.; Hartmann, W.; Büttner, R.; Müller, S.C.; Von Ruecker, A.; Ellinger, J. Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU Int. 2012, 109, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global Levels of Histone Modifications Predict Prognosis in Different Cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger, J.; Kahl, P.; Mertens, C.; Rogenhofer, S.; Hauser, S.; Hartmann, W.; Bastian, P.J.; Büttner, R.; Müller, S.C.; von Ruecker, A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int. J. Cancer 2010, 127, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Minardi, D.; Lucarini, G.; Filosa, A.; Milanese, G.; Zizzi, A.; Di Primio, R.; Montironi, R.; Muzzonigro, G. Prognostic role of global DNA-methylation and histone acetylation in pT1a clear cell renal carcinoma in partial nephrectomy specimens. J. Cell. Mol. Med. 2009, 13, 2115–2121. [Google Scholar] [CrossRef]
- Mosashvilli, D.; Kahl, P.; Mertens, C.; Holzapfel, S.; Rogenhofer, S.; Hauser, S.; Büttner, R.; Von Ruecker, A.; Müller, S.C.; Ellinger, J. Global histone acetylation levels: Prognostic relevance in patients with renal cell carcinoma. Cancer Sci. 2010, 101, 2664–2669. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, G.; Yu, Z. Computational prediction and analysis of histone H3k27me1-associated miRNAs. Biochim. Biophys. Acta BBA Proteins Proteom. 2021, 1869, 140539. [Google Scholar] [CrossRef]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef]
- Henrique, R.; Luís, A.S.; Jerónimo, C. The Epigenetics of Renal Cell Tumors: From Biology to Biomarkers. Front. Genet. 2012, 3, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.-P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-Dependent Histone H3K36 Trimethylation Is Required for Homologous Recombination Repair and Genome Stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valera, V.A.; Walter, B.A.; Linehan, W.M.; Merino, M.J. Regulatory Effects of microRNA-92 (miR-92) on VHL Gene Expression and the Hypoxic Activation of miR-210 in Clear Cell Renal Cell Carcinoma. J. Cancer 2011, 2, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Reisman, D.; Glaros, S.; Thompson, E.A. The SWI/SNF complex and cancer. Oncogene 2009, 28, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, B.; Porter, E.G.; Stewart, J.C.; Ferreira, C.R.; Schipma, M.J.; Dykhuizen, E.C. PBRM1 Regulates the Expression of Genes Involved in Metabolism and Cell Adhesion in Renal Clear Cell Carcinoma. PLoS ONE 2016, 11, e0153718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapur, P.; Peña-Llopis, S.; Christie, A.; Zhrebker, L.; Pavía-Jiménez, A.; Rathmell, W.K.; Xie, X.-J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.L.; Kaelin, W.C. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/- clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, E.G.; Dykhuizen, E.C. Individual Bromodomains of Polybromo-1 Contribute to Chromatin Association and Tumor Suppression in Clear Cell Renal Carcinoma. J. Biol. Chem. 2017, 292, 2601–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Su, L.; Liao, L.; Liu, Z.Z.; Langbein, L.; Dulaimi, E.; Testa, J.R.; Uzzo, R.G.; Zhong, Z.; Jiang, W.; et al. PBRM1 acts as a p53 lysine-acetylation reader to suppress renal tumor growth. Nat. Commun. 2019, 10, 5800. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K.; et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef]
- Rondinelli, B.; Rosano, D.; Antonini, E.; Frenquelli, M.; Montanini, L.; Huang, D.; Segalla, S.; Yoshihara, K.; Amin, S.B.; Lazarevič, D.; et al. Histone demethylase JARID1C inactivation triggers genomic instability in sporadic renal cancer. J. Clin. Investig. 2016, 126, 4387. [Google Scholar] [CrossRef] [Green Version]
- Arseneault, M.; Monlong, J.; Vasudev, N.S.; Laskar, R.S.; Safisamghabadi, M.; Harnden, P.; Egevad, L.; Nourbehesht, N.; Panichnantakul, P.; Holcatova, I.; et al. Loss of chromosome Y leads to down regulation of KDM5D and KDM6C epigenetic modifiers in clear cell renal cell carcinoma. Sci. Rep. 2017, 7, 44876. [Google Scholar] [CrossRef]
