
Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains—Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
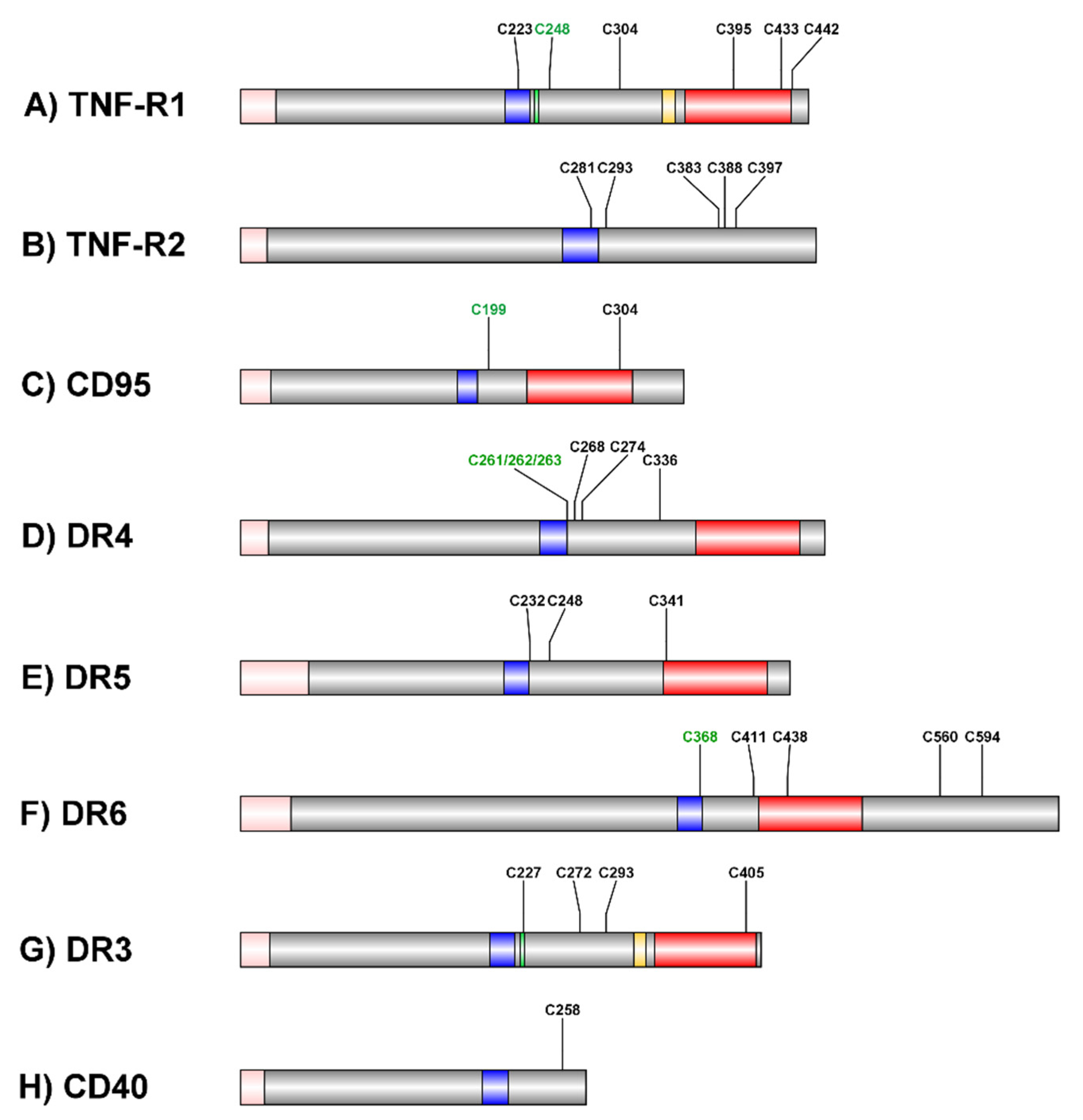
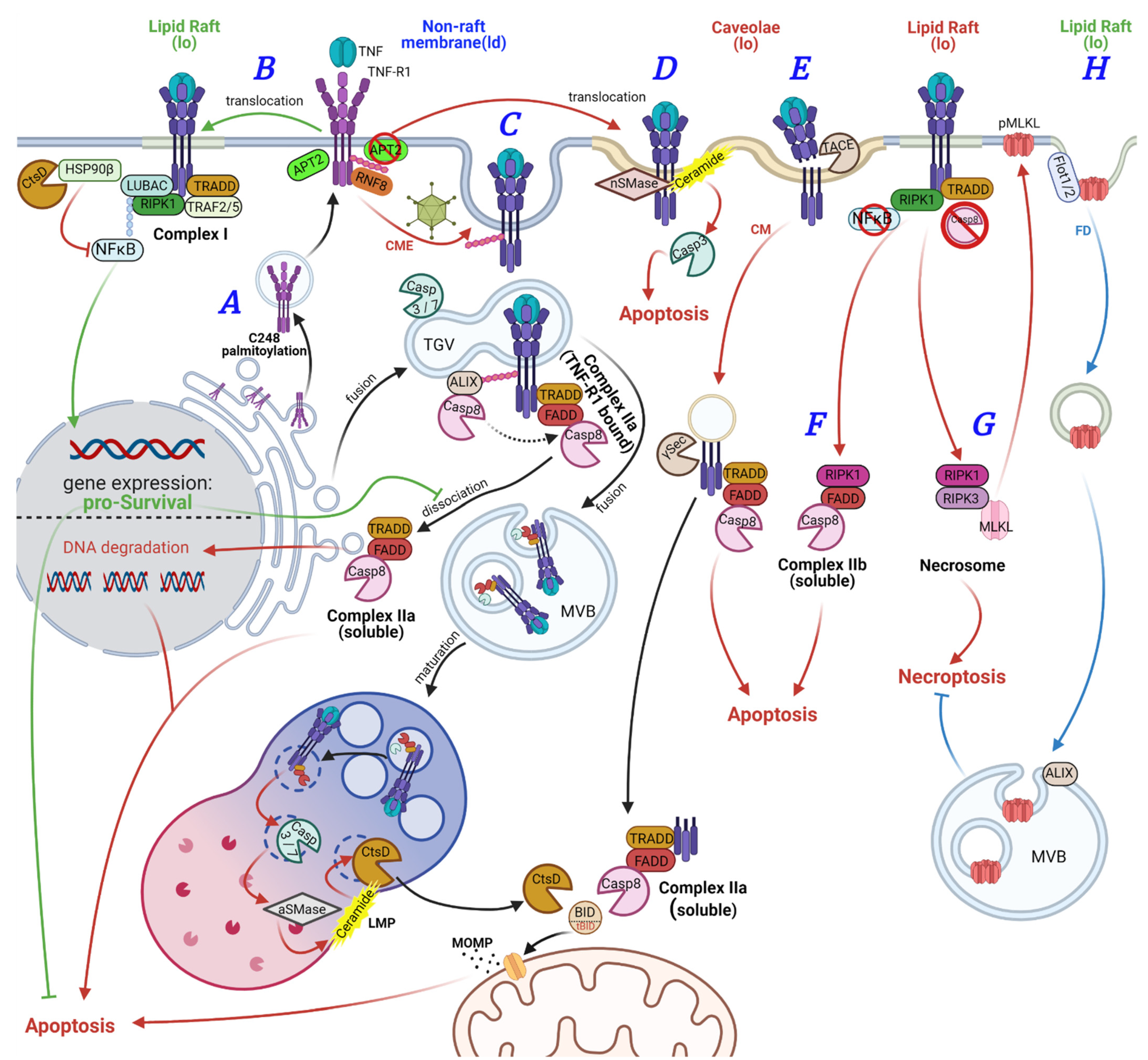
2. Roles of Palmitoylation and Lipid Rafts in TNF-R1 Signaling
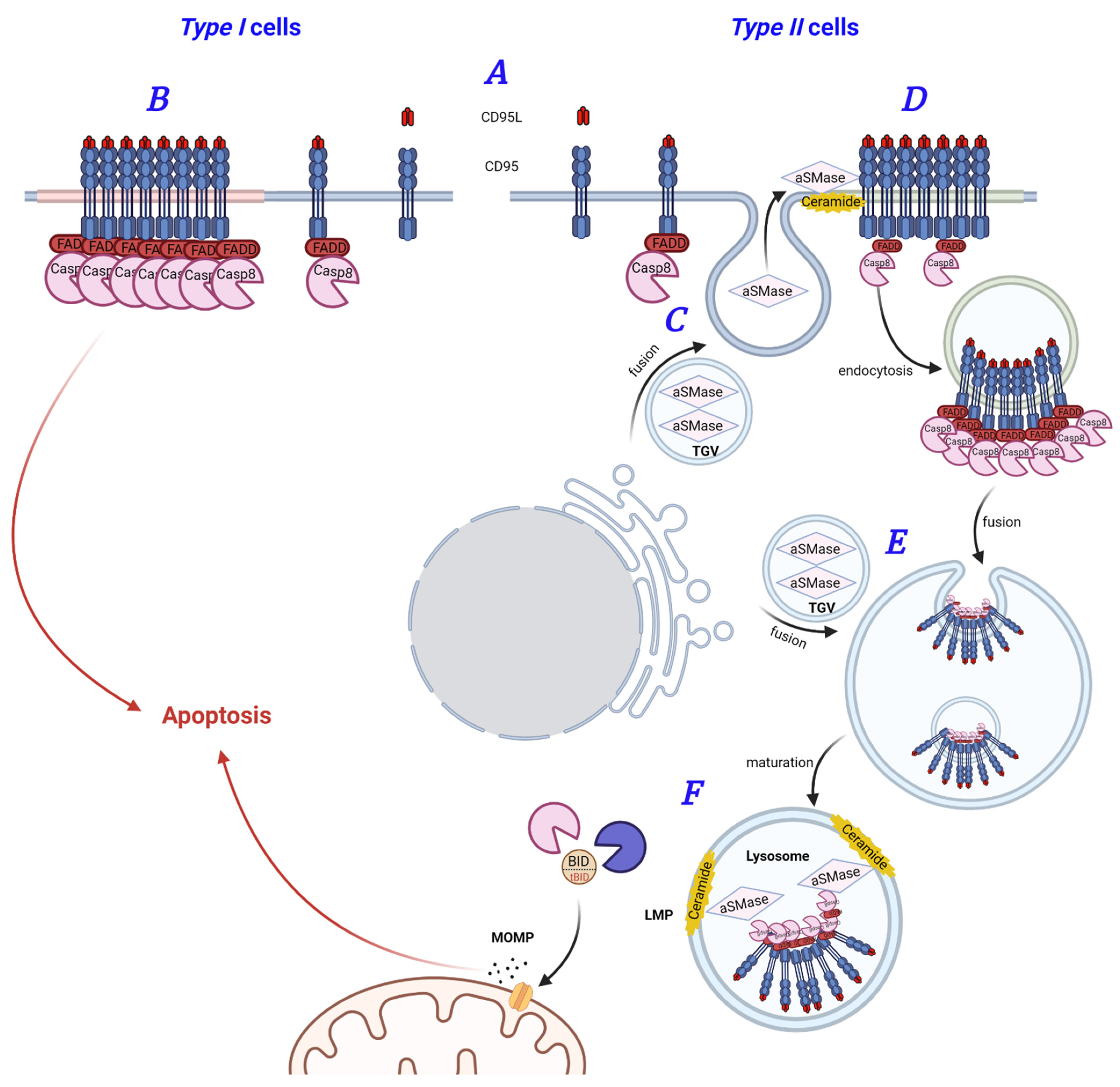
3. Roles of Palmitoylation and Lipid Rafts in CD95 Signaling
4. Roles of Palmitoylation and Lipid Rafts in TRAIL-R Signaling
5. Palmitoylation in DR6 Signaling
6. Palmitoylation and Lipid Raft Association of Other Proteins in Death-Receptor-Mediated Signal Transduction
7. Death Receptor Signaling from the Nucleus
8. Myristoylation in Cell Death
9. Alterations in the Lipid Composition of Membranes—Opportunities for Clinical Exploitation
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Goldschneider, D.; Mehlen, P. Dependence receptors: A new paradigm in cell signaling and cancer therapy. Oncogene 2010, 29, 1865–1882. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Fritsch, J.; Zingler, P.; Särchen, V.; Heck, A.L.; Schütze, S. Role of ubiquitination and proteolysis in the regulation of pro- and anti-apoptotic TNF-R1 signaling. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 2138–2146. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, D.; Lang, I.; Wajant, H. Cell death-independent activities of the death receptorsCD95, TRAILR1, and TRAILR2. FEBS J. 2016, 284, 1131–1159. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, A.; Zahedi, R.P.; Ahrends, R. Protein lipid modifications-More than just a greasy ballast. Proteomics 2016, 16, 759–782. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The ins and outs of lipid rafts: Functions in intracellular cholesterol homeostasis, microparticles, and cell membranes. J. Lipid Res. 2020, 61, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.A.; Kenworthy, A.K. Functions of cholera toxin B-subunit as a raft cross-linker. Essays Biochem. 2015, 57, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Muszbek, L.; Haramura, G.; Cluette-Brown, J.E.; Van Cott, E.M.; Laposata, M. The pool of fatty acids covalently bound to platelet proteins by thioester linkages can be altered by exogenously supplied fatty acids. Lipids 1999, 34, S331–S337. [Google Scholar] [CrossRef]
- Greaves, J.; Munro, K.R.; Davidson, S.C.; Riviere, M.; Wojno, J.; Smith, T.K.; Tomkinson, N.; Chamberlain, L.H. Molecular basis of fatty acid selectivity in the zDHHC family of S-acyltransferases revealed by click chemistry. Proc. Natl. Acad. Sci. USA 2017, 114, E1365–E1374. [Google Scholar] [CrossRef] [Green Version]
- Won, S.J.; Kit, M.C.S.; Martin, B.R. Protein depalmitoylases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 83–98. [Google Scholar] [CrossRef]
- Tabaczar, S.; Czogalla, A.; Podkalicka, J.; Biernatowska, A.; Sikorski, A.F. Protein palmitoylation: Palmitoyltransferases and their specificity. Exp. Biol. Med. 2017, 242, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, L.H.; Shipston, M.J. The Physiology of Protein S-acylation. Physiol. Rev. 2015, 95, 341–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Fang, C. Methodology for Detecting Protein Palmitoylation. Adv. Exp. Med. Biol. 2020, 1248, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Stevens, T.; Munro, S. A Comprehensive Comparison of Transmembrane Domains Reveals Organelle-Specific Properties. Cell 2010, 142, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Ubarretxena-Belandia, I.; Taguchi, T.; Warren, G.; Engelman, D.M. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc. Natl. Acad. Sci. USA 2004, 101, 4083–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, S.D.; Smith, D.A. The atomic force microscope as a tool for studying phase separation in lipid membranes (Review). Mol. Membr. Biol. 2006, 23, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Cornell, C.E.; Mileant, A.; Thakkar, N.; Lee, K.K.; Keller, S.L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2020, 117, 19713–19719. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Moss, M.L.; Jin, S.-L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Utsumi, T.; Takeshige, T.; Tanaka, K.; Takami, K.; Kira, Y.; Klostergaard, J.; Ishisaka, R. Transmembrane TNF (pro-TNF) is palmitoylated. FEBS Lett. 2001, 500, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Poggi, M.; Kara, I.; Brunel, J.-M.; Landrier, J.-F.; Govers, R.; Bonardo, B.; Fluhrer, R.; Haass, C.; Alessi, M.-C.; Peiretti, F. Palmitoylation of TNF alpha is involved in the regulation of TNF receptor 1 signalling. Biochim. Biophys. Acta Bioenerg. 2013, 1833, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, T.; Liang, H.; Zhang, H.; Yan, D.; Wang, N.; Jiang, X.; Feng, W.; Wang, J.; Li, P.; et al. Lipid rafts uncouple surface expression of transmembrane TNF-α from its cytotoxicity associated with ICAM-1 clustering in Raji cells. Mol. Immunol. 2009, 46, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, M.; Hu, X.; Han, L.; Yang, K.; Ba, H.; Zhang, Z.; Yin, B.; Yang, X.-P.; Li, Z.; et al. STAT1 mediates transmembrane TNF-alpha-induced formation of death-inducing signaling complex and apoptotic signaling via TNFR1. Cell Death Differ. 2017, 24, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Zingler, P.; Särchen, V.; Glatter, T.; Caning, L.; Saggau, C.; Kathayat, R.S.; Dickinson, B.C.; Adam, D.; Schneider-Brachert, W.; Schütze, S.; et al. Palmitoylation is required for TNF-R1 signaling. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Al-Lamki, R.S.; Zhang, H.; Kirkiles-Smith, N.; Gaeta, M.L.; Thiru, S.; Pober, J.S.; Bradley, J.R. Histamine Antagonizes Tumor Necrosis Factor (TNF) Signaling by Stimulating TNF Receptor Shedding from the Cell Surface and Golgi Storage Pool. J. Biol. Chem. 2003, 278, 21751–21760. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.J.; Ledgerwood, E.; Prins, J.B.; Galbraith, J.; Johnson, D.R.; Pober, J.S.; Bradley, J.R. TNF recruits TRADD to the plasma membrane but not the trans-Golgi network, the principal subcellular location of TNF-R1. J. Immunol. 1999, 162, 1042–1048. [Google Scholar]
- Gaeta, M.L.; Johnson, D.R.; Kluger, M.S.; Pober, J.S. The Death Domain of Tumor Necrosis Factor Receptor 1 Is Necessary but Not Sufficient for Golgi Retention of the Receptor and Mediates Receptor Desensitization. Lab. Investig. 2000, 80, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Gaeta, M.L.; Madge, L.A.; Yang, J.-H.; Bradley, J.R.; Pober, J.S. Caveolin-1 Associates with TRAF2 to Form a Complex That Is Recruited to Tumor Necrosis Factor Receptors. J. Biol. Chem. 2001, 276, 8341–8349. [Google Scholar] [CrossRef] [Green Version]
