
Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis


1
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
2
NanoVelos S.A., Rakowiecka 36, 02-532 Warsaw, Poland
*
Author to whom correspondence should be addressed.
†
Contributed equally.
Catalysts 2021, 11(8), 972; https://doi.org/10.3390/catal11080972
Submission received: 22 July 2021
/
Revised: 11 August 2021
/
Accepted: 13 August 2021
/
Published: 14 August 2021
(This article belongs to the Special Issue Gold, Silver and Copper Catalysis)
Abstract
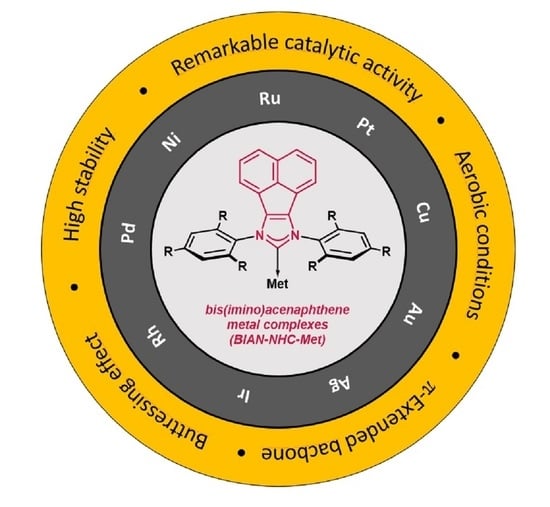
:N-Heterocyclic carbene (NHC) ligands have become a privileged structural motif in modern homogenous and heterogeneous catalysis. The last two decades have brought a plethora of structurally and electronically diversified carbene ligands, enabling the development of cutting-edge transformations, especially in the area of carbon-carbon bond formation. Although most of these were accomplished with common imidazolylidene and imidazolinylidene ligands, the most challenging ones were only accessible with the acenaphthylene-derived N-heterocyclic carbene ligands bearing a π-extended system. Their superior σ-donor capabilities with simultaneous ease of modification of the rigid backbone enhance the catalytic activity and stability of their transition metal complexes, which makes BIAN-NHC (BIAN—bis(imino)acenaphthene) ligands an attractive tool for the development of challenging reactions. The present review summarizes synthetic efforts towards BIAN-NHC metal complexes bearing acenaphthylene subunits and their applications in modern catalysis, with special emphasis put on recently developed enantioselective processes.
1. Introduction
N-Heterocyclic carbene ligands (NHC), since first being isolated in a stable form by Arduengo [1] in 1991, have revolutionized the field of modern metal catalysis [2,3,4,5,6,7,8,9], as well as organocatalysis [10,11,12,13,14]. The unique stereoelectronic characteristics (strong σ-donor and weak π-acceptor properties) are mainly responsible for the formation of well-defined, thermally stable metal complexes, and hence, widespread application for the development of new efficient catalytic processes. To date, many structurally diversified NHC ligands have been developed, varying in ring size, aryl or alkyl substituents attached to the nitrogen atom, the heteroatom incorporated in the ring core, or cyclic and acyclic backbone (for selected examples, see Figure 1). All these structural modifications allow for straightforward tuning of steric and electronic properties of a given ligand, critical for the development of new catalytic processes or the further improvement of existing ones.
The control of the activity of the catalytic pocket by modulating the electronic and steric properties is easily achieved in the case of NHC ligands. Although much research is limited to the classical imidazolium or imidazolinium ligands, further structural tuning of the imidazole core has brought a breakthrough for catalysis [15,16]. In this respect, NHC-BIAN-type ligands [BIAN–bis(imino)acenaphthene] bearing a π-extended system have found tremendous applications in the area of C-C or C-heteroatom bond formation, as well as in enantioselective catalysis in the last decade. Their key structural feature arises from the perfect combination of the electronic and steric properties of NHC-BIAN type carbenes. Regarding electronic properties, the π-extended polyaromatic system slightly increases σ-donor properties with respect to the classical imidazolium and imidazolium ligands. Plenio and co-workers [17] have investigated the electron donor ability of NHC-BIAN-Rh complexes by infrared spectroscopy and electrochemical methods (Figure 2). It was found that CO stretching frequencies (average value) of the respective NHC-Rh(CO)2Cl complexes are very similar to common SIMes or IMes carbene ligands. Due to the small difference in νav value (around 4 cm−1), it is difficult to estimate and directly compare the electronic character of NHC-BIAN-type ligands with others [18]. However, one can assume that NHC-BIAN ligands are slightly stronger donors than commonly used SIMes or IMes, on the basis of infrared spectroscopic data. Furthermore, the electrochemical studies confirmed that the redox potential is slightly lower (in comparison with SIMes or IMes), also indicating a slightly stronger donor ability (Figure 2).
Apart from the electronic properties, steric effects seem to add to the excellent reactivity of NHC-BIAN-type ligands in catalysis. This could be attributed to the “buttressing effect” of the acenaphthylene backbone [19]. The close proximity of the rigid polyaromatic skeleton would seem to push the N-aryl substituent closer to the metallic center, enabling better control of the catalytic pocket under homogenous conditions. Unfortunately, careful analysis of the available crystallographic data of the metal complexes did not confirm this assumption. Rather surprisingly (Figure 3), the angles between N-aryl substituents of NHC-BIAN-IPrAgCl [20] and NHC-BIAN-IPrAuCl [20] are higher (155°, and 149°, respectively) in comparison to their congeners possessing structurally identical N-wingtip substituents, such IPrAgCl [21] (137°) and IPrAuCl [22] (138°) derivatives.
Furthermore, the %Vbur parameter (describing the steric properties of NHC ligands [23,24]), estimated on the basis of X-ray data, has indicated much the same steric hindrance of NHC-BIAN-type ligands as for other ligands commonly used in catalysis. Unfortunately, the real situation under homogenous conditions in solution is unknown, and further evidence is required to understand the unique reactivity of NHC-BIAN metal complexes.
The aim of the present article is to provide a comprehensive overview of the methods for the synthesis of NHC-BIAN-type ligands and their metal complexes, with special emphasis on the practical aspects (such as accessibility of the substrate, scalability, etc.). To the best of our knowledge, no similar review has been published to date. The catalytic properties of NHC-BIAN type complexes will also be briefly discussed, since an excellent review on the catalytic applications of NHC-BIAN-type complexes has recently been published by Szostak and co-workers [19].
2. Synthetic Routes to BIAN-Type NHC Precursors
N-Heterocyclic carbene precursors containing an acenaphthylene element (NHC-BIAN) have found widespread application in the synthesis of many metal complexes, and hence allowed for the development of many catalytic reactions due to the unique electronic and, even more so, steric properties. In the present chapter, the synthetic efforts towards NHC precursors will be discussed to provide a comprehensive overview of the most common synthetic approaches in a comparative fashion. The structure of the present chapter is governed by the symmetry of NHC-BIAN-type ligands, including chiral NHC-BIAN-type ligands, which will also be presented in the final part.
