
Combining APR-246 and HDAC-Inhibitors: A Novel Targeted Treatment Option for Neuroblastoma

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals
2.3. Western Blot
2.4. Real-Time PCR
2.5. Flow Cytometry Analysis
2.6. Metabolic Activity Assays
2.7. Trypan Blue Assay
2.8. Zebrafish Lines
2.9. Cell Preparation for Zebrafish Embryo Xenotransplantation
2.10. Zebrafish Embryo Drug Toxicity Assay
2.11. Zebrafish Embryo Drug Treatment and Efficiency Evaluation
2.12. In Silico Data Analysis and Genetic Signature Generation
2.13. Stastical Analyses
3. Results
3.1. Pure TP53 Mutation Status Is Not a Sufficient Biomarker for APR-246 Sensitivity of Pediatric Tumors of the Nervous System
3.2. APR-246 Impairs Oxygene Species Elimination and Basal Reactive Oxygen Species Level Indicate APR-246 Sensitivity
3.3. A Novel Biomarker Signature Improves Response Predicition for Neuroblastoma Cell Lines and Patients
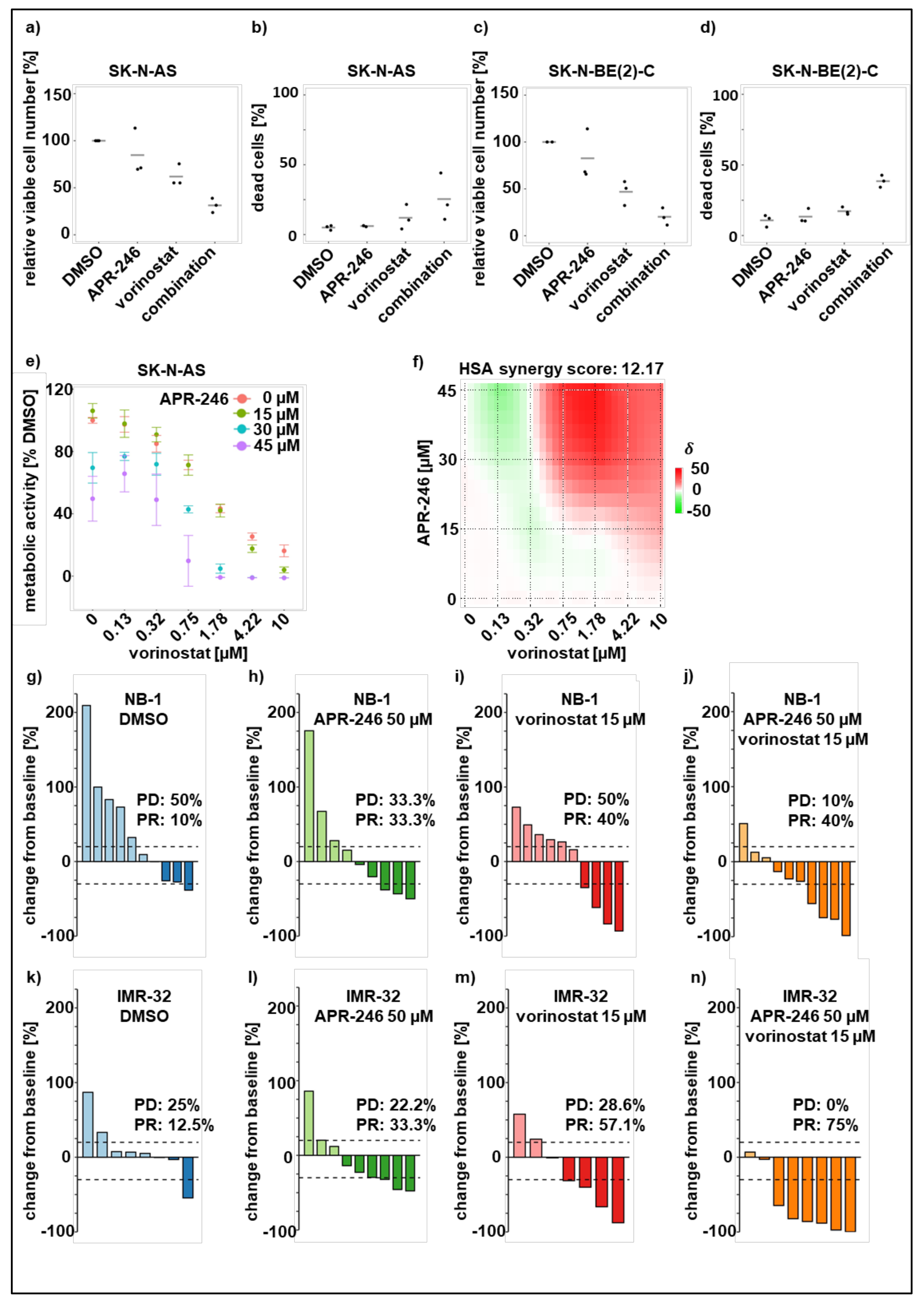
3.4. Sensitizing Low-Responsive Neuroblastoma Cells to APR-246 Using HDAC-Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG task force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Neuroblastoma Treatment (PDQ®)—Health Professional Version. Available online: https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq#_534_toc (accessed on 1 February 2021).
