
Possible Therapeutic Strategy Involving the Purine Synthesis Pathway Regulated by ITK in Tongue Squamous Cell Carcinoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Immunohistochemistry (IHC)
2.3. Cell Lines, Antibodies, and Drugs
2.4. Establishment of Stably Infected Cells
2.5. Western Blot Analysis
2.6. In Vitro Cell Growth Assay
2.7. Animal Experiments
2.8. Preparation and Digestion of Protein Lysate
2.9. Mass Spectrometry Analysis for Quantitative Phosphotyrosine Proteomics
2.10. Identification and Quantification of Phosphopeptides
2.11. Bioluminescence Resonance Energy Transfer (BRET) Assay
2.12. Immunofluorescence Cytochemistry
2.13. Metabolomic Analysis
2.14. Ion Chromatography-Tandem Mass Spectrometry for Anionic Metabolites
2.15. Growth Inhibition Analysis with ITK Inhibitor
2.16. Statistical Analysis
3. Results
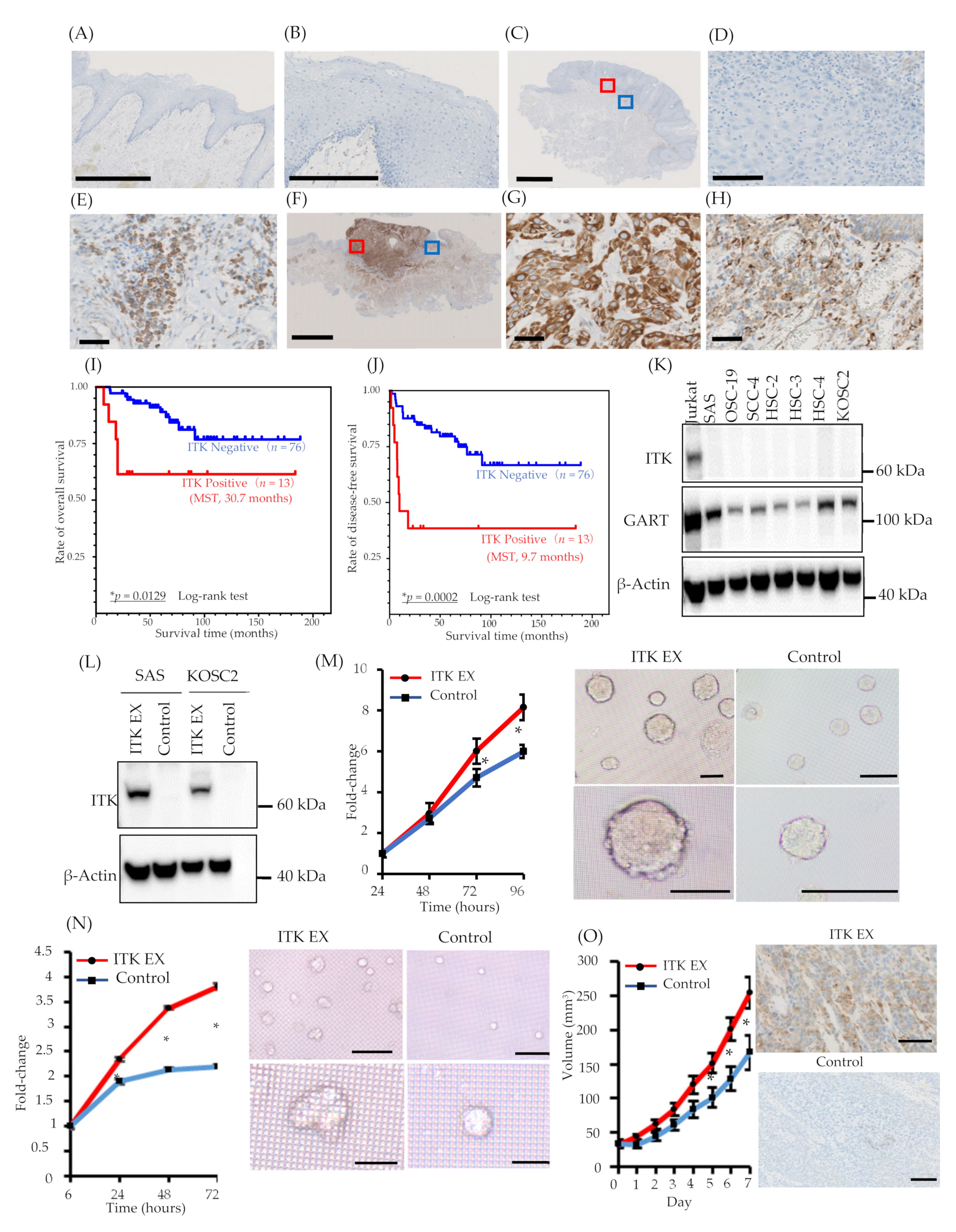
3.1. ITK Protein Expression in Patients with TSCC
3.2. Hazard Ratios (HRs) for Death and Prognostic Significance of ITK Protein Expression in TSCC
3.3. Role of ITK in Cancer Cell Proliferation In Vitro and In Vivo
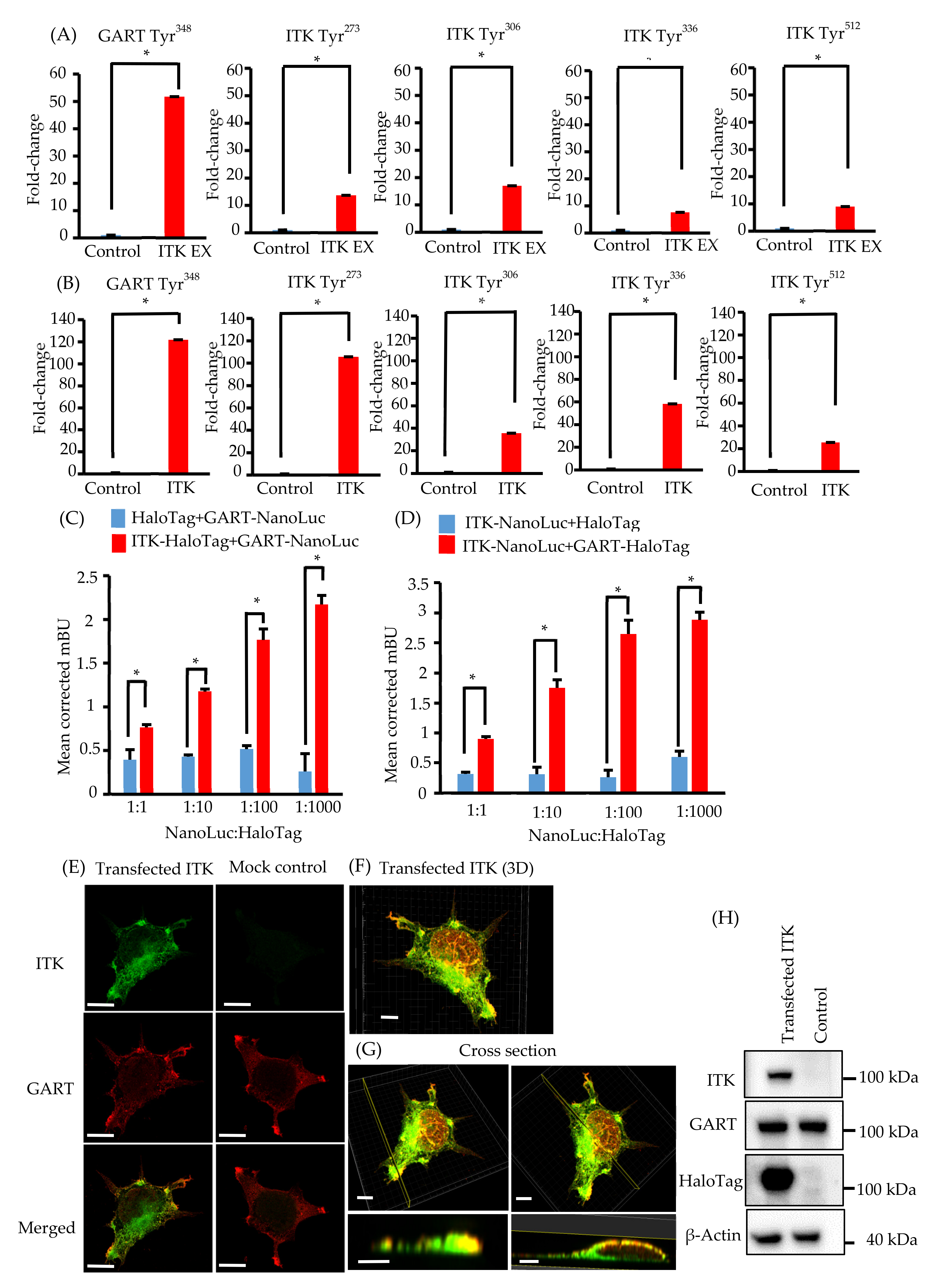
3.4. Proteomic Analysis of ITK Tyrosine Phosphorylation
3.5. Elucidation of the Association between ITK and GART
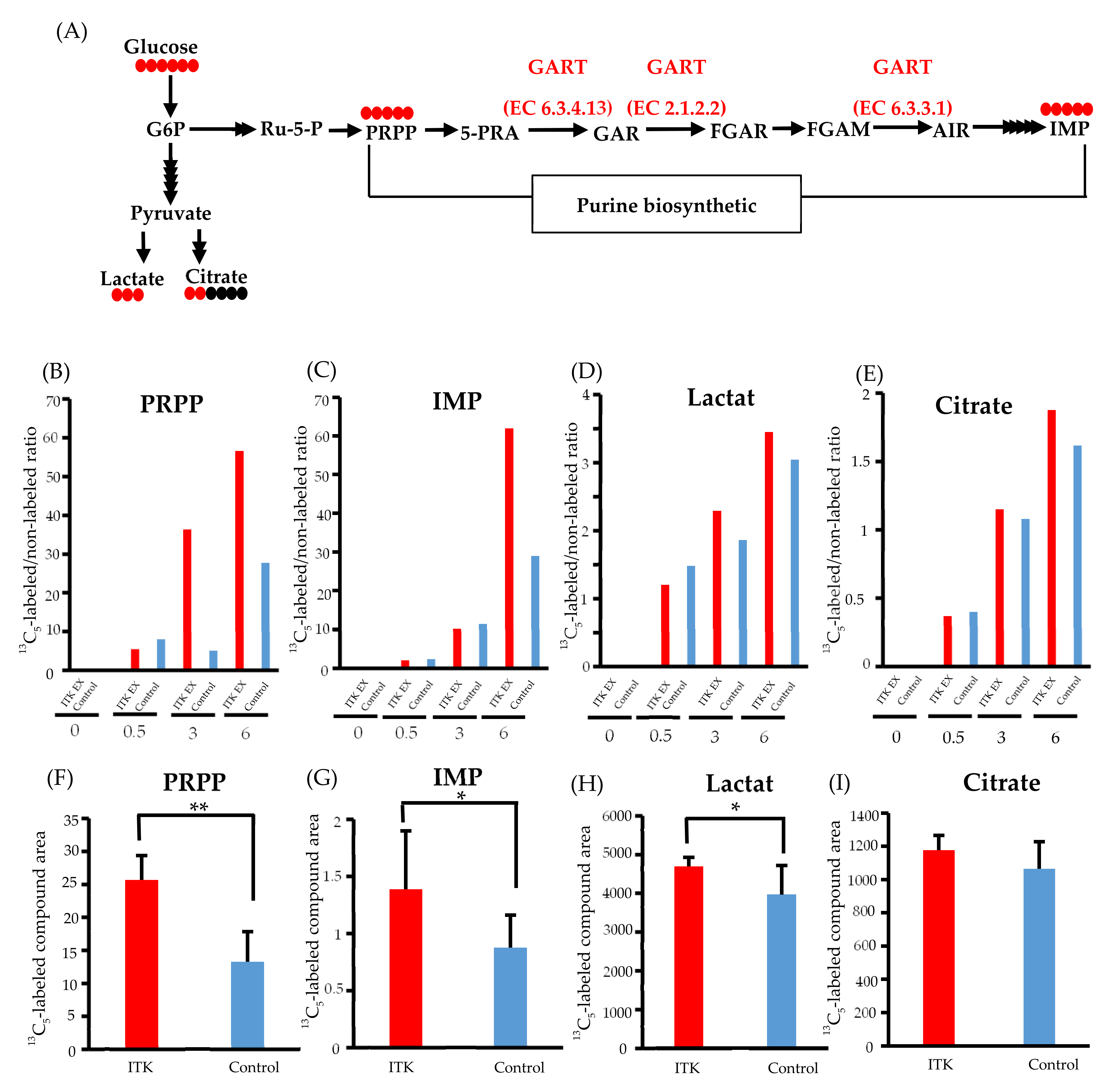
3.6. Assessment of De Novo Synthesis of Purines in ITK-Expressing Cells
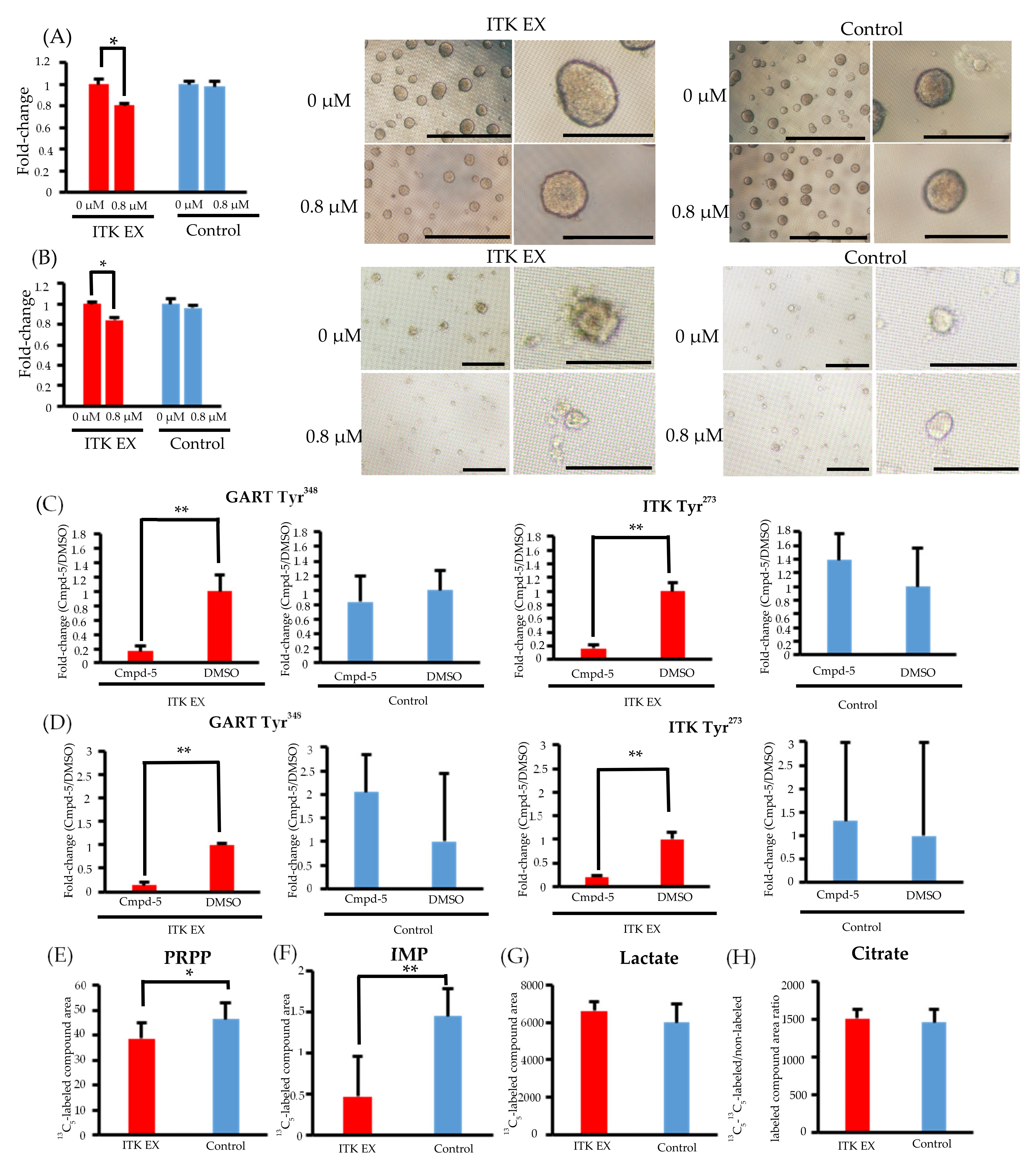
3.7. Inhibition of CellG and Suppression of GART Phosphorylation and Purine Metabolism Using an ITK Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect. Biol. 2010, 2, a002287. