
Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles

Department of Physiology and Biophysics, University of Washington School of Medicine, Seattle, WA 98195, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(22), 5650; https://doi.org/10.3390/cancers13225650
Submission received: 16 October 2021
/
Revised: 9 November 2021
/
Accepted: 10 November 2021
/
Published: 12 November 2021
(This article belongs to the Special Issue Microtubule Dynamics and Cancer)
{kind=link}
{kind=link}
Abstract
:Simple Summary
Microtubules (MTs) are highly conserved proteins present in all eukaryotic organisms. They form the cell cytoskeleton, and its function is essential for a large number of biological processes. Drugs against MTs (i.e., microtubule-targeting agents or MTAs) have been used for centuries to treat arthritis and gout. In the last 100 years, new MTAs either isolated from natural sources or synthesized in labs have been used to treat a great variety of human illnesses, from cancer and neurodegenerative diseases to the elimination of parasites. In this review, we analyze how MTAs bind to MTs, and the molecular mechanisms behind MTAs function, and we describe the last and novel roles of these drugs.
Abstract
Microtubule-targeting agents (MTAs) represent one of the most successful first-line therapies prescribed for cancer treatment. They interfere with microtubule (MT) dynamics by either stabilizing or destabilizing MTs, and in culture, they are believed to kill cells via apoptosis after eliciting mitotic arrest, among other mechanisms. This classical view of MTA therapies persisted for many years. However, the limited success of drugs specifically targeting mitotic proteins, and the slow growing rate of most human tumors forces a reevaluation of the mechanism of action of MTAs. Studies from the last decade suggest that the killing efficiency of MTAs arises from a combination of interphase and mitotic effects. Moreover, MTs have also been implicated in other therapeutically relevant activities, such as decreasing angiogenesis, blocking cell migration, reducing metastasis, and activating innate immunity to promote proinflammatory responses. Two key problems associated with MTA therapy are acquired drug resistance and systemic toxicity. Accordingly, novel and effective MTAs are being designed with an eye toward reducing toxicity without compromising efficacy or promoting resistance. Here, we will review the mechanism of action of MTAs, the signaling pathways they affect, their impact on cancer and other illnesses, and the promising new therapeutic applications of these classic drugs.
1. Microtubules and Tubulin
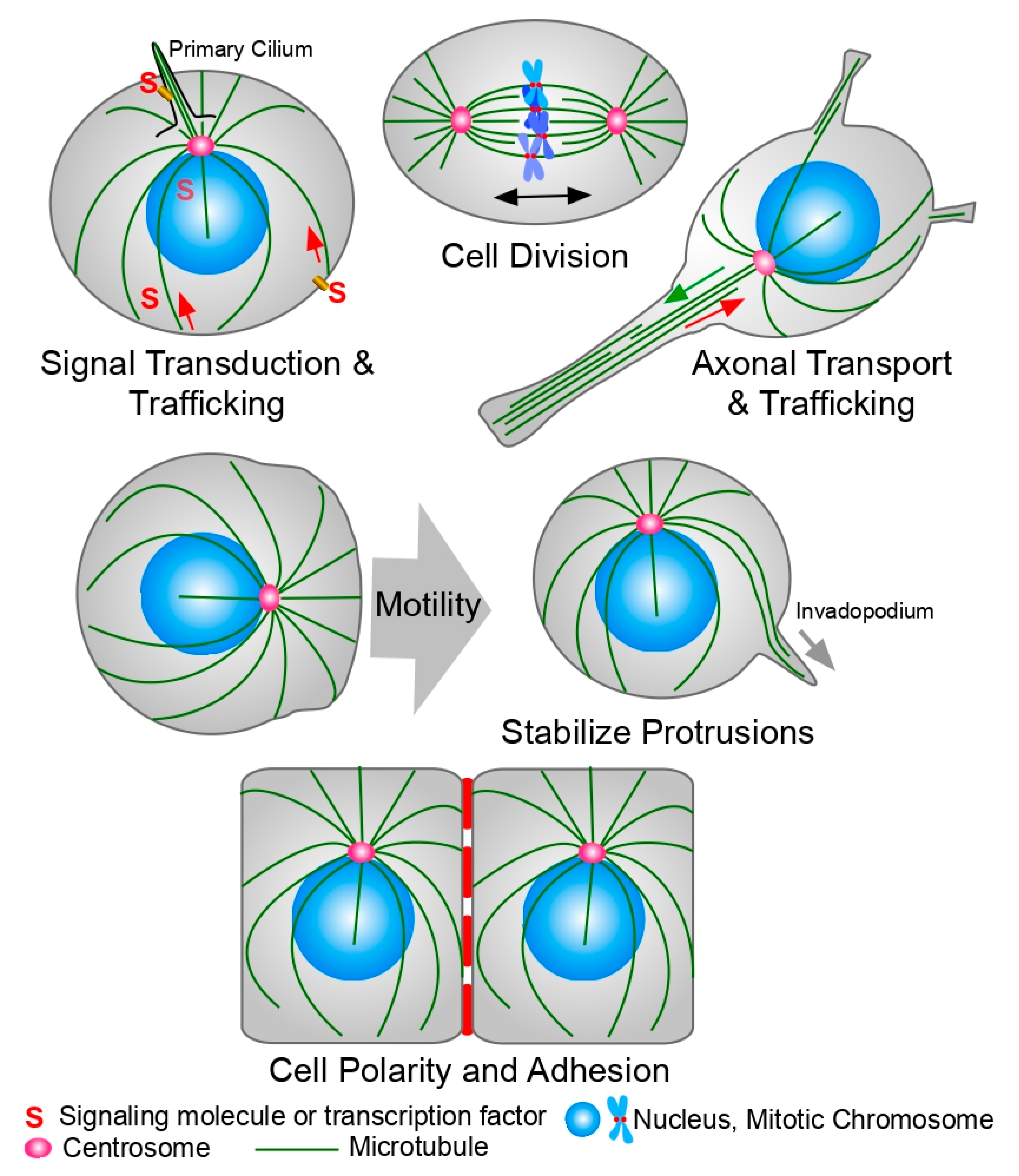
Microtubules (MTs) are hollow cylindrical polymers composed of tubulin dimers. They play vital roles in a wide variety of cellular functions: intracellular trafficking, cell shape and morphology, positioning of organelles inside the cell, cell motility and migration, and the assembly of the mitotic spindle, which is the machinery responsible for chromosome segregation during mitosis [1] (Figure 1). Tubulin is a 50 kDa globular GTP-binding protein that is found in all eukaryotic cells. There are six members of the tubulin family in eukaryotic cells: alpha (α), beta (β), gamma (ɣ), delta (δ), epsilon (ε), and zeta (ζ) [1,2]. However, not all organisms possess all the tubulin genes. For example, zeta tubulin is not present in humans. The widely distributed α and β tubulins assemble to form heterodimers that will ultimately constitute the cytoplasmic microtubules, and they have been found in all eukaryotic cells studied thus far. Both of them bind GTP, but only β tubulin is able to hydrolyze GTP during microtubule polymerization. ɣ tubulin is part of the machinery that nucleates microtubule growth. δ, ε, and ζ tubulins are part of cilia, flagella, and basal bodies, and they are generally specific to organisms that possess these structures. In humans, there are 23 functional genes coding for tubulin [2], with α and β constituting the elementary building blocks of MTs and ɣ, δ, and ε localizing to the centrosome. The α-β tubulin heterodimers polymerize into a linear protofilament, which associates laterally with more protofilaments to form the pseudo-helical structure that is the MT. Most MTs are composed of 13 protofilaments, but MTs with different numbers of protofilaments have been observed in different species [3].
MTs present with a polarity consisting of a plus-end where tubulin dimers are preferentially added, and a minus-end often embedded within the centrosome, the chief microtubule organizing center (MTOC) of most human cells. Polarity emerges due to the head-to-tail orientation of the α-β tubulin heterodimers inside the MT lattice, with the α subunit pointing towards the minus-end of the MT and the β subunit towards the plus-end. Of the two, the β-tubulin subunit hydrolyzes GTP after the incorporation of the tubulin dimer to the microtubule [4]. Prior to this hydrolysis event, GTP-β-tubulin forms a structural cap that stabilizes the MT. As long as there is enough GTP-β-tubulin in the vicinity of the MT tip, more dimers will continue to be added to the tip and the MT will continue growing. However, if the growth rate falls below the rate of GTP-β-tubulin hydrolysis, loss of the stabilizing cap triggers the disassembly of the MT in a process called MT catastrophe. Once a new cap is formed, the MT can begin growing again. Periods of growth and shrinking alternate in a cycle known as dynamic instability [5]. In cells, an ample cohort of microtubule-associated proteins like the end-binding proteins EB1, EB2, and EB3 regulate this process [6,7,8]. This dynamic turnover of MT polymer is essential for virtually all MT-dependent biological processes.
2. Microtubule-Targeting Agents (MTAs)
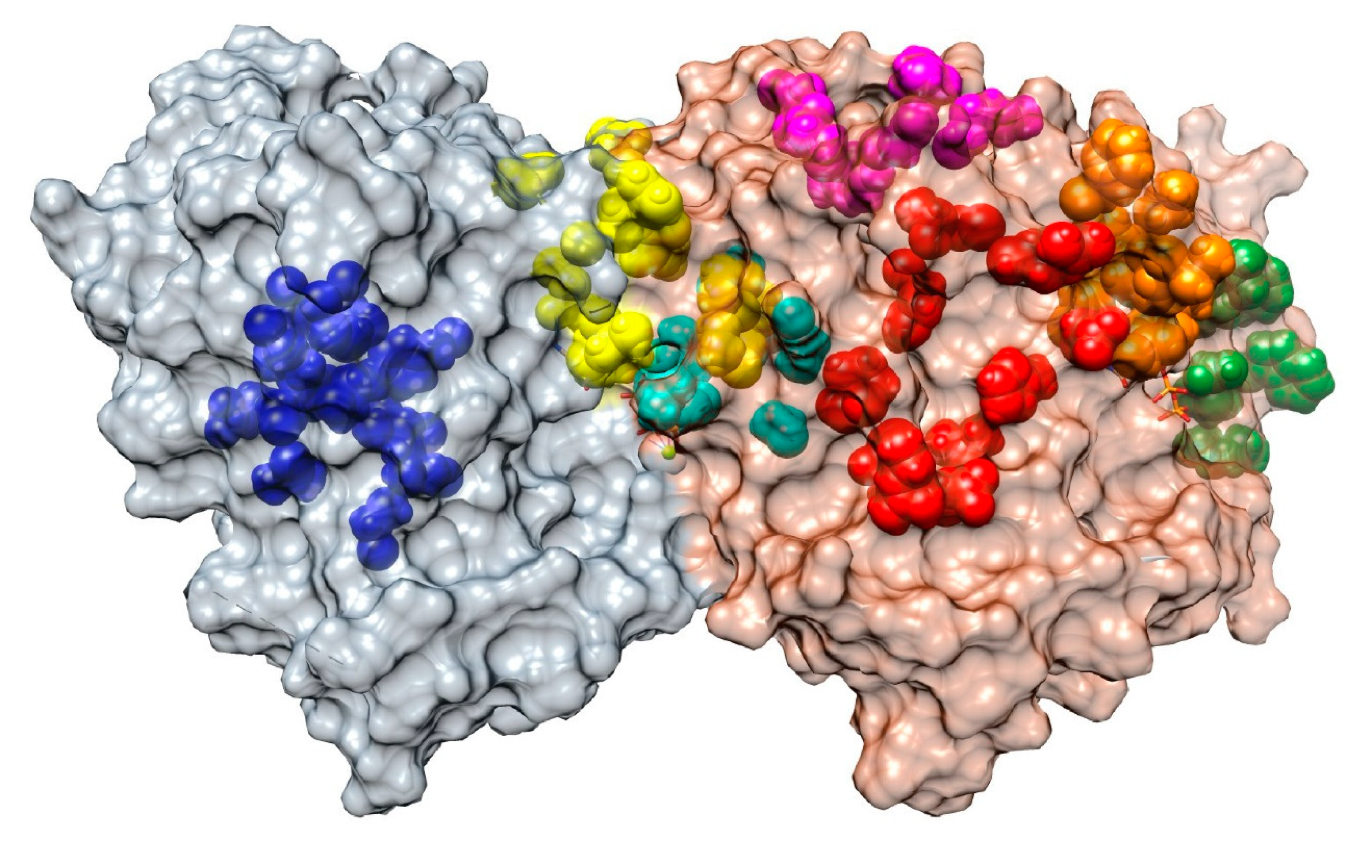
MTAs are a large and diverse family of chemical compounds that are able to bind tubulin and interfere with the dynamic behavior of MTs, either by stabilizing or destabilizing the MT polymer. Accordingly, the simplest classification of these drugs is into two groups: MT-stabilizing agents (MSAs) and MT-destabilizing agents (MDAs). All MTAs bind to one of these seven sites in the tubulin dimers [9,10,11] (Figure 2).
- −
- Vinca domain (MDA, orange in Figure 2): this domain is located in the β tubulin monomer at the inter-dimer interface between two longitudinally aligned tubulin dimers. Drugs that bind to this domain inhibit the assembly of tubulin by sequestering tubulin into paracrystalline aggregates. Examples of these drugs are vincristine, vinblastine, and vindesine [12,13,14,15].
- −
- Colchicine site (MDA, cyan in Figure 2): this site is in the β tubulin monomer in a deep pocket space between the αβ-tubulin dimer itself. Binding to this site inhibits microtubule formation by preventing a conformational change in tubulin required for polymerization. Colchicine, benzimidazoles (e.g., nocodazole), and combretastatins are among the drugs that bind to this site [16,17].
