Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol


Department of Chemical and Biochemical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(7), 726; https://doi.org/10.3390/catal10070726
Submission received: 28 May 2020
/
Revised: 22 June 2020
/
Accepted: 25 June 2020
/
Published: 29 June 2020
(This article belongs to the Special Issue Structure and Function of Ceria-Based Mixed Metal Oxides and Supported Transition Metal Oxide Catalysts)
Abstract
:Understanding the catalytic behavior of sulfated metal oxides has been the topic of several research studies in the past few decades. Their apparent super-acidic behavior has been correlated with the molecular structure of the surface sulfate species. Herein, we couple FTIR and Raman spectroscopies to study the molecular structural evolution of surface sulfate species on mixed metal hydroxides as well as calcined oxides. We show that on the surface of hydroxides, monodentate and possibly bidentate species are dominant, while for SnO2-rich samples, clusters of polymeric sulfate species may also be present. After calcination, sulfate species bind strongly on the surface of mixed oxides, and different configurations can be seen with a range of S=O functionalities of varying strength. Through comparison of the catalytic performance of all sulfate oxides in the tert-butylation of phenol, it was found that SnO2-rich samples show high TBA conversion, with monoalkylated phenols as the primary product.
1. Introduction
Solid acid materials, encompassing such materials as aluminosilicates and metal oxides, have received considerable attention over the past three decades for their promise in replacing traditional homogeneous acid catalysts. These homogenous acids present a number of problems, ranging from the difficulty of separation to corrosivity, that usually do not exist for heterogenous acid materials, which provide equal or greater activity while benefitting from generally mild reaction conditions and relatively facile methods of separation from reaction solutions [1,2,3]. Among solid acid materials, sulfated metal oxides represent a promising means towards achieving the activity necessary for catalyzing isomerization, alkylation, acylation, and oxidative dehydrogenation reactions.
Hino and Arata demonstrated the superacidity that is generated by sulfate-loading for a range of metal oxides by use of a Hammett indicator, while also reporting that these materials were active for butane isomerization [4,5]. Further studies elucidated that a significant portion of the potential associated with these materials stems from the numerous opportunities that exist to selectively tune reactivity through catalyst properties optimization during synthesis and post-treatment [3]. Tunable properties include the relative ratios of the Brønsted and Lewis acidity, the strength and quantity of the acidic sites, and such physical parameters as pore size and surface area. It is predicted that by altering these properties, the selectivity of the catalyst may be optimized towards reducing undesirable side-products that often occur on solid catalyst surfaces.
The most accessible means to alter reactivity stems from the selection of metal oxide support, which has been demonstrated to affect the strength of the Brønsted acidity [4]. Within this family of solid superacids, while tin oxide has been reported to exhibit the highest acidity without further doping, sulfated zirconia has been by far the most studied as well as applied sulfated metal oxide to date. However, the limited molecular level understanding as to the behavior of such catalysts under reaction conditions has stalled attempts to mitigate the deleterious effects of coking that have been observed thus far and hindered further application.
One route that has been investigated to increase the robustness of sulfated zirconia catalysts during reaction has been to dope the bulk structure with another metal oxide, creating a mixed metal oxide which exhibits improved chemical and structural properties than either metal oxide alone. This field has experienced rapid growth recently, as several authors have reported that enhanced reactivities as well as improved stabilities can be achieved through bulk doping. One potential route of stabilization stems from the formation of a mixed crystalline phase that is distinctly different from the parent oxides, as reported by Reddy et al. in their observation of ZrTiO4 after calcination of amorphous TiO2/ZrO2 [6]. Improved activity was also observed in Pt-doped ZrO2/SO4, although this is potentially attributed to the Pt-promoted hydrogenation of carbon deposits in the pores that would otherwise cause deactivation [7,8,9].
The surfaces of these bulk mixed metal oxides are of particular interest, as a discrepancy continues to exist between the current understanding of these interfaces and the conclusions that are drawn from long-range crystallographic data [10]. This is particularly important for sulfated materials, as the outermost surface composition may play a crucial role in determining structure and reactivity of these species. Many investigators have shown that mixed oxide surfaces are not simply terminations of the proposed bulk crystal structure, but rather preferentially enrich in one component to reduce surface energy [9,11]. Thoughtful and precise characterization is necessary to determine the possible implications of this effect on sulfated mixed metal oxides, as current theories regarding the Lewis and Brønsted acidity point towards the support-specific withdrawal of electron density by S=O. In the case that one metal is enriched on the surface, reactivity could tend towards that of a one-component oxide rather than true bulk behavior, or induction effects from neighboring species may lead to novel reactivities.
Research interest regarding the effects of surface composition on the activity of supported species is accompanied by questions on which surface molecular structures control catalytic activity for sulfated metal oxides. While numerous spectroscopic and theoretical investigations have been undertaken, the conclusions drawn rarely coincide and are occasionally contradictory [12,13,14,15,16,17]. Authors generally concur on the existence of three sulfate species, observed spectroscopically and thermogravimetrically across a number of support oxides: bidentate, tridentate, and polymeric sulfates; though the debate begins at the exact chelation structure of these species, as well as the conditions under which they are formed, particularly in regard to hydration.
Findings by Morterra et al. using IR spectroscopy suggest that the activity of sulfated zirconia is highest when the sample is mostly dehydrated, coinciding with the lowest Brønsted acidity according to pyridine adsorption [7]. This study was followed by another research article that questioned the superacidity of sulfated zirconia, instead attributing the activity of the dehydrated sulfated zirconia to a butene activation mechanism, which may result from Lewis acidity rather than Brønsted [18]. The results of these studies seem to suggest that the reactivity of sulfated metal oxides can be attributed to the electron deficiency of the support due to the sulfate species, rather than any superacidity caused by sulfur–oxygen bonds themselves.
This conclusion makes sulfated tin oxide a rather interesting system to study. Though receiving less attention than sulfated zirconium, Arata identified sulfated tin oxide as the strongest superacid of the metal oxides reported [4]. This has led to its application as a catalyst for liquid phase reactions, such as the dehydration of xylose and the production of biodiesel [19,20,21]. In the xylose dehydration reaction, sulfated tin oxide was more active than concentrated H2SO4, seemingly indicating a higher Brønsted acidity which would infer superacidic behavior. However, it was also found through CO adsorption that strong Lewis acid sites existed, which were believed to play a role in the dehydration as well.
Unveiling the molecular structure of sulfated species on the surface of various metal oxides has been a subject of intense research efforts in recent decades. Among the techniques that have been utilized, the importance of vibrational spectroscopies, such as Raman and transmission IR, is unquestionable. The use of ATR-FTIR spectroscopy, especially for the study of sulfated species in aqueous suspensions (in the presence of oxides), has proven to be an alternative characterization method, considering the improved resolution between the sulfate and oxide vibrational bands in the mid-infrared region.
Taking the state of the field into account and considering the gaps that exist in the collective understanding of the behavior of these materials, it is proposed herein that a bulk mixed tin and zirconium oxide provides an intriguing system to study the effects of support composition on sulfate reactivity and structure. Thus, this work takes aim at two topics that have received little attention in literature to this point: the characteristics—both chemical and physical—of intimately mixed bulk oxides of tin and zirconium at various ratios, and the variation of the predominant structure of surface sulfates on these materials over a range of environmental conditions.
2. Results and Discussion
2.1. Physicochemical Properties of SnO2/ZrO2 and S-SnO2/ZrO2 Mixed Metal Oxide Catalysts
Table 1 summarizes the properties of all synthesized mixed metal oxides; the measured specific surface areas along with available literature information on similar materials are shown for comparison. The BET specific surface areas of all sulfated mixed metal oxides were observed to be higher than those of the non-sulfated parent materials that were calcined under identical conditions. It is generally accepted that sulfated metal oxides show higher surface area than their non-sulfated counterparts because the dispersed surface sulfate species prevent sintering of metal oxide particles and hence reduce particle size, which in turn increases the surface area. The tabulated results generally indicate a good agreement with the broad trends reported in literature, that is, that sulfated zirconias tend to exhibit higher surface areas than sulfated tin oxides, and that in both cases sulfated oxides have higher surface areas than non-sulfated oxides. It can be seen that reported values for sulfated tin oxide and sulfated zirconium oxide indicate that sulfated zirconium oxide exhibits a higher surface area, consistent with what has been reported in other works [17,22,23,24,25,26,27]. Very few literature reports exist for direct comparison of the mixed oxides reported here; however, a comparison has been made for materials that concerning similar (but not identical) composition and/or sulfate process with this work. Herein, it can be seen that the aforementioned sulfation effect on the improved surface area is general and applicable, not only to single metal oxides, but also to mixed metal oxides, though to different extents, as shall be discussed later.
