Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells


{kind=link}
Abstract
:1. Introduction
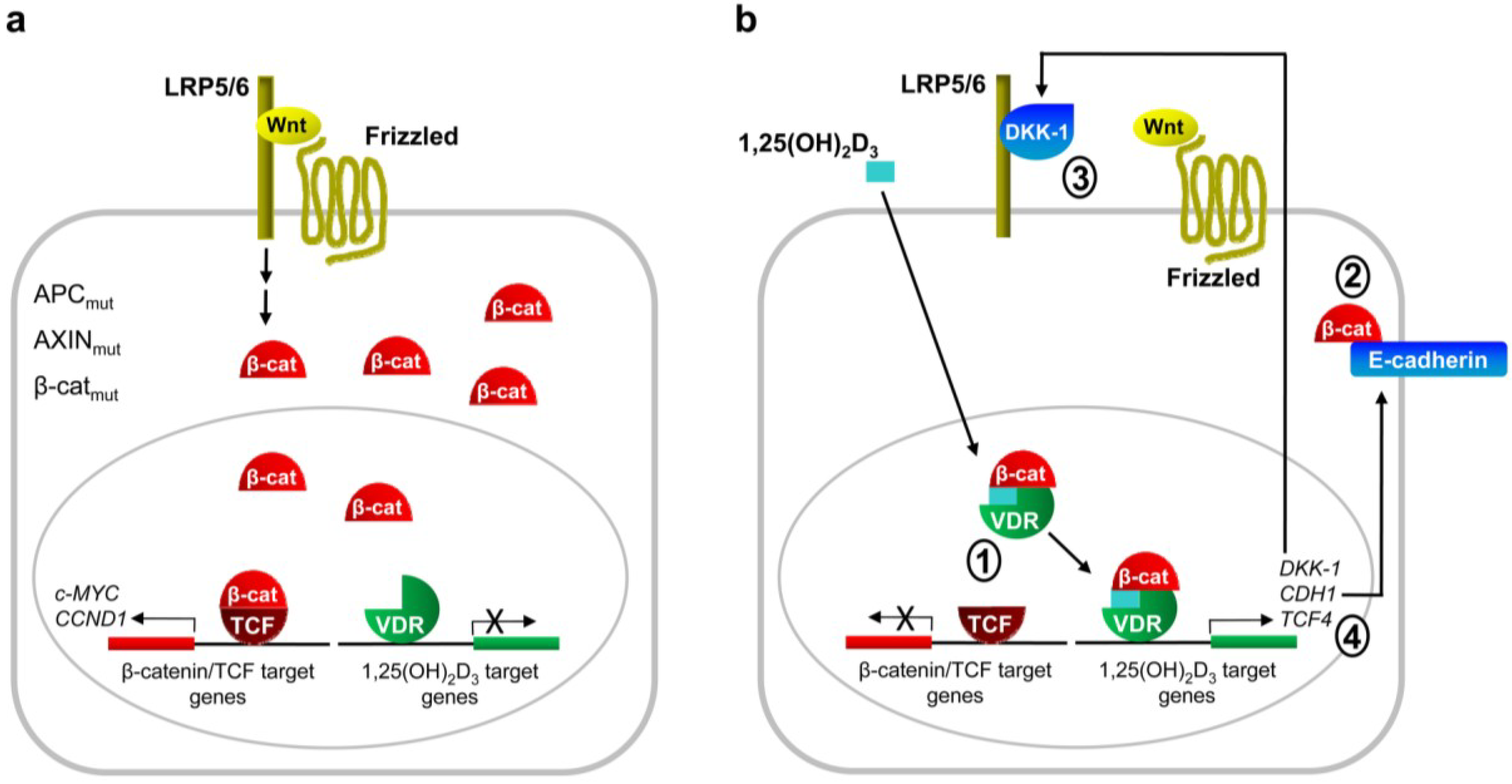
2. Antagonism of Wnt/β-Catenin Signaling by 1,25(OH)2D3
2.1. Studies in Cultured Cells

2.2. Studies in Animal Models
2.3. Studies in Patients
3. Cooperation Between VDR and Wnt/β-Catenin Signaling
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar]
- Pereira, F.; Larriba, M.J.; Muñoz, A. Vitamin D and colon cancer. Endocr. Relat. Cancer 2012, 19, R51–R71. [Google Scholar] [Green Version]
- Leyssens, C.; Verlinden, L.; Verstuyf, A. Antineoplastic effects of 1,25(OH)2D3 and its analogs in breast, prostate and colorectal cancer. Endocr. Relat. Cancer 2013, 20, R31–R47. [Google Scholar]
- IARC. Vitamin D and cancer. In IARC Working Group Reports Vol. 5; International Agency for Research on Cancer: Lyon, France, 2008. [Google Scholar]
- Giovannucci, E. Epidemiology of vitamin D and colorectal cancer. Anticancer Agents Med. Chem. 2013, 13, 11–19. [Google Scholar] [CrossRef]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2012, 92, 77–98. [Google Scholar]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)2vitamin D3: Genomic and non-genomic mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar]
- Ordóñez-Morán, P.; Larriba, M.J.; Pálmer, H.G.; Valero, R.A.; Barbáchano, A.; Duñach, M.; García de Herreros, A.; Villalobos, C.; Berciano, M.T.; Lafarga, M.; et al. RhoA-ROCK and p38MAPK-MSK1 mediate vitamin D effects on gene expression, phenotype, and Wnt pathway in colon cancer cells. J. Cell Biol. 2008, 183, 697–710. [Google Scholar]
- Willert, K.; Nusse, R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Rasola, A.; Fassetta, M.; de Bacco, F.; D’Alessandro, L.; Gramaglia, D.; di Renzo, M.F.; Comoglio, P.M. A positive feedback loop between hepatocyte growth factor receptor and β-catenin sustains colorectal cancer cell invasive growth. Oncogene 2007, 26, 1078–1087. [Google Scholar]
- Korkaya, H.; Wicha, M.S. Cancer stem cells: Nature versus nurture. Nat. Cell Biol. 2010, 12, 419–421. [Google Scholar]
- Vermeulen, L.; de Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar]
- Singh, A.; Sweeney, M.F.; Yu, M.; Burger, A.; Greninger, P.; Benes, C.; Haber, D.A.; Settleman, J. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012, 148, 639–650. [Google Scholar]
- Zucman-Rossi, J.; Jeannot, E.; Van Nhieu, J.T.; Scoazec, J.-Y.; Guettier, C.; Rebouissou, S.; Bacq, Y.; Leteurtre, E.; Paradis, V.; Michalak, S.; et al. Genotype-phenotype correlation in hepatocellular adenoma: New classification and relationship with HCC. Hepatology 2006, 43, 515–524. [Google Scholar]
- Austinat, M.; Dunsch, R.; Wittekind, C.; Tannapfel, A.; Gebhardt, R.; Gaunitz, F. Correlation between β-catenin mutations and expression of Wnt-signaling target genes in hepatocellular carcinoma. Mol. Cancer 2008. [Google Scholar] [CrossRef]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar]