- Deberge, M.P.; Ely, K.H.; Wright, P.F.; Thorp, E.; Enelow, R.I. Shedding of TNF receptor 2 by effector CD8+T cells by ADAM17 is important for regulating TNF-α availability during influenza infection. J. Leukoc. Biol. 2015, 98, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.; Reading, S.J.; Jones, S.; Fitchett, C.J.; Howl, J.; Martin, A.; Longland, C.L.; Michelangeli, F.; Dubrova, Y.E.; Brown, C.A. Critical evaluation of ECV304 as a human endothelial cell model defined by genetic analysis and functional responses: A comparison with the human bladder cancer derived epithelial cell line T24/83. Lab. Investig. 2000, 80, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Doan, J.E.S.; Riches, D.W.H. Restricted Localization of the TNF Receptor CD120a to Lipid Rafts: A Novel Role for the Death Domain. J. Immunol. 2002, 168, 4095–4102. [Google Scholar] [CrossRef] [Green Version]
- Legler, D.F.; Micheau, O.; Doucey, M.A.; Tschopp, J.; Bron, C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003, 18, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Lotocki, G.; Alonso, O.F.; Dietrich, W.D.; Keane, R.W. Tumor Necrosis Factor Receptor 1 and Its Signaling Intermediates Are Recruited to Lipid Rafts in the Traumatized Brain. J. Neurosci. 2004, 24, 11010–11016. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, J.; Stephan, M.; Tchikov, V.; Winoto-Morbach, S.; Gubkina, S.; Kabelitz, D.; Schütze, S. Cell Fate Decisions Regulated by K63 Ubiquitination of Tumor Necrosis Factor Receptor. Mol. Cell. Biol. 2014, 34, 3214–3228. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Zhang, D.; Song, Y.; Liu, S.; Long, Q.; Yao, L.; Li, W.; Duan, Z.; Wu, D.; Liu, L. HRG switches TNFR1-mediated cell survival to apoptosis in Hepatocellular Carcinoma. Theranostics 2020, 10, 10434–10447. [Google Scholar] [CrossRef]
- Gould, N.S.; Evans, P.; Martínez-Acedo, P.; Marino, S.M.; Gladyshev, V.N.; Carroll, K.S.; Ischiropoulos, H. Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chem. Biol. 2015, 22, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; De Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Dreschers, S.; Gille, C.; Haas, M.; Grosse-Ophoff, J.; Schneider, M.; Leiber, A.; Bühring, H.-J.; Orlikowsky, T.W. Infection–induced Bystander-Apoptosis of Monocytes Is TNF-alpha-mediated. PLoS ONE 2013, 8, e53589. [Google Scholar] [CrossRef] [PubMed]
- Dreschers, S.; Gille, C.; Haas, M.; Seubert, F.; Platen, C.; Orlikowsky, T.W. Reduced internalization of TNF-ɑ/TNFR1 down-regulates caspase dependent phagocytosis induced cell death (PICD) in neonatal monocytes. PLoS ONE 2017, 12, e0182415. [Google Scholar] [CrossRef] [Green Version]
- Kontchou, C.W.; Tzivelekidis, T.; Gentle, I.E.; Häcker, G. Infection of epithelial cells with Chlamydia trachomatis inhibits TNF-induced apoptosis at the level of receptor internalisation while leaving non-apoptotic TNF-signalling intact. Cell. Microbiol. 2016, 18, 1583–1595. [Google Scholar] [CrossRef]
- Imre, G. The involvement of regulated cell death forms in modulating the bacterial and viral pathogenesis. Int. Rev. Cell Mol. Biol. 2020, 353, 211–253. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Brachert, W.; Tchikov, V.; Merkel, O.; Jakob, M.; Hallas, C.; Kruse, M.-L.; Groitl, P.; Lehn, A.; Hildt, E.; Held-Feindt, J.; et al. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J. Clin. Investig. 2006, 116, 2901–2913. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Meng, J.; Cheng, J.; Fan, Z.; Chen, P.; Ruan, H.; Tu, Z.; Kang, N.; Li, N.; Xu, Y.; et al. Pseudomonas aeruginosa quorum-sensing metabolite induces host immune cell death through cell surface lipid domain dissolution. Nat. Microbiol. 2019, 4, 97–111. [Google Scholar] [CrossRef]
- Ko, Y.G.; Lee, J.S.; Kang, Y.S.; Ahn, J.H.; Seo, J.S. TNF-alpha-mediated apoptosis is initiated in caveolae-like domains. J. Immunol. 1999, 162, 7217–7223. [Google Scholar]
- Omran, O.M.; Saqr, H.E.; Yates, A.J. Molecular Mechanisms of GD3-Induced Apoptosis in U-1242 MG Glioma Cells. Neurochem. Res. 2006, 31, 1171–1180. [Google Scholar] [CrossRef]
- Saqr, H.E.; Omran, O.M.; Oblinger, J.L.; Yates, A.J. TRAIL-Induced Apoptosis in U-1242 MG Glioma Cells. J. Neuropathol. Exp. Neurol. 2006, 65, 152–161. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, A.; Kluger, M.S.; Li, J.H.; Al-Lamki, R.; Bradley, J.R.; Pober, J.S. Targeting of Tumor Necrosis Factor Receptor 1 to Low Density Plasma Membrane Domains in Human Endothelial Cells. J. Biol. Chem. 2010, 285, 23868–23879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhibber-Goel, J.; Coleman-Vaughan, C.; Agrawal, V.; Sawhney, N.; Hickey, E.; Powell, J.C.; McCarthy, J.V. γ-Secretase Activity Is Required for Regulated Intramembrane Proteolysis of Tumor Necrosis Factor (TNF) Receptor 1 and TNF-mediated Pro-apoptotic Signaling. J. Biol. Chem. 2016, 291, 5971–5985. [Google Scholar] [CrossRef] [Green Version]
- Schneider-Brachert, W.; Tchikov, V.; Neumeyer, J.; Jakob, M.; Winoto-Morbach, S.; Held-Feindt, J.; Heinrich, M.; Merkel, O.; Ehrenschwender, M.; Adam, D.; et al. Compartmentalization of TNF Receptor 1 Signaling: Internalized TNF Receptosomes as Death Signaling Vesicles. Immunity 2004, 21, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Schütze, S.; Machleidt, T.; Adam, D.; Schwandner, R.; Wiegmann, K.; Kruse, M.-L.; Heinrich, M.; Wickel, M.; Krönke, M. Inhibition of Receptor Internalization by Monodansylcadaverine Selectively Blocks p55 Tumor Necrosis Factor Receptor Death Domain Signaling. J. Biol. Chem. 1999, 274, 10203–10212. [Google Scholar] [CrossRef] [Green Version]
- Sosna, J.; Philipp, S.; Chico, J.F.; Saggau, C.; Fritsch, J.; Föll, A.; Plenge, J.; Arenz, C.; Pinkert, T.; Kalthoff, H.; et al. Differences and Similarities in TRAIL- and Tumor Necrosis Factor-Mediated Necroptotic Signaling in Cancer Cells. Mol. Cell. Biol. 2016, 36, 2626–2644. [Google Scholar] [CrossRef] [Green Version]
- Holdbrooks, A.T.; Britain, C.M.; Bellis, S.L. ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 2018, 293, 1610–1622. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Swindall, A.F.; Kesterson, R.A.; Schoeb, T.R.; Bullard, D.C.; Bellis, S.L. ST6Gal-I Regulates Macrophage Apoptosis via α2-6 Sialylation of the TNFR1 Death Receptor. J. Biol. Chem. 2011, 286, 39654–39662. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Fritsch, J.; Zigdon, H.; Pewzner-Jung, Y.; Schütze, S.; Futerman, A.H. Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis. 2013, 4, e929. [Google Scholar] [CrossRef]