- Lee, K.-H.; Kim, B.-C.; Jeong, S.-H.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Histone Demethylase LSD1 Regulates Kidney Cancer Progression by Modulating Androgen Receptor Activity. Int. J. Mol. Sci. 2020, 21, 6089. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumari, N.; Nallabelli, N.; Sharma, U.; Rai, A.; Singh, S.K.; Kakkar, N.; Prasad, R. Expression profile of H3K4 demethylases with their clinical and pathological correlation in patients with clear cell renal cell carcinoma. Gene 2020, 739, 144498. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Weikert, S.; Magheli, A.; Hoffmann, M.; Engers, R.; Miller, K.; Kempkensteffen, C. Expression Profile of the Polycomb Group Protein Enhancer of Zeste Homologue 2 and its Prognostic Relevance in Renal Cell Carcinoma. J. Urol. 2009, 182, 2920–2925. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Z.; Zhong, L.; Wang, H.; Jiang, S.; Long, Q.; Xu, J.; Guo, J. Prognostic Value of EZH2 Expression and Activity in Renal Cell Carcinoma: A Prospective Study. PLoS ONE 2013, 8, e81484. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Q.; Zhang, L.; Gao, B.S.; Wan, Y.G.; Zhang, X.H.; Chen, B.; Wang, Y.T.; Sun, N.; Fu, Y.W. EZH2 promotes tumor progression by increasing VEGF expression in clear cell renal cell carcinoma. Clin. Transl. Oncol. 2015, 17, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Z.; Zhong, L.; Wang, H.; Jiang, S.; Long, Q.; Xu, J.; Guo, J. Enhancer of zeste homolog 2 (EZH2) promotes tumour cell migration and invasion via epigenetic repression of E-cadherin in renal cell carcinoma. BJU Int. 2016, 117, 351–362. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Ostrovnaya, I.; Reva, B.; Schultz, N.; Chen, Y.-B.; Gonen, M.; Liu, H.; Takeda, S.; Voss, M.H.; Tickoo, S.K.; et al. Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clin. Cancer Res. 2013, 19, 3259–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, A.A.; Chen, Y.-B.; Wren, J.; Gonen, M.; Abdel-Wahab, O.; Heguy, A.; Liu, H.; Takeda, S.; Tickoo, S.K.; Reuter, V.E.; et al. Clinical and Pathologic Impact of Select Chromatin-modulating Tumor Suppressors in Clear Cell Renal Cell Carcinoma. Eur. Urol. 2013, 63, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Outeiro-Pinho, G.; Barros-Silva, D.; Correia, M.P.; Henrique, R.; Jerónimo, C. Renal Cell Tumors: Uncovering the Biomarker Potential of ncRNAs. Cancers 2020, 12, 2214. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated Profiling of MicroRNAs and mRNAs: MicroRNAs Located on Xq27.3 Associate with Clear Cell Renal Cell Carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Brannon, A.R.; Reddy, A.R.; Alexe, G.; Seiler, M.W.; Arreola, A.; Oza, J.H.; Yao, M.; Juan, D.; Liou, L.S.; et al. Identifying mRNA targets of microRNA dysregulated in cancer: With application to clear cell Renal Cell Carcinoma. BMC Syst. Biol. 2010, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zabirnyk, O.; Wang, H.; Shiao, Y.-H.; Nickerson, M.L.; Khalil, S.; Anderson, L.M.; O Perantoni, A.; Phang, J.M. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 2010, 29, 4914–4924. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wu, S.; Shen, X.; Shi, Z.; Wu, D.; Yuan, Y.; Jiang, W.; Wang, Q.; Ke, Q.; Mao, Q.; et al. Methylation-mediated miR-214 regulates proliferation and drug sensitivity of renal cell carcinoma cells through targeting LIVIN. J. Cell. Mol. Med. 2020, 24, 6410–6425. [Google Scholar] [CrossRef]
- Wulfken, L.M.; Moritz, R.; Ohlmann, C.; Holdenrieder, S.; Jung, V.; Becker, F.; Herrmann, E.; Walgenbach-Brünagel, G.; Von Ruecker, A.; Müller, S.C.; et al. MicroRNAs in Renal Cell Carcinoma: Diagnostic Implications of Serum miR-1233 Levels. PLoS ONE 2011, 6, e25787. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.; Wulfken, L.M.; Holdenrieder, S.; Moritz, R.; Ohlmann, C.-H.; Jung, V.; Becker, F.; Herrmann, E.; Walgenbach-Brünagel, G.; Von Ruecker, A.; et al. Analysis of serum microRNAs (miR-26a-2*, miR-191, miR-337-3p and miR-378) as potential biomarkers in renal cell carcinoma. Cancer Epidemiol. 2012, 36, 391–394. [Google Scholar] [CrossRef]
