
The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis

 , , , , , , ,
, , , , , , ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
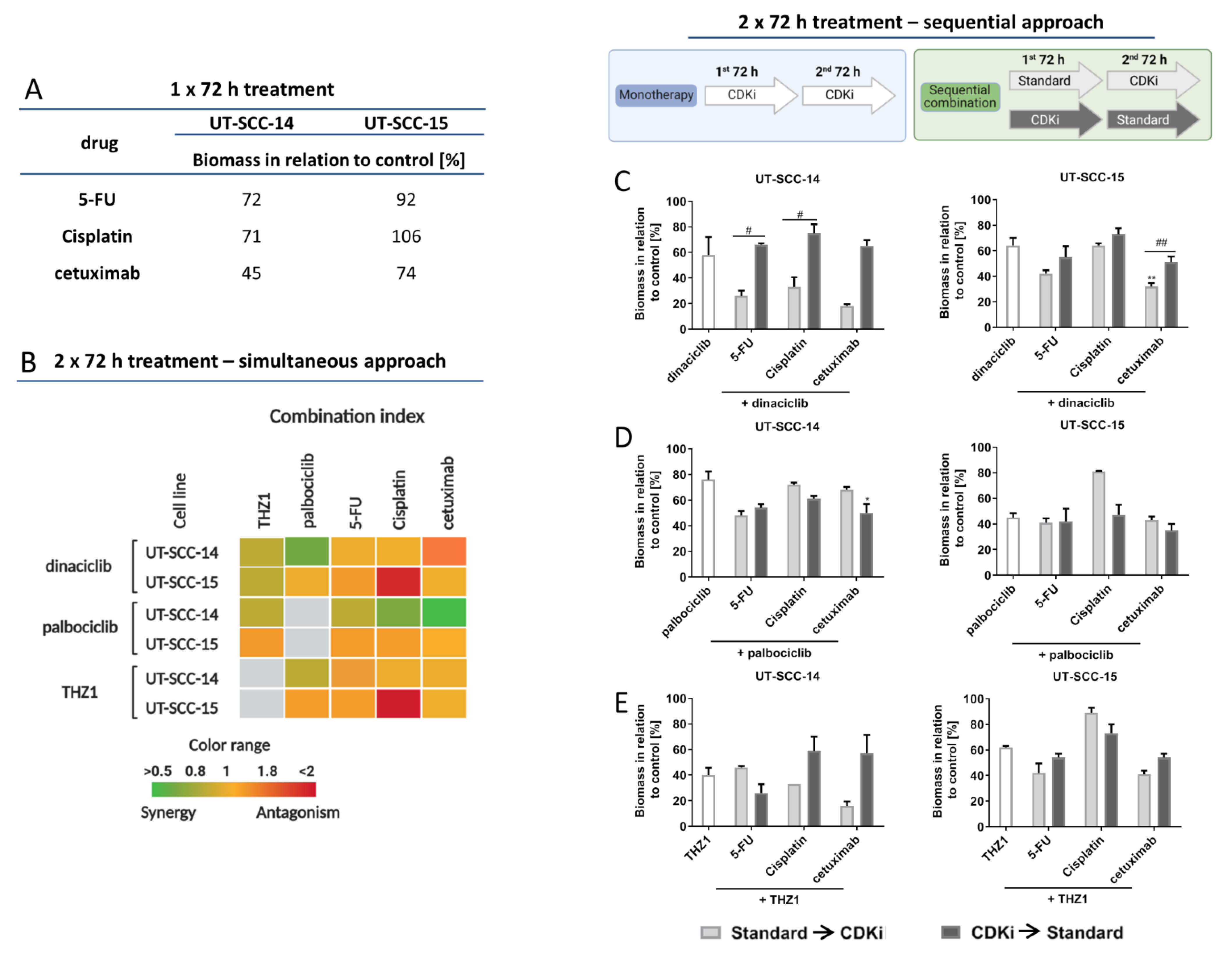
2.1. CDKi Treatment Impairs Viability and Exerts Synergistic Effects in Combination Therapy
2.2. CDKi’s Induce Apoptotic and Necrotic Cell Death and Mediate Calreticulin Translocation
2.3. CDKi Induce Cell Cyclce Arrest
2.4. CDKi’s Have Minor Impact on Double-Strand Breaks and Radiosensitivity
2.5. CDKi’s Remodel the Actin Filament
2.6. Influence on Mitochondria, Lysosomes, the Endoplasmatic Reticulum and Vacuole Formation
2.6.1. CDKi’s Reduce Invasiveness and Migratory Potential
2.6.2. In Vivo Results
3. Discussion
4. Materials and Methods
4.1. Tumor Cell Lines and Culture Conditions
4.2. Cytostatic Drugs and Targeted Substances
4.3. Dose Response Curves and Combination Therapy
4.4. γ-H2AX Staining
4.5. Apoptosis-Necrosis Assay, Phenotyping, and Immunogenic Cell Death
4.6. Cell Cycle Assay
4.7. Influence on Mitochondria, Lysosomes, ER, and Vacuole Formation
4.8. Senescence
4.9. Impedance Measurement and Actin Staining
4.10. Wound Healing and Invasion Assay
4.11. In Vivo Study
4.11.1. Ethical Statement
4.11.2. Experimental Protocol
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schafer, K.A. The Cell Cycle: A review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef]
- Jingwen, B.; Yaochen, L.; Guojun, Z. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorgan. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Jakus, J.; Harris, C.M.; Ensley, J.F.; Robbins, K.C.; Yeudall, W.A. Altered expression and activity of G1/S cyclins and cyclin-dependent kinases characterize squamous cell carcinomas of the head and neck. Int. J. Cancer 1997, 73, 551–555. [Google Scholar] [CrossRef]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Schoninger, S.F.; Blain, S.W. The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol. Cancer Ther. 2020, 19, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, B.J.; Perez-Bertoldi, J.M.; Lim, K.; Iavarone, A.T.; Toso, D.B.; Nogales, E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc. Natl. Acad. Sci. USA 2020, 117, 22849–22857. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, V.; Chirovsky, D.; Cheung, W.Y.; Bertolini, F.; Ahn, M.J.; Yang, M.H.; Castro, G.; Berrocal, A.; Sjoquist, K.; Kuyas, H.; et al. Global treatment patterns and outcomes among patients with recurrent and/or metastatic head and neck squamous cell carcinoma: Results of the GLANCE H&N study. Oral Oncol. 2020, 102, 104526. [Google Scholar] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Gebre-Medhin, M.; Brun, E.; Engström, P.; Haugen Cange, H.; Hammarstedt-Nordenvall, L.; Reizenstein, J.; Nyman, J.; Abel, E.; Friesland, S.; Sjödin, H.; et al. ARTSCAN III: A Randomized Phase III Study Comparing Chemoradiotherapy With Cisplatin Versus Cetuximab in Patients With Locoregionally Advanced Head and Neck Squamous Cell Cancer. J. Clin. Oncol. 2021, 39, 38–47. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.D.; Bowles, D.W. Epidemiology of Head and Neck Squamous Cell Carcinomas: Impact on Staging and Prevention Strategies. Curr. Treat. Options Oncol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Salakova, M.; Koslabova, E.; Grega, M.; Smahelova, J.; Klozar, J.; Vencalek, O.; Tachezy, R. Mucosal and skin HPV types in tumour-free tonsils and tonsillar tumours. Neoplasma 2018, 65, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: A multicentre, multigroup, phase 2 trial. Lancet Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef]
