
A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment


1
Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA
2
Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Atlanta, GA 30322, USA
3
Winship Cancer Institute, Emory University, Atlanta, GA 30322, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(18), 4650; https://doi.org/10.3390/cancers13184650
Submission received: 18 August 2021
/
Revised: 11 September 2021
/
Accepted: 13 September 2021
/
Published: 16 September 2021
(This article belongs to the Special Issue The Tumor Microenvironment of Neuroblastoma)
Abstract
:Simple Summary
Neuroblastoma is the most common extra-cranial solid tumor of childhood arising from the developing sympathetic neuroblast. Despite intense multimodal therapy, more than half of patients with high-risk neuroblastoma relapse with incurable disease. The Yes-Associated Protein (YAP) has been shown to play a critical role in many types of cancers, including neuroblastoma. YAP has also been recently highlighted as an important regulator of the tumor microenvironment (TME) that can affect cancer growth and response to therapies. Here, we focus on YAP and its role in neuroblastoma and the TME that underscores the therapeutic potential of inhibiting YAP in this highly aggressive pediatric solid tumor.
Abstract
Neuroblastoma is the most common extra-cranial pediatric solid tumor that accounts for more than 15% of childhood cancer-related deaths. High risk neuroblastomas that recur during or after intense multimodal therapy have a <5% chance at a second sustained remission or cure. The solid tumor microenvironment (TME) has been increasingly recognized to play a critical role in cancer progression and resistance to therapy, including in neuroblastoma. The Yes-Associated Protein (YAP) in the Hippo pathway can regulate cancer proliferation, tumor initiation, and therapy response in many cancer types and as such, its role in the TME has gained interest. In this review, we focus on YAP and its role in neuroblastoma and further describe its demonstrated and potential effects on the neuroblastoma TME. We also discuss the therapeutic strategies for inhibiting YAP in neuroblastoma.
1. Introduction
Neuroblastoma is the most common extra-cranial solid tumor of childhood arising from the developing sympathetic nervous system [1], with approximately 800 cases per year identified in the United States [2]. Neuroblastoma is both a clinically and biologically heterogeneous cancer, with some children presenting with isolated or metastatic self-resolving disease, while other children present with widespread aggressive disease that is fraught with high morbidity and mortality [3]. Despite intensive therapy, including chemotherapy, surgery, myeloablative chemotherapy followed by autologous stem cell rescue, radiation, and immunotherapy, survival for patients with high-risk neuroblastoma remains poor [2], with approximately half of patients relapsing with aggressive chemotherapy-resistant disease [4]. Advances in understanding the genomic landscape and the tumor microenvironment (TME) in recurrent and newly diagnosed high-risk neuroblastomas has identified novel targets with therapeutic relevance in neuroblastoma patients [5,6]. For example, next-generation sequencing of paired diagnostic and recurrent high-risk neuroblastoma identified an increase in activating mutations in the RAS/RAF/MAPK pathway at relapse [7,8]. In addition to genomic alterations, gene set enrichment analysis of relapsed neuroblastoma tumors identified the downregulation of genes transcriptionally silenced by the Yes-Associated Protein (YAP), a core effector of the Hippo signaling pathway, compared to diagnostic tumors, suggesting increased YAP transcriptional activity at relapse [9]. In this particular study, increased YAP activation was noted to be the only event significantly associated with relapsed neuroblastoma [9]. The YAP/Hippo pathway has been well described to crosstalk with growth-promoting tumor-specific mutations, such as hyperactivating mutations in RAS/RAF/MAPK, which is further described in this review in the context of neuroblastoma. Therefore, the YAP/Hippo pathway may be a crucial target driving high risk neuroblastoma recurrence. Importantly, the complex neuroblastoma TME contributes to cancer progression [5,10,11,12,13,14,15,16,17] and YAP has been shown to regulate various aspects of the TME in many other cancer types [18,19,20,21,22,23,24,25,26]. Herein, we highlight YAP and its role in the neuroblastoma TME and how it may regulate therapy resistance as well as highlight opportunities for therapeutic YAP inhibition in this highly aggressive pediatric solid tumor.
2. YAP and Its Role in High-Risk Neuroblastoma
YAP is a transcriptional co-activator that binds with the TEA Domain (TEAD) family of transcription factors to initiate transcription of downstream target genes critical to organ growth and development [27,28,29,30,31,32]. The transcriptional co-activator with a PDZ-binding motif (TAZ) is a paralog of YAP. Both YAP and TAZ activity are regulated by the Hippo pathway kinases [32]. Hippo pathway proteins such as LATS1/2 and MST1/2 phosphorylate YAP and/or TAZ, leading to their cytoplasmic retention and ultimate degradation [29,32]. When the Hippo pathway is inactivated, YAP/TAZ can translocate into the nucleus and bind TEAD to regulate the transcription of effector genes (Figure 1). YAP has been reported to contribute to cell identity and tumor initiation, metastasis, angiogenesis, and resistance to chemotherapy in many solid tumors, such as head and neck, lung, colon, pancreas, and ovarian cancer [33,34,35,36,37,38,39,40]. YAP has also been reported to play a role in pediatric and young adult cancers, including rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, and neuroblastoma [41,42,43,44,45,46].
In certain tumor types upon YAP inactivation, TAZ can independently bind to TEAD family members to compensate for YAP loss and regulate transcription [47,48]. Genetic inhibition of TAZ alone in neuroblastoma cells showed that TAZ may affect cell proliferation, self-renewal, and cell cycle progression independent of YAP expression in certain neuroblastoma models [49,50]. More recently, however, we and others have found no compensatory upregulation of TAZ expression or activity in response to YAP genetic inhibition [51,52]. Specifically, RNA sequencing of two neuroblastoma cell lines, SK-N-AS and NLF, both before and after YAP shRNA stable knockdown, showed that the expression of WWTR1, the gene encoding TAZ, decreases with YAP genetic inhibition. YAP/TAZ target genes follow a similar trend, confirming that TAZ is not being upregulated to balance the loss of YAP, supporting TAZ as an unlikely influence in neuroblastoma biology [51,52].
Research regarding YAP’s role in neuroblastoma supports that this Hippo pathway protein plays a role in almost every element of the “Hallmarks of Cancer”, many of which support a role for YAP in the TME (Table 1).
2.1. Tumorigenesis
YAP and TAZ in normal tissues transcriptionally control organ size and growth under normal physiologic conditions [60,61]. The Hippo proteins serve as an intrinsic regulator of organ growth such that when an organ reaches its optimal size, the Hippo cascade activates to sequester YAP/TAZ in the cytosol, leading to cessation of unfettered organ growth [30,60,61]. For example, in Drosophila, Yorkie (Yki; homolog of YAP in fruit flies) constitutive activation by inactivation of the Hippo cascade led to Yki nuclear accumulation and organ overgrowth, which was reversible when Yki expression was turned off [60,61]. Organ overgrowth occurred in murine models with similar unrestrained YAP activity that was reversed upon Hippo pathway activation [61]. Under malignant conditions, solid tumors capitalize on structural environmental cues normally upregulated during organogenesis or wound healing that downregulate Hippo proteins and upregulate YAP/TAZ; yet, concomitant oncogenic mutations in those tumors prevent the usual feedback inhibition of YAP, leading to uninhibited cell proliferation and tumor growth [62].
2.1.1. Cell Proliferation
Multiple groups have investigated YAP and its effect on cell proliferation in neuroblastoma [51,53,54]. The results from these studies were mixed. Our group found no significant effect of YAP knockdown or YAP overexpression on neuroblastoma cell line proliferation in vitro [51]. Yang et al. and Shen et al. showed that siRNA and shRNA knockdown of YAP, respectively, decreased both cell viability and cell proliferation in neuroblastoma cells (SK-N-SH and SH-SY5Y) [53,54]. These conflicting results may be secondary to cell line characteristic/genetic differences and conditional properties such as metabolism and cell density that are known to influence YAP expression, especially in the setting of transient or incomplete YAP genetic inhibition [24,27,31,63]. For instance, the Hippo pathway downregulates YAP or inactivates YAP under high cell density conditions, leading to contact inhibition [31]. Moreover, glucose metabolism, which is very variable in high density culture conditions, has been shown to regulate YAP/TAZ interaction with TEAD, mediating YAP/TAZ/TEAD transcriptional activity [24,63]. Despite some inconsistency for its role in cell proliferation, a consensus finding is a decrease in colony formation in response to YAP knockdown, highlighting the clonogenic potential of cells with high YAP expression [51,53].
2.1.2. Tumor Growth
YAP’s effect on neuroblastoma tumor growth in vivo seems to be significant and consistent, highlighting the collaborative role of YAP and the in situ tumor environment [51,55]. We have demonstrated that when SK-N-AS cells with stable YAP knockdown were injected subcutaneously into immunodeficient Nod scid gamma (NSG) mice, there was a significant delay in early tumor formation compared to control YAP-expressing SK-N-AS xenografts [51]. Seong et al. developed a metastatic neuroblastoma mouse model by injecting SK-N-AS cells into NSG mice via intracardiac injection and selected metastatic cells from the brain and bone marrow sites. Interestingly, the cells harvested from metastatic sites expressed higher YAP/TAZ protein compared to the parental cell line [55]. Importantly, shRNA knockdown of YAP/TAZ in the neuroblastoma cell lines derived from these metastatic subpopulations exhibited decreased tumor growth and prolonged disease-free survival in mice after intracardiac injection [55]. These results support that YAP may regulate unique cues in the TME that promote tumor survival as well as growth in neuroblastoma.