- Tsiakanikas, P.; Giaginis, C.; Kontos, C.K.; Scorilas, A. Clinical utility of microRNAs in renal cell carcinoma: Current evidence and future perspectives. Expert Rev. Mol. Diagn. 2018, 18, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Redova, M.; Poprach, A.; Nekvindova, J.; Iliev, R.; Radova, L.; Lakomy, R.; Svoboda, M.; Vyzula, R.; Slaby, O. Circulating miR-378 and miR-451 in serum are potential biomarkers for renal cell carcinoma. J. Transl. Med. 2012, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Zhao, A.; Li, G.; Péoc’H, M.; Genin, C.; Gigante, M. Serum miR-210 as a novel biomarker for molecular diagnosis of clear cell renal cell carcinoma. Exp. Mol. Pathol. 2013, 94, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mollenkopf, H.-J.; Grimm, C.; Wagner, I.; Albrecht, M.; Waller, T.; Pilarsky, C.; Johannsen, M.; Stephan, C.; Lehrach, H.; et al. MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J. Cell. Mol. Med. 2009, 13, 3918–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Santos, R.M.; Costapinheiro, P.; De Luis, A.; Antunes, L.; Lobo, F.D.A.; De Oliveira, J.M.P.F.; Henrique, R.D.S.; Jeronimo, C. MicroRNA profile: A promising ancillary tool for accurate renal cell tumour diagnosis. Br. J. Cancer 2013, 109, 2646–2653. [Google Scholar] [CrossRef] [Green Version]
- Petillo, D.; Kort, E.J.; Anema, J.; Furge, K.A.; Yang, X.J.; Teh, B.T. MicroRNA profiling of human kidney cancer subtypes. Int. J. Oncol. 2009, 35, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, A.L.; Ferreira, M.; Silva, J.; Gomes, M.; Dias, F.; Santos, J.I.; Maurício, J.; Lobo, F.; Medeiros, R. Higher circulating expression levels of miR-221 associated with poor overall survival in renal cell carcinoma patients. Tumor Biol. 2014, 35, 4057–4066. [Google Scholar] [CrossRef]
- Outeiro-Pinho, G.; Barros-Silva, D.; Aznar, E.; Sousa, A.-I.; Vieira-Coimbra, M.; Oliveira, J.; Gonçalves, C.S.; Costa, B.M.; Junker, K.; Henrique, R.; et al. MicroRNA-30a-5pme: A novel diagnostic and prognostic biomarker for clear cell renal cell carcinoma in tissue and urine samples. J. Exp. Clin. Cancer Res. 2020, 39, 98. [Google Scholar] [CrossRef]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; Van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef]
- Mitsui, Y.; Hirata, H.; Arichi, N.; Hiraki, M.; Yasumoto, H.; Chang, I.; Fukuhara, S.; Yamamura, S.; Shahryari, V.; Deng, G.; et al. Inactivation of bone morphogenetic protein 2 may predict clinical outcome and poor overall survival for renal cell carcinoma through epigenetic pathways. Oncotarget 2015, 6, 9577–9591. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, Q.-L.; Zhai, T.-S.; Lu, J.; Dong, Y.-Z.; Xu, Y.-F. Silencing miR-454 suppresses cell proliferation, migration and invasion via directly targeting MECP2 in renal cell carcinoma. Am. J. Transl. Res. 2020, 12, 4277–4289. [Google Scholar]
- White, N.M.A.; Khella, H.W.Z.; Grigull, J.; Adzovic, S.; Youssef, Y.M.; Honey, R.J.; Stewart, R.; Pace, K.T.; Bjarnason, G.A.; Jewett, M.A.S.; et al. miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br. J. Cancer 2011, 105, 1741–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrie, C.H.; Larrea, E.; Larrinaga, G.; Goicoechea, I.; Arestin, M.; Fernandez-Mercado, M.; Hes, O.; Cáceres, F.; Manterola, L.; López, J.I. Targeted next-generation sequencing and non-coding RNA expression analysis of clear cell papillary renal cell carcinoma suggests distinct pathological mechanisms from other renal tumour subtypes. J. Pathol. 2014, 232, 32–42. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, J.; Chen, Z.; Cheng, A.S.-L. Understanding the epigenetic regulation of tumours and their microenvironments: Opportunities and problems for epigenetic therapy. J. Pathol. 2017, 241, 10–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Yao, W.; Wang, J.; Ma, X.; Xiao, W.; Li, H.; Xia, D.; Yang, Y.; Deng, K.; Xiao, H.; et al. LncRNAs Expression Signatures of Renal Clear Cell Carcinoma Revealed by Microarray. PLoS ONE 2012, 7, e42377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.; Han, Z.; Qian, J.; Bao, M.; Li, P.; Ju, X.; Zhang, S.; Zhang, L.; Li, S.; Cao, Q.; et al. Expression Pattern of Long Non-Coding RNAs in Renal Cell Carcinoma Revealed by Microarray. PLoS ONE 2014, 9, e99372. [Google Scholar] [CrossRef] [Green Version]