- Swindall, A.F.; Bellis, S.L. Sialylation of the Fas Death Receptor by ST6Gal-I Provides Protection against Fas-mediated Apoptosis in Colon Carcinoma Cells. J. Biol. Chem. 2011, 286, 22982–22990. [Google Scholar] [CrossRef] [Green Version]
- Martinez, T.N.; Chen, X.; Bandyopadhyay, S.; Merrill, A.H.; Tansey, M.G. Ceramide sphingolipid signaling mediates Tumor Necrosis Factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol. Neurodegener. 2012, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Airola, M.V.; Hannun, Y.A. Sphingolipid Metabolism and Neutral Sphingomyelinases. Organotypic Models Drug Dev. 2013, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Philipp, S.; Puchert, M.; Adam-Klages, S.; Tchikov, V.; Winoto-Morbach, S.; Mathieu, S.; Deerberg, A.; Kolker, L.; Marchesini, N.; Kabelitz, D.; et al. The Polycomb group protein EED couples TNF receptor 1 to neutral sphingomyelinase. Proc. Natl. Acad. Sci. USA 2010, 107, 1112–1117. [Google Scholar] [CrossRef] [Green Version]
- Adam-Klages, S.; Adam, D.; Wiegmann, K.; Struve, S.; Kolanus, W.; Schneider-Mergener, J.; Krönke, M. FAN, a Novel WD-Repeat Protein, Couples the p55 TNF-Receptor to Neutral Sphingomyelinase. Cell 1996, 86, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Guthrie, J.M.; Hannun, Y.A. Regulation of Neutral Sphingomyelinase-2 (nSMase2) by Tumor Necrosis Factor-α Involves Protein Kinase C-δ in Lung Epithelial Cells. Mol. Pharmacology 2008, 74, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Truong, T.-G.; Hannun, Y.A. Role for Neutral Sphingomyelinase-2 in Tumor Necrosis Factor α-Stimulated Expression of Vascular Cell Adhesion Molecule-1 (VCAM) and Intercellular Adhesion Molecule-1 (ICAM) in Lung Epithelial Cells. J. Biol. Chem. 2007, 282, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Neumeyer, J.; Hallas, C.; Merkel, O.; Winoto-Morbach, S.; Jakob, M.; Thon, L.; Adam, D.; Schneider-Brachert, W.; Schütze, S. TNF-receptor I defective in internalization allows for cell death through activation of neutral sphingomyelinase. Exp. Cell Res. 2006, 312, 2142–2153. [Google Scholar] [CrossRef]
- Dargelos, E.; Renaud, V.; Decossas, M.; Bure, C.; Lambert, O.; Poussard, S. Caveolae-mediated effects of TNF-α on human skeletal muscle cells. Exp. Cell Res. 2018, 370, 623–631. [Google Scholar] [CrossRef]
- Yazdanpanah, B.; Wiegmann, K.; Tchikov, V.; Krut, O.; Pongratz, C.; Schramm, M.; Kleinridders, A.; Wunderlich, T.; Kashkar, H.; Utermöhlen, O.; et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nat. Cell Biol. 2009, 460, 1159–1163. [Google Scholar] [CrossRef]
- Tani, M.; Hannun, Y.A. Neutral Sphingomyelinase 2 Is Palmitoylated on Multiple Cysteine Residues. J. Biol. Chem. 2007, 282, 10047–10056. [Google Scholar] [CrossRef] [Green Version]
- Tani, M.; Kuge, O. Sphingomyelin synthase 2 is palmitoylated at the COOH-terminal tail, which is involved in its localization in plasma membranes. Biochem. Biophys. Res. Commun. 2009, 381, 328–332. [Google Scholar] [CrossRef]
- Goswami, R.; Ahmed, M.; Kilkus, J.; Han, T.; Dawson, S.; Dawson, G. Differential regulation of ceramide in lipid-rich microdomains (rafts): Antagonistic role of palmitoyl:protein thioesterase and neutral sphingomyelinase. J. Neurosci. Res. 2005, 81, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Sabourdy, F.; Garcia, V.; Jalanko, A.; Therville, N.; Levade, T.; Andrieu-Abadie, N. Palmitoyl protein thioesterase 1 modulates tumor necrosis factor α-induced apoptosis. Biochim. Biophys. Acta Bioenerg. 2009, 1793, 1250–1258. [Google Scholar] [CrossRef]
- Heck, A.; Mishra, S.; Prenzel, T.; Feulner, L.; Achhammer, E.; Särchen, V.; Blagg, B.; Schneider-Brachert, W.; Schütze, S.; Fritsch, J. Selective HSP90β inhibition results in TNF and TRAIL mediated HIF1α degradation. Immunobiology 2021, 226, 152070. [Google Scholar] [CrossRef]
- Li, R.; Li, K.; Yang, Y.; Sun, P.-B.; Chen, A.-J.; Ni, Y. Palmitoylation of heat shock protein 90 in mouse sperm. Sheng Li Xue Bao 2017, 69, 298–304. [Google Scholar]
- Chandra, G.; Bagh, M.B.; Peng, S.; Saha, A.; Sarkar, C.; Moralle, M.; Zhang, Z.; Mukherjee, A.B. Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum. Mol. Genet. 2015, 24, 5416–5432. [Google Scholar] [CrossRef] [Green Version]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Zheng, H.; Zhao, C.; Soherwardy, A.; Moore, D.F.; Lobel, P. Analysis of Brain and Cerebrospinal Fluid from Mouse Models of the Three Major Forms of Neuronal Ceroid Lipofuscinosis Reveals Changes in the Lysosomal Proteome. Mol. Cell. Proteom. 2019, 18, 2244–2261. [Google Scholar] [CrossRef]
- Gorenberg, E.L.; Zhao, H.R.; Bishai, J.; Chou, V.; Wirak, G.S.; Lam, T.T.; Chandra, S.S. Identification of palmitoyl protein thioesterase 1 substrates defines roles for synaptic depalmitoylation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Segal-Salto, M.; Sapir, T.; Reiner, O. Reversible Cysteine Acylation Regulates the Activity of Human Palmitoyl-Protein Thioesterase 1 (PPT1). PLoS ONE 2016, 11, e0146466. [Google Scholar] [CrossRef] [Green Version]
- Krut, O.; Wiegmann, K.; Kashkar, H.; Yazdanpanah, B.; Krönke, M. Novel Tumor Necrosis Factor-responsive Mammalian Neutral Sphingomyelinase-3 Is a C-tail-anchored Protein. J. Biol. Chem. 2006, 281, 13784–13793. [Google Scholar] [CrossRef] [Green Version]
- Moylan, J.S.; Smith, J.D.; Horrell, E.M.W.; McLean, J.B.; Deevska, G.M.; Bonnell, M.R.; Nikolova-Karakashian, M.N.; Reid, M.B. Neutral sphingomyelinase-3 mediates TNF-stimulated oxidant activity in skeletal muscle. Redox Biol. 2014, 2, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Düppe, M.; Baumecker, L.; Kordowski, F.; Büch, J.; Chico, J.F.; Fritsch, J.; Schütze, S.; Adam, D.; Sperrhacke, M.; et al. Extracellular sphingomyelinase activity impairs TNF-alpha-indiced endothelial cell death via ADAM17 activation and TNF receptor 1 shedding. Oncotarget 2017, 8, 72584. [Google Scholar] [CrossRef] [Green Version]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Esposito, B.; Giampietri, C.; Ziparo, E.; Pober, J.S.; Filippini, A. Plasma membrane microdomains regulate TACE-dependent TNFR1 shedding in human endothelial cells. J. Cell. Mol. Med. 2011, 16, 626–635. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Caveolae Participate in Tumor Necrosis Factor Receptor 1 Signaling and Internalization in a Human Endothelial Cell Line. Am. J. Pathol. 2005, 166, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Doan, J.E.S.; Windmiller, D.A.; Riches, D.W.H. Differential Regulation of TNF-R1 Signaling: Lipid Raft Dependency of p42mapk/erk2 Activation, but Not NF-κB Activation. J. Immunol. 2004, 172, 7654–7660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, I.; Nixon, G.F. Spatial Compartmentalization of Tumor Necrosis Factor (TNF) Receptor 1-dependent Signaling Pathways in Human Airway Smooth Muscle Cells. J. Biol. Chem. 2006, 281, 34705–34715. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, B.P.; Chern, Y.; He, J.; Chan, C. The ubiquitin ligase RNF8 regulates Rho GTPases and promotes cytoskeletal changes and motility in triple-negative breast cancer cells. FEBS Lett. 2021, 595, 241–252. [Google Scholar] [CrossRef]
