
The Evolving Role of Ferroptosis in Breast Cancer: Translational Implications Present and Future
by
 ,
,
Hung-Yu Lin
1,2,† ,
,
Hui-Wen Ho
2,†,
Yen-Hsiang Chang
3,4,
Chun-Jui Wei
5,* and
Pei-Yi Chu
1,5,6,7,8,* 1
Graduate Institute of Biomedical Engineering, National Chung Hsing University, Taichung 402, Taiwan
2
Research Assistant Center, Show Chwan Memorial Hospital, Changhua 500, Taiwan
3
Department of Nuclear Medicine, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung 833, Taiwan
4
Center for Mitochondrial Research and Medicine, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung 833, Taiwan
5
Department of Pathology, Show Chwan Memorial Hospital, Changhua 500, Taiwan
6
School of Medicine, College of Medicine, Fu Jen Catholic University, New Taipei City 242, Taiwan
7
Department of Health Food, Chung Chou University of Science and Technology, Changhua 510, Taiwan
8
National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan
*
Authors to whom correspondence should be addressed.
†
These authors contribute equally to this work.
Cancers 2021, 13(18), 4576; https://doi.org/10.3390/cancers13184576
Submission received: 18 August 2021
/
Revised: 10 September 2021
/
Accepted: 10 September 2021
/
Published: 12 September 2021
(This article belongs to the Special Issue Interconnectivity of Cell Death Pathways in Cancer)
Abstract
:Simple Summary
Despite decades of extensive study into targeting cell death in breast cancer, including apoptosis, the clinical treatment remains challenging due to its high probability of recurrence. As an emerging form of cell death, ferroptosis features suppression of drug resistance and augmentation of antitumor immunity. The advances in the development of clinical drugs targeting ferroptosis provide growing silver linings for breast cancer treatment. Research into biomarkers to precisely trace ferroptosis in patients with cancer, and the development and subsequent application of novel ferroptosis-based therapies will be of critical importance in the next few years.
Abstract
Breast cancer (BC) is the most common malignancy among women worldwide. The discovery of regulated cell death processes has enabled advances in the treatment of BC. In the past decade, ferroptosis, a new form of iron-dependent regulated cell death caused by excessive lipid peroxidation has been implicated in the development and therapeutic responses of BC. Intriguingly, the induction of ferroptosis acts to suppress conventional therapy-resistant cells, and to potentiate the effects of immunotherapy. As such, pharmacological or genetic modulation targeting ferroptosis holds great potential for the treatment of drug-resistant cancers. In this review, we present a critical analysis of the current understanding of the molecular mechanisms and regulatory networks involved in ferroptosis, the potential physiological functions of ferroptosis in tumor suppression, its potential in therapeutic targeting, and explore recent advances in the development of therapeutic strategies for BC.
1. Introduction
Breast cancer (BC) is the most commonly diagnosed cancer among women, and is the fourth leading cause of cancer deaths worldwide, according to a status report on the global cancer burden provided by GLOBOCAN 2020 [1]. To date, the standard treatments for patients with BC include surgery, radiation therapy, hormone therapy, and chemotherapy [2,3]. The cause of death in patients with BC is primarily related to cancer metastasis and relapse, which are associated with metabolic reprogramming that fosters a corrupted tumor microenvironment (TME) to counteract therapy-induced cell death [4]. Regulated cell death (RCD) is an autonomous and orderly death. In addition to apoptosis and necroptosis, recent studies have revealed new modes of RCD, including pyroptosis and ferroptosis [5,6,7,8]. All of these death modes present distinct features in terms of cellular morphology, biochemistry, and signaling pathways (Table 1). Despite decades of extensive study into targeting cancer cell death, such as approaches targeting caspases and BCL-2 families in apoptosis, the clinical implementation of related therapeutic agents remains challenging [9]. Indeed, cancer cells present resistance against apoptotic cellular death [10]. Therefore, targeting a nonapoptotic RCD may offer an alternative path to the development of effective cancer therapeutics.
Apoptosis can be triggered by extrinsic (also known as death receptor-activated) and intrinsic (also known as mitochondrial or BCL-2 regulated) pathways. The extrinsic pathway can be activated by the ligation of tumor necrosis factor receptor (TNFR) superfamily members, which promotes adaptor proteins (e.g., FADD) to activate caspase-8 and then the downstream effector caspase-3 and -7 [11]. The intrinsic pathway can be induced by intrinsic stress (growth factor deprivation, DNA damage, and endoplasmic reticulum stress), and BH3-only proteins (PUMA, NOXA, BIM, BID, BAD) [12,13]. For example, p53-upregulated PUMA can bind with a high affinity to BCL-2, thereby liberating BAX/BAK to the mitochondria. This results in the formation of mitochondrial outer membrane permeabilization (MOMP) and the released cytochrome c binding to APAF-1 to form an apoptosome, leading to apoptosis. Under the induction of endoplasmic reticulum stress, the conformational activation of BAX/BAK acts at the mitochondrial membrane, thereby relaying the signaling for the assembly of the apoptosome [14]. In necroptosis, tumor necrosis factor α (TNFα), the CD95 receptor/Fas ligand complex, and other members of the TNF superfamily were identified as inducers [15]. Receptor-interacting protein kinase 1 (RIPK1), RIPK3 and the mixed lineage kinase domain-like pseudokinase (MLKL) are required proteins for the activation of necroptosis. In response to death receptor activation, the binding of RIPK1 to RIPK3 triggers the formation of necrosomes, resulting in MLKL activation [8]. As a necroptotic effector, the activated MLKL translocates to the plasma membrane, causing permeabilization and subsequent cell death. The predominant hallmarks of pyroptosis are the activation of an inflammasome, a cytosolic multiprotein complex accounting for the release of interleukin-1β (IL-1β) and IL-18, the formation of an apoptosis-associated speck-like protein containing a CARD (ASC), and the activation of proinflammatory cascades [16]. Generally, pattern recognition receptors (PRRs, e.g., nod-like receptor 3 (NLRP3) and absent in melanoma-like receptor 2 (AIM2)) first recognize a variety of dangerous signals, then activate procaspase-1 cleavage and ASC recruitment to assemble inflammasomes. Activated caspase-1 acts to cleave the pyroptosis executor gasdemin D (GSDMD) at the Asp275 site to free the N-terminal domain (GSDMD-NT) and generate nonselective pores on the cell membrane. Meanwhile, caspase-1 cleaves and activates the precursors of IL-1β and IL-18 to produce mature IL-1β and IL-18. The intracellular contents are then released through pores caused by GSDMD-NT, leading to pyroptosis [16]. In addition, an inflammasome-independent, non-canonical pathway mediated by a caspase-1/4/5/11-cleaved GSDMD-NT was recognized [17,18,19]. Furthermore, caspase-3, an iconic apoptosis-related caspase, was revealed to trigger gasdemin E (GSDME)-dependent pyroptosis under the scenario of chemotherapy drug treatment [20,21]. Ferroptosis is characterized by an iron-dependent manner of RCD, described in detail below. Table 1 summarizes the basic features and representative signaling molecules of the four RCDs.
In terms of anticancer immunity, apoptosis has mainly been considered as tolerogenic cell death (TCD), while some reports suggested that apoptosis is involved in immunogenic cell death (ICD) [15,22]. In contrast, mounting evidence has revealed that ICD can be mediated by the activation of ferroptosis, necroptosis, and pyroptosis [22]. The existence and clarification of the interconnectivity of various RCDs renders growing silver linings for the development of anticancer therapeutics. For example, signatures of pro-ferroptosis, pro-necroptosis, and pro-pyroptosis were reported to be associated with CD8+ T cell infiltration across seven common cancers [22]. The sensitivity of the genes involved in ferroptosis, necroptosis and pyroptosis were shown to be positively correlated with microsatellite instability (MSI) and tumor mutation burden (TMB) in a portion of cancers [22]. Therapeutically, clinically approved antitumor drugs that target immunity are reported to induce ICD by way of combined RCDs. For instance, artesunate was shown to simultaneously elicit ferroptosis and necroptosis in cancer cells [23] and exert a potentiation effect on antitumor immunity [24,25,26]. Doxorubicin was demonstrated to enhance antitumor immunity through the induction of both ferroptosis and pyroptosis. In a mechanistic regard, necroptosis acts to interconnectedly facilitate pyroptosis via activation of the receptor-interacting protein kinase 1 (RIPK1) pathway, which then activates the Nod-like receptor 3 (NLRP3)-caspase-1 pathway [27]. The concomitant induction of ferroptosis and pyroptosis was reported to be mediated by the perturbation of glutathione (GSH) and the activation of gasdermin E (GSDME) in the context of cisplatin exposure [28,29].