2.1. Synthesis of Monodentate Carbene Precursors
Monodentate precursors of N-heterocyclic carbenes constitute the most numerous group among all salts possessing acenaphthylene elements in their structure. In this chapter, the presented synthetic methods are classified in terms of ligand symmetry, and C2-symmetric achiral ligands will be discussed first, followed by C1-symmetric ones.
N-Heterocyclic carbene precursors with low steric congestion are easily accessed following a two-step pathway (Scheme 1). The first step leads to the formation of the corresponding bisimine 3, by the condensation of aniline derivative 1 with acenaphthoquinone (2) in the presence of a Lewis acid, usually ZnCl2, and AcOH as the solvent. In some cases, bisimine 3 formation could be accomplished without Lewis acid in AcOH, or in a mixture of MeCN and AcOH [17,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. An interesting “green” alternative to the classical condensation leading to 3n, w, x has been proposed by Garcia [26,39]. The authors have proved that effective formation of bisimine 3n, w, x could be attained under mechanochemical conditions (in a ball mill).
To complete NHC skeleton formation, bisimines 3 are subjected to a cyclization reaction, usually performed with chloromethyl methyl ether (MOMCl) or its less volatile homolog, ethyl chloromethyl ether (EOMCl), as the source of the precarbenic carbon atom. It should be noted that the strong alkylating agents (MOMCl or EOMCl) are usually used as the solvents in the ring closure step, affording imdazolium salts 4 with excellent yields. All examples of bisimines 3 and NHC precursors 4 described in the literature are depicted in Scheme 1. As it was shown, significantly more examples of bisimine 3 formation could be found in the literature data than for the related NHC precursor 4. This results from the fact that bisimines 3 possessing an acenaphthylene subunit serve as superior N,N-ligands for metal complexes, beyond the main subject of this review, NHC-BIAN-type complexes.
Sterically more hindered NHC precursors 6 containing benzhydryl substituents at positions 2 and 6 relative to the aniline nitrogen could also be obtained by the pathway described above using similar reaction conditions (Scheme 2). However, bisimine 5 formation step required the presence of Lewis acids, such as ZnCl2 or AlMe3 (TMA) [40,41,42,43,44]. It should be noted that bisimine formation could be conducted on a large scale. Bisimine 5i was synthesized on 100 g scale in 75% yield. All bisimines 5 bearing benzhydryl subunits and individual salts 6 are presented in Scheme 2.
Acenaphthoquinone (1) could also be used as the platform for the synthesis of unsymmetrical NHC precursors 9 [44,45,46]. The synthetic pathway commenced with the addition of one molar equivalent of aniline 1 to form monoimine 7 under acidic conditions (AcOH or p-TSA had to be used, Scheme 3). The incorporation of the second aniline subunit needs the strongly acidic p-TSA in boiling toluene to reach bisimine 8 in the yield range 31–81%. The final ring closure was performed with chloromethyl ethyl ether to afford NHC precursor 9 in excellent yield, usually above 80%. It should be noted that sequential formation of bisimine (via initial formation of keto imine) was also applied for the synthesis of symmetric salts 9a,b with excellent results [46].
Among the sterically hindered NHC precursors, special attention was given to pentipticene derivatives. Plenio and co-workers [47] have developed a scalable synthetic route to those derivatives bearing a unique rigid skeleton (Scheme 4). The resulting salt 13 could be easily accessed via bisimine formation starting from aminophenol 10 (available via double cycloaddition of hydroquinone and anthracene on a multigram scale), and subsequent O-alkylation and ring closure, performed in the usual manner. It should be noted that bisimine 11 plays a dual role—a protecting group for the amine function in the O-alkylation step and a direct structural element for the construction of the imidazole ring.
2.2. Synthesis of Polidentate Carbene Precursors
Polyfunctional carbene ligands are particularly attractive due to their potential for use in catalysis, as they lead to metal complexes [48] often exhibiting higher catalytic activity compared to their monofunctional analogues [49].
Methods for the preparation of polyfunctional precursors of N-heterocyclic carbenes are identical to those applied for the synthesis of their monofunctional counterparts. A precursor of this type was first described by Cowley and co-workers [50]. The authors used mesityl amine to convert tetraketopyracene 14 into tetraimine 16a in the presence of ZnCl2. Unfortunately, isolation of free tetraimine 16a required decomplexation using potassium oxalate. A few years later, Alcarazo and co-workers [51] synthesized NHC precursor based on 2,6-diisopropylamine (DIPP). This time, the condensation was performed in a mixture of AcOH and MeCN without any Lewis acid additive to provide tetraimine 16b with an excellent 90% yield. The final cyclization with MOMCl afforded NHC precursor 17 in good yield (Scheme 5). Some applications of polyfunctional NHC ligands for catalytic processes are discussed in Section 3.1.5.
2.3. Synthesis of Chiral Carbene Precursors
Rather rare, but very promising from the point of view of enantioselective catalysis are chiral salts of N-heterocyclic carbenes based on the acenaphthylene framework. The rigidity of the polyaromatic skeleton could directly influence the N-wingtip substituent, creating a chiral pocket around the metallic centre more efficiently than simple imidazolium or imidazolinium ligands.
In 2018, Cramer and co-workers [52] obtained a series of chiral NHC precursors bearing C2 symmetry (Scheme 6). The synthesis of salts 20 commenced with the addition of aniline 18 to acenaphthoquinone (1), followed by cyclization with EOMCl (Scheme 6). It should be noted that the substitution of the methyl substituent in the para position relative to the amino group with a sterically hindered tert-butyl group (Table 1, entry 2) afforded bisimine 19b and the chiral precursor NHC 20b in low yields. Similarly, the introduction of a xylyl group (3,5-dimethylbenzyl), possessing two electron-donating methyl groups, in place of the phenyl substituent in the side-arm, led to the chiral salt 20c in higher yield after two steps (Table 1, entry 3). Further attempts to introduce substituents with higher steric hindrance, namely, the 3,5-di-tert-butylphenyl group (Table 1, entry 4), led to a significantly decreased yield. It should be emphasized that bisimine 19d was prepared under slightly different conditions where the use of strongly acidic p-TSA in place of ZnCl2 was required.
Further research of Shi and co-workers [53] has proved that the yield of bisimine formation could be greatly improved by the exclusion of Lewis or Brönsted acids (Scheme 7). Condensation of aniline 18a with acenaphthoquinone (1) conducted in a mixture of MeCN and AcOH afforded cleanly product 19a in higher 88% yield (in comparison to 70%, reported by Cramer [52], Scheme 6 and Scheme 7). Rather surprisingly, increasing the reaction temperature to 100 °C and extension the cyclization time to 24 h afforded salt 20a with a two-fold better yield than that in Cramer’s work.