2.2. Metastasis
Anoikis is a form of programmed cell death that occurs as cells remove themselves by apoptosis when they are not in the correct context of development within a tissue [64,65]. Cell detachment leads to Hippo pathway activation and YAP inactivation, eventually inducing anoikis [66]. Impaired anoikis with tumor cell survival can lead to enhanced tumor metastatic potential [64,65]. Anchorage-independent growth is a hallmark of cancer cells that have overcome anoikis [64,65]. YAP has been shown to contribute to anoikis resistance and metastasis in many cancers [36,40,67,68,69,70]. Indeed, in vitro studies showed impaired anchorage-independent growth and diminished invasion/migration abilities in neuroblastoma cells with YAP knockdown [56]. Moreover, YAP transcriptional activity and protein expression were increased in neuroblastoma patient-derived xenografts (PDXs) from patient tumors at relapse and from metastatic sites [51]. Furthermore, gene expression profiling from parental and metastatic subtypes of neuroblastoma cells developed in vivo from a metastatic mouse model revealed the Hippo signaling pathway to be enriched with YAP expression upregulated in metastatic populations [55]. YAP protein expression was elevated in metastatic cells compared to the parental cells and in PDX tumors derived from non-primary relapsed tumor compared to the diagnostic tumor [51,55]. Accordingly, YAP/TAZ knockdown suppressed the metastatic phenotype in vivo [55].
2.3. Therapy Resistance
The description of the detailed mechanisms underlying YAP-induced therapy resistance vary but they converge on one common theme: YAP/TAZ-mediated transcription is upregulated and responsible—either by the inactivation of upstream Hippo proteins or other negative YAP regulators or by direct YAP/TAZ activation [71,72,73,74]. While mutations in negative YAP regulators such as PTPN14 in relapsed neuroblastomas can lead to upregulated YAP transcriptional activity at relapse [9], not all YAP-expressing neuroblastomas harbor PTPN14 mutations and the exact mechanism as to how YAP induces therapy resistance in neuroblastoma may be context-dependent based on the type of chemotherapy or targeted therapy used [71,72].
2.3.1. Cytotoxic Therapy Resistance Imparted by YAP Differs In Vitro Versus In Vivo
We have shown that YAP expression and transcriptional activity immediately increase following one cycle of standard high risk neuroblastoma therapy, topotecan and cyclophosphamide, when given to mice harboring established neuroblastoma PDXs [51]. The tumors continued to grow through the treatment without significant regression. Given the brief exposure to therapy and sustained tumor growth, we posited that increased YAP expression post-therapy may represent a cell intrinsic response to the therapy itself rather than the selection of a YAP-expressing drug-resistant clone [51]. For instance, DNA-damaging agents, such as alkylating agents or topoisomerase inhibitors, have been reported to downregulate MST1 due to Hsp70-mediated degradation, or to downregulate micro RNAs that inhibit YAP translation [74,75,76,77]. By downregulating MST1 through Hsp-70-mediated degradation, LATS1/2 is inactivated, leading to YAP activation as YAP remains in its dephosphorylated state to enter the nucleus (Figure 1) [72]. In a hepatocellular carcinoma model, miR-590-50 was identified as a functional modulator of YAP, and YAP promoted chemotherapy resistance through the upregulation of genes involved in drug efflux pumps and stemness [76]. Thus, both Hippo-dependent and Hippo-independent mechanisms seem to affect therapy-induced expression of YAP in cancer.
In other cancer models, YAP has been shown to transcriptionally upregulate Bcl2 family pro-survival proteins, such as Bcl2 and Bcl-XL, to promote therapy resistance [78,79]. However, YAP knockdown does not affect Bcl2 family pro-survival protein expression in neuroblastoma [51]. Bim binding patterns to different pro-survival Bcl2 members determine Bcl2- or Mcl1-mediated apoptosis resistance in neuroblastoma [80]. Although we found that YAP knockdown upregulates the expression of the pro-apoptotic BH3 protein Bim, Bim remained sequestered to and inactivated by either Mcl1 or Bcl2, depending on the cell line tested—suggesting an alternative mechanism of cytotoxic therapy resistance by YAP occurs in neuroblastoma [51].
In a study exploring YAP and chemotherapy resistance in neuroblastoma, Yang et al. derived cisplatin-resistant SH-SY5Y cells and demonstrated that YAP siRNA inhibition led to reduced proliferation and colony formation in vitro while cells were still incubated in low dose cisplatin [53]. The cisplatin-resistant cells were likely re-sensitized to low dose cisplatin upon YAP knockdown, supporting YAP’s effect on cytotoxic therapy resistance. In contrast, we found that YAP knockdown or overexpression does not lead to significant sensitization or resistance, respectively, to cytotoxic agents such as melphalan in vitro [51]. Further, we noted no significant increase in apoptosis in response to etoposide in the setting of YAP knockdown in vitro. Contrastingly, SK-N-AS murine xenografts harboring shYAP tumors had significant tumor regression when treated with cyclophosphamide compared to YAP-expressing control tumors [51]. These findings support that YAP plays more of a critical role in therapy resistance within the TME verses 2D culture setting.
2.3.2. Tyrosine Kinase Inhibitor Resistance Due to YAP
The dysregulation of Hippo signaling and subsequent activation of YAP/TAZ is a major resistance mechanism identified in response to multiple targeted therapies [71,72,73,74]. Two of the most studied targeted therapies in the context of YAP deregulation in cancers is EGFR and MAPK pathway inhibitors. Both EGFR and MAPK pathway signaling are critical for cell proliferation and survival; thus, oncogenic mutations in these pathways can lead to tumorigenesis [73]. YAP expression in lung cancer has been identified as the cause for drug resistance and tumor recurrence in response to EGFR tyrosine kinase inhibitors (TKI) [71,81]. Lee et al. noted upregulated YAP expression and activation in long-term TKI-induced resistant lung cancer cells and YAP inhibition of TKI-resistant cells restored sensitivity to TKI treatment [81]. The precise mechanism of how YAP drives TKI resistance in many cancers remains poorly understood, yet is thought to be secondary to YAP transcriptional target upregulation, such as AXL, AREG, ERBB3, and CTGF [82,83,84]. The RAS/MAPK pathway is frequently deregulated in many cancer types, including neuroblastoma, due to activating mutations in ALK/RAS/RAF or inhibitory NF1 mutations or deletions [8,85,86,87]. Lin et al. reported that YAP promotes resistance to RAF- and MEK-inhibitors potentially through upregulation of the pro-survival gene BCL-XL and that combined YAP and RAF or MEK inhibition leads to synthetic lethality in BRAF- and RAS-mutant tumor types [88]. In rhabdomyosarcoma, YAP and oncogenic RAS cooperate in tumorigenesis, suggesting the importance of co-targeting these pathways [89]. Studies have shown that there may be cross-talk between YAP and the RAS/MAPK pathway: RAS pathway proteins stabilize YAP protein to prevent turnover and induction of the YAP downstream target AREG leads to EGFR/RAS pathway activation, forming a positive feedback loop [90,91].
Relapsed neuroblastoma tumors harbor increased RAS/MAPK pathway mutations compared to paired diagnostic tumors [8]. Neuroblastoma cells with RAS/MAPK pathway aberrations have shown sensitivity to MEK inhibitors trametinib and binimetinib in vitro [8,92]. However, in vivo treatment with single-agent binimetinib in various neuroblastoma cell line-derived xenograft models demonstrated inhibition of tumor growth and extended survival in NRAS or NF1 mutated xenografts while ALK mutated tumors did not respond, likely due to persistent or alternative tyrosine kinase pathway activation [8,93]. Moreover, there may be a role for microRNAs such as the let-7 microRNA family for contributing to ALK inhibitor therapy resistance by regulating RAS expression [94,95]. Therefore, these findings suggest the need for combined targeting of pathways. Dual inhibition strategies, such as MEK inhibition in combination with BRAF, BET, and CDK4/6 inhibitors have been employed in neuroblastoma, but studies have reported limited anti-tumor activity or concerns of eventual escape and resistance [96,97,98,99].
Given the interaction between YAP and oncogenic RAS, Coggins et al. and our group investigated the combined effects of YAP genetic inhibition and MEK inhibition [51,52]. Coggins showed that the MEK inhibitor trametinib induces YAP nuclear translocation while reducing cytoplasmic YAP in RAS- or NF1-mutated neuroblastoma cell lines, suggesting resistance to MEK inhibitor therapy via YAP activation [52]. CRISPR-Cas9 knockout of YAP and constitutively active YAP expression promoted sensitization and resistance, respectively, to trametinib in neuroblastoma cell lines with RAS hyperactivation [52]. RNA sequencing of the MYCN-amplified NLF cells with and without YAP knockout treated with vehicle or trametinib showed that YAP mediates trametinib resistance through the transcriptional activation of E2F and MYCN, allowing the maintenance of the proliferative capacity of neuroblastoma cells [52]. Our studies expanded on this data, showing enhanced tumor regression in response to trametinib in an MYCN non-amplified NRAS-mutated SK-N-AS xenograft when YAP is genetically knocked down [51]. We further defined the mechanism for enhanced in vivo sensitivity to MEK inhibition, independent of MYCN amplification, which is further explained in Section 3.1.1 of this review.
