
Identification of a Chemotherapeutic Lead Molecule for the Potential Disruption of the FAM72A-UNG2 Interaction to Interfere with Genome Stability, Centromere Formation, and Genome Editing

 ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection—Sequence and Structure Details
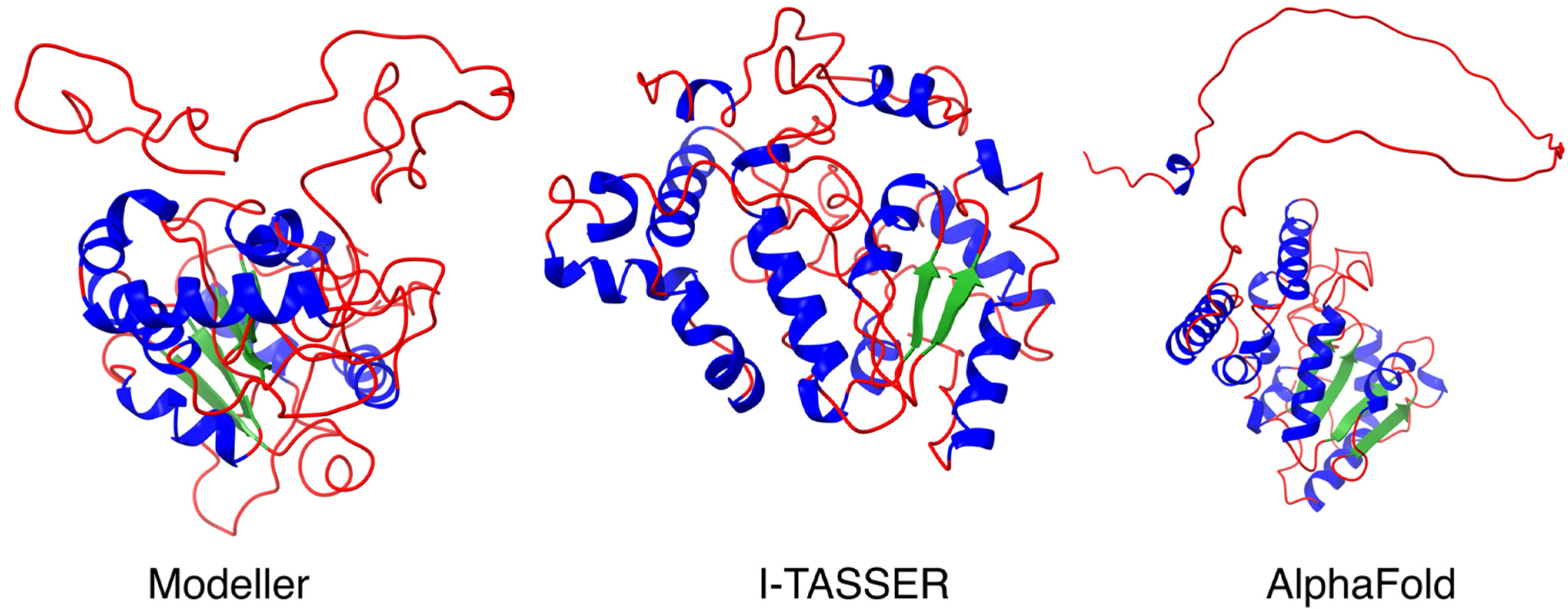
2.2. Homology Modeling and Protein Structure Validation of UNG2 by Modeller, I-TASSER and AlphaFold
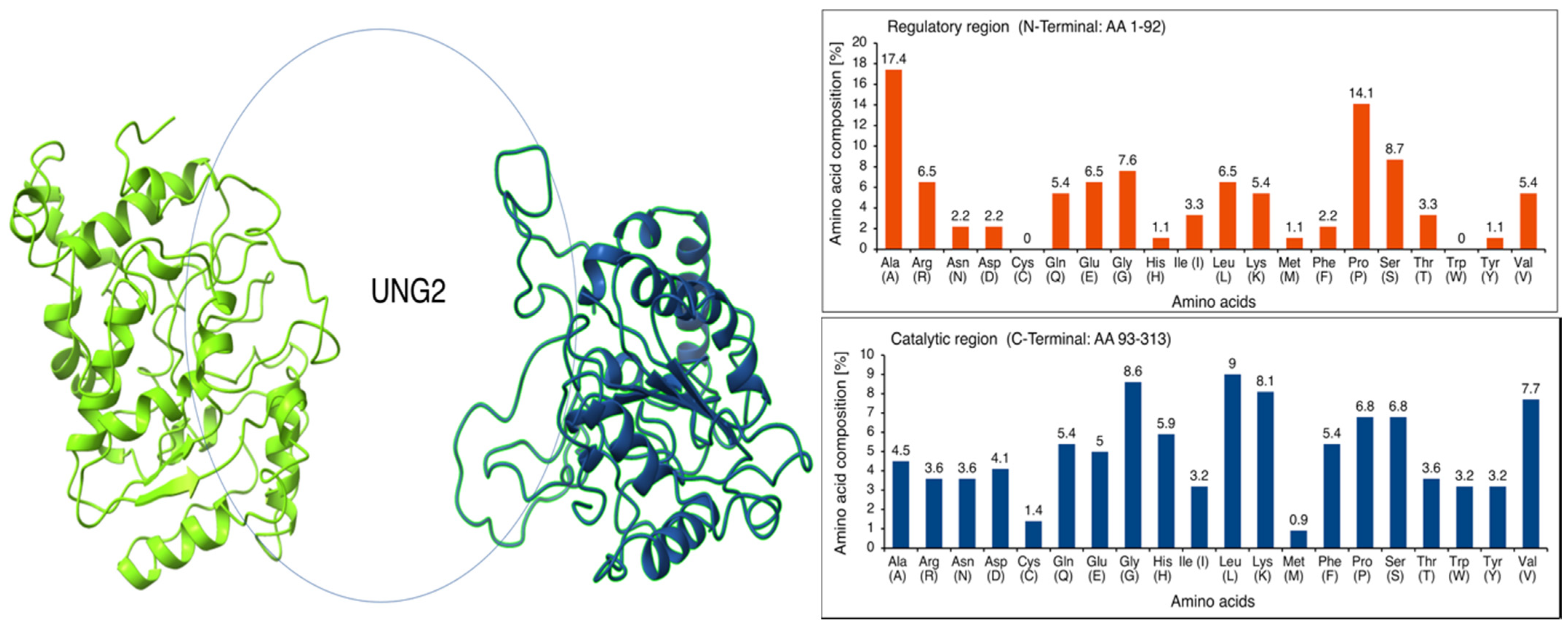
2.3. Intrinsically Disordered Region in UNG2 (AA 1–92)
2.4. Molecular Docking of FAM72A Protein and UNG2 Peptide (AA 1–45) by HPEPDOCK
2.5. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) Calculation
2.6. Carbon Distribution (CARd) Analysis
2.7. Amino Acid-Specific Mutations in FAM72A Protein-UNG2 (AA 1–45) Peptide Heterodimer (FAM72A F104A, F104R, F104N, F104G, and F104S)
2.8. Molecular Dynamics Simulation by GROMACS
2.9. Virtual Screening for Lead Molecule Identification against FAM72A Protein and UNG2 (AA 1–45) Peptide Mono/Heterodimer
2.10. Molecular Docking of FAM72A Protein and UNG2 (AA 1–45) Peptide Heterodimer and FAM72A Monomer with Small Chemical Molecules
3. Results and Discussion
3.1. Homology Modeling and Protein Structure Validation of UNG2 by Modeller, I-TASSER and AlphaFold
3.2. Intrinsically Disordered Region in N-Terminal UNG2 (AA 1–92)
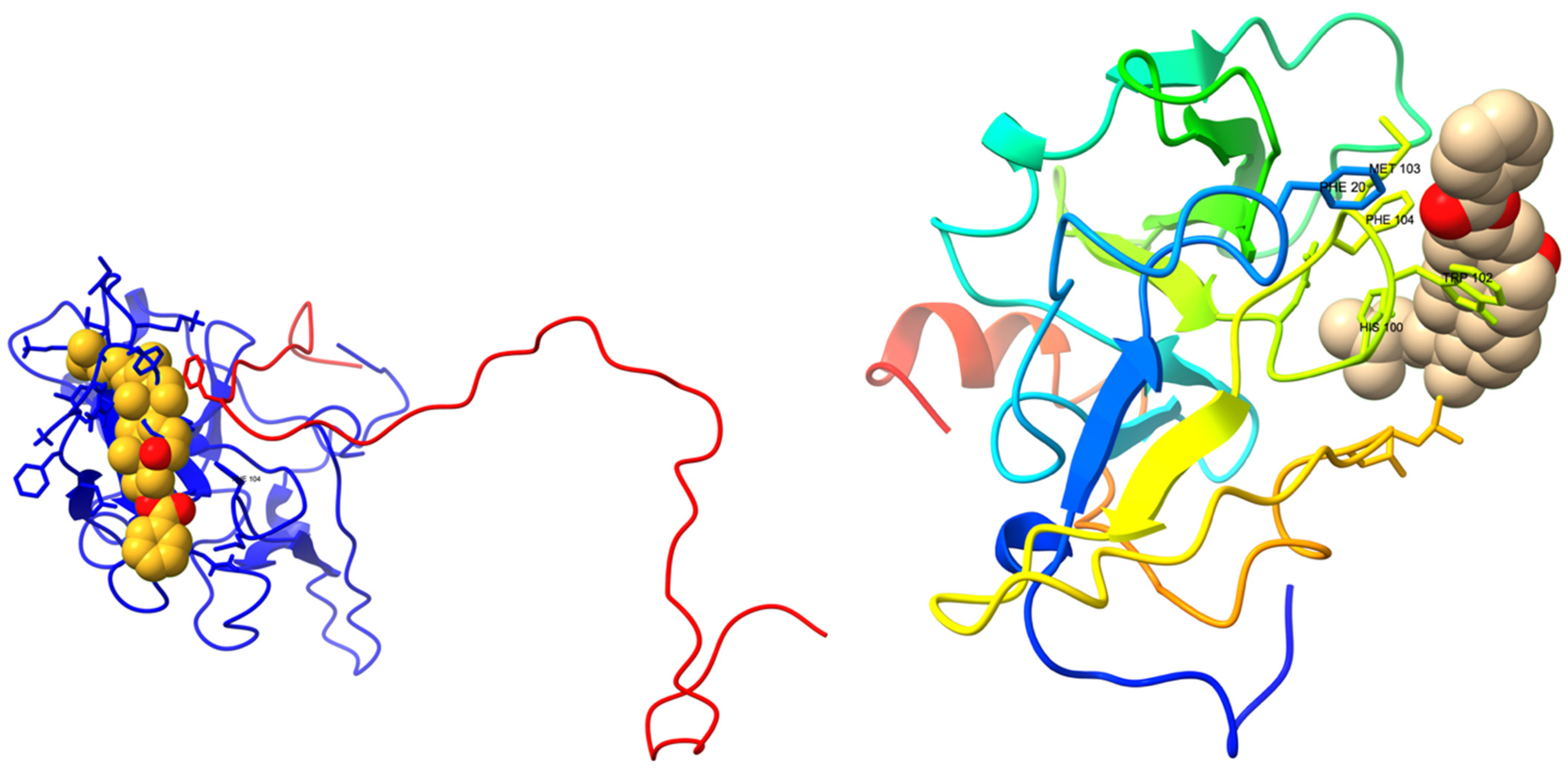
3.3. FAM72A-UNG2 Interaction and Molecular Docking Study of FAM72A Protein and UNG2 (AA 1–45) Peptide by HPEPDOCK
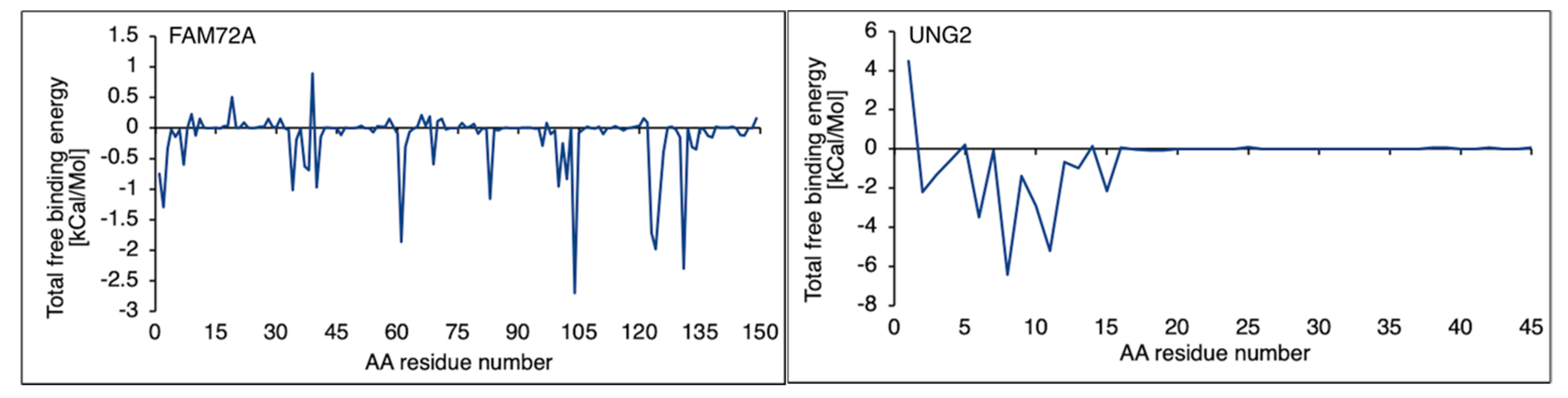
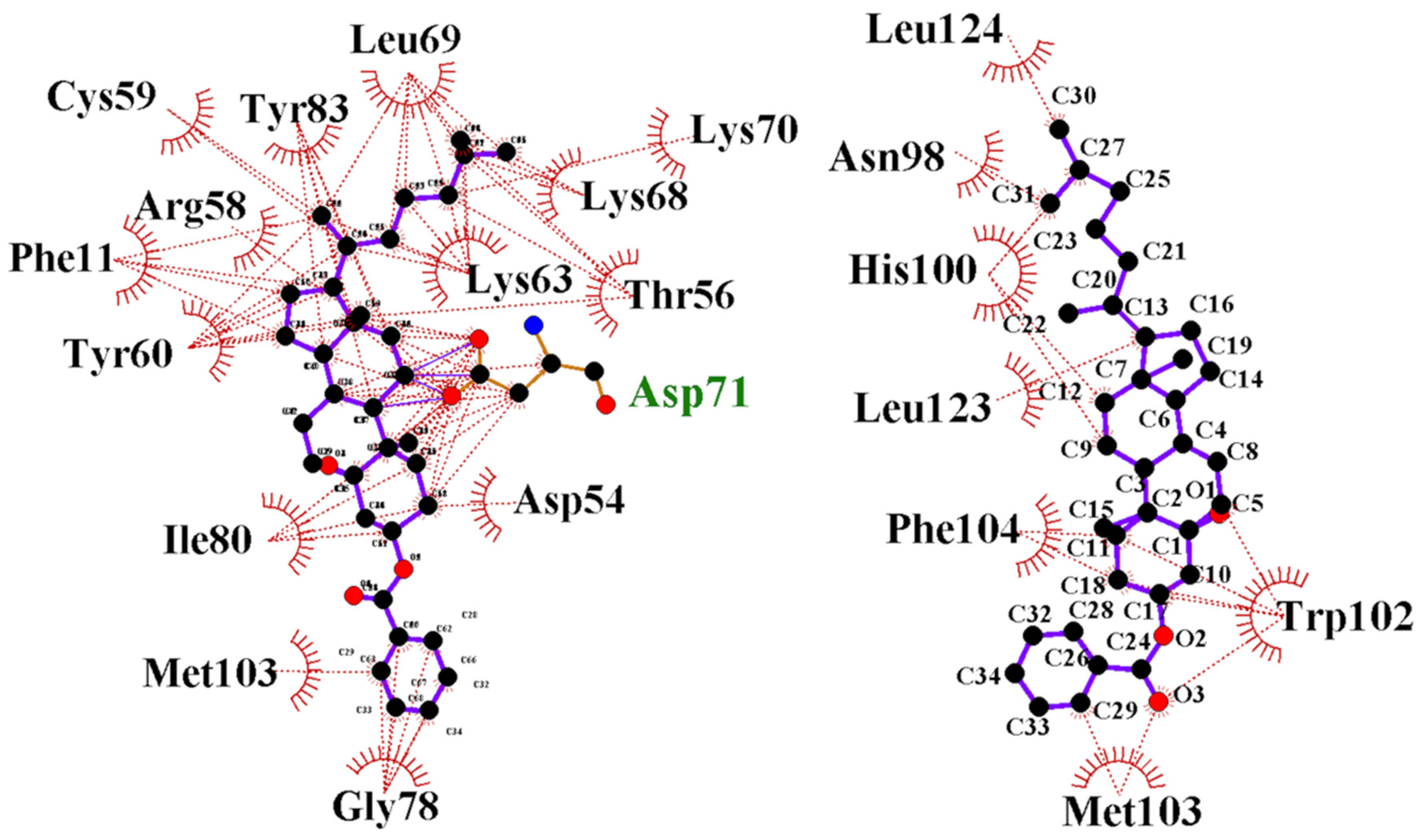
3.4. Free Binding Energy Prediction on FAM72A Protein and UNG2 (AA 1–45) Peptide Heterodimer
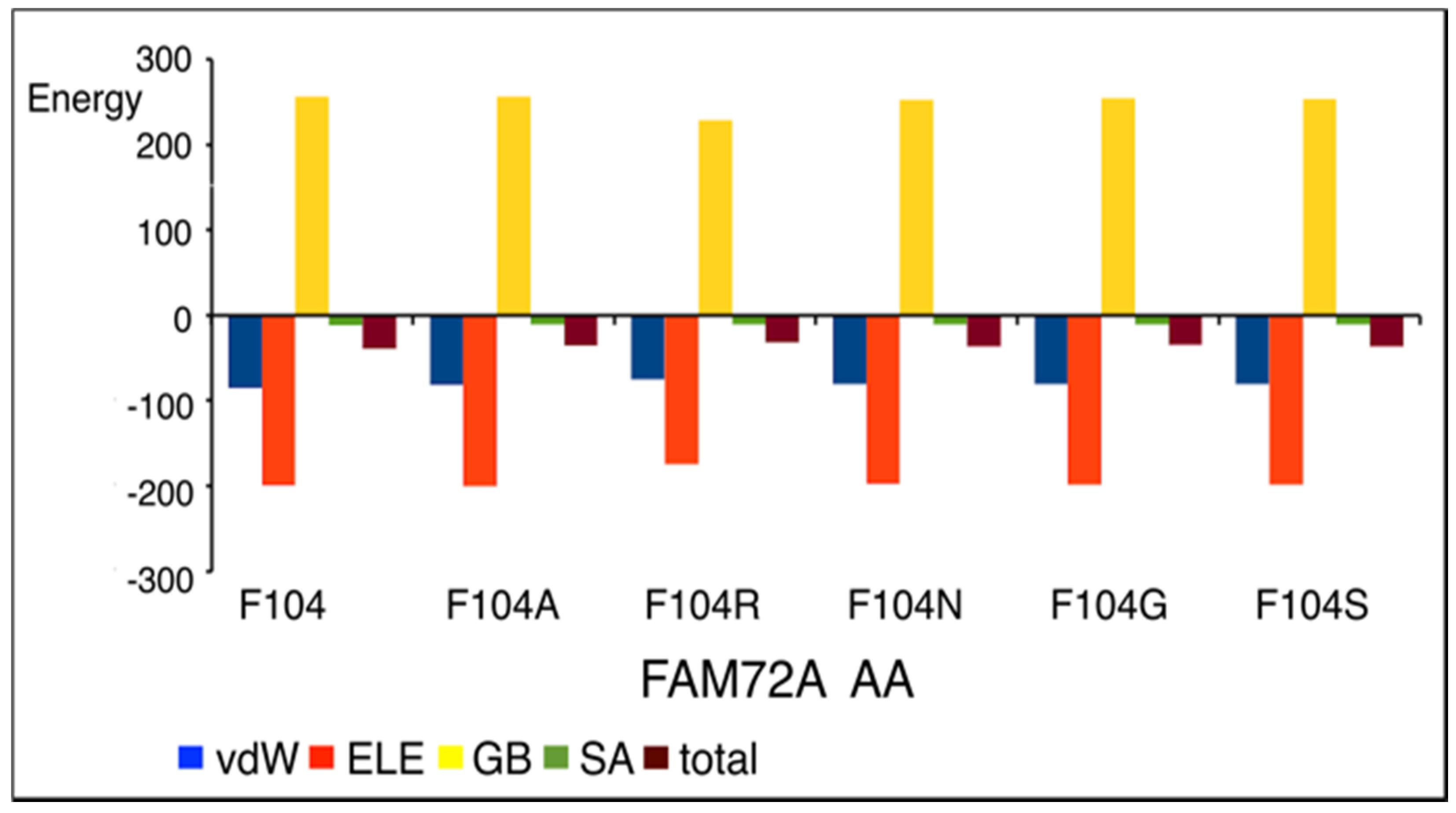
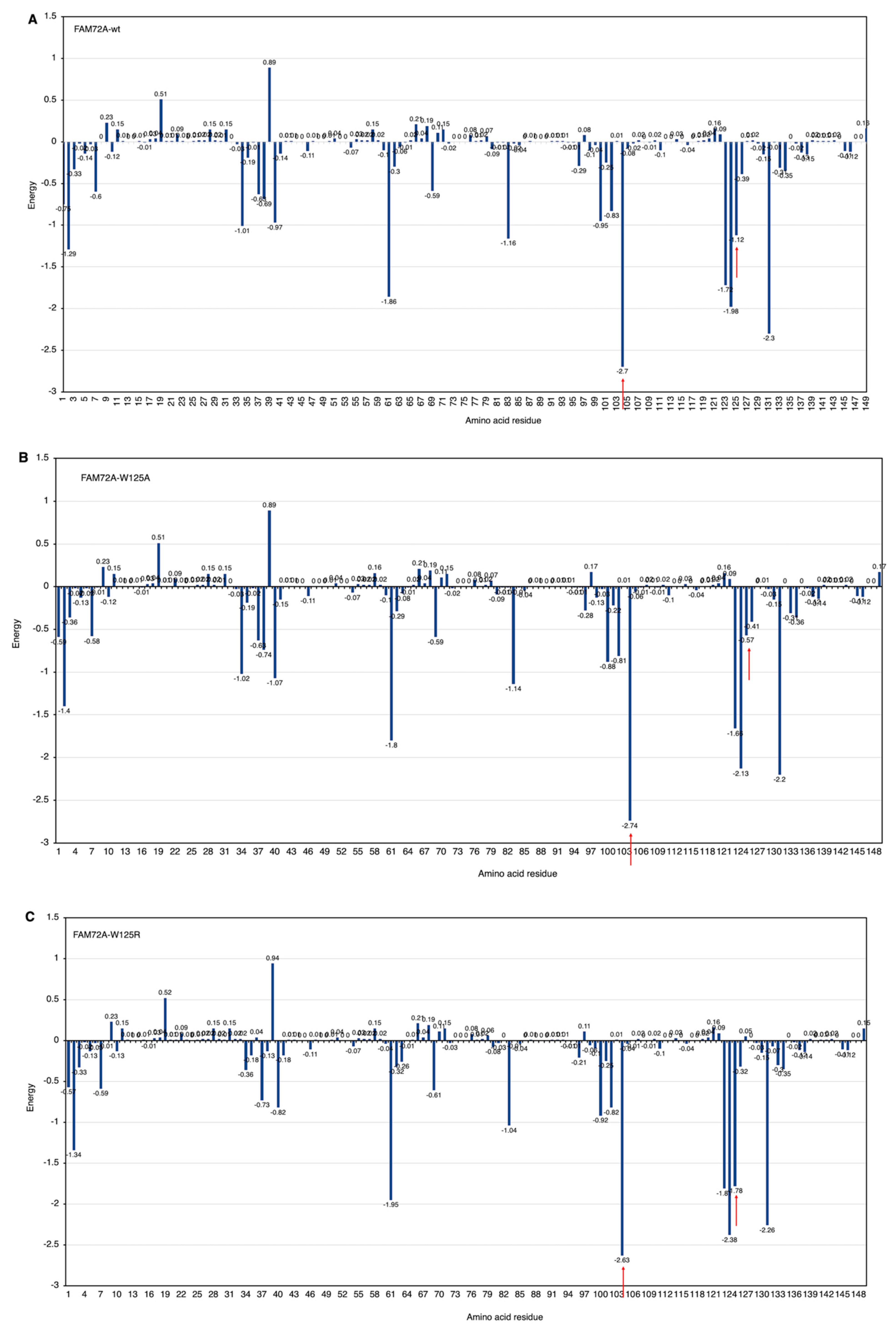
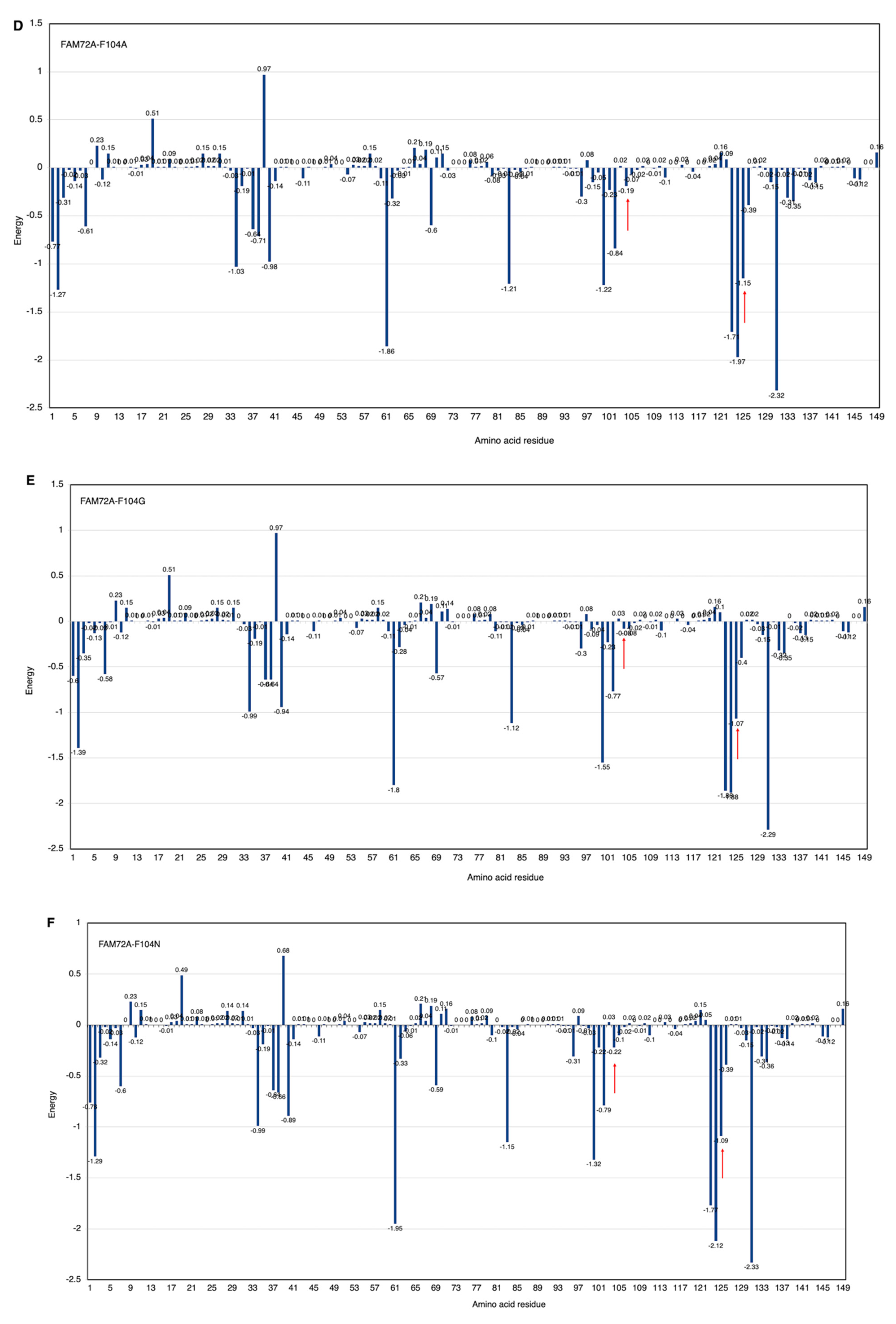
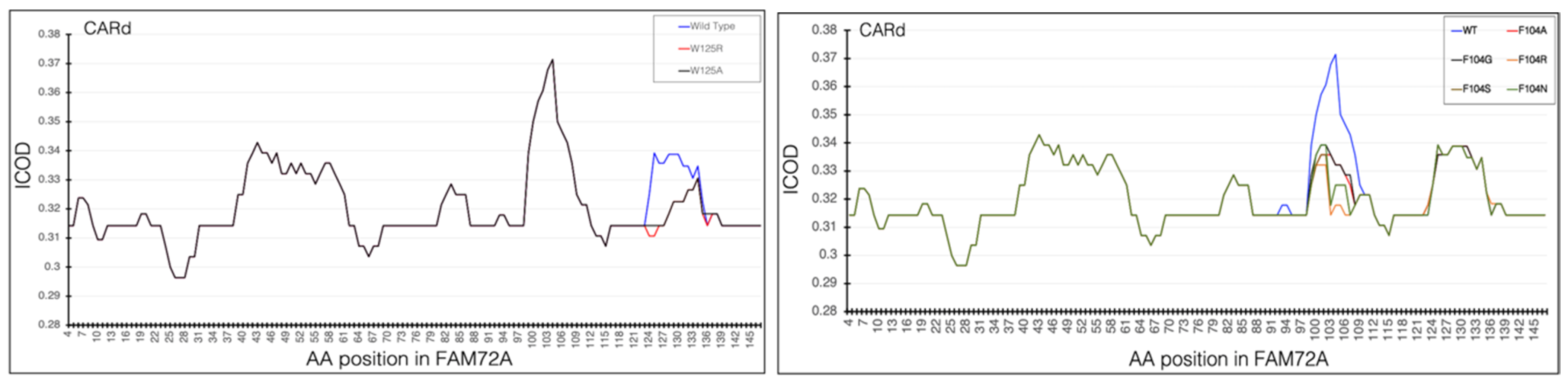
3.5. AA-Specific Mutations in the FWMF Motif (AA 101–104) of FAM72A Affecting the FAM72A Protein and UNG2 (AA 1–45) Peptide Heterodimer Binding
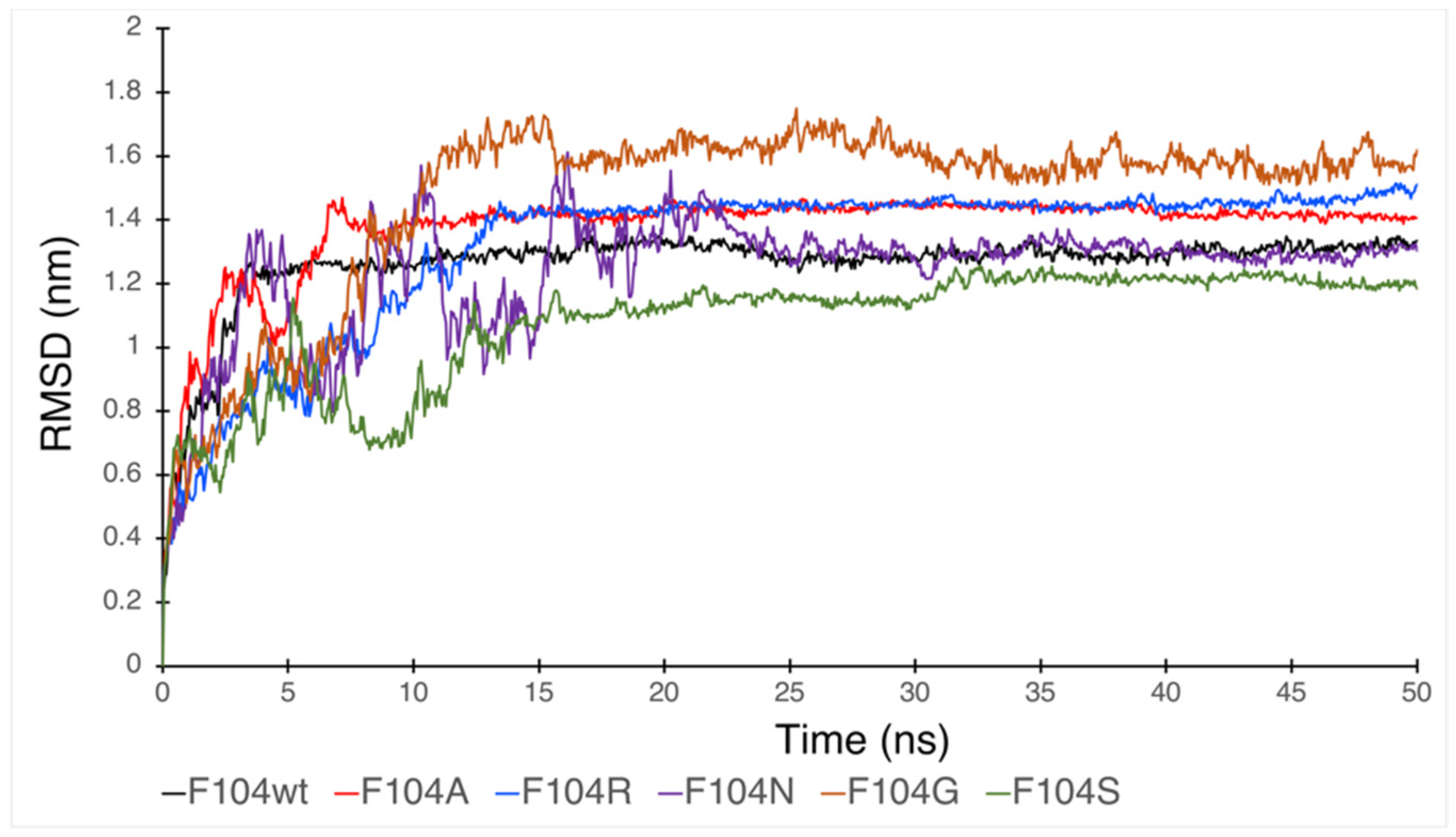
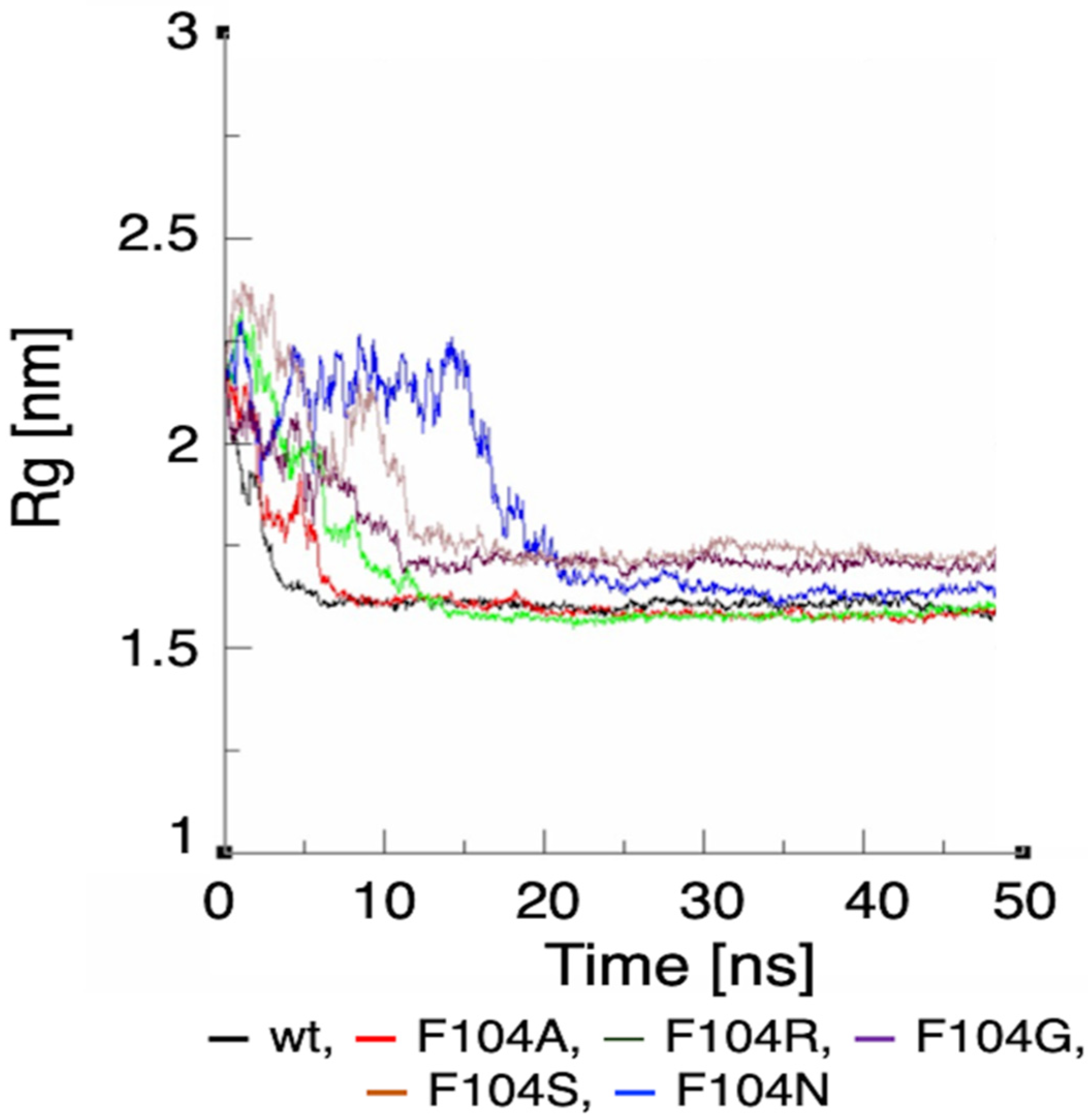