Chiral NHC salts described above appear to be excellent ligands for metal-catalyzed enantioselective formation of indole [54], pyrrole [54], and pyridone derivatives [52] (vide infra), often found in drugs and natural products. A key structural element for their successful application were readily available chiral anilines, originally reported by Gawley and co-workers, and accessible via the resolution of the complicated mixture on chiral preparative HPLC (Scheme 8) [55]. Further research by Gawley and Pfaltz has revealed that chiral aniline 18 could be easily accessed by the initial formation of bis-styrene derivative 23, and subsequent enantioselective hydrogenation of 1,1-diaryl-substituted alkene [56]. Finally, aniline 18b was obtained on a 5 g scale with excellent selectivity (98:0.2:1.8, mixture of R,R:S,S:meso product).
In 2018, Cramer and co-workers [52] implemented Gawley and Pfaltz’s protocol based on enantioselective reduction for the preparation of anilines 18b,c bearing more bulky aryl substituents. The authors also proposed synthetic routes to bisalkenyl aniline derivative 23. In the first step, terminal alkyne 21 was converted to vinyl borate 24 through hydroboration catalyzed by nickel-bisphosphine complex (Scheme 8). The respective vinyl borate 24 was subjected to Suzuki coupling with 2,6-dibromoaniline to furnish the desired alkenylaniline 23. It should be mentioned that an increase in the steric hindrance of aryl substituent (23a vs. 23b) decreased the yield of the enantioselective reduction (Scheme 8).
3. Preparation of Metal Complexes and Their Application in Catalysis
3.1. Synthesis of Palladium Complexes and Their Applications
Palladium complexes constitute the most abundant group among metal complexes based on the acenaphthylene skeleton. To provide a systematic overview of the synthesis and applications of palladium complexes, methods for their preparation were divided into a few categories, including symmetry of BIAN-NHC ligands, their structural modifications, and the character of the ligand coordinated to the palladium center. In the first subsection, the methods for PEPPSI complexes preparation as well as modifications of NHC ligands and ligands coordinated to palladium (including chelating ones) will be discussed. In the final part of the subsection, the application of complexes with a π-extended NHC-backbone will be briefly presented, and the possibility of using palladium complexes under heterogeneous conditions will be commented on as well.
3.1.1. NHC-BIAN-PEPPSI-Type Complexes
PEPPSI-type complexes (Pyridine-Enhanced Precatalyst Preparation Stabilization, and Initiation) were first described in 2006 by Organ and co-workers [16,57,58], who proved their utility in cross-coupling reactions due to excellent catalytic activity and stability. The key structural element, namely pyridine or its substituted analogues (usually 3-chloropyridine) coordinated to palladium could be easily introduced by the treatment of palladium complexes with an excess of ligand at elevated temperature. Organ’s method appeared suitable for a plethora of C2-symmetric BIAN-NHC-PdCl2Py complexes, and their synthesis could be conducted on a few hundred milligram scale, highlighting the practical aspect for further applications in catalysis (Scheme 9).
A significant number of palladium(II) complexes bearing C2 symmetry have been synthesized by the Tu and Liu research groups, independently (Scheme 9). First, complexes possessing only simple alkyl substituents in the ortho position of the aniline, such as mesityl or DIPP derivatives (25a–c [35,59,60,61], 25d–g [62] and 25h–i [43,63]) were obtained in a moderate 48–68% yield. Further research in this area resulted in the synthesis of the highly sterically hindered palladium(II) complexes 25j–n [46] and 25n [41], containing benzhydryl substituents, in comparable yields. Finally, Liu and co-workers synthesized interesting examples of C2-symmetric complexes 37 [64] with diverse substituents in the ortho position of the aniline ring. The synthesis of all mentioned PEPPSI-Pd complexes appeared to be easily scalable, delivering 100 milligram quantities of the respective metal complexes for further catalytic applications.
Bearing in mind that the catalytic activity of palladium(II) complexes strictly depends on even small structural changes in the ligand, Liu has developed a series of C1-symmetric complexes 26 (Scheme 10) exhibiting remarkable reactivity in cross-coupling reactions (see, vide infra) [45]. It should be noted that this is one of the very rare examples of successful synthesis of N-heterocyclic carbene precursors and metal complexes bearing an electron-withdrawing substituent [65,66,67], in this particular case a fluorine. The syntheses of complexes of general structure 26 were conducted on a large scale, providing easy access to more than 0.5 g of the metal complex in each case.
NHC-BIAN-Pd palladium complexes have proved to be the most prevalent in catalysis among metal complexes possessing an acenaphthylene structural subunit and have found widespread applications in cross-coupling reactions. Among cross-coupling reactions, Suzuki and Buchwald-Hartwig reactions are the most explored ones with PEPPSI-type palladium complexes.
One of the early examples of the Suzuki reaction was described by Tu and co-workers in 2012 (Scheme 11) [35]. Authors proved the usefulness of complex 25a for the coupling of boronic acids with a wide array of aryl bromides and more challenging aryl chlorides. The respective products were isolated with excellent yields, also in the case of heteroaromatic components, using 0.5 mol% of 25a and KOtBu as base. Further improvement for the Suzuki reaction has been achieved by the Liu group [68]. The authors decorated the acenaphthylene backbone with the bulky tert-Bu group (in anti-orientation) by the addition reaction with tert-BuLi. The respective complex 27a appeared to be extremely active for the coupling of low reactive aryl chloride without inert gas atmosphere. The most important feature of Liu’s protocol is the use of weakly basic conditions (K3PO4 instead of KOtBu) and low catalyst loading, in the range 0.05–0.1 mol%.
Further investigations of Liu and co-workers furnished complexes 25h–i and 25o–s, catalytically active in the Suzuki reaction under aerobic conditions. Sterically hindered PEPPSI-IPent-Pd complex 25i proved to be very effective for the synthesis of sterically hindered biaryls, including 2,6,2′,6′-tetrasubstituted ones from a wide range of aryl and especially heteroaryl chlorides and boronic acids [43]. Comparable reactivity of complex 25i has been reported in reactions catalyzed by complex 25q [64]. The most significant advantage of the developed protocol is a further decrease in catalyst loading, being as low as 0.025 mol%. Moreover, the application of K2CO3 as the base furnished heterobiaryls and polyheteroarenes with excellent functional group tolerance from a variety of heterocycle building blocks, including pyridine, furan, and thiophene derivatives, as well as much more challenging thiazoles and pyrazines (usually effecting deactivation of the palladium catalyst).
As an alternative to the Suzuki reaction, biaryl systems can be efficiently accessed via the Negishi reaction, especially when boronic acids (or their analogues) are difficult to prepare. Tu and co-workers have developed the coupling of aromatic and aliphatic organozinc compounds with both aryl chlorides and bromides at room temperature (Scheme 12) [60]. The respective products 30 were obtained with excellent yield, even for secondary organozinc compounds, prone to undergo β-elimination. It should be noted that 0.25 mol% of complex 25a was needed to achieve the high yield, also in the case of heteroaromatic compounds, including thiophene, furan, as well as pyridine and pyridazine derivatives.