In 2012, Dixon et al. first described the concept of ferroptosis [30], which refers to an iron-dependent cell death caused by lipid peroxidation and subsequent plasma membrane rupture [31]. Differing from other types of RCD, ferroptosis does not present the cellular swelling observed in necroptosis and pyroptosis, nor the cellular shrinkage and formation of apoptotic bodies exhibited in apoptosis (Table 1). In terms of organellar morphology, ferroptosis does not exhibit chromatin condensation in the nucleus or cytoskeletal disintegration; it does, however, manifest a distinct disorganization of mitochondria including mitochondrial shrinkage, vanishing of mitochondrial cristae, and rupture of the outer mitochondrial membrane (OMM). A growing body of study has led to the identification of an intricate signaling pathway that controls ferroptosis by inducing iron accumulation and lipid peroxidation or perturbing the antioxidant defensive system. Importantly, recent reports have revealed that cancer cells which are resistant to conventional therapy or harbor a propensity to metastasize are vulnerable to ferroptosis, and that immunotherapeutic effects can be potentiated by ferroptosis [32,33,34,35]. In light of this, we herein aimed to review the latest research on ferroptosis to further the understanding of its pathogenesis and to propose new targets for the treatment of BC (Table 2).

{kind=link}
{kind=link}
Table 1.
Characteristics of ferroptosis, apoptosis, necroptosis, and pyroptosis.
| Characteristics | Ferroptosis | Apoptosis | Necroptosis | Pyroptosis |
|---|---|---|---|---|
| Morphological features | small mitochondria, | unaltered mitochondria | swollen mitochondria | unaltered mitochondria |
| vanishing mitochondrial cristae | apoptotic bodies | release of cytoplasmic constituents | Pore formation on plasma membrane | |
| OMM rupture | cytoskeletal disintegration | plasma membrane rupture | Inflammasome formation | |
| normal nucleus | chromatin condensation | chromatin condensation | Chromatin condensation | |
| normal cell size | shrinkage of cell | swollen cell | swollen cell | |
| Biochemical hallmarks | iron accumulation | PS exposure | No PS exposure * | secretion of IL-18 and IL-1β |
| lipid peroxidation | DNA fragmentation | depletion of ATP | ||
| Primary immune features | ICD | TCD | ICD | ICD |
| Signaling pathways | TFRC IREB/SLC11A2 | TNFRSF1A/FADD TRAILR1/2 Caspases-9/-3/-7 P53/PUMA BCL-2/BAX/BAK cytochrome c/APAF-1 endoplasmic reticulum | TNFR1/RIPK1 RIPK1/RIPK3/MLKL PKC-MAPK-AP-1 | release of mtDNA in cytosol NLRP3/AIM2 caspase-1/ IL-18/IL-1β capase-1/4/5/11/GSDMD caspase-3/ GSDME |
| Haem/HO-1 FTH1/FTL/Prominin 2 NCOA4/ferrotinophagy CISD1/2/Fe-S ACSLs/LPCAT3/ALOXs | ||||
| GSH/GPX4, MVA/HMGCR | ||||
| FSP1/CoQ10 DHODH/CoQ10 |
AIM2, absent in melanoma like receptor 2; AP-1, activator protein 1; APAF-1, apoptotic peptidase activating factor 1; BAK, Bcl-2 homologous antagonist/killer; BAX, Bcl-2-associated X; BCL2, B-cell lymphoma 2; FADD, Fas-associated protein with death domain; GSDMD, gasdermin D; GSDME, gasdermin E; ICD, immunogenic cell death; MAPK, mitogen-activated protein kinase; MLKL, mixed lineage kinase domain-like pseudokinase; NLRP3, Nod-like receptor 3; OMM, outer membrane of mitochondria; PKC, protein kinase C; PS, phosphatidylserine; PUMA, p53 upregulated modulator of apoptosis; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; TCD, tolerogenic cell death; TNFR1, tumor necrosis factor receptor 1; TNFRSF1A, tumor necrosis factor receptor superfamily member 1A; TRAILR, tumor necrosis factor-related apoptosis-inducing ligand receptor-1. * While Annexin V assay remains the most widely used assay for apoptosis detection based on PS exposure, several reports identified this phenomenon in necroptosis as well. Wang et al., in this regard, demonstrated a necroptotic phenotype with an increased PS exposure proportion, activated RIPK1/MLKL axis, and damaged plasma membrane [36].
Table 2.
Updated therapeutic approaches for targeting of ferroptotic pathways in BC.
| Target | Approaches | Phase of Clinical Development | Reference |
|---|---|---|---|
| Iron activators | |||
| ↑Transferrin | lapatinib | in vitro model | 28827805 [37] |
| ↑Transferrin | lapatinib | in vitro model | 27441659 [38] |
| ↓SLC40A1 | lapatinib | Marketed | NCT00667251 [38] |
| ↑Iron | UV light | Preclinical animal model | 32804509 [39] |
| ↑Iron | MOF-Fe2+ | Preclinical animal model | 32944722 [40] |
| ↑Iron | Fe3O4 nanocapcules | in vitro model | 34225869 [41] |
| ↑Iron | neratinib | Preclinical animal model | 31409375 [42] |
| ↑Iron | neratinib | Marketed | NCT04366713 |
| ↑Iron | neratinib | Marketed | NCT03377387 |
| ↑Iron | Fe3+-PDA NP | in vitro model | 33808898 [43] |
| ↓CISD1/2, ↑Iron | MAD-28 | Preclinical animal model | 25762074 [44] |
| ↑HO-1 | MI-463 | in vitro model | 32945449 [45] |
| ↑HO-1 | SGNI | Preclinical animal model | 33827043 [46] |
| Ferritinophagy activators | |||
| ↑Iron | artesunate | Phase I trials | NCT00764036 [47] |
| Lipid peroxides activators | |||
| ↑ACSL1 | α-eleostearic acid | Preclinical animal model | 33854057 [48] |
| ↑ACSL4 | Polyphyllin III | Preclinical animal model | 34040532 [49] |
| System xc- inhibitors | |||
| ↓SLC7A11 | Erastin | in vitro model | 33672555 [50] |
| ↓SLC7A11 | Lidocaine | in vitro model | 34122108 [51] |
| ↓SLC7A11 | miR-106a-5p | in vitro model | 33686957 [52] |
| ↓SLC7A11 | sulfasalazine | Phase I trials | NCT03847311 |
| ↓SLC7A11 | metformin, sulfasalazine | in vitro model | 34162423 [53] |
| ↓SLC7A11 | 18-β-glycyrrhetinic acid | in vitro model | 34271106 [54] |
| GPX4 pathway inhibitors | |||
| ↓GPX4 | JQ1+BTZ | Preclinical animal model | 32937365 [55] |
| ↓NRF2 | overexpression of GSK-3β | Preclinical animal model | 32642794 [56] |
| ↓GPX4 | RSL-3 | Preclinical animal model | 34170581 [57] |
| ↓GPX4 | metformin | Preclinical animal model | 33522578 [58] |
| ↓GPX4 | DMOCPTL | Preclinical animal model | 33472669 [59] |
| ↓GPX4, MDM2 | Compound 3d | Preclinical animal model | 33725632 [60] |
| ↓TYRO3 | LDC1267 | Preclinical animal model | 33855973 [32] |
| ↓DHODH | leflunomide | Preclinical animal model | 32034120 [61] |
| ↓DHODH | BQR, leflunomide, 4SC-101 | in vitro model | 28196676 [62] |
| HMGCR inhibitor | |||
| ↓HMGCR | fluvastatin | Phase I trials | 19728082 [63] |
| ↓HMCGR | atorvastatin | Phase II trials | NCT00816244 [64] |
↑, promote; ↓, inhibit; ACSL1/4, Acyl-CoA synthetase long chain family members; BQR, brequinar sodium; CISD1/2, CDGSH iron sulfur domain 1/2; DHODH, dihydroorotate dehydrogenase; GPX4, glutathione peroxidase 4; GSK-3β, glycogen synthase kinase 3 beta; HMGCR, HMG-CoA reductase; HO-1, heme oxygenase-1; MDM2, mouse double minute 2; MOF-Fe2+, metal-organic framework-based Fe2+ delivery; MI-463, menin-mixed-lineage leukemia (MLL) inhibitor-463; NRF2, nuclear factor, erythroid 2-like 2; PDA NPs, polydopamine nanoparticles; SGNI, Shuganning injection; SLC40A1, solute carrier family 40 member 1; SLC7A11, solute carrier family 7 member 11; TYRO3, tyrosine protein kinase receptor 3.