- Von Mässenhausen, A.; Tonnus, W.; Himmerkus, N.; Parmentier, S.; Saleh, D.; Rodriguez, D.; Ousingsawat, J.; Ang, R.L.; Weinberg, J.M.; Sanz, A.B.; et al. Phenytoin inhibits necroptosis. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Moerke, C.; Jaco, I.; Dewitz, C.; Müller, T.; Jacobsen, A.V.; Gautheron, J.; Fritsch, J.; Schmitz, J.; Bräsen, J.H.; Günther, C.; et al. The anticonvulsive Phenhydan® suppresses extrinsic cell death. Cell Death Differ. 2018, 26, 1631–1645. [Google Scholar] [CrossRef]
- Ali, M.; Roback, L.; Mocarski, E.S. Herpes simplex virus 1 ICP6 impedes TNF receptor 1–induced necrosome assembly during compartmentalization to detergent-resistant membrane vesicles. J. Biol. Chem. 2019, 294, 991–1004. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Guo, J.; Gao, B.; Zhang, W.; Ling, L.; Xu, T.; Pan, C.; Li, L.; Chen, S.; Wang, H.; et al. Flotillin-mediated endocytosis and ALIX–syntenin-1–mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci. Signal. 2019, 12, eaaw3423. [Google Scholar] [CrossRef]
- Romancino, D.P.; Buffa, V.; Caruso, S.; Ferrara, I.; Raccosta, S.; Notaro, A.; Campos, Y.; Noto, R.; Martorana, V.; Cupane, A.; et al. Palmitoylation is a post-translational modification of Alix regulating the membrane organization of exosome-like small extracellular vesicles. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nat. Cell Biol. 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Le Gallo, M.; Poissonnier, A.; Blanco, P.; Legembre, P. CD95/Fas, Non-Apoptotic Signaling Pathways, and Kinases. Front. Immunol. 2017, 8, 1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardiola-Serrano, F.; Rossin, A.; Cahuzac, N.; Lückerath, K.; Melzer, I.; Mailfert, S.; Marguet, D.; Zörnig, M.; Hueber, A.-O. Palmitoylation of human FasL modulates its cell death-inducing function. Cell Death Dis. 2010, 1, e88. [Google Scholar] [CrossRef] [Green Version]
- Ebsen, H.; Lettau, M.; Kabelitz, D.; Janssen, O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Mol. Immunol. 2015, 65, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Tchikov, V.; Schütze, S.; Peter, M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2006, 26, 221–231. [Google Scholar] [CrossRef]
- Chakrabandhu, K.; Hérincs, Z.; Huault, S.; Dost, B.; Peng, L.; Conchonaud, F.; Marguet, D.; He, H.-T.; Hueber, A.-O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 2006, 26, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Sargiacomo, M.; Ricci-Vitiani, L.; Todaro, M.; Stassi, G.; Messina, C.G.M.; Parolini, I.; Lotti, F.; Sette, G.; Peschle, C.; et al. CD95 death-inducing signaling complex formation and internalization occur in lipid rafts of type I and type II cells. Eur. J. Immunol. 2004, 34, 1930–1940. [Google Scholar] [CrossRef]
- Cruz, A.C.; Ramaswamy, M.; Ouyang, C.; Klebanoff, C.A.; Sengupta, P.; Yamamoto, T.N.; Meylan, F.; Thomas, S.K.; Richoz, N.; Eil, R.; et al. Fas/CD95 prevents autoimmunity independently of lipid raft localization and efficient apoptosis induction. Nat. Commun. 2016, 7, 13895. [Google Scholar] [CrossRef]
- Rossin, A.; Durivault, J.; Chakhtoura-Feghali, T.; Lounnas, N.; Gagnoux-Palacios, L.; Hueber, A.-O. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates Fas stability. Cell Death Differ. 2015, 22, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Eisinger, K.R.T.; Woolfrey, K.M.; Swanson, S.P.; Schnell, S.A.; Meitzen, J.; Dell’Acqua, M.; Mermelstein, P.G. Palmitoylation of caveolin-1 is regulated by the same DHHC acyltransferases that modify steroid hormone receptors. J. Biol. Chem. 2018, 293, 15901–15911. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.M.; de Almeida, R.; Goormaghtigh, E.; Fedorov, A.; Prieto, M. Organization and Dynamics of Fas Transmembrane Domain in Raft Membranes and Modulation by Ceramide. Biophys. J. 2011, 101, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Haynes, A.P.; Daniels, I.; Abhulayha, A.M.; Carter, G.I.; Metheringham, R.; Gregory, C.D.; Thomson, B.J. CD95 (Fas) expression is regulated by sequestration in the Golgi complex in B-cell lymphoma. Br. J. Haematol. 2002, 118, 488–494. [Google Scholar] [CrossRef]
- Lincoln, J.E.; Boling, M.; Yeh, Y.; Gilchrist, D.G.; Morse, L.S.; Parikh, A.N. Fas Signaling Induces Raft Coalescence That Is Blocked by Cholesterol Depletion in Human RPE Cells Undergoing Apoptosis. Investig. Opthalmology Vis. Sci. 2006, 47, 2172–2178. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.-L. Lipid Raft Clustering and Redox Signaling Platform Formation in Coronary Arterial Endothelial Cells. Hypertension. 2005, 47, 74–80. [Google Scholar] [CrossRef]
- Jin, S.; Yi, F.; Li, P.-L. Contribution of Lysosomal Vesicles to the Formation of Lipid Raft Redox Signaling Platforms in Endothelial Cells. Antioxid. Redox Signal. 2007, 9, 1417–1426. [Google Scholar] [CrossRef]
- Stephan, M.; Edelmann, B.; Winoto-Morbach, S.; Janssen, O.; Bertsch, U.; Perrotta, C.; Schütze, S.; Fritsch, J. Role of caspases in CD95-induced biphasic activation of acid sphingomyelinase. Oncotarget 2017, 8, 20067–20085. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gulbins, E.; Zhang, Y. Oxidative stress triggers Ca-dependent lysosome trafficking and activation of acid sphingomyelinase. Cell. Physiol. Biochem. 2012, 30, 815–826. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Cytoskeleton-mediated Death Receptor and Ligand Concentration in Lipid Rafts Forms Apoptosis-promoting Clusters in Cancer Chemotherapy. J. Biol. Chem. 2005, 280, 11641–11647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2006, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Involvement of Raft Aggregates Enriched in Fas/CD95 Death-Inducing Signaling Complex in the Antileukemic Action of Edelfosine in Jurkat Cells. PLoS ONE 2009, 4, e5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Futur. Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.-T. Cisplatin-Induced CD95 Redistribution into Membrane Lipid Rafts of HT29 Human Colon Cancer Cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, D.; Rébé, C.; Lacour, S.; Filomenko, R.; Athias, A.; Gambert, P.; Cherkaoui-Malki, M.; Jannin, B.; Dubrez-Daloz, L.; Latruffe, N.; et al. Resveratrol-induced Apoptosis Is Associated with Fas Redistribution in the Rafts and the Formation of a Death-inducing Signaling Complex in Colon Cancer Cells. J. Biol. Chem. 2003, 278, 41482–41490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmas, D.; Rébé, C.; Micheau, O.; Athias, A.; Gambert, P.; Grazide, S.; Laurent, G.; Latruffe, N.; Solary, E. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene 2004, 23, 8979–8986. [Google Scholar] [CrossRef] [Green Version]