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.D.; Pan, Y.H.; Liu, J. The relevance between the immune response- related gene module and clinical traits in head and neck squamous cell carcinoma. Cancer Manag. Res. 2019, 11, 7455–7472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, C.C.; Moschos, S.J.; Edmiston, S.N.; Darr, D.B.; Nikolaishvili-Feinberg, N.; Groben, P.A.; Zhou, X.; Kuan, P.F.; Pandey, S.; Chan, K.T.; et al. IL2 Inducible T-cell Kinase, a Novel Therapeutic Target in Melanoma. Clin. Cancer Res. 2015, 21, 2167–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, A.; Saha, A.; Chan, A.; Casteel, D.E.; Pilz, R.B.; Boss, G.R. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 2013, 454, 91–99. [Google Scholar] [CrossRef]
- Kakuya, T.; Mori, T.; Yoshimoto, S.; Watabe, Y.; Miura, N.; Shoji, H.; Onidani, K.; Shibahara, T.; Honda, K. Prognostic significance of gene amplification of ACTN4 in stage I and II oral tongue cancer. Int. J. Oral. Maxillofac. Surg. 2017, 46, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Hishiki, T.; Yamamoto, S.; Yamamoto, T.; Miura, N.; Kubo, A.; Itoh, M.; Chen, W.Y.; Takano, M.; Yoshikawa, T.; et al. On-tissue polysulfide visualization by surface-enhanced Raman spectroscopy benefits patients with ovarian cancer to predict post-operative chemosensitivity. Redox Biol. 2021, 41, 101926. [Google Scholar] [CrossRef]
- Abe, Y.; Nagano, M.; Kuga, T.; Tada, A.; Isoyama, J.; Adachi, J.; Tomonaga, T. Deep Phospho- and Phosphotyrosine Proteomics Identified Active Kinases and Phosphorylation Networks in Colorectal Cancer Cell Lines Resistant to Cetuximab. Sci. Rep. 2017, 7, 10463. [Google Scholar] [CrossRef] [Green Version]
- Ansarin, M.; Bruschini, R.; Navach, V.; Giugliano, G.; Calabrese, L.; Chiesa, F.; Medina, J.E.; Kowalski, L.P.; Shah, J.P. Classification of GLOSSECTOMIES: Proposal for tongue cancer resections. Head Neck 2019, 41, 821–827. [Google Scholar] [CrossRef]