- −
- Taxane site (MSA, red in Figure 2): it is located at the β tubulin monomer in the lumen of microtubules. Binding to this site stabilizes the MT lattice. Interestingly, MT-stabilizing drugs, such as paclitaxel and epothilone, achieve their microtubule-stabilizing effect through pharmacologically distinct mechanisms despite binding competitively to the same taxane site [18].
- −
- −
- Laulimalide/peloruside site (MSA, magenta in Figure 2): this site is located in a pocket of the β-tubulin that faces the outside of the MT. Studies suggest that they inhibit microtubule disassembly by acting as molecular ‘clamps’ that hold together protofilaments. The drugs Laulimalide and Peloruside isolated from marine sponges gave name to this group [22,23,24].
- −
- −
- Gatorbulin site (MDA, yellow in Figure 2): this new site has been described in 2021, is located in the α-tubulin subunit (between α- and β-tubulin) close to the colchicine site, and it functions in a similar way to the vinca site, creating a wedge between two longitudinally aligned tubulin dimers at the end of MTs [10]. Recently, a new compound called Cevipabulin has been described to also bind to this pocket [11].
For an excellent in-depth review of MTA binding sites and their mechanism of action, see Steinmetz and Prota in 2018 [9]. MSAs and MDAs exhibit opposite effects on MT polymer mass at high concentrations, inducing either depolymerization or stabilization of the MTs, and changing the ratio of monomer and polymer. At low concentrations, however, they can influence MT dynamics to affect cellular functions without appreciable changes in the total amount of MT polymer. Drugs binding to the vinca, colchicine, and taxane sites comprise the most well-studied MTAs, and they have been used as medicines for a long time. Colchicine, for example, originally extracted from the plant autumn crocus (Colchicum autumnale), is probably the oldest and first reported MTA used as a therapeutic drug. The Egyptian medical text known as the “Ebers papyrus”, dated from 1500 BC, described the use of the plant autumn crocus as a remedy for inflammation and rheumatism [29]. Colchicine has also been used for centuries to treat gout [29,30]. However, in the last century, MTAs have been also used as herbicides, anti-parasitics and anti-fungal agents, to treat neurodegenerative disease and for cancer treatment.
3. MTAs and Pathogens
Tubulin is conserved among all eukaryotic cells, from Homo sapiens to the deeply rooted supergroup Excavata [2]. However, there are differences in the tubulin sequence between humans, parasites, and fungi that make this protein an appropriate target for antifungal and antiparasitic drugs [31,32,33]. There are, in fact, several MTAs used as antiparasitic drugs that bind more efficiently to pathogen tubulin than to human tubulin [34].
Benzimidazole is an aromatic organic compound resulting from the fusion of imidazole and benzene. Through modifications of the benzimidazole group, chemists have generated a vast group of compounds that have been used successfully to affect a variety of biological activities. It has been shown that compounds derived from benzimidazole have antitumor activity, antiviral activity, and anti-inflammatory, antihypertensive, and anticoagulant properties [35]. A group of benzimidazole compounds that bind close to the colchicine binding site of β-tubulin have been used as antihelminthics to treat a great variety of parasitic worm infections. Examples of these compounds are albendazole, mebendazole, triclabendazole, and thiabendazole. In vitro studies of benzimidazole compounds show that they bind with greater affinity to the β-tubulin site of worms than the equivalent site in humans, effectively blocking MT polymerization at lower drug concentrations in the parasites [36]. However, the therapeutic efficiency of benzimidazoles is based on their poor solubility in water and poor absorption in the intestines. For example, the absorption rate for mebendazole by the human intestine is between 1 and 5% unless it is administered with high-fat foods. Thus, mebendazole affects the parasite in the human intestine, sparing the host cells [37,38,39,40]. Benzimidazole compounds carbendazim, benomyl, thiophanate-methyl, and thiabendazole have also been used as fungicides based, also, on their lack of a measurable interaction with mammalian tubulin [41,42,43].
MTAs have also been used in agriculture for weed control [44]. Dinitroanilines are a class of chemicals derived from either dinitrobenzene or aniline that are used as dyes, pesticides, and herbicides. They are also used as antiparasitic drugs as they bind to α-tubulin in parasites, while exhibiting a lack of measurable affinity for vertebrate α-tubulin [45,46,47,48,49]. Among these compounds, we have trifluralin (herbicide and antiparasitic) [47] and oryzalin (herbicide and antiparasitic) [50].
4. MTAs and Neurodegenerative Diseases
The brain is critically dependent on MTs for function. Neurons possess long axons full of parallel MTs that act as tracks for the transport of neurotransmitter from the cell body to the synaptic cleft (Figure 1). This transport is mediated by kinesins and dyneins, which serve as MT-dependent molecular motors [52]. In most neurodegenerative diseases, neuronal MTs are disrupted and the transport of neurotransmitters via MTs in the axon is impaired. Thus, MSAs have the potential to treat the symptoms of neurodegenerative diseases like Parkinson’s and Alzheimer’s, protecting the MTs in the axons and restoring synaptic transmission [53,54,55,56,57,58,59].
Tauopathies
Tau proteins are a group of microtubule-associated proteins that originate by alternative splicing from the MAPT gene in the chromosome 17 in humans. There are six isoforms expressed in the human adult brain, where they play an essential role stabilizing the MTs in the axons of neurons and are involved in neural transport and axonal growth. Defects in the phosphorylation levels of Tau proteins lead to the formation of fibrillar inclusions composed of hyperphosphorylated tau protein [60,61,62]. Neurodegenerative disorders affecting Tau proteins are called tauopathies, with Alzheimer’s and Parkinson’s diseases being the most well-known and most studied [63,64].
Alzheimer’s disease is one of the most prevalent tauopathies in the world. In Alzheimer’s disease, a hyper-phosphorylated form of tau self-assembles into polymers to create insoluble deposits in the neurons. The creation of these aggregates decreases the ability of tau to bind and stabilize MTs, resulting in axonal and neural dysfunction [65,66]. Alzheimer’s disease is also characterized by the accumulation of β-amyloid plaques in the brain, and most of the research in Alzheimer’s disease has been focused on decreasing the levels of amyloid-β (Aβ) peptides in the brain. However, the lack of success using drugs to target the Aβ peptides changed the focus toward reducing the accumulation of Tau protein, and the development of molecules that are able to compensate the lack of normal Tau function [59,67]. Tau, like paclitaxel, binds to β-tubulin and stabilizes MT polymer. Thus, there exists a formal possibility that MSAs could stabilize MTs in situations where hyperphosphorylated aggregations of Tau cannot properly exert their function [53,56,59,68,69,70]. In support of this idea, treatment of a tau transgenic mouse model (mice with a pre-existing tau pathology) with paclitaxel restored fast axonal transport in spinal axons and improved motor impairment. At the cellular level, it was seen an increase in MT number and stable (detyrosinated) MTs [71]. The MT-stabilizing triazolopyrimidine CNDR-51657 significantly lowered tau pathology and improved cognitive function in transgenic mouse models of tauopathy [72]. One of the drugs that has been especially successful in treating tau transgenic mouse models is the MSA epothilone [73]. Paclitaxel and derivatives cannot cross the blood–brain barrier (BBB). Epothilone D was identified as a brain penetrant MSA, and when used at even lower doses than the ones required for cancer treatment, it was able to improve the axonal MT density and transport, decrease axonal dystrophy, and improve the cognitive deficits in a transgenic Tau mouse model [56,74]. These improvements appear to be related to the recovery of normal MT dynamics. In a different study, two different tau transgenic mouse models exhibited age-dependent increases in MT dynamics that were dependent on tau expression levels. Treatment of these mice with epothilone returned MT dynamics levels to basal levels, and had beneficial effects on tau pathology and neurodegeneration [75]. In all these cases, the treatment used a low concentration of drugs and recorded no other adverse effects in the mice. A good review about Alzheimer’s disease, tau protein aggregation, and different therapeutic agents was published in 2017 [66].
There is growing evidence that MT alteration is also a component of Parkinson’s disease. MT defects induced with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice were rescued with Epothilone D treatment, suggesting that MSAs could be employed in the treatment of Parkinson’s disease [55,76].
MSAs may also improve axonal growth and recovery after a stroke in mouse models. The use of Epothilone increases the number and length of neural projections from peri-infarct tissue, and improves forelimb motor function [77]. Epothilone B also promotes axon regeneration and fibroblast migration after spinal cord injury [78].
Therefore, MSAs may constitute a treatment option for neurodegenerative illness. However, there are two drawbacks when using MSAs to treat the nervous system. The first one is peripheral neurotoxicity. MTAs disrupt the MT network in the axon, which allows kinesin molecular motors to transport organelles and vesicles with neurotransmitters to the synaptic cleft, leading to muscle pain and weakness, numbness, and sensory defects [79,80,81]. To address this problem, new taxanes formulations have been developed to improve delivery and reduce side effects [82,83]. For example, the epothilone B analogue ixabepilone has shown some potential to reduce peripheral neurotoxicity without affecting therapeutic capacity [84,85]. Different pharmacological strategies like the use of antidepressants and anticonvulsants, and the use of glutathione, glutamine, and amifostine have also been employed to treat neuropathy, but their benefits are limited [82,84,86]. As today, there are several approaches at different clinical trial phases to address the neuropathy problem related to MTAs [86]. Nevertheless, dose reduction and treatment delay are the most used management tools for MTAs-induced neuropathy [84]. The second drawback is that most of MSAs are not able to cross the BBB as the brain tissue contains a high number of P-glycoprotein drug efflux pumps to remove most chemicals [87]. While paclitaxel and its derivates cannot cross the BBB, Sagopilone and other Epothilone derivates are able to cross the BBB [76,88].
5. MTAs and Cancer
Cancer is the second leading cause of death in the world [89]. The last decade has seen almost 600,000 deaths per year just in the US [90]. Accordingly, MTAs have been used since the mid-1960s as a first-line therapy for cancer treatment [91,92,93,94,95]. While vinca alkaloid and taxane compounds are broadly used for cancer treatment, colchicine use has been limited to the treatment of gout due to its toxicity [30,96,97]. It is challenging to achieve a safe dose of colchicine, and unintentional toxicity can lead to multiorgan failure and death [98]. Despite having different binding sites, the majority of MTAs share a similar mechanism: they affect MT dynamics by increasing, decreasing, or pausing MT polymerization rates. MTs play essential roles in most cell physiological events and the dynamic turnover of MTs is, in turn, essential to their proper function. Thus, altering MT dynamics usually has extremely deleterious effects on cells and often leads to cell death. The most obvious and well-studied defect in cells treated with MTAs is the disruption of the mitotic spindle. The mitotic spindle is composed of microtubules and it is the cell machinery responsible for the proper segregation of chromosomes during mitosis [99]. MTAs disrupt MT dynamics in the spindle, eliciting the mitotic checkpoint, arresting cells in metaphase, and triggering cell death [96]. This led to the conclusion that these drugs kill tumor cells while they are in mitosis. This spurred the development of new chemotherapeutic drugs specifically targeting mitotic proteins, especially mitotic kinases like Aurora and Polo kinases [100,101,102,103,104]. These mitotic-specific drugs have had some success, but they have never attained the tumor killing efficiency of MTAs. Furthermore, most human tumors exhibit slow doubling times, with a mitotic index between 1 and 3% at any given time that cannot account for the tumor shrinkage rate triggered by MTAs [105,106,107]. This demands further investigation of the mechanism of action of MTAs, as most probably they exert their function through a combination of mitotic and interphase effects [108]. Moreover, the possibility exists that the effect of MTAs during mitosis could even be detrimental for cancer treatment. Changes of MT dynamics caused by MTAs also alter the MT–chromosome attachments, which ultimately can lead to chromosome missegregation during anaphase. The increased rate of chromosome missegregation in mitosis is known as chromosome instability (CIN), and it is the cause of aneuploidy. CIN can drive tumorigenesis and cancer development, and has been correlated with drug resistance, therapy failure, metastasis, and poor prognosis (studied and reviewed in [109,110,111,112,113,114,115,116,117,118,119]). Interestingly, while moderate levels of CIN can promote tumorigenesis by promoting karyotype evolution toward the goal of increasing cell survival and growth, high levels of CIN often induced by MTAs kill tumor cells [120]. Importantly, arresting cells in mitosis can be a double-edged sword as these arrested cells can either die or exit mitosis without completing chromosome segregation or cytokinesis. Some of these cells will die in the subsequent cell cycle, but some are able to continue living as polyploid cells, becoming polyploid giant cells (PGCCs) [121,122,123,124,125]. These PGCCs are seen as cancer stem-like cells and reportedly exhibit increased metastasis, increased invasive phenotypes, increased migratory capabilities, and the expression of epithelial to mesenchymal transition markers [121,122,126]. Cells can manifest a wide variation in outcomes after treatment with different MTAs [124]. Even cells from the same cell line can present a variety of cell fates: dying in the first mitosis after MTA treatment, exiting mitosis to arrest in interphase, entering mitosis two or three times after treatment and then dying in interphase or mitosis, dying in interphase after the first mitotic arrest, and finally, dying in interphase without ever entering mitosis [124]. Moreover, the heterogeneity in the cell death in response to MTAs within the same cell line does not appear to be genetically predeterminated. Thus, there is a fine balance between MTAs’ effect on mitosis and cell fate. Clearly, MTAs can kill cells in mitosis and interphase; however, the molecular pathways behind the ability of these drugs to kill cells in interphase remain unclear.