- Reis-Sobreiro, M.; Gajate, C.; Mollinedo, F. Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions. Oncogene 2009, 28, 3221–3234. [Google Scholar] [CrossRef] [Green Version]
- Ewaschuk, J.B.; Newell, M.; Field, C.J. Docosahexanoic Acid Improves Chemotherapy Efficacy by Inducing CD95 Translocation to Lipid Rafts in ER− Breast Cancer Cells. Lipids 2012, 47, 1019–1030. [Google Scholar] [CrossRef]
- Lin, M.-L.; Chen, S.-S.; Wu, T.-S. Synthetic Bichalcone TSWU-BR23 Induces Apoptosis of Human Colon Cancer HT-29 Cells by p53-Mediated Mitochondrial Oligomerization of BAX/BAK and Lipid Raft Localization of CD95/FADD. Anticancer. Res. 2015, 35, 5407–5416. [Google Scholar] [PubMed]
- DeMorrow, S.; Glaser, S.; Francis, H.; Venter, J.; Vaculin, B.; Vaculin, S.; Alpini, G. Opposing Actions of Endocannabinoids on Cholangiocarcinoma Growth. J. Biol. Chem. 2007, 282, 13098–13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Kung, F.-L.; Tsai, I.-L.; Chou, T.-H.; Chen, I.-S.; Guh, J.-H. Cryptocaryone, a Natural Dihydrochalcone, Induces Apoptosis in Human Androgen Independent Prostate Cancer Cells by Death Receptor Clustering in Lipid Raft and Nonraft Compartments. J. Urol. 2010, 183, 2409–2418. [Google Scholar] [CrossRef]
- Gmeiner, W.H.; Jennings-Gee, J.; Stuart, C.H.; Pardee, T.S. Thymineless death in F10-treated AML cells occurs via lipid raft depletion and Fas/FasL co-localization in the plasma membrane with activation of the extrinsic apoptotic pathway. Leuk. Res. 2015, 39, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.-S.; Choo, H.-J.; Cho, B.-R.; Kim, H.-M.; Kim, Y.-N.; Ham, Y.-M.; Ko, Y.-G. Ginsenoside Rh2 induces ligand-independent Fas activation via lipid raft disruption. Biochem. Biophys. Res. Commun. 2009, 385, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Gniadecki, R. Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem. Biophys. Res. Commun. 2004, 320, 165–169. [Google Scholar] [CrossRef]
- Moretti, S.; Procopio, A.; Lazzarini, R.; Rippo, M.R.; Testa, R.; Marra, M.; Tamagnone, L.; Catalano, A. Semaphorin3A signaling controls Fas (CD95)-mediated apoptosis by promoting Fas translocation into lipid rafts. Blood 2008, 111, 2290–2299. [Google Scholar] [CrossRef]
- Lepelletier, Y.; Smaniotto, S.; Hadj-Slimane, R.; Villa-Verde, D.M.S.; Nogueira, A.C.; Dardenne, M.; Hermine, O.; Savino, W. Control of human thymocyte migration by Neuropilin-1/Semaphorin-3A-mediated interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 5545–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, M.; Cruz, A.C.; Cleland, S.Y.; Deng, M.; Price, S.; Rao, V.K.; Siegel, R.M. Specific elimination of effector memory CD4+ T cells due to enhanced Fas signaling complex formation and association with lipid raft microdomains. Cell Death Differ. 2010, 18, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Muppidi, J.R.; Siegel, R.M. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat. Immunol. 2004, 5, 182–189. [Google Scholar] [CrossRef]
- Elyassaki, W.; Wu, S. Lipid Rafts Mediate Ultraviolet Light–induced Fas Aggregation in M624 Melanoma Cells. Photochem. Photobiol. 2006, 82, 787–792. [Google Scholar] [CrossRef]
- George, K.S.; Elyassaki, W.; Wu, Q.; Wu, S. The Role of Cholesterol in UV Light B-induced Apoptosis†. Photochem. Photobiol. 2011, 88, 1191–1197. [Google Scholar] [CrossRef]
- Chatterjee, M.; Wu, S. Involvement of Fas receptor and not tumor necrosis factor-? receptor in ultraviolet-induced activation of acid sphingomyelinase. Mol. Carcinog. 2001, 30, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, J.A.; Zhang, J.; Donepudi, M.; Lee, H.; Fuks, Z.; Kolesnick, R. Caspase-dependent and -independent Activation of Acid Sphingomyelinase Signaling. J. Biol. Chem. 2005, 280, 26425–26434. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.C.; Sharma, K.; Wei, L.X.; Fujita, T.; Shi, Y.F. A novel role for sphingolipid intermediates in activation-induced cell death in T cells. Cell Death Differ. 2003, 10, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmé, H.; Cremesti, A.; Kolesnick, R.; Gulbins, E. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 2003, 22, 5457–5470. [Google Scholar] [CrossRef] [Green Version]
- Grassme, H.; Jekle, A.; Riehle, A.; Schwarz, H.; Berger, J.; Sandhoff, K.; Kolesnick, R.; Gulbins, E. CD95 Signaling via Ceramide-rich Membrane Rafts. J. Biol. Chem. 2001, 276, 20589–20596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, I.; Fick, A.; Schäfer, V.; Giner, T.; Siegmund, D.; Wajant, H. Signaling Active CD95 Receptor Molecules Trigger Co-translocation of Inactive CD95 Molecules into Lipid Rafts. J. Biol. Chem. 2012, 287, 24026–24042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, C.; Bizzozero, L.; Cazzato, D.; Morlacchi, S.; Assi, E.; Simbari, F.; Zhang, Y.; Gulbins, E.; Bassi, M.T.; Rosa, P.; et al. Syntaxin 4 Is Required for Acid Sphingomyelinase Activity and Apoptotic Function*. J. Biol. Chem. 2010, 285, 40240–40251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Lipid raft-mediated Fas/CD95 apoptotic signaling in leukemic cells and normal leukocytes and therapeutic implications. J. Leukoc. Biol. 2015, 98, 739–759. [Google Scholar] [CrossRef]
- Gilbert, S.; Loranger, A.; Omary, M.B.; Marceau, N. Keratin impact on PKCδ- and ASMase-mediated regulation of hepatocyte lipid raft size—implication for FasR-associated apoptosis. J. Cell Sci. 2016, 129, 3262–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyrek, K.; Lavrik, I.N. Modulation of CD95-mediated signaling by post-translational modifications: Towards understanding CD95 signaling networks. Apoptosis 2019, 24, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Moosmayer, D.; Wüest, T.; Bartke, T.; Gerlach, E.; Schönherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossin, A.; Derouet, M.; Abdel-Sater, F.; Hueber, A.-O. Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. Biochem. J. 2009, 419, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linderoth, E.; Pilia, G.; Mahajan, N.P.; Ferby, I. Activated Cdc42-associated Kinase 1 (Ack1) Is Required for Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) Receptor Recruitment to Lipid Rafts and Induction of Cell Death. J. Biol. Chem. 2013, 288, 32922–32931. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, K.; Nakano, A.; Miki, H.; Oda, A.; Amou, H.; Takeuchi, K.; Nakamura, S.; Harada, T.; Fujii, S.; Yata, K.; et al. Inhibition of TACE Activity Enhances the Susceptibility of Myeloma Cells to TRAIL. PLoS ONE 2012, 7, e31594. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.; Shi, J.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Death receptor 5-recruited raft components contributes to the sensitivity of Jurkat leukemia cell lines to TRAIL-induced cell death. IUBMB Life 2009, 61, 261–267. [Google Scholar] [CrossRef]