- Blondeau, J.J.; Deng, M.; Syring, I.; Schrödter, S.; Schmidt, D.; Perner, S.; Müller, S.C.; Ellinger, J. Identification of novel long non-coding RNAs in clear cell renal cell carcinoma. Clin. Epigenetics 2015, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Liu, J.; Zheng, Y.; You, L.; Kuang, D.; Liu, T. Suppressed expression of long non-coding RNA HOTAIR inhibits proliferation and tumourigenicity of renal carcinoma cells. Tumor Biol. 2014, 35, 11887–11894. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yao, L.; Zhang, Q.; Wang, F.; Mei, H.; Guo, X.; Huang, W. Long noncoding RNA HOTAIR promotes metastasis of renal cell carcinoma by up-regulating histone H3K27 demethylase JMJD3. Oncotarget 2017, 8, 19795–19802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Niu, S.; Wang, Y.; Duan, L.; Pan, Y.; Tong, Z.; Zhang, X.; Yang, Z.; Peng, B.; Wang, X.; et al. DMDRMR-Mediated Regulation of m6A-Modified CDK4 by m6A Reader IGF2BP3 Drives ccRCC Progression. Cancer Res. 2020, 81, 923–934. [Google Scholar] [CrossRef]
- Xin, Z.; Soejima, H.; Higashimoto, K.; Yatsuki, H.; Zhu, X.; Satoh, Y.; Masaki, Z.; Kaneko, Y.; Jinno, Y.; Fukuzawa, R.; et al. A Novel Imprinted Gene, KCNQ1DN, within the WT2 Critical Region of Human Chromosome 11p15.5 and Its Reduced Expression in Wilms’ Tumors. J. Biochem. 2000, 128, 847–853. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enroth, C.; Poulsen, L.D.; Iversen, S.; Kirpekar, F.; Albrechtsen, A.; Vinther, J. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 2019, 47, e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiao, J.; Bai, J.; Tian, Y.; Qu, Y.; Chen, X.; Wang, Q.; Li, X.; Zhang, Y.; Xu, J. Molecular characterization and clinical relevance of m6A regulators across 33 cancer types. Mol. Cancer 2019, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Jing, C.; Xiao, C.; Li, T.; Wang, Y. Prognostic risk signature based on the expression of three m6A RNA methylation regulatory genes in kidney renal papillary cell carcinoma. Aging 2020, 12, 22078–22094. [Google Scholar] [CrossRef]
- Lobo, J.; Barros-Silva, D.; Henrique, R.; Jerónimo, C. The Emerging Role of Epitranscriptomics in Cancer: Focus on Urological Tumors. Genes 2018, 9, 552. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.-P.J.; Kantarjian, H.M. Targeting DNA Methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—Miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Marques-Magalhães, Â.; Graca, I.; Henrique, R.; Jerónimo, C. Targeting DNA Methyltranferases in Urological Tumors. Front. Pharmacol. 2018, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Faleiro, I.; Leão, R.; Binnie, A.; De Mello, R.A.; Maia, A.-T.; Castelo-Branco, P. Epigenetic therapy in urologic cancers: An update on clinical trials. Oncotarget 2017, 8, 12484–12500. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epigenetics in Breast and Prostate Cancer. Methods Mol. Biol. 2015, 1238, 425–466. [Google Scholar] [CrossRef] [Green Version]
- Pili, R.; Liu, G.; Chintala, S.; Verheul, H.; Rehman, S.; Attwood, K.; Lodge, M.A.; Wahl, R.; Martin, J.I.; Miles, K.M.; et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 2017, 116, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zibelman, M.; Wong, Y.-N.; Devarajan, K.; Malizzia, L.; Corrigan, A.; Olszanski, A.J.; Denlinger, C.S.; Roethke, S.K.; Tetzlaff, C.H.; Plimack, E.R. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Investig. New Drugs 2015, 33, 1040–1047. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Investig. 2011, 29, 451–455. [Google Scholar] [CrossRef]
- Wood, A.; George, S.; Adra, N.; Chintala, S.; Damayanti, N.; Pili, R. Phase I study of the mTOR inhibitor everolimus in combination with the histone deacetylase inhibitor panobinostat in patients with advanced clear cell renal cell carcinoma. Investig. New Drugs 2020, 38, 1108–1116. [Google Scholar] [CrossRef]