5.1. How Can MTAs Kill Cancer Cells through Mitosis-Independent Mechanisms?
It is possible that MTAs are killing cells in interphase because they interfere with processes like vesicular transport, cell morphology and migration, and/or cell signaling pathways [106]. For example, MTAs are very effective in cancer treatment when used together with DNA-damaging agents or in conjunction with DNA damage induced by radiotherapy. This could be explained, in part, by the fact that MTs are essential for the transport of DNA repair proteins to the nucleus, and that MTAs interfere with and block this transport [127]. MTAs could also directly trigger apoptosis. For example, Taxanes kill cells through a different mechanism depending on the concentration. At high concentrations, they completely suppress MT dynamics, resulting in necrosis. However, at low concentrations, taxanes are able to trigger caspase-induced apoptosis [128]. Finally, MTAs may disturb the vascular tissue around the tumor that provides the cancer cells with oxygen and nutrients [129,130]. Independently of the precise mechanism triggered by MTAs, it appears that these drugs often affect cancer cells more severely than normal cells. Cancer cells walk a tightrope between proliferation, cell cycle arrest, genome instability, and unfavorable growth conditions [107,131,132]. It has been proposed that cancer cells operate very close to their cell death threshold at all times and that MTAs can disrupt this tenuous balance to specifically trigger cancer cell death.
5.2. MTAs and Intracellular Trafficking
MTs are the cellular tracks for the transport of vesicles, proteins, mRNAs, and organelles inside the cell. For example, the transport of proteins to the nucleus and the internalization of receptors requires MTs [133,134]. The tumor suppressor protein p53 requires both intact MTs and the molecular motor dynein to be transported to the nucleus. High doses of MTAs that completely disassemble the MT network will impede the transport of p53 to the nucleus. Paradoxically, it has been shown that subtly altering MT dynamics with low concentrations of MTAs can increase the amount of p53 inside the nucleus, which can sensitize cells to p53-dependent apoptotic cell death [135,136,137]. Another protein that requires MTs for the transport to the nucleus is the parathyroid hormone (PTH)-related protein [138]. Paclitaxel treatment also affects EGFR internalization and endocytic trafficking [139]. MTAs disturb hypoxia-inducible factor 1α (HIF-1α) protein trafficking and function, impairing tumor angiogenesis [140,141,142].
Taxanes are among the most successful drugs used as cancer treatments. Taxol (the commercial brand name of Paclitaxel) has been used for ovarian cancer since 1992 and breast cancer since 1994 [143,144,145]. Additionally, Docetaxel (commercially branded as Taxotere) is used for breast, lung, and prostate cancer [146]. In melanoma cells, paclitaxel reduces invasiveness, disrupting the transport and exocytosis of the metalloproteases necessary to break and degrade the extracellular matrix component [147]. Taxanes also represent the best agents for hormone-refractory prostate cancer as they prevent the transport of the androgen receptor to the nucleus, a necessary step for tumor progression [148,149]. Paclitaxel treatment of the prostate cancer cell line CRPC (castration-resistant prostate cancer) inhibits androgen receptor (AR) activity. Taxanes decreased the expression of androgen receptor (AR)-activated genes, such as prostate-specific antigen (PSA), and increases the expression of the AR repression gene maspin, resulting in global inhibition of AR activity. Paclitaxel also induces nuclear accumulation of the transcription factor FOXO1, a known AR suppressive nuclear factor [150,151].
Another example of the use of MTAs to disturb intracellular trafficking with therapeutic potential is the localization of the Hedgehog (Hh) signaling pathway components to the primary cilium. The Hedgehog (Hh) signaling pathway is essential for embryonic development and it is involved in the morphogenesis of organs like the lungs, brains, skeleton, and the gastrointestinal tract. In adults, it is involved in the self-renewal of stem cells and tissue regeneration [152,153,154]. Uncontrolled activation of this signaling pathway has been involved in the development of several cancers like basal cell carcinoma, glioblastomas, melanomas, and carcinomas of the ovary, prostate, breast, lung, and pancreas [155]. Thus, inhibition of this pathway shows promise as a cancer treatment [156,157,158]. The primary cilium is an MT-based organelle found in eukaryotic cells that acts as a communication hub to couple extracellular signals with intracellular responses [159,160]. The primary cilium plays an essential role in Hh signaling in mammals [161]. Some known drugs that affect the Hh pathway have also been shown to affect MT dynamics, leading to disruptions in the proper localization of the pathway components [162,163]. Increasing evidence supports a role for the primary cilium in cancer progression and development, making the cilium a candidate target for cancer treatment (reviewed in [164]).
5.3. MTAs and Cell Death by Apoptosis
Taxanes can induce mitotic catastrophe in cancer cells [165]; cell death via proteases, such as cathepsins [166]; and cell death through autophagy [167]. Furthermore, paclitaxel can also promote cell death by affecting the signaling pathways responsible for preventing the build-up of reactive oxygen species (ROS) [168]. However, as described above, taxanes are mostly known for their ability to trigger caspase-induced apoptosis [128,169]. In fact, the first studies of cell death triggered by paclitaxel revealed that the principal antitumor effect was through apoptosis rather than mitotic arrest [170]. Taxanes induce apoptosis through both the intrinsic and extrinsic pathways. The intrinsic pathway, also known as the mitochondrial pathway, is activated by intracellular signals after detecting cell damage while the extrinsic pathway is initiated by extracellular ligands binding to death receptor. The intrinsic pathway activates caspase 9 and the extrinsic pathway the caspase 8, but both of them converge in the activation of caspase 3 [171,172]. Taxanes activate the initiator caspases 8 and 9, and the executioner caspases 3, 6, and 7. In fact, the siRNA inactivation of caspase 3 increases the cell survival of taxane-treated cells [173]. Taxanes bind to the anti-apoptotic BCL2 family proteins and block their activity [174]. Treatment with taxanes also seems to downregulate protein levels of anti-apoptotic players like Bcl-2 and Bcl-xL, and upregulate pro-apoptotic ones like BAD and BAX [175,176]. In fact, MEFs lacking pro-apoptotic factors like BAX and BAK were resistant to apoptosis induced by taxanes [177,178].
5.4. MTAs and Effects on the Tumor Vascular Tissue
Tumor cells require oxygen and nutrients to survive, and tumor-infiltrating blood vessels transport these resources to the tumor cells. In addition, tumor blood vessels are also required for metastasis initiation. When the tumor vasculature is disturbed and cannot exert this function, tumor cells begin to die, and the tumor becomes necrotic. Thus, targeting tumor vasculature represents another pathway to treat cancer, reduce the tumor mass, and block the pathway to metastasis. There are two ways to target the tumor vasculature: by blocking the formation of new blood vessels (antiangiogenic effects) or by disrupting the already established vasculature (vasculature-disrupting agents). MTAs have both antiangiogenic and vascular-disrupting properties because they inhibit endothelial cell proliferation, migration, and morphology. Early studies from 1993 showed that the vinca alkaloids, vincristine and vinblastine, decrease the blood flow in tumors and induce necrosis of the tumor cells [179]. Interestingly, most of these effects are seen after treating cells with low concentrations of MTAs. These low concentrations do not elicit the typical cytotoxic effects, such as mitotic arrest and apoptosis, seen at high concentrations commensurate with high levels of MT polymer loss. Rather, these lower concentrations alter MT dynamics enough to affect signaling pathways and the proper formation of focal adhesions and adherents junctions required for cell–cell interactions with fewer overtly toxic effects [180].
5.4.1. Antiangiogenic Effects
The formation of new blood vessels requires the proliferation and migration of endothelial cells. Dynamic MTs are essential for both functions, and any alteration of the MT’s dynamicity elicits cellular defects that ultimately lead to reduced proliferation, migration, and angiogenic capacity. For example, concentrations of paclitaxel that are found to be antiangiogenic increase MT dynamics in endothelial cells [181,182]. MTAs like docetaxel, epothilone B, and vinblastine prevent angiogenesis through the inhibition of the small GTPases Rac1 and Cdc42, proteins involved in cytoskeleton rearrangements required for cell motility and migration [183]. Taxane increases the levels of acetylated tubulin in HUVECs (human umbilical vein endothelial cells) with a correlative inhibition cell motility [184]. Interestingly, in this same study, the authors found that MTAs caused translocation to the nucleus of the transcription factor FOXO3, a negative regulator of cell motility and a positive regulator of cell cycle arrest, senescence, and cell death [185].
Focal adhesions are multiprotein assemblies that are essential for cell migration and facilitate the interaction between the extracellular matrix (ECM) and the cellular cytoskeleton. Focal adhesion formation and turnover is regulated by different extracellular factors, among them the vascular endothelial growth factor (VEGF). Laulimalide is a natural MTA that inhibits tubule formation and VEGF-induced cell migration of HUVECs [186]. The inhibition of these two processes occurred at substantially lower concentrations than that required to inhibit proliferation. At low concentrations, MTAs inhibit cell migration by reducing the number of peripheral MTs, affecting cell polarity formation, and disturbing the maintenance of the cell’s leading edge. Changes in MT dynamics reduce the total number of focal adhesions and disrupt its highly regulated turnover [187,188]. MTAs like paclitaxel can inhibit cell migration by impacting the repositioning of the microtubule-organizing center during migration [189].
HIF-1α (hypoxia-inducible factor 1-alpha) is a transcription factor that controls the transcription of VEGF [190,191], and its overexpression induces tumor angiogenesis and increases the survival of cancer cells [190,192,193]. MTAs can disrupt the transport of HIF-1α mRNA and releases it from polysomes, impeding its translation [140]. Moreover, HIF-1α is transported to the nucleus by the motor dynein, and taxane treatments that alter the MT cytoskeleton inhibit the transport of HIF-1α to the nucleus and impede its function [141]. MTAs like taxotere, epothilone B, discodermolide, vincristine, and colchicine have been shown to inhibit HIF-1α nuclear accumulation and activity by disrupting MT function [194]. Finally, 2-methoxyestradiol (2ME2) is a natural molecule derived from the hormone estradiol that also alters MT dynamics and has antitumor and antiangiogenic properties [195,196,197]. 2ME2 impairs HIF-1α function and therefore inhibits angiogenesis [194,198].
It has been shown that the vinca alkaloid vinorelbine has antiangiogenic effects in human endothelial cells [199]. MTAs like the taxanes downregulate the expression of Angiopoietin-1 (Ang-1), a protein required for vascular development and angiogenesis [200], and upregulate the expression and increased secretion of Thrombospondin 1 in the tumor microenvironment, a protein that is able to inhibit the formation of new blood vessels [201,202].
5.4.2. Vasculature-Disrupting Effects
The antiangiogenic activity of some drugs is often ineffective as tumors find alternative mechanisms for angiogenesis activation. Therefore, a useful alternative is to target existing tumor vasculature using vascular-disrupting agents (VDAs). Interestingly, the tumor vasculature is different from the blood vessels in normal tissues, suggesting that it would be possible to specifically target the tumor vasculature while leaving the normal blood vessels intact. There are three classes of VDAs: MTAs, flavonoids, and vascular targeted drugs based on endothelial cell receptors [180,203,204,205,206,207].
The most highly studied MTAs used as VDAs are combretastatin molecules. Combretastatins are small molecules found in the bark of the Combretum caffrum and Combretum leprosum trees. They inhibit angiogenesis, cell proliferation, and cell migration (reviewed in [208]). The natural form known as Combretastatin A-4 (CA4) has low solubility in water, necessitating the synthesis of several water-soluble options: a phosphorylated form (CA4P) and a version with increased cytotoxic activity against tumor cells (CA1P) [209,210]. Combretastatins bind to the colchicine binding site of β-tubulin, and induce cell cycle arrest in G2/M, and cell death through apoptosis and mitotic catastrophe [211,212]. Killing the endothelial cells in blood vessels in this manner leads to vascular leakage, blockage of blood flow, poor transport of nutrients to cancer cells, and subsequent necrosis of the tumor [203,213,214]. Fortunately, combretastatins are more efficient at disturbing endothelial tumor cells than normal cells, but the exact molecular mechanisms behind this effect are not well-known [215,216,217]. One possibility is that the blood vessels newly formed around a tumor are still immature, unstable, and not properly formed [218]. Cells on new formed blood vessels rely more on the MT cytoskeleton to provide rigidity and shape to the vessel, while already mature vasculature relies more on the actin cytoskeleton. Thus, small changes in MT dynamics could affect the cell cytoskeleton and polarization and migration of new endothelial cells, which would contribute specifically to tumor vessel destruction. Combretastatins can also reduce the blood flow in normal blood vessels, but this reduction is temporary and the morphology and functionality of normal vessels remains intact [219].
Vinblastine and vincristine also possess anti-vascular properties, but they act at high concentrations, close or even higher than the maximum tolerated dose [220,221]. Plinabulin binds to the colchicine site and causes similar MT depolymerization and similar anti-vascular effects as other MTAs [222]. The tubulin polymerization inhibitor CDK-516 also has anti-vascular properties. It induces apoptosis, inhibits tumor growth in mice, and is especially effective in combination with other treatments like doxorubicin [223,224,225].
5.5. MTAs, Metastasis, and Cell Migration
One of the most dangerous characteristics of cancer cells is their ability to spread outside of the main tumor mass, travel through the body, and establish new tumors in other organs. This process, known as metastasis, is dependent on cell migration (reviewed in [226]). Initially cells lose their cell-to-cell adhesion capabilities and acquire a migratory and invasive phenotype. This is known as the epithelial to mesenchymal transition. Following this transition, the new cells invade the stroma, enter in the blood and lymphatic vessels, and begin circulating throughout the body. Finally, the metastatic cells leave the vessels in a process known as extravasation, and colonize new tissues. All these steps during the metastatic process require a functional MT and actin cytoskeleton for proper cell movement, changes in cell morphology, and cell migration.