- Marconi, M.; Ascione, B.; Ciarlo, L.; Vona, R.; Garofalo, T.; Sorice, M.; Gianni, A.M.; Locatelli, S.L.; Carlo-Stella, C.; Malorni, W.; et al. Constitutive localization of DR4 in lipid rafts is mandatory for TRAIL-induced apoptosis in B-cell hematologic malignancies. Cell Death Dis. 2013, 4, e863. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Ishdorj, G.; Sun, J.; Johnston, J.B.; Gibson, S.B. Death receptor 4 is preferentially recruited to lipid rafts in chronic lymphocytic leukemia cells contributing to tumor necrosis related apoptosis inducing ligand-induced synergistic apoptotic responses. Leuk. Lymphoma 2011, 52, 1290–1301. [Google Scholar] [CrossRef]
- Song, J.H.; Tse, M.C.; Bellail, A.; Phuphanich, S.; Khuri, F.; Kneteman, N.M.; Hao, C. Lipid Rafts and Nonrafts Mediate Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Induced Apoptotic and Nonapoptotic Signals in Non–Small Cell Lung Carcinoma Cells. Cancer Res. 2007, 67, 6946–6955. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Ouyang, W.; Yang, C.; Liu, Y.; Xiong, J.; Zhang, J.; Zhong, Y.; Zhou, F. Redistribution of DR4 and DR5 in lipid rafts accounts for the sensitivity to TRAIL in NSCLC cells. Int. J. Oncol. 2011, 39, 1577–1586. [Google Scholar] [CrossRef]
- Bellail, A.C.; Tse, M.C.L.; Song, J.H.; Phuphanich, S.; Olson, J.J.; Sun, S.Y.; Hao, C. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J. Cell. Mol. Med. 2009, 14, 1303–1317. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tomiyama, A.; Sasaki, N.; Yamaguchi, H.; Shirakihara, T.; Nakashima, K.; Kumagai, K.; Takeuchi, S.; Toyooka, T.; Otani, N.; et al. Intracellular cholesterol level regulates sensitivity of glioblastoma cells against temozolomide-induced cell death by modulation of caspase-8 activation via death receptor 5-accumulation and activation in the plasma membrane lipid raft. Biochem. Biophys. Res. Commun. 2018, 495, 1292–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-C.; Duong, H.-Q.; Choi, J.E.; Lee, T.-B.; Kang, J.-H.; Oh, S.H.; Han, S.I. Lipid raft-dependent death receptor 5 (DR5) expression and activation are critical for ursodeoxycholic acid-induced apoptosis in gastric cancer cells. Carcinogensis 2011, 32, 723–731. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, L.; Qu, X.; Luo, Y.; Zhang, Y. Epirubicin enhances TRAIL-induced apoptosis in gastric cancer cells by promoting death receptor clustering in lipid rafts. Mol. Med. Rep. 2011, 4, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, Y.; Liu, J.; Qu, J.; Hu, X.; Zhang, F.; Zheng, H.; Qu, X.; Liu, Y. TRAIL-activated EGFR by Cbl-b-regulated EGFR redistribution in lipid rafts antagonises TRAIL-induced apoptosis in gastric cancer cells. Eur. J. Cancer 2012, 48, 3288–3299. [Google Scholar] [CrossRef]
- Xu, L.; Qu, X.; Li, H.; Li, C.; Liu, J.; Zheng, H.; Liu, Y. Src/caveolin-1-regulated EGFR activation antagonizes TRAIL-induced apoptosis in gastric cancer cells. Oncol. Rep. 2014, 32, 318–324. [Google Scholar] [CrossRef]
- Xu, L.; Qu, X.; Hu, X.; Zhu, Z.; Li, C.; Li, E.; Ma, Y.; Song, N.; Liu, Y. Lipid raft-regulated IGF-1R activation antagonizes TRAIL-induced apoptosis in gastric cancer cells. FEBS Lett. 2013, 587, 3815–3823. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Han, W.-Q.; Boini, K.M.; Xia, M.; Zhang, Y.; Li, P.-L. TRAIL death receptor 4 signaling via lysosome fusion and membrane raft clustering in coronary arterial endothelial cells: Evidence from ASM knockout mice. J. Mol. Med. 2012, 91, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Lafont, E. Stress Management: Death Receptor Signalling and Cross-Talks with the Unfolded Protein Response in Cancer. Cancers 2020, 12, 1113. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, D.; Jeltsch, A.; Rehm, M. TRAIL receptor signaling: From the basics of canonical signal transduction toward its entanglement with ER stress and the unfolded protein response. Int. Rev. Cell Mol. Biol. 2020, 351, 57–99. [Google Scholar] [CrossRef]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nat. Cell Biol. 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klima, M.; Zájedová, J.; Doubravská, L.; Anděra, L. Functional analysis of the posttranslational modifications of the death receptor. Biochim. Biophys. Acta Bioenerg. 2009, 1793, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nat. Cell Biol. 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Xu, K.; Olsen, O.; Tzvetkova-Robev, D.; Tessier-Lavigne, M.; Nikolov, D.B. The crystal structure of DR6 in complex with the amyloid precursor protein provides insight into death receptor activation. Genes Dev. 2015, 29, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, R.; Fenn, R.H.; Barren, C.; Tanzi, R.E.; Kovacs, D.M. Palmitoylated APP Forms Dimers, Cleaved by BACE1. PLoS ONE 2016, 11, e0166400. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.Y.; Kwon, O.-H.; Chung, S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 2020, 25, 5490. [Google Scholar] [CrossRef]
- Fröhlich, M.; Dejanovic, B.; Kashkar, H.; Schwarz, G.; Nussberger, S. S-palmitoylation represents a novel mechanism regulating the mitochondrial targeting of BAX and initiation of apoptosis. Cell Death Dis. 2014, 5, e1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Rotolo, J.A.; Mesicek, J.; Penate-Medina, T.; Rimner, A.; Liao, W.-C.; Yin, X.; Ragupathi, G.; Ehleiter, D.; Gulbins, E.; et al. Mitochondrial Ceramide-Rich Macrodomains Functionalize Bax upon Irradiation. PLoS ONE 2011, 6, e19783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 2004, 11, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, J.; Weiler, S.; Oh, K.J.; Wei, M.C.; Korsmeyer, S.J. Posttranslational N-Myristoylation of BID as a Molecular Switch for Targeting Mitochondria and Apoptosis. Science 2000, 290, 1761–1765. [Google Scholar] [CrossRef]