- Watabe, Y.; Mori, T.; Yoshimoto, S.; Nomura, T.; Shibahara, T.; Yamada, T.; Honda, K. Copy number increase of ACTN4 is a prognostic indicator in salivary gland carcinoma. Cancer Med. 2014, 3, 613–622. [Google Scholar] [CrossRef]
- Watanabe, T.; Ueno, H.; Watabe, Y.; Hiraoka, N.; Morizane, C.; Itami, J.; Okusaka, T.; Miura, N.; Kakizaki, T.; Kakuya, T.; et al. ACTN4 copy number increase as a predictive biomarker for chemoradiotherapy of locally advanced pancreatic cancer. Br. J. Cancer 2015, 112, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Noro, R.; Honda, K.; Tsuta, K.; Ishii, G.; Maeshima, A.M.; Miura, N.; Furuta, K.; Shibata, T.; Tsuda, H.; Ochiai, A.; et al. Distinct outcome of stage I lung adenocarcinoma with ACTN4 cell motility gene amplification. Ann. Oncol. 2013, 24, 2594–2600. [Google Scholar] [CrossRef]
- Miura, N.; Kamita, M.; Kakuya, T.; Fujiwara, Y.; Tsuta, K.; Shiraishi, H.; Takeshita, F.; Ochiya, T.; Shoji, H.; Huang, W.; et al. Efficacy of adjuvant chemotherapy for non-small cell lung cancer assessed by metastatic potential associated with ACTN4. Oncotarget 2016, 7, 33165–33178. [Google Scholar] [CrossRef] [Green Version]
- Shiokawa, D.; Sato, A.; Ohata, H.; Mutoh, M.; Sekine, S.; Kato, M.; Shibata, T.; Nakagama, H.; Okamoto, K. The Induction of Selected Wnt Target Genes by Tcf1 Mediates Generation of Tumorigenic Colon Stem Cells. Cell Rep. 2017, 19, 981–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Nagano, M.; Tada, A.; Adachi, J.; Tomonaga, T. Deep Phosphotyrosine Proteomics by Optimization of Phosphotyrosine Enrichment and MS/MS Parameters. J. Proteome Res. 2017, 16, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Thean, D.; Ebo, J.S.; Luxton, T.; Lee, X.C.; Yuen, T.Y.; Ferrer, F.J.; Johannes, C.W.; Lane, D.P.; Brown, C.J. Enhancing Specific Disruption of Intracellular Protein Complexes by Hydrocarbon Stapled Peptides Using Lipid Based Delivery. Sci. Rep. 2017, 7, 1763. [Google Scholar] [CrossRef]
- Hayashida, Y.; Honda, K.; Idogawa, M.; Ino, Y.; Ono, M.; Tsuchida, A.; Aoki, T.; Hirohashi, S.; Yamada, T. E-cadherin regulates the association between beta-catenin and actinin-4. Cancer Res. 2005, 65, 8836–8845. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Yamada, T.; Hayashida, Y.; Idogawa, M.; Sato, S.; Hasegawa, F.; Ino, Y.; Ono, M.; Hirohashi, S. Actinin-4 increases cell motility and promotes lymph node metastasis of colorectal cancer. Gastroenterology 2005, 128, 51–62. [Google Scholar] [CrossRef]
- Kunisawa, J.; Sugiura, Y.; Wake, T.; Nagatake, T.; Suzuki, H.; Nagasawa, R.; Shikata, S.; Honda, K.; Hashimoto, E.; Suzuki, Y.; et al. Mode of Bioenergetic Metabolism during B Cell Differentiation in the Intestine Determines the Distinct Requirement for Vitamin B1. Cell Rep. 2015, 13, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, M.; Zhang, B.; Sugiura, Y.; Sonomura, K.; Guerrini, M.M.; Tsutsui, Y.; Maruya, M.; Vogelzang, A.; Chamoto, K.; Honda, K.; et al. Metabolic shift induced by systemic activation of T cells in PD-1-deficient mice perturbs brain monoamines and emotional behavior. Nat. Immunol. 2017, 18, 1342–1352. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Monti, S.; Aires, D.J.; Duvic, M.; Golub, T.; Jones, D.A.; Kupper, T.S. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood 2007, 110, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Liu, R.; Ono, Y.; Ma, A.H.; Martinez, A.; Sanchez, E.; Wang, Y.; Huang, W.; Mazloom, A.; Li, J.; et al. Molecular characteristics of CTA056, a novel interleukin-2-inducible T-cell kinase inhibitor that selectively targets malignant T cells and modulates oncomirs. Mol. Pharm. 