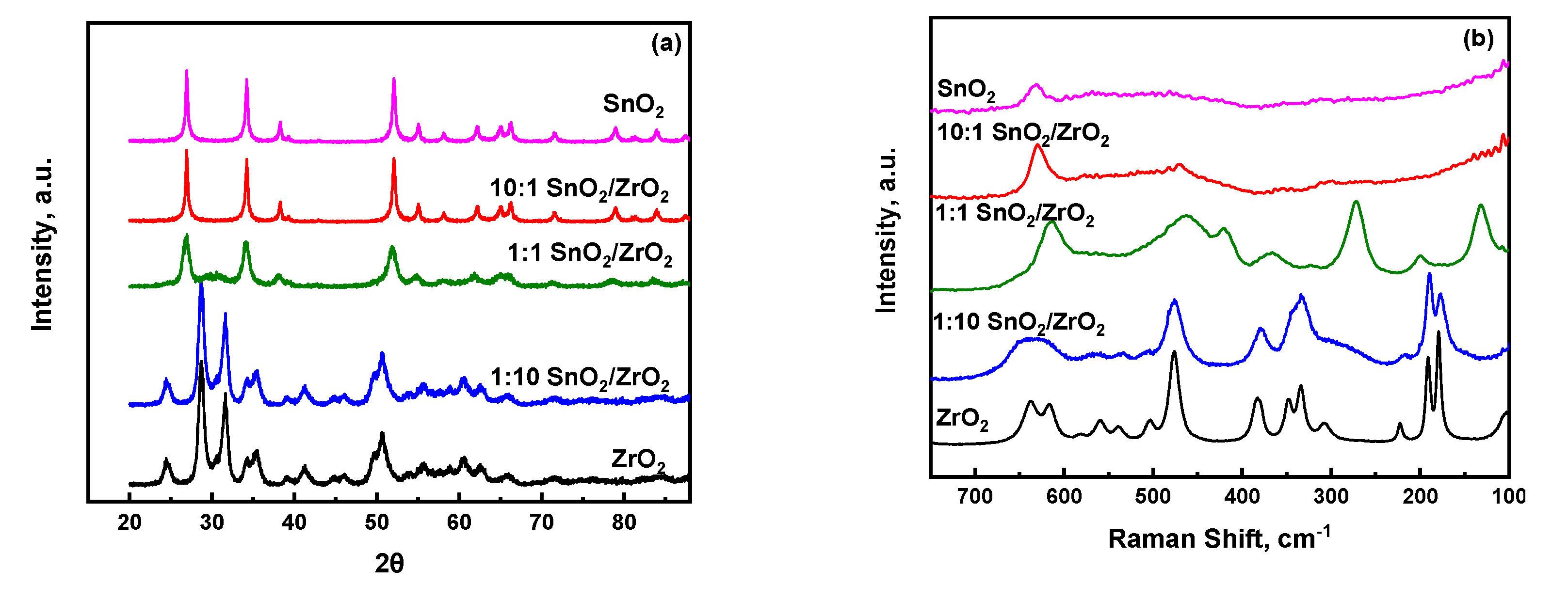
Information regarding the major crystalline phases detected from the XRD and Raman analysis are also shown in Table 1. Figure 1a shows that, for the parent ZrO2, all the dominant diffraction patterns pertain to monoclinic zirconia, characterized by the strong reflections of the (111) and (11) planes at 2 = 28.69°, 31.62°, respectively [28]. For the case of 1:10 SnO2/ZrO2, the XRD results reveal that the monoclinic phase of ZrO2 is maintained without any significant change in the position of the diffraction peaks. Upon further increase of the Sn content to 1:1 SnO2/ZrO2, the main diffraction peaks observed at 2 = 26.92°, 34.22° are assigned to the (110) and (101) planes of tetragonal SnO2 [29]. The only additional distinguishable band that can clearly be seen is at 30.22°, corresponding to the highest intensity peak of tetragonal zirconia [28], thus highlighting the potential of lattice matching between ZrO2 units and the tetragonal phase of SnO2. Further addition of SnO2 in both the 10:1 SnO2/ZrO2 and neat SnO2 materials results in nearly identical diffraction patterns, both of which clearly indicate the formation of tetragonal SnO2 with no crystalline ZrO2 phase able to be detected.
The structural trends indicated by XRD for the 1:1 SnO2/ZrO2 show a non-monotonic behavior. Whereas the 1:10 SnO2/ZrO2 and 10:1 SnO2/ZrO2 materials can be seen as being dominated by the most thermodynamically-stable phase of the high composition oxide, this argument cannot be leveraged to understand the diffraction pattern of 1:1 SnO2/ZrO2. In this case, though both oxides are present in equimolar amounts, the diffraction pattern is dominated by tetragonal tin oxide. However, upon further analysis and comparison with the 10:1 SnO2/ZrO2 and SnO2 materials, it can be seen that the tetragonal tin oxide peaks for 1:1 SnO2/ZrO2 are much broader, indicated the presence of less crystalline or smaller SnO2 domains. This is agreement with the specific surface areas reported in Table 1. Additionally, the diffraction pattern of 1:1 SnO2/ZrO2 exhibits a very broad and weak feature centered at approximately 30.22°, in a region where there is no reported feature for tetragonal SnO2. However, this region does resemble the highest intensity peak for tetragonal ZrO2. It is known that these broad background features can be attributed to amorphous or sub-XRD sensitive crystallites. Considering this, we propose that there are two mechanisms at play which explains the absence of ZrO2 features in the 1:1 SnO2/ZrO2 material: exchange and size effects. First, it has been reported that for a ternary SnO2/ZrO2/TiO2 system [30], ZrO2 exhibits significant ability to dissolve into SnO2 phase without significant disruption. Therefore, it is reasonable to believe that ZrO2 is able to, at some extent, exist within the tetragonal SnO2 without lattice disruption. Secondly, and more importantly, the ZrO2 is likely present as nanoscale ZrO2 crystallites smaller than 3 nm, which is accepted as the rule-of-thumb limit for when peak broadening causes XRD diffraction peaks to be indistinguishable from the background signal [31]. The Raman spectroscopic data in Figure 1b further corroborate these results. The Raman spectrum for pure ZrO2 is indicative of monoclinic ZrO2, as is the Raman spectrum of 1:10 SnO2/ZrO2, which is consistent with the XRD observations. However, whereas ZrO2 is invisible in the XRD of 1:1 SnO2/ZrO2, the Raman spectrum of this material exhibits a strong band located at ~270 cm−1, which is characteristic of tetragonal ZrO2. In this case, Raman is able to elucidate characteristic bands for a material that is unable to be observed in XRD. This behavior has been also reported for other systems where the studied tungstated zirconia catalysts present sub-nanometer crystallites that were observed by Raman and confirmed by HAADF-STEM, but otherwise invisible to XRD [31].
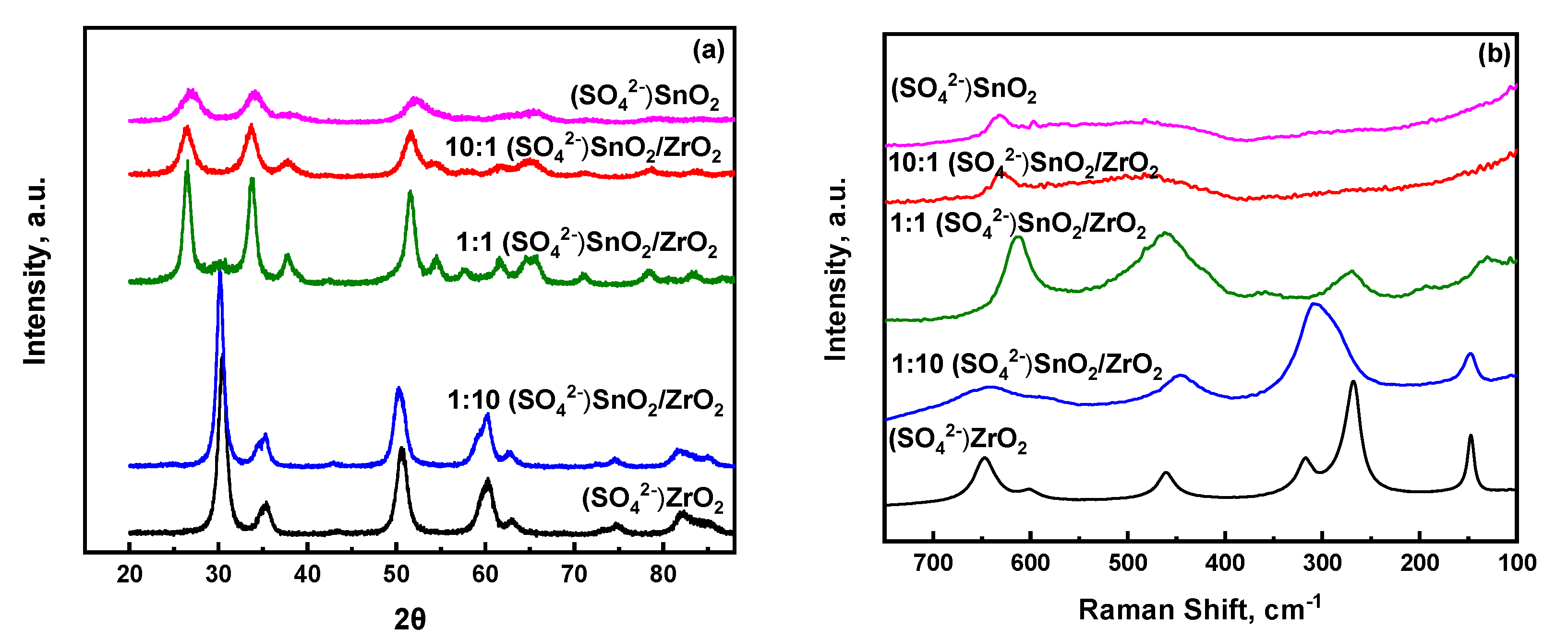
Figure 2a,b show the X-ray diffraction patterns and ambient Raman spectra, respectively, of all the sulfated metal and mixed metal oxides. The XRD peaks of S-ZrO2 are characteristic of the tetragonal crystalline phase of ZrO2, a result that is also corroborated by its corresponding Raman spectrum. Upon incorporation of a small amount of SnO2, i.e., 1:10 S-SnO2/ZrO2, the tetragonal crystalline phase of ZrO2 is maintained. However, it is worth mentioning here that a shift to lower angles is observed, indicating changes in the lattice parameters of the tetragonal ZrO2, possibly due to doping of Sn in the structure of ZrO2. In the Raman spectrum of 1:10 S-SnO2/ZrO2, the Raman bands of tetragonal ZrO2 have broadened significantly, an expected behaviour in highly doped metal oxide samples. Considering that Raman spectroscopy provides information from both the surface as well as bulk of the material tested, one cannot exclude the possibility that the surface sulfate species present result also in broadening the spectral features of the mixed metal oxides. At higher Sn content, Raman spectra of the sulfated mixed metal oxides appear to resemble those of their non-sulfated counterparts, thus underscoring that sulfation does not significantly alter their molecular structure. A closer look to the XRD spectra reveals that the 1:1 S-SnO2/ZrO2 catalyst is more crystalline than the 10:1 S-SnO2/ZrO2 and S-SnO2, thus suggesting that, at intermediate loadings, sulfation of the mixed metal oxides does not hamper (as was anticipated) a possible aggregation of the mixed metal oxide particles. This result is consistent with the specific surface areas reported in Table 1, where a change from 54 m2/g (1:1 SnO2/ZrO2) to 74 m2/g (1:1 S-SnO2/ZrO2) was observed. It is worth mentioning here that, at this intermediate composition, the surface area of the 1:1 S-SnO2/ZrO2 catalyst was the least among the sulfated mixed metal oxides. For higher Sn content, the changes in surface areas were much larger as compared to those of the non-sulfated metal oxides, which is consistent with the observed broadening in the XRD peaks upon sulfation, as shown in Figure 2a. Next, a comprehensive analysis from the perspective of the dispersed surface sulfate species is made by means of ambient and in-situ ATR-FTIR as well as Raman spectroscopies in an effort to enable molecular level understanding of their structural evolution upon temperature elevation (before and after calcination).