- Prakash, N.; Wurst, W. A Wnt signal regulates stem cell fate and differentiation in vivo. Neurodegener. Dis. 2007, 4, 333–338. [Google Scholar]
- Tanaka, S.; Terada, K.; Nohno, T. Canonical Wnt signaling is involved in switching from cell proliferation to myogenic differentiation of mouse myoblast cells. J. Mol. Signal. 2011. [Google Scholar] [CrossRef]
- Regard, J.B.; Zhong, Z.; Williams, B.O.; Yang, Y. Wnt signaling in bone development and disease: Making stronger bone with Wnts. Cold Spring Harb. Perspect. Biol. 2012, 4, a007997. [Google Scholar] [CrossRef]
- Sharma, A.R.; Choi, B.S.; Park, J.M.; Lee, D.H.; Lee, J.E.; Kim, H.S.; Yoon, J.K.; Song, D.K.; Nam, J.S.; Lee, S.S. Rspo 1 promotes osteoblast differentiation via Wnt signaling pathway. Indian J. Biochem. Biophys. 2013, 50, 19–25. [Google Scholar]
- Webster, M.R.; Weeraratna, A.T. A Wnt-er migration: The confusing role of β-catenin in melanoma metastasis. Sci. Signal. 2013. [Google Scholar] [CrossRef]
- Biechele, T.L.; Kulikauskas, R.M.; Toroni, R.A.; Lucero, O.M.; Swift, R.D.; James, R.G.; Robin, N.C.; Dawson, D.W.; Moon, R.T.; Chien, A.J. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci. Signal. 2012. [Google Scholar] [CrossRef]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar]
- Gao, B. Wnt regulation of planar cell polarity (PCP). Curr. Top. Dev. Biol. 2012, 101, 263–295. [Google Scholar]
- Ying, J.; Li, H.; Yu, J.; Ng, K.M.; Poon, F.F.; Wong, S.C.; Chan, A.T.; Sung, J.J.; Tao, Q. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/β-catenin signaling, and is frequently methylated in colorectal cancer. Clin. Cancer Res. 2008, 14, 55–61. [Google Scholar]
- Sugimura, R.; Li, L. Noncanonical Wnt signaling in vertebrate development, stem cells, and diseases. Birth Defects Res. C Embryo Today 2010, 90, 243–256. [Google Scholar]
- Easwaran, V.; Pishvaian, M.; Salimuddin; Byers, S. Cross-regulation of β-catenin-LEF/TCF and retinoid signaling pathways. Curr. Biol. 1999, 9, 1415–1418. [Google Scholar]
- Truica, C.I.; Byers, S.; Gelmann, E.P. β-Catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000, 60, 4709–4713. [Google Scholar]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; García de Herreros, A.; Lafarga, M.; et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of β-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the Wnt/β-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar]
- Beildeck, M.E.; Gelmann, E.P.; Byers, S.W. Cross-regulation of signaling pathways: An example of nuclear hormone receptors and the canonical Wnt pathway. Exp. Cell Res. 2010, 316, 1763–1772. [Google Scholar]
- Aguilera, O.; Peña, C.; García, J.M.; Larriba, M.J.; Ordóñez-Morán, P.; Navarro, D.; Barbáchano, A.; López de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1α,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007, 28, 1877–1884. [Google Scholar]
- Larriba, M.J.; Valle, N.; Pálmer, H.G.; Ordóñez-Morán, P.; Álvarez-Díaz, S.; Becker, K.F.; Gamallo, C.; García de Herreros, A.; González-Sancho, J.M.; Muñoz, A. The inhibition of Wnt/β-catenin signalling by 1α,25-dihydroxyvitamin D3 is abrogated by Snail1 in human colon cancer cells. Endocr. Relat. Cancer 2007, 14, 141–151. [Google Scholar]