- Hung, C.-L.; Chang, H.-H.; Lee, S.W.; Chiang, Y.-W. Stepwise activation of the pro-apoptotic protein Bid at mitochondrial membranes. Cell Death Differ. 2021, 1–16. [Google Scholar] [CrossRef]
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef] [Green Version]
- Sorice, M.; Garofalo, T.; Misasi, R.; Manganelli, V.; Vona, R.; Malorni, W. Ganglioside GD3 as a Raft Component in Cell Death Regulation. Anti-Cancer Agents Med. Chem. 2012, 12, 376–382. [Google Scholar] [CrossRef]
- Sorice, M.; Matarrese, P.; Manganelli, V.; Tinari, A.; Giammarioli, A.M.; Mattei, V.; Misasi, R.; Garofalo, T.; Malorni, W. Role of GD3-CLIPR-59 Association in Lymphoblastoid T Cell Apoptosis Triggered by CD95/Fas. PLoS ONE 2010, 5, e8567. [Google Scholar] [CrossRef]
- Sorice, M.; Matarrese, P.; Tinari, A.; Giammarioli, A.M.; Garofalo, T.; Manganelli, V.; Ciarlo, L.; Gambardella, L.; Maccari, G.; Botta, M.; et al. Raft component GD3 associates with tubulin following CD95/Fas ligation. FASEB J. 2009, 23, 3298–3308. [Google Scholar] [CrossRef]
- Sorice, M.; Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; et al. Dynamics of mitochondrial raft-like microdomains in cell life and death. Commun. Integr. Biol. 2012, 5, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; Malorni, W.; et al. Recruitment of cellular prion protein to mitochondrial raft-like microdomains contributes to apoptosis execution. Mol. Biol. Cell 2011, 22, 4842–4853. [Google Scholar] [CrossRef] [PubMed]
- Skotte, N.H.; Sanders, S.S.; Singaraja, R.R.; Ehrnhoefer, D.E.; Vaid, K.; Qiu, X.; Kannan, S.; Verma, C.; Hayden, M.R. Palmitoylation of caspase-6 by HIP14 regulates its activation. Cell Death Differ. 2016, 24, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.; David, F.; Abrami, L.; Migliozzi, D.; Armand, F.; Bürgi, J.; Van Der Goot, F.G. SwissPalm: Protein Palmitoylation database. F1000Research 2015, 4, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesavardhana, S.; Malireddi, R.S.; Kanneganti, T.-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Watanabe, M.; Kitano, T.; Kondo, T.; Yabu, T.; Taguchi, Y.; Tashima, M.; Umehara, H.; Domae, N.; Uchiyama, T.; Okazaki, T. Increase of Nuclear Ceramide through Caspase-3-Dependent Regulation of the “Sphingomyelin Cycle” in Fas-Induced Apoptosis. Cancer Res. 2004, 64, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Mahul-Mellier, A.-L.; Strappazzon, F.; Petiot, A.; Chatellard-Causse, C.; Torch, S.; Blot, B.; Freeman, K.; Kuhn, L.; Garin, J.; Verna, J.-M.; et al. Alix and ALG-2 Are Involved in Tumor Necrosis Factor Receptor 1-induced Cell Death. J. Biol. Chem. 2008, 283, 34954–34965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahul-Mellier, A.-L.; Hemming, F.J.; Blot, B.; Fraboulet, S.; Sadoul, R. Alix, Making a Link between Apoptosis-Linked Gene-2, the Endosomal Sorting Complexes Required for Transport, and Neuronal Death In Vivo. J. Neurosci. 2006, 26, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Takahashi, Y.; Chen, L.; Tang, Z.; Wills, C.A.; Liang, X.; Wang, H.-G. Targeting the ESCRT-III component CHMP2A for noncanonical Caspase-8 activation on autophagosomal membranes. Cell Death Differ. 2021, 28, 657–670. [Google Scholar] [CrossRef]
- Dowlatshahi, D.P.; Sandrin, V.; Vivona, S.; Shaler, T.A.; Kaiser, S.E.; Melandri, F.; Sundquist, W.I.; Kopito, R.R. ALIX Is a Lys63-Specific Polyubiquitin Binding Protein that Functions in Retrovirus Budding. Dev. Cell 2012, 23, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chatterjee, V.; Ma, Y.; Zheng, E.; Yuan, S.Y. Protein Palmitoylation in Leukocyte Signaling and Function. Front. Cell Dev. Biol. 2020, 8, 600368. [Google Scholar] [CrossRef]
- Akimzhanov, A.M.; Boehning, D. Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 11876–11880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-C.; Tsai, H.-F.; Tzeng, H.-T.; Liao, H.-J.; Hsu, P.-N. Lipid Raft Assembly and Lck Recruitment in TRAIL Costimulation Mediates NF-κB Activation and T Cell Proliferation. J. Immunol. 2010, 186, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, I.; Obata, Y.; Kasahara, K.; Nakayama, Y.; Fukumoto, Y.; Yamasaki, T.; Yokoyama, K.K.; Saito, T.; Yamaguchi, N. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 2009, 122, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, K.; Matveichuk, O.V.; Fronk, J.; Ciesielska, A. Flotillins: At the Intersection of Protein S-Palmitoylation and Lipid-Mediated Signaling. Int. J. Mol. Sci. 2020, 21, 2283. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb-Abraham, E.; Gutman, O.; Pai, G.M.; Rubio, I.; Henis, Y.I. The residue at position 5 of the N-terminal region of Src and Fyn modulates their myristoylation, palmitoylation, and membrane interactions. Mol. Biol. Cell 2016, 27, 3926–3936. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Mullen, T.; Senkal, C.; Lu, P.; Ogretmen, B.; Obeid, L.; Voelkel-Johnson, C. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 2009, 28, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verlekar, D.; Wei, S.-J.; Cho, H.; Yang, S.; Kang, M.H. Ceramide synthase-6 confers resistance to chemotherapy by binding to CD95/Fas in T-cell acute lymphoblastic leukemia. Cell Death Dis. 2018, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.-J. Receptor Tyrosine Kinases in the Nucleus. Cold Spring Harb. Perspect. Biol. 2013, 5, a008979. [Google Scholar] [CrossRef] [Green Version]
- Bertsch, U.; Röder, C.; Kalthoff, H.; Trauzold, A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: Emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014, 5, e1390. [Google Scholar] [CrossRef]
- Mert, U.; Adawy, A.; Scharff, E.; Teichmann, P.; Willms, A.; Haselmann, V.; Colmorgen, C.; Lemke, J.; Von Karstedt, S.; Fritsch, J.; et al. TRAIL Induces Nuclear Translocation and Chromatin Localization of TRAIL Death Receptors. Cancers 2019, 11, 1167. [Google Scholar] [CrossRef] [Green Version]
- Bamias, G.; Evangelou, K.; Vergou, T.; Tsimaratou, K.; Kaltsa, G.; Antoniou, C.; Kotsinas, A.; Kim, S.; Gorgoulis, V.; Stratigos, A.J.; et al. Upregulation and nuclear localization of TNF-like Cytokine 1A (TL1A) and its receptors DR3 and DcR3 in psoriatic skin lesions. Exp. Dermatol. 2011, 20, 725–731. [Google Scholar] [CrossRef]
- Lin-Lee, Y.-C.; Pham, L.V.; Tamayo, A.T.; Fu, L.; Zhou, H.-J.; Yoshimura, L.C.; Decker, G.L.; Ford, R.J. Nuclear Localization in the Biology of the CD40 Receptor in Normal and Neoplastic Human B Lymphocytes. J. Biol. Chem. 2006, 281, 18878–18887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadiri, A.; Polyak, M.J.; Jundi, M.; Alturaihi, H.; Reyes-Moreno, C.; Hassan, G.S.; Mourad, W. CD40 translocation to lipid rafts: Signaling requirements and downstream biological events. Eur. J. Immunol. 2011, 41, 2358–2367. [Google Scholar] [CrossRef]