2.4. Mesenchymal Properties
Cells with mesenchymal phenotypes exhibit extreme therapy resistance in cancer [100,101]. High YAP/TAZ activity has been observed in progenitor, or self-renewing, embryonic stem cells and is involved in the embryonic development of tissues and organs. YAP activation has also been shown to impair normal cell differentiation [102,103]. In the context of cancer, cancer stem cells (CSC) have been identified in neoplastic tissues and those that contain self-renewing and stem-like properties [103,104]. YAP/TAZ have been shown to participate in epithelial mesenchymal transition (EMT) and promote CSC self-renewal [37,103,105]. Moreover, the Hippo pathway is involved in developing neural tissue by preventing YAP/TAZ-driven neural progenitor growth and proliferation [106,107]. In fact, YAP promotes an early neural crest phenotype and displays a mesenchymal morphology [59]. Early neural crest cells are highly migratory with multipotential progenitor features, and therefore can give rise to sympatho-adrenal precursors as well as neuroblasts [108]. Thus, as a potential driver of the mesenchymal stem-like cell, YAP may induce therapy resistance.
2.4.1. Neurosphere Formation
In pediatric and young adult cancer rhabdomyosarcoma, Slemmons et al. demonstrated that when rhabdomyosarcoma cells were cultured as “rhabdospheres” in neurobasal media, YAP and Notch expression were upregulated [44]. They further found that YAP suppression decreased expression of downstream stemness genes, OCT4 and SOX2, in cells grown as rhabdospheres, suggesting the role of YAP in mesenchymal properties of embryonic tumors [44]. To investigate the mesenchymal properties of YAP in neuroblastoma, our group derived neurospheres from neuroblastoma cell lines in neurobasal media, mirroring the stem-like environment [51,109]. We noted increased YAP transcriptional activity along with increased OCT4 and SOX2 expression. YAP knockdown led to the suppression of mesenchymal gene expression despite neurobasal conditions, suggesting that YAP helps mediate the mesenchymal state [51]. We also noted fewer and an increased number of neurospheres in YAP knockdown and overexpressing neuroblastoma cells, respectively. To further support our findings, RNA sequencing of SK-N-AS cells with YAP shRNA knockdown verses control revealed the downregulation of genes involved in mesenchymal states in other tissue types, such as JAK1, IL6ST, and TBX3 [110,111,112].
2.4.2. Mesenchymal Phenotype
Neuroblastoma tumor cells demonstrate phenotypic heterogeneity. An increasing focus has been to fully characterize the mesenchymal and adrenergic lineages of neuroblastoma and to understand cell plasticity and the epigenetic regulation of these states [57,58]. Isogenic cell lines from the same patient distinguished solely by CD133+ (mesenchymal) and CD133- (adrenergic) phenotypes demonstrated extremely divergent mRNA profiles [58]. YAP and TAZ protein expression were consistently increased in the mesenchymal neuroblastoma cells and absent in the adrenergic populations [58]. In this study, the adrenergic and mesenchymal phenotypes were driven by distinct super-enhancer landscapes and super-enhancer-related transcription factors (TF), such as PRRX1, a TF that they mechanistically showed induces a mesenchymal state. These two states are able to interconvert, and YAP protein expression increased consistently following PRRX1-induced overexpression [57,58]. Importantly, the mesenchymal neuroblastoma cell types are more resistant to standard cytotoxic therapy compared to their adrenergic counterparts [58]. While recent studies have implicated that the PRRX1 super-enhancer TF drives the mesenchymal state, in our hands, genetic knockdown of YAP alone leads to increased expression of adrenergic genes/proteins and decreased mesenchymal proteins in mesenchymal neuroblastoma cells. Reciprocally, YAP overexpression in an adrenergic neuroblastoma cell line leads to increase in SNAI2 and PRRX1 with a concomitant decrease in PHOX2B, GATA3, and DBH (data unpublished). Therefore, further exploration for whether YAP alone can drive the neuroblastoma mesenchymal phenotype is underway and may further support YAP as a critical therapeutic target in neuroblastoma.
3. YAP and the Tumor Microenvironment in Neuroblastoma
3.1. Current Knowledge and Potential Contributions
Given the lack of recurrent driver mutations in diagnostic high-risk neuroblastoma, going beyond the genetic events and further understanding the neuroblastoma TME has been a crucial focus to identify novel therapies [16]. The TME in neuroblastoma has been extensively reviewed, pointing to roles for immune cells, non-immune cells, and MYCN amplification, influencing therapy response and survival [5,16,17]. Many studies highlight YAP as a regulator of key aspects of the TME that impact therapeutic response in cancers [23]. YAP tumor expression interacts with the TME to influence stress-induced apoptosis, tumor hypoxia, angiogenesis, extracellular matrix (ECM) remodeling, and the stromal and immune cell networks (Figure 2). These interactions ultimately impact tumorigenesis, metastasis, therapy resistance, and mesenchymal properties [23,113]. Therefore, further understanding the mechanisms underlying Hippo/YAP signaling in the TME in neuroblastoma may also provide therapeutic opportunities.
The role of YAP in normal tissue wound healing and tissue regeneration is an important concept to understand how YAP may influence or be influenced by the neuroblastoma tumor environment [28,40,114]. We and others have shown that YAP protein expression and transcriptional activity increase as a response to chemotherapy in neuroblastoma tumors or in cells derived from metastatic sites [51,55]. Mutations in YAP/TAZ itself are rare and limited to certain cancer types; thus, recent investigations correlate YAP/TAZ oncogenic activation in solid tumors to “wounds that never heal”, as YAP is activated following extensive damage (due to radiation and/or chemotherapy), and in cooperation with oncogenic mutations driving proliferative pathways (such as RAS/MAPK, etc.), cooperate to drive a chronic regenerative response [40]. The lack of genomic YAP alterations (i.e., mutations, amplification) and our finding of increased YAP expression following a single cycle of cytotoxic therapy in RAS-mutated neuroblastoma in vivo, support that chemotherapy-induced damage may upregulate YAP in the tumor and may also explain the presence of increased YAP in post-chemotherapy relapsed primary neuroblastoma [51].
The chronic regenerative response includes YAP activation and pathways that suppress apoptosis, promote neo-angiogenesis, remodel the ECM, and recruit cancer-associated immune cells, forming a niche for cancer cell survival and proliferation. In the next section of the review, we will focus on the current literature highlighting YAP and its role in the TME and discuss potential contributing aspects of YAP in the neuroblastoma TME, especially in the context of stress-induced apoptosis, tumor hypoxia and vasculature, ECM remodeling, and the immune milieu.
3.1.1. Tumor Environmental Stress-Induced Apoptosis
A tumor-promoting environment imparts signals that suppress stress responses to prevent cancer cell death in the face of nutrient deprivation or hypoxia. We found that YAP indeed can suppress stress-induced apoptosis in neuroblastoma [51]. Due to the significant impact of YAP inhibition in neuroblastoma xenografts that suppressed tumor growth and therapy responses in vivo, we investigated downstream targets regulated by YAP that might contribute to in situ tumor responses [51]. RNA sequencing data obtained from SK-N-AS cells with and without YAP knockdown showed that pathways involved in apoptosis, metabolism, and ECM remodeling, all processes important for the TME, were significantly affected. When evaluating the differential expression of genes in gene set enrichment analyses, we noted that HRK, a gene that encodes the protein Harakiri, was significantly upregulated in the cells with YAP knockdown [51].
Harakiri (HRK) is a BH3-only pro-death protein that activates the intrinsic or mitochondrial apoptosis pathway in the setting of cytokine deprivation and hypoxia, both properties in the solid tumor environment shown to promote therapy resistance [115,116]. We functionally validated HRK suppression by YAP in neuroblastoma cell lines, showed that HRK is suppressed when YAP is increased in relapsed tumors, and demonstrated that following chemotherapy treatment of PDXs in vivo, YAP expression increases while HRK expression decreases [51]. We were also able to restore cytotoxic therapy response in vitro in neuroblastoma cells by serum starvation, but apoptosis only occurred when YAP was genetically inhibited to allow for HRK expression and activation in response to serum deprivation and chemotherapy. Therefore, we identified HRK as a novel tumor suppressor in neuroblastoma that is negatively regulated by YAP to prevent therapy induced apoptosis in the in situ TME [51].
3.1.2. Hypoxia and Angiogenesis
Hypoxia has been shown to induce YAP nuclear translocation and activation through inhibition of Hippo signaling [117,118]. In relation to the hypoxia-inducible factor 1 (HIF1) and its ability to drive glycolysis in the setting of hypoxia, YAP binds to HIF1 alpha, forming a complex that both sustains HIF1α stability and promotes glycolysis in hepatocellular carcinoma [26,117]. The hypoxic environment also leads to angiogenesis and YAP signaling is involved in tumor vasculature development [119]. Nuclear YAP/TAZ (active state) is expressed in developing endothelial cells (ECs) and the remodeling vasculature [120,121]. Moreover, recent studies demonstrated that YAP activation is crucial for angiogenesis regulated by VEGF signaling and cytoskeletal remodeling [122]. Active EC YAP induces a downstream transcriptional program which regulates further ECM remodeling for a tumor niche [20]. Thus, therapeutic targeting of YAP may be important for vascular normalization to improve cancer-directed therapies.
In neuroblastoma, hypoxic conditions can shift cells into a de-differentiated or stem-like phenotype [123]. Jogi et al. showed that neuroblastoma cells in hypoxic conditions were shifted towards an immature, neural crest-like phenotype with an increase in gene markers of neural crest sympathetic progenitors [124]. Patients with hypoxic tumors were predicted to have an unfavorable prognosis with tumors associated with telomerase activation and a more immunosuppressive, poorly differentiated, and apoptosis-resistant TME [125]. Angiogenesis occurs through angiogenic factors such as vascular endothelial cell growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF) and are influenced by hypoxia and inflammation [5,16]. HIF1 transcription factors and MYCN have been shown to be involved in both processes [16,126]. In addition to hypoxia, increased VEGF expression and tumor angiogenesis correlates with more aggressive disease and poor outcomes in neuroblastoma [127]. Therefore, the role for YAP as well as HRK in hypoxia and angiogenesis in neuroblastoma deserve further exploration.