- Larriba, M.J.; Ordóñez-Morán, P.; Chicote, I.; Martín-Fernández, G.; Puig, I.; Muñoz, A.; Pálmer, H.G. Vitamin D receptor deficiency enhances Wnt/β-catenin signaling and tumor burden in colon cancer. PLoS One 2011, 6, e23524. [Google Scholar]
- Pálmer, H.G.; Larriba, M.J.; García, J.M.; Ordóñez-Morán, P.; Peña, C.; Peiró, S.; Puig, I.; Rodríguez, R.; de la Fuente, R.; Bernad, A.; et al. The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat. Med. 2004, 10, 917–919. [Google Scholar]
- Larriba, M.J.; Martín-Villar, E.; García, J.M.; Pereira, F.; Peña, C.; García de Herreros, A.; Bonilla, F.; Muñoz, A. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis 2009, 30, 1459–1468. [Google Scholar]
- Shah, S.; Islam, M.N.; Dakshanamurthy, S.; Rizvi, I.; Rao, M.; Herrell, R.; Zinser, G.; Valrance, M.; Aranda, A.; Moras, D.; et al. The molecular basis of vitamin D receptor and β-catenin crossregulation. Mol. Cell 2006, 21, 799–809. [Google Scholar]
- Byers, S.W.; Rowlands, T.; Beildeck, M.; Bong, Y.-S. Mechanism of action of vitamin D and the vitamin D receptor in colorectal cancer prevention and treatment. Rev. Endocr. Metab. Disord. 2012, 13, 31–38. [Google Scholar]
- Egan, J.B.; Thompson, P.A.; Vitanov, M.V.; Bartik, L.; Jacobs, E.T.; Haussler, M.R.; Gerner, E.W.; Jurutka, P.W. Vitamin D receptor ligands, adenomatous polyposis coli, and the vitamin D receptor FokI polymorphism collectively modulate β-catenin activity in colon cancer cells. Mol. Carcinog. 2010, 49, 337–352. [Google Scholar]
- Pereira, F.; Barbáchano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Muñoz, A.; Larriba, M.J. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665. [Google Scholar]
- Xu, H.; McCann, M.; Zhang, Z.; Posner, G.H.; Bingham, V.; El-Tanani, M.; Campbell, F.C. Vitamin D receptor modulates the neoplastic phenotype through antagonistic growth regulatory signals. Mol. Carcinog. 2009, 48, 758–772. [Google Scholar]
- Semenov, M.V.; Tamai, K.; Brott, B.K.; Kuhl, M.; Sokol, S.; He, X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 2001, 11, 951–961. [Google Scholar]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nature 2002, 417, 664–667. [Google Scholar]
- Aguilera, O.; Fraga, M.F.; Ballestar, E.; Paz, M.F.; Herranz, M.; Espada, J.; García, J.M.; Muñoz, A.; Esteller, M.; González-Sancho, J.M. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 2006, 25, 4116–4121. [Google Scholar]
- Pendás-Franco, N.; García, J.M.; Peña, C.; Valle, N.; Pálmer, H.G.; Heinaniemi, M.; Carlberg, C.; Jiménez, B.; Bonilla, F.; Muñoz, A.; et al. DICKKOPF-4 is induced by TCF/β-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1α,25-dihydroxyvitamin D3. Oncogene 2008, 27, 4467–4477. [Google Scholar]