3.6. Molecular Dynamic Simulation by GROMACS Validates AA-specific Mutations in the FWMF Motif (AA 101–104) of FAM72A Affecting FAM72A-UNG2 Heterodimer Binding
3.7. CARd Analysis
3.8. Lead Discovery and Chemical Docking—Interference with FAM72A-UNG2 Interaction and Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chastain, P.D., II; Nakamura, J.; Rao, S.; Chu, H.; Ibrahim, J.G.; Swenberg, J.A.; Kaufman, D.G. Abasic sites preferentially form at regions undergoing DNA replication. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3674–3680. [Google Scholar] [CrossRef] [Green Version]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA glycosylases-structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014, 23, 1667–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Otterlei, M.; Peña-Diaz, J.; Krokan, H.E. Different organization of base excision repair of uracil in DNA in nuclei and mitochondria and selective upregulation of mitochondrial uracil-DNA glycosylase after oxidative stress. Neuroscience 2007, 145, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Otterlei, M.; Nilsen, H.; Kavli, B.; Skorpen, F.; Andersen, S.; Skjelbred, C.; Akbari, M.; Aas, P.A.; Slupphaug, G. Properties and functions of human uracil-DNA glycosylase from the UNG gene. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 365–386. [Google Scholar] [CrossRef]
- Akbari, M.; Otterlei, M.; Peña-Diaz, J.; Aas, P.A.; Kavli, B.; Liabakk, N.B.; Hagen, L.; Imai, K.; Durandy, A.; Slupphaug, G.; et al. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res. 2004, 32, 5486–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.B.; Bianchet, M.A.; Krosky, D.J.; Friedman, J.I.; Amzel, L.M.; Stivers, J.T. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 2007, 449, 433–437. [Google Scholar] [CrossRef]
- Wyatt, M.D. Advances in understanding the coupling of DNA base modifying enzymes to processes involving base excision repair. Adv. Cancer Res. 2013, 119, 63–106. [Google Scholar] [CrossRef]