2.2. Insights into the Molecular Structure of Surface Sulfate Species on Mixed SnO2/ZrO2 Hydroxides
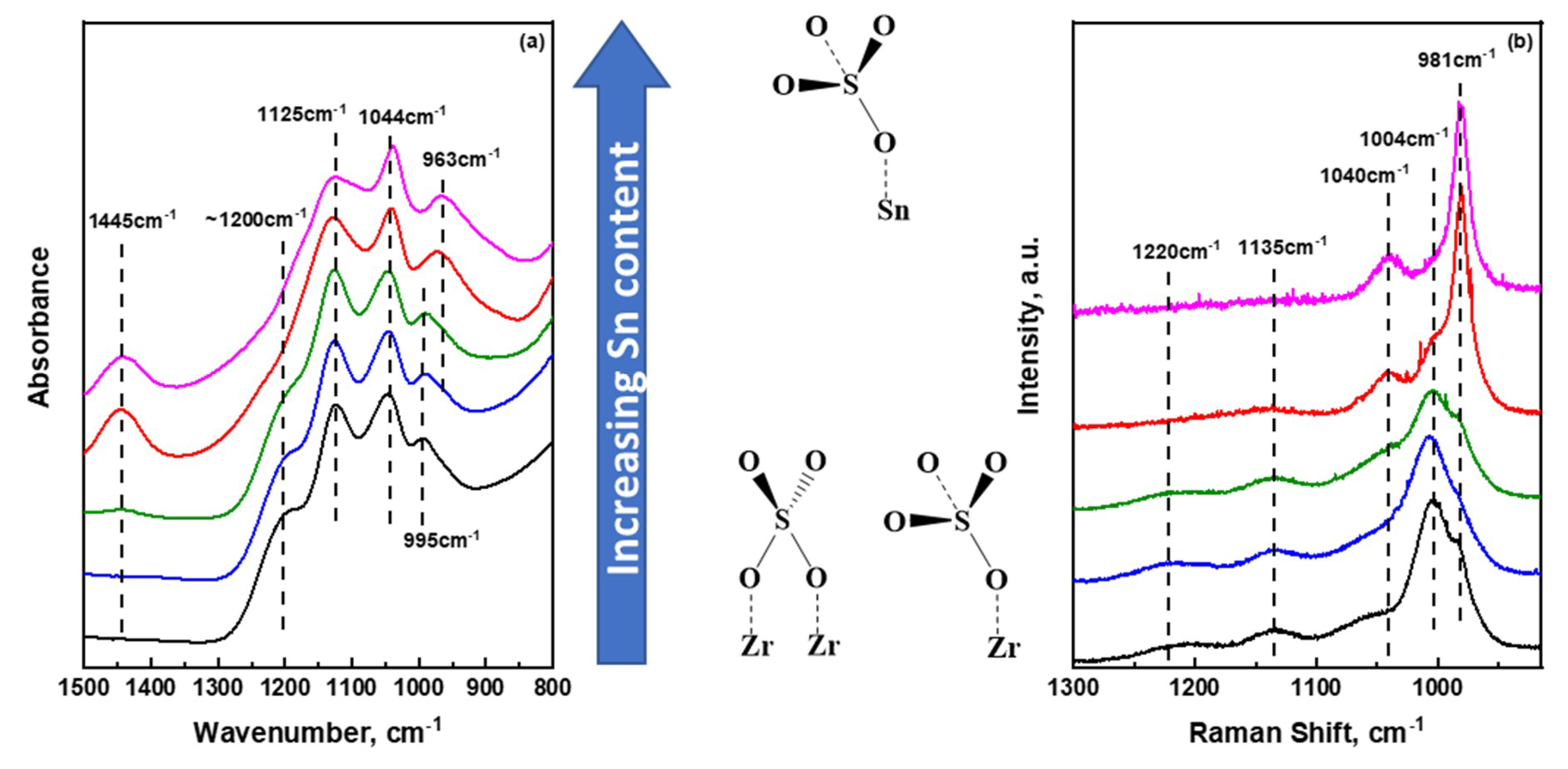
The sulfate species that are formed and evolved on the catalyst support have proven to play important roles in modulating the nature of acid sites present, which in turn affect catalytic reactivity. Hence, understanding the nature and molecular structure of these surface species is of utmost importance. It is also important to notice that efforts need also to be placed on following structural changes of the surface species occurring during the synthesis of final sulfate oxides. In Figure 3a, the ATR-FTIR spectra of all sulfate metal and mixed metal hydroxides (prior to any thermal treatment) are presented at the wavenumber regime where vibrational modes of bound sulfate units are expected. The ‘ideal-case’ of the free sulfate ion, belongs to a Td symmetry where a symmetric S–O stretching vibration (ν1) at 983 cm−1 and an asymmetric, triply degenerate S–O stretching vibration (ν3) at 1105 cm−1 are expected in the 800–1500 cm−1 range [32]. The former is IR inactive while the latter is IR active; both are Raman active. The data shown in Figure 3a show a very rich spectral envelope in vibrational bands which is associated with sulfate species with reduced symmetry, usually C3v and C2v, due to strong chemical bonding with the surface [33]. Upon reducing the symmetry of the sulfate species, the ν1 band becomes IR active and depends strongly on the metal cation that is coordinated with the sulfate species [34]. At the same time, the asymmetric ν3 vibrational mode splits into two or three bands, and as a result, multiple overlapping peaks, like those shown in Figure 3a, are observed. At a first glance, the spectra of all hydrous sulfated metal oxides look similar, indicating common surface species present on the different surfaces. In the case of S-/ZrO2, the ν1 mode is observed at 995 cm−1 while the ν3 degenerate mode after splitting give rise to the 1044, 1125, and 1200 cm−1 vibrational bands which are consistent with previous report [15]. Upon incorporating a small amount of SnO2 in the mixed support, no changes were observed. Increasing Sn content further, the symmetric ν1 mode is red shifted gradually to 970 and 963 cm−1 for the 10:1 S-SnO2/ZrO2 and S-SnO2 hydroxides, respectively. As has been highlighted by Hug [33], Lefevre [35] and reviewed also by Lavalley [36], the position of the ν1 vibrational mode is sensitive to the nature of surface sulfate species, i.e., monodentate (C3v symmetry), bidentate binuclear (C2v symmetry) and bidentate mononuclear (C2v symmetry). For the monodentate C3v, the ν1 mode is expected to appear at lower wavenumbers while the ν3 will appear as two bands. With a careful look at the Sn rich samples, the presence of only two bands at 1043 cm−1 and 1125 cm−1 is observed, while the 1200 cm−1 was observed only in Zr-rich samples. This result underscores that, for hydrous oxides, the prevalent surface sulfate species on Sn-rich hydroxides resemble monodentate units, while in ZrO2-rich samples, bidentate structures may dominate the surface [32,37]. These results are further corroborated by the Raman data shown in Figure 3b. For the sulfated zirconium hydroxide, a well-defined strong peak at 1004 cm−1, indicative of the ν1 mode, is observed. However, a shoulder at 985 cm−1 can be clearly seen that matches very well with the main symmetric ν1 peak of sulfated tin hydroxide (981 cm−1). The Raman results, in conjunction with the aforementioned ATR-IR discussion on the sulfate species, suggest that, although the main sulfate species in tin hydroxide appear to resemble solely monodentate configurations, a mixture of both monodentate and bidentate configurations are present in zirconium as well as mixed metal hydroxides. By observing the 981–985 and 1004 cm−1 bands, a clear transition can be seen from Zr-rich to Sn-rich mixed metal hydroxides.
Slightly distorted Td symmetry has also been reported for weakly bonding anions that are represented as ‘outer-sphere surface complexes. One can assume that since the present spectra shown in Figure 3 pertain to highly hydrated samples (hydroxides prior to calcination), there should exist the possibility of creating outer-sphere sulfate complexes where SO42− units are bound very weakly with the surface, thus inducing a slightly distorted Td symmetry. A study by Muller and Lefevre [38] on the ATR-IR spectroscopic signatures of several hydroxides in the presence of SO42− in aqueous environment has shown that the outer-sphere complexes do not strongly depend on the nature of the hydroxide present, and the only peak observed (ν3 mode) was at an almost identical position at ~1100 cm−1. The absence of a strong peak at 1100 cm−1 in the spectra reported herein exclude the possibility of this type of weakly bound sulfated species (reduced Td) on the surface of the single as well as mixed hydroxides.
In addition, a band at 1445 cm−1 in the IR spectra can be seen rising from 1:1 S-SnO2/ZrO2 hydroxide, with its intensity reaching a maximum in pure sulfated tin hydroxide. This band has been ascribed to the S=O stretching mode of polymeric-like sulfate units that can be present on the surface of Sn-rich hydroxides and is readily visible due to the high ionic character of the bond [39,40,41]. Therefore, one can hypothesize that with increase in tin content, the possibility of inducing polymeric sulfate species in the hydroxide framework increases. However, the position of this band is absent in spectroscopic studies concerning pyrosulfate oxo-complexes [42,43,44] and thus concrete evidence regarding the possibility of these type of species cannot be provided here. Due to the high surface area of zirconium hydroxide, sulfate species are well distributed on the surface (or bulk [17]) of the hydroxide, thus hampering the presence of potential polymeric species on Zr-rich samples. It should be mentioned that a peak at ~1400 cm−1 has been also reported to be characteristic of a ν3 split arising from the interaction of the sulfate group with dehydrated SnO2 [45]. However, Raman spectra of the hydrous tin oxide, shown in the Supplementary Materials, do not show any crystalline domains of SnO2 prior to calcination, thus excluding this possibility. Next, we discuss the structural evolution of the sulfate species upon increasing temperature.