- Wasylishen, A.R.; Lozano, G. Attenuating the p53 pathway in human cancers: Many means to the same end. Cold Spring Harb Perspect. Med. 2016, 6, a026211. [Google Scholar] [CrossRef]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweddle, D.A.; Malcolm, A.J.; Cole, M.; Pearson, A.D.; Lunec, J. p53 cellular localization and function in neuroblastoma: Evidence for defective G(1) arrest despite WAF1 induction in MYCN-amplified cells. Am. J. Pathol. 2001, 158, 2067–2077. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Hardman, R.; Carman, T.A.; Barrett, J.C.; Weissman, B.E. Differential subcellular p53 localization and function in N- and S-type neuroblastoma cell lines. Cell Growth Differ. 1998, 9, 545–555. [Google Scholar] [PubMed]
- Moll, U.M.; LaQuaglia, M.; Bénard, J.; Riou, G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl. Acad. Sci. USA 1995, 92, 4407–4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Aprea Therapeutics Website: Our Clinical Trials. Available online: https://www.aprea.com/clinical-trials/ (accessed on 1 February 2021).
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andrén, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system x(C)(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC family members intertwined in the regulation of autophagy: A druggable vulnerability in aggressive tumor entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef] [Green Version]
- Fabian, J.; Lodrini, M.; Oehme, I.; Schier, M.C.; Thole, T.M.; Hielscher, T.; Kopp-Schneider, A.; Opitz, L.; Capper, D.; von Deimling, A.; et al. GRHL1 acts as tumor suppressor in neuroblastoma and is negatively regulated by MYCN and HDAC3. Cancer Res. 2014, 74, 2604–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, J.; Oehme, I.; Mazitschek, R.; Korshunov, A.; Kool, M.; Hielscher, T.; Kiss, J.; Selt, F.; Konrad, C.; Lodrini, M.; et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 2015, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Lodrini, M.; Brady, N.R.; Witt, O. Histone deacetylase 10-promoted autophagy as a druggable point of interference to improve the treatment response of advanced neuroblastomas. Autophagy 2013, 9, 2163–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Linke, J.P.; Bock, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Witt, O.; Deubzer, H.E.; Lodrini, M.; Milde, T.; Oehme, I. Targeting histone deacetylases in neuroblastoma. Curr. Pharm. Des. 2009, 15, 436–447. [Google Scholar] [CrossRef]
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Muller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K.; et al. The HDAC6/8/10 inhibitor TH34 induces DNA damage-mediated cell death in human high-grade neuroblastoma cell lines. Arch. Toxicol. 2018, 92, 2649–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, e510. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Witt, O.; Milde, T.; Deubzer, H.E.; Oehme, I.; Witt, R.; Kulozik, A.; Eisenmenger, A.; Abel, U.; Karapanagiotou-Schenkel, I. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin. Padiatr. 2012, 224, 398–403. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 322968, Prima-1. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Prima-1 (accessed on 8 May 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 52918385, Prima-1met. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Prima-1met (accessed on 8 May 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 322973, 2-Methylenequinuclidin-3-one. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2-Methylenequinuclidin-3-one (accessed on 8 May 2021).