Another efficient protocol for the synthesis of biaryl compounds was developed by Organ, Feringa, and co-workers in 2019 [69]. The authors achieved for the first time the Murahashi coupling of an organolithium reagent with aryl bromide (Scheme 13) at low temperature (−62 °C). The coupling reaction proved to be highly chemoselective towards aryl bromide in the presence of chloride, making sequential selective Br/Cl coupling possible. A wide range of aryl and alkyl lithium reagents 31 were successfully coupled with bromides, including heteroaromatic ones. Unfortunately, the reaction requires high catalyst loading (5.0 mol% of 25t) to maintain the high yields.
The second extensively investigated group of reactions catalyzed by palladium complexes, besides the formation of biaryls, is the Buchwald-Hartwig amination. Indeed, PEPPSI-Pd type complexes have shown remarkable activity for C-N bond formation. The first literature report came from the Tu group [59] and concerned the use of complex 25a for the synthesis of secondary and tertiary amines from low-reactivity aryl chlorides with virtually quantitative yields (Scheme 14). Further research of the same group has led to the development of complexes 26c bearing C1 symmetry, which drastically influenced the reactivity [45]. Tu and co-workers have demonstrated their remarkable activity for the coupling of ortho- and more sterically hindered bis-ortho-substituted chlorides with primary and secondary amines. Importantly, complex 26c exhibits perfect stability under the reaction conditions, and an inert atmosphere was not required for high efficiency. Similar catalytic activity and scope of applicability were reported for the more sterically hindered complexes 25h [63] and 25m [46]. It should be noted that the evaluation of the catalytic activity of complexes presented in Scheme 14 was performed on model Buchwald-Hartwig reactions with slightly structurally different substrate and conditions, preventing a direct comparison. Furthermore, no data are available for comparison with other PEPPSI-type complexes, in particular with IPr or IMes derivatives.
Palladium complexes based on the acenaphthylene scaffold were also applied in the Sonogashira reaction (Scheme 15) [70]. Here, the combination of two carbene complexes, namely palladium complex 25a and IPrCuCl (35a) enabled the reaction between terminal alkynes and aromatic bromides. Regrettably, the reaction requires a relatively high temperature of 120 °C. However, it could be performed with very low palladium catalyst loading 25a, as little as 0.01 mol%.
The modified-PEPPSI-Pd complexes were also successfully applied in azole arylation [62]. The sequential C-H activation/arylation was described by Liu and co-workers with complex 25g. The key to successful coupling was the introduction of a benzhydryl substituent into the aniline subunit of complex 25g, increasing the σ-donor properties of NHC ligand, and hence facilitating the oxidative addition step. This structural modification enabled highly regioselective coupling of a wide range of azoles, including imidazole, triazole, oxazole, and thiazole derivatives under aerobic conditions at very low catalyst loading of complex 25g, in the range 0.05–0.5 mol% (Scheme 16).
3.1.2. π-Allyl NHC-BIAN-Pd Complexes
Palladium π-allyl complexes are usually extremely active in many catalytic transformations, but less stable than the respective NHCPdCl2 derivatives (or their congeners). Taking into account the excellent activity, it is rather surprising that palladium π-allyl complexes bearing acenaphthylene backbone were marginally reported [71,72,73]. Their preparation directly from an NHC-BIAN-type precursor required a palladium dimer and a strong base (38a, Scheme 17) [72]. It should be noted that the introduction of a phenyl-substituted π-allyl moiety (38b) was achieved at elevated temperatures with rather moderate yields.
The above-mentioned π-allyl complexes 38b were first applied as catalysts in the aminocarbonylation reaction of iodoarenes (Scheme 18) [72]. Tu and co-workers demonstrated that the reaction can be carried out with carbon monoxide under relatively low pressure of the gas (1 atm.). The developed procedure was applied to a wide range of primary and secondary amines, and finally for the synthesis of tamiboratene on a gram scale. Similar conditions have been successfully used for the aminocarbonylation of ortho-diiodoarenes, providing access to imides [73]. In this case, slightly higher temperature and DABCO as base were needed.
An interesting application of a π-allyl BIAN-NHC palladium complex was also reported by the Tu group who described for the first time the sulfonation of boronic acids [72]. The authors developed a unique method for the preparation of aryl-alkyl sulfones from boronic acids and α-bromoesters. The sulfone formation was efficiently catalyzed by a cinnamyl derivative of palladium complex 38b where potassium metabisulfite (K2S2O5) served as the source of the sulfone group. Moreover, the authors proposed, based on several control experiments, a reaction catalytic cycle comprising transmetalation, SO2 insertion, and final alkylation (Scheme 19). They gave particular attention to the role of TBAF in the alkylation step of the dimeric palladium compound, most likely facilitating the sulfinate formation.
3.1.3. Chelate Complexes
An interesting equilibrium between the catalytic activity and stability of the palladium complex was presented by Tu and co-workers [74]. Conducting studies on the coupling of amines and heteroaryl chlorides, the authors demonstrated that palladacycle 38b exhibits much better activity in comparison to standard PEPPSI-Pd complexes. In this manner, a number of secondary and tertiary amines were obtained from challenging chloropyridines with excellent yields. The corresponding series of palladacycles 38 were synthesized from the carbene precursor 44 in the usual manner, in the presence of K2CO3. Moreover, the coordination between palladium and the amine was unambiguously confirmed by X-ray crystallography (Scheme 20).
3.1.4. Skeleton Modifications of NHC-BIAN-Pd Complexes and Their Catalytic Activity
Further research in the area of palladium catalysis has led to the development of PEPPSI-type complexes with a modified acenaphthylene structural element. Liu and co-workers have synthetized carbene precursors 45 possessing two tert-butyl groups in an anti-relationship (Scheme 21) [68]. These salts were used for the synthesis of palladium complexes 27 using PdCl2 and pyridine as the solvent. The catalytic activity was evaluated in the Suzuki reaction (for details, see Scheme 11).
3.1.5. Polyfunctional NHC-BIAN-Pd Complexes
As mentioned earlier (Scheme 5), polyfunctional metal complexes based on polyaromatic systems usually exhibit different catalytic properties. To date, a single example of polyfunctional palladium complexes has been described by Peris and co-workers [49]. The catalytic activity of the respective bispalladium complexes 49, bearing acenaphthylene scaffold, was studied in the acylation of aryl chloride and the Suzuki coupling (Scheme 22). Unfortunately, bispalladium 49 shows similar catalytic activity in comparison to monofunctional complexes.
3.1.6. Heterogenization of NHC-BIAN-Pd Complexes
The methods discussed so far have concerned only homogeneous palladium catalysis. Unfortunately, there are many drawbacks associated with homogeneous catalysis, especially on an industrial scale. Catalytic homogeneous processes yield large amounts of waste, contributing to environmental pollution. In addition, low atom economy and difficult catalyst recovery are common drawbacks. Thus, heterogenization of homogenous catalysts on solid support proved useful in many processes to date, allowing for the straightforward recovery of the usually expensive metal complex.