- Lindahl, T. DNA repair enzymes. Annu. Rev. Biochem. 1982, 51, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Loeb, L.A. Apurinic sites as mutagenic intermediates. Cell 1985, 40, 483–484. [Google Scholar] [CrossRef]
- Slupphaug, G.; Markussen, F.H.; Olsen, L.C.; Aasland, R.; Aarsaether, N.; Bakke, O.; Krokan, H.E.; Helland, D.E. Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res. 1993, 21, 2579–2584. [Google Scholar] [CrossRef] [Green Version]
- Haug, T.; Skorpen, F.; Kvaløy, K.; Eftedal, I.; Lund, H.; Krokan, H.E. Human uracil-DNA glycosylase gene: Sequence organization, methylation pattern, and mapping to chromosome 12q23-q24.1. Genomics 1996, 36, 408–416. [Google Scholar] [CrossRef]
- Nilsen, H.; Otterlei, M.; Haug, T.; Solum, K.; Nagelhus, T.A.; Skorpen, F.; Krokan, H.E. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997, 25, 750–755. [Google Scholar] [CrossRef] [Green Version]
- Otterlei, M.; Haug, T.; Nagelhus, T.A.; Slupphaug, G.; Lindmo, T.; Krokan, H.E. Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 1998, 26, 4611–4617. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, G.; Orris, B.; Majumdar, A.; Bhat, S.; Stivers, J.T. Macromolecular crowding induces compaction and DNA binding in the disordered N-terminal domain of hUNG2. DNA Repair 2020, 86, 102764. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.L.; Zhao, L. The N-terminal domain of uracil-DNA glycosylase: Roles for disordered regions. DNA Repair 2021, 101, 103077. [Google Scholar] [CrossRef] [PubMed]
- Weiser, B.P.; Rodriguez, G.; Cole, P.A.; Stivers, J.T. N-terminal domain of human uracil DNA glycosylase (hUNG2) promotes targeting to uracil sites adjacent to ssDNA-dsDNA junctions. Nucleic Acids Res. 2018, 46, 7169–7178. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, S.G.; Chapados, B.R.; Baker, N.M.; Tai, C.; Slupphaug, G.; Wang, J.Y. Uracil DNA N-glycosylase promotes assembly of human centromere protein A. PLoS ONE 2011, 6, e17151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehar, S.; Mishra, M.; Heese, K. Identification and characterisation of the novel amyloid-beta peptide-induced protein p17. FEBS Lett. 2009, 583, 3247–3253. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.T.T.; Kutzner, A.; Heese, K. Brain plasticity, cognitive functions and neural stem cells: A pivotal role for the brain-specific neural master gene |-SRGAP2-FAM72-|. Biol. Chem. 2017, 399, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.T.; Kim, P.S.; Kutzner, A.; Heese, K. Cognitive Functions: Human vs. Animal-4:1 Advantage |-FAM72-SRGAP2-|. J. Mol. Neurosci. 2017, 61, 603–606. [Google Scholar] [CrossRef]
- Kutzner, A.; Pramanik, S.; Kim, P.S.; Heese, K. All-or-(N)One—an epistemological characterization of the human tumorigenic neuronal paralogous FAM72 gene loci. Genomics 2015, 106, 278–285. [Google Scholar] [CrossRef]
- Rahane, C.S.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet 2019, 230, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rahane, C.S.; Kutzner, A.; Heese, K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J. Neurooncol. 2019, 141, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhang, X.; Fink, S.P.; Platzer, P.; Wilson, K.; Willson, J.K.; Wang, Z.; Markowitz, S.D. Ugene, a newly identified protein that is commonly overexpressed in cancer and binds uracil DNA glycosylase. Cancer Res. 2008, 68, 6118–6126. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Li, C.; Stewart, J.; Barbulescu, P.; Desivo, N.S.; Álvarez-Quilón, A.; Pezo, R.C.; Perera, M.L.W.; Chan, K.; Tong, A.H.Y.; et al. FAM72A antagonizes UNG2 to promote mutagenic uracil repair during antibody maturation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rogier, M.; Moritz, J.; Robert, I.; Lescale, C.; Heyer, V.; Thomas-Claudepierre, A.-S.; Abello, A.; Deriano, L.; Reina-San-Martin, B. Fam72a controls the balance between error-prone and error-free DNA repair during antibody diversification. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ho, N.T.T.; Rahane, C.S.; Pramanik, S.; Kim, P.S.; Kutzner, A.; Heese, K. FAM72, Glioblastoma Multiforme (GBM) and Beyond. Cancers 2021, 13, 1025. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, S.; Kutzner, A.; Heese, K. Lead discovery and in silico 3D structure modeling of tumorigenic FAM72A (p17). Tumour Biol. 2015, 36, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Mol, C.D.; Slupphaug, G.; Bharati, S.; Krokan, H.E.; Tainer, J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Pramanik, S.; Kutzner, A.; Heese, K. 3D Structure, Dimerization Modeling, and Lead Discovery by Ligand-protein Interaction Analysis of p60 Transcription Regulator Protein (p60TRP). Mol. Inform. 2016, 35, 99–108. [Google Scholar] [CrossRef]
- Pramanik, S.; Thaker, M.; Perumal, A.G.; Ekambaram, R.; Poondla, N.; Schmidt, M.; Kim, P.S.; Kutzner, A.; Heese, K. Proteomic Atomics Reveals a Distinctive Uracil-5-Methyltransferase. Mol. Inform. 2020, 39, e1900135. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 2.9.1–2.9.37. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Buchinger, E.; Wiik, S.A.; Kusnierczyk, A.; Rabe, R.; Aas, P.A.; Kavli, B.; Slupphaug, G.; Aachmann, F.L. Backbone (1)H, (13)C and (15)N chemical shift assignment of full-length human uracil DNA glycosylase UNG2. Biomol. NMR Assign 2018, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, E. CARd: Carbon distribution analysis program for protein sequences. Bioinformation 2012, 8, 508–512. [Google Scholar] [CrossRef]
- Kemmish, H.; Fasnacht, M.; Yan, L. Fully automated antibody structure prediction using BIOVIA tools: Validation study. PLoS ONE 2017, 12, e0177923. [Google Scholar] [CrossRef] [Green Version]
- Kalathiya, U.; Padariya, M.; Baginski, M. Structural, functional, and stability change predictions in human telomerase upon specific point mutations. Sci. Rep. 2019, 9, 8707. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Lomb, L.; Barends, T.R.; Kassemeyer, S.; Aquila, A.; Epp, S.W.; Erk, B.; Foucar, L.; Hartmann, R.; Rudek, B.; Rolles, D.; et al. Radiation damage in protein serial femtosecond crystallography using an x-ray free-electron laser. Phys. Review B Condens. Matter Mater. Phys. 2011, 84, 214111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancourt, M.R.; Skolnick, J. Universal similarity measure for comparing protein structures. Biopolymers 2001, 59, 305–309. [Google Scholar] [CrossRef]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavruša, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.