- Eelen, G.; Valle, N.; Sato, Y.; Rochel, N.; Verlinden, L.; De Clercq, P.; Moras, D.; Bouillon, R.; Muñoz, A.; Verstuyf, A. Superagonistic fluorinated vitamin D3 analogs stabilize helix 12 of the vitamin D receptor. Chem. Biol. 2008, 15, 1029–1034. [Google Scholar]
- Beildeck, M.E.; Islam, M.; Shah, S.; Welsh, J.; Byers, S.W. Control of TCF-4 expression by VDR and vitamin D in the mouse mammary gland and colorectal cancer cell lines. PLoS One 2009, 4, e7872. [Google Scholar] [CrossRef]
- Tang, W.; Dodge, M.; Gundapaneni, D.; Michnoff, C.; Roth, M.; Lum, L. A genome-wide RNAi screen for Wnt/β-catenin pathway components identifies unexpected roles for TCF transcription factors in cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 9697–9702. [Google Scholar]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2012, 26, 37–51. [Google Scholar]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar]
- Morrish, F.; Isern, N.; Sadilek, M.; Jeffrey, M.; Hockenbery, D.M. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene 2009, 28, 2485–2491. [Google Scholar]
- Toropainen, S.; Väisänen, S.; Heikkinen, S.; Carlberg, C. The down-regulation of the human MYC gene by the nuclear hormone 1α,25-dihydroxyvitamin D3 is associated with cycling of corepressors and histone deacetylases. J. Mol. Biol. 2010, 400, 284–294. [Google Scholar]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar]
- Salehi-Tabar, R.; Nguyen-Yamamoto, L.; Tavera-Mendoza, L.E.; Quail, T.; Dimitrov, V.; An, B.S.; Glass, L.; Goltzman, D.; White, J.H. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc. Natl. Acad. Sci. USA 2012, 109, 18827–18832. [Google Scholar]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar]
- Wend, P.; Runke, S.; Wend, K.; Anchondo, B.; Yesayan, M.; Jardon, M.; Hardie, N.; Loddenkemper, C.; Ulasov, I.; Lesniak, M.S.; et al. WNT10B/β-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. MBO Mol. Med. 2013, 5, 264–279. [Google Scholar] [Green Version]
- Pendás-Franco, N.; González-Sancho, J.M.; Suárez, Y.; Aguilera, O.; Steinmeyer, A.; Gamallo, C.; Berciano, M.T.; Lafarga, M.; Muñoz, A. Vitamin D regulates the phenotype of human breast cancer cells. Differentiation 2007, 75, 193–207. [Google Scholar]
- He, W.; Kang, Y.S.; Dai, C.; Liu, Y. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J. Am. Soc. Nephrol. 2011, 22, 90–103. [Google Scholar]
- Martínez-Moreno, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; Montes de Oca, A.; Estepa, J.C.; Canalejo, R.; Rodríguez-Ortiz, M.E.; Pérez-Martínez, P.; Aguilera-Tejero, E.; Canalejo, A.; et al. In vascular smooth muscle cells paricalcitol prevents phosphate-induced Wnt/β-catenin activation. Am. J. Physiol. Renal Physiol. 2012, 303, F1136–F1144. [Google Scholar]
- Ordóñez-Morán, P.; Larriba, M.J.; Pendás-Franco, N.; Aguilera, O.; González-Sancho, J.M.; Muñoz, A. Vitamin D and cancer: An update of in vitro and in vivo data. Front. Biosci. 2005, 10, 2723–2749. [Google Scholar]
- Deeb, K.K.; Trump, D.L.; Johnson, C.S. Vitamin D signalling pathways in cancer: Potential for anticancer therapeutics. Nat. Rev. Cancer 2007, 7, 684–700. [Google Scholar]