- Pili, R.; Salumbides, B.; Zhao, M.; Altiok, S.; Qian, D.; Zwiebel, J.; Carducci, M.A.; Rudek, M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer 2012, 106, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pili, R.; Quinn, D.I.; Hammers, H.J.; Monk, P.; George, S.; Dorff, T.B.; Olencki, T.; Shen, L.; Orillion, A.; LaMonica, D.; et al. Immunomodulation by Entinostat in Renal Cell Carcinoma Patients Receiving High-Dose Interleukin 2: A Multicenter, Single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin. Cancer Res. 2017, 23, 7199–7208. [Google Scholar] [CrossRef] [Green Version]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A Phase II Study of Depsipeptide in Refractory Metastatic Renal Cell Cancer. Clin. Genitourin. Cancer 2006, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, R.P.; Rankin, C.; Hoff, H.M.G.; Gold, P.J.; Billingsley, K.G.; Chapman, R.A.; Wong, L.; Ward, J.H.; Abbruzzese, J.L.; Blanke, C.D. Phase II Trial of depsipeptide (NSC-630176) in previously treated colorectal cancer patients with advanced disease: A Southwest Oncology Group Study (S0336). Investig. New Drugs 2009, 27, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.L.; Plumb, J.A.; Vidal, L.; Tjørnelund, J.; Knoblauch, P.; Rasmussen, A.; Ooi, C.E.; Buhl-Jensen, P.; Brown, R.; Evans, T.J.; et al. A Phase 1 Pharmacokinetic and Pharmacodynamic Study of the Histone Deacetylase Inhibitor Belinostat in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braiteh, F.; Soriano, A.O.; Garcia-Manero, G.; Hong, D.; Johnson, M.M.; Silva, L.D.P.; Yang, H.; Alexander, S.; Wolff, J.; Kurzrock, R. Phase I Study of Epigenetic Modulation with 5-Azacytidine and Valproic Acid in Patients with Advanced Cancers. Clin. Cancer Res. 2008, 14, 6296–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.J.; Issa, J.-P.; Kurzrock, R.; Nunez, M.I.; Jelinek, J.; Hong, D.; Oki, Y.; Guo, Z.; Gupta, S.; Wistuba, I.I. Decitabine Effect on Tumor Global DNA Methylation and Other Parameters in a Phase I Trial in Refractory Solid Tumors and Lymphomas. Clin. Cancer Res. 2009, 15, 3881–3888. [Google Scholar] [CrossRef] [Green Version]
- Gollob, J.A.; Sciambi, C.J.; Peterson, B.L.; Richmond, T.; Thoreson, M.; Moran, K.; Dressman, H.K.; Jelinek, J.; Issa, J.-P.J. Phase I Trial of Sequential Low-Dose 5-Aza-2′-Deoxycytidine Plus High-Dose Intravenous Bolus Interleukin-2 in Patients with Melanoma or Renal Cell Carcinoma. Clin. Cancer Res. 2006, 12, 4619–4627. [Google Scholar] [CrossRef] [Green Version]
- Winquist, E.; Knox, J.; Ayoub, J.-P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New Drugs 2006, 24, 159–167. [Google Scholar] [CrossRef]
- Amato, R.J. Inhibition of DNA Methylation by Antisense Oligonucleotide MG98 as Cancer Therapy. Clin. Genitourin. Cancer 2007, 5, 422–426. [Google Scholar] [CrossRef]
- Vassilakos, A.; Lee, Y.; Viau, S.; Feng, N.; Jin, H.; Chai, V.; Wang, M.; Avolio, T.; Wright, J.; Young, A.; et al. GTI-2040 displays cooperative anti-tumor activity when combined with interferon α against human renal carcinoma xenografts. Int. J. Oncol. 2009, 34, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Margolin, K.; Synold, T.W.; Lara, P.; Frankel, P.; Lacey, S.F.; Quinn, D.I.; Baratta, T.; Dutcher, J.P.; Xi, B.; Diamond, D.J.; et al. Oblimersen and α-interferon in metastatic renal cancer: A phase II study of the California Cancer Consortium. J. Cancer Res. Clin. Oncol. 2007, 133, 705–711. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Roboz, G.J.; Kantarjian, H.M.; Yee, K.W.L.; Kropf, P.L.; O’Connell, C.L.; Griffiths, E.A.; Stock, W.; Daver, N.G.; Jabbour, E.; Ritchie, E.K.; et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer 2018, 124, 325–334. [Google Scholar] [CrossRef]