MTs are essential for cell polarization and cell migration [227]. For example, T cells have the ability to polarize their actin and MT cytoskeleton to be able to orient themselves toward the antigen-presenting cells and to migrate [228]. The MSA Zampanolide inhibits cell migration [229], and Epothilone B is able to inhibit cell migration of glioblastoma cells without overtly affecting MT dynamic criteria, such as the accumulation of EB1 at microtubule plus ends or the MT growth rate [230]. However, most cell migration studies have been done in traditional 2D tissue culture systems. New cell migration studies in organoids and 3D cell culture systems seem to indicate that MTAs more profoundly affect the cell migration of cells in 3D systems than in 2D cultures. For example, paclitaxel is 100-fold more effective at blocking migration in a 3D matrix than on a 2D matrix [231]. A good review to understand the role of MTs in 3D cell motility was published in 2017 [232]. Notably, the concentrations of MTAs required to affect cell migration are much lower than the concentrations required to trigger a cytotoxic effect. This is probably because low concentrations may be capable of significantly altering the behavior of the MT network close to the cell membrane without triggering apoptosis [233].
As in the case of the tumor vasculature, focal adhesion turnover is also essential during metastasis. Proper MT dynamics is required for the focal adhesions’ assembly-disassembly cycle necessary for cell migration. Disturbing this cycle with MTAs like nocodazole inhibits cell migration required for metastasis [188,234].
In cancer cells, invadopodia are small evaginations of the cell membrane that are required to contact the extracellular matrix (ECM) and start the invasion of adjacent tissues. These invadopodia grow to form long membrane protrusions that promote invasion. In a 3D model for cell invasion, where cells were placed in contact with native collagen type-I matrix overlaid with a thin basement membrane equivalent, paclitaxel was able to inhibit the formation of long membrane protrusions that are required for efficient invasion [235]. Interestingly, paclitaxel did not affect the initial formation of invadopodia but instead prevented the recruitment of MTs to complete the assembly of long and functionally effective membrane protrusions [236].
6. MTAs and Drug Resistance
MTAs have been used to treat different pathologies for decades. However, treatment with MTAs for long periods of time can trigger resistance to therapy.
The most common method of drug resistance is to expel the therapeutic compounds out of the cell through cellular transporters. The ATP-binding cassette (ABC) transporters is a family of transmembrane proteins that transport molecules across the cell membrane. At least 11 ABC transporters are involved in multidrug resistance (MDR) development to different treatments. Examples of these transporters are the P-glycoprotein 1 transporter (P-GP/ABCB1), the breast cancer resistance protein (BCRP/ABCG2), and the multidrug resistance-associated proteins (MRPs/ABCCs) [237]. The P-glycoprotein 1 transporter (ABCB1) is the most well-studied, the most prevalent drug resistance system, and the most important transporter from the clinical point of view [238,239,240]. This transporter can move taxanes out of cancer cells, rendering taxane therapy ineffective over time [241,242,243]. Another taxane transporter is the member of the same family ABCC3 [244]. One factor that contributes to the cell’s ability to expel these drugs is based on the fact that most MTA-tubulin binding events are transitory and reversible. There are some MSAs that are able to bind in a covalent manner to the tubulin, making these compounds very useful to avoid drug resistance [245,246].
Another method to generate drug resistance is the expression of different isotypes of β-tubulin to desensitize cells to MTAs [247]. Cancer cells favor the expression of βIII-tubulin, which interferes with the suppression of dynamic instability by taxanes [248,249,250,251]. In fact, inhibiting βIII-tubulin by siRNA renders cells more sensitive to taxanes and triggers cell death by apoptosis [252,253]. βIII-tubulin expression may influence MT dynamics [254] or, alternatively, may influence signaling pathways important for tumorigenesis [255]. For these reasons, new MTAs and combination therapies are being designed and tested to overcome βIII-tubulin-dependent drug resistance [256,257,258,259].
Finally, we would like to mention another shortcoming of most MTAs. Similar to the limitations described above in treating neurodegenerative diseases of the brain, many MTAs do not cross the BBB. This limits their ability to target brain cancers, such as gliomas. Accordingly, there is considerable interest in adapting Epothilone derivatives [260] and in developing novel MTAs [261,262] that do cross the BBB to target these devastating cancers.
7. Conclusions
Housekeeping proteins are essential proteins that are involved in the basic functioning of a cell. Tubulin is, therefore, a housekeeping protein, as MTs participate in a wide variety of essential cellular activities including but not limited to cell division. This presents both opportunities and challenges for the use of MTAs in the treatment of abnormal cell growth associated with cancer and other cell physiological pathologies. Because MTs are broadly essential for cellular functions, MTAs exhibit well-documented toxic effects against normal tissue. However, proper MT function is exquisitely dependent on MT dynamics and turnover, the key parameters of which are often tissue, cell, or activity specific. This has contributed to the surprising therapeutic efficacy of many MTAs. For example, even mild modulation of MT dynamics can, often fortuitously, direct the efficacy of these drugs toward specific tissues, unique signaling processes, or even exclusively to cancerous tissue. More research into how dynamic MTs impact broad cellular activities will enable greater effective utilization of MTAs against human disease.
Author Contributions
Writing—original draft preparation, L.W. and J.J.V.; writing—review and editing, L.W. and J.J.V. All authors have read and agreed to the published version of the manuscript.
Funding
L.W. and J.J.V. have been supported by N.I.H. grant GM069429. J.J.V. is also supported by University of Washington Royalty Research Fund.
Acknowledgments
We thank Mike Wagenbach for the design and composition of the tubulin figure.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goodson, H.V.; Jonasson, E.M. Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a022608. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, P.; Mühlhausen, S.; Dempewolf, S.; Hertzog, J.; Zietlow, A.; Carlomagno, T.; Kollmar, M. Six Subgroups and Extensive Recent Duplications Characterize the Evolution of the Eukaryotic Tubulin Protein Family. Genome Biol. Evol. 2014, 6, 2274–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaaban, S.; Brouhard, G.J. A Microtubule Bestiary: Structural Diversity in Tubulin Polymers. Mol. Biol. Cell 2017, 28, 2924–2931. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, T.J. Localization of an Exchangeable GTP Binding Site at the plus End of Microtubules. Science 1993, 261, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.; Kirschner, M. Dynamic Instability of Microtubule Growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Nehlig, A.; Molina, A.; Rodrigues-Ferreira, S.; Honoré, S.; Nahmias, C. Regulation of End-Binding Protein EB1 in the Control of Microtubule Dynamics. Cell. Mol. Life Sci. 2017, 74, 2381–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudimchuk, N.B.; McIntosh, J.R. Regulation of Microtubule Dynamics, Mechanics and Function through the Growing Tip. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wittmann, T. +TIPs: SxIPping along Microtubule Ends. Trends Cell Biol. 2012, 22, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Matthew, S.; Chen, Q.-Y.; Ratnayake, R.; Fermaintt, C.S.; Lucena-Agell, D.; Bonato, F.; Prota, A.E.; Lim, S.T.; Wang, X.; Díaz, J.F.; et al. Gatorbulin-1, a Distinct Cyclodepsipeptide Chemotype, Targets a Seventh Tubulin Pharmacological Site. Proc. Natl. Acad. Sci. USA 2021, 118, e2021847118. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Y.; Li, Y.; Yan, W.; Ye, H.; Niu, L.; Tang, M.; Wang, Z.; Yang, Z.; Pei, H.; et al. Cevipabulin-Tubulin Complex Reveals a Novel Agent Binding Site on α-Tubulin with Tubulin Degradation Effect. Sci. Adv. 2021, 7, eabg4168. [Google Scholar] [CrossRef]
- Johnson, I.S.; Armstrong, J.G.; Gorman, M.; Burnett, J.P. The vinca alkaloids: A new class of oncolytic agents. Cancer Res. 1963, 23, 1390–1427. [Google Scholar] [PubMed]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Binding of Dolastatin 10 to Tubulin at a Distinct Site for Peptide Antimitotic Agents near the Exchangeable Nucleotide and Vinca Alkaloid Sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a Powerful Cytostatic Peptide Derived from a Marine Animal. Inhibition of Tubulin Polymerization Mediated through the Vinca Alkaloid Binding Domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Hamel, E. Natural Products Which Interact with Tubulin in the Vinca Domain: Maytansine, Rhizoxin, Phomopsin A, Dolastatins 10 and 15 and Halichondrin B. Pharmacol. Ther. 1992, 55, 31–51. [Google Scholar] [CrossRef]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Uppuluri, S.; Knipling, L.; Sackett, D.L.; Wolff, J. Localization of the Colchicine-Binding Site of Tubulin. Proc. Natl. Acad. Sci. USA 1993, 90, 11598–11602. [Google Scholar] [CrossRef] [Green Version]
- Nogales, E.; Wolf, S.G.; Downing, K.H. Structure of the Alpha Beta Tubulin Dimer by Electron Crystallography. Nature 1998, 391, 199–203. [Google Scholar] [CrossRef]
- Mandelbaum-Shavit, F.; Wolpert-DeFilippes, M.K.; Johns, D.G. Binding of Maytansine to Rat Brain Tubulin. Biochem. Biophys. Res. Commun. 1976, 72, 47–54. [Google Scholar] [CrossRef]
- Takahashi, M.; Iwasaki, S.; Kobayashi, H.; Okuda, S.; Murai, T.; Sato, Y. Rhizoxin Binding to Tubulin at the Maytansine-Binding Site. Biochim. Biophys. Acta 1987, 926, 215–223. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A New Tubulin-Binding Site and Pharmacophore for Microtubule-Destabilizing Anticancer Drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [Green Version]
- Hood, K.A.; West, L.M.; Rouwé, B.; Northcote, P.T.; Berridge, M.V.; Wakefield, S.J.; Miller, J.H. Peloruside A, a Novel Antimitotic Agent with Paclitaxel-like Microtubule- Stabilizing Activity. Cancer Res. 2002, 62, 3356–3360. [Google Scholar]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and Isolaulimalide, New Paclitaxel-like Microtubule-Stabilizing Agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.-H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural Basis of Microtubule Stabilization by Laulimalide and Peloruside A. Angew. Chem. Int. Ed. 2014, 53, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Tsuchiya, K.; Harada, T.; Nishide, M.; Kurokawa, T.; Nakagawa, T.; Shimada, N.; Kobayashi, K. Pironetin, a Novel Plant Growth Regulator Produced by Streptomyces Sp. NK10958. I. Taxonomy, Production, Isolation and Preliminary Characterization. J. Antibiot. (Tokyo) 1994, 47, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Tsuchiya, K.; Kurokawa, T.; Nakagawa, T.; Shimada, N.; Iitaka, Y. Pironetin, a Novel Plant Growth Regulator Produced by Streptomyces Sp. NK10958. II. Structural Elucidation. J. Antibiot. (Tokyo) 1994, 47, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Tsuchiya, K.; Nishide, M.; Nishikiori, T.; Nakagawa, T.; Shimada, N. Pironetin, a Novel Plant Growth Regulator Produced by Streptomyces Sp. NK10958. III. Biosynthesis. J. Antibiot. (Tokyo) 1995, 48, 893–895. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Watanabe, H.; Usui, T.; Kondoh, M.; Osada, H.; Kitahara, T. Synthesis of Pironetin and Related Analogs: Studies on Structure-Activity Relationships as Tubulin Assembly Inhibitors. J. Antibiot. (Tokyo) 2000, 53, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Graham, W.; Roberts, J.B. Intravenous Colchicine in the Management of Gouty Arthritis. Ann. Rheum. Dis. 1953, 12, 16–19. [Google Scholar] [CrossRef] [Green Version]
- Massarotti, A.; Coluccia, A.; Silvestri, R.; Sorba, G.; Brancale, A. The Tubulin Colchicine Domain: A Molecular Modeling Perspective. ChemMedChem 2012, 7, 33–42. [Google Scholar] [CrossRef]