-Y.; Chang, K.-W.; Chen, C.Y.-C. iScreen: World’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J. Comput.-Aided Mol. Des. 2011, 25, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. S9), 114–122. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Senthil, R.; Sakthivel, M.; Usha, S. Structure-based drug design of peroxisome proliferator-activated receptor gamma inhibitors: Ferulic acid and derivatives. J. Biomol. Struct. Dyn. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. On the roles of intrinsically disordered proteins and regions in cell communication and signaling. Cell Commun. Signal 2021, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Intrinsically Disord Proteins 2013, 1, e24684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berná, L.; Alvarez-Valin, F. Evolutionary volatile Cysteines and protein disorder in the fast evolving tunicate Oikopleura dioica. Mar. Genom. 2015, 24 Pt 1, 47–54. [Google Scholar] [CrossRef]
- Mejia, J.S.; Arthun, E.N.; Titus, R.G. Cysteine-free proteins in the immunobiology of arthropod-borne diseases. J. Biomed. Biotechnol. 2010, 2010, 171537. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [CrossRef]
- Schröder, J.; Klinger, A.; Oellien, F.; Marhöfer, R.J.; Duszenko, M.; Selzer, P.M. Docking-based virtual screening of covalently binding ligands: An orthogonal lead discovery approach. J. Med. Chem. 2013, 56, 1478–1490. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Orry, A.J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M. Docking and Virtual Screening in Drug Discovery. Methods Mol. Biol. 2017, 1647, 255–266. [Google Scholar] [CrossRef]