- Wali, R.K.; Khare, S.; Tretiakova, M.; Cohen, G.; Nguyen, L.; Hart, J.; Wang, J.; Wen, M.; Ramaswamy, A.; Joseph, L.; et al. Ursodeoxycholic acid and F6-D3 inhibit aberrant crypt proliferation in the rat azoxymethane model of colon cancer: Roles of cyclin D1 and E-cadherin. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1653–1662. [Google Scholar]
- Fichera, A.; Little, N.; Dougherty, U.; Mustafi, R.; Cerda, S.; Li, Y.C.; Delgado, J.; Arora, A.; Campbell, L.K.; Joseph, L.; et al. A vitamin D analogue inhibits colonic carcinogenesis in the AOM/DSS model. J. Surg. Res. 2007, 142, 239–245. [Google Scholar]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar]
- Huerta, S.; Irwin, R.W.; Heber, D.; Go, V.L.W.; Koeffler, H.P.; Uskokovic, M.R.; Harris, D.M. 1α,25-(OH)2D3 and its synthetic analogue decrease tumor load in the Apcmin mouse. Cancer Res. 2002, 62, 741–746. [Google Scholar]
- Xu, H.; Posner, G.H.; Stevenson, M.; Campbell, F.C. ApcMIN modulation of vitamin D secosteroid growth control. Carcinogenesis 2010, 31, 1434–1441. [Google Scholar]
- Irving, A.A.; Halberg, R.B.; Albrecht, D.M.; Plum, L.A.; Krentz, K.J.; Clipson, L.; Drinkwater, N.; Amos-Landgraf, J.M.; Dove, W.F.; DeLuca, H.F. Supplementation by vitamin D compounds does not affect colonic tumor development in vitamin D sufficient murine models. Arch. Biochem. Biophys. 2011, 515, 64–71. [Google Scholar]
- Yang, K.; Yang, W.; Mariadason, J.; Velcich, A.; Lipkin, M.; Augenlicht, L. Dietary components modify gene expression: Implications for carcinogenesis. J. Nutr. 2005, 135, 2710–2714. [Google Scholar]
- Yang, K.; Kurihara, N.; Fan, K.; Newmark, H.; Rigas, B.; Bancroft, L.; Corner, G.; Livote, E.; Lesser, M.; Edelmann, W.; et al. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008, 68, 7803–7810. [Google Scholar]
- Wang, D.; Peregrina, K.; Dhima, E.; Lin, E.Y.; Mariadason, J.M.; Augenlicht, L.H. Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 10272–10277. [Google Scholar]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Kállay, E.; Pietschmann, P.; Toyokuni, S.; Bajna, E.; Hahn, P.; Mazzucco, K.; Bieglmayer, C.; Kato, S.; Cross, H.S. Characterization of a vitamin D receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis 2001, 22, 1429–1435. [Google Scholar] [CrossRef]
- Kállay, E.; Bareis, P.; Bajna, E.; Kriwanek, S.; Bonner, E.; Toyokuni, S.; Cross, H.S. Vitamin D receptor activity and prevention of colonic hyperproliferation and oxidative stress. Food Chem. Toxicol. 2002, 40, 1191–1196. [Google Scholar] [CrossRef]
- Zheng, W.; Wong, K.E.; Zhang, Z.; Dougherty, U.; Mustafi, R.; Kong, J.; Deb, D.K.; Zheng, H.; Bissonnette, M.; Li, Y.C. Inactivation of the vitamin D receptor in APCmin/+ mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int. J. Cancer 2012, 130, 10–19. [Google Scholar] [CrossRef]
- Garland, C.F.; Garland, F.C. Do sunlight and vitamin D reduce the likelihood of colon cancer? Int. J. Epidemiol. 1980, 9, 65–71. [Google Scholar] [CrossRef]