2012, 82, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Hashiguchi, K.; Nagano, M.; Sato, M.; Sato, A.; Fukamizu, K.; Ishihama, Y.; Tomonaga, T. Improved Proteome and Phosphoproteome Analysis on a Cation Exchanger by a Combined Acid and Salt Gradient. Anal. Chem. 2016, 88, 7899–7903. [Google Scholar] [CrossRef]
- Abe, Y.; Hirano, H.; Shoji, H.; Tada, A.; Isoyama, J.; Kakudo, A.; Gunji, D.; Honda, K.; Boku, N.; Adachi, J.; et al. Comprehensive characterization of the phosphoproteome of gastric cancer from endoscopic biopsy specimens. Theranostics 2020, 10, 2115–2129. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneja, A.; Johnson, H.; Gabrovsek, L.; Lauffenburger, D.A.; White, F.M. Qualitatively different T cell phenotypic responses to IL-2 versus IL-15 are unified by identical dependences on receptor signal strength and duration. J. Immunol. 2014, 192, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Kyoung, M.; Russell, S.J.; Kohnhorst, C.L.; Esemoto, N.N.; An, S. Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry 2015, 54, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chiaro, C.R.; Zhang, L.; Smith, P.B.; Chan, C.Y.; Pedley, A.M.; Pugh, R.J.; French, J.B.; Patterson, A.D.; Benkovic, S.J. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 2015, 290, 6705–6713. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Keating, M.; Wierda, W.; Estrov, Z.; Ferrajoli, A.; Jain, N.; George, B.; James, D.; Kantarjian, H.; Burger, J.; et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 2015, 125, 2062–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bonin, A.; Rausch, A.; Mengel, A.; Hitchcock, M.; Kruger, M.; von Ahsen, O.; Merz, C.; Rose, L.; Stock, C.; Martin, S.F.; et al. Inhibition of the IL-2-inducible tyrosine kinase (Itk) activity: A new concept for the therapy of inflammatory skin diseases. Exp. Derm. 2011, 20, 41–47. [Google Scholar] [CrossRef]
- Kaur, M.; Bahia, M.S.; Silakari, O. Inhibitors of interleukin-2 inducible T-cell kinase as potential therapeutic candidates for the treatment of various inflammatory disease conditions. Eur. J. Pharm. Sci. 2012, 47, 574–588. [Google Scholar] [CrossRef]
- Sahu, N.; August, A. ITK inhibitors in inflammation and immune-mediated disorders. Curr. Top. Med. Chem. 2009, 9, 690–703. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Sharifnia, T.; Rusu, V.; Piccioni, F.; Bagul, M.; Imielinski, M.; Cherniack, A.D.; Pedamallu, C.S.; Wong, B.; Wilson, F.H.; Garraway, L.A.; et al. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 18661–18666. [Google Scholar] [CrossRef] [Green Version]
- Conley, J.M.; Gallagher, M.P.; Berg, L.J. T Cells and Gene Regulation: The Switching on and Turning Up of Genes after T Cell Receptor Stimulation in CD8 T Cells. Front. Immunol. 2016, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, J.P.; McGee, M.C.; Nidetz, N.F.; Anannya, O.; Huang, W.; August, A. Tuning T helper cell differentiation by ITK. Biochem. Soc. Trans. 2020, 48, 179–185. [Google Scholar] [CrossRef]
- Shan, X.; Czar, M.J.; Bunnell, S.C.; Liu, P.; Liu, Y.; Schwartzberg, P.L.; Wange, R.L. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol. 2000, 20, 6945–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchihara, T.; Miyake, K.; Yonemura, A.; Komohara, Y.; Itoyama, R.; Koiwa, M.; Yasuda, T.; Arima, K.; Harada, K.; Eto, K.; et al. Extracellular Vesicles from Cancer-Associated Fibroblasts Containing Annexin A6 Induces FAK-YAP Activation by Stabilizing beta1 Integrin, Enhancing Drug Resistance. Cancer Res. 2020, 80, 3222–3235. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Cases (%) | ITK-Negative | ITK-Positive | p-value a | |
|---|---|---|---|---|
| Total | 86 | 73 | 13 | |
| Age (years) | 0.3655 | |||
| <64 | 43 (50.0) | 38 | 5 | |
| ≥64 | 43 (50.0) | 35 | 8 | |
| Sex | 0.1715 | |||
| Male | 48 (55.8) | 43 | 5 | |
| Female | 38 (44.2) | 30 | 8 | |
| Stage b | 0.9776 | |||
| I | 46 (53.5) | 39 | 7 | |
| II | 40(46.5) | 34 | 6 | |
| Histologic differentiation | 0.1760 | |||
| Poor/Moderate | 25 (29.1) | 18 | 6 | |
| Well | 61 (70.9) | 55 | 7 | |
| Mode of invasion c | 0.0000 * | |||
| 1,2 | 58 (67.4) | 56 | 2 | |
| 3,4 | 28 (32.6) | 17 | 11 | |
| Late metastasis of cervical lymph nodes | 0.0006 * | |||
| Negative | 68 (79.1) | 63 | 5 | |
| Positive | 18 (20.9) | 10 | 8 | |
| Lymphovascular invasion | 0.0101 * | |||
| Negative | 75 (87.2) | 67 | 8 | |
| Positive | 11 (12.8) | 6 | 5 | |
| Perineural invasion | ||||
| Negative | 82 (95.3) | 71 | 11 | 0.1067 |
| Positive | 4 (4.7) | 2 | 2 | |
| Covariate | Univariate Analysis (n = 86) | Multivariate Analysis (n = 86) | ||||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |||
| Age (years) | ||||||||
| <64 | Reference | Reference | ||||||
| ≥64 | 2.866 | 0.923 | 8.894 | 0.068 | 2.611 | 0.836 | 8.153 | 0.099 |
| ITK protein expression | ||||||||
| Negative | Reference | Reference | ||||||
| Positive | 3.544 | 1.225 | 10.247 | 0.020 * | 3.167 | 1.089 | 9.215 | 0.034 * |
| Sex | ||||||||
| Male | Reference | |||||||