- Ji, D.; Xu, M.; Udenigwe, C.C.; Agyei, D. Physicochemical characterisation, molecular docking, and drug-likeness evaluation of hypotensive peptides encrypted in flaxseed proteome. Curr. Res. Food Sci. 2020, 3, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Opo, F.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Author Correction: Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 19106. [Google Scholar] [CrossRef]
- Renganathan, S.; Manokaran, S.; Vasanthakumar, P.; Singaravelu, U.; Kim, P.S.; Kutzner, A.; Heese, K. Phytochemical Profiling in Conjunction with In Vitro and In Silico Studies to Identify Human α-Amylase Inhibitors in Leucaena leucocephala (Lam.) De Wit for the Treatment of Diabetes Mellitus. ACS Omega 2021, 6, 19045–19057. [Google Scholar] [CrossRef]
- Kaileh, M.; Vanden Berghe, W.; Heyerick, A.; Horion, J.; Piette, J.; Libert, C.; De Keukeleire, D.; Essawi, T.; Haegeman, G. Withaferin a strongly elicits IkappaB kinase beta hyperphosphorylation concomitant with potent inhibition of its kinase activity. J. Biol. Chem. 2007, 282, 4253–4264. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.K.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.N.; Rangnekar, V.M.; Gupta, R.C.; Bondada, S. Anti-cancer activity of withaferin A in B-cell lymphoma. Cancer Biol. Ther. 2015, 16, 1088–1098. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.K.; Kumar, S.; Saloni, H.S.; Kim, M.H.; Sharma, P.; Misra, S.; Khan, F. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Des. Devel Ther. 2017, 11, 1859–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dom, M.; Vanden Berghe, W.; Van Ostade, X. Broad-spectrum antitumor properties of Withaferin A: A proteomic perspective. RSC Med. Chem. 2020, 11, 30–50. [Google Scholar] [CrossRef]
- Chirumamilla, C.S.; Pérez-Novo, C.; Van Ostade, X.; Vanden Berghe, W. Molecular insights into cancer therapeutic effects of the dietary medicinal phytochemical withaferin A. Proc. Nutr. Soc. 2017, 76, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Mori, M.; Infante, P.; Basili, I.; Di Marcotullio, L.; Calcaterra, A.; Catti, F.; Botta, B.; Passarella, D. Design and Synthesis of New Withaferin A Inspired Hedgehog Pathway Inhibitors. Chemistry 2021, 27, 8350–8357. [Google Scholar] [CrossRef]
