
Role of Splicing Regulatory Elements and In Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. In Silico Variant Annotation and Analysis for All Datasets
2.3. Statistical Analysis
2.4. Experimental RNA Analysis in Patient’s Data Set
2.4.1. Reverse Transcription-PCR (RT-PCR) and Sanger Sequencing
2.4.2. Qualitative Analysis by Capillary Electrophoresis of Fluorescent Amplicons
2.5. Editorial Policies and Ethical Considerations
3. Results
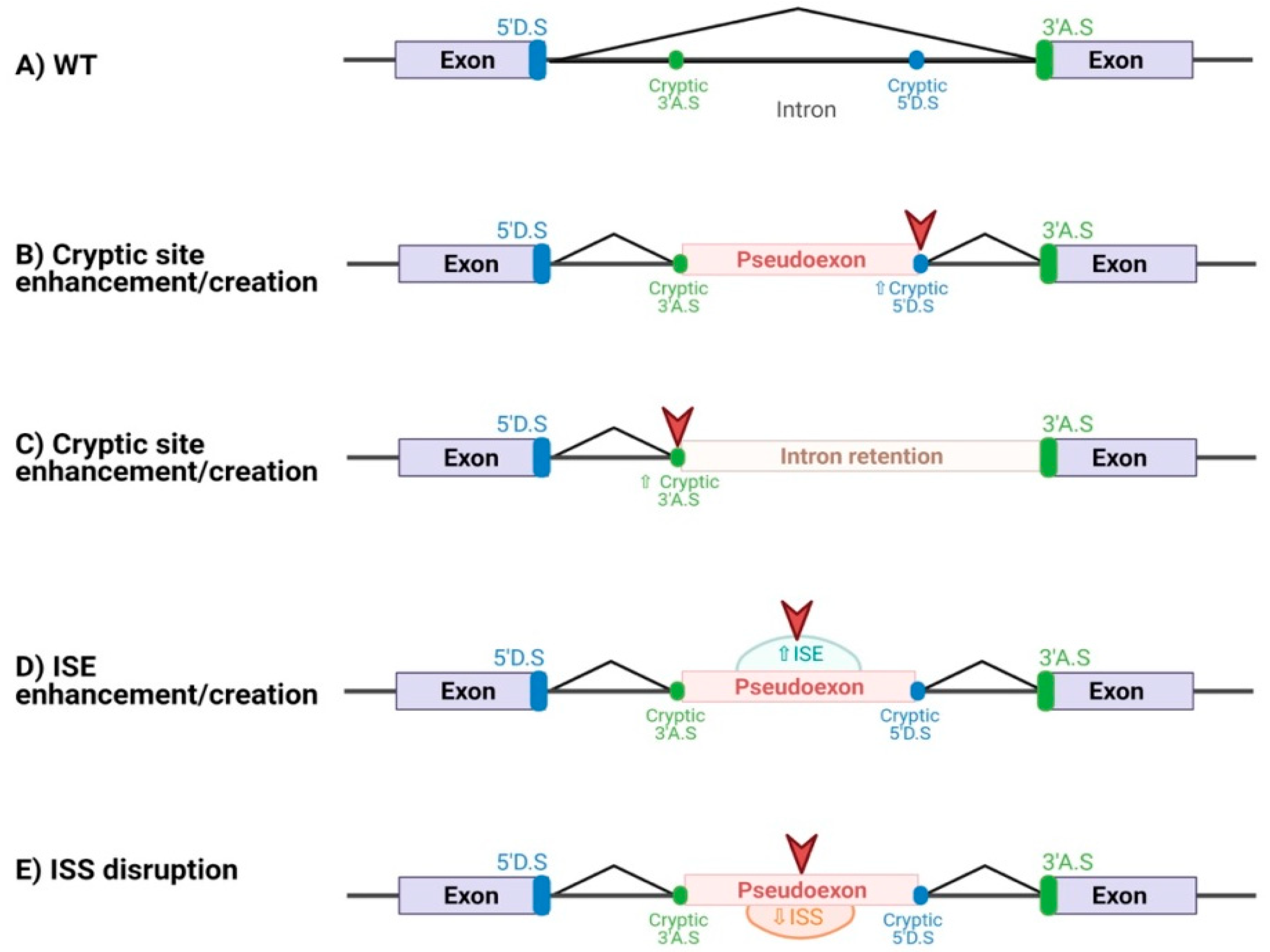
3.1. SpliceAI Optimally Predicts Deep Intronic Splice-Altering Variants but with Less Sensitivity Those Affecting Splicing by Altering Regulatory Elements
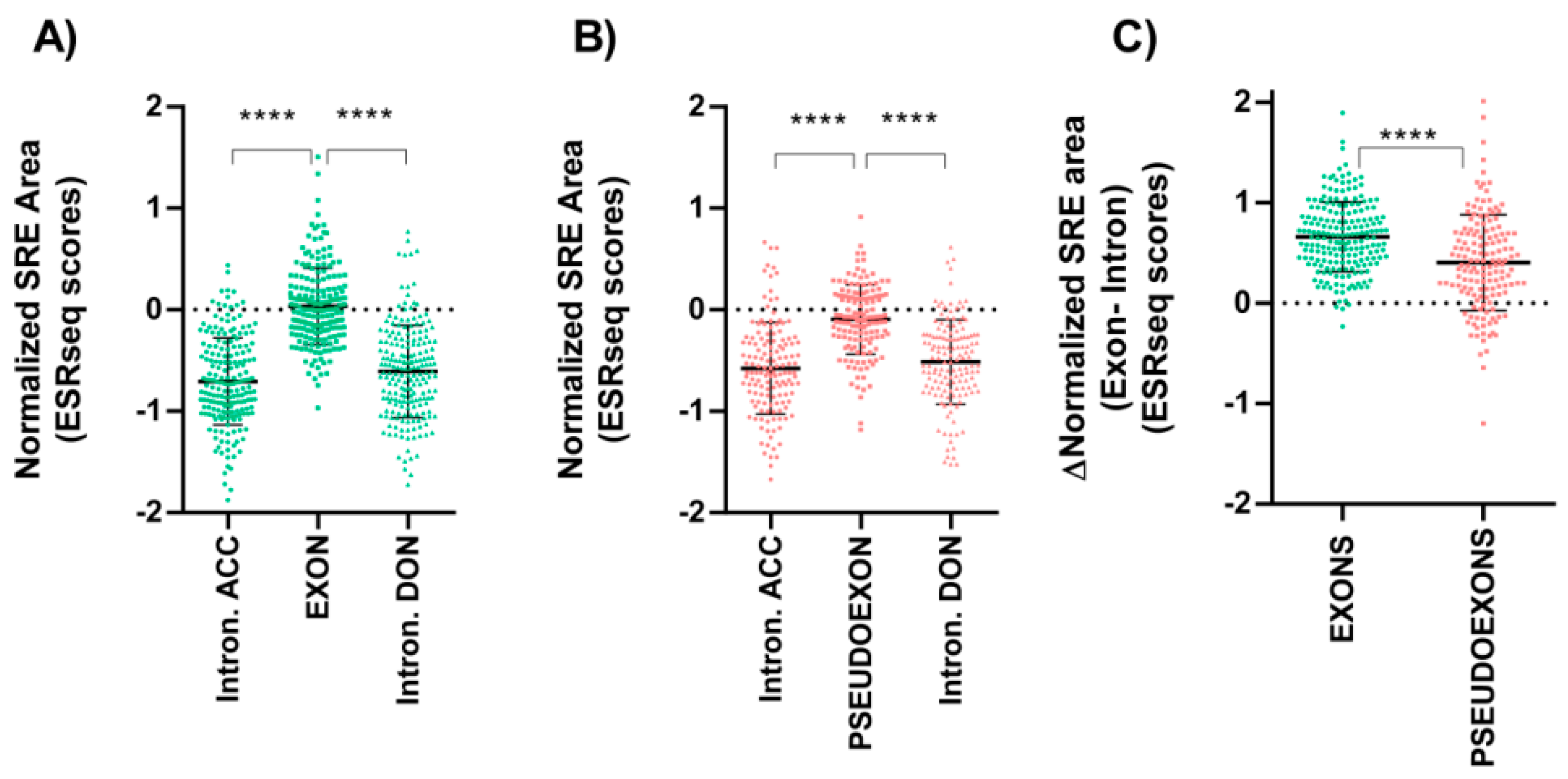
3.2. Splicing Regulatory Elements Balance Is Similar between Pseudoexons and Canonical Exons
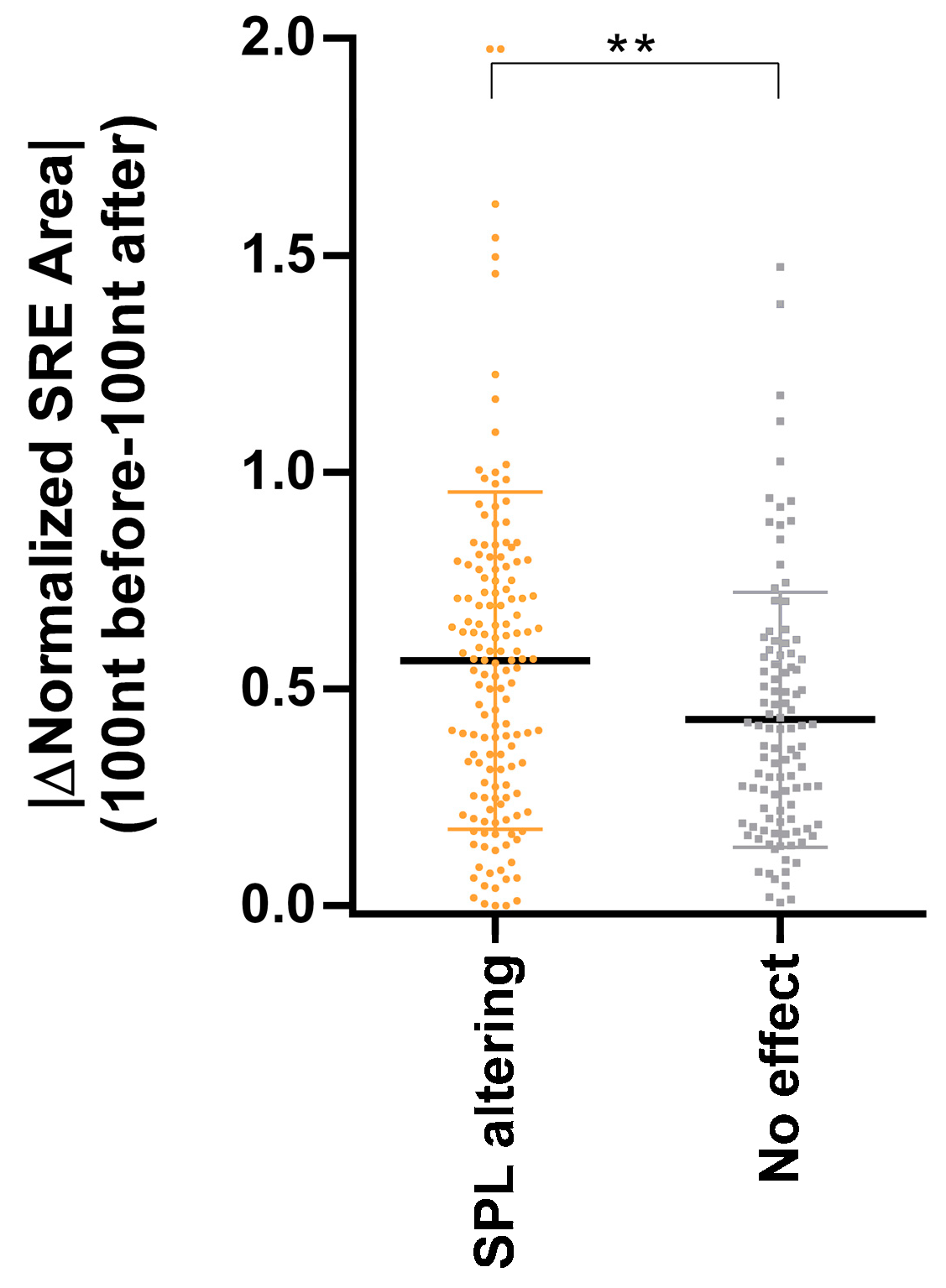
3.3. Inclusion of SRE Landscape in the In Silico Detection of Deep Intronic Splice-Altering Variants
3.4. Experimental Analysis of Hereditary Cancer Gene Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Hasson, S.P.; Menes, T.; Sonnenblick, A. Comparison of patient susceptibility genes across breast cancer: Implications for prognosis and therapeutic outcomes. Pharmgenomics Personal. Med. 2020, 13, 227–238. [Google Scholar] [CrossRef]
- Bonache, S.; Esteban, I.; Moles-Fernández, A.; Tenés, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; López-Fernández, A.; et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef] [PubMed]
- Feliubadaló, L.; López-Fernández, A.; Pineda, M.; Díez, O.; del Valle, J.; Gutiérrez-Enríquez, S.; Teulé, A.; González, S.; Stjepanovic, N.; Salinas, M.; et al. Opportunistic testing of BRCA1, BRCA2 and mismatch repair genes improves the yield of phenotype driven hereditary cancer gene panels. Int. J. Cancer 2019, 145, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef] [Green Version]