2.3. Temperature Dependent Evolution of Surface Sulfate Species on Mixed Oxides
The generation of stable sulfate species on the surface of metal oxides usually requires treatment at relatively high temperature. It is also underscored in the open literature that there is an intrinsic sensitivity of SO42− adsorbed species to moisture which in turns causes an apparent reversible behavior between hydrated, slightly dehydrated, and fully dehydrated sulfate species on the surface of ZrO2 [7,15,39,41]. Similar observations have been reported for various metal oxide ions that have been anchored of the surface of TiO2, where reversibility has been correlated with the extent of hydroxylation of the support [46,47,48,49,50]. The structural evolution of sulfate species on the surface of ZrO2 has been the subject of several publications [12,15,51,52] and is thus compared here with the evolution observed in pure SnO2 as well as 1:1 S-SnO2/ZrO2. To the best of our knowledge, the evolution of sulfate species in mixed SnO2/ZrO2 samples has not been discussed, and only limited information is available regarding the sulfate units present at high temperature via in situ IR pyridine adsorption studies [25].
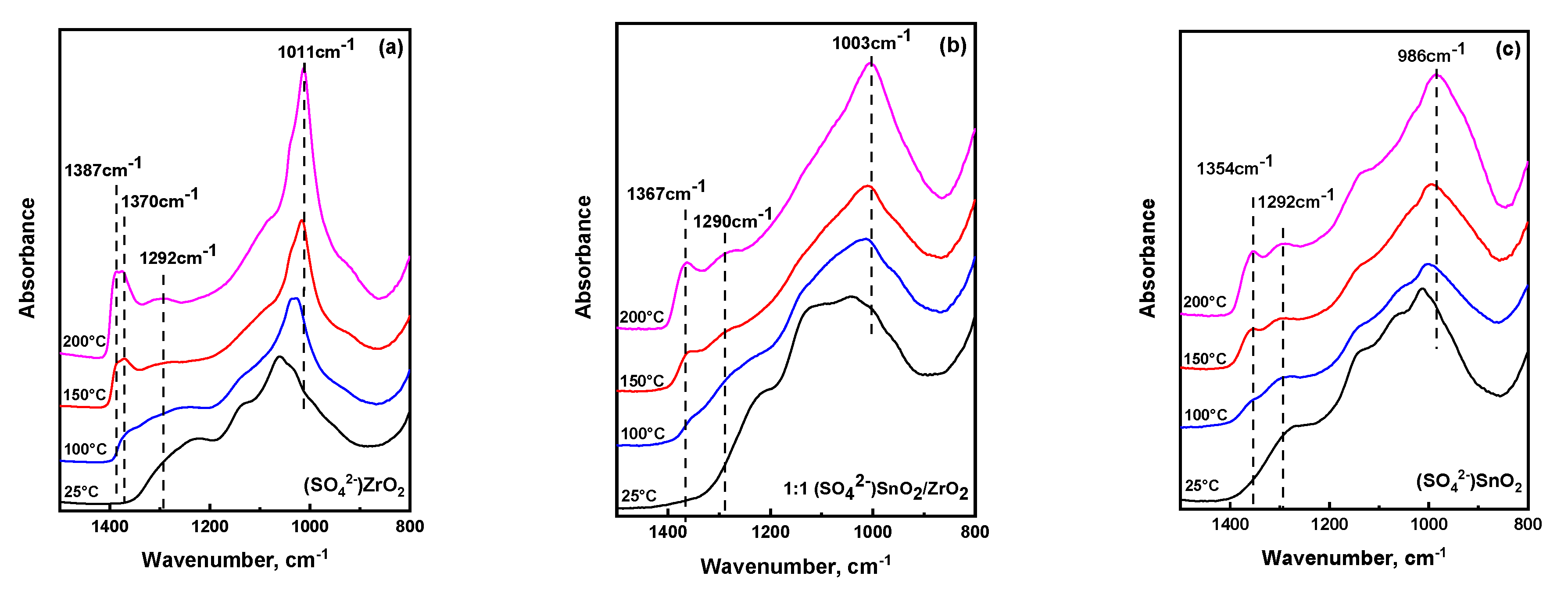
Figure 4 shows temperature dependent ATR-IR spectra of the S-ZrO2, 1:1 S-SnO2/ZrO2, and S-SnO2 up to 200 °C (close to the temperature limit of the ATR setup). The corresponding spectra for 1:10 S-SnO2/ZrO2 and 10:1 S-SnO2/ZrO2 catalysts are shown in the Supporting Information. The spectra shown in Figure 4 pertain to materials that have been calcined at 500–600 °C and then left at ambient conditions. By comparing the spectra of Figure 3a and Figure 4a–c at 25 °C, it is observed that, after calcination, the main peaks are shifted to higher wavenumbers due to the formation of covalent bonds. In addition, these spectral changes can be also ascribed to the much smaller extent of available surface hydroxyl groups (after calcination) that directly interact with sulfate species, thus weakening the S–O bonds and consequently shifting the bands to lower wavenumbers [41,53]. Upon heating, in the case of S–ZrO2, a gradual evolution of a band at 1010 cm−1 is observed, indicative of the S–O…Zr stretching vibration of anchoring bonds [7]. However, the broad character of this band underscores that a range of S-O bonds exists with varying length, strength, and bond order [39]. In addition, characteristic S=O stretches of sulfate species are exhibited within the 1360–1390 cm−1 range, with a clear contribution of two distinct peaks located at 1370 and 1387 cm−1; the 1387 cm−1 band evolves at the expense of that at 1370 cm−1 upon heating, which indicates a plausible transformation between sulfate species with similar molecular structure. The increase in intensity of the 1387 cm−1 S=O stretching band is accompanied by a gradual increase of a band around ~1290 cm−1, which has been previously ascribed to the possibility of the di-oxo O=S=O symmetric stretch of bidentate sulfate species [25,35,39] or the S=O stretching mode of S2O72− units adsorbed on a slightly hydrated surface [39,41]. At low temperatures, according to a DFT study, di-oxo type sulfate units may exist [40], but clear evidence for such species cannot be derived from these experiments.
For the Sn-rich sulfated oxides, the main S–O peak appears at much lower frequency than those in zirconia. This shift is ascribed to the consecutive anchoring of sulfate species to more Sn sites [54]. A careful look shows that, besides the main S–O band, there is a gradual shift to lower frequencies of the main S=O stretching mode of adsorbed sulfate species from 1387 cm−1 (S–ZrO2) to 1365 cm−1 and 1354 cm−1 for the 1:1 S-SnO2/ZrO2 and S-SnO2 catalysts, respectively. An earlier study on similar systems [25] has shown a similar shift of the S=O band upon Sn addition, however the extent of the shift was much less than what is reported herein. Herein, it is proposed that the reason for this discrepancy lies behind the synthesis of sulfated metal oxides. As opposed to the earlier study where a 0.1 M H2SO4 was used, the samples in this work were sulfated with 1 M H2SO4, since it has been suggested that successful sulfation of SnO2 usually requires higher acid concentration [54,55,56,57]. As a consequence, while also considering the very broad spectral envelope in this wavenumber regime, more sulfate species are expected to be anchored on Sn-rich sites, and a larger spectral contribution towards lower wavenumbers is expected. In addition, the position of the broad band centered around 1290 cm−1 was found to be independent of the Sn content in mixed metal oxides. However, the relative intensity of this peak increases significantly with the Sn content as compared to the main S=O stretching mode observed in the 1354–1390 cm−1 range. If the pair of the high frequency S=O (1354–1390 cm−1) and the low frequency (1290 cm−1) peaks was due to the asymmetric and symmetric stretches of di-oxo O=S=O units respectively, one should expect that their relative intensities should be consistent among different materials. Contrarily, intensity of the 1290 cm−1 band is observed to increase with increasing Sn content. These results exclude the possibility of the presence of di-oxo bidentate units in Sn rich oxides and suggest that the prevalent structure of sulfate species resembles polymeric S2O72− units.