- Yoon, N.; Vander Velde, R.; Marusyk, A.; Scott, J.G. Optimal Therapy Scheduling Based on a Pair of Collaterally Sensitive Drugs. Bull. Math. Biol. 2018, 80, 1776–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Chang, L.; Li, W.H.; Xiao, M.Y.; Wang, Y.; He, W.J.; Xia, Y.X.; Wang, L.; Chen, Y. Radiotherapy concurrent versus sequential with endocrine therapy in breast cancer: A meta-analysis. Breast 2016, 27, 93–98. [Google Scholar] [CrossRef]
- Robinson, A.M.; Rathore, R.; Redlich, N.J.; Adkins, D.R.; VanArsdale, T.; Van Tine, B.A.; Michel, L.S. Cisplatin exposure causes c-Myc-dependent resistance to CDK4/6 inhibition in HPV-negative head and neck squamous cell carcinoma. Cell Death Dis. 2019, 10, 867. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling stress: The mechanics of cancer progression and aggression. Front. Cell Dev. Biol. 2018, 6, 1–12. [Google Scholar] [CrossRef]
- Humphries, W.H.; Szymanski, C.J.; Payne, C.K. Endo-lysosomal vesicles positive for rab7 and lamp1 are terminal vesicles for the transport of dextran. PLoS ONE 2011, 6, e26626. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Bloomquist, E.; Tang, S.; Fu, W.; Bi, Y.; Liu, Q.; Yu, J.; Zhao, P.; Palmby, T.R.; Goldberg, K.B.; et al. FDA approval: Ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin. Cancer Res. 2018, 24, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Groenland, S.L.; Martínez-Chávez, A.; van Dongen, M.G.J.; Beijnen, J.H.; Schinkel, A.H.; Huitema, A.D.R.; Steeghs, N. Clinical Pharmacokinetics and Pharmacodynamics of the Cyclin-Dependent Kinase 4 and 6 Inhibitors Palbociclib, Ribociclib, and Abemaciclib. Clin. Pharmacokinet. 2020, 59, 1501–1520. [Google Scholar] [CrossRef] [PubMed]
- Göttgens, E.L.; Bussink, J.; Leszczynska, K.B.; Peters, H.; Span, P.N.; Hammond, E.M. Inhibition of CDK4/CDK6 Enhances Radiosensitivity of HPV Negative Head and Neck Squamous Cell Carcinomas. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 548–558. [Google Scholar] [CrossRef]
- Billard-Sandu, C.; Tao, Y.G.; Sablin, M.P.; Dumitrescu, G.; Billard, D.; Deutsch, E. CDK4/6 inhibitors in P16/HPV16-negative squamous cell carcinoma of the head and neck. Eur. Arch. Oto-Rhino-Laryngol. 2020, 277, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Agarwal, R. New combination therapies with cell-cycle agents. Curr. Opin. Investig. Drugs 2008, 9, 591–604. [Google Scholar]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar] [PubMed]
- Weiss, J.M.; Csoszi, T.; Maglakelidze, M.; Hoyer, R.J.; Beck, J.T.; Domine Gomez, M.; Lowczak, A.; Aljumaily, R.; Rocha Lima, C.M.; Boccia, R.V.; et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: A phase Ib/randomized phase II trial. Ann. Oncol. 2019, 30, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with Trilaciclib in Patients Receiving Topotecan for Small Cell Lung Cancer: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Study. Adv. Ther. 2021, 38, 350–365. [Google Scholar] [CrossRef]