- Jemaà, M.; Sime, W.; Abassi, Y.; Lasorsa, V.A.; Bonne Køhler, J.; Michaelis, M.; Cinatl, J., Jr.; Capasso, M.; Massoumi, R. Gene expression signature of acquired chemoresistance in neuroblastoma cells. Int. J. Mol. Sci. 2020, 21, 5811. [Google Scholar] [CrossRef] [PubMed]
- Sapio, R.T.; Nezdyur, A.N.; Krevetski, M.; Anikin, L.; Manna, V.J.; Minkovsky, N.; Pestov, D.G. Inhibition of post-transcriptional steps in ribosome biogenesis confers cytoprotection against chemotherapeutic agents in a p53-dependent manner. Sci. Rep. 2017, 7, 9041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalized medicine for high-risk pediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.L.; Monje, M. A protocol for rapid post-mortem cell culture of Diffuse Intrinsic Pontine Glioma (DIPG). J. Vis. Exp. 2017, 9, 5233–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of pediatric solid tumors. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Witt, O.; Monkemeyer, S.; Ronndahl, G.; Erdlenbruch, B.; Reinhardt, D.; Kanbach, K.; Pekrun, A. Induction of fetal hemoglobin expression by the histone deacetylase inhibitor apicidin. Blood 2003, 101, 2001–2007. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, J.K.; Najafi, S.; Ayhan, S.; Gatzweiler, C.; Krunic, D.; Ridinger, J.; Milde, T.; Westermann, F.; Peterziel, H.; Meder, B.; et al. Rapid in vivo validation of HDAC inhibitor-based treatments in neuroblastoma zebrafish xenografts. Pharmaceuticals 2020, 13, 345. [Google Scholar] [CrossRef]
- Aksoy, B.A.; Dancík, V.; Smith, K.; Mazerik, J.N.; Ji, Z.; Gross, B.; Nikolova, O.; Jaber, N.; Califano, A.; Schreiber, S.L.; et al. CTD2 Dashboard: A searchable web interface to connect validated results from the Cancer Target Discovery and Development Network. Database 2017, 11, 7400. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zou, Y.; Xu, G.; Potter, J.A.; Taylor, G.L.; Duan, Q.; Yang, Q.; Xiong, H.; Qiu, H.; Ye, D.; et al. PRIMA-1Met suppresses colorectal cancer independent of p53 by targeting MEK. Oncotarget 2016, 7, 83017–83030. [Google Scholar] [CrossRef] [Green Version]
- Aryee, D.N.T.; Niedan, S.; Ban, J.; Schwentner, R.; Muehlbacher, K.; Kauer, M.; Kofler, R.; Kovar, H. Variability in functional p53 reactivation by PRIMA-1Met/APR-246 in Ewing sarcoma. Br. J. Cancer 2013, 109, 2696–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depmap Release: Broad, D. Public_21q1. 2021. Available online: https://doi.org/10.6084/m9.figshare.13681534.v1 (accessed on 5 January 2021). [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, F.; Manasa, K.L.; Shaik, S.P.; Bonam, S.R.; Kamal, A. Zinc dependent histone deacetylase inhibitors in cancer therapeutics: Recent update. Curr. Med. Chem. 2019, 26, 7212–7280. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Jurkovic Mlakar, S.; Lesne, L.; Marino, D.; Rathi, K.S.; Maris, J.M.; Ansari, M.; Gumy-Pause, F. PRIMA-1(MET)-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J. Exp. Clin. Cancer Res. 2019, 38, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demir, S.; Boldrin, E.; Sun, Q.; Hampp, S.; Tausch, E.; Eckert, C.; Ebinger, M.; Handgretinger, R.; Kronnie, G.T.; Wiesmüller, L.; et al. Therapeutic targeting of mutant p53 in pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, A.; Fiveash, J.B.; Yang, E.S. Combined targeting of mutant p53 and Jumonji family histone demethylase augments therapeutic efficacy of radiation in H3K27M DIPG. Int. J. Mol. Sci. 2020, 21, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Rosa, J.; Urdiciain, A.; Zelaya, M.V.; Zazpe, I.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. APR-246 combined with 3-deazaneplanocin A, panobinostat or temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2021, 58, 312–330. [Google Scholar] [CrossRef]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef] [PubMed]
- Groningen, T.; Koster, J.; Valentijn, L.; Zwijnenburg, D.; Akogul, N.; Hasselt, N.; Broekmans, M.; Haneveld, F.; Nowakowska, N.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Cen, J.; Zhang, L.; Liu, F.; Zhang, F.; Ji, B.S. Long-term alteration of reactive oxygen species led to multidrug resistance in MCF-7 cells. Oxid Med. Cell Longev. 2016, 2016, 7053451. [Google Scholar] [CrossRef]
- Waghela, B.N.; Vaidya, F.U.; Pathak, C. Upregulation of NOX-2 and Nrf-2 promotes 5-Fluorouracil resistance of human colon carcinoma (HCT-116) cells. Biochemistry 2021, 86, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Quan, J.; Tan, Y.; Liu, Y.; Liao, C.; Li, Z.; Liao, W.; Liu, J.; Cao, Y.; Luo, X. RIP3 mediates TCN-induced necroptosis through activating mitochondrial metabolism and ROS production in chemotherapy-resistant cancers. Am. J. Cancer Res. 2021, 11, 729–745. [Google Scholar] [PubMed]
- Halliwell, B. Cell culture, oxidative stress, and antioxidants: Avoiding pitfalls. Biomed. J. 2014, 37, 99–105. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Company | Stock Conc., Diluent |
|---|---|---|
| APR-246 | Selleck Chemicals, Munich, Germany | 50 mM, DMSO |
| doxorubicin | Biozol, Eching, Germany | 10 mM, H2O |
| vincristine | Enzo Life Sciences, Lörrach, Germany | 10 mg/mL, DMSO |
| vorinostat | Selleck Chemicals, Munich, Germany | 100 mM, DMSO |
| entinostat | Biomol GmbH, Hamburg, Germany | 10 mM, DMSO |
| abexinostat | Selleck Chemicals, Munich, Germany | 10 mM, DMSO |
| Panobinostat | Cayman Chemical, Hamburg, Germany | 0.5 mM, DMSO |
| romidepsin | Biovision Inc., Milpitas, CA, USA | 1 mM, DMSO |
| H2O2 | Sigma-Aldrich, Munich, Germany | 30% w/w H2O |
| DMSO | Sigma-Aldrich, Munich, Germany | - |
| Cell Line | Entity/TP53 Status | IC50/95% CI [µM] |
|---|---|---|
| SJ-GBM2 | pHGG 1/mut, p.R273C | 71.8/69.3–74.4 |
| SF188 | pHGG/mut, p.G266E | 43.9/39.4–48.8 |
| KNS-42 | pHGG/mut, p.R342 *,2 | 27.8/22.8–33.7 |
| SK-N-BE(2)-C | neuroblastoma/mut, p.C135F | 24.7/22.0–27.7 |