- Keeling, P.J.; Doolittle, W.F. Alpha-Tubulin from Early-Diverging Eukaryotic Lineages and the Evolution of the Tubulin Family. Mol. Biol. Evol. 1996, 13, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlind, T.D.; Li, J.; Visvesvara, G.S.; Vodkin, M.H.; McLaughlin, G.L.; Katiyar, S.K. Phylogenetic Analysis of Beta-Tubulin Sequences from Amitochondrial Protozoa. Mol. Phylogenet. Evol. 1996, 5, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F. A Hypothesis on the Origin and Evolution of Tubulin. Int. Rev. Cell Mol. Biol. 2013, 302, 41–185. [Google Scholar] [CrossRef] [PubMed]
- Fennell, B.; Naughton, J.; Barlow, J.; Brennan, G.; Fairweather, I.; Hoey, E.; McFerran, N.; Trudgett, A.; Bell, A. Microtubules as Antiparasitic Drug Targets. Expert Opin. Drug Discov. 2008, 3, 501–518. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The Therapeutic Journey of Benzimidazoles: A Review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Lacey, E. The Role of the Cytoskeletal Protein, Tubulin, in the Mode of Action and Mechanism of Drug Resistance to Benzimidazoles. Int. J. Parasitol. 1988, 18, 885–936. [Google Scholar] [CrossRef]
- Chai, J.-Y.; Jung, B.-K.; Hong, S.-J. Albendazole and Mebendazole as Anti-Parasitic and Anti-Cancer Agents: An Update. Korean J. Parasitol. 2021, 59, 189–225. [Google Scholar] [CrossRef] [PubMed]
- Dayan, A.D. Albendazole, Mebendazole and Praziquantel. Review of Non-Clinical Toxicity and Pharmacokinetics. Acta Trop. 2003, 86, 141–159. [Google Scholar] [CrossRef]
- Pawluk, S.A.; Roels, C.A.; Wilby, K.J.; Ensom, M.H.H. A Review of Pharmacokinetic Drug-Drug Interactions with the Anthelmintic Medications Albendazole and Mebendazole. Clin. Pharmacokinet. 2015, 54, 371–383. [Google Scholar] [CrossRef]
- Edwards, G.; Breckenridge, A.M. Clinical Pharmacokinetics of Anthelmintic Drugs. Clin. Pharmacokinet. 1988, 15, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Hawkins, N.J.; Fraaije, B.A. The Evolution of Fungicide Resistance. Adv. Appl. Microbiol. 2015, 90, 29–92. [Google Scholar] [CrossRef]
- Davidse, L.C. Benzimidazole Fungicides: Mechanism of Action and Biological Impact. Annu. Rev. Phytopathol. 1986, 24, 43–65. [Google Scholar] [CrossRef]
- Ware, G.W.; Whitacre, D.M. The Pesticide Book; Meister Media Worldwide: Willoughby, OH, USA, 2004; ISBN 1-892829-11-8. [Google Scholar]
- Anthony, R.; Hussey, P. Dinitroaniline Herbicide Resistance and the Microtubule Cytoskeleton. Trends Plant Sci. 1999, 4, 112–116. [Google Scholar] [CrossRef]
- Morrissette, N.S.; Mitra, A.; Sept, D.; Sibley, L.D. Dinitroanilines Bind Alpha-Tubulin to Disrupt Microtubules. Mol. Biol. Cell 2004, 15, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Sant’anna, V.; de Souza, W.; Vommaro, R.C. Anthelmintic Effect of Herbicidal Dinitroanilines on the Nematode Model Caenorhabditis Elegans. Exp. Parasitol. 2016, 167, 43–49. [Google Scholar] [CrossRef]
- Chan, M.M.; Fong, D. Inhibition of Leishmanias but Not Host Macrophages by the Antitubulin Herbicide Trifluralin. Science 1990, 249, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Hugdahl, J.D.; Morejohn, L.C. Rapid and Reversible High-Affinity Binding of the Dinitroaniline Herbicide Oryzalin to Tubulin from Zea Mays L. Plant Physiol. 1993, 102, 725–740. [Google Scholar] [CrossRef] [Green Version]
- Murthy, J.V.; Kim, H.H.; Hanesworth, V.R.; Hugdahl, J.D.; Morejohn, L.C. Competitive Inhibition of High-Affinity Oryzalin Binding to Plant Tubulin by the Phosphoric Amide Herbicide Amiprophos-Methyl. Plant Physiol. 1994, 105, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.M.; Triemer, R.E.; Fong, D. Effect of the Anti-Microtubule Drug Oryzalin on Growth and Differentiation of the Parasitic Protozoan Leishmania Mexicana. Differentiation 1991, 46, 15–21. [Google Scholar] [CrossRef]
- Chatterji, B.P.; Jindal, B.; Srivastava, S.; Panda, D. Microtubules as Antifungal and Antiparasitic Drug Targets. Expert Opin. Ther. Pat. 2011, 21, 167–186. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatore, C.; Brunden, K.R.; Huryn, D.M.; Trojanowski, J.Q.; Lee, V.M.-Y.; Smith, A.B. Microtubule Stabilizing Agents as Potential Treatment for Alzheimer’s Disease and Related Neurodegenerative Tauopathies. J. Med. Chem. 2012, 55, 8979–8996. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.; Daughenbaugh, R.; Trojanowski, J.Q. Microtubule Stabilizing Drugs for the Treatment of Alzheimer’s Disease. Neurobiol. Aging 1994, 15 (Suppl. S2), S87–S89. [Google Scholar] [CrossRef]
- Cartelli, D.; Casagrande, F.; Busceti, C.L.; Bucci, D.; Molinaro, G.; Traficante, A.; Passarella, D.; Giavini, E.; Pezzoli, G.; Battaglia, G.; et al. Microtubule Alterations Occur Early in Experimental Parkinsonism and the Microtubule Stabilizer Epothilone D Is Neuroprotective. Sci. Rep. 2013, 3, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunden, K.R.; Ballatore, C.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q. Brain-Penetrant Microtubule-Stabilizing Compounds as Potential Therapeutic Agents for Tauopathies. Biochem. Soc. Trans. 2012, 40, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballatore, C.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y.; Smith, A.B.; Huryn, D.M. Modulation of Protein-Protein Interactions as a Therapeutic Strategy for the Treatment of Neurodegenerative Tauopathies. Curr. Top. Med. Chem. 2011, 11, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, M.R.; Kovalevich, J.; Lee, V.M.-Y.; Trojanowski, J.Q.; Brunden, K.R. Therapeutic Strategies for the Treatment of Tauopathies: Hopes and Challenges. Alzheimer’s Dement. 2016, 12, 1051–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the Dynamic Instability of Tubulin Assembly by the Microtubule-Associated Protein Tau. Mol. Biol. Cell. 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-Based Classification of Tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau Protein in Neurodegenerative Diseases—A Review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.-S. Tau Protein Aggregation in Alzheimer’s Disease: An Attractive Target for the Development of Novel Therapeutic Agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Advances in Tau-Focused Drug Discovery for Alzheimer’s Disease and Related Tauopathies. Nat. Rev. Drug Discov. 2009, 8, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, S.; Florence, G.J.; Paterson, I.; Amos, L.A. Discodermolide Interferes with the Binding of Tau Protein to Microtubules. FEBS Lett. 2003, 539, 34–36. [Google Scholar] [CrossRef] [Green Version]
- Das, V.; Miller, J.H. Microtubule Stabilization by Peloruside A and Paclitaxel Rescues Degenerating Neurons from Okadaic Acid-Induced Tau Phosphorylation. Eur. J. Neurosci. 2012, 35, 1705–1717. [Google Scholar] [CrossRef]
- Das, V.; Miller, J.H. Non-Taxoid Site Microtubule-Stabilizing Drugs Work Independently of Tau Overexpression in Mouse N2a Neuroblastoma Cells. Brain Res. 2012, 1489, 121–132. [Google Scholar] [CrossRef]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-Binding Drugs Offset Tau Sequestration by Stabilizing Microtubules and Reversing Fast Axonal Transport Deficits in a Tauopathy Model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yao, Y.; Cornec, A.-S.; Oukoloff, K.; James, M.J.; Koivula, P.; Trojanowski, J.Q.; Smith, A.B.; Lee, V.M.-Y.; Ballatore, C.; et al. A Brain-Penetrant Triazolopyrimidine Enhances Microtubule-Stability, Reduces Axonal Dysfunction and Decreases Tau Pathology in a Mouse Tauopathy Model. Mol. Neurodegener. 2018, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Brandt, R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.-M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The Microtubule-Stabilizing Agent, Epothilone D, Reduces Axonal Dysfunction, Neurotoxicity, Cognitive Deficits, and Alzheimer-like Pathology in an Interventional Study with Aged Tau Transgenic Mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Fanara, P.; Andorfer, C.; Hoque, N.; Wong, P.Y.A.; Husted, K.H.; Cadelina, G.W.; Decarr, L.B.; Yang, L.; Liu, V.; et al. Hyperdynamic Microtubules, Cognitive Deficits, and Pathology Are Improved in Tau Transgenic Mice with Low Doses of the Microtubule-Stabilizing Agent BMS-241027. J. Neurosci. 2012, 32, 7137–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning Microtubule Stabilizing Drugs for Brain Disorders. Front. Cell. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef]
- Kugler, C.; Thielscher, C.; Tambe, B.A.; Schwarz, M.K.; Halle, A.; Bradke, F.; Petzold, G.C. Epothilones Improve Axonal Growth and Motor Outcomes after Stroke in the Adult Mammalian CNS. Cell Rep. Med. 2020, 1, 100159. [Google Scholar] [CrossRef]
- Duan, Y.-Y.; Chai, Y.; Zhang, N.-L.; Zhao, D.-M.; Yang, C. Microtubule Stabilization Promotes Microcirculation Reconstruction After Spinal Cord Injury. J. Mol. Neurosci. 2021, 71, 583–595. [Google Scholar] [CrossRef]
- Gornstein, E.; Schwarz, T.L. The Paradox of Paclitaxel Neurotoxicity: Mechanisms and Unanswered Questions. Neuropharmacology 2014, 76 (Pt A), 175–183. [Google Scholar] [CrossRef]
- Shemesh, O.A.; Spira, M.E. Paclitaxel Induces Axonal Microtubules Polar Reconfiguration and Impaired Organelle Transport: Implications for the Pathogenesis of Paclitaxel-Induced Polyneuropathy. Acta Neuropathol. 2010, 119, 235–248. [Google Scholar] [CrossRef]
- Klein, I.; Lehmann, H.C. Pathomechanisms of Paclitaxel-Induced Peripheral Neuropathy. Toxics 2021, 9, 229. [Google Scholar] [CrossRef]
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A Review of Adverse Toxicities and Novel Delivery Strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Yared, J.A.; Tkaczuk, K.H.R. Update on Taxane Development: New Analogs and New Formulations. Drug Des. Dev. Ther. 2012, 6, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Carlson, K.; Ocean, A.J. Peripheral Neuropathy with Microtubule-Targeting Agents: Occurrence and Management Approach. Clin. Breast Cancer 2011, 11, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, N.K. Ixabepilone: Overview of Effectiveness, Safety, and Tolerability in Metastatic Breast Cancer. Front. Oncol. 2021, 11, 617874. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lustberg, M.B.; Hu, S. Emerging Pharmacological and Non-Pharmacological Therapeutics for Prevention and Treatment of Chemotherapy-Induced Peripheral Neuropathy. Cancers 2021, 13, 766. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-Resistance Gene (P-Glycoprotein) Is Expressed by Endothelial Cells at Blood-Brain Barrier Sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.; Fichtner, I.; Lemm, M.; Lienau, P.; Hess-Stumpp, H.; Rotgeri, A.; Hofmann, B.; Klar, U. Sagopilone Crosses the Blood-Brain Barrier in Vivo to Inhibit Brain Tumor Growth and Metastases. Neuro-Oncol. 2009, 11, 158–166. [Google Scholar] [CrossRef] [Green Version]
- GBD 2017 Causes of Death Collaborators Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980-2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [CrossRef] [Green Version]
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of Chance Observations in Chemotherapy: Vinca rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef]
- Cutts, J.H.; Beer, C.T.; Noble, R.L. Biological Properties of Vincaleukoblastine, an Alkaloid in Vinca rosea Linn, with Reference to Its Antitumor Action. Cancer Res. 1960, 20, 1023–1031. [Google Scholar]
- Warwick, O.H.; Darte, J.M.; Brown, T.C.; Beer, C.T.; Cutts, J.H.; Noble, R.L. Some Biological Effects of Vincaleukoblastine, an Alkaloid in Vinca rosea Linn in Patients with Malignant Disease. Cancer Res. 1960, 20, 1032–1040. [Google Scholar]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Further Biological Activities of Vincaleukoblastine—An Alkaloid Isolated from Vinca rosea (L.). Biochem. Pharmacol. 1959, 1, 347–348. [Google Scholar] [CrossRef]
- Johnson, I.S.; Wright, H.F.; Svoboda, G.H.; Vlantis, J. Antitumor Principles Derived from Vinca rosea Linn. I. Vincaleukoblastine and Leurosine. Cancer Res. 1960, 20, 1016–1022. [Google Scholar] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer. 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.A. What Tubulin Drugs Tell Us about Microtubule Structure and Dynamics. Semin. Cell Dev. Biol. 2011, 22, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Aks, S.E.; Hutson, J.R.; Juurlink, D.N.; Nguyen, P.; Dubnov-Raz, G.; Pollak, U.; Koren, G.; Bentur, Y. Colchicine Poisoning: The Dark Side of an Ancient Drug. Clin. Toxicol. (Phila) 2010, 48, 407–414. [Google Scholar] [CrossRef]
- Vicente, J.J.; Wordeman, L. The Quantification and Regulation of Microtubule Dynamics in the Mitotic Spindle. Curr. Opin. Cell Biol. 2019, 60, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Bastians, H. Mitotic Drug Targets and the Development of Novel Anti-Mitotic Anticancer Drugs. Drug Resist. Update 2007, 10, 162–181. [Google Scholar] [CrossRef]