- Stubbins, R.E.; Hakeem, A.; Núñez, N.P. Using components of the vitamin D pathway to prevent and treat colon cancer. Nutr. Rev. 2012, 70, 721–729. [Google Scholar] [CrossRef]
- Plum, L.A.; DeLuca, H.F. Vitamin D, disease and therapeutic opportunities. Nat. Rev. Drug Discov. 2010, 9, 941–955. [Google Scholar] [CrossRef]
- Ahearn, T.U.; Shaukat, A.; Flanders, W.D.; Rutherford, R.E.; Bostick, R.M. A randomized clinical trial of the effects of supplemental calcium and vitamin D3 on the APC/β-catenin pathway in the normal mucosa of colorectal adenoma patients. Cancer Prev. Res. 2012, 5, 1247–1256. [Google Scholar] [CrossRef]
- Cross, H.S.; Kállay, E.; Khorchide, M.; Lechner, D. Regulation of extrarenal synthesis of 1,25-dihydroxyvitamin D3-relevance for colonic cancer prevention and therapy. Mol. Aspects Med. 2003, 24, 459–465. [Google Scholar] [CrossRef]
- Larriba, M.J.; Muñoz, A. SNAIL vs. vitamin D receptor expression in colon cancer: Therapeutics implications. Br. J. Cancer 2005, 92, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Matusiak, D.; Murillo, G.; Carroll, R.E.; Mehta, R.G.; Benya, R.V. Expression of vitamin D receptor and 25-hydroxyvitamin D3-1α-hydroxylase in normal and malignant human colon. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 2370–2376. [Google Scholar] [CrossRef]
- Anderson, M.G.; Nakane, M.; Ruan, X.; Kroeger, P.E.; Wu-Wong, J.R. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother. Pharmacol. 2006, 57, 234–240. [Google Scholar] [CrossRef]
- Cross, H.S.; Bajna, E.; Bises, G.; Genser, D.; Kállay, E.; Pötzi, R.; Wenzl, E.; Wrba, F.; Roka, R.; Peterlik, M. Vitamin D receptor and cytokeratin expression may be progression indicators in human colon cancer. Anticancer Res. 1996, 16, 2333–2337. [Google Scholar]
- Evans, S.R.T.; Nolla, J.; Hanfelt, J.; Shabahang, M.; Nauta, R.J.; Shchepotin, I.B. Vitamin D receptor expression as a predictive marker of biological behavior in human colorectal cancer. Clin. Cancer Res. 1998, 4, 1591–1595. [Google Scholar]
- Peña, C.; García, J.M.; Silva, J.; García, V.; Rodríguez, R.; Alonso, I.; Millán, I.; Salas, C.; García de Herreros, A.; Muñoz, A.; et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: Clinicopathological correlations. Hum. Mol. Genet. 2005, 14, 3361–3370. [Google Scholar] [CrossRef]
- Larriba, M.J.; Bonilla, F.; Muñoz, A. The transcription factors Snail1 and Snail2 repress vitamin D receptor during colon cancer progression. J. Steroid Biochem. Mol. Biol. 2010, 121, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Fearon, E.R.; Weiss, S.J. Wnt-dependent regulation of the E-cadherin repressor Snail. J. Biol. Chem. 2005, 280, 11740–11748. [Google Scholar]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef]
- Ten Berge, D.; Koole, W.; Fuerer, C.; Fish, M.; Eroglu, E.; Nusse, R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 2008, 3, 508–518. [Google Scholar] [CrossRef]
- Wu, Z.-Q.; Brabletz, T.; Fearon, E.; Willis, A.L.; Hu, C.Y.; Li, X.-Y.; Weiss, S.J. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11312–11317. [Google Scholar]