- Venturelli, S.; Berger, A.; Weiland, T.; Essmann, F.; Waibel, M.; Nuebling, T.; Häcker, S.; Schenk, M.; Schulze-Osthoff, K.; Salih, H.R.; et al. Differential Induction of Apoptosis and Senescence by the DNA Methyltransferase Inhibitors 5-Azacytidine and 5-Aza-2′-Deoxycytidine in Solid Tumor Cells. Mol. Cancer Ther. 2013, 12, 2226–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.-W.C.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Morris, M.R.; Gentle, D.; Shuib, S.; Brown, M.; Clarke, N.; Wei, W.; Nathan, P.; Latif, F.; Maher, E.R. Methylation profiling and evaluation of demethylating therapy in renal cell carcinoma. Clin. Epigenetics 2013, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, D.; Han, T.; Xu, X.; Liu, Y. Decitabine induces G2/M cell cycle arrest by suppressing p38/NF-κB signaling in human renal clear cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11140–11148. [Google Scholar] [PubMed]
- Konac, E.; Varol, N.; Yilmaz, A.; Menevse, S.; Sozen, S. DNA methyltransferase inhibitor-mediated apoptosis in the Wnt/β-catenin signal pathway in a renal cell carcinoma cell line. Exp. Biol. Med. Maywood 2013, 238, 1009–1016. [Google Scholar] [CrossRef]
- Xi, W.; Chen, X.; Sun, J.; Wang, W.; Huo, Y.; Zheng, G.; Wu, J.; Li, Y.; Yang, A.; Wang, T. Combined Treatment with Valproic Acid and 5-Aza-2′-Deoxycytidine Synergistically Inhibits Human Clear Cell Renal Cell Carcinoma Growth and Migration. Med Sci. Monit. 2018, 24, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Reu, F.J.; Bae, S.I.; Cherkassky, L.; Leaman, D.W.; Lindner, D.; Beaulieu, N.; MacLeod, A.R.; Borden, E.C. Overcoming Resistance to Interferon-Induced Apoptosis of Renal Carcinoma and Melanoma Cells by DNA Demethylation. J. Clin. Oncol. 2006, 24, 3771–3779. [Google Scholar] [CrossRef]
- Reu, F.J.; Leaman, D.W.; Maitra, R.R.; Bae, S.I.; Cherkassky, L.; Fox, M.W.; Rempinski, D.R.; Beaulieu, N.; MacLeod, A.R.; Borden, E.C. Expression of RASSF1A, an epigenetically silenced tumor suppressor, overcomes resistance to apoptosis induction by interferons. Cancer Res. 2006, 66, 2785–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Yu, L.; Qin, Z.; Chen, L.; Hu, H.; Zheng, X.; Zeng, S. Regulation of OCT2 transcriptional repression by histone acetylation in renal cell carcinoma. Epigenetics 2019, 14, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Stephenson, J.; Hotte, S.; Nemunaitis, J.; Bélanger, K.; Reid, G.; Martell, R.E. MG98, a Second-Generation DNMT1 Inhibitor, in the Treatment of Advanced Renal Cell Carcinoma. Cancer Investig. 2012, 30, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Dasari, A.; Gore, L.; Messersmith, W.A.; Diab, S.; Jimeno, A.; Weekes, C.D.; Lewis, K.D.; Drabkin, H.A.; Flaig, T.W.; Camidge, D.R. A phase I study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Investig. New Drugs 2013, 31, 115–125. [Google Scholar] [CrossRef]
- Morgan, S.S.; Cranmer, L.D. Vorinostat synergizes with ridaforolimus and abrogates the ridaforolimus-induced activation of AKT in synovial sarcoma cells. BMC Res. Notes 2014, 7, 812. [Google Scholar] [CrossRef] [Green Version]
- Cha, T.-L.; Chuang, M.-J.; Wu, S.-T.; Sun, G.-H.; Chang, S.-Y.; Yu, D.-S.; Huang, S.-M.; Huan, S.K.-H.; Cheng, T.-C.; Chen, T.-T.; et al. Dual Degradation of Aurora A and B Kinases by the Histone Deacetylase Inhibitor LBH589 Induces G2-M Arrest and Apoptosis of Renal Cancer Cells. Clin. Cancer Res. 2009, 15, 840–850. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, M.; Derenzini, E.; Buglio, D.; Medeiros, L.J.; Davis, R.E.; Zhang, J.; Ji, Y.; Younes, A. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood 2012, 119, 4017–4025. [Google Scholar] [CrossRef]
- Beider, K.; Bitner, H.; Voevoda-Dimenshtein, V.; Rosenberg, E.; Sirovsky, Y.; Magen, H.; Canaani, J.; Ostrovsky, O.; Shilo, N.; Shimoni, A.; et al. The mTOR inhibitor everolimus overcomes CXCR4-mediated resistance to histone deacetylase inhibitor panobinostat through inhibition of p21 and mitotic regulators. Biochem. Pharmacol. 2019, 168, 412–428. [Google Scholar] [CrossRef]