The first example of heterogenization of BIAN-NHC-Pd complexes was given by Bazzi and co-workers [38]. The authors synthetized PEPPSI-Pd-type complexes covalently bonded to a polymeric support via long nonpolar chains containing 18 isobutylene units. Pleasingly, the polyisobutylene polymer (PIB) attached to complex 50 increased its solubility in a nonpolar solvent. This allowed for easy separation of amines from the palladium catalyst by polar solvent extraction, without the need for time-consuming and expensive column chromatography and, accordingly, enabled the efficient recovery of the catalyst. The respective palladium complex 50 was obtained under standard conditions (PdCl2 and K2CO3) from NHC precursor 4f with moderate 68% yield (Scheme 23), and its catalytic activity was investigated using the Buchwald-Hartwig amination. The authors observed remarkably high catalytic activity in non-polar media, such as n-heptane, for the coupling of aryl halides with secondary amines and anilines.
Considering the drawbacks associated with the recovery of the catalyst under homogenous conditions, Tu and co-workers described the heterogenization of BIAN-NHC-Pd complexes on magnetic nanoparticles, easily sparable by external magnetic field [75]. The immobilization of palladium complex 54 was achieved via a two-step protocol. First, palladacycle 54 was synthesized by the reaction of salt 4g with silane 53 and PdCl2 in the presence of K2CO3. Then, palladacycle 54, possessing a long-chain triethylsilyl tail was anchored on the surface of a magnetic nanoparticle coated with silica (SMNPs) by silyl ether formation. The immobilized catalyst 55 proved its high catalytic activity in the Suzuki-Miyaury reaction of 9-chloroacridine (51) with phenylboronic acid (48) (Scheme 24).
Apart from the palladium complexes immobilized on solid support, other unconventional approaches to reactions under heterogeneous conditions have been described. An interesting option for carrying out palladium-catalyzed reactions under mechanochemical conditions was presented by Browne (Scheme 25) [76]. Mechanochemistry is a relatively new field of organic synthesis, in which the energy required for the reaction is supplied by collisions, and the chemical reactions are usually carried out in ball mills. The major advantages of this innovative approach, in comparison to the conventional reaction in solution, are higher reaction rates, greater selectivity, and reduced usage of organic solvents, which are not environmentally neutral.
The first examples of a cross-coupling reaction under mechanochemical conditions was described by Browne’s group in 2018 (Scheme 25) [76]. The respective Negishi coupling of a zinc compound with bromobenzene was efficiently catalyzed by PEPPSI-type complex 25a, previously reported by Tu and co-workers [60] (Scheme 9). It should be mentioned that organozinc compound 57 was also generated in situ, providing ester 58 with 64% yield after two steps (Scheme 25). Further utility of palladium catalyst 25a under mechanochemical conditions was exemplified also by Browne and co-workers [77] in the Buchwald-Hartwig reaction of chlorobenzene 29a with morpholine 32b. The reaction was carried out in ball mills with sand as the grinding medium, in the presence of strong KOtBu base, providing tertiary amines with low yield. Notably, the above-described reactions under mechanochemical conditions do not require anhydrous conditions or inert gas atmosphere, providing a practical alternative to classical, strictly anhydrous conditions.
3.2. NHC-BIAN-Ni Complexes in Catalysis
NHC-Ni complexes have found many applications in catalysis and can be viewed as an inexpensive replacement for palladium. Currently, NHC-Ni-catalyzed transformations are the subject of growing intensive research, including NHC-BIAN-Ni catalysis. The following subsection covering NHC-BIAN-Ni catalysis was divided into two subgroups, regarding the structure of NHC precursors for the formation of achiral and chiral compounds.
Usually, nickel complexes are generated in situ due to their inherent instability. To date, there is only one example of the synthesis of an N-heterocyclic nickel complex bearing an acenaphthylene backbone in isolable form. In 2018, Cramer and co-workers [52] synthesized this type of complex following the procedure previously reported by Nolan [78]. Therefore, NHC precursor 20a was reacted with nickelocene 59 under microwave conditions for 16 h at 75 °C, affording nickel complex 60 in high yield (Scheme 26).
Regarding the catalytic applications, the majority of the NHC-BIAN-Ni-catalyzed reactions include transformations where the active nickel complex is generated in situ, as mentioned. The first example of a Ni-catalyzed transformation was reported by Newman’s group [79] and concerns amide bond formation in the presence of Ni(COD)2 and salt 4g. In this approach, methyl benzoate (61) underwent a reaction with aniline 39a in the presence of strong KOtBu base to give benzanilide 39a, although in low yield (Scheme 27). It should be noted that NHC precursor 4g was the only one examined during the optimization process.
The following year, Sun and co-workers [80] presented the reductive coupling of 1,2-diphenylacetylene (63a) with imine 62a in the presence of Ni(COD)2 catalyst and NHC precursor 4c with low steric hindrance. In the coupling reaction, i-PrOH was used as a hydrogen source for the hydrogen transfer reduction. More sterically hindered salt 4g with isopropyl groups afforded allyl amines 64a in much higher yields than the less active NHC precursor 4c bearing methyl groups (Scheme 28).
Further research by Tu’s group [61] proved that palladium could be efficiently replaced by nickel for Buchwald-Hartwig-type amination. The authors applied the catalytic system composed of NiCl2-(DME) and NHC precursor 4g for the coupling reaction of 2-naphthyl p-toluenesulfonate (65) with morpholine (51), in the presence of phenylboronic acid pinacol ester. The reaction afforded tertiary amines in varying yields, depending on the base and solvent used (Scheme 29).
The last example of an achiral transformation, proving the usefulness of NHC nickel complexes for [3+2] cross-dimerization, was developed by Huang and Ho in 2020 [81]. The authors described the unique reactivity of unactivated olefins 67 and methylenecyclopropanes 66, leading to cyclopentanes bearing exocyclic double bonds (Scheme 30). The method exhibited high chemoselectivity and regioselectivity. The nickel complexes used in the studies were prepared in situ by the reaction of Ni(COD)2 and allyl chloride with the corresponding NHC, also generated in situ through the treatment of the NHC salt with KHMDS. Thus, the authors obtained [Ni(allyl)]Cl (70a) and [Ni(Phallyl)]Cl (70b) complexes in high yields, 82% and 71%, respectively. The highest reaction yields and selectivity of cross-dimerization reaction towards alkene insertion were obtained when the less hindered Ni-complex 70a was used. The authors created a vast scope of cyclopentenes 68, including spiro and fused ring systems.