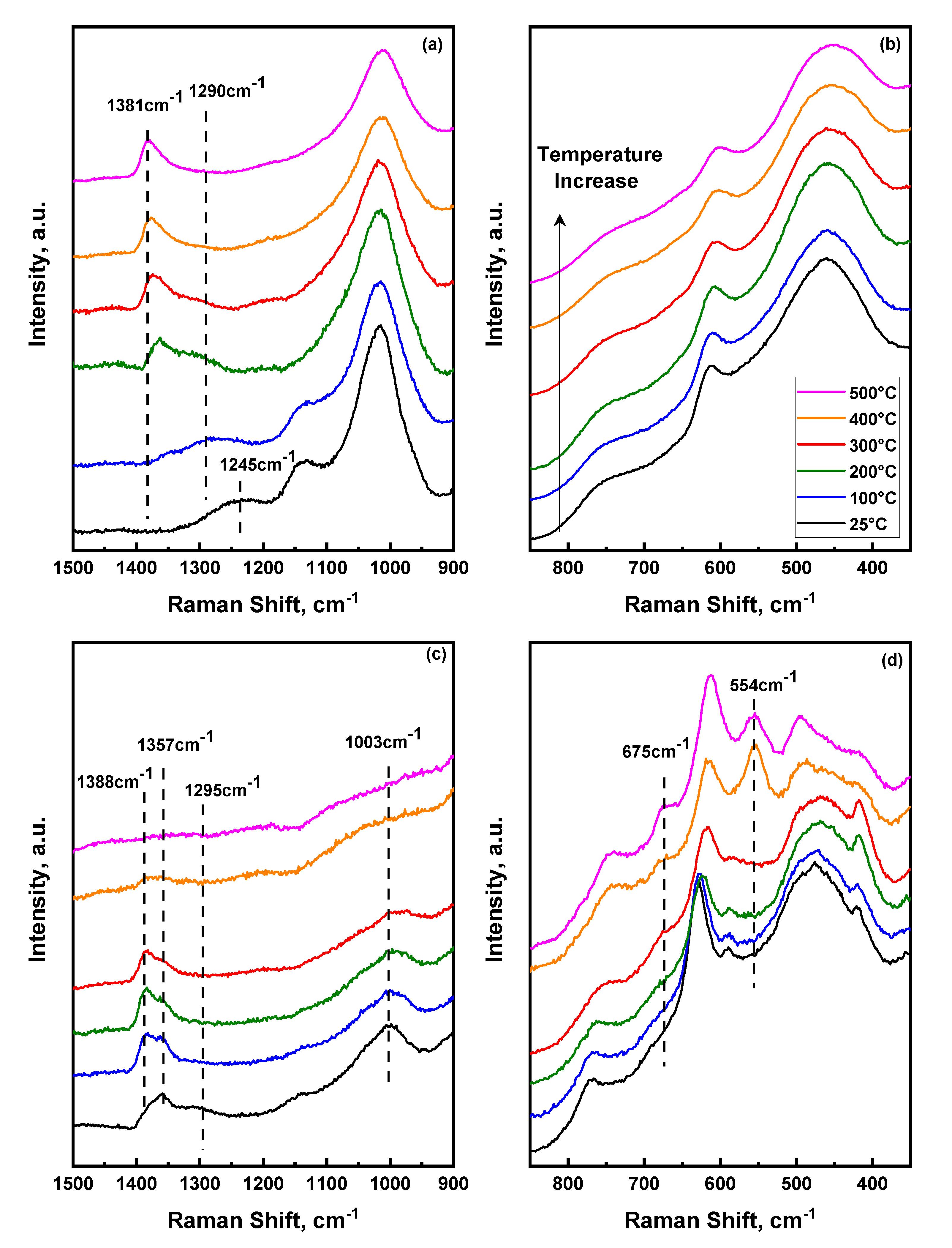
Additional insights into the structural changes that occur upon heating sulfated mixed metal oxides can be drawn by utilizing in-situ Raman spectroscopy. Raman spectra were acquired for the 1:1 S-SnO2/ZrO2 and S-SnO2 catalysts at 25–500 °C in the presence of air, as shown in Figure 5. Due to severe fluorescence (consistent with a previous report [58]), the corresponding Raman spectrum of (SO42−)ZrO2 was impossible to be collected with our experimental setup. In the 350–850 cm−1 range, the peaks in Figure 5b,d pertain to the vibrational modes of the main support, while in the 850–1500 cm−1 regime (Figure 5a,c), the observed peaks correspond to surface sulfate species. It is expected that the symmetric O=S=O stretch (hypothesized to appear at ~1290 cm−1) should be very strong in Raman spectroscopy while weak (as observed herein) in IR. The Raman spectra of both materials did not show any strong vibrational band at ~1290 cm−1 at any temperature. A closer look at the spectra of 1:1 S-SnO2/ZrO2 shows a broad band at 1245–1290 cm−1 that shifts with temperature up to 200 °C and disappears at higher temperatures. This observation agrees with the in-situ IR spectra discussed earlier, thus corroborating our suggestion of the presence of polymeric-like surface units at low temperatures. Upon heating to 200 °C, a clear peak at 1366 cm−1 is observed at an almost identical position to the peak observed in the ATR-IR spectra at the same temperature. This coincidence of the IR and Raman bands underscores the prevalence of mono-oxo type surface species with only one S=O stretching mode. Increasing temperature further up to 500 °C, the S=O stretching mode shifts to higher frequencies indicating possible transformation to tridentate mono-oxo species while the 1290 cm−1 of the dimeric sulfate species disappears, thus suggesting their low temperature stability. This result is consistent with previous Raman spectroscopic measurements where tridentate sulfate species were dominant on the surface of rich zirconia catalysts [59].
Focusing on the S-SnO2 catalyst, a broad band at ~1290 cm−1 can be seen at ambient temperature (previously ascribed to dimeric sulfates) that coincides with the corresponding ATR-IR presented earlier. In addition, a clear peak at 1357 cm−1 within the S=O regime is observed with an asymmetric high frequency shoulder located around 1388 cm−1. Interestingly, a similar peak on the corresponding FTIR spectra was not observed until heating the material up to 200 °C. Although the spectra have been taken with low laser power intensity (10–25% neutral density filter), in comparing the Raman and IR of the S-SnO2 catalyst, one could also argue that partial dehydration could occur due to laser induced heating of the surface, intrinsically affecting the vibrational fingerprint of moisture sensitive peaks, such as those of surface sulfate species. This effect was not observed in the case of the mixed metal oxides and could be ascribed to the tendency of SnO2 to be dehydrated due to its relatively low affinity for surface H2O molecules [60,61]. Upon heating to 300 °C, a clear transition between the 1357 cm−1 and 1388 cm−1 peaks is observed due to the higher degree of dehydration of the surface [61]. Both peaks lie within the expected S=O stretching regime. Interestingly, and contrary to what was observed in the 1:1 S-SnO2/ZrO2 material, further increase in the temperature to 400 °C revealed a dramatic decrease in all sulfate-related peaks, while at 500 °C, no peaks were observed for S–SnO2. At the same temperature range, significant changes were observed in the low frequency regime, where only vibrational modes of the support are present. The high temperature spectra of Figure 5d show the appearance of new bands located at 495, 554, and 675 cm−1. These vibrational modes have been observed before and ascribed to surface defects on SnO2 or to the formation of large nanoparticles that can be induced upon heating, doping, and/or surface dihydroxylation [62,63]. It is noteworthy that upon cooling and collecting the Raman spectrum of S–SnO2 at room temperature (Supporting Information), the sulfate species can be clearly seen again while the main peaks of SnO2 resemble those shown in Figure 5d. This result suggests that, at relatively high temperatures, S–SnO2 undergoes surface reconstruction that induces defects, while no surface sulfate species can be observed. The reasons behind the reversibility of the surface sulfate species on SnO2 cannot be unravelled from the endeavours of this work. However, future research on unveiling the molecular structure of sulfate species on complex mixed metal oxides will require the persistent use of advanced spectroscopic characterization techniques under dynamic conditions at a wide range of temperatures in conjunction with computational efforts to guide spectral interpretation.
2.4. Catalytic Activity of S-SnO2/ZrO2 on Tert-Butylation of Phenol
Among a plethora of building block molecules, phenol as well as phenol-derived compounds have an undoubtable importance [64]. Current production of alkylated phenolics, such as tert-butyl phenols (TBPs), relies mainly on the use of homogeneous mineral acids that raise environmental concerns. Although heterogeneous alternatives such as zeolites have been considered, the utilization of sulfated metal oxides on the tert-butylation of phenol is a topic that has received little attention. The synthesized sulfated catalysts were evaluated for the solvent-free alkylation of phenol with tert-butyl alcohol (TBA) in batch reactor.
Screening results of all catalysts at a phenol:TBA ratio equal to 10:1, 4 h reaction time and 120 °C are summarized in Table 2. The S-SnO2 catalyst shows the highest reactivity, where a TBA conversion of close to 90% is achieved, while the primary products are the mono-alkylated tert-butylated phenols, i.e., 2-TBP and 4-TBP. The 10:1 S-SnO2/ZrO2 presented a conversion/product distribution behavior similar to that of S-SnO2. ZrO2-rich catalysts exhibited lower conversion, while a significant amount of tert-butyl-ether (TBPE) was also observed, indicating promotion of the neutral alkylation pathway. Usually, TBPE is generated in significant concentrations via O-alkylation at early reaction times. This can be attributed to the lower activation energy for alkylation at the oxygen position as opposed directly to the carbon of the ring, thus making TBPE the kinetically favorable product [65]. The formation of TBPE has been reported to occur at relatively less acidic conditions. It is worth mentioning here that, although the addition of minute concentrations of SnO2 significantly improves the catalytic activity of 1:10 S-SnO2/ZrO2 (as compared to S-ZrO2), at intermediate composition, i.e., 1:1 S-SnO2/ZrO2, TBA conversion, as well as selectivity to 2-TBP and 4-TBP, decrease with a simultaneous increase in the selectivity to TBPE. This behavior can be in part attributed to the smaller surface area of the 1:1 S-SnO2/ZrO2 catalyst due to the presence of more crystalline mixed oxide. In addition, as shown in Figure 4 and Figure 5, at low temperatures—i.e., 100–200 °C—the S=O functionalities have not been developed since no intense peaks were observed in the 1350–1400 cm−1 spectral range. The absence of well-defined S=O bands can be associated with surface species that are still hydrated to some extent, thus inhibiting the formation of acidic functionalities. However, the possibility that changes in crystallinity upon SnO2 addition might also affect the dominant exposed facets of the individual oxides (or new mixed phase [30]), thus promoting the formation of surface species with different intrinsic reactivities, cannot be excluded. Further studies via advanced microscopy and spectroscopic characterization can provide invaluable insights into this research direction. Future endeavours toward evaluating kinetically these materials at different reaction conditions (e.g., temperature, phenol:TBA ratio, nature of solvent, etc.) can shed more light into the robustness of these materials at industrially relevant conditions. In addition, it is worth mentioning here that any attempts to evaluate the overall catalytic performance and stability of sulfated metal oxides should be done in light of thorough acidity measurements, where information regarding the Bronsted/Lewis acid sites ratio as well as availability and accessibility of these sites is investigated. Towards this direction, the utilization of advanced in situ spectroscopic techniques that can be coupled with NH3 (or pyridine) and 2,6-dimethylpyridine temperature programmed desorption can help to establish structure–function relationships at the molecular level.
3. Materials and Methods
3.1. Materials and Synthesis of Catalysts
Zirconium oxychloride (ZrOCl2.8H2O), Tin(IV) chloride pentahydrate (SnCl4.5H2O), aqueous ammonia solution (NH4OH) and concentrated sulfuric acid (5M H2SO4) were purchased from Sigma Aldrich (St. Louis, MO, USA) and used without further purification as precursors, precipitating agents and SO42− source respectively.
The mixed hydroxides of zirconium and tin were prepared by a co-precipitation method. In a typical experiment, both the precursor salts were separately dissolved in water. The concentration of the salts in aqueous solutions was maintained at 0.1M. Appropriate volumes of solution were stirred in two separate beakers with magnetic stirrers before mixing. The mixed solution containing ZrOCl2∙8H2O (0.1M) and SnCl4∙5H2O (0.1M) was hydrolyzed with ammonia solution via dropwise addition and vigorously stirred until the pH reached a value of 8. The resulting white precipitates were allowed to age for an hour and then washed thoroughly with deionized water to ensure that chloride ions from the precursors do not get incorporated into the hydroxides. The filtered solid was thereafter dried in a vacuum oven overnight at 85 °C.