- Sudakin, V.; Yen, T.J. Targeting Mitosis for Anti-Cancer Therapy. BioDrugs 2007, 21, 225–233. [Google Scholar] [CrossRef]
- Kaestner, P.; Bastians, H. Mitotic Drug Targets. J. Cell. Biochem. 2010, 111, 258–265. [Google Scholar] [CrossRef]
- Keen, N.; Taylor, S. Aurora-Kinase Inhibitors as Anticancer Agents. Nat. Rev. Cancer 2004, 4, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K. Multifaceted Polo-like Kinases: Drug Targets and Antitargets for Cancer Therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.L.; Fojo, A.T. Inhibitors Targeting Mitosis: Tales of How Great Drugs against a Promising Target Were Brought down by a Flawed Rationale. Clin. Cancer Res. 2012, 18, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis Is Not a Key Target of Microtubule Agents in Patient Tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.J. The Proliferation Rate Paradox in Antimitotic Chemotherapy. Mol. Biol. Cell 2012, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-Targeting Agents Are Clinically Successful Due to Both Mitotic and Interphase Impairment of Microtubule Function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.M.; Cimini, D. Link between Aneuploidy and Chromosome Instability. Int. Rev. Cell Mol. Biol. 2015, 315, 299–317. [Google Scholar] [CrossRef]
- Giam, M.; Rancati, G. Aneuploidy and Chromosomal Instability in Cancer: A Jackpot to Chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansregret, L.; Swanton, C. The Role of Aneuploidy in Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potapova, T.; Gorbsky, G.J. The Consequences of Chromosome Segregation Errors in Mitosis and Meiosis. Biology 2017, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Bastians, H. Causes of Chromosomal Instability. Recent Results Cancer Res. 2015, 200, 95–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Chen, R.; Tian, Z.; Zhai, Y.; Janz, S.; Gu, C.; Yang, Y. Chromosomal Instability and Acquired Drug Resistance in Multiple Myeloma. Oncotarget 2017, 8, 78234–78244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClelland, S.E. Role of Chromosomal Instability in Cancer Progression. Endocr. Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targa, A.; Rancati, G. Cancer: A CINful Evolution. Curr. Opin. Cell Biol. 2018, 52, 136–144. [Google Scholar] [CrossRef]
- Tanaka, K.; Hirota, T. Chromosomal Instability: A Common Feature and a Therapeutic Target of Cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [CrossRef]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer Chromosomal Instability: Therapeutic and Diagnostic Challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and Clinical Implications of Chromosomal Instability in Cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, L.L.; Jeusset, L.M.-P.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The Number of Polyploid Giant Cancer Cells and Epithelial-Mesenchymal Transition-Related Proteins Are Associated with Invasion and Metastasis in Human Breast Cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of Cancer Stem-like Cells through the Formation of Polyploid Giant Cancer Cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Kohler, R.H.; Foijer, F.; Sorger, P.K.; Weissleder, R.; Mitchison, T.J. Analysis of Mitosis and Antimitotic Drug Responses in Tumors by in Vivo Microscopy and Single-Cell Pharmacodynamics. Cancer Res. 2011, 71, 4608–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. Cancer Cells Display Profound Intra- and Interline Variation Following Prolonged Exposure to Antimitotic Drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of Paclitaxel in Breast Cancer Is Due to Chromosome Missegregation on Multipolar Spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Svanvik, J.; Lindström, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of Cell Fusion, Hybridization and Polyploid Cell Formation in Cancer Metastasis. World J. Clin. Oncol. 2020, 11, 121–135. [Google Scholar] [CrossRef]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Maity, T.K.; Robey, R.; Burotto, M.; et al. Microtubule-Targeting Agents Augment the Toxicity of DNA-Damaging Agents by Disrupting Intracellular Trafficking of DNA Repair Proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.K.; Germond, C.; Chen, X.; Wang, Z. The Mode of Action of Taxol: Apoptosis at Low Concentration and Necrosis at High Concentration. Biochem. Biophys. Res. Commun. 1999, 263, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Kanthou, C.; Tozer, G.M. Microtubule Depolymerizing Vascular Disrupting Agents: Novel Therapeutic Agents for Oncology and Other Pathologies. Int. J. Exp. Pathol. 2009, 90, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; André, N.; Braguer, D. Targeting Microtubules to Inhibit Angiogenesis and Disrupt Tumour Vasculature: Implications for Cancer Treatment. Curr. Cancer Drug Targets 2007, 7, 566–581. [Google Scholar] [CrossRef]
- Hahn, W.C. Cancer: Surviving on the Edge. Cancer Cell 2004, 6, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Ogden, A.; Rida, P.C.G.; Reid, M.D.; Kucuk, O.; Aneja, R. Die-Hard Survivors: Heterogeneity in Apoptotic Thresholds May Underlie Chemoresistance. Expert Rev. Anticancer Ther. 2015, 15, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, D.M.; Moseley, G.W.; Glover, D.; Pouton, C.W.; Jans, D.A. A Microtubule-Facilitated Nuclear Import Pathway for Cancer Regulatory Proteins. Traffic 2007, 8, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Kornilova, E.S. Receptor-Mediated Endocytosis and Cytoskeleton. Biochemistry (Mosc) 2014, 79, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. P53 Is Associated with Cellular Microtubules and Is Transported to the Nucleus by Dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Giannakakou, P.; Nakano, M.; Nicolaou, K.C.; O’Brate, A.; Yu, J.; Blagosklonny, M.V.; Greber, U.F.; Fojo, T. Enhanced Microtubule-Dependent Trafficking and P53 Nuclear Accumulation by Suppression of Microtubule Dynamics. Proc. Natl. Acad. Sci. USA 2002, 99, 10855–10860. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 Status Determines the Role of Autophagy in Pancreatic Tumour Development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.H.C.; Thomas, R.J.; Loveland, K.L.; Schilders, S.; Gu, M.; Martin, T.J.; Gillespie, M.T.; Jans, D.A. Nuclear Transport of Parathyroid Hormone (PTH)-Related Protein Is Dependent on Microtubules. Mol. Endocrinol. 2002, 16, 390–401. [Google Scholar] [CrossRef]
- Li, H.; Duan, Z.-W.; Xie, P.; Liu, Y.-R.; Wang, W.-C.; Dou, S.-X.; Wang, P.-Y. Effects of Paclitaxel on EGFR Endocytic Trafficking Revealed Using Quantum Dot Tracking in Single Cells. PLoS ONE 2012, 7, e45465. [Google Scholar] [CrossRef]
- Carbonaro, M.; O’Brate, A.; Giannakakou, P. Microtubule Disruption Targets HIF-1alpha MRNA to Cytoplasmic P-Bodies for Translational Repression. J. Cell Biol. 2011, 192, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Carbonaro, M.; Escuin, D.; O’Brate, A.; Thadani-Mulero, M.; Giannakakou, P. Microtubules Regulate Hypoxia-Inducible Factor-1α Protein Trafficking and Activity: Implications for Taxane Therapy. J. Biol. Chem. 2012, 287, 11859–11869. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-P.H.; Horwitz, S.B. Taxol®: The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnst, J. When Taxol Met Tubulin. J. Biol. Chem. 2020, 295, 13994–13995. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/Paclitaxel Kills Cancer Cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Engels, F.K.; Sparreboom, A.; Mathot, R.a.A.; Verweij, J. Potential for Improvement of Docetaxel-Based Chemotherapy: A Pharmacological Review. Br. J. Cancer 2005, 93, 173–177. [Google Scholar] [CrossRef]
- Schnaeker, E.-M.; Ossig, R.; Ludwig, T.; Dreier, R.; Oberleithner, H.; Wilhelmi, M.; Schneider, S.W. Microtubule-Dependent Matrix Metalloproteinase-2/Matrix Metalloproteinase-9 Exocytosis: Prerequisite in Human Melanoma Cell Invasion. Cancer Res. 2004, 64, 8924–8931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darshan, M.S.; Loftus, M.S.; Thadani-Mulero, M.; Levy, B.P.; Escuin, D.; Zhou, X.K.; Gjyrezi, A.; Chanel-Vos, C.; Shen, R.; Tagawa, S.T.; et al. Taxane-Induced Blockade to Nuclear Accumulation of the Androgen Receptor Predicts Clinical Responses in Metastatic Prostate Cancer. Cancer Res. 2011, 71, 6019–6029. [Google Scholar] [CrossRef] [Green Version]
- Thadani-Mulero, M.; Nanus, D.M.; Giannakakou, P. Androgen Receptor on the Move: Boarding the Microtubule Expressway to the Nucleus. Cancer Res. 2012, 72, 4611–4615. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Chen, S.; Wang, Y.; Watahiki, A.; Bohrer, L.; Sun, Z.; Wang, Y.; Huang, H. Inhibition of the Androgen Receptor as a Novel Mechanism of Taxol Chemotherapy in Prostate Cancer. Cancer Res. 2009, 69, 8386–8394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.-L.; Horbinski, C.M.; Garzotto, M.; Qian, D.Z.; Beer, T.M.; Kyprianou, N. Tubulin-Targeting Chemotherapy Impairs Androgen Receptor Activity in Prostate Cancer. Cancer Res. 2010, 70, 7992–8002. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical Mechanisms of Vertebrate Hedgehog Signaling. Development 2019, 146, dev166892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T.C.L. A Highlight on Sonic Hedgehog Pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Jacob, L.; Lum, L. Hedgehog Signaling Pathway. Sci. STKE 2007, 2007, cm6. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Xin, M.; Ji, X.; De La Cruz, L.K.; Thareja, S.; Wang, B. Strategies to Target the Hedgehog Signaling Pathway for Cancer Therapy. Med. Res. Rev. 2018, 38, 870–913. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gutzmer, R.; Kieran, M.W.; Solomon, J.A. Hedgehog Signaling Inhibitors in Solid and Hematological Cancers. Cancer Treat. Rev. 2019, 76, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglio, D.; Infante, P.; Di Marcotullio, L.; Botta, B.; Mori, M. Hedgehog Signaling Pathway Inhibitors: An Updated Patent Review (2015-Present). Expert Opin. Ther. Pat. 2020, 30, 235–250. [Google Scholar] [CrossRef]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalan, J.; Wordeman, L.; Scott, J.D. Kinase-Anchoring Proteins in Ciliary Signal Transduction. Biochem. J. 2021, 478, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Khatra, H.; Khan, P.P.; Pattanayak, S.; Bhadra, J.; Rather, B.; Chakrabarti, S.; Saha, T.; Sinha, S. Hedgehog Antagonist Pyrimidine-Indole Hybrid Molecule Inhibits Ciliogenesis through Microtubule Destabilisation. Chembiochem 2018, 19, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.R.; Bai, R.-Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the Antihelmintic Mebendazole as a Hedgehog Inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, L.; Bost, F.; Mazure, N.M. Primary Cilium in Cancer Hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, D.L.; Gray, H.; Payne, C.M.; Gillies, R.J. Docetaxel Induces Cell Death through Mitotic Catastrophe in Human Breast Cancer Cells. Mol. Cancer Ther. 2005, 4, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mediavilla-Varela, M.; Pacheco, F.J.; Almaguel, F.; Perez, J.; Sahakian, E.; Daniels, T.R.; Leoh, L.S.; Padilla, A.; Wall, N.R.; Lilly, M.B.; et al. Docetaxel-Induced Prostate Cancer Cell Death Involves Concomitant Activation of Caspase and Lysosomal Pathways and Is Attenuated by LEDGF/P75. Mol. Cancer 2009, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, R.; Wang, L.; Liu, P.; Zhao, Y.; Zhang, C.; Bai, B.; Liu, X.; Shi, C.; Wei, S.; Zhang, H. Cabazitaxel-Induced Autophagy via the PI3K/Akt/MTOR Pathway Contributes to A549 Cell Death. Mol. Med. Rep. 2016, 14, 3013–3020. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.J.; Hwang, S.; Cho, K.Y.; Kim, D.K.; Chay, K.-O.; Kim, J.-K. Taxol Induces Oxidative Neuronal Cell Death by Enhancing the Activity of NADPH Oxidase in Mouse Cortical Cultures. Neurosci. Lett. 2008, 443, 17–22. [Google Scholar] [CrossRef]
- Lan, Y.-Y.; Chen, Y.-H.; Liu, C.; Tung, K.-L.; Wu, Y.-T.; Lin, S.-C.; Wu, C.-H.; Chang, H.-Y.; Chen, Y.-C.; Huang, B.-M. Role of JNK Activation in Paclitaxel-Induced Apoptosis in Human Head and Neck Squamous Cell Carcinoma. Oncol. Lett. 2021, 22, 705. [Google Scholar] [CrossRef]