- Rhine, C.L.; Cygan, K.J.; Soemedi, R.; Maguire, S.; Murray, M.F.; Monaghan, S.F.; Fairbrother, W.G. Hereditary cancer genes are highly susceptible to splicing mutations. PLoS Genet. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening To Silence and Understanding Nonsense: Exonic Mutations That Affect Splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Chasin, L.A. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004, 18, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Ke, S.; Shang, S.; Kalachikov, S.M.; Morozova, I.; Yu, L.; Russo, J.J.; Ju, J.; Chasin, L.A. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011, 21, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhir, A.; Buratti, E. Alternative splicing: Role of pseudoexons in human disease and potential therapeutic strategies: Minireview. FEBS J. 2010, 277, 841–855. [Google Scholar] [CrossRef]
- Sironi, M.; Menozzi, G.; Riva, L.; Cagliani, R.; Comi, G.P.; Bresolin, N.; Giorda, R.; Pozzoli, U. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004, 32, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Pozzoli, U.; Sironi, M. Silencers regulate both constitutive and alternative splicing events in mammals. Cell. Mol. Life Sci. 2005, 62, 1579–1604. [Google Scholar] [CrossRef]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef]
- Romano, M.; Buratti, E.; Baralle, D. Role of Pseudoexons and Pseudointrons in Human Cancer. Int. J. Cell Biol. 2013, 2013, 810572. [Google Scholar] [CrossRef] [PubMed]
- Montalban, G.; Bonache, S.; Moles-fernández, A.; Gisbert-beamud, A.; Tenés, A.; Bach, V.; Carrasco, E.; López-fernández, A.; Stjepanovic, N.; Balmaña, J.; et al. Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet. 2018, 56, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Buratti, E.; Stuani, C.; Bendix, R.; Dörk, T.; Baralle, F.E. A new type of mutation causes a splicing defect in ATM. Nat. Genet. 2002, 30, 426–429. [Google Scholar] [CrossRef]