For the synthesis of sulfated mixed metal oxides, the dried hydroxides were mixed with appropriate amount of 1 M H2SO4 to ensure anchoring of sulfate species on the surface. The sample was subsequently extracted via washing with deionized water under vacuum filtration and then dried in a vacuum oven overnight at 85 °C. Finally, the oven-dried sulfated hydrous tin-zirconium hydroxides were calcined for 3 h at 600 °C (or 500 °C for the sulfated and non-sulfated hydrous tin oxides) with a ramp rate of 2 °C/min in the presence of air flow of 100 mL/min. The various samples reported here are denoted as X:Y SnO2/ZrO2 for the non-sulfated catalysts or X:Y S-SnO2/ZrO2 for the sulfated catalysts, where X:Y represents the molar ratio of SnO2/ZrO2. S is used to denote sulfation treatment for the reported materials only and does not represent any structural or stoichiometric information about the sulfates
3.2. Physicochemical Characterization
BET surface area measurements were conducted by using a Quantachrome Autosorb-I system (Boynton Beach, FL, USA). In a typical measurement, 200 mg of catalyst was loaded into a BET tube and degassed at 300 °C for 3 h prior to BET analysis in order to completely remove chemisorbed water from sample surface. A total of 13 points were collected for each measurement within the 0.05 to 0.3 P/P0 range with 0.02 increment.
XRD patterns of all the materials of this work were collected with a PANalytical Philips X’Pert X-ray diffractometer (Almelo, The Netherlands) to determine crystallinity and phase composition changes. The XRD instrument is equipped with a CuKα source at 40 kV and 40 mA and angular incidence 2θ between 20° and 90° with 0.05° step and 4.0 s/step. Phase composition was analysed by whole pattern fitting (WPF) refinement 2-phase analysis with relative error R % targeted below 15%. Silicon was used as an external standard reference to determine possible peak shifts.
Ex situ, as well as in situ, Raman spectra of all samples were collected by utilizing a Horiba Scientific LabRam HR Evolution Raman spectrometer (HYJ, Arras, France) cooled with a Synapse CCD detector (−70 °C). A 532 nm solid-state laser line was used (80 mw) was directed on the sample and focused by using a 10X long working distance objective. The power of the laser was kept at a low value to avoid overheating by using a neutral density filter (10–25%). The acquisition time was 30 s with a total of twenty accumulations on average. FTIR spectra were collected using Nicolet iS50 FT-IR spectrometer (Thermo-Fisher, Waltham, MA, USA) equipped with DLaTGS and MCT-A detectors. The FTIR was connected to a Parker-Balston 75–45 purge unit to provide a purified purge gas and air bearing gas from compressed air. A Specac ATR high-performance single reflection monolithic diamond ATR was utilized for the collection of all infrared spectra. All spectra have been baseline corrected according to previous reported methods [66,67,68,69] and normalized with respect to the total area of each spectrum.
3.3. Catalytic Measurements
Tert-butylation of phenol was carried out in batch using a 15 mL pressure vessel from Ace Glass Incorporated (Vineland, NJ, USA), equipped with a thermowell to accommodate a thermocouple for precise control of reaction temperature. In a typical reaction, a mixture of tert-butyl alcohol (TBA) and phenol in a 1:10 molar ratio, respectively, was added to the reaction vessel with an appropriate stir bar; the reactions were carried out in solvent-free conditions. Approximately 0.313 g of catalyst, corresponding to 3 wt % catalyst loading, was carefully added to the vessel, which was subsequently sealed tightly with the provided bushing and O-ring. A thermocouple was placed in the thermowell with silicon oil; the vessel was submerged into an oil bath on top of a heating plate. The reaction was then carried out with 1050 rpm mixing at 120 °C for 4 h. Initial samples were taken prior to addition of catalyst and analyzed by gas chromatography (Agilent Technologies 7890B GC System, Palo Alto, CA, USA), equipped with a flame ionization detector (FID) and capillary column (HP-5, 30 m × 0.320 mm × 0.25 μm), to confirm initial concentration. Toluene was used as a solvent throughout GC analysis. After reaction, the mixture underwent centrifugation to separate the liquid products from solid catalyst particles; final samples were subsequently taken to confirm final concentration and product distribution by GC. TBA conversion as well as product distribution has been evaluated as .
4. Conclusions
In this work, focus has been given to understanding the molecular structural implications of surface sulfate species upon changing the composition of SnO2/ZrO2 mixtures. By utilizing FTIR spectroscopy, it was found that the surface sulfate species on mixed hydroxides (prior to any calcination) strongly depend on the nature of the hydroxide, and their structure lies between the formation of monodentate to bidentate units. For SnO2-rich samples, the existence of small polymeric species cannot be excluded. By coupling Raman and FTIR spectroscopy for the evaluation of calcined materials, the temperature-dependent evolution of surface species was investigated, where a transition between bidentate to tridentate species was found for SnO2-rich samples at high temperature. The terminal S=O peak varies significantly as a function of the nature of the support (sulfate species anchored on Sn or Zr) which is suggested to further influence the catalytic reactivity of the studied materials. The sulfated SnO2-rich samples present higher reactivity than ZrO2-rich samples for the tert-butylation of phenol, a result which is consistent with reported acidity measurements for sulfated SnO2 catalysts.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/7/726/s1, Figure S1: Raman spectra of (a) precursor solution, hydrous zirconia and calcined ZrO2 and (b) precursor solution, tin hydroxide and calcined SnO2; Figure S2: Temperature dependent ATR-IR spectra of the (a) 1:10 (SO42−)SnO2/ZrO2, and (b) 10:1 (SO42−)SnO2/ZrO2, up to 200 °C. Spectral resolution was set to 4 cm−1 while 32 accumulations have been used to increase signal/noise ratio; Figure S3: Temperature dependent Raman spectra of the (SO42−)SnO2 up to 500 °C. The blue spectrum pertains to room temperature Raman after heat treatment.
Author Contributions
Conceptualization, G.T.; Methodology, S.A., J.M., and G.T.; Data curation, J.M. and G.T.; Writing—original draft preparation, J.M., S.A., A.Z., and G.T.; Writing—review and editing J.M., A.Z., and G.T.; Supervision, G.T.; Funding acquisition, G.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the American Chemical Society, Petroleum Research Fund (Doctoral New Investigator Award 58542-DNI5) and startup funds granted to George Tsilomelekis.
Acknowledgments
The authors would like to thank A. Neimark for helping with the BET measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tanabe, K.; Holderich, W.F. Industrial application of solid acid-base catalysts. Appl. Catal. A Gen. 1999, 181, 399–434. [Google Scholar] [CrossRef]
- Okuhara, T. Water-tolerant solid acid catalysts. Chem. Rev. 2002, 102, 3641–3665. [Google Scholar] [CrossRef] [PubMed]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Arata, K.; Matsuhashi, H.; Hino, M.; Nakamura, H. Synthesis of solid superacids and their activities for reactions of alkanes. Catal. Today 2003, 81, 17–30. [Google Scholar] [CrossRef]
- Arata, K.; Hino, M. Preparation of Superacids by Metal-Oxides and Their Catalytic Action. Mater Chem. Phys. 1990, 26, 213–237. [Google Scholar] [CrossRef]
- Reddy, B.M.; Sreekanth, P.M.; Yamada, Y.; Xu, X.; Kobayashi, T. Surface characterization of sulfate, molybdate, and tungstate promoted TiO2-ZrO2 solid acid catalysts by XPS and other techniques. Appl. Catal. A Gen. 2002, 228, 269–278. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Pinna, F.; Signoretto, M.; Strukul, G. On the Acid-Catalyzed Isomerization of Light Paraffins over a Zro2/So4 System: The Effect of Hydration. J. Catal. 1994, 149, 181–188. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Signoretto, M. On the role of the calcination step in the preparation of active (superacid) sulfated zirconia catalysts. Catal. Lett. 1996, 41, 101–109. [Google Scholar] [CrossRef]
- Signoretto, M.; Pinna, F.; Strukul, G.; Cerrato, G.; Morterra, C. Platinum promoted zirconia-sulfate catalysts: One-pot preparation, physical properties and catalytic activity. Catal. Lett. 1996, 36, 129–133. [Google Scholar] [CrossRef]
- Wachs, I.E.; Routray, K. Catalysis Science of Bulk Mixed Oxides. ACS Catal. 2012, 2, 1235–1246. [Google Scholar] [CrossRef]
- Rosenberg, D.J.; Coloma, F.; Anderson, J.A. Modification of the acid properties of silica-zirconia aerogels by in situ and ex situ sulfation. J. Catal. 2002, 210, 218–228. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.C.; Morrow, B.A. An Infrared Study of Sulfated Zirconia. Mater. Chem. Phys. 1988, 19, 147–156. [Google Scholar] [CrossRef]