- Milross, C.G.; Mason, K.A.; Hunter, N.R.; Chung, W.K.; Peters, L.J.; Milas, L. Relationship of Mitotic Arrest and Apoptosis to Antitumor Effect of Paclitaxel. J. Natl. Cancer Inst. 1996, 88, 1308–1314. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowski, S. Apoptosis: Its Origin, History, Maintenance and the Medical Implications for Cancer and Aging. Phys. Biol. 2016, 13, 031001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelínek, M.; Balušíková, K.; Schmiedlová, M.; Němcová-Fürstová, V.; Šrámek, J.; Stančíková, J.; Zanardi, I.; Ojima, I.; Kovář, J. The Role of Individual Caspases in Cell Death Induction by Taxanes in Breast Cancer Cells. Cancer Cell Int. 2015, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlini, C.; Cicchillitti, L.; Raspaglio, G.; Bartollino, S.; Cimitan, S.; Bertucci, C.; Mozzetti, S.; Gallo, D.; Persico, M.; Fattorusso, C.; et al. Paclitaxel Directly Binds to Bcl-2 and Functionally Mimics Activity of Nur77. Cancer Res. 2009, 69, 6906–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi, S.; Barar, J.; Hejazi, M.S.; Samadi, N. Roles of the Bcl-2/Bax Ratio, Caspase-8 and 9 in Resistance of Breast Cancer Cells to Paclitaxel. Asian Pac. J. Cancer Prev. 2014, 15, 8617–8622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Wang, C.; Xing, Y.; Chen, S.; Meng, T.; You, H.; Ojima, I.; Dong, Y. SB-T-121205, a next-Generation Taxane, Enhances Apoptosis and Inhibits Migration/Invasion in MCF-7/PTX Cells. Int. J Oncol. 2017, 50, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Fauzee, N.J.S.; Wang, Y.-L.; Dong, Z.; Li, Q.-G.; Wang, T.; Mandarry, M.T.; Xu, L.; Pan, J. Novel Hydrophilic Docetaxel (CQMU-0519) Analogue Inhibits Proliferation and Induces Apoptosis in Human A549 Lung, SKVO3 Ovarian and MCF7 Breast Carcinoma Cell Lines. Cell Prolif. 2012, 45, 352–364. [Google Scholar] [CrossRef]
- Janssen, K.; Pohlmann, S.; Jänicke, R.U.; Schulze-Osthoff, K.; Fischer, U. Apaf-1 and Caspase-9 Deficiency Prevents Apoptosis in a Bax-Controlled Pathway and Promotes Clonogenic Survival during Paclitaxel Treatment. Blood 2007, 110, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.A.; Lonergan, S.J.; Denekamp, J.; Chaplin, D.J. Vinca Alkaloids: Anti-Vascular Effects in a Murine Tumour. Eur. J. Cancer 1993, 29, 1320–1324. [Google Scholar] [CrossRef]
- Schwartz, E.L. Antivascular Actions of Microtubule-Binding Drugs. Clin. Cancer Res. 2009, 15, 2594–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, G.; Di Paolo, A.; Danesi, R. The Pharmacological Bases of the Antiangiogenic Activity of Paclitaxel. Angiogenesis 2013, 16, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, E.; Honore, S.; Pourroy, B.; Jordan, M.A.; Lehmann, M.; Briand, C.; Braguer, D. Antiangiogenic Concentrations of Paclitaxel Induce an Increase in Microtubule Dynamics in Endothelial Cells but Not in Cancer Cells. Cancer Res. 2005, 65, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Bijman, M.N.A.; van Nieuw Amerongen, G.P.; Laurens, N.; van Hinsbergh, V.W.M.; Boven, E. Microtubule-Targeting Agents Inhibit Angiogenesis at Subtoxic Concentrations, a Process Associated with Inhibition of Rac1 and Cdc42 Activity and Changes in the Endothelial Cytoskeleton. Mol. Cancer Ther. 2006, 5, 2348–2357. [Google Scholar] [CrossRef] [Green Version]
- Bonezzi, K.; Belotti, D.; North, B.J.; Ghilardi, C.; Borsotti, P.; Resovi, A.; Ubezio, P.; Riva, A.; Giavazzi, R.; Verdin, E.; et al. Inhibition of SIRT2 Potentiates the Anti-Motility Activity of Taxanes: Implications for Antineoplastic Combination Therapies. Neoplasia 2012, 14, 846–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Fan, L.Y.-N.; Lam, E.W.-F. The FOXO3-FOXM1 Axis: A Key Cancer Drug Target and a Modulator of Cancer Drug Resistance. Semin. Cancer Biol. 2018, 50, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Murtagh, J.; Schwartz, E.L. The Microtubule Binding Drug Laulimalide Inhibits Vascular Endothelial Growth Factor-Induced Human Endothelial Cell Migration and Is Synergistic When Combined with Docetaxel (Taxotere). Mol. Pharmacol. 2006, 69, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, K.; Smiyun, G.; Wilson, L.; Jordan, M.A. Mechanisms of Inhibition of Endothelial Cell Migration by Taxanes. Cytoskeleton 2014, 71, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-Induced Focal Adhesion Disassembly Is Mediated by Dynamin and Focal Adhesion Kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef]
- Hotchkiss, K.A.; Ashton, A.W.; Mahmood, R.; Russell, R.G.; Sparano, J.A.; Schwartz, E.L. Inhibition of Endothelial Cell Function in Vitro and Angiogenesis in Vivo by Docetaxel (Taxotere): Association with Impaired Repositioning of the Microtubule Organizing Center. Mol. Cancer Ther. 2002, 1, 1191–1200. [Google Scholar]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Levy, A.P.; Levy, N.S.; Wegner, S.; Goldberg, M.A. Transcriptional Regulation of the Rat Vascular Endothelial Growth Factor Gene by Hypoxia. J. Biol. Chem. 1995, 270, 13333–13340. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Escuin, D.; Kline, E.R.; Giannakakou, P. Both Microtubule-Stabilizing and Microtubule-Destabilizing Drugs Inhibit Hypoxia-Inducible Factor-1alpha Accumulation and Activity by Disrupting Microtubule Function. Cancer Res. 2005, 65, 9021–9028. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, N.J.; Sarkar, M.A.; Venitz, J.; Figg, W.D. 2-Methoxyestradiol, a Promising Anticancer Agent. Pharmacotherapy 2003, 23, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brueggemeier, R.W.; Bhat, A.S.; Lovely, C.J.; Coughenour, H.D.; Joomprabutra, S.; Weitzel, D.H.; Vandre, D.D.; Yusuf, F.; Burak, W.E. 2-Methoxymethylestradiol: A New 2-Methoxy Estrogen Analog That Exhibits Antiproliferative Activity and Alters Tubulin Dynamics. J. Steroid Biochem. Mol. Biol. 2001, 78, 145–156. [Google Scholar] [CrossRef]
- Mercier, A.E.; Prudent, R.; Pepper, M.S.; De Koning, L.; Nolte, E.; Peronne, L.; Nel, M.; Lafanechère, L.; Joubert, A.M. Characterization of Signalling Pathways That Link Apoptosis and Autophagy to Cell Death Induced by Estrone Analogues Which Reversibly Depolymerize Microtubules. Molecules 2021, 26, 706. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 Inhibits Tumor Growth and Angiogenesis by Disrupting Microtubules and Dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Biziota, E.; Briasoulis, E.; Mavroeidis, L.; Marselos, M.; Harris, A.L.; Pappas, P. Cellular and Molecular Effects of Metronomic Vinorelbine and 4-O-Deacetylvinorelbine on Human Umbilical Vein Endothelial Cells. Anti-Cancer Drugs 2016, 27, 216–224. [Google Scholar] [CrossRef]
- Hata, K.; Osaki, M.; Dhar, D.K.; Nakayama, K.; Fujiwaki, R.; Ito, H.; Nagasue, N.; Miyazaki, K. Evaluation of the Antiangiogenic Effect of Taxol in a Human Epithelial Ovarian Carcinoma Cell Line. Cancer Chemother. Pharmacol. 2004, 53, 68–74. [Google Scholar] [CrossRef]
- Zhang, M.; Tao, W.; Pan, S.; Sun, X.; Jiang, H. Low-Dose Metronomic Chemotherapy of Paclitaxel Synergizes with Cetuximab to Suppress Human Colon Cancer Xenografts. Anti-Cancer Drugs 2009, 20, 355–363. [Google Scholar] [CrossRef]
- Jiang, H.; Tao, W.; Zhang, M.; Pan, S.; Kanwar, J.R.; Sun, X. Low-Dose Metronomic Paclitaxel Chemotherapy Suppresses Breast Tumors and Metastases in Mice. Cancer Investig. 2010, 28, 74–84. [Google Scholar] [CrossRef]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting Tumour Blood Vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef]
- Hinnen, P.; Eskens, F.a.L.M. Vascular Disrupting Agents in Clinical Development. Br. J. Cancer 2007, 96, 1159–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolarczyk, R.; Czapla, J.; Jarosz-Biej, M.; Czerwinski, K.; Cichoń, T. Vascular Disrupting Agents in Cancer Therapy. Eur. J. Pharmacol. 2021, 891, 173692. [Google Scholar] [CrossRef] [PubMed]
- Lippert, J.W. Vascular Disrupting Agents. Bioorg. Med. Chem. 2007, 15, 605–615. [Google Scholar] [CrossRef]
- Kanthou, C.; Tozer, G.M. Tumour Targeting by Microtubule-Depolymerizing Vascular Disrupting Agents. Expert Opin. Ther. Targets 2007, 11, 1443–1457. [Google Scholar] [CrossRef]
- Sherbet, G.V. Suppression of Angiogenesis and Tumour Progression by Combretastatin and Derivatives. Cancer Lett. 2017, 403, 289–295. [Google Scholar] [CrossRef]
- Pettit, G.R.; Temple, C.; Narayanan, V.L.; Varma, R.; Simpson, M.J.; Boyd, M.R.; Rener, G.A.; Bansal, N. Antineoplastic Agents 322. Synthesis of Combretastatin A-4 Prodrugs. Anti-Cancer Drug Des. 1995, 10, 299–309. [Google Scholar]
- Mao, J.; Wang, D.; Wang, Z.; Tian, W.; Li, X.; Duan, J.; Wang, Y.; Yang, H.; You, L.; Cheng, Y.; et al. Combretastatin A-1 Phosphate, a Microtubule Inhibitor, Acts on Both Hepatocellular Carcinoma Cells and Tumor-Associated Macrophages by Inhibiting the Wnt/β-Catenin Pathway. Cancer Lett. 2016, 380, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than Just Vascular Targeting Agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Porcù, E.; Salvador, A.; Primac, I.; Mitola, S.; Ronca, R.; Ravelli, C.; Bortolozzi, R.; Vedaldi, D.; Romagnoli, R.; Basso, G.; et al. Vascular Disrupting Activity of Combretastatin Analogues. Vasc. Pharmacol. 2016, 83, 78–89. [Google Scholar] [CrossRef]
- Weber, A.; Iturri, J.; Benitez, R.; Zemljic-Jokhadar, S.; Toca-Herrera, J.L. Microtubule Disruption Changes Endothelial Cell Mechanics and Adhesion. Sci. Rep. 2019, 9, 14903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemann, D.W.; Chaplin, D.J.; Horsman, M.R. Vascular-Targeting Therapies for Treatment of Malignant Disease. Cancer 2004, 100, 2491–2499. [Google Scholar] [CrossRef]
- Lunt, S.J.; Akerman, S.; Hill, S.A.; Fisher, M.; Wright, V.J.; Reyes-Aldasoro, C.C.; Tozer, G.M.; Kanthou, C. Vascular Effects Dominate Solid Tumor Response to Treatment with Combretastatin A-4-Phosphate. Int. J. Cancer 2011, 129, 1979–1989. [Google Scholar] [CrossRef] [Green Version]
- Tozer, G.M.; Prise, V.E.; Wilson, J.; Locke, R.J.; Vojnovic, B.; Stratford, M.R.; Dennis, M.F.; Chaplin, D.J. Combretastatin A-4 Phosphate as a Tumor Vascular-Targeting Agent: Early Effects in Tumors and Normal Tissues. Cancer Res. 1999, 59, 1626–1634. [Google Scholar] [PubMed]
- Siemann, D.W. The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Tozer, G.M.; Akerman, S.; Cross, N.A.; Barber, P.R.; Björndahl, M.A.; Greco, O.; Harris, S.; Hill, S.A.; Honess, D.J.; Ireson, C.R.; et al. Blood Vessel Maturation and Response to Vascular-Disrupting Therapy in Single Vascular Endothelial Growth Factor-A Isoform-Producing Tumors. Cancer Res. 2008, 68, 2301–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Saito, S. Microvascular Mechanisms by Which the Combretastatin A-4 Derivative AC7700 (AVE8062) Induces Tumour Blood Flow Stasis. Br. J. Cancer 2003, 89, 1334–1344. [Google Scholar] [CrossRef]
- Clémenson, C.; Chargari, C.; Deutsch, E. Combination of Vascular Disrupting Agents and Ionizing Radiation. Crit. Rev. Oncol./Hematol. 2013, 86, 143–160. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, L.B.; Shen, Y.Y.; Nielsen, T.; Stødkilde-Jørgensen, H.; Lloyd, G.K.; Siemann, D.W.; Horsman, M.R. Vascular Effects of Plinabulin (NPI-2358) and the Influence on Tumour Response When given Alone or Combined with Radiation. Int. J. Radiat. Biol. 2011, 87, 1126–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, S.J.; Choi, H.; Kim, Y.H.; Lim, I.T.; Yang, H.; Lee, C.S.; Kang, H.R.; Ahn, S.K.; Moon, S.K.; et al. Identification of CKD-516: A Potent Tubulin Polymerization Inhibitor with Marked Antitumor Activity against Murine and Human Solid Tumors. J. Med. Chem. 2010, 53, 6337–6354. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.J.; Lee, M.; Kim, S.J.; Kim, Y.K.; Won, J.Y.; Chung, J.W. Chemoembolization with Vascular Disrupting Agent CKD-516 Dissolved in Ethiodized Oil in Combination with Doxorubicin: A VX2 Tumor Model Study. J. Vasc. Interv. Radiol. 2018, 29, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.H.; Lee, S.J.; Lee, H.Y.; Dung, L.T.K.; Cho, W.J.; Cha, H.; Park, J.W.; Min, Y.J. CKD-516 Displays Vascular Disrupting Properties and Enhances Anti-Tumor Activity in Combination with Chemotherapy in a Murine Tumor Model. Investig. New Drugs 2014, 32, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Aguet, M.; Stamenkovic, I. Cancer Metastasis: A Reappraisal of Its Underlying Mechanisms and Their Relevance to Treatment. Annu. Rev. Pathol. 2018, 13, 117–140. [Google Scholar] [CrossRef] [PubMed]