- Ling, Y.; Liu, J.; Qian, J.; Meng, C.; Guo, J.; Gao, W.; Xiong, B.; Ling, C.; Zhang, Y. Recent Advances in Multi-target Drugs Targeting Protein Kinases and Histone Deacetylases in Cancer Therapy. Curr. Med. Chem. 2020, 27, 7264–7288. [Google Scholar] [CrossRef]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.-W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.F.; Qian, D.Z.; Ren, M.; Kato, Y.; Wei, Y.; Zhang, L.; Fansler, Z.; Clark, D.; Nakanishi, O.; Pili, R. Epigenetic modulation of retinoic acid receptor beta2 by the histone deacetylase inhibitor MS-275 in human renal cell carcinoma. Clin. Cancer Res. 2005, 11, 3535–3542. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Chan, K.K.; Bakhtiar, R.; Jiang, C. Depsipeptide (FR901228, NSC-630176) pharmacokinetics in the rat by LC/MS/MS. Investig. New Drugs 1997, 15, 195–206. [Google Scholar] [CrossRef]
- Sborov, D.W.; Canella, A.; Hade, E.M.; Mo, X.; Khountham, S.; Wang, J.; Ni, W.; Poi, M.; Coss, C.; Liu, Z.; et al. A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 2017, 58, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Wang, W.-H.; Wu, W.-Y.; Hsu, C.-C.; Wei, L.-R.; Wang, S.-F.; Hsu, Y.-W.; Liaw, C.-C.; Tsai, W.-C. Novel histone deacetylase inhibitor AR-42 exhibits antitumor activity in pancreatic cancer cells by affecting multiple biochemical pathways. PLoS ONE 2017, 12, e0183368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.-S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2020, 28, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.D.; Dai, J.; Eschwege, P.; Goldberg, J.S.; Duggan, B.P.; Williamson, K.E.; Bander, N.H.; Nanus, D.M. Downregulation of Bcl-2 sensitises interferon-resistant renal cancer cells to Fas. Br. J. Cancer 2004, 91, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Vassilakos, A.; Feng, N.; Lam, V.; Xie, H.; Wang, M.; Jin, H.; Xiong, K.; Liu, C.; Wright, J.; et al. GTI-2040, an antisense agent targeting the small subunit component (R2) of human ribonucleotide reductase, shows potent antitumor activity against a variety of tumors. Cancer Res. 2003, 63, 2802–2811. [Google Scholar]
- Stadler, W.M.; Desai, A.A.; Quinn, D.I.; Bukowski, R.; Poiesz, B.; Kardinal, C.G.; Lewis, N.; Makalinao, A.; Murray, P.; Torti, F.M. A Phase I/II study of GTI-2040 and capecitabine in patients with renal cell carcinoma. Cancer Chemother. Pharmacol. 2008, 61, 689–694. [Google Scholar] [CrossRef]
- Tan, X.; Zhang, Z.; Liu, P.; Yao, H.; Shen, L.; Tong, J.-S. Inhibition of EZH2 enhances the therapeutic effect of 5-FU via PUMA upregulation in colorectal cancer. Cell Death Dis. 2020, 11, 1061. [Google Scholar] [CrossRef] [PubMed]
- Dockerill, M.; Gregson, C.; Donovan, D.H.O. Targeting PRC2 for the treatment of cancer: An updated patent review (2016–2020). Expert Opin. Ther. Pat. 2021, 31, 1–17. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.M.; Béguelin, W.; Koche, R.P.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- López, J.I.; Pulido, R.; Cortés, J.M.; Angulo, J.C.; Lawrie, C.H. Potential impact of PD-L1 (SP-142) immunohistochemical heterogeneity in clear cell renal cell carcinoma immunotherapy. Pathol. Res. Pract. 2018, 214, 1110–1114. [Google Scholar] [CrossRef]
- Liu, S.; Yang, Z.; Li, G.; Li, C.; Luo, Y.; Gong, Q.; Wu, X.; Li, T.; Zhang, Z.; Xing, B.; et al. Multi-omics Analysis of Primary Cell Culture Models Reveals Genetic and Epigenetic Basis of Intratumoral Phenotypic Diversity. Genom. Proteom. Bioinform. 2019, 17, 576–589. [Google Scholar] [CrossRef]
- Ortega, M.A.; Poirion, O.; Zhu, X.; Huang, S.; Wolfgruber, T.K.; Sebra, R.; Garmire, L.X. Using single-cell multiple omics approaches to resolve tumor heterogeneity. Clin. Transl. Med. 2017, 6, 46. [Google Scholar] [CrossRef]
- López-Fernández, E.; López, J.I. The Impact of Tumor Eco-Evolution in Renal Cell Carcinoma Sampling. Cancers 2018, 10, 485. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Bao, Y.; Gan, X.; Wang, J.; Chen, Q.; Dai, Z.; Liu, B.; Wang, A.; Sun, S.; Yang, F.; et al. DNA methylation-regulated QPCT promotes sunitinib resistance by increasing HRAS stability in renal cell carcinoma. Theranostics 2019, 9, 6175–6190. [Google Scholar] [CrossRef]