- Reyes-Moreno, C.; Sharif-Askari, E.; Girouard, J.; Léveillé, C.; Jundi, M.; Akoum, A.; Lapointe, R.; Darveau, A.; Mourad, W. Requirement of Oxidation-dependent CD40 Homodimers for CD154/CD40 Bidirectional Signaling*. J. Biol. Chem. 2007, 282, 19473–19480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsumi, T.; Sakurai, N.; Nakano, K.; Ishisaka, R. C-terminal 15 kDa fragment of cytoskeletal actin is posttranslationally N-myristoylated upon caspase-mediated cleavage and targeted to mitochondria. FEBS Lett. 2003, 539, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, N.; Utsumi, T. Posttranslational N-Myristoylation Is Required for the Anti-apoptotic Activity of Human tGelsolin, the C-terminal Caspase Cleavage Product of Human Gelsolin. J. Biol. Chem. 2006, 281, 14288–14295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.D.O.; Ahpin, C.Y.; Heit, R.J.; Perinpanayagam, M.A.; Yap, M.C.; Veldhoen, R.A.; Goping, I.S.; Berthiaume, L.G. Tandem reporter assay for myristoylated proteins post-translationally (TRAMPP) identifies novel substrates for post-translational myristoylation: PKC∊, a case study. FASEB J. 2011, 26, 13–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.; Nichols, A.; Song, J.C.; Mammen, J.; Calvo, I.; Worrell, R.T.; Cuppoletti, J.; Matlin, K.; Matthews, J.B. Bryostatin-1 attenuates TNF-induced epithelial barrier dysfunction: Role of novel PKC isozymes. Am. J. Physiol. Liver Physiol. 2003, 284, G703–G712. [Google Scholar] [CrossRef] [Green Version]
- Schütze, S.; Nottrott, S.; Pfizenmaier, K.; Krönke, M. Tumor necrosis factor signal transduction. Cell-type-specific activation and translocation of protein kinase C. J. Immunol. 1990, 144, 2604–2608. [Google Scholar]
- Harper, N.; Hughes, M.A.; Farrow, S.N.; Cohen, G.M.; MacFarlane, M. Protein Kinase C Modulates Tumor Necrosis Factor-related Apoptosis-inducing Ligand-induced Apoptosis by Targeting the Apical Events of Death Receptor Signaling. J. Biol. Chem. 2003, 278, 44338–44347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, P.; Resh, M.D. Myristoylation and Membrane Binding Regulate c-Src Stability and Kinase Activity. Mol. Cell. Biol. 2010, 30, 4094–4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powley, I.R.; Hughes, M.A.; Cain, K.; Macfarlane, M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing procaspase-8 maturation and cycling within the complex. Oncogene 2016, 35, 5629–5640. [Google Scholar] [CrossRef] [Green Version]
- Perinpanayagam, M.A.; Beauchamp, E.; Martin, D.D.O.; Sim, J.Y.W.; Yap, M.C.; Berthiaume, L.G. Regulation of co- and post-translational myristoylation of proteins during apoptosis: Interplay of N -myristoyltransferases and caspases. FASEB J. 2013, 27, 811–821. [Google Scholar] [CrossRef]
- Di Bello, E.; Zwergel, C.; Mai, A.; Valente, S. The Innovative Potential of Statins in Cancer: New Targets for New Therapies. Front. Chem. 2020, 8, 516. [Google Scholar] [CrossRef]
- Dehnavi, S.; Sohrabi, N.; Sadeghi, M.; Lansberg, P.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Statins and autoimmunity: State-of-the-art. Pharmacol. Ther. 2020, 214, 107614. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [Green Version]
- Bojková, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary Fat and Cancer—Which Is Good, Which Is Bad, and the Body of Evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef]
- Greenlee, J.D.; Subramanian, T.; Liu, K.; King, M.R. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2020, 81, 5–17. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Mukhamedova, N.; Sviridov, D. Lipid rafts and pathogens: The art of deception and exploitation. J. Lipid Res. 2020, 61, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Suzuki, Y. Virus Infection and Lipid Rafts. Biol. Pharm. Bull. 2006, 29, 1538–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.; Hiller, N.L.; Harrison, T.; Lomasney, J.W.; Mohandas, N.; Haldar, K. Lipid rafts and malaria parasite infection of erythrocytes (Review). Mol. Membr. Biol. 2006, 23, 81–88. [Google Scholar] [CrossRef]
- Fozo, E.; Rucks, E. The Making and Taking of Lipids. Adv. Microb. Physiol. 2016, 69, 51–155. [Google Scholar] [CrossRef]
- Audi, A.; Soudani, N.; Dbaibo, G.; Zaraket, H. Depletion of Host and Viral Sphingomyelin Impairs Influenza Virus Infection. Front. Microbiol. 2020, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.J.; Sani, M.-A.; Parker, M.W.; Separovic, F. Cholesterol-Dependent Cytolysins: Membrane and Protein Structural Requirements for Pore Formation. Chem. Rev. 2019, 119, 7721–7736. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Lippincott-Schwartz, J. Revisiting Membrane Microdomains and Phase Separation: A Viral Perspective. Viruses 2020, 12, 745. [Google Scholar] [CrossRef] [PubMed]
- Dadhich, R.; Kapoor, S. Various Facets of Pathogenic Lipids in Infectious Diseases: Exploring Virulent Lipid-Host Interactome and Their Druggability. J. Membr. Biol. 2020, 253, 399–423. [Google Scholar] [CrossRef]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef]
- Fritsch, J.; Tchikov, V.; Hennig, L.; Lucius, R.; Schütze, S. A toolbox for the immunomagnetic purification of signaling organelles. Traffic 2018, 20, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Azizi, S.-A.; Lan, T.; Delalande, C.; Kathayat, R.; Dickinson, B. Development of an Acrylamide-Based Inhibitor of Protein S-Acylation. 2021. Available online: https://chemrxiv.org/articles/preprint/Development_of_an_Acrylamide-Based_Inhibitor_of_Protein_S-Acylation/13755763 (accessed on 20 March 2021).
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritsch, J.; Särchen, V.; Schneider-Brachert, W. Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains—Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation. Cancers 2021, 13, 2513. https://doi.org/10.3390/cancers13112513
Fritsch J, Särchen V, Schneider-Brachert W. Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains—Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation. Cancers. 2021; 13(11):2513. https://doi.org/10.3390/cancers13112513
Chicago/Turabian StyleFritsch, Jürgen, Vinzenz Särchen, and Wulf Schneider-Brachert. 2021. "Regulation of Death Receptor Signaling by S-Palmitoylation and Detergent-Resistant Membrane Micro Domains—Greasing the Gears of Extrinsic Cell Death Induction, Survival, and Inflammation" Cancers 13, no. 11: 2513. https://doi.org/10.3390/cancers13112513