3.1.3. Extracellular Matrix Remodeling
The ECM affects and is affected by tumor cells bidirectionally, acting as the signaling hub and organizer for tissue homeostasis [113,128]. The ECM is comprised of a complex nest of structural proteins, such as collagen, hyaluronic acid, elastin, matricellular proteins, glycoproteins, proteoglycans, and polysaccharides. These molecules form a dynamic and versatile network of a cell-matrix environment that form the structural foundation for tissue function and mechanical sustainability [128,129]. Abnormal ECM dynamics can contribute to cancer development and progression by promoting a niche for cancer cells to metastasize and invade surrounding tissues [129,130,131]. In cancers, hypoxia and inflammation in the TME can promote ECM stiffness due to an increase in collagen deposition, leading to upregulation of integrin signaling and various pathways including PI3K/AKT to promote cell proliferation and anti-apoptotic effects [131]. YAP/TAZ and their role in mechanotransduction is well described [20,132]. YAP and TAZ act as mechanotransducers and sensors of mechanical cues from the ECM and receive and communicate those signals in a bidirectional manner. YAP and TAZ can be induced by increased ECM rigidity or stiffness in the setting of inflammation and tissue damage [20,23]. Cells spreading over a surface can activate YAP/TAZ as well. Furthermore, YAP/TAZ activity requires the GTPase Rho, which regulates the actin cytoskeleton and cytoskeletal tension induced by the pulling forces of the ECM [20,132]. Most importantly, YAP and TAZ are required mediators of ECM elasticity and cell geometry, as alterations of YAP/TAZ levels can overrule cell mechanophysical behavior [20]. YAP/TAZ activation can remodel the ECM itself through complex pathways to promote cancer aggressiveness, metastasis, and therapy response [23]. A recent study by Jang et al. demonstrated that matrix stiffness epigenetically regulates YAP activation in gastric cancer through DNA methylation modifiers leading to YAP promoter hypomethylation, proposing that epigenetic reprogramming of the ECM properties in solid tumors may be a potential therapeutic strategy [133].
In neuroblastoma, Lam et al. artificially increased the rigidity of polyacrylamide hydrogels on which neuroblastoma cells were seeded to mimic increased stiffness of the ECM. They showed that increased ECM rigidity enhanced neuritogenesis (measurement of differentiation), decreased proliferation, and reduced expression of MYCN [134]. Additionally, retinoic acid, which is a differentiating agent currently used in the clinical setting of high risk neuroblastoma therapy, potentiated neuroblastoma differentiation with increasing ECM stiffness [134]. More studies are needed to validate these findings in the context of YAP. If changes in ECM stiffness influences differentiation, MYCN expression, and YAP expression, then the use of therapies focused on remodeling the ECM may have therapeutic gain in neuroblastoma. Indeed, therapies targeting ECM components are heavily under investigation as a novel anticancer approach [128,135]. Unpublished data from our laboratory demonstrate that neuroblastoma cells cultured in low density express increased YAP compared to high density states, suggesting YAP is influenced by contact inhibition in addition to ECM rigidity states. Therefore, YAP and its role in mechano-sensing is complex and ECM remodeling strategies to target structural components or signaling molecules and remodeling enzymes deserve further investigation in the context of YAP in neuroblastoma [130,135].
3.1.4. Immune Milieu
In the era of immunotherapy, understanding the immune milieu in the TME has been important to strategize ways to improve immunotherapy response. As described in depth by S. Joshi and Blavier et al., stromal cells (cancer-associated fibroblasts (CAFs) and mesenchymal stromal cells (MSCs)) and immune cells (tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor infiltrating lymphocytes (TILs)) are all key players of the neuroblastoma TME [5,17]. YAP’s role in the immune environment in other cancers has been described. YAP can activate CAFs, establishing a feed-forward loop, leading to a cancer-friendly niche with more ECM rigidity [23,136]. Through the transcriptional regulation of cytokines and chemokines, YAP has been shown to influence the phenotype of tumor-resident immune cells in favor of an inhibitory environment [23,137]. For example, YAP activation is associated with TAM recruitment and M2 phenotype polarization, leading to a pro-tumorigenic or immune suppressive environment, potentially via C-C motif chemokine ligand 2 (CCL2 or MCP1) activation, as shown in hepatocellular carcinoma [138,139,140]. YAP also contributes to the immune-suppressive environment further by recruiting MDSCs to suppress cytotoxic T cells through the production of cytokines such as IL-6, macrophage colony-stimulating factor (CSF1), and granulocyte–macrophage colony-stimulating factor (GM–CSF) in pancreatic and prostate cancers [141,142]. Additionally, Stampouloglou et al. and Lebid et al. evaluated YAP expression in activated CD4+ and CD8+ T cells and found that the loss of YAP in T cells resulted in enhanced T cell activation, differentiation, and function, leading to improved T cell infiltration in tumors, signifying that YAP therapeutic inhibition in immune cells themselves may contribute to improved immunotherapy responses [143,144]. Tregs are important for immune homeostasis, and YAP expression in these cells has shown to be essential to suppress anti-tumor immunity [21]. Recent studies have also reported that YAP affects immune check points, by upregulating programmed cell death ligand 1 (PDL1) expression on tumor cells to turn off tumor-specific effector T cells and escape antitumor immunity [145,146]. Overall, YAP’s role specifically in the pediatric cancer immune environment remains to be explored.
3.2. Future Investigations
YAP has been shown to promote a tumor permissive environment through inhibiting TME stress-induced apoptosis, remodeling the ECM and vasculature, and suppressing the immune response in other cancers. Although we have identified a novel interaction in neuroblastoma—YAP-mediated repression of the tumor suppressor HRK—that could explain one of the roles for YAP in the context of tumor environmental stress-induced apoptosis, further studies are needed to fully understand the breadth of YAP’s regulation in the neuroblastoma TME. Figure 2 summarizes the ways in which YAP contributes to the general TME (highlighted in the blue box) and in neuroblastoma specifically (highlighted in the yellow box) and areas in which further investigations are warranted based on the role of YAP in other cancers. We understand that all tumors and their genetic/epigenetic environment and the interplay amongst signaling pathways are complex and different, supporting these interactions cannot be inferred but must be explored in neuroblastoma specifically.
Importantly, the neuroblastoma TME is dynamic and complex [5,17]. As shown in Figure 2, YAP is involved in almost every aspect of the solid tumor environment that composes the neuroblastoma TME. We also note that the interaction between the TME and YAP is bidirectional, suggesting the oncogenic role of YAP in promoting a pro-tumorigenic environment can further potentiate YAP activation or related pathways. For instance, YAP is activated by the stiff and rigid ECM formed by CAFs, which in turn leads to downstream signaling of further ECM remodeling, creating a cancer niche suitable for metastasis and tumor growth [23,132].
While our studies have identified YAP’s role in suppressing HRK to promote neuroblastoma tumor growth and resistance to chemotherapy and targeted therapy in the in situ tumor environment [51], further investigations in our laboratory are underway to understand the actual mechanism for how YAP regulates HRK and other tumor suppressor genes to inhibit their expression and activity. Whether this mechanism or others leads to YAP influences on immunotherapy responses in neuroblastoma is also a topic of heavy exploration in our laboratory.
YAP has been in the spotlight over the past few years for its role in the ECM organization and mechanotransduction, especially in the context of cancer [20,132]. Therefore, further studies using models that recapitulate the neuroblastoma ECM in the context of YAP may help delineate other pathways and signaling events that promote the tumor niche for growth and metastasis. In addition to its direct role in ECM organization and mechano-sensing, YAP can promote CAFs to induce a stiffer ECM for tumor growth. CAFs have been identified in neuroblastoma tumors as well and are associated with a poor outcome due to a more therapy-resistant phenotype [17,147], supporting the need to explore YAP’s contributions to neuroblastoma CAF formation.
YAP and its role in the cancer immune milieu has been surfacing with advances in immunotherapy. The emerging role of YAP in the cancer immune environment is an ongoing area of research to improve immunotherapy approaches. Thus, further understanding YAP’s role in the neuroblastoma immune environment may provide the opportunity to improve targeted immunotherapy responses. Many groups have begun to characterize the neuroblastoma immune landscape and ongoing studies continue to demonstrate low immunogenicity, low T and NK cell tumor infiltration, and immune evasion strategies [148,149]. Further investigations in our laboratory are being pursued to understand YAP’s impact on the neuroblastoma immune environment and response to immunotherapies. Collectively, studies exploring the mechanisms behind YAP and neuroblastoma TME regulation will be essential to identifying therapeutic targets and pathways that either cooperate with or are able to antagonize YAP’s oncogenic effects in neuroblastoma.
Finally, we would like to emphasize and highlight the importance of using models that closely recapitulate the neuroblastoma TME, given the strong influence each aspect of the TME (ECM stiffness, cellular contact, EC interactions) has on YAP expression. A variety of pre-clinical models for studying neuroblastoma exist, ranging from in vitro 2D culture to 3D bioprinted models that strive to recapitulate the in vivo setting. Multiple recent publications and reviews have illustrated the emerging development and use of 3D in vitro models for pre-clinical studies in neuroblastoma [150,151,152,153,154]. In fact, 3D bioprinted models have the advantage of high tunability with the addition or removal of TME components such as immune or stromal cells and manipulation of vasculature or ECM properties. However, studies have shown that in vivo models, especially PDX models and immunocompetent transgenic or humanized mouse models, most closely resemble the neuroblastoma TME, and as such serve as an important tool for validating in vitro findings [150,155]. For example, future studies exploring YAP genetic knockout in the TH-MYCN transgenic mouse or MYCN amplified zebrafish models of neuroblastoma may contribute to our understanding of YAP and MYCN and their TME effects [156,157].