- Odongo, R.; Demiroglu-Zergeroglu, A.; Çakır, T. A systems pharmacology approach based on oncogenic signalling pathways to determine the mechanisms of action of natural products in breast cancer from transcriptome data. BMC Complement Med. Ther. 2021, 21, 181. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Shi, H.; Du, Y.; Ou, J. Withaferin A inhibits proliferation of human endometrial cancer cells via transforming growth factor-β (TGF-β) signalling. 3 Biotech 2021, 11, 323. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, Z.; Zheng, Q.; Li, J. FAM72 serves as a biomarker of poor prognosis in human lung adenocarcinoma. Aging 2021, 13, 8155–8176. [Google Scholar] [CrossRef]
- Zhang, T.; Nie, Y.; Gu, J.; Cai, K.; Chen, X.; Li, H.; Wang, J. Identification of Mitochondrial-Related Prognostic Biomarkers Associated With Primary Bile Acid Biosynthesis and Tumor Microenvironment of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 587479. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renganathan, S.; Pramanik, S.; Ekambaram, R.; Kutzner, A.; Kim, P.-S.; Heese, K. Identification of a Chemotherapeutic Lead Molecule for the Potential Disruption of the FAM72A-UNG2 Interaction to Interfere with Genome Stability, Centromere Formation, and Genome Editing. Cancers 2021, 13, 5870. https://doi.org/10.3390/cancers13225870
Renganathan S, Pramanik S, Ekambaram R, Kutzner A, Kim P-S, Heese K. Identification of a Chemotherapeutic Lead Molecule for the Potential Disruption of the FAM72A-UNG2 Interaction to Interfere with Genome Stability, Centromere Formation, and Genome Editing. Cancers. 2021; 13(22):5870. https://doi.org/10.3390/cancers13225870
Chicago/Turabian StyleRenganathan, Senthil, Subrata Pramanik, Rajasekaran Ekambaram, Arne Kutzner, Pok-Son Kim, and Klaus Heese. 2021. "Identification of a Chemotherapeutic Lead Molecule for the Potential Disruption of the FAM72A-UNG2 Interaction to Interfere with Genome Stability, Centromere Formation, and Genome Editing" Cancers 13, no. 22: 5870. https://doi.org/10.3390/cancers13225870