- Özkan, S.; Padilla, N.; Moles-Fernández, A.; Diez, O.; Gutiérrez-Enríquez, S.; de la Cruz, X. The computational approach to variant interpretation: Principles, results, and applicability. In Clinical DNA Variant Interpretation: Theory and Practice; Lázaro, C., Lerner-Ellis, J., Spurdle, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 1, ISBN 9780128205198. [Google Scholar]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Moles-Fernández, A.; Duran-Lozano, L.; Montalban, G.; Bonache, S.; López-Perolio, I.; Menéndez, M.; Santamariña, M.; Behar, R.; Blanco, A.; Carrasco, E.; et al. Computational tools for splicing defect prediction in breast/ovarian cancer genes: How efficient are they at predicting RNA alterations? Front. Genet. 2018, 9, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, A.B.; Patwardhan, R.P.; Shendure, J.; Seelig, G. Learning the Sequence Determinants of Alternative Splicing from Millions of Random Sequences. Cell 2015, 163, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Theiss, S.; Otte, M.; Widera, M.; Peter, J.O.; Schaal, H. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014, 42, 10681–10697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubeuf, H.; Charbonnier, C.; Soukarieh, O.; Blavier, A.; Lefebvre, A.; Dauchel, H.; Frebourg, T.; Gaildrat, P.; Martins, A. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mutat. 2020, 41, 1811–1829. [Google Scholar] [CrossRef]
- Canson, D.; Glubb, D.; Spurdle, A.B. Variant effect on splicing regulatory elements, branchpoint usage, and pseudoexonization: Strategies to enhance bioinformatic prediction using hereditary cancer genes as exemplars. Hum. Mutat. 2020, 41, 1705–1721. [Google Scholar] [CrossRef]