4. Therapeutic Targeting of YAP in Neuroblastoma
There are various published studies and reviews regarding the Hippo pathway and YAP/TAZ targeting [114,158]. Many direct and indirect inhibitors targeting YAP and related pathways have been described. We have summarized potential therapies to target YAP and related pathways in Table 2. Verteporfin, a photodynamic therapy that is FDA-approved for macular degeneration, has been shown to disrupt the YAP-TEAD complex by directly inhibiting YAP and has been the most widely used “YAP inhibitor” in pre-clinical studies of YAP driven cancers [159,160]. Unfortunately, we and other groups have found off-target and non-specific cytotoxicity induced by verteporfin specifically in YAP null neuroblastoma cells, likely through the activation of reactive oxygen species [161]. Other groups have looked at cyclic peptides that disrupt the YAP-TEAD binding pocket, yet no significant pre-clinical evidence has validated their clinical use [162,163]. Additionally, we have found that YAP mimetic peptides are ineffective due to poor cell membrane penetrability and high potential for being protein-bound (data unpublished). Small molecules that inhibit TEAD auto-palmitoylation, a post-translational modification essential for TEAD activation and binding to YAP, pre-clinically show efficacy in NF2-mutated malignant mesothelioma and meningiomas or schwannomas, leading to a first in human phase 1 clinical trial in those tumor types, supporting the need to assess their efficacy in YAP-driven neuroblastoma [164,165,166].
In addition to YAP-TEAD directed therapy, combination therapies targeting YAP downstream effectors and its cooperative pathways are critical avenues to explore. Currently, how YAP regulates HRK expression in neuroblastoma is unknown. HRK is inactivated in other cancers via epigenetic silencing through DNA promoter hypermethylation or histone modifications [167,168,169,170]. YAP-TEAD has also been shown to impart chromatin alterations on target gene loci to induce or repress target gene expression in other cancers [171,172,173,174]. Therefore, epigenetic modifying agents may have therapeutic potential in the context of HRK upregulation to restore stress-induced apoptosis in neuroblastoma. Specifically, the histone deacetylase (HDAC) inhibitor vorinostat has already shown pre-clinical efficacy in potentiating anti-tumor therapy effects in early-phase clinical trials in combination with isotretinoin or I-131 MIBG therapy in neuroblastoma [175,176]. In RAS-mutated melanoma and non-small cell lung cancer, where YAP also plays a significant role, the combination of HDAC inhibitors and MEK inhibitors has shown in vivo antitumor effects [177,178]. Thus, considering combination therapies with epigenetic modifying agents (DNA demethylating agents or HDAC inhibitors) and those that target YAP-cooperating pathways in the TME (MEK inhibitors or BET inhibitors) could be considered. Investigations are underway in our laboratory to define YAP’s method for HRK and other gene suppression and preclinically target those pathways with similar novel combinations. Until a sensitive and specific YAP inhibitor is developed and identified for its therapeutic potential in neuroblastoma, alternative methods to exploit targets and signaling pathways downstream of YAP should continue to be explored.
5. Conclusions and Future Directions
In this review, we have provided a comprehensive summary of the oncogenic role for YAP in neuroblastoma. Further, we outlined YAP and its influence on surrounding cells and stroma that sculpt the complex TME, and the potential effects that YAP may impart on the high risk neuroblastoma TME, specifically in the setting of stress-induced apoptosis, neo-angiogenesis, ECM remodeling, and the immune milieu. Most importantly, we propose ways to further investigate YAP in the neuroblastoma TME for the discovery of novel therapeutic opportunities. Overall, the presented data and literature support YAP as a logical therapeutic target in high risk neuroblastoma. While the implementation of the first phase 1 trial of a high affinity TEAD inhibitor for adult cancers gives promise that targeting YAP/TEAD therapeutically now has higher potential, results from this review support that a combinatorial approach will be most optimal for YAP-driven heterogeneous tumors, such as neuroblastoma, that carry cooperating alterations that may attenuate single-agent TEAD inhibitor potency. Therefore, future investigations are critical to understanding the mechanisms underlying the role of YAP in the neuroblastoma TME and to identify optimal therapeutic strategies to target YAP directly or indirectly in novel combinations to improve outcomes for patients with high risk and relapsed neuroblastoma.
Author Contributions
Conceptualization, J.S., K.C.G. Writing—original draft preparation, J.S.; writing—review and editing, J.S., K.C.G. Funding acquisition, J.S., K.C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Hyundai Hope on Wheels, Young Investigator Grant, grant number 639727 to J.S. and Hyundai Hope on Wheels, Scholars Award, grant to K.C.G.
Acknowledgments
We would like to thank Hunter C. Jonus for her kind and thorough review of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.K.; Hogarty, M.; Committee, C.O.G.N. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S. Targeting the Tumor Microenvironment in Neuroblastoma: Recent Advances and Future Directions. Cancers 2020, 12, 57. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Padovan-Merhar, O.M.; Raman, P.; Ostrovnaya, I.; Kalletla, K.; Rubnitz, K.R.; Sanford, E.M.; Ali, S.M.; Miller, V.A.; Mosse, Y.P.; Granger, M.P.; et al. Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet. 2016, 12, e1006501. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Koster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, E.L.; Mosse, Y.P. Targeting ALK in neuroblastoma--preclinical and clinical advancements. Nat. Rev. Clin. Oncol. 2012, 9, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.H.; Voss, S.D.; Hall, D.C.; Minard, C.G.; Balis, F.M.; Wilner, K.; Berg, S.L.; Fox, E.; Adamson, P.C.; Blaney, S.; et al. Activity of Crizotinib in Patients with ALK-aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin. Cancer Res. 2021, 27, 3543–3548. [Google Scholar] [CrossRef] [PubMed]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mosse, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Trigg, R.M.; Turner, S.D. ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Borriello, L.; Seeger, R.C.; Asgharzadeh, S.; DeClerck, Y.A. More than the genes, the tumor microenvironment in neuroblastoma. Cancer Lett. 2016, 380, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players, New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat. Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The role of YAP/TAZ activity in cancer metabolic reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, T.; Zhang, J.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. Emerging roles of Hippo signaling in inflammation and YAP-driven tumor immunity. Cancer Lett. 2018, 426, 73–79. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, I.M.; Halder, G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Li, L.; Zhao, B. The regulation and function of YAP transcription co-activator. Acta Biochim. Biophys. Sin. 2015, 47, 16–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Zhang, W.; Pan, Y.; Gao, Y.; Deng, L.; Li, F.; Li, F.; Ma, X.; Hou, S.; Xu, J.; et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63-GPX2 Axis and ROS Accumulation. Cancer Res. 2017, 77, 5769–5781. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Eisenbarth, D.; Choi, W.; Kim, H.; Choi, C.; Lee, D.; Lim, D.S. YAP and AP-1 Cooperate to Initiate Pancreatic Cancer Development from Ductal Cells in Mice. Cancer Res. 2020, 80, 4768–4779. [Google Scholar] [CrossRef]
- Qu, Y.; Zhang, L.; Wang, J.; Chen, P.; Jia, Y.; Wang, C.; Yang, W.; Wen, Z.; Song, Q.; Tan, B.; et al. Yes-associated protein (YAP) predicts poor prognosis and regulates progression of esophageal squamous cell cancer through epithelial-mesenchymal transition. Exp. Med. 2019, 18, 2993–3001. [Google Scholar] [CrossRef] [Green Version]
- Azar, W.J.; Christie, E.L.; Mitchell, C.; Liu, D.S.; Au-Yeung, G.; Bowtell, D.D.L. Noncanonical IL6 Signaling-Mediated Activation of YAP Regulates Cell Migration and Invasion in Ovarian Clear Cell Cancer. Cancer Res. 2020, 80, 4960–4971. [Google Scholar] [CrossRef]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122 e112. [Google Scholar] [CrossRef]
- Ling, H.H.; Kuo, C.C.; Lin, B.X.; Huang, Y.H.; Lin, C.W. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp. Cell Res. 2017, 350, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Mohamed, A.D.; Gener, M.; Li, W.; Taboada, E. YAP and the Hippo pathway in pediatric cancer. Mol. Cell. Oncol. 2017, 4, e1295127. [Google Scholar] [CrossRef] [Green Version]
- Morice, S.; Danieau, G.; Redini, F.; Brounais-Le-Royer, B.; Verrecchia, F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers 2020, 12, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.H.; Lawlor, E.R. BMI-1 suppresses contact inhibition and stabilizes YAP in Ewing sarcoma. Oncogene 2011, 30, 2077–2085. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Crose, L.E.S.; Riedel, S.; Sushnitha, M.; Belyea, B.; Linardic, C.M. A Novel Notch-YAP Circuit Drives Stemness and Tumorigenesis in Embryonal Rhabdomyosarcoma. Mol. Cancer Res. 2017, 15, 1777–1791. [Google Scholar] [CrossRef] [Green Version]