The next subsection highlights enantioselective transformations affording chiral compounds, which are often a structural element found in biologically active compounds. Enantioselectivity of the reaction is achieved by the design of a chiral space in the catalyst area, which determines the orientation of the substrate during the catalyst-substrate bonding. In the examples discussed herein, such an effect could be achieved by modifying the steric crowding around the nickel atom by NHC. The acenaphthylene-based ligand allowed for subtle structural changes to create a chiral pocket, providing excellent performance in enantioselective Ni-catalyzed reactions.
In 2018, Cramer and co-workers [52] reported the enantioselective cyclization of 2-pyridone and its isomer 4-pyridone. In the first example, 2-pyridone 71 was subjected to cyclization catalyzed by NHC-Ni(COD)2 complex in the presence of a Lewis acid—MAD (Scheme 31). Notably, substitution of the methyl group in NHC ligand 20 by the tert-butyl group (20c) significantly decreased the yield and enantioselectivity of product 72. Replacement of the phenyl groups with 3,5-xylyl substituent in the side chains attached to the aromatic rings (20b) led to product 72 with higher enantiomeric excess and yield (Scheme 31). Further attempts to introduce more crowded substituents did not provide improved results.
Moreover, the cyclization reaction of 2-pyridone derivative 73 bearing trisubstituted alkenes, catalyzed by the N-heterocyclic carbene 20d with a nickel complex, allowed to extend the scope of chiral quinolizidines 74. The corresponding reaction conditions appeared to be effective for 4-pyridone derivatives 76 as well, leading to a product with high efficiency.
In the following year, Cramer’s group [54] carried out the enantioselective cyclization of indole 77 catalyzed by NHC-Ni(COD)2 complex 20c (generated in situ). Although the cyclization reaction was performed under relatively mild conditions, indole derivative 78 was obtained in 25% yield and with very low enantiomeric excess, namely 4% (Scheme 32).
In 2019, Shi and co-workers [82] reported the first example of enantioselective cyclization of alkenylpyridine 79, resulting in the formation of a chiral isoquinoline derivative 80 (Scheme 32). Direct activation of the C-H bond of the pyridine ring in positions 3 and 4 was accomplished by a chiral N-heterocyclic carbene ligand with diverse steric crowding, leading to product 80 in high yield and with high enantiomeric excess. Here, the pyridine derivative 79 was cyclized in the presence of Ni(COD)2 and the N-heterocyclic carbene, with MAD in cyclopropyl methyl ether (CPME) (Scheme 32). It should be mentioned that the replacement of the methyl or benzyl groups with 3,5-xylyl group (20b) in the wingtip substituent of the NHC ligand led to the product with higher enantiomeric excess, up to 86%.
Sun and co-workers [80] have developed an enantioselective reductive coupling reaction of 1,2-diphenylacetylene (63a) with tosyl hydrazones 62b (Scheme 33). The authors investigated a variety of NHC ligand structures featuring diverse steric hindrance. All the NHC-BIAN ligands afforded allylic sulfonamides 64b in good yields with S absolute configuration, confirmed by X-ray structural analysis. Notably, the authors observed very high enantiomeric excess in all cases, irrespective of the ligand structure.
Further synthetic potential of chiral NHC-BIAN type ligands was explored by Shi and co-workers (Scheme 34). The authors proved the excellent performance of ligand 20c for the arylation of ketones 81 with specially designed boronic ester 82 (Bneo) [83]. The developed Ni-catalyzed protocol appeared to be applicable for a broad range of aromatic and heteroaromatic ketones, providing easy access to a variety of tertiary alcohols 83 with excellent enantioselectivity and perfect tolerance of functional groups. The same research group have developed a Ni-catalyzed protocol for the synthesis of chiral N-arylated amines 85 and unprotected amines 86. A series of chiral tetrahydroquinolines 85 and 86 were synthesized via kinetic Buchwald-Hartwig coupling with aryl chloride. The key to highly enantioselective kinetic resolution was a low-temperature procedure, allowing for excellent control of stereoselectivity.
3.3. NHC-BIAN-Ru Complexes
One of the most important applications of N-heterocyclic carbene ruthenium complexes as catalysts is the olefin metathesis reaction. The utility of this reaction in biologically active structure synthesis has proven to be invaluable, therefore it is a trending subject in organometallic catalysis [84]. This subsection covers the synthesis of second generation Hoveyda-type ruthenium complexes containing an acenaphthylene-based N-heterocyclic carbene ligand. Applications of the corresponding complexes in ring-closing metathesis (RCM) and ring-opening metathesis polymerization (ROMP) are also highlighted.
Merino and co-workers [30] obtained, for the first time, an N-heterocyclic ruthenium complex 92 with acenaphthylene-containing structure. The two-step synthetic pathway involved deprotonation of 4c salt with a strong base (KOtAmyl) in n-hexane, and subsequent exchange of the phosphine ligand for the NHC ligand in first generation Hoveyda-type ruthenium complex 91. The reaction was carried out in the presence of CuCl as a phosphine scavenger, resulting in the corresponding ruthenium complex 92 in 76% yield. The ruthenium complex 92 exhibits high catalytic activity in RCM of model reagents (Scheme 35), leading to formation of cyclopentene 88 and dihydropyrrole 90 derivatives.
Shortly after, Bazzi and co-workers reported the synthesis of an N-heterocyclic ruthenium complex attached to polyisobutene (17 isobutylene units) [85]. The respective ruthenium complex 95 was synthesized by the initial deprotonation of salt 4f with KOtBu, and the resulting free carbene was further reacted with first generation Hoveyda-type complex 95 to give ruthenium complex 95 in moderate yield (Scheme 36). The catalytic activity of complex 95 was investigated in ring-closing metathesis reaction (RCM) of N,N′-diallyl-4-methylbenzenesulfonamide (89), yielding tosyl-protected pyrrolidine (90) and ring-opening metathesis polymerization (ROMP) of norbornene derivatives (93). It should be mentioned that the RCM reactions were performed in n-heptane, allowing for easy separation of the respective products by simple filtration. The respective mother liquor containing the active catalyst was reused for up to eight cycles, affording pyrrolidine derivatives 90 without a decrease in yield.
3.4. NHC-BIAN-Au Complexes
N-Heterocyclic carbene gold(I) complexes are a relatively numerous group of carbene metal complexes. For this reason, the material reported herein has been divided into two sections describing the synthesis and application of the complexes in catalysis. The first section is focused on the synthetic methods reported in the literature [17,20,47,86,87,88,89]. Notably, not all complexes have proven catalytic activity, although the authors did not exclude the possibility of their application in catalysis [17,47].
In 2016, Plenio and co-workers reported the synthesis of bulky gold(I) complex 96, pentiptycene derivatives, and complexes 97 with less steric hindrance (Scheme 37) [24,31]. The preparation procedure involves the reaction between NHC precursor (13 or 4) and AuCl•Me2S, in the presence of an excess (3–7 equiv) of K2CO3 in acetone as the solvent. This method was independently developed by the groups of Gimeno [90] and Nolan [91].