- Rowlands, C.F.; Baralle, D.; Ellingford, J.M. Machine Learning Approaches for the Prioritization of Genomic Variants Impacting Pre-mRNA Splicing. Cells 2019, 8, 1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Tang, L.; Xie, Z.; Sun, C.; Shuai, H.; Zhou, C.; Liu, Y.; Yu, M.; Zheng, Y.; Meng, L.; et al. Splicing characteristics of dystrophin pseudoexons and identification of a novel pathogenic intronic variant in the DMD gene. Genes 2020, 11, 1180. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Buratti, E.; Van Santen, M.A.; Lührmann, R.; Baralle, F.E. The intronic splicing code: Multiple factors involved in ATM pseudoexon definition. EMBO J. 2010, 29, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Lee, K.S.; Choi, J.K. Comprehensive characterisation of intronic mis-splicing mutations in human cancers. Oncogene 2021, 40, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, M.T.; Beemon, K.L. Splicing of internal large exons is defined by novel cis -acting sequence elements. Nucleic Acids Res. 2012, 40, 9244–9254. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, N.; Suyama, M. In silico identification of pseudo-exon activation events in personal genome and transcriptome data. RNA Biol. 2021, 18, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases. Front. Genet. 2021, 12, 647400. [Google Scholar] [CrossRef]
- Riepe, T.V.; Khan, M.; Roosing, S.; Cremers, F.P.M.; Hoen, P.A.C. Benchmarking deep learning splice prediction tools using functional splice assays. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Wai, H.A.; Lord, J.; Lyon, M.; Gunning, A.; Kelly, H.; Cibin, P.; Seaby, E.G.; Spiers-Fitzgerald, K.; Lye, J.; Ellard, S.; et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet. Med. 2020, 22, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Baeza-Centurion, P.; Miñana, B.; Valcárcel, J.; Lehner, B. Mutations primarily alter the inclusion of alternatively spliced exons. eLife 2020, 9, 1–74. [Google Scholar] [CrossRef]