2. Inducing Ferroptosis by Iron Toxicity and Lipid Peroxides
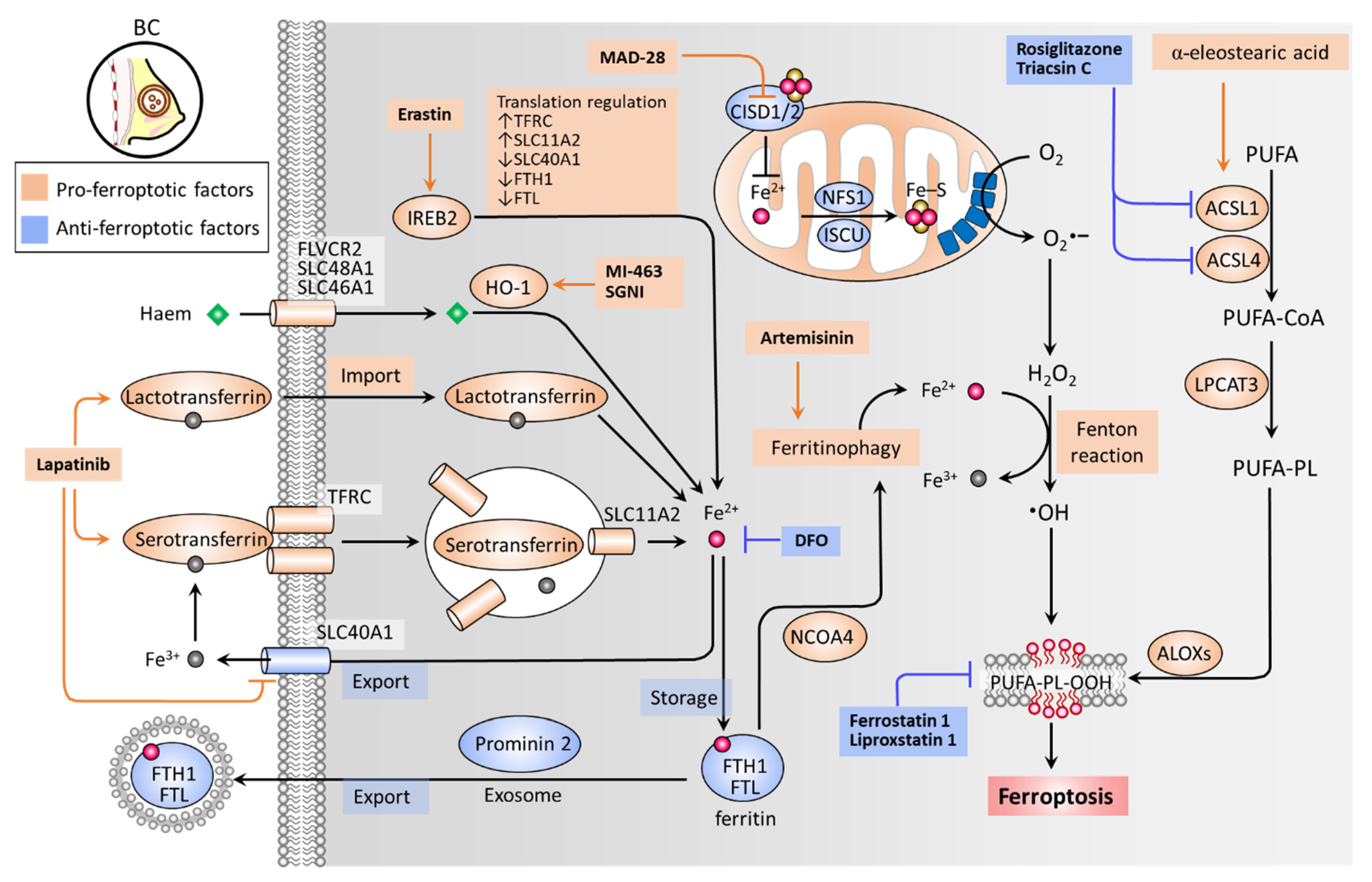
Iron accumulation and lipid peroxidation are two key hallmarks of ferroptosis [30]. Iron is an important trace element, while an aberrant distribution or content of iron in the body can lead to physiological disorders. Iron imported into a cell can be mediated by serotransferrin through the transferrin receptor (TFRC) (Figure 1) [65]. Iron-loaded serotransferrin-TFRC complexes are internalized through endosomes, where they release iron (Fe2+) into the cytoplasm through solute carrier family 11 member 2 (SLC11A2), leading to increased iron accumulation and subsequent induction of ferroptosis [65] (Figure 1). Lactotransferrin and heme provide additional sources of iron through differing import mechanisms in the cell membrane [31]. On the other hand, iron export mediated by solute carrier family 40 member 1 (SLC40A1) inhibits ferroptosis [66]. Knockdown of TFRC can inhibit erastin-induced ferroptosis [67], while heme oxygenase-1 (HO-1) can accelerate erastin-induced ferroptosis by supplementing iron [68]. Ferritin acts as an iron storage protein complex, which is composed of ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL) [69]. Brown et al. reported that prominin-2 acts to form ferritin-containing exosomes, which are exported out of the cell to prevent ferroptosis [70]. As a transcription factor, iron response element binding protein 2 (IREB2) acts to mediate the pro-ferroptosis effect of erastin by increasing expression levels of TFRC and SLC11A2, while decreasing those of SLC40A1, FTH1, and FTL [71]. Ferritinophagy (an autophagic degradation of ferritin), mediated by nuclear receptor coactivator 4 (NCOA4), can enhance intracellular iron (Fe2+) levels and ultimately result in ferroptosis [72,73] (Figure 1). In mitochondria, proteins involved in the utilization of iron for iron-sulfur cluster biogenesis, including cysteine desulfurase (NFS1), iron-sulfur cluster assembly enzyme (ISCU), CDGSH iron sulfur domain 1 (CISD1, also known as mitoNEET), and CISD2 (also known as nutrient-deprivation autophagy factor-1 (NAF-1)), inhibit ferroptosis by increasing the biosynthesis of iron-sulfur clusters (Fe-S), thereby reducing intracellular iron levels [74,75,76,77]. Intracellular iron excess can promote subsequent lipid peroxidation by way of at least two mechanisms: (1) the iron-dependent Fenton reaction that produces reactive oxygen species (ROS); and (2) the activation of iron-containing enzymes such as lipoxygenases (ALOXs) [31,78,79]. In the process of ferroptosis, polyunsaturated fatty acids (PUFAs) are most susceptible to lipid peroxidation, which can lead to a damaged membrane structure [80]. Acyl-CoA synthetase long chain family members (ACSLs) and lysophospholipid acyltransferase 3 (LPCAT3) promote the incorporation of polyunsaturated fatty acids (PUFAs) into phospholipids (PLs) to form polyunsaturated fatty acid-containing phospholipids (PUFA-PLs), which are sensitive to ROS-initiated oxidation mediated by ALOXs, leading to the formation of lipid peroxides (PUFA-PL-OOH), and ultimately ferroptosis [81,82,83,84] (Figure 1).