- Garcin, C.; Straube, A. Microtubules in Cell Migration. Essays Biochem. 2019, 63, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Gomez, T.S.; Billadeau, D.D. T Cell Activation and the Cytoskeleton: You Can’t Have One without the Other. Adv. Immunol. 2008, 97, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Field, J.J.; Northcote, P.T.; Paterson, I.; Altmann, K.-H.; Díaz, J.F.; Miller, J.H. Zampanolide, a Microtubule-Stabilizing Agent, Is Active in Resistant Cancer Cells and Inhibits Cell Migration. Int. J. Mol. Sci. 2017, 18, 971. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Honoré, S.; Mohan, R.; Berges, R.; Akhmanova, A.; Braguer, D. Epothilone B Inhibits Migration of Glioblastoma Cells by Inducing Microtubule Catastrophes and Affecting EB1 Accumulation at Microtubule plus Ends. Biochem. Pharmacol. 2012, 84, 432–443. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Giri, A.; Karl, M.; Aifuwa, I.; Trenton, N.J.; Phillip, J.M.; Khatau, S.; Wirtz, D. EB1 and Cytoplasmic Dynein Mediate Protrusion Dynamics for Efficient 3-Dimensional Cell Migration. FASEB J. 2018, 32, 1207–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet, B.P.; Akhmanova, A. Microtubules in 3D Cell Motility. J. Cell Sci. 2017, 130, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ganguly, A.; Cabral, F. Inhibition of Cell Migration and Cell Division Correlates with Distinct Effects of Microtubule Inhibiting Drugs. J. Biol. Chem. 2010, 285, 32242–32250. [Google Scholar] [CrossRef] [Green Version]
- Broussard, J.A.; Webb, D.J.; Kaverina, I. Asymmetric Focal Adhesion Disassembly in Motile Cells. Curr. Opin. Cell Biol. 2008, 20, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Takahashi, K. WAVE2- and Microtubule-Dependent Formation of Long Protrusions and Invasion of Cancer Cells Cultured on Three-Dimensional Extracellular Matrices. Cancer Sci. 2008, 99, 2252–2259. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, Microtubules, and Vimentin Intermediate Filaments Cooperate for Elongation of Invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X. ABC Family Transporters. Adv. Exp. Med. Biol. 2019, 1141, 13–100. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Menefee, M. Mechanisms of Multidrug Resistance: The Potential Role of Microtubule-Stabilizing Agents. Ann. Oncol. 2007, 18 (Suppl. S5), v3–v8. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M. Microtubules and Resistance to Tubulin-Binding Agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Dostál, V.; Libusová, L. Microtubule Drugs: Action, Selectivity, and Resistance across the Kingdoms of Life. Protoplasma 2014, 251, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Aldonza, M.B.D.; Hong, J.-Y.; Alinsug, M.V.; Song, J.; Lee, S.K. Multiplicity of Acquired Cross-Resistance in Paclitaxel-Resistant Cancer Cells Is Associated with Feedback Control of TUBB3 via FOXO3a-Mediated ABCB1 Regulation. Oncotarget 2016, 7, 34395–34419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S.N.; Westergaard, D.; Thomsen, M.B.H.; Vistesen, M.; Do, K.N.; Fogh, L.; Belling, K.C.; Wang, J.; Yang, H.; Gupta, R.; et al. Acquisition of Docetaxel Resistance in Breast Cancer Cells Reveals Upregulation of ABCB1 Expression as a Key Mediator of Resistance Accompanied by Discrete Upregulation of Other Specific Genes and Pathways. Tumour Biol. 2015, 36, 4327–4338. [Google Scholar] [CrossRef]
- Jelínek, M.; Balušíková, K.; Daniel, P.; Němcová-Fürstová, V.; Kirubakaran, P.; Jaček, M.; Wei, L.; Wang, X.; Vondrášek, J.; Ojima, I.; et al. Substituents at the C3′ and C3′N Positions Are Critical for Taxanes to Overcome Acquired Resistance of Cancer Cells to Paclitaxel. Toxicol. Appl. Pharmacol. 2018, 347, 79–91. [Google Scholar] [CrossRef]
- Němcová-Fürstová, V.; Kopperová, D.; Balušíková, K.; Ehrlichová, M.; Brynychová, V.; Václavíková, R.; Daniel, P.; Souček, P.; Kovář, J. Characterization of Acquired Paclitaxel Resistance of Breast Cancer Cells and Involvement of ABC Transporters. Toxicol. Appl. Pharmacol. 2016, 310, 215–228. [Google Scholar] [CrossRef]
- Field, J.J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodríguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S.J.; et al. Zampanolide, a Potent New Microtubule-Stabilizing Agent, Covalently Reacts with the Taxane Luminal Site in Tubulin α,β-Heterodimers and Microtubules. Chem. Biol. 2012, 19, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Li, J.; Bennett, M.J.; Rohena, C.C.; Peng, J.; Schriemer, D.C.; Mooberry, S.L. Taccalonolide Binding to Tubulin Imparts Microtubule Stability and Potent in Vivo Activity. Cancer Res. 2013, 73, 6780–6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.L.; Teo, W.S.; McCarroll, J.A.; Kavallaris, M. An Emerging Role for Tubulin Isotypes in Modulating Cancer Biology and Chemotherapy Resistance. Int. J. Mol. Sci. 2017, 18, 1434. [Google Scholar] [CrossRef]
- Kamath, K.; Wilson, L.; Cabral, F.; Jordan, M.A. BetaIII-Tubulin Induces Paclitaxel Resistance in Association with Reduced Effects on Microtubule Dynamic Instability. J. Biol. Chem. 2005, 280, 12902–12907. [Google Scholar] [CrossRef] [Green Version]
- Person, F.; Wilczak, W.; Hube-Magg, C.; Burdelski, C.; Möller-Koop, C.; Simon, R.; Noriega, M.; Sauter, G.; Steurer, S.; Burdak-Rothkamm, S.; et al. Prevalence of ΒIII-Tubulin (TUBB3) Expression in Human Normal Tissues and Cancers. Tumour Biol. 2017, 39, 1010428317712166. [Google Scholar] [CrossRef] [Green Version]
- Sève, P.; Mackey, J.; Isaac, S.; Trédan, O.; Souquet, P.-J.; Pérol, M.; Lai, R.; Voloch, A.; Dumontet, C. Class III Beta-Tubulin Expression in Tumor Cells Predicts Response and Outcome in Patients with Non-Small Cell Lung Cancer Receiving Paclitaxel. Mol. Cancer Ther. 2005, 4, 2001–2007. [Google Scholar] [CrossRef] [Green Version]
- Kanakkanthara, A.; Miller, J.H. ΒIII-Tubulin Overexpression in Cancer: Causes, Consequences, and Potential Therapies. Biochim Biophys. Acta Rev. Cancer 2021, 1876, 188607. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Thorne, R.F.; de Bock, C.E.; Zhang, X.D.; Hersey, P. Melanoma Cell Sensitivity to Docetaxel-Induced Apoptosis Is Determined by Class III Beta-Tubulin Levels. FEBS Lett. 2008, 582, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.P.; McCarroll, J.A.; Po’uha, S.T.; Kamath, K.; Jordan, M.A.; Kavallaris, M. Microtubule Dynamics, Mitotic Arrest, and Apoptosis: Drug-Induced Differential Effects of BetaIII-Tubulin. Mol. Cancer Ther. 2010, 9, 1339–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchet, B.P.; Puisieux, A.; Galmarini, C.M. ΒIII-Tubulin Is Required for Interphase Microtubule Dynamics in Untransformed Human Mammary Epithelial Cells. Eur. J. Cell Biol. 2011, 90, 872–878. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Gan, P.P.; Erlich, R.B.; Liu, M.; Dwarte, T.; Sagnella, S.S.; Akerfeldt, M.C.; Yang, L.; Parker, A.L.; Chang, M.H.; et al. TUBB3/ΒIII-Tubulin Acts through the PTEN/AKT Signaling Axis to Promote Tumorigenesis and Anoikis Resistance in Non-Small Cell Lung Cancer. Cancer Res. 2015, 75, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Matesanz, R.; Trigili, C.; Rodríguez-Salarichs, J.; Zanardi, I.; Pera, B.; Nogales, A.; Fang, W.-S.; Jímenez-Barbero, J.; Canales, A.; Barasoain, I.; et al. Taxanes with High Potency Inducing Tubulin Assembly Overcome Tumoural Cell Resistances. Bioorg. Med. Chem. 2014, 22, 5078–5090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, A.; Sun, L.; Zanardi, I.; Wu, X.; Ferlini, C.; Fontana, G.; Bombardelli, E.; Ojima, I. Novel C-Seco-Taxoids Possessing High Potency against Paclitaxel-Resistant Cancer Cell Lines Overexpressing Class III Beta-Tubulin. Bioorg. Med. Chem. Lett. 2009, 19, 3300–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlini, C.; Raspaglio, G.; Mozzetti, S.; Cicchillitti, L.; Filippetti, F.; Gallo, D.; Fattorusso, C.; Campiani, G.; Scambia, G. The Seco-Taxane IDN5390 Is Able to Target Class III Beta-Tubulin and to Overcome Paclitaxel Resistance. Cancer Res. 2005, 65, 2397–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Rodríguez-Salarichs, J.; Zhao, Y.; Cai, P.; Estévez-Gallego, J.; Balaguer-Pérez, F.; Redondo Horcajo, M.; Lucena-Agell, D.; Barasoain, I.; Díaz, J.F.; et al. Modification of C-Seco Taxoids through Ring Tethering and Substituent Replacement Leading to Effective Agents against Tumor Drug Resistance Mediated by ΒIII-Tubulin and P-Glycoprotein (P-Gp) Overexpressions. Eur J. Med. Chem. 2017, 137, 488–503. [Google Scholar] [CrossRef]
- Oehler, C.; Frei, K.; Rushing, E.J.; McSheehy, P.M.J.; Weber, D.; Allegrini, P.R.; Weniger, D.; Lütolf, U.M.; Knuth, A.; Yonekawa, Y.; et al. Patupilone (Epothilone B) for Recurrent Glioblastoma: Clinical Outcome and Translational Analysis of a Single-Institution Phase I/II Trial. Oncology 2012, 83, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horne, E.A.; Diaz, P.; Cimino, P.J.; Jung, E.; Xu, C.; Hamel, E.; Wagenbach, M.; Kumasaka, D.; Wageling, N.B.; Azorín, D.D.; et al. A Brain-Penetrant Microtubule-Targeting Agent That Disrupts Hallmarks of Glioma Tumorigenesis. Neurooncol. Adv. 2021, 3, vdaa165. [Google Scholar] [CrossRef]
- Cherry, A.E.; Haas, B.R.; Naydenov, A.V.; Fung, S.; Xu, C.; Swinney, K.; Wagenbach, M.; Freeling, J.; Canton, D.A.; Coy, J.; et al. ST-11: A New Brain-Penetrant Microtubule-Destabilizing Agent with Therapeutic Potential for Glioblastoma Multiforme. Mol. Cancer Ther. 2016, 15, 2018–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Microtubules (MTs) functions in the cell. MTs play roles in a variety of cellular functions. Top row, left: MTs are involved in the transport of a variety of molecules, vesicles, and organelles inside the cell, affecting signaling transduction pathways. They are also essential in the formation and maintenance of the primary cilium, a signaling hub for the cell. Top row, center: MTs form the mitotic spindle, the machinery in charge of chromosome segregation during mitosis. Top row, right: MTs form the tracks used by molecular motors to transport neurotransmitters through the axon to the synaptic cleft. Center row: Proper organization of the MT cytoskeleton and centrosome localization is important to stabilize the migrating leading edge, for cell motility and for the formation of the invadopodium, a cellular extension of the cell membrane required for invasion of adjacent tissues. Bottom row: MTs are also important during the adhesion and the basal-to-lumen cell polarity establishment of epithelial cells. They also play an important role during the formation and turnover of focal adhesions. See the text for details.
Figure 1.
Microtubules (MTs) functions in the cell. MTs play roles in a variety of cellular functions. Top row, left: MTs are involved in the transport of a variety of molecules, vesicles, and organelles inside the cell, affecting signaling transduction pathways. They are also essential in the formation and maintenance of the primary cilium, a signaling hub for the cell. Top row, center: MTs form the mitotic spindle, the machinery in charge of chromosome segregation during mitosis. Top row, right: MTs form the tracks used by molecular motors to transport neurotransmitters through the axon to the synaptic cleft. Center row: Proper organization of the MT cytoskeleton and centrosome localization is important to stabilize the migrating leading edge, for cell motility and for the formation of the invadopodium, a cellular extension of the cell membrane required for invasion of adjacent tissues. Bottom row: MTs are also important during the adhesion and the basal-to-lumen cell polarity establishment of epithelial cells. They also play an important role during the formation and turnover of focal adhesions. See the text for details.

Figure 2.
Microtubule-targeting agents (MTAs) biding sites on the tubulin dimer. The αβ-tubulin dimer is shown in gray and light brown with the α subunit on the left and the β subunit on the right. Colored representative structures of the ligands are located on their binding sites. Color code: blue, Pironetin; yellow, Gatorbulin; cyan, colchicine; red, taxane; orange, vinca; magenta, laulimalide/peloruside; green, Maytansine. See text for details.
Figure 2.
Microtubule-targeting agents (MTAs) biding sites on the tubulin dimer. The αβ-tubulin dimer is shown in gray and light brown with the α subunit on the left and the β subunit on the right. Colored representative structures of the ligands are located on their binding sites. Color code: blue, Pironetin; yellow, Gatorbulin; cyan, colchicine; red, taxane; orange, vinca; magenta, laulimalide/peloruside; green, Maytansine. See text for details.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wordeman, L.; Vicente, J.J. Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles. Cancers 2021, 13, 5650. https://doi.org/10.3390/cancers13225650
AMA Style
Wordeman L, Vicente JJ. Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles. Cancers. 2021; 13(22):5650. https://doi.org/10.3390/cancers13225650
Chicago/Turabian StyleWordeman, Linda, and Juan Jesus Vicente. 2021. "Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles" Cancers 13, no. 22: 5650. https://doi.org/10.3390/cancers13225650
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.