- Slemmons, K.K.; Yeung, C.; Baumgart, J.T.; Juarez, J.O.M.; McCalla, A.; Helman, L.J. Targeting Hippo-Dependent and Hippo-Independent YAP1 Signaling for the Treatment of Childhood Rhabdomyosarcoma. Cancer Res. 2020, 80, 3046–3056. [Google Scholar] [CrossRef]
- LaQuaglia, M.J.; Grijalva, J.L.; Mueller, K.A.; Perez-Atayde, A.R.; Kim, H.B.; Sadri-Vakili, G.; Vakili, K. YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 30238. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., 3rd; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, Y.; Zou, J.; Yang, R.; Xuan, F.; Wang, Y.; Gao, N.; Cui, H. Transcriptional co-activator TAZ sustains proliferation and tumorigenicity of neuroblastoma by targeting CTGF and PDGF-beta. Oncotarget 2015, 6, 9517–9530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Huang, M.; Tan, J.; Hou, J.; He, J.; Wang, F.; Cui, H.; Yi, L. Transcriptional co-activator with PDZ-binding motif overexpression promotes cell proliferation and transcriptional co-activator with PDZ-binding motif deficiency induces cell cycle arrest in neuroblastoma. Oncol. Lett. 2017, 13, 4295–4301. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.; Lee, J.Y.; Jonus, H.C.; Arnold, A.; Schnepp, R.W.; Janssen, K.M.; Maximov, V.; Goldsmith, K.C. YAP-Mediated Repression of HRK Regulates Tumor Growth, Therapy Response, and Survival Under Tumor Environmental Stress in Neuroblastoma. Cancer Res. 2020, 80, 4741–4753. [Google Scholar] [CrossRef]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Tan, J.; Zhu, J.; Wang, S.; Wei, G. YAP promotes tumorigenesis and cisplatin resistance in neuroblastoma. Oncotarget 2017, 8, 37154–37163. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Xu, X.; Xie, C.; Liu, H.; Yang, D.; Zhang, J.; Wu, Q.; Feng, W.; Wang, L.; Du, L.; et al. YAP promotes the proliferation of neuroblastoma cells through decreasing the nuclear location of p27(Kip1) mediated by Akt. Cell Prolif. 2020, 53, e12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, B.K.; Fathers, K.E.; Hallett, R.; Yung, C.K.; Stein, L.D.; Mouaaz, S.; Kee, L.; Hawkins, C.E.; Irwin, M.S.; Kaplan, D.R. A Metastatic Mouse Model Identifies Genes That Regulate Neuroblastoma Metastasis. Cancer Res. 2017, 77, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Chen, K.; Cheng, C.; Xu, Y.; Cheng, Q.; Xu, G.; Wu, Y.; Wu, Z. Prp19 Is an Independent Prognostic Marker and Promotes Neuroblastoma Metastasis by Regulating the Hippo-YAP Signaling Pathway. Front. Oncol. 2020, 10, 575366. [Google Scholar] [CrossRef] [PubMed]
- van Groningen, T.; Akogul, N.; Westerhout, E.M.; Chan, A.; Hasselt, N.E.; Zwijnenburg, D.A.; Broekmans, M.; Stroeken, P.; Haneveld, F.; Hooijer, G.K.J.; et al. A NOTCH feed-forward loop drives reprogramming from adrenergic to mesenchymal state in neuroblastoma. Nat. Commun. 2019, 10, 1530. [Google Scholar] [CrossRef]
- van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Hindley, C.J.; Condurat, A.L.; Menon, V.; Thomas, R.; Azmitia, L.M.; Davis, J.A.; Pruszak, J. The Hippo pathway member YAP enhances human neural crest cell fate and migration. Sci. Rep. 2016, 6, 23208. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12 (Suppl. 2), 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, L.; Szeto, P.; Abali, G.K.; Zhang, Y.; Kulkarni, A.; Amarasinghe, K.; Li, J.; Vergara, I.A.; Molania, R.; et al. The Hippo pathway oncoprotein YAP promotes melanoma cell invasion and spontaneous metastasis. Oncogene 2020, 39, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yang, X.; Huang, X.; Yao, Y.; Wei, X.; Yang, S.; Zhou, D.; Zhang, W.; Long, Z.; Xu, X.; et al. 2-Hydroxylation of Fatty Acids Represses Colorectal Tumorigenesis and Metastasis via the YAP Transcriptional Axis. Cancer Res. 2021, 81, 289–302. [Google Scholar] [CrossRef]
- Zhao, B.; Xie, J.; Zhou, X.; Zhang, L.; Cheng, X.; Liang, C. YAP activation in melanoma contributes to anoikis resistance and metastasis. Exp. Biol. Med. (Maywood) 2020, 246, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Ambrosetti, D.C.; Sancisi, V. Multiple roles and context-specific mechanisms underlying YAP and TAZ-mediated resistance to anti-cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188341. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Role of YAP/TAZ transcriptional regulators in resistance to anti-cancer therapies. Cell. Mol. Life Sci. 2017, 74, 1457–1474. [Google Scholar] [CrossRef]
- Zeng, R.; Dong, J. The Hippo Signaling Pathway in Drug Resistance in Cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef]
- Gujral, T.S.; Kirschner, M.W. Hippo pathway mediates resistance to cytotoxic drugs. Proc. Natl. Acad. Sci. USA 2017, 114, E3729–E3738. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wu, L.; Tu, J.; Zhao, Z.; Fan, X.; Mao, J.; Weng, Q.; Wu, X.; Huang, L.; Xu, M.; et al. miR-590-5p suppresses hepatocellular carcinoma chemoresistance by targeting YAP1 expression. EBioMedicine 2018, 35, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wu, B.; Zhong, Z. Downregulation of YAP inhibits proliferation, invasion and increases cisplatin sensitivity in human hepatocellular carcinoma cells. Oncol. Lett. 2018, 16, 585–593. [Google Scholar] [CrossRef]
- Chen, X.; Gu, W.; Wang, Q.; Fu, X.; Wang, Y.; Xu, X.; Wen, Y. C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Oncotarget 2018, 9, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; He, F.; Zhao, X.; Zhang, Y.; Chu, X.; Hua, C.; Qu, Y.; Duan, Y.; Ming, L. YAP Inhibits the Apoptosis and Migration of Human Rectal Cancer Cells via Suppression of JNK-Drp1-Mitochondrial Fission-HtrA2/Omi Pathways. Cell. Physiol. Biochem. 2017, 44, 2073–2089. [Google Scholar] [CrossRef]
- Goldsmith, K.C.; Gross, M.; Peirce, S.; Luyindula, D.; Liu, X.; Vu, A.; Sliozberg, M.; Guo, R.; Zhao, H.; Reynolds, C.P.; et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012, 72, 2565–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.F.; Tseng, Y.C.; Chang, W.C.; Chen, Y.C.; Kao, Y.R.; Chou, T.Y.; Ho, C.C.; Wu, C.W. YAP1 is essential for tumor growth and is a potential therapeutic target for EGFR-dependent lung adenocarcinomas. Oncotarget 2017, 8, 89539–89551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Park, H.S.; Lee, D.; Yoo, G.; Kim, T.; Jeon, H.; Yeo, M.K.; Lee, C.S.; Moon, J.Y.; Jung, S.S.; et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem. Biophys. Res. Commun. 2016, 474, 154–160. [Google Scholar] [CrossRef]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef]
- Hsu, P.C.; You, B.; Yang, Y.L.; Zhang, W.Q.; Wang, Y.C.; Xu, Z.; Dai, Y.; Liu, S.; Yang, C.T.; Li, H.; et al. YAP promotes erlotinib resistance in human non-small cell lung cancer cells. Oncotarget 2016, 7, 51922–51933. [Google Scholar] [CrossRef] [Green Version]
- Ney, G.M.; McKay, L.; Koschmann, C.; Mody, R.; Li, Q. The Emerging Role of Ras Pathway Signaling in Pediatric Cancer. Cancer Res. 2020, 80, 5155–5163. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Slemmons, K.K.; Crose, L.E.; Rudzinski, E.; Bentley, R.C.; Linardic, C.M. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS ONE 2015, 10, e0140781. [Google Scholar] [CrossRef]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.A.; Liu, Y.; Zage, P.E. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [Green Version]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK inhibitor trametinib does not prevent the growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas. Sci. Signal. 2017, 10, eaam7550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misiak, D.; Hagemann, S.; Bell, J.L.; Busch, B.; Lederer, M.; Bley, N.; Schulte, J.H.; Huttelmaier, S. The MicroRNA Landscape of MYCN-Amplified Neuroblastoma. Front. Oncol. 2021, 11, 647737. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.A.; Lu, C.Y.; Cheng, T.Y.; Pan, S.H.; Chen, H.F.; Chang, N.S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Jiang, D.; Chi, F.; Zhao, B. The Hippo pathway regulates stem cell proliferation, self-renewal, and differentiation. Protein Cell 2012, 3, 291–304. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Mishima, T.; Takano, S.; Yoshitomi, H.; Furukawa, K.; Takayashiki, T.; Kuboki, S.; Takada, M.; Miyazaki, M.; Ohtsuka, M. The Expression of Yes-Associated Protein (YAP) Maintains Putative Cancer Stemness and Is Associated with Poor Prognosis in Intrahepatic Cholangiocarcinoma. Am. J. Pathol. 2019, 189, 1863–1877. [Google Scholar] [CrossRef] [Green Version]
- Lavado, A.; Park, J.Y.; Pare, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591 e578. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Pfaff, S.L.; Gage, F.H. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008, 22, 3320–3334. [Google Scholar] [CrossRef] [Green Version]
- Maguire, L.H.; Thomas, A.R.; Goldstein, A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015, 244, 311–322. [Google Scholar] [CrossRef]
- Bate-Eya, L.T.; Ebus, M.E.; Koster, J.; den Hartog, I.J.; Zwijnenburg, D.A.; Schild, L.; van der Ploeg, I.; Dolman, M.E.; Caron, H.N.; Versteeg, R.; et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur. J. Cancer 2014, 50, 628–637. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Lyu, X.; Faleti, O.D.; He, M.L. The special stemness functions of Tbx3 in stem cells and cancer development. Semin. Cancer Biol. 2019, 57, 105–110. [Google Scholar] [CrossRef]