- Mitri, Z.; Karakas, C.; Wei, C.; Briones, B.; Simmons, H.; Ibrahim, N.; Alvarez, R.; Murray, J.L.; Keyomarsi, K.; Moulder, S. A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest. New Drugs 2015, 33, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K.; et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef]
- Riess, C.; Irmscher, N.; Salewski, I.; Strüder, D.; Classen, C.F.; Große-Thie, C.; Junghanss, C.; Maletzki, C. Cyclin-dependent kinase inhibitors in head and neck cancer and glioblastoma—backbone or add-on in immune-oncology? Cancer Metastasis Rev. 2020, 40, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Md Sakib Hossain, D.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti- PD1–mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Bandyopadhyay, D.; Sharma, K.; Allen, A.J.; Kmieciak, M.; Grant, S. The covalent CDK7 inhibitor THz1 potently induces apoptosis in multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2019, 25, 6195–6205. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.H.; Chen, C.C.; Leu, Y.L.; Lee, Y.S.; Lian, J.H.; Hsieh, H.L.; Chen, C.Y. Palbociclib induces DNA damage and inhibits DNA repair to induce cellular senescence and apoptosis in oral squamous cell carcinoma. J. Formos. Med. Assoc. 2020. [Google Scholar] [CrossRef]
- Wagner, V.; Gil, J. Senescence as a therapeutically relevant response to CDK4/6 inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the cancer microenvironment as a target for therapeutic interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [Green Version]
- Brückner, B.R.; Nöding, H.; Skamrahl, M.; Janshoff, A. Mechanical and morphological response of confluent epithelial cell layers to reinforcement and dissolution of the F-actin cytoskeleton. Prog. Biophys. Mol. Biol. 2019, 144, 77–90. [Google Scholar] [CrossRef]
- Shannon, S.; Jia, D.; Entersz, I.; Beelen, P.; Yu, M.; Carcione, C.; Carcione, J.; Mahtabfar, A.; Vaca, C.; Weaver, M.; et al. Inhibition of glioblastoma dispersal by the MEK inhibitor PD0325901. BMC Cancer 2017, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Yaromina, A.; Zips, D.; Thames, H.D.; Eicheler, W.; Krause, M.; Rosner, A.; Haase, M.; Petersen, C.; Raleigh, J.A.; Quennet, V.; et al. Pimonidazole labelling and response to fractionated irradiation of five human squamous cell carcinoma (hSCC) lines in nude mice: The need for a multivariate approach in biomarker studies. Radiother. Oncol. 2006, 81, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.; Paluch, E.K. The actin cortex at a glance. J. Cell Sci. 2018, 131, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Ann. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-filho, O.; Roberts, T.M.; Kim, H.; Mcallister, S.S.; Jean, J. CDK4/6 inhibition triggers anti-tumor immunity. Nature 2017, 548, 471–475. [Google Scholar]
- Wang, J.; Yang, T.; Xu, G.; Liu, H.; Ren, C.; Xie, W.; Wang, M. Cyclin-dependent kinase 2 promotes tumor proliferation and induces radio resistance in glioblastoma. Transl. Oncol. 2016, 9, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Zheng, W.; Chen, T.; Lin, W.; Liao, Z.; Liu, J.; Ding, Y. CDK4/6 inhibitor palbociclib amplifies the radiosensitivity to nasopharyngeal carcinoma cells via mediating apoptosis and suppressing DNA damage repair. Onco. Targets. Ther. 2019, 12, 11107–11117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Düster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019, 26, 792–803.e10. [Google Scholar] [CrossRef]