- Xiong, Z.; Yuan, C.; Shi, J.; Xiong, W.; Huang, Y.; Xiao, W.; Yang, H.; Chen, K.; Zhang, X. Restoring the epigenetically silenced PCK2 suppresses renal cell carcinoma progression and increases sensitivity to sunitinib by promoting endoplasmic reticulum stress. Theranostics 2020, 10, 11444–11461. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Jerónimo, C.; Henrique, R. Targeting the Immune system and Epigenetic Landscape of Urological Tumors. Int. J. Mol. Sci. 2020, 21, 829. [Google Scholar] [CrossRef] [Green Version]
- Dunn, J.; Rao, S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-D.; Kong, W.; Peterson, C.B.; McGrail, D.J.; Hoang, A.; Zhang, X.; Lam, T.; Pilie, P.G.; Zhu, H.; Beckermann, K.E.; et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat. Commun. 2020, 11, 2135. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bossé, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGoverne, I.; Dunn, J.; Batham, J.; Tu, W.J.; Chrisp, J.; Rao, S. Epitherapy and immune checkpoint blockade: Using epigenetic reinvigoration of exhausted and dysfunctional T cells to reimburse immunotherapy response. BMC Immunol. 2020, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Goldsamt, A.; Damayanti, N.P.; De Nigris, F.; Pili, R. Epigenetic dysregulation in advanced kidney cancer: Opportunities for therapeutic interventions. Cancer J. 2020, 26, 399–406. [Google Scholar] [CrossRef]


{kind=link}
| Epigenetic Drug | Combined Therapy | Phase | Trial Registry | Ref. |
|---|---|---|---|---|
| HDAC Inhibition | ||||
| Vorinostat | - | II | NCT00278395 | - |
| Vorinostat | Isotretinoin | I/II | NCT00324740 | - |
| Vorinostat | Bevacizumab | I/II | NCT00324870 | [177] |
| Vorinostat | Sirolimus | I | NCT01087554 | [178] |
| Vorinostat | Ridaforolimus | I | - | [179] |
| Vorinostat | Pembrolizumab | I | NCT02619253 | - |
| Panobinostat | Sorafenib | I | NCT01005797 | - |
| Panobinostat | - | II | NCT00550277 | [180] |
| Panobinostat | Everolimus | I/II | NCT01582009 | [181] |
| Entinostat | Isotretinoin | I | - | [182] |
| Entinostat | IL-2 | I/II | NCT01038778 | [183] |
| Entinostat | IL-2 | I/II | NCT03501381 | - |
| Entinostat | Atezolizumab plus Bevacizumab | I/II | NCT03024437 | - |
| Entinostat | Nivolumab plus Ipilimumab | II | NCT03552380 | - |
| Depsipeptide | - | II | - | [184] |
| Romidepsin | - | I | NCT01638533 | - |
| Romidepsin | - | II | NCT00106613 | [185] |
| Belinostat | - | I | NCT00413075 | [186] |
| DNMT Inhibition | ||||
| Azacytidine | IFN-α | I | NCT00217542 | - |
| Azacytidine | Valproic Acid | I | - | [187] |
| Azacytidine | Bevacizumab | I/II | NCT00934440 | - |
| Decitabine | - | I | - | [188] |
| Decitabine | IL-2 | I | - | [189] |
| Decitabine | IFN-α | II | NCT00561912 | - |
| Decitabine | Anti-PD-1 | I/II | NCT02961101 | - |
| Decitabine | MBG453 | I | NCT02608268 | - |
| Decitabine | Oxaliplatin | II | NCT04049344 | - |
| Oligonucleotide MG98 | - | I/II | NCT00003890 | [190] |
| Oligonucleotide MG98 | IFN-α | I/II | - | [191] |
| Other Therapeutic Strategies | ||||
| miRNA MRX34 | - | I | NCT01829971 | - |
| Oligonucleotide GTI-2040 | Capecitabine | I/II | NCT00056173 | [192] |
| Oligonucleotide Oblimersen | IFN-α | II | NCT00059813 | [193] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angulo, J.C.; Manini, C.; López, J.I.; Pueyo, A.; Colás, B.; Ropero, S. The Role of Epigenetics in the Progression of Clear Cell Renal Cell Carcinoma and the Basis for Future Epigenetic Treatments. Cancers 2021, 13, 2071. https://doi.org/10.3390/cancers13092071
Angulo JC, Manini C, López JI, Pueyo A, Colás B, Ropero S. The Role of Epigenetics in the Progression of Clear Cell Renal Cell Carcinoma and the Basis for Future Epigenetic Treatments. Cancers. 2021; 13(9):2071. https://doi.org/10.3390/cancers13092071
Chicago/Turabian StyleAngulo, Javier C., Claudia Manini, Jose I. López, Angel Pueyo, Begoña Colás, and Santiago Ropero. 2021. "The Role of Epigenetics in the Progression of Clear Cell Renal Cell Carcinoma and the Basis for Future Epigenetic Treatments" Cancers 13, no. 9: 2071. https://doi.org/10.3390/cancers13092071