In contrast to Plenio’s results, Cowley and co-workers [20,86] have reported the indirect synthesis of gold(I) complexes from NHC precursors (Scheme 38). This approach commenced with silver(I) complex (98a) formation by the reaction of salt 4g with Ag2O in the mixture of DCM and THF. Subsequent transmetalation of silver(I) complex 98a with (AuCl•tht) allowed to isolate gold(I) complex 99a in high 80% yield, after two steps (Scheme 38). Apart from the catalytic activity, gold(I) complexes described above, including gold(I) complex 99a, have proven antifungal and antibacterial properties [86,87,88].
Belpassi’s group [89] shortened the two-step synthetic route, previously proposed by Cowley, to one step by eliminating transmetalation (Scheme 37). The gold(I) complex (99a) was obtained directly after the reaction of the NHC precursor 4g and AuCl•tht in the presence of KHCO3 (Scheme 39). Although the reaction time increased from 12 h to 72 h, this method afforded gold(I) complex 99a in higher yield, compared to Cowley’s method (Scheme 38) [20].
The second section covers gold(I) complexes with confirmed catalytic activity and their applications in catalysis [92,93]. Plenio’s group [92] have obtained a series of gold(I) complexes bearing a pentiptycene subunit and proved their catalytic activity (Scheme 40). It is worth to mention that gold(I) complex 96a with acenaphthylene-based ligand structure was prepared according to the method described in Plenio’s earlier work [17,47]. In the following years, the authors reported the application of a gold(I) complex as a catalyst in the hydration of 1-phenylacetylene (21a) (Scheme 40). The reaction resulted in 99% conversion of the substrate after 21 h, affording acetophenone 100a as the product. It should be mentioned that the loading of gold complex 99a could be reduced to 0.01 mol%. However, additives such as AgOTf and TfOH were still required to keep the high yield.
Zuccacia’s team have also investigated an alkyne hydration catalyzed by gold(I) complexes [94]. In contrast to Plenio’s research, the main goal was to develop an efficient procedure under silver-, acid-, and even solvent-free conditions. The authors selected the hydration of hex-3-yne as the model reaction to optimize the reaction conditions. The ligand structure and the type of counterion appeared to have great influence on the catalytic activity of the gold complex. NBu4OTf was selected as the superior additive for gold(I) complex activation (Scheme 40). Many gold complexes have been tested, including a complex containing an acenaphthylene fragment in the ligand structure 99. The synthesis of complex 99a was achieved by the transmetalation route, as described by Cowley [20] (Scheme 38). The suitable combination of ligand and counterion allowed to obtain 99% substrate conversion in 2 h and TOF equal to 495. Among the gold complexes tested, the highest yield was achieved for complex 99c, possessing an acenaphthylene subunit with OTf- as the counterion. It should be mentioned that the addition of silver salts did not influence the reaction outcome.
Tu’s group has presented an interesting application of gold complexes for sulfone synthesis [93,95]. The gold(I) chloride complexes (99) were synthesized by the method of Nolan [91] and Gimeno [90], and further transformed into the respective hydroxides 104a by the treatment of complexes 99 with KOH in a mixture of toluene and THF (Scheme 41). The aforementioned gold(I) complexes 99a,b were initially used as catalysts in the alkylsulfonylation reaction of boronic acid 40, with K2S2O5 as the source of the sulfonyl group [93]. Equal activity was observed with hydroxide 104a, whereas the common IPrAuCl provided the sulfone with reduced yield.
Two years later, the same research group developed the arylsulfonylation reaction of aryl boronic acid (40) with diaryliodonium salts 102, providing access to diaryl sulfones 103 [95]. The gold(I) chloride complex 99a exhibited the best catalytic activity, leading to a broad range of sulfones 103, including heterocyclic derivatives as well. This easily scalable protocol offers a straightforward approach to diaryl sulfones bearing sterically hindered aryl groups, such as mesityl, which can be found in drugs and biologically active compounds [96].
Plenio’s group have reported good catalytic properties of complex 96b in enyne cyclization (Scheme 42) [47]. The preliminary catalytic study of 96b has led to a mixture of five- (106) and six-membered (107) rings as products in a 72/28 ratio with excellent yield. In contrast, a regioselectivity switch in the reaction catalyzed by hindered IMesAuCl was noted, affording cyclohexene derivative 107 preferentially, although with diminished total yield.
3.5. NHC-BIAN-Cu and NHC-BIAN-Pt
N-Heterocyclic carbene complexes of copper and platinum possessing an acenaphthylene backbone subunit have been marginally reported to date [53,70,97], but have found catalytic applications.
In 2018, Shi and co-workers [53] developed an efficient synthesis of chiral NHC copper complex 111 by conducting the reaction of chiral NHC precursor 20a with CuCl (Scheme 43). The respective copper complex 111 was applied in the asymmetric hydroboration reaction, proceeding according to Markovnikov’s regioselectivity rule, and providing a plethora of secondary alkylboronic esters as the major products (109) [53]. It should be mentioned that the application of classic achiral imidazolylidene-based NHC ligand afforded the product with much lower regioselectivity, demonstrating the superior properties of BIAN-type derivatives.
In the same year, Tu’s group [70] proved the high catalytic activity of N-heterocyclic carbene copper complexes in the Sonogashira reaction. Copper complex 36b and palladium complex 25a were used as the catalytic system in the coupling reaction of 1-phenylacetylene (21) with bromoanisole 29b during the optimization studies. The reaction led to disubstituted alkyne 34 with varying yields, depending on the base and solvent used (Scheme 44). The highest efficiency of alkyne formation was achieved with the application of the K2CO3 base and DMSO as solvent. It turned out that the solvent had a crucial role in the presented reaction, since the replacement of DMSO with THF caused significant decrease in the yield of alkyne 34 (Table 2).
Literature reports on NHC-BIAN-Pt complexes cover only a single example. In 2016, Tobisu and co-workers [97] published a synthesis of the platinum N-heterocyclic carbene complex 113 by the reaction of NHC precursor 4g with Karstedt’s complex, yielding complex 113 in 36% yield (Scheme 45). The authors reported high catalytic activity of platinum complex 113 in the C-H activation/borylation reaction of 1,3-dimethyl-5-tert-butylbenzene (112), affording arylboronic ester 40b in 72% yield.
3.6. Synthesis of Other Metal Complexes (Rh, Ir, Ag)
N-Heterocyclic carbene complexes of other metals have been marginally reported in the literature and have not found practical application in catalysis. In particular, synthetic approaches to rhodium, iridium, and silver complexes will be presented in this subsection.
Rhodium complexes could be synthesised directly from the corresponding salts in the presence of a strong base, e.g., a metal alkoxide, and a dimer of rhodium(I) chloride [RhCl(COD)]2 [17]. Plenio and co-workers unequivocally proved that the replacement of alkoxide by K2CO3 and the use of acetone as solvent allow for the preparation of rhodium complexes 114 under mild conditions (Scheme 46) [98]. It should be noted that the combinations of K2CO3/acetone for the synthesis of NHC metal complexes, originally developed by Nolan and co-workers for the synthesis of gold(I) complexes, could be successfully applied for much more sensitive rhodium compounds [91].