- de Jong, L.C.; Cree, S.; Lattimore, V.; Wiggins, G.A.R.; Spurdle, A.B.; Miller, A.; Kennedy, M.A.; Walker, L.C. Nanopore sequencing of full-length BRCA1 mRNA transcripts reveals co-occurrence of known exon skipping events. Breast Cancer Res. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anczuków, O.; Buisson, M.; Léońe, M.; Coutanson, C.; Lasset, C.; Calender, A.; Sinilnikova, O.M.; Mazoyer, S. BRCA2 deep intronic mutation causing activation of a cryptic exon: Opening toward a new preventive therapeutic strategy. Clin. Cancer Res. 2012, 18, 4903–4909. [Google Scholar] [CrossRef] [Green Version]
- Montalban, G.; Bonache, S.; Moles-Fernández, A.; Gadea, N.; Tenés, A.; Torres-Esquius, S.; Carrasco, E.; Balmaña, J.; Diez, O.; Gutiérrez-Enríquez, S. Incorporation of semi-quantitative analysis of splicing alterations for the clinical interpretation of variants in BRCA1 and BRCA2 genes. Hum. Mutat. 2019, 40, 2296–2317. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Splicing Altering Variants | No Splicing Altering Variants | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|---|
| All variants | 133 | 100 | 93.99 | 92.00 | 93.13 | 0.86 |
| Cryptic splice | 117 | 100 | 95.73 | 92.00 | 94.01 | 0.88 |
| SREs altering | 16 | 100 | 81.25 | 92.00 | 90.52 | 0.66 |
| Pipeline | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| (1) ∆ SpliceAI ≥ 0.05 | 93.98 | 92.00 | 93.13 | 0.86 |
| (2) ∆ SpliceAI ≥ 0.05 + SpliceAI < 0.05 and ∆ESRseq ≥ 0.63 | 96.24 | 69.00 | 84.55 | 0.69 |
| (3) ∆ SpliceAI ≥ 0.05 + SpliceAI < 0.05 and ∆ESRseq ≥ 0.63 and Abs. Dif. 0.51 | 95.49 | 86.00 | 91.42 | 0.83 |
| Gene * | cNomenclature ** | Intron | SpliceAI † (Position of Predicted Splice site) | ∆ESRseq ‡ | ABS dif. | Splicing Outcome § | Population Variant Frequencies (GnomAD) | ClinVar Review Status ¥ |
|---|---|---|---|---|---|---|---|---|
| ATM | c.1899-123A > G | 12 | AG 0.15 (−51 bp) and AG 0.74 (−90 bp)/DG 0.71 (−1 bp) | 0.633 | 1.217 | Three pseudoexons: ▼12A.1 (r.1899_1900ins1899-174_1899-124), ▼12A.2 (r.1899_1900ins1899-177_1899-124) ▼12A.3 (r.1899_1900ins1899-213_1899-124) | 0.000032 | NR |