Zhu et al. have reported that irradiation-activated azobenzene combretastatin A4 (Azo-CA4)-loaded nanocarriers promote the ferroptosis of TNBC cells [39]. A UV light-triggered reduction of Fe3+ to Fe2+ induces ferroptosis, while the photoisomerization of Azo-CA4 elicits apoptosis. Xu et al. designed a metal–organic framework (MOF)-based Fe2+ delivery system to increase intracellular iron toxicity, whereby BC cells and in vivo tumors undergo ferroptosis [40]. Nieto et al. revealed that Fe3+ loaded-polydopamine nanoparticles induce ferroptosis by increasing intracellular levels of iron, exerting a synergetic effect on the combination of doxorubicin [43]. Antoniak et al. demonstrated that nanocapsules containing Fe3O4 induce BC cell ferroptosis [41]. Notably, TNBC (MDA-MB-231) cells were more susceptible to Fe3O4 nanocapsules than estrogen receptor (ER)-positive (MCF7) cells. Kato et al. reported that menin-mixed-lineage leukemia inhibitor MI-463 induces ferroptosis in an HO-1 activity-dependent manner [45]. Du et al. have shown that Shuganning injection (SGNI), a traditional Chinese patent medicine, can induce ferroptosis and in vivo tumor growth in TNBC cells by inducing HO-1, which acts to promote intracellular iron accumulation [46]. Ma et al. reported that lapatinib promotes the accumulation of intracellular iron, ROS expression levels, and ultimately ferroptosis by increasing expression levels of transferrin and decreasing SLC40A1 (also known as ferroportin-1) [37,38]. Nagpal et al. revealed that neratinib induces ferroptosis by increasing intracellular iron levels [42]. Induced ferritinophagy has been reported to mediate ferroptosis and the antitumor effect of artemisinins [85,86,87], which are derived from the Chinese herb Artemisia annua, categorized as a class of antimalarial drugs [88]. An artemisinin derivative, artesunate, was demonstrated as being safe and well-tolerated at an oral dose of up to 200 mg when applied to patients with metastatic BC (NCT00764036; completed phase I clinical trial) [47]. Bai et al. revealed that a derivative of mitocan (small molecules selectively targeting mitochondria), MAD-28, acts to promote ferroptosis by inhibiting CISD1 and CISD2, and increasing mitochondrial iron levels [44]. Zhou et al. demonstrated that Polyphyllin III, a major saponin extracted from Paris polyphylla rhizomes, promotes ferroptosis of TNBC cells through ACSL4-mediated lipid peroxidation [49]. The combination of Polyphyllin III and sulfasalazine (an inhibitor of system xc-) exhibited a potentiation effect in an in vivo tumor growth model. Beatty et al. reported that α-eleostearic acid acts toward an increase in lipid peroxidation and ferroptosis of BC cells by promoting ACSL1 [48]. The molecular basis and drugs targeting iron toxicity and lipid peroxidation of ferroptosis in BC are summarized in Figure 1, and the current therapeutic approaches aiming to induce iron accumulation and lipid peroxidation are summarized in Table 2.
3. Inducing Ferroptosis by Inhibiting Antioxidant Defense
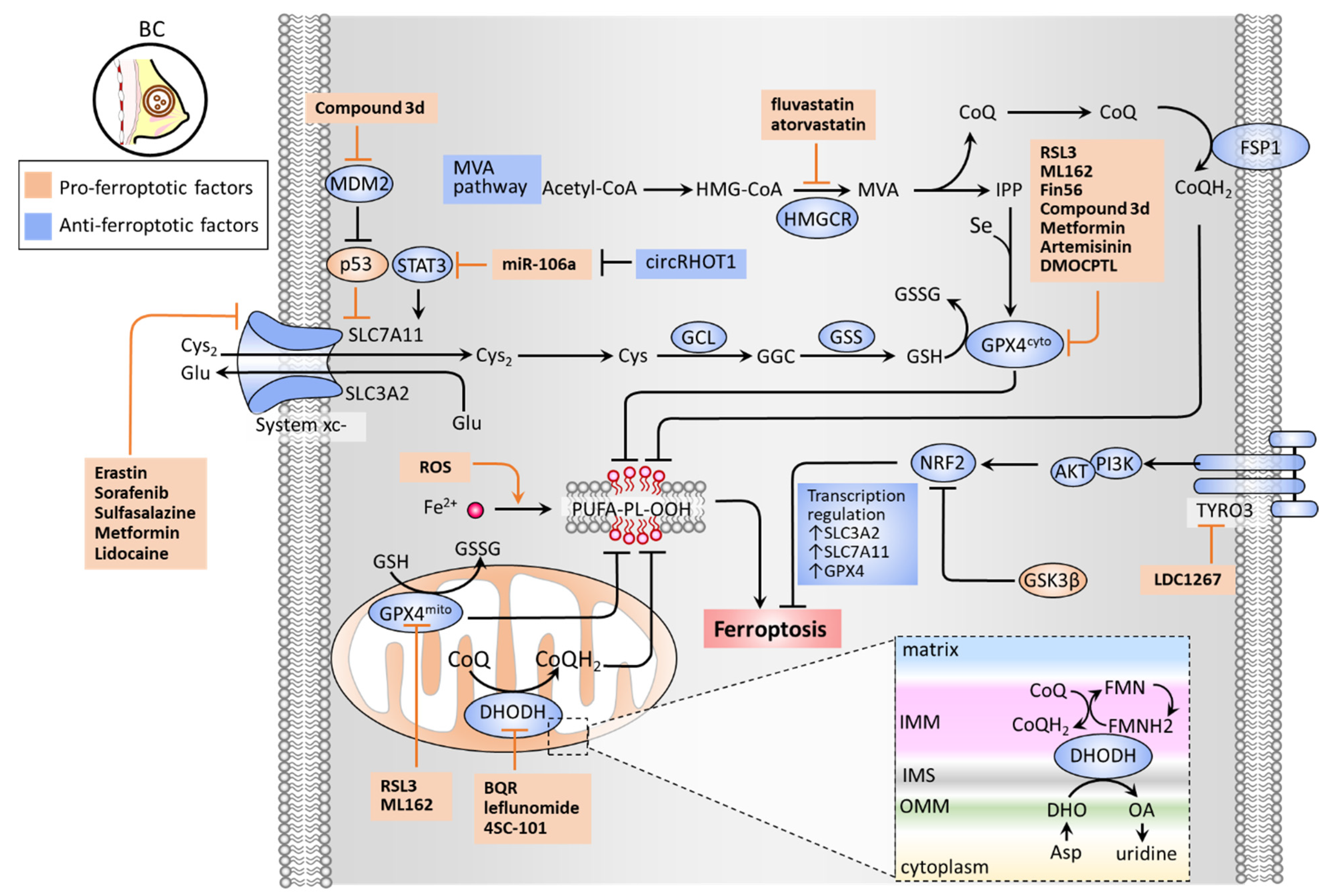
There are several classes of antioxidant pathways that counteract ferroptosis. These include the glutathione (GSH)-dependent phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) pathway in the cytosol (GPX4cyto) and mitochondria (GPX4mito), while the GSH-independent coenzyme Q10 (CoQ10, also known as ubiquinone) pathway is underpinned by ferroptosis suppressor protein 1 (FSP1, also known as apoptosis-inducing factor mitochondrial 2 (AIF-M2)) at the plasma membrane (FSP1-CoQ10 axis) and dihydroorotate dehydrogenase (DHODH) in the mitochondrial inner membrane (DHODH-CoQ10 axis) (Figure 2) [89,90].