- Veo, B.; Danis, E.; Pierce, A.; Wang, D.; Fosmire, S.; Sullivan, K.D.; Joshi, M.; Khanal, S.; Dahl, N.; Karam, S.; et al. Transcriptional control of DNA repair networks by CDK7 regulates sensitivity to radiation in Myc-driven medulloblastoma. Cell Rep. 2021, 35, 109013. [Google Scholar] [CrossRef]
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019, 21, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [Green Version]
- Maskey, R.S.; Wang, F.; Lehman, E.; Wang, Y.; Emmanuel, N.; Zhong, W.; Jin, G.; Abraham, R.T.; Arndt, K.T.; Myers, J.S.; et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle 2020, 20, 65–80. [Google Scholar] [CrossRef]
- Yu, Y.; Liao, H.; Xie, R.; Zhang, Y.; Zheng, R.; Chen, J.; Zhang, B. Overexpression of miRNA-3613-3p Enhances the Sensitivity of Triple Negative Breast Cancer to CDK4/6 Inhibitor Palbociclib. Front. Oncol. 2020, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin e negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-Fluorouracil on Morphology, Cell Cycle, Proliferation, Apoptosis, Autophagy and ROS Production in Endothelial Cells and Cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef]
- Tokunaga, E.; Oda, S.; Fukushima, M.; Maehara, Y.; Sugimachi, K. Differential growth inhibition by 5-fluorouracil in human colorectal carcinoma cell lines. Eur. J. Cancer 2000, 36, 1998–2006. [Google Scholar] [CrossRef]
- Schachter, M.M.; Fisher, R.P. The CDK-activating kinase Cdk7. Cell Cycle 2013, 12, 3239–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, H.; Suzuki-Takahashi, I.; Saitoh, S.; Segawa, K.; Taya, Y.; Okuyama, A.; Nishimura, S.; Kitagawa, M. Cyclin-dependent kinase-2 (Cdk2) forms an inactive complex with cyclin D1 since Cdk2 associated with cyclin D1 is not phosphorylated by Cdk7-cyclin-H. Eur. J. Biochem. 1996, 237, 460–467. [Google Scholar] [CrossRef]
- Riess, C.; Koczan, D.; Schneider, B.; Linke, C.; del Moral, K.; Classen, C.F.; Maletzki, C. Cyclin-dependent kinase inhibitors exert distinct effects on patient-derived 2D and 3D glioblastoma cell culture models. Cell Death Discov. 2021, 7, 54. [Google Scholar] [CrossRef]
- Maltese, W.A.; Overmeyer, J.H. Methuosis: Nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 2014, 184, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.X.; Xie, F.F.; Zhu, X.J.; Lin, F.; Pan, S.S.; Gong, L.H.; Qiu, J.G.; Zhang, W.J.; Jiang, Q.W.; Mei, X.L.; et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget 2015, 6, 14926–14939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoenwaelder, N.; Salewski, I.; Engel, N.; Krause, M.; Schneider, B.; Müller, M.; Riess, C.; Lemcke, H.; Skorska, A.; Grosse-Thie, C.; et al. The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis. Cancers 2021, 13, 2396. https://doi.org/10.3390/cancers13102396
Schoenwaelder N, Salewski I, Engel N, Krause M, Schneider B, Müller M, Riess C, Lemcke H, Skorska A, Grosse-Thie C, et al. The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis. Cancers. 2021; 13(10):2396. https://doi.org/10.3390/cancers13102396
Chicago/Turabian StyleSchoenwaelder, Nina, Inken Salewski, Nadja Engel, Mareike Krause, Björn Schneider, Michael Müller, Christin Riess, Heiko Lemcke, Anna Skorska, Christina Grosse-Thie, and et al. 2021. "The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis" Cancers 13, no. 10: 2396. https://doi.org/10.3390/cancers13102396