- Mittal, M.K.; Myers, J.N.; Misra, S.; Bailey, C.K.; Chaudhuri, G. In vivo binding to and functional repression of the VDR gene promoter by SLUG in human breast cells. Biochem. Biophys. Res. Commun. 2008, 372, 30–34. [Google Scholar] [CrossRef]
- De Frutos, C.A.; Dacquin, R.; Vega, S.; Jurdic, P.; Machuca-Gayet, I.; Nieto, M.A. Snail1 controls bone mass by regulating Runx2 and VDR expression during osteoblast differentiation. EMBO J. 2009, 28, 686–696. [Google Scholar] [CrossRef]
- Bai, S.; Wang, H.; Shen, J.; Zhou, R.; Bushinsky, D.A.; Favus, M.J. Elevated vitamin D receptor levels in genetic hypercalciuric stone-forming rats are associated with downregulation of Snail. J. Bone Miner. Res. 2010, 25, 830–840. [Google Scholar]
- Yang, H.; Zhang, Y.; Zhou, Z.; Jiang, X.; Shen, A. Snail-1 regulates VDR signaling and inhibits 1,25(OH)-D3 action in osteosarcoma. Eur. J. Pharmacol. 2011, 670, 341–346. [Google Scholar] [CrossRef]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Peña, C.; García, J.M.; García, V.; Silva, J.; Domínguez, G.; Rodríguez, R.; Maximiano, C.; García de Herreros, A.; Muñoz, A.; Bonilla, F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 2006, 119, 2098–2104. [Google Scholar] [CrossRef]
- González-Sancho, J.M.; Aguilera, O.; García, J.M.; Pendás-Franco, N.; Peña, C.; Cal, S.; García de Herreros, A.; Bonilla, F.; Muñoz, A. The Wnt antagonist DICKKOPF-1 gene is a downstream target of β-catenin/TCF and is downregulated in human colon cancer. Oncogene 2005, 24, 1098–1103. [Google Scholar] [CrossRef]
- Rawson, J.B.; Sun, Z.; Dicks, E.; Daftary, D.; Parfrey, P.S.; Green, R.C.; Gallinger, S.; McLaughlin, J.R.; Wang, P.P.; Knight, J.A.; et al. Vitamin D intake is negatively associated with promoter methylation of the Wnt antagonist gene DKK1 in a large group of colorectal cancer patients. Nutr. Cancer 2012, 64, 919–928. [Google Scholar] [CrossRef]
- You, J.; Nguyen, A.V.; Albers, C.G.; Lin, F.; Holcombe, R.F. Wnt pathway-related gene expression in inflammatory bowel disease. Dig. Dis. Sci. 2008, 53, 1013–1019. [Google Scholar] [CrossRef]
- Matsui, A.; Yamaguchi, T.; Maekawa, S.; Miyazaki, C.; Takano, S.; Uetake, T.; Inoue, T.; Otaka, M.; Otsuka, H.; Sato, T.; et al. DICKKOPF-4 and -2 genes are upregulated in human colorectal cancer. Cancer Sci. 2009, 100, 1923–1930. [Google Scholar] [CrossRef]
- Kennell, J.A.; MacDougald, O.A. Wnt signaling inhibits adipogenesis through β-catenin-dependent and -independent mechanisms. J. Biol. Chem. 2005, 280, 24004–24010. [Google Scholar] [CrossRef]
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; Macdougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-bindingprotein α and peroxisome proliferator-activated receptor γ. J. Biol. Chem. 2007, 282, 14515–14524. [Google Scholar] [CrossRef]