The synthesis of GSH relies mainly on the import of cystine (Cys2). System xc− is a cystine/glutamate antiporter widely distributed in phospholipid bilayers, acting to import Cys2 into cells with a 1:1 counter-transport of glutamate [6,30] and preserve the homeostasis of the antioxidant system in cells. System xc− is a heterodimer composed of two subunits: solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2). The Cys2 taken up into cells can then be oxidized to cysteine (Cys), which is required for the synthesis of GSH in a reaction catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS) [31]. GSH functions to reduce reactive oxygen species (ROS) and reactive nitrogen under the action of glutathione peroxidases (GPXs). Among the GPX family, GPX4 plays a critical role in regulating the occurrence of ferroptosis. GPX4 can convert GSH into oxidized glutathione (GSSG) and reduce cytotoxic lipid peroxides (L-OOH) to the corresponding alcohols (L-OH), thereby inhibiting the formation of lipid peroxides (Figure 2). RSL3 and ML162 can serve as GPX4 inhibitors to promote cell ferroptosis [90]. The mevalonate (MVA) pathway counteracts ferroptosis by generating anti-ferroptotic biomolecules, including isopentenyl-pyrophosphate (IPP) and CoQ10. The synthetic processes of the two molecules require a rate-limiting enzyme, HMG-CoA reductase (HMGCR), which is also an inhibitory target of statins (a class of cholesterol-lowering drugs) [35]. IPP acts to stabilize selenocysteine tRNA, which is required for the synthesis of GPX4 [91]. Concerning the GPX4-independent CoQ10 pathway, Bersuker et al. first identified that FSP-1, a flavoprotein formerly known as AIF-M2 (apoptosis-inducing factor mitochondrial 2), exhibits a protective effect against ferroptosis, as induced by GPX4 deletion [92]. At the plasma membrane, FSP-1 acts as an oxidoreductase that reduces CoQ10 to generate CoQH2 (also known as ubiquinol) which can repair lipid peroxides [93]. More recently, Mao et al. reported that the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH) acts to coordinate with GPX4mito to prevent ferroptosis by detoxifying accumulated lipid peroxides in mitochondria [89]. DHODH has been recognized as an iron-containing flavin-dependent enzyme, and is involved in the de novo synthesis of pyrimidines in mitochondria [94]. Further study revealed that DHODH generates CoQH2 by reducing CoQ10 through a uridine-synthesizing redox reaction that catalyzed dihydroorotate to orotate [89]. DHODH inhibitors have previously been used in the treatment of autoimmune diseases, such as multiple sclerosis and rheumatoid arthritis [90]. The findings of Mao et al. regarding the role of DHODH in ferroptosis could exploit the synthetic lethal (SL) concept, by which DHODH inhibitors exert an anticancer effect in GPX4low cells; meanwhile, the combination of DHODH inhibitors with sulfasalazine (an inhibitor of system xc−) can be applied to GPXhigh cells [89].
Wen et al. reported that an active compound from the medicinal herbal licorice, 18-β-glycyrrhetinic acid, could induce ferroptosis in TNBC cells by downregulating the expression of SLC7A11 of system xc-, the GSH level, the GPX activity, and upregulating the ROS and lipid peroxidation [54]. Sun et al. reported that lidocaine exerts a pro-ferroptosis effect in BC and ovarian cancer by targeting the miR-382-5p/SLC7A11 axis [51]. Zhang et al. revealed that microRNA(miR)-106a-5p promotes ferroptosis via the suppression of the signal transducer and activator of transcription 3 (STAT3), whereby SLC7A11 levels are downregulated in BC cells [52]. Circular RNA RHOT1 (circRHOT1) has been identified as a ferroptosis inhibitor by sponging miR-106-5a [52]. Liu et al. revealed a nitroisoxazole-containing compound (3d) that can effectively induce ferroptosis of MCF-7 both in vitro and in vivo [60]. As a GPX4/mouse double minute 2 (MDM2) dual inhibitor, 3d presents dual inhibitory activities with the suppression of GPX4 levels and MDM2-mediated degradation of p53 [60]. Wu et al. demonstrated that overexpression of glycogen synthase kinase-3β (GSK-3β) sensitizes erastin-induced ferroptosis by inhibition of a nuclear factor, erythroid 2-like 2 (NFE2L2, also known as NRF2), thereby downregulating GPX4 levels [56]. It is worth noting that upregulation of NRF2 can lead to ferroptosis resistance. Qiao et al. demonstrated that upregulated NRF2 contributed to BET inhibitor-induced ferroptosis in BC cancer cells [95]. siRNA-based knockdown of nuclear receptor subfamily 5 group A member 2 (NR5A2) and nuclear receptor coactivator 3 (NCOA3) caused a decrease in NRF2 expression levels, thereby counteracting BET inhibitor-induced ferroptosis. Verma et al. demonstrated a high-throughput screen reliant on the concept of SL which reported that a combination of BET inhibitor JQ1 and proteasome inhibitor bortezomib (BTZ) (JQ1+BTZ) considerably induced TNBC ferroptosis by inhibiting GPX4 expression levels [55]. Ding et al. demonstrated that DMOCPTL, a derivative of the natural product parthenolide, exhibited a pro-ferroptosis effect on TNBC cell growth by inducing GPX4 ubiquitination [59]. Lee et al. reported that erastin induces TNBC cell ferroptosis by inhibiting system xc− and depleting GPX4 levels, despite the resistance against oxidative stress [50]. Jiang et al. reported that tyrosine-protein kinase receptor 3 (TYRO3) plays a negative role on tumor ferroptosis, and induces resistance to anti-PD1/PD-L1 treatment [32]. TYRO3 overexpression activates the PI3K/AKT pathway to increase NRF2 transcriptional activity that is responsible for the transcription of ferroptosis-inhibitory genes, including SLC3A2, SLC7A11, FTL, FTH1, GPX4, SLC40A1, and biliverdin reductase A/B (BLVRA/B) [32]. Further study showed that the TYRO3 inhibitor, LDC1267, can elicit ferroptosis and potentiate immunotherapy in a 4T1 BC cell inoculation mouse model [32]. Song et al. developed acidity-activatable dynamic nanoparticles encapsulating RSL-3 to target GPX4, unveiling a strategy for ferroptosis-inducing tumor specific delivery. This nanoparticle approach acts to potentiate immunotherapy by recruiting tumor-infiltrating T lymphocytes for interferon gamma (IFNγ) secretion [57]. Hou et al. demonstrated that metformin induces TNBC cell ferroptosis via an upregulation of miR-324-3p, by which GPX4 expression is inhibited by targeting its 3′UTR [58]. In addition, Yang et al. revealed that antidiabetic metformin acts as an anti-BC agent by reducing the protein stability of SLC7A11 of system xc-, increasing intracellular Fe2+ and lipid ROS levels [53]. Furthermore, the combination of metformin with sulfasalazine, the system xc- inhibitor, can cause a potentiation effect to induce ferroptosis [53]. Concerning the blocking of the MVA pathway as a targeting strategy, two clinical trials involving BC patients have indicated that fluvastatin and atorvastatin might have antiproliferative effects in tumors overexpressing HMGCR [63,64]. Hubackova et al. reported that the DHODH inhibitor leflunomide exerts a potentiation effect on inducing TNBC cell death in vitro, and tumor growth in vivo, when combined with a checkpoint kinase 1 (Chk1) inhibitor [61]. Mohama Fairus et al. demonstrated that DHODH inhibitors, including brequinar sodium (BQR), leflunomide, and 4SC-101 provoked ROS generation and ATP depletion by a p53-mediated route, thereby suppressing BC cell proliferation [62]. The molecular basis and drugs targeting the antioxidant defense systems of ferroptosis in BC, including GPX4/GSH, MVA, FSP1-CoQ10, and DHODH-CoQ10, are illustrated in Figure 2. The current therapeutic approaches for targeting these pathways are summarized in Table 2.