Analogous conditions could be used for the preparation of iridium complexes. Cowley [20] and Plenio [47] described an efficient approach to sterically hindered complexes via in situ generation of carbenes in the presence of a suitable alkoxide, and their further reaction with iridium dimer [IrCl(COD)]2 to provide complexes 116 in very high yield (Scheme 47). Further ligand exchange with gaseous carbon monoxide furnished NHCIr(CO)3 complexes in virtually quantitative yield. Identical reaction conditions were successfully applied for the synthesis of bifunctional iridium and rhodium complexes 118 by Alcarazo and co-workers (Scheme 48) [51]. In contrast, Liu and co-workers [46] proved that NHC-Ir(CO)3 complexes 117 syntheses could be simplified to one-pot protocol applying Nolan’s method (K2CO3/acetone) under low pressure of carbon monoxide (Scheme 47). Unfortunately, none of the presented NHC-BIAN-Ir or NHC-BIAN-Rh complexes have found a practical application. It should be noted that the partially reduced NHC-BIAN-Ir complex (not shown), developed by Dastgir and Green [99] was investigated in the hydroformylation of 1-octene. However, a mixture of regioisomeric alcohols and aldehydes was obtained.
The last group of complexes, deserving special attention due to the popularity in the synthesis of other metal complexes by transmetalation, are the silver complexes [5]. The most common conditions for the synthesis of silver complexes involve a stoichiometric amount of Ag2O (in DCM with exclusion of light), acting as a base and metal source simultaneously. A series of NHC-BIAN-AgCl complexes was obtained under those classical conditions at rt (Scheme 49, structures 98a [20] and 98c [41]). However, a higher temperature was required to secure the full conversion of the starting NHC precursors in the reaction of sterically hindered derivatives leading to complexes 98d,e [17] and 98b [47] (Scheme 49).
3.7. Influence of Backbone Modification on Catalytic Activity
As mentioned in the introduction, direct comparison of electronic and steric properties of NHC-BIAN type ligands has revealed that the %Vbur parameter (describing the steric properties), as well as electronic properties have similar values as their congener, namely, IPr* ligand (and its derivatives). In addition, comparative analysis of selected X-ray crystallographic data for IPr*-BIAN-AgCl, IPr*-BIAN-AuCl, IPr*-BIAN-AgCl and IPr*-BIAN-AuCl (for details, see Figure 3) has confirmed that the respective bonds distances, such as Ccarbene-metal are also similar. Furthermore, the structural analysis has proved that the angles between N-wingtip substituents are higher in the case of IPr*-BIAN-MetCl complexes (around 150°) than in the case of IPr*MetCl complexes (around 139°). These data contradict some suggestions that the π-expanded rigid acenaphthylene backbone pushes N-wingtip aryl substituents closer to the metallic center [19]. In fact, analysis of X-ray crystallographic data has shown that the situation in the solid state is reversed. For these reasons, we could expect that the excellent activity of NHC-BIAN-MetX complexes arises from subtle conformational changes in solution, and in-depth theoretical and experimental studies are highly desirable to shed some light on this unclear aspect. Further studies should also attempt to correlate the parameters that determine the stereoelectronic properties of the ligands with their reactivities. Unfortunately, no systematic investigations on the effect of the backbone modification have appeared. Even worse, direct comparisons of catalytic activity with commonly applied ligands such as IMes, IPr or IPr* are rarely provided. To exemplify the significant role of the backbone modification, some selected examples will be shown in the present chapter.
The first examples of differential reactivity come from Tu’s [60] and Organ’s [15] work. The authors have investigated palladium complexes 25a and 120 in the Negishi coupling (Scheme 50). Although a direct comparison is not possible due to different solvents used and the steric hindrance around the palladium center, NHC-BIAN-Pd 25a afforded the product with comparable yield with four-fold lower loading than for the dichloro derivative 120. Irrespective of the ligand structure, the selectivity of the reaction (branched vs. linear product) remained at a very high level. Further studies of Tu’s [60] group have shown comparable yields in the case of Negishi reaction with cyclic organozinc compounds.
A clear trend in reactivity with regard to the NHC ligand backbone modifications has been described by Organ and Feringa in the Murahashi cross-coupling (Scheme 51) [69]. The authors observed that placing chloride atoms on the NHC core (structure 120), and further extension of the skeleton to an acenaphthylene subunit (structure 25t) improved significantly the yield of the substituted naphthalene 30e. This trend is especially visible at −78 °C, where complex 121 is unreactive whereas NHC-PEPPSI-Pd 25t provided the product in 37% yield.
An interesting example of the sensitivity towards backbone modification, and its direct influence on catalytic activity has been presented by Liu and co-workers [68] (Scheme 52). During optimization studies on the Suzuki reaction, the authors have noted that extension of the NHC core provided product 30f with slightly better 56% yield. Rather unexpectedly, introduction of two tert-butyl groups in the remote position of the acenaphthylene skeleton improved significantly the yield of the biphenyl derivative 30f. Bearing in mind that the Suzuki cross-coupling was conducted in aerobic conditions, the remarkable activity of 27 was explained by preventing the palladium catalytic species to form the catalytically inactive peroxo NHC-Pd(O-O) complex. In particular, the authors suggested that the presence of bulky tert-butyl groups in anti-orientation inhibits CAr-N bond rotation and prevents the oxidation of the palladium center.
A detailed study focused on the modification of NHC skeleton has been performed by Ye and co-workers in the reductive coupling of imines 62a with alkynes 63a (Scheme 53) [80]. The authors investigated a selected group of phosphines and NHC ligands for this transformation in the optimization studies. They unambiguously proved that phosphines, including electron-rich ones such as Cy3P or (tert-Bu)3P, were ineffective in this reaction, whereas the yield of the allylic amines was strictly dependent on the NHC ligand used. More electron-rich carbene ligands afforded the product with a higher yield (IPr vs. SIPr), while electron-deficient ligands such as naphthoquinone-derived carbene 126 gave amine 64a a low 15% yield. The best results in terms of yield were achieved with IPr-acenaphthylene ligand 4g. The authors also mentioned that less sterically-hindered IMes derivatives (not shown) were less effective [80].
Similar conclusions could be drawn for Ni-catalyzed [3+2] cross-dimerization of methylene cyclopropanes with alkynes, developed by Ho and co-workers (Scheme 54) [81]. Among NHC ligands selected in the initial optimization, the authors reported a higher yield for more electron-rich ligands (IPrCl 123b vs. IPr 123a). Also in this case, acenaphthylene-derived ligand 123d provided methylene cyclopentene 68 with the best yield. It should be mentioned that no clear influence of NHC ligand