| Female | 1.235 | 0.463 | 3.296 | 0.673 | ||||
| Stage a | ||||||||
| Stage I | Reference | |||||||
| Stage II | 1.519 | 0.567 | 4.068 | 0.405 | ||||
| Histologic differentiation | ||||||||
| Well | Reference | |||||||
| Poor and moderate | 3.401 | 1.264 | 9.149 | 0.015 * | ||||
| Mode of invasion b | ||||||||
| 1, 2 | Reference | |||||||
| 3, 4 | 5.112 | 1.768 | 14.783 | 0.003 * | ||||
| Late metastasis of cervical lymph nodes | ||||||||
| Negative | Reference | |||||||
| Positive | 4.497 | 1.684 | 12.010 | 0.003 * | ||||
| Lymphovascular invasion | ||||||||
| Negative | Reference | |||||||
| Positive | 3.497 | 1.210 | 10.110 | 0.021 * | ||||
| Perineural invasion | ||||||||
| Negative | Reference | |||||||
| Positive | N.A.c | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onidani, K.; Miura, N.; Sugiura, Y.; Abe, Y.; Watabe, Y.; Kakuya, T.; Mori, T.; Yoshimoto, S.; Adachi, J.; Kiyoi, T.; et al. Possible Therapeutic Strategy Involving the Purine Synthesis Pathway Regulated by ITK in Tongue Squamous Cell Carcinoma. Cancers 2021, 13, 3333. https://doi.org/10.3390/cancers13133333
Onidani K, Miura N, Sugiura Y, Abe Y, Watabe Y, Kakuya T, Mori T, Yoshimoto S, Adachi J, Kiyoi T, et al. Possible Therapeutic Strategy Involving the Purine Synthesis Pathway Regulated by ITK in Tongue Squamous Cell Carcinoma. Cancers. 2021; 13(13):3333. https://doi.org/10.3390/cancers13133333
Chicago/Turabian StyleOnidani, Kaoru, Nami Miura, Yuki Sugiura, Yuichi Abe, Yukio Watabe, Takanori Kakuya, Taisuke Mori, Seiichi Yoshimoto, Jun Adachi, Takao Kiyoi, and et al. 2021. "Possible Therapeutic Strategy Involving the Purine Synthesis Pathway Regulated by ITK in Tongue Squamous Cell Carcinoma" Cancers 13, no. 13: 3333. https://doi.org/10.3390/cancers13133333