| c.2466+1552G > C | 16 | AG 0.93 (3 bp)/DG 0.69 (97 bp) | −0.144 | 1.816 | Pseudoexon ▼16A (r.2466_2467ins2466 + 1555_2466 + 1650) | NR | Likely pathogenic |1| | |
| c.8850+2029A > G | 61 | AG 0.22(1 bp)/DG 0.16 (102 bp) | −0.187 | 0.765 | Pseudoexon▼61A (r.8850_8851ins8850 + 2030_8850 + 2131) | NR | NR | |
| FAM175A | c.476+158G > T | 5 | AG 0.17 (2 bp)/DG 0.22 (−94 bp) | 0.559 | 0.591 | Pseudoexon ▼5A (r.476_477ins476 + 156_476 + 252 | 0.000446 | NR |
| MUTYH | c.998-27G > A | 11 | AG 0.41 (−4 bp)/DG 0.05 (−215 bp) | 0.344 | 1.000 | Intron retention ▼12A (r.997_998ins998-23 + 998) | 0.001192 | Likely benign|Likely benign 1|1 |
| Pipeline | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|
| Splice ≥ 0.05 | 100 | 71.42 | 75.75 | 0.62 |
| Splice ≥ 0.05; ESRseq ≥ 0.63; Abs. Dif. 0.51 | 100 | 50.00 | 57.57 | 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moles-Fernández, A.; Domènech-Vivó, J.; Tenés, A.; Balmaña, J.; Diez, O.; Gutiérrez-Enríquez, S. Role of Splicing Regulatory Elements and In Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes. Cancers 2021, 13, 3341. https://doi.org/10.3390/cancers13133341
Moles-Fernández A, Domènech-Vivó J, Tenés A, Balmaña J, Diez O, Gutiérrez-Enríquez S. Role of Splicing Regulatory Elements and In Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes. Cancers. 2021; 13(13):3341. https://doi.org/10.3390/cancers13133341
Chicago/Turabian StyleMoles-Fernández, Alejandro, Joanna Domènech-Vivó, Anna Tenés, Judith Balmaña, Orland Diez, and Sara Gutiérrez-Enríquez. 2021. "Role of Splicing Regulatory Elements and In Silico Tools Usage in the Identification of Deep Intronic Splicing Variants in Hereditary Breast/Ovarian Cancer Genes" Cancers 13, no. 13: 3341. https://doi.org/10.3390/cancers13133341