- Chen, X.; Yang, W.; Deng, X.; Ye, S.; Xiao, W. Interleukin-6 promotes proliferative vitreoretinopathy by inducing epithelial-mesenchymal transition via the JAK1/STAT3 signaling pathway. Mol. Vis. 2020, 26, 517–529. [Google Scholar]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling Components of the Tumor Microenvironment to Enhance Cancer Therapy. Front. Oncol. 2015, 5, 214. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Pike, L.R.; Phadwal, K.; Simon, A.K.; Harris, A.L. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol. Biol. Rep. 2012, 39, 10811–10822. [Google Scholar] [CrossRef]
- Nakamura, M.; Shimada, K.; Konishi, N. The role of HRK gene in human cancer. Oncogene 2008, 27 (Suppl. 1), S105–S113. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zeng, C.; Ye, S.; Dai, X.; He, Q.; Yang, B.; Zhu, H. Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ): A nexus between hypoxia and cancer. Acta Pharm. Sin. B 2020, 10, 947–960. [Google Scholar] [CrossRef]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Hooglugt, A.; van der Stoel, M.M.; Boon, R.A.; Huveneers, S. Endothelial YAP/TAZ Signaling in Angiogenesis and Tumor Vasculature. Front. Oncol. 2020, 10, 612802. [Google Scholar] [CrossRef]
- Neto, F.; Klaus-Bergmann, A.; Ong, Y.T.; Alt, S.; Vion, A.C.; Szymborska, A.; Carvalho, J.R.; Hollfinger, I.; Bartels-Klein, E.; Franco, C.A.; et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 2018, 7, e31037. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Pahlman, S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Jogi, A.; Ora, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Pahlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [Green Version]
- Cangelosi, D.; Morini, M.; Zanardi, N.; Sementa, A.R.; Muselli, M.; Conte, M.; Garaventa, A.; Pfeffer, U.; Bosco, M.C.; Varesio, L.; et al. Hypoxia Predicts Poor Prognosis in Neuroblastoma Patients and Associates with Biological Mechanisms Involved in Telomerase Activation and Tumor Microenvironment Reprogramming. Cancers 2020, 12, 2343. [Google Scholar] [CrossRef]
- Pahlman, S.; Mohlin, S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res. 2018, 372, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Meitar, D.; Crawford, S.E.; Rademaker, A.W.; Cohn, S.L. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J. Clin. Oncol. 1996, 14, 405–414. [Google Scholar] [CrossRef]
- Giussani, M.; Triulzi, T.; Sozzi, G.; Tagliabue, E. Tumor Extracellular Matrix Remodeling: New Perspectives as a Circulating Tool in the Diagnosis and Prognosis of Solid Tumors. Cells 2019, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Jang, M.; An, J.; Oh, S.W.; Lim, J.Y.; Kim, J.; Choi, J.K.; Cheong, J.H.; Kim, P. Matrix stiffness epigenetically regulates the oncogenic activation of the Yes-associated protein in gastric cancer. Nat. Biomed. Eng. 2021, 5, 114–123. [Google Scholar] [CrossRef]
- Lam, W.A.; Cao, L.; Umesh, V.; Keung, A.J.; Sen, S.; Kumar, S. Extracellular matrix rigidity modulates neuroblastoma cell differentiation and N-myc expression. Mol. Cancer 2010, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Abyaneh, H.S.; Regenold, M.; McKee, T.D.; Allen, C.; Gauthier, M.A. Towards extracellular matrix normalization for improved treatment of solid tumors. Theranostics 2020, 10, 1960–1980. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- White, S.M.; Murakami, S.; Yi, C. The complex entanglement of Hippo-Yap/Taz signaling in tumor immunity. Oncogene 2019, 38, 2899–2909. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189.e5. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Liu, Y.; Xu, R.; Park, O.; He, Y.; Cha, B.; Gao, B.; Yang, Y. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut 2018, 67, 1692–1703. [Google Scholar] [CrossRef]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef]
- Lebid, A.; Chung, L.; Pardoll, D.M.; Pan, F. YAP Attenuates CD8 T Cell-Mediated Anti-tumor Response. Front. Immunol. 2020, 11, 580. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Hsu, P.C.; Yang, Y.L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Zeine, R.; Salwen, H.R.; Peddinti, R.; Tian, Y.; Guerrero, L.; Yang, Q.; Chlenski, A.; Cohn, S.L. Presence of cancer-associated fibroblasts inversely correlates with Schwannian stroma in neuroblastoma tumors. Mod. Pathol. 2009, 22, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Wienke, J.; Dierselhuis, M.P.; Tytgat, G.A.M.; Kunkele, A.; Nierkens, S.; Molenaar, J.J. The immune landscape of neuroblastoma: Challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur. J. Cancer 2021, 144, 123–150. [Google Scholar] [CrossRef]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Nolan, J.C.; Frawley, T.; Tighe, J.; Soh, H.; Curtin, C.; Piskareva, O. Preclinical models for neuroblastoma: Advances and challenges. Cancer Lett. 2020, 474, 53–62. [Google Scholar] [CrossRef]
- Corallo, D.; Frabetti, S.; Candini, O.; Gregianin, E.; Dominici, M.; Fischer, H.; Aveic, S. Emerging Neuroblastoma 3D In Vitro Models for Pre-Clinical Assessments. Front. Immunol. 2020, 11, 584214. [Google Scholar] [CrossRef]
- Ornell, K.J.; Coburn, J.M. Developing preclinical models of neuroblastoma: Driving therapeutic testing. BMC Biomed. Eng. 2019, 1, 33. [Google Scholar] [CrossRef]
- Monferrer, E.; Martin-Vano, S.; Carretero, A.; Garcia-Lizarribar, A.; Burgos-Panadero, R.; Navarro, S.; Samitier, J.; Noguera, R. A three-dimensional bioprinted model to evaluate the effect of stiffness on neuroblastoma cell cluster dynamics and behavior. Sci. Rep. 2020, 10, 6370. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.H.; Beierle, A.M.; Beierle, E.A. Artificial Tumor Microenvironments in Neuroblastoma. Cancers 2021, 13, 1629. [Google Scholar] [CrossRef]
- Braekeveldt, N.; Wigerup, C.; Tadeo, I.; Beckman, S.; Sanden, C.; Jonsson, J.; Erjefalt, J.S.; Berbegall, A.P.; Borjesson, A.; Backman, T.; et al. Neuroblastoma patient-derived orthotopic xenografts reflect the microenvironmental hallmarks of aggressive patient tumours. Cancer Lett. 2016, 375, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Zhu, S.; Thomas Look, A. Neuroblastoma and Its Zebrafish Model. Adv. Exp. Med. Biol. 2016, 916, 451–478. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Brodowska, K.; Al-Moujahed, A.; Marmalidou, A.; Meyer Zu Horste, M.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp. Eye Res. 2014, 124, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, F.; Wang, Y.; Li, T.; Xiu, P.; Zhong, J.; Sun, X.; Li, J. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017, 108, 478–487. [Google Scholar] [CrossRef]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Qiao, S.; Post, L. Targeting the Hippo-YAP pathway with novel small-molecule inhibitors of the YAP-TEAD transcription activity [abstract]. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Laraba, L.; de Guibert, J.G.; Tang, T.T.; Post, L.; Parkinson, D.B. TEAD inhibition reduces tumor proliferation in Merlin null meningioma and schwannoma [abstract]. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.X.; Guan, K.L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer 2021, 20, 986–998. [Google Scholar] [CrossRef]
- Higuchi, T.; Nakamura, M.; Shimada, K.; Ishida, E.; Hirao, K.; Konishi, N. HRK inactivation associated with promoter methylation and LOH in prostate cancer. Prostate 2008, 68, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Toyota, M.; Satoh, A.; Sasaki, Y.; Ogi, K.; Akino, K.; Suzuki, H.; Murai, M.; Kikuchi, T.; Mita, H.; et al. Identification of HRK as a target of epigenetic inactivation in colorectal and gastric cancer. Clin. Cancer Res. 2003, 9, 6410–6418. [Google Scholar] [PubMed]
- Li, H.; Cai, Q.; Wu, H.; Vathipadiekal, V.; Dobbin, Z.C.; Li, T.; Hua, X.; Landen, C.N.; Birrer, M.J.; Sanchez-Beato, M.; et al. SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Mol. Cancer Res. 2012, 10, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Chen, X.; Chen, N.; Nie, L.; Li, X.; Li, Q.; Zeng, H.; Zhou, Q. Synergistic silencing by promoter methylation and reduced AP-2alpha transactivation of the proapoptotic HRK gene confers apoptosis resistance and enhanced tumor growth. Am. J. Pathol. 2013, 182, 84–95. [Google Scholar] [CrossRef]
- Hillmer, R.E.; Link, B.A. The Roles of Hippo Signaling Transducers Yap and Taz in Chromatin Remodeling. Cells 2019, 8, 502. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Mao, B.; Cheng, C.; Zou, Z.; Gao, J.; Yang, Y.; Lei, T.; Qi, X.; Yuan, Z.; Xu, W.; et al. YAP promotes breast cancer metastasis by repressing growth differentiation factor-15. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Sun, S.; Chen, X.; Zhang, H.; Li, B.; Sun, S. YAP/TAZ-mediated activation of serine metabolism and methylation regulation is critical for LKB1-deficient breast cancer progression. Biosci. Rep. 2017, 37, BSR20171072. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phimmachanh, M.; Han, J.Z.R.; O’Donnell, Y.E.I.; Latham, S.L.; Croucher, D.R. Histone Deacetylases and Histone Deacetylase Inhibitors in Neuroblastoma. Front. Cell Dev. Biol. 2020, 8, 578770. [Google Scholar] [CrossRef] [PubMed]
- Jubierre, L.; Jimenez, C.; Rovira, E.; Soriano, A.; Sabado, C.; Gros, L.; Llort, A.; Hladun, R.; Roma, J.; Toledo, J.S.; et al. Targeting of epigenetic regulators in neuroblastoma. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Faiao-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019, 25, 5686–5701. [Google Scholar] [CrossRef]
- Yamada, T.; Amann, J.M.; Tanimoto, A.; Taniguchi, H.; Shukuya, T.; Timmers, C.; Yano, S.; Shilo, K.; Carbone, D.P. Histone Deacetylase Inhibition Enhances the Antitumor Activity of a MEK Inhibitor in Lung Cancer Cells Harboring RAS Mutations. Mol. Cancer 2018, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef]
Figure 1.