- Jin, T.; Yamaguchi, T.; Tanabe, K. Mechanism of Acidity Generation on Sulfur-Promoted Metal-Oxides. J. Phys. Chem. 1986, 90, 4794–4796. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Jin, T.; Tanabe, K. Structure of Acid Sites on Sulfur-Promoted Iron-Oxide. J. Phys. Chem. 1986, 90, 3148–3152. [Google Scholar] [CrossRef]
- Babou, F.; Coudurier, G.; Vedrine, J.C. Acidic Properties of Sulfated Zirconia: An Infrared Spectroscopic Study. J. Catal. 1995, 152, 341–349. [Google Scholar] [CrossRef]
- Saur, O.; Bensitel, M.; Saad, A.B.M.; Lavalley, J.C.; Tripp, C.P.; Morrow, B.A. The Structure and Stability of Sulfated Alumina and Titania. J. Catal. 1986, 99, 104–110. [Google Scholar] [CrossRef]
- Ward, D.A.; Ko, E.I. One-Step Synthesis and Characterization of Zirconia-Sulfate Aerogels as Solid Superacids. J. Catal. 1994, 150, 18–33. [Google Scholar] [CrossRef]
- Tabora, J.E.; Davis, R.J. On the superacidity of sulfated zirconia catalysts for low-temperature isomerization of butane. J. Am. Chem. Soc. 1996, 118, 12240–12241. [Google Scholar] [CrossRef]
- Suzuki, T.; Yokoi, T.; Otomo, R.; Kondo, J.N.; Tatsumi, T. Dehydration of xylose over sulfated tin oxide catalyst: Influences of the preparation conditions on the structural properties and catalytic performance. Appl. Catal. A Gen. 2011, 408, 117–124. [Google Scholar] [CrossRef]
- Furuta, S.; Matsuhashi, H.; Arata, K. Biodiesel fuel production with solid superacid catalysis in fixed bed reactor under atmospheric pressure. Catal. Commun. 2004, 5, 721–723. [Google Scholar] [CrossRef]
- Furuta, S.; Matsuhashi, H.; Arata, K. Catalytic action of sulfated tin oxide for etherification and esterification in comparison with sulfated zirconia. Appl Catal. A Gen. 2004, 269, 187–191. [Google Scholar] [CrossRef]
- Varala, R.; Narayana, V.; Kulakarni, S.R.; Khan, M.; Alwarthan, A.; Adil, S.F. Sulfated tin oxide (STO)—Structural properties and application in catalysis: A review. Arab. J. Chem. 2016, 9, 550–573. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.I.; El-Hakam, S.A.; Khder, A.S.; El-Yazeed, W.S.A. Nanostructure sulfated tin oxide as an efficient catalyst for the preparation of 7-hydroxy-4-methyl coumarin by Pechmann condensation reaction. J. Mol. Catal. A Chem. 2013, 366, 99–108. [Google Scholar] [CrossRef]
- Wang, G.W.; Hattori, H.; Tanabe, K. Acid-Base and Catalytic Properties of ZrO2-SnO2. Bull. Chem. Soc. Jpn. 1983, 56, 2407–2410. [Google Scholar] [CrossRef]
- Patel, A.; Coudurier, G.; Essayem, N.; Vedrine, J.C. Effect of the addition of Sn to zirconia on the acidic properties of the sulfated mixed oxide. J. Chem. Soc. Faraday Trans. 1997, 93, 347–353. [Google Scholar] [CrossRef]
- Chen, W.H.; Ko, H.H.; Sakthivel, A.; Huang, S.J.; Liu, S.H.; Lo, A.Y.; Tsai, T.C.; Liu, S.B. A solid-state NMR, FT-IR and TPD study on acid properties of sulfated and metal-promoted zirconia: Influence of promoter and sulfation treatment. Catal. Today 2006, 116, 111–120. [Google Scholar] [CrossRef]
- Stichert, W.; Schuth, F.; Kuba, S.; Knozinger, H. Monoclinic and tetragonal high surface area sulfated zirconias in butane isomerization: CO adsorption and catalytic results. J. Catal. 2001, 198, 277–285. [Google Scholar] [CrossRef]
- Occhiuzzi, M.; Cordischi, D.; Dragone, R. Manganese ions in the monoclinic, tetragonal and cubic phases of zirconia: An XRD and EPR study. Phys. Chem. Chem. Phys. 2003, 5, 4938–4945. [Google Scholar] [CrossRef]
- Jiang, L.H.; Sun, G.Q.; Zhou, Z.H.; Sun, S.G.; Wang, Q.; Yan, S.Y.; Li, H.Q.; Tian, J.; Guo, J.S.; Zhou, B.; et al. Size-controllable synthesis of monodispersed SnO2 nanoparticles and application in electrocatalysts. J. Phys. Chem. B 2005, 109, 8774–8778. [Google Scholar] [CrossRef]
- Wilson, G.; Glasser, F.P. Solid-Solution in the ZrO2-Sno2-TiO2 System. Br. Ceram. Trans. J. 1989, 88, 69–74. [Google Scholar]
- Zhou, W.; Soultanidis, N.; Xu, H.; Wong, M.S.; Neurock, M.; Kiely, C.J.; Wachs, I.E. Nature of Catalytically Active Sites in the Supported WO3/ZrO2 Solid Acid System: A Current Perspective. ACS Catal. 2017, 7, 2181–2198. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, K. Applications in Inorganic Chemistry. In Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2008; pp. 149–354. [Google Scholar] [CrossRef]
- Hug, S.J. In situ Fourier transform infrared measurements of sulfate adsorption on hematite in aqueous solutions. J. Colloid. Interface Sci. 1997, 188, 415–422. [Google Scholar] [CrossRef]
- Ben Mabrouk, K.; Kauffmann, T.H.; Aroui, H.; Fontana, M.D. Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions. J. Raman Spectrosc. 2013, 44, 1603–1608. [Google Scholar] [CrossRef]
- Lefevre, G. In situ Fourier-transform infrared spectroscopy studies of inorganic ions adsorption on metal oxides and hydroxides. Adv. Colloid. Interface 2004, 107, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Lavalley, J.C. Infrared spectrometric studies of the surface basicity of metal oxides and zeolites using adsorbed probe molecules. Catal. Today 1996, 27, 377–401. [Google Scholar] [CrossRef]
- Tabora, J.E.; Davis, R.J. Structure of Fe, Mn-Promoted Sulfated Zirconia Catalyst by X-ray and in Absorption Spectroscopies. J. Chem. Soc. Faraday Trans. 1995, 91, 1825–1833. [Google Scholar] [CrossRef]
- Muller, K.; Lefevre, G. Vibrational Characteristics of Outer-Sphere Surface Complexes: Example of Sulfate Ions Adsorbed onto Metal (Hydr)oxides. Langmuir 2011, 27, 6830–6835. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.X.; Wang, A.Q.; Wachs, I.E.; Baltrusaitis, J. Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production. Appl. Catal. A Gen. 2019, 572, 210–225. [Google Scholar] [CrossRef]
- Haase, F.; Sauer, J. The surface structure of sulfated zirconia: Periodic ab initio study of sulfuric acid adsorbed on ZrO2(101) and ZrO2(001). J. Am. Chem. Soc. 1998, 120, 13503–13512. [Google Scholar] [CrossRef]
- Klose, B.S.; Jentoft, F.C.; Schlogl, R. In situ diffuse-reflectance infrared spectroscopic investigation of promoted sulfated zirconia catalysts during n-butane isomerization. J. Catal. 2005, 233, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Kalampounias, A.G.; Tsilomelekis, G.; Berg, R.W.; Boghosian, S. Molybdenum(VI) Oxosulfato Complexes in MoO3-K2S2O7-K2SO4 Molten Mixtures: Stoichiometry, Vibrational Properties, and Molecular Structures. J. Phys. Chem. A 2012, 116, 8861–8872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalampounias, A.G.; Tsilomelekis, G.; Boghosian, S. Liquid phase dynamics of molten M2S2O7 (M = K, Cs): A temperature dependent Raman spectroscopic study. Vib. Spectrosc. 2013, 65, 66–73. [Google Scholar] [CrossRef]
- Kalampounias, A.G.; Tsilomelekis, G.; Boghosian, S. Molten and glassy tellurium(IV) oxosulfato complexes in the TeO2-K2S2O7 system studied by Raman spectroscopy: Stoichiometry, vibrational properties and molecular structure. Vib. Spectrosc. 2018, 97, 85–90. [Google Scholar] [CrossRef]
- Dong, J.L.; Li, X.H.; Zhao, L.J.; Xiao, H.S.; Wang, F.; Guo, X.; Zhang, Y.H. Raman observation of the interactions between NH4+, SO42-, and H2O in supersaturated (NH4)(2)SO4 droplets. J. Phys. Chem. B 2007, 111, 12170–12176. [Google Scholar] [CrossRef]
- Andriopoulou, C.; Boghosian, S. Heterogeneity of deposited phases in supported transition metal oxide catalysts: Reversible temperature-dependent evolution of molecular structures and configurations. Phys. Chem. Chem. Phys. 2018, 20, 1742–1751. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Tribalis, A.; Kalampounias, A.G.; Boghosian, S.; Panagiotou, G.D.; Bourikas, K.; Kordulis, C.; Lycourghiotis, A. Temperature-dependent evolution of molecular configurations of oxomolybdenum species on MoO3/TiO2 catalysts monitored by in situ Raman spectroscopy. Stud. Surf. Sci. Catal. 2010, 175, 613–616. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Boghosian, S. On the configuration, molecular structure and vibrational properties of MoOx sites on alumina, zirconia, titania and silica. Catal. Sci. Technol. 2013, 3, 1869–1888. [Google Scholar] [CrossRef]
- Tribalis, A.; Panagiotou, G.D.; Tsilomelekis, G.; Kalampounias, A.G.; Bourikas, K.; Kordulis, C.; Boghosian, S.; Lycourghiotis, A. Temperature-Dependent Evolution of the Molecular Configuration of Oxo-Tungsten(VI) Species Deposited on the Surface of Titania. J. Phys. Chem. C 2014, 118, 11319–11332. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Panagiotou, G.D.; Stathi, P.; Kalampounias, A.G.; Bourikas, K.; Kordulis, C.; Deligiannakis, Y.; Boghosian, S.; Lycourghiotis, A. Molybdena deposited on titania by equilibrium deposition filtration: Structural evolution of oxo-molybdenum(VI) sites with temperature. Phys. Chem. Chem. Phys. 2016, 18, 23980–23989. [Google Scholar] [CrossRef]
- Anderson, B.G.; Dang, Z.; Morrow, B.A. Silica-Supported Zirconia. 2. Effect of Sulfation on the Surface-Acidity and Its Potential as a Catalyst for Methane-Olefin Coupling. J. Phys. Chem. 1995, 99, 14444–14449. [Google Scholar] [CrossRef]
- Platero, E.E.; Mentruit, M.P.; Arean, C.O.; Zecchina, A. FTIR studies on the acidity of sulfated zirconia prepared by thermolysis of zirconium sulfate. J. Catal. 1996, 162, 268–276. [Google Scholar] [CrossRef]