4. Conclusions
There exist several tumor subtypes of BC, which are highly heterogenous malignancies, and considered particularly challenging to treat due to their resilience to standard therapeutic agents and high probability of recurrence. The past five years have seen a growing number of studies focused on the role of ferroptosis in BC. These investigations have further clarified our understanding of ferroptosis, which involves the integration of a highly organized system that regulates iron metabolism, lipid peroxidation, and anti-oxidant defense, relying on specialized mechanisms in the plasma membrane, mitochondria, and cytosol. The accumulated knowledge has been exploited to further improve the prevention and treatment of this disease. For instance, mounting preclinical evidence indicates that the induction of ferroptosis may be an effective therapeutic strategy to prevent acquired resistance to several cancer therapies, and to exert a potentiation effect on immunotherapy [32,42,49]. More importantly, clinical trials that apply ferroptosis-inducing agents to patients with BC are ongoing. As such, it is inevitable that continuing research in this field will further elucidate the physiological and pathological roles of ferroptosis, leading to the development of translational anticancer strategies. Research into biomarkers to precisely trace ferroptosis in patients with cancer, and the development and subsequent application of novel ferroptosis-based therapies will be of critical importance in the next few years.
Author Contributions
Conceptualization, H.-Y.L. and H.-W.H.; methodology, Y.-H.C.; software, H.-W.H.; validation, C.-J.W. and P.-Y.C.; formal analysis, H.-W.H.; investigation, H.-Y.L. and Y.-H.C.; resources, C.-J.W. and P.-Y.C.; data curation, H.-W.H.; writing—original draft preparation, H.-Y.L.; writing—review and editing, C.-J.W. and P.-Y.C.; visualization, H.-W.H. and Y.-H.C.; supervision, P.-Y.C.; project administration, H.-Y.L., H.-W.H. and P.-Y.C.; funding acquisition, H.-W.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology, Taiwan (MOST 106-2314-B-442-001-MY3, MOST 109-2314-B-442-001- and MOST 109-2314-B-075B-002), National Health Research Institutes, Taiwan (NHRI-109BCCO-MF-202015-01), and Show Chwan Memorial Hospital, Taiwan (SRD-109035, SRD-110016 and SRD-110017).
Acknowledgments
The authors would like to thank James Waddell for his assistance with the proofreading of this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wen, X.F.; Guo, Y.X.; Zhang, G.J. Targeting Notch degradation system provides promise for breast cancer therapeutics. Crit. Rev. Oncol. Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Ogilvie, J.M.; Wilson, J.S.; Green, H.J.; Chambers, S.K.; Ownsworth, T.; Shum, D.H. A meta-analysis of cognitive impairment and decline associated with adjuvant chemotherapy in women with breast cancer. Front. Oncol. 2015, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ansari, R.; McIntyre, A.; Craze, M.L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Altered glutamine metabolism in breast cancer; subtype dependencies and alternative adaptations. Histopathology 2018, 72, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A new frontier in cancer. Biomed. Pharmacother 2020, 121, 109595. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef]
- Khan, I.; Yousif, A.; Chesnokov, M.; Hong, L.; Chefetz, I. A decade of cell death studies: Breathing new life into necroptosis. Pharmacol Ther. 2021, 220, 107717. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Puthalakath, H.; Strasser, A. Keeping killers on a tight leash: Transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002, 9, 505–512. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, T.; Gao, J.; Li, P.; Wang, Y.; Qi, Q.; Liu, X.; Li, J.; Wang, C.; Du, L. Pyroptosis, metabolism, and tumor immune microenvironment. Clin. Transl. Med. 2021, 11, e492. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Gasdermin D: The long-awaited executioner of pyroptosis. Cell Res. 2015, 25, 1183–1184. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.X.; Liu, Z.N.; Ye, J.; Sha, M.; Qian, H.; Bu, X.H.; Luan, Z.Y.; Xu, X.L.; Huang, A.H.; Yuan, D.L.; et al. Artesunate exerts an anti-immunosuppressive effect on cervical cancer by inhibiting PGE2 production and Foxp3 expression. Cell Biol. Int. 2014, 38, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Feng, H.; Shi, X.; Wang, Y.; Feng, Z.; Liu, J.; Han, Z.; Fu, J.; Fu, Z.; Tong, H. Artesunate down-regulates immunosuppression from colorectal cancer Colon26 and RKO cells in vitro by decreasing transforming growth factor beta1 and interleukin-10. Int. Immunopharmacol. 2015, 27, 110–121. [Google Scholar] [CrossRef]
- Qian, P.; Zhang, Y.W.; Zhou, Z.H.; Liu, J.Q.; Yue, S.Y.; Guo, X.L.; Sun, L.Q.; Lv, X.T.; Chen, J.Q. Artesunate enhances gammadelta T-cell-mediated antitumor activity through augmenting gammadelta T-cell function and reversing immune escape of HepG2 cells. Immunopharmacol. Immunotoxicol. 2018, 40, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Li, C.G.; Wang, Y.F.; Xu, L.H.; He, X.H.; Zeng, Q.Z.; Zeng, C.Y.; Mai, F.Y.; Hu, B.; Ouyang, D.Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Jiang, Z.; Lim, S.O.; Yan, M.; Hsu, J.L.; Yao, J.; Wei, Y.; Chang, S.S.; Yamaguchi, H.; Lee, H.H.; Ke, B.; et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Investig. 2021, 131, e139434. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.J.; Meng, X.Y.; Chen, J.F.; Wang, K.Y.; Zhou, C.; Yu, R.; Ma, Q. Emodin Induced Necroptosis and Inhibited Glycolysis in the Renal Cancer Cells by Enhancing ROS. Oxid. Med. Cell Longev. 2021, 2021, 8840590. [Google Scholar]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Dai, P.; Liu, F.; Li, Y.; Qin, Y.; Yang, Q.; Tian, R.; Fan, A.; Medeiros, S.F.; Wang, Z.; et al. Upconverting Nanocarriers Enable Triggered Microtubule Inhibition and Concurrent Ferroptosis Induction for Selective Treatment of Triple-Negative Breast Cancer. Nano Lett. 2020, 20, 6235–6245. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Y.; Zhang, Y.; Yao, Y.; Ji, P. Highly stable and biocompatible hyaluronic acid-rehabilitated nanoscale MOF-Fe(2+) induced ferroptosis in breast cancer cells. J. Mater. Chem B 2020, 8, 9129–9138. [Google Scholar] [CrossRef]
- Antoniak, M.A.; Pazik, R.; Bazylinska, U.; Wiwatowski, K.; Tomaszewska, A.; Kulpa-Greszta, M.; Adamczyk-Grochala, J.; Wnuk, M.; Mackowski, S.; Lewinska, A.; et al. Multimodal polymer encapsulated CdSe/Fe3O4 nanoplatform with improved biocompatibility for two-photon and temperature stimulated bioapplications. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 127, 112224. [Google Scholar] [CrossRef]
- Nagpal, A.; Redvers, R.P.; Ling, X.; Ayton, S.; Fuentes, M.; Tavancheh, E.; Diala, I.; Lalani, A.; Loi, S.; David, S.; et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019, 21, 94. [Google Scholar] [CrossRef]
- Nieto, C.; Vega, M.A.; Martin Del Valle, E.M. Tailored-Made Polydopamine Nanoparticles to Induce Ferroptosis in Breast Cancer Cells in Combination with Chemotherapy. Int. J. Mol. Sci. 2021, 22, 3161. [Google Scholar] [CrossRef]
- Bai, F.; Morcos, F.; Sohn, Y.S.; Darash-Yahana, M.; Rezende, C.O.; Lipper, C.H.; Paddock, M.L.; Song, L.; Luo, Y.; Holt, S.H.; et al. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, I.; Kasukabe, T.; Kumakura, S. MeninMLL inhibitors induce ferroptosis and enhance the antiproliferative activity of auranofin in several types of cancer cells. Int. J. Oncol. 2020, 57, 1057–1071. [Google Scholar] [PubMed]
- Du, J.; Wang, L.; Huang, X.; Zhang, N.; Long, Z.; Yang, Y.; Zhong, F.; Zheng, B.; Lan, W.; Lin, W.; et al. Shuganning injection, a traditional Chinese patent medicine, induces ferroptosis and suppresses tumor growth in triple-negative breast cancer cells. Phytomedicine 2021, 85, 153551. [Google Scholar] [CrossRef] [PubMed]