YAP/Hippo Signaling Pathway. Adapted from “Hippo Pathway in Mammals” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 8 September 2021).
Figure 1.
YAP/Hippo Signaling Pathway. Adapted from “Hippo Pathway in Mammals” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 8 September 2021).

Figure 2.
Potential roles for YAP in the neuroblastoma TME and areas for further investigation. YAP plays a role in every aspect of the TME from angiogenesis, ECM remodeling, immune evasion, and regulation of TME-related target genes, such as HRK. We have published on the novel relationship between YAP and HRK in neuroblastoma leading to regulation of stress-induced apoptosis in the tumor environment (yellow highlighted box). Areas of YAP TME regulation in other cancers that have also been described as TME factors in neuroblastoma support that further investigation should be pursued (blue boxes). Adapted from “The Tumor Microenvironment: Overview of Cancer-Associated Changes”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 8 September 2021).
Figure 2.
Potential roles for YAP in the neuroblastoma TME and areas for further investigation. YAP plays a role in every aspect of the TME from angiogenesis, ECM remodeling, immune evasion, and regulation of TME-related target genes, such as HRK. We have published on the novel relationship between YAP and HRK in neuroblastoma leading to regulation of stress-induced apoptosis in the tumor environment (yellow highlighted box). Areas of YAP TME regulation in other cancers that have also been described as TME factors in neuroblastoma support that further investigation should be pursued (blue boxes). Adapted from “The Tumor Microenvironment: Overview of Cancer-Associated Changes”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates (accessed on 8 September 2021).


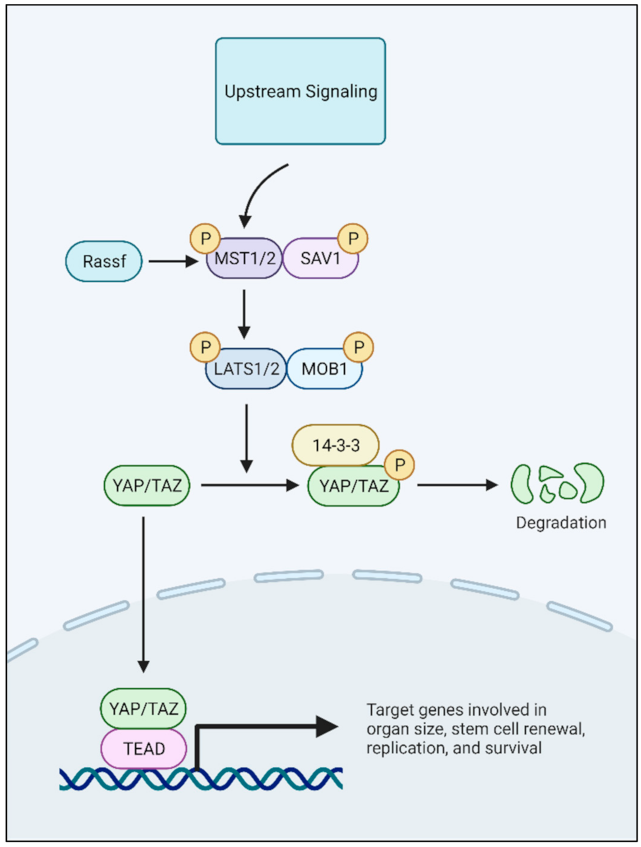{kind=link}
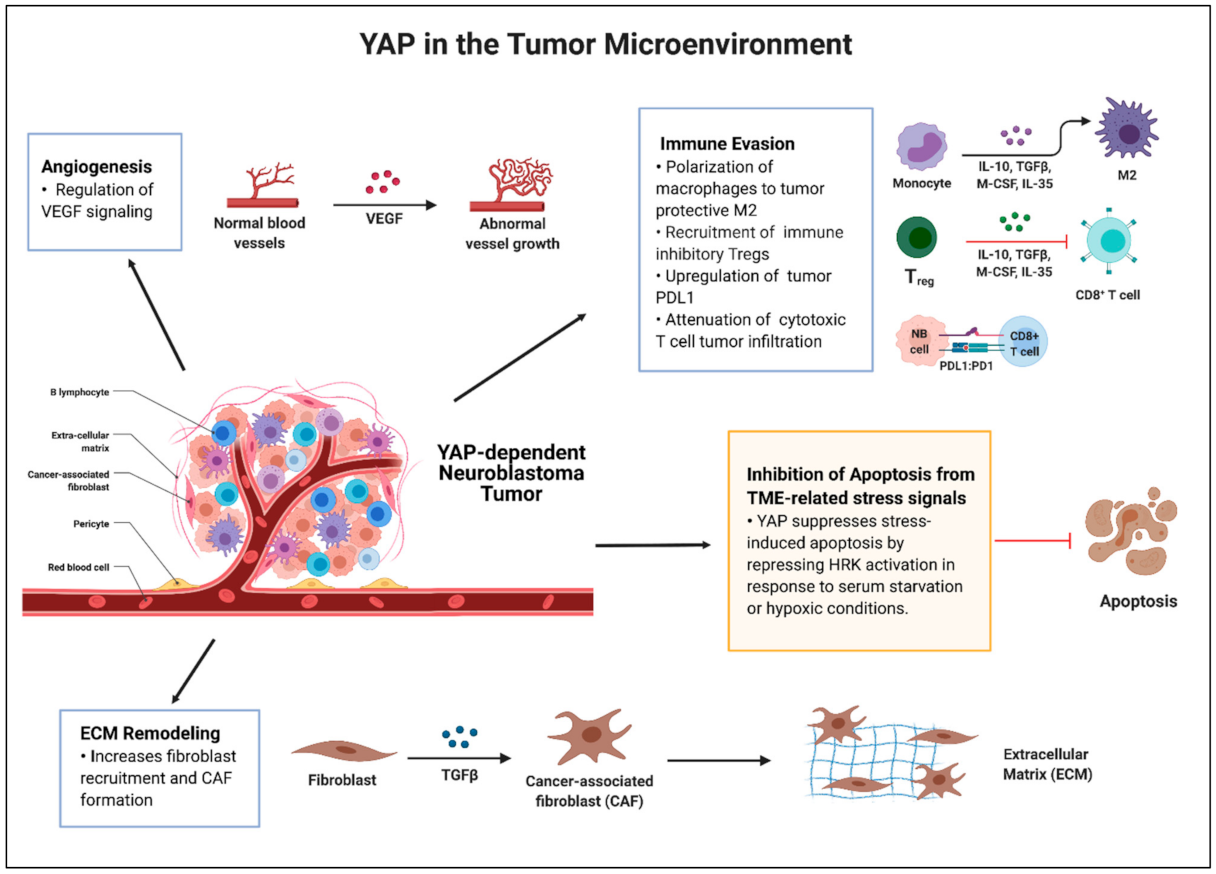{kind=link}
Table 1.
YAP in neuroblastoma: summary of the literature.
| Focus | Comments | References |
|---|---|---|
| Tumorigenesis |
| [51,53,54,55] |
| Metastasis |
| [51,55,56] |
| Therapy Resistance |
| [51,52,53] |
| Mesenchymal Properties |
| [51,57,58,59] |
The table summarizes a review of the recent literature describing the function of YAP in neuroblastoma.
Table 2.
Potential therapies to target YAP and related pathways.
| Therapy | Mechanism and Effects | References |
|---|---|---|
| Verteporfin |
| [159,160,161] |
| Cyclic Peptides (i.e., Peptide 17) |
| [162,163] |
| TEAD auto-palmitoylation inhibitors |
| [166,179] |
| Combination therapies |
| [113,135,175,176,177,178] |
The table summarizes potential YAP/TEAD inhibitors or related YAP pathway targeted therapy approaches.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shim, J.; Goldsmith, K.C. A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers 2021, 13, 4650. https://doi.org/10.3390/cancers13184650
AMA Style
Shim J, Goldsmith KC. A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment. Cancers. 2021; 13(18):4650. https://doi.org/10.3390/cancers13184650
Chicago/Turabian StyleShim, Jenny, and Kelly C. Goldsmith. 2021. "A New Player in Neuroblastoma: YAP and Its Role in the Neuroblastoma Microenvironment" Cancers 13, no. 18: 4650. https://doi.org/10.3390/cancers13184650
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.