- Breitkopf, C.; Papp, H.; Li, X.; Olindo, R.; Lercher, J.A.; Lloyd, R.; Wrabetz, S.; Jentoft, F.C.; Meinel, K.; Forster, S.; et al. Activation and isomerization of n-butane on sulfated zirconia model systems—An integrated study across the materials and pressure gaps. Phys. Chem. Chem. Phys. 2007, 9, 3600–3618. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-F.; Yan, P.; Hao, X.-Y.; Wang, Z.-Z. Influences of introducing Al on the solid super acid SO42−/SnO2. Mater. Chem. Phys. 2008, 112, 1065–1068. [Google Scholar] [CrossRef]
- Matsuhashi, H.; Miyazaki, H.; Kawamura, Y.; Nakamura, H.; Arata, K. Preparation of a solid superacid of sulfated tin oxide with acidity higher than that of sulfated zirconia and its applications to aldol condensation and benzoylation. Chem. Mater. 2001, 13, 3038–3042. [Google Scholar] [CrossRef]
- Cui, X.; Ma, H.Z.; Wang, B.; Chen, H.W. Direct oxidation of n-heptane to ester over modified sulfated SnO2 catalysts under mild conditions. J. Hazard. Mater. 2007, 147, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, J.; Zhang, W.; Tan, Y.; Tang, T.; Wang, S.; Tang, B.; Zhao, J. SO42-/SnO2 as a new electrode for electrochemical supercapacitors. Ceram. Int. 2014, 40, 8925–8929. [Google Scholar] [CrossRef]
- Li, C.; Stair, P.C. Ultraviolet Raman spectroscopy characterization of sulfated zirconia catalysts: Fresh, deactivated and regenerated. Catal. Lett. 1996, 36, 119–123. [Google Scholar] [CrossRef]
- Due-Hansen, J.; Boghosian, S.; Kustov, A.; Fristrup, P.; Tsilomelekis, G.; Stahl, K.; Christensen, C.H.; Fehrmann, R. Vanadia-based SCR catalysts supported on tungstated and sulfated zirconia: Influence of doping with potassium. J. Catal. 2007, 251, 459–473. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Castro, R.H.R.; Zhou, W.; Navrotsky, A. Surface enthalpy and enthalpy of water adsorption of nanocrystalline tin dioxide: Thermodynamic insight on the sensing activity. J. Mater. Res. 2011, 26, 848–853. [Google Scholar] [CrossRef]
- Zuo, J.; Xu, C.Y.; Liu, X.M.; Wang, C.S.; Wang, C.Y.; Hu, Y.; Qian, Y.T. Study of the Raman-Spectrum of Nanometer SnO2. J. Appl. Phys. 1994, 75, 1835–1836. [Google Scholar] [CrossRef]
- Aragon, F.H.; Coaquira, J.A.H.; Hidalgo, P.; da Silva, S.W.; Brito, S.L.M.; Gouvea, D.; Moraisa, P.C. Evidences of the evolution from solid solution to surface segregation in Ni-doped SnO2 nanoparticles using Raman spectroscopy. J. Raman Spectrosc. 2011, 42, 1081–1086. [Google Scholar] [CrossRef]
- Wang, H.W.; Wesolowski, D.J.; Proffen, T.E.; Vlcek, L.; Wang, W.; Allard, L.F.; Kolesnikov, A.I.; Feygenson, M.; Anovitz, L.M.; Paul, R.L. Structure and Stability of SnO2 Nanocrystals and Surface-Bound Water Species. J. Am. Chem. Soc. 2013, 135, 6885–6895. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.J. A review of refinery complexity applications. Petrol Sci. 2017, 14, 167–194. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.S.; Chakraborty, D.; Faglioni, F.; Muller, R.P.; Goddard, W.A.; Harris, T.; Campbell, C.; Tang, Y.C. Alkylation of phenol: A mechanistic view. J. Phys. Chem. A 2006, 110, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Colon-Ortiz, J.; Ramesh, P.; Tsilomelekis, G.; Neimark, A.V. Permeation dynamics of dimethyl methylphosphonate through polyelectrolyte composite membranes by in-situ Raman spectroscopy. J. Membr. Sci. 2020, 595. [Google Scholar] [CrossRef]
- Ramesh, P.; Kritikos, A.; Tsilomelekis, G. Effect of metal chlorides on glucose mutarotation and possible implications on humin formation. React. Chem. Eng. 2019, 4, 273–277. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Boghosian, S. In Situ Raman and FTIR Spectroscopy of Molybdenum(VI) Oxide Supported on Titania Combined with O-18/O-16 Exchange: Molecular Structure, Vibrational Properties, and Vibrational Isotope Effects. J. Phys. Chem. C 2011, 115, 2146–2154. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Josephson, T.R.; Nikolakis, V.; Caratzoulas, S. Origin of 5-Hydroxymethylfurfural Stability in Water/Dimethyl Sulfoxide Mixtures. ChemSusChem 2014, 7, 117–126. [Google Scholar] [CrossRef]
Figure 1.
(a) X-ray diffraction patterns and (b) ambient temperature Raman spectra of non-sulfated single as well as mixed SnO2/ZrO2 oxides.
Figure 1.
(a) X-ray diffraction patterns and (b) ambient temperature Raman spectra of non-sulfated single as well as mixed SnO2/ZrO2 oxides.

Figure 2.
(a) X-ray diffraction patterns and (b) ambient temperature Raman spectra of sulfated single as well as mixed (SO42−) SnO2/ZrO2 oxides.
Figure 2.
(a) X-ray diffraction patterns and (b) ambient temperature Raman spectra of sulfated single as well as mixed (SO42−) SnO2/ZrO2 oxides.

Figure 3.
Ambient temperature (a) FTIR and (b) Raman spectra of sulfated single as well as mixed (SO42−) SnO2/ZrO2 oxides at the spectral regime where characteristic vibrational modes of sulfate species can be observed. The spectra have been organized from ZrO2-rich (bottom) to SnO2-rich (top) samples.
Figure 3.
Ambient temperature (a) FTIR and (b) Raman spectra of sulfated single as well as mixed (SO42−) SnO2/ZrO2 oxides at the spectral regime where characteristic vibrational modes of sulfate species can be observed. The spectra have been organized from ZrO2-rich (bottom) to SnO2-rich (top) samples.

Figure 4.
Temperature dependent ATR-IR spectra of the (a) (SO42−) ZrO2, (b) 1:1 (SO42−)SnO2/ZrO2, and (c) (SO42−)SnO2 up to 200 °C. Spectral resolution was set to 4 cm−1, while 32 accumulations have been used to increase signal/noise ratio.
Figure 4.
Temperature dependent ATR-IR spectra of the (a) (SO42−) ZrO2, (b) 1:1 (SO42−)SnO2/ZrO2, and (c) (SO42−)SnO2 up to 200 °C. Spectral resolution was set to 4 cm−1, while 32 accumulations have been used to increase signal/noise ratio.

Figure 5.
Temperature dependent Raman spectra of the (a,b) 1:1 (SO42−)SnO2/ZrO2, and (c,d) (SO42−)SnO2 up to 500 °C. Acquisition time was set to 60 secs, while 32 accumulations have been used to increase signal/noise ratio. A 10X long working distance objective was used at a slight defocused mode to avoid local overheating.
Figure 5.
Temperature dependent Raman spectra of the (a,b) 1:1 (SO42−)SnO2/ZrO2, and (c,d) (SO42−)SnO2 up to 500 °C. Acquisition time was set to 60 secs, while 32 accumulations have been used to increase signal/noise ratio. A 10X long working distance objective was used at a slight defocused mode to avoid local overheating.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sulfation parameters, calcination temperature conditions, and physicochemical properties of all catalysts synthesized. Comparison of surface areas with relevant literature resources is also shown.
Table 1.
Sulfation parameters, calcination temperature conditions, and physicochemical properties of all catalysts synthesized. Comparison of surface areas with relevant literature resources is also shown.
| Catalyst | Calcination Temperature | Concentration of H2SO4 | Major Crystalline Phases Detected | Surface Area (S.A.) (m2/g) | Literature (S.A.) (m2/g) |
|---|---|---|---|---|---|
| SnO2 | 500 °C | - | Tetragonal SnO2 | 43 | 30.5 [22] |
| S-SnO2 | 500 °C | 1M | Tetragonal SnO2 | 91 | 66–93 [22,23] |
| 10:1 SnO2/ZrO2 | 600 °C | - | Tetragonal SnO2 | 34 | 37.4 [24] |
| 10:1 S- SnO2/ZrO2 | 600 °C | 1M | Tetragonal SnO2 | 81 | N/A |
| 1:1 SnO2/ZrO2 | 600 °C | - | Tetragonal SnO2 Monoclinic ZrO2 | 54 | 31.4 [24] |
| 1:1 S-SnO2/ZrO2 | 600 °C | 1M | Tetragonal SnO2 Monoclinic ZrO2 | 74 | N/A |
| 1:10 SnO2/ZrO2 | 600 °C | - | Monoclinic ZrO2 | 55 | 35.8 [24] |
| 1:10 S-SnO2/ZrO2 | 600 °C | 1M | Tetragonal ZrO2 | 129 | 53 [25] |
| ZrO2 | 600 °C | - | Monoclinic ZrO2 | 45 | 30–40 [26,27] |
| S-ZrO2 | 600 °C | 1M | Tetragonal ZrO2 | 149 | 90–150 [17,26,27] |
Table 2.
Catalytic performance data of all synthesized catalysts for the tert-butylation of phenol reaction. Reaction conditions: t = 4 h, T = 120 °C; phenol:TBA = 10:1; catalyst loading = 3.0 wt %.
Table 2.
Catalytic performance data of all synthesized catalysts for the tert-butylation of phenol reaction. Reaction conditions: t = 4 h, T = 120 °C; phenol:TBA = 10:1; catalyst loading = 3.0 wt %.
| Conversion, % | Product Distribution, % | ||||||
|---|---|---|---|---|---|---|---|
| Catalyst | TBA | 2-TBP | 4-TBP | 2,4-TBP | 2,6-TBP | 2,4,6-TBP | TBPE |
| S-SnO2 | 89.9 | 62.0 | 33.1 | 4.4 | 0.3 | 0.1 | 0 |
| 10:1 S- SnO2/ZrO2 | 88.0 | 59.2 | 30.9 | 5.2 | 0.7 | 0.2 | 3.7 |
| 1:1 S-SnO2/ZrO2 | 47.6 | 34.9 | 19.6 | 0.3 | 0.0 | 0 | 45.1 |
| 1:10 S-SnO2/ZrO2 | 66.4 | 54.2 | 28.6 | 2.9 | 0.3 | 0.1 | 14.0 |
| S-ZrO2 | 55.2 | 45.2 | 25.5 | 0.9 | 0.0 | 0 | 28.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marlowe, J.; Acharya, S.; Zuber, A.; Tsilomelekis, G. Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol. Catalysts 2020, 10, 726. https://doi.org/10.3390/catal10070726
AMA Style
Marlowe J, Acharya S, Zuber A, Tsilomelekis G. Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol. Catalysts. 2020; 10(7):726. https://doi.org/10.3390/catal10070726
Chicago/Turabian StyleMarlowe, Justin, Shreyas Acharya, Adam Zuber, and George Tsilomelekis. 2020. "Characterization of Sulfated SnO2-ZrO2 Catalysts and Their Catalytic Performance on the Tert-Butylation of Phenol" Catalysts 10, no. 7: 726. https://doi.org/10.3390/catal10070726
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.