- von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsasser, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res. Treat. 2017, 164, 359–369. [Google Scholar] [CrossRef]
- Beatty, A.; Singh, T.; Tyurina, Y.Y.; Tyurin, V.A.; Samovich, S.; Nicolas, E.; Maslar, K.; Zhou, Y.; Cai, K.Q.; Tan, Y.; et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat. Commun. 2021, 12, 2244. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, J.; Chen, C.; Li, Z.; Chen, Y.; Zhang, X.; Wang, L.; Zhou, J. Polyphyllin -Induced Ferroptosis in MDA-MB-231 Triple-Negative Breast Cancer Cells can Be Protected Against by KLF4-Mediated Upregulation of xCT. Front. Pharmacol. 2021, 12, 670224. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Carlisle, A.E.; Peppers, A.; Park, S.J.; Doshi, M.B.; Spears, M.E.; Kim, D. xCT-Driven Expression of GPX4 Determines Sensitivity of Breast Cancer Cells to Ferroptosis Inducers. Antioxidants 2021, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Y.C.; Zhang, X.Y. Lidocaine Promoted Ferroptosis by Targeting miR-382-5p /SLC7A11 Axis in Ovarian and Breast Cancer. Front. Pharmacol. 2021, 12, 681223. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, Z.; Wang, Z.; Gao, Y.; Wang, Y.; Qu, X. Circular RNA RHOT1 promotes progression and inhibits ferroptosis via mir-106a-5p/STAT3 axis in breast cancer. Aging (Albany NY) 2021, 13, 8115–8126. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Xie, S.; Wang, J.; Li, Z.; Chen, L.; Mao, M.; Chen, C.; Huang, A.; Chen, Y.; et al. Metformin induces Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 206. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, H.; Zhang, L.; Wu, M.; Zhang, F.; Yang, D.; Shen, J.; Chen, J. Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple-negative breast cancer cells. Free Radic. Biol. Med. 2021, 173, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, eaba8968. [Google Scholar] [CrossRef]
- Wu, X.; Liu, C.; Li, Z.; Gai, C.; Ding, D.; Chen, W.; Hao, F.; Li, W. Regulation of GSK3beta/Nrf2 signaling pathway modulated erastin-induced ferroptosis in breast cancer. Mol. Cell Biochem. 2020, 473, 217–228. [Google Scholar] [CrossRef]
- Song, R.; Li, T.; Ye, J.; Sun, F.; Hou, B.; Saeed, M.; Gao, J.; Wang, Y.; Zhu, Q.; Xu, Z.; et al. Acidity-Activatable Dynamic Nanoparticles Boosting Ferroptotic Cell Death for Immunotherapy of Cancer. Adv. Mater. 2021, 33, e2101155. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Cai, S.; Yu, S.; Lin, H. Metformin induces ferroptosis by targeting miR-324-3p/GPX4 axis in breast cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2021, 53, 333–341. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, X.; Liu, C.; Ge, W.; Wang, Q.; Hao, X.; Wang, M.; Chen, Y.; Zhang, Q. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J. Hematol. Oncol. 2021, 14, 19. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhao, Q.; Peng, C.; Mao, Q.; Wu, F.; Zhang, F.H.; Feng, Q.S.; He, G.; Han, B. Design, synthesis, and biological evaluation of nitroisoxazole-containing spiro[pyrrolidin-oxindole] derivatives as novel glutathione peroxidase 4/mouse double minute 2 dual inhibitors that inhibit breast adenocarcinoma cell proliferation. Eur. J. Med. Chem. 2021, 217, 113359. [Google Scholar] [CrossRef]
- Hubackova, S.; Davidova, E.; Boukalova, S.; Kovarova, J.; Bajzikova, M.; Coelho, A.; Terp, M.G.; Ditzel, H.J.; Rohlena, J.; Neuzil, J. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 2020, 11, 110. [Google Scholar] [CrossRef]
- Mohamad Fairus, A.K.; Choudhary, B.; Hosahalli, S.; Kavitha, N.; Shatrah, O. Dihydroorotate dehydrogenase (DHODH) inhibitors affect ATP depletion, endogenous ROS and mediate S-phase arrest in breast cancer cells. Biochimie 2017, 135, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Garwood, E.R.; Kumar, A.S.; Baehner, F.L.; Moore, D.H.; Au, A.; Hylton, N.; Flowers, C.I.; Garber, J.; Lesnikoski, B.A.; Hwang, E.S.; et al. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res. Treat. 2010, 119, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjarnadottir, O.; Romero, Q.; Bendahl, P.O.; Jirstrom, K.; Ryden, L.; Loman, N.; Uhlen, M.; Johannesson, H.; Rose, C.; Grabau, D.; et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res. Treat. 2013, 138, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.; Devraj, K.; Ingram, J.; Slagle-Webb, B.; Madhankumar, A.B.; Liu, X.; Klinger, M.; Simpson, I.A.; Connor, J.R. Ferritin: A novel mechanism for delivery of iron to the brain and other organs. Am. J. Physiol. Cell Physiol. 2007, 293, C641–C649. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxid. Med. Cell Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Zhang, Z.; Chen, L.; Zhou, Y.; Zou, P.; Feng, C.; Wang, L.; Liang, G. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016, 381, 165–175. [Google Scholar] [CrossRef]
- Ooko, E.; Saeed, M.E.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Klayman, D.L. Qinghaosu (artemisinin): An antimalarial drug from China. Science 1985, 228, 1049–1055. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Wang, F.; Min, J. DHODH tangoing with GPX4 on the ferroptotic stage. Signal. Transduct. Target. Ther. 2021, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bermudez, J.; Birsoy, K. A mitochondrial gatekeeper that helps cells escape death by ferroptosis. Nature 2021, 593, 514–515. [Google Scholar] [CrossRef]
- Qiao, J.; Chen, Y.; Mi, Y.; Jin, H.; Huang, T.; Liu, L.; Gong, L.; Wang, L.; Wang, Q.; Zou, Z. NR5A2 synergizes with NCOA3 to induce breast cancer resistance to BET inhibitor by upregulating NRF2 to attenuate ferroptosis. Biochem. Biophys. Res. Commun. 2020, 530, 402–409. [Google Scholar] [CrossRef]
Figure 1.
Iron metabolism and lipid peroxides in ferroptosis. Ferroptosis is primarily triggered by iron accumulation-mediated lipid peroxidation, which is determined by key factors contributing to the import, export, and metabolism of iron and the formation of PUFA-PL-OOH. Serotransferrin-TFRC complexes load iron and facilitate its import through endosome internalization, by which iron can be released via SLC11A2. IREB2 transcriptional activity, heme/HO-1, and lactotransferrin provide additional routes of intracellular iron import. DFO acts as a scavenger of intracellular iron. FTH1 and FTL together form an iron storage protein complex (ferritin), preventing Fe2+ from being oxidized to Fe3+ by the Fenton reaction. Prominin-2 acts to form FTH1/FTL/iron complex-containing exosomes, which are then exported to the extracellular space. NCOA4 plays a role in inducing autophagic degradation of FTH1/FTL (ferritinophagy), leading to an increase in intracellular iron. ACSLs and LPCAT3 facilitate the formation of PUFA-PLs, which are susceptible to •OH-catalyzed oxidation that is mediated by ALOXs. Mitochondria serve as a main source of intracellular superoxide (O2•−), which can be reduced to H2O2 by dismutase. The partially reduced H2O2 by iron generates •OH, a process called the Fenton reaction. •OH can act to abstract hydrogen atoms from PUFA-PLs to activate lipid peroxidation and the accumulation of PUFA-PL-OOH, ultimately causing ferroptosis. •OH, hydroxyl radical; ACSL, acyl-CoA synthetase long chain family members; ALOXs, lipoxygenases; CISD1/2, CDGSH iron sulfur domain 1/2; DFO, desferoxamine; FTH1, ferritin heavy chain 1; FTL, ferritin light chain; H2O2, hydrogen peroxide; HO-1, heme oxygenase-1; IREB2, iron response element binding protein 2; ISCU, iron-sulfur cluster assembly enzyme; LPCAT3, lysophospholipid acyl-transferase 3; NCOA4, nuclear receptor co-activator 4; NFS1, NFS1 cysteine desulfurase; PUFA-PL-OOH, lipid peroxides generated from polyunsaturated fatty acid-containing phospholipids; SGNI, Shuganning injection; SLC11A2, solute carrier family 11 member 2; SLC40A1, solute carrier family 40 member 1; TFRC, transferrin receptor.