| IMR-32 NIH-3T3 C5-GL (TP53 ko) NIH-3T3 Δ6RT (TP53 wt) | neuroblastoma/wt murine embryo fibroblasts murine embryo fibroblasts | 4.0/3.2–4.8 18.4/15.8–21.3 18.7/17.6–19.9 |
| Cell Line | Entity | IC50/95% CI [µM] | SLC7A11 Expression 3 |
|---|---|---|---|
| Kelly | Neuroblastoma | 8.48/6.8–10.0 | 1.43 |
| NB-1 | Neuroblastoma | 4.83/4.22–5.42 | 1.35 |
| SH-EP | Neuroblastoma | 44.5/40.1–49.5 | - |
| SK-N-FI | Neuroblastoma | 9.21/8.02–10.1 | 0.88 |
| NALM-6 | ALL 1 | 4.56/3.09–6.85 | 2.51 |
| REH | ALL 1 | 4.39/4.0–4.84 | 1.41 |
| HuT 78 | NHL 2 | 9.47/8.23–10.74 | 1.45 |
| Model Type | Name/Entity | Biomarker | IC50/95% CI [µM] |
|---|---|---|---|
| Established cell lines | SK-N-AS/neuroblastoma | 1.03 | 35.1/29.2–43.1 |
| SIMA/neuroblastoma | 1.80 | 2.04/1.74–2.4 | |
| SH-SY5Y/neuroblastoma | 1.68 | 19.7/16.8–22.4 | |
| KNS-42/pHGG | 0.90 | 27.8/22.8–33.7 | |
| Jurkat/ALL | 3.20 | 1.46/1.38–1.54 | |
| JeKo-1/NHL | 5.78 | 1.44/1.32–1.58 | |
| INFORM long-term cultures INFORM-PDX long-term culture | INF_R_1288_LTC/rhabdoid tumor | 1.09 | 19.7/14.9–25.8 |
| INF_R_1467_LTC/soft tissue sarcoma | 1.58 | 18.4/16.9–20.0 | |
| INF_R_1490_LTC/osteosarcoma | 1.19 | 28.7/25.5–32.0 | |
| INF_R_1632_PDX_LTC/neuroblastoma | 3.66 | 12.2/8.7–16.6 |
| Cell Line | HDAC Inhibitor | IC50 [nM] | 95% CI [nM] | + APR-246 [µM] | IC50 [nM] | 95% CI [nM] |
|---|---|---|---|---|---|---|
| SK-N-AS | abexinostat | 46.3 | 29.0–69.8 | 20 | 15.4 | 9.7–23.8 |
| romidepsin | 5.7 | 4.9–6.6 | 20 | 1.8 | 1.5–2.2 | |
| Panobinostat | 2.2 | 1.6–2.9 | 20 | 1.0 | 0.6–1.4 | |
| SK-N-BE(2)-C | abexinostat | 172.1 | 142.2–207.5 | 10 | 37.5 | 22.5–58.9 |
| romidepsin | 26.9 | 15.2–140.9 | 10 | 9.9 | 3.6–52.9 | |
| Panobinostat | 40.2 | 29.4–54.6 | 20 | 3.1 | 0.3–12.2 | |
| IMR-32 | abexinostat | 100.0 | 56.3–169.7 | 2 | 30.4 | 6.6–48.9 |
| romidepsin | 0.73 | 0.64–0.83 | 1 | 0.24 | 0.13–0.33 | |
| Panobinostat | 35.6 | 29.0–43.7 | 2 | 13.9 | 11.0–17.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, M.; Rösch, L.; Najafi, S.; Gatzweiler, C.; Ridinger, J.; Gerloff, X.F.; Jones, D.T.W.; Baßler, J.; Kreth, S.; Stainczyk, S.; et al. Combining APR-246 and HDAC-Inhibitors: A Novel Targeted Treatment Option for Neuroblastoma. Cancers 2021, 13, 4476. https://doi.org/10.3390/cancers13174476
Müller M, Rösch L, Najafi S, Gatzweiler C, Ridinger J, Gerloff XF, Jones DTW, Baßler J, Kreth S, Stainczyk S, et al. Combining APR-246 and HDAC-Inhibitors: A Novel Targeted Treatment Option for Neuroblastoma. Cancers. 2021; 13(17):4476. https://doi.org/10.3390/cancers13174476
Chicago/Turabian StyleMüller, Michael, Lisa Rösch, Sara Najafi, Charlotte Gatzweiler, Johannes Ridinger, Xenia F. Gerloff, David T. W. Jones, Jochen Baßler, Sina Kreth, Sabine Stainczyk, and et al. 2021. "Combining APR-246 and HDAC-Inhibitors: A Novel Targeted Treatment Option for Neuroblastoma" Cancers 13, no. 17: 4476. https://doi.org/10.3390/cancers13174476