- Fretz, J.A.; Zella, L.A.; Kim, S.; Shevde, N.K.; Pike, J.W. 1,25-Dihydroxyvitamin D3 regulates the expression of low-density lipoprotein receptor-related protein 5 via deoxyribonucleic acid sequence elements located downstream of the start site of transcription. Mol. Endocrinol. 2006, 20, 2215–2230. [Google Scholar] [CrossRef]
- Fretz, J.A.; Zella, L.A.; Kim, S.; Shevde, N.K.; Pike, J.W. 1,25-Dihydroxyvitamin D3 induces expression of the Wnt signaling co-regulator LRP5 via regulatory elements located significantly downstream of the gene’s transcriptional start site. J. Steroid Biochem. Mol. Biol. 2007, 103, 440–445. [Google Scholar] [CrossRef]
- Cianferotti, L.; Demay, M.B. VDR-mediated inhibition of DKK1 and SFRP2 suppresses adipogenic differentiation of murine bone marrow stromal cells. J. Cell Biochem. 2007, 101, 80–88. [Google Scholar] [CrossRef]
- Sakai, Y.; Kishimoto, J.; Demay, M.B. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J. Clin. Invest. 2001, 107, 961–966. [Google Scholar] [CrossRef]
- Cianferotti, L.; Cox, M.; Skorija, K.; Demay, M.B. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc. Natl. Acad. Sci. USA 2007, 104, 9428–9433. [Google Scholar] [CrossRef]
- Pálmer, H.G.; Anjos-Afonso, F.; Carmeliet, G.; Takeda, H.; Watt, F.M. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS One 2008, 3, e1483. [Google Scholar]
- Luderer, H.F.; Gori, F.; Demay, M.B. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. J. Biol. Chem. 2011, 286, 18444–18451. [Google Scholar] [CrossRef]
- Van Genderen, C.; Okamura, R.M.; Farinas, I.; Quo, R.G.; Parslow, T.G.; Bruhn, L.; Grosschedl, R. Development of several organs that require inductive epithelial-mesenchymal interactions is impaired in LEF-1-deficient mice. Genes Dev. 1994, 8, 2691–2703. [Google Scholar] [CrossRef]
- Niemann, C.; Owens, D.M.; Hülsken, J.; Birchmeier, W.; Watt, F.M. Expression of ΔNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development 2002, 129, 95–109. [Google Scholar]
- Bikle, D.D. The vitamin D receptor: A tumor suppressor in skin. Discov. Med. 2011, 11, 7–17. [Google Scholar]
- Jiang, Y.J.; Teichert, A.E.; Fong, F.; Oda, Y.; Bikle, D.D. 1α,25(OH)2-Dihydroxyvitamin D3/VDR protects the skin from UVB-induced tumor formation by interacting with the β-catenin pathway. J. Steroid Biochem. Mol. Biol. 2012, 136, 229–232. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Larriba, M.J.; González-Sancho, J.M.; Barbáchano, A.; Niell, N.; Ferrer-Mayorga, G.; Muñoz, A. Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells. Cancers 2013, 5, 1242-1260. https://doi.org/10.3390/cancers5041242
Larriba MJ, González-Sancho JM, Barbáchano A, Niell N, Ferrer-Mayorga G, Muñoz A. Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells. Cancers. 2013; 5(4):1242-1260. https://doi.org/10.3390/cancers5041242
Chicago/Turabian StyleLarriba, María Jesús, José Manuel González-Sancho, Antonio Barbáchano, Núria Niell, Gemma Ferrer-Mayorga, and Alberto Muñoz. 2013. "Vitamin D Is a Multilevel Repressor of Wnt/b-Catenin Signaling in Cancer Cells" Cancers 5, no. 4: 1242-1260. https://doi.org/10.3390/cancers5041242