Figure 1.
Iron metabolism and lipid peroxides in ferroptosis. Ferroptosis is primarily triggered by iron accumulation-mediated lipid peroxidation, which is determined by key factors contributing to the import, export, and metabolism of iron and the formation of PUFA-PL-OOH. Serotransferrin-TFRC complexes load iron and facilitate its import through endosome internalization, by which iron can be released via SLC11A2. IREB2 transcriptional activity, heme/HO-1, and lactotransferrin provide additional routes of intracellular iron import. DFO acts as a scavenger of intracellular iron. FTH1 and FTL together form an iron storage protein complex (ferritin), preventing Fe2+ from being oxidized to Fe3+ by the Fenton reaction. Prominin-2 acts to form FTH1/FTL/iron complex-containing exosomes, which are then exported to the extracellular space. NCOA4 plays a role in inducing autophagic degradation of FTH1/FTL (ferritinophagy), leading to an increase in intracellular iron. ACSLs and LPCAT3 facilitate the formation of PUFA-PLs, which are susceptible to •OH-catalyzed oxidation that is mediated by ALOXs. Mitochondria serve as a main source of intracellular superoxide (O2•−), which can be reduced to H2O2 by dismutase. The partially reduced H2O2 by iron generates •OH, a process called the Fenton reaction. •OH can act to abstract hydrogen atoms from PUFA-PLs to activate lipid peroxidation and the accumulation of PUFA-PL-OOH, ultimately causing ferroptosis. •OH, hydroxyl radical; ACSL, acyl-CoA synthetase long chain family members; ALOXs, lipoxygenases; CISD1/2, CDGSH iron sulfur domain 1/2; DFO, desferoxamine; FTH1, ferritin heavy chain 1; FTL, ferritin light chain; H2O2, hydrogen peroxide; HO-1, heme oxygenase-1; IREB2, iron response element binding protein 2; ISCU, iron-sulfur cluster assembly enzyme; LPCAT3, lysophospholipid acyl-transferase 3; NCOA4, nuclear receptor co-activator 4; NFS1, NFS1 cysteine desulfurase; PUFA-PL-OOH, lipid peroxides generated from polyunsaturated fatty acid-containing phospholipids; SGNI, Shuganning injection; SLC11A2, solute carrier family 11 member 2; SLC40A1, solute carrier family 40 member 1; TFRC, transferrin receptor.

Figure 2.
Antioxidant defense system that regulates ferroptosis. GSH-dependent pathways (GSH/GPX4 and MVA) and GSH-independent pathways (FSP1/CoQ10 and DHODH/CoQ10) constitute an antioxidant defense system. System xc− composed of SLC3A2 and SLC7A11 is a cystine/glutamate antiporter that imports Cys2 into cells with an equal amount of counter-transport of glutamate. The Cys2 is oxidized to cysteine (Cys), which contributes to the synthesis of GSH in a reaction catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). Cytosolic and mitochondrial GPX4 (GPX4cyto and GPX4mito) eliminates ROS by converting GSH to GSSG. The MVA pathway requires HMGCR as a critical rate-limiting enzyme to generate IPP and CoQ10. IPP is involved in the synthesis of GPX4 in the presence of Se, while CoQ10 plays an important role in suppressing lipid peroxidation. FSP1 repairs lipid peroxidation on the plasma membrane by converting CoQ10 to CoQH2. By contrast, DHODH generates CoQH2 by reducing CoQ10 through a uridine-synthesizing redox reaction that catalyzes DHO to OA to exert a protective effect on lipid peroxidation at the mitochondrial membrane. TYRO3 acts toward anti-ferroptosis by the PI3K/AKT/NRF2 axis to bolster the activity of GSH/GPX4. Abbreviations: BQR, brequinar sodium; circRHOT1, circular RNA RHOT1; CoQ10, coenzyme Q10; Cys, cysteine; Cys2, cystine; DHO, dihydroorotate; DHODH, dihydroorotate dehydrogenase; FSP1, ferroptosis suppressor protein 1; GCL, glutamate-cysteine ligase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSK3B, glycogen synthase kinase 3 beta; GSS, glutathione synthetase; HMGCR, HMG-CoA reductase; IMM, inner membrane of mitochondria; IMS, intermembrane space; IPP, isopentenyl-pyrophosphate; MDM2, mouse double minute 2; MVA, mevalonate; NRF2, nuclear factor, erythroid 2-like 2; OA, orotate; ROS, reactive oxygen species; OMM, outer membrane of mitochondria; Se, selenium; SLC3A2, solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; STAT3, signal transducer and activator of transcription 3; TYRO3, tyrosine-protein kinase receptor 3.
Figure 2.
Antioxidant defense system that regulates ferroptosis. GSH-dependent pathways (GSH/GPX4 and MVA) and GSH-independent pathways (FSP1/CoQ10 and DHODH/CoQ10) constitute an antioxidant defense system. System xc− composed of SLC3A2 and SLC7A11 is a cystine/glutamate antiporter that imports Cys2 into cells with an equal amount of counter-transport of glutamate. The Cys2 is oxidized to cysteine (Cys), which contributes to the synthesis of GSH in a reaction catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). Cytosolic and mitochondrial GPX4 (GPX4cyto and GPX4mito) eliminates ROS by converting GSH to GSSG. The MVA pathway requires HMGCR as a critical rate-limiting enzyme to generate IPP and CoQ10. IPP is involved in the synthesis of GPX4 in the presence of Se, while CoQ10 plays an important role in suppressing lipid peroxidation. FSP1 repairs lipid peroxidation on the plasma membrane by converting CoQ10 to CoQH2. By contrast, DHODH generates CoQH2 by reducing CoQ10 through a uridine-synthesizing redox reaction that catalyzes DHO to OA to exert a protective effect on lipid peroxidation at the mitochondrial membrane. TYRO3 acts toward anti-ferroptosis by the PI3K/AKT/NRF2 axis to bolster the activity of GSH/GPX4. Abbreviations: BQR, brequinar sodium; circRHOT1, circular RNA RHOT1; CoQ10, coenzyme Q10; Cys, cysteine; Cys2, cystine; DHO, dihydroorotate; DHODH, dihydroorotate dehydrogenase; FSP1, ferroptosis suppressor protein 1; GCL, glutamate-cysteine ligase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSK3B, glycogen synthase kinase 3 beta; GSS, glutathione synthetase; HMGCR, HMG-CoA reductase; IMM, inner membrane of mitochondria; IMS, intermembrane space; IPP, isopentenyl-pyrophosphate; MDM2, mouse double minute 2; MVA, mevalonate; NRF2, nuclear factor, erythroid 2-like 2; OA, orotate; ROS, reactive oxygen species; OMM, outer membrane of mitochondria; Se, selenium; SLC3A2, solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; STAT3, signal transducer and activator of transcription 3; TYRO3, tyrosine-protein kinase receptor 3.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, H.-Y.; Ho, H.-W.; Chang, Y.-H.; Wei, C.-J.; Chu, P.-Y. The Evolving Role of Ferroptosis in Breast Cancer: Translational Implications Present and Future. Cancers 2021, 13, 4576. https://doi.org/10.3390/cancers13184576
AMA Style
Lin H-Y, Ho H-W, Chang Y-H, Wei C-J, Chu P-Y. The Evolving Role of Ferroptosis in Breast Cancer: Translational Implications Present and Future. Cancers. 2021; 13(18):4576. https://doi.org/10.3390/cancers13184576
Chicago/Turabian StyleLin, Hung-Yu, Hui-Wen Ho, Yen-Hsiang Chang, Chun-Jui Wei, and Pei-Yi Chu. 2021. "The Evolving Role of Ferroptosis in Breast Cancer: Translational Implications Present and Future" Cancers 13, no. 18: 4576. https://doi.org/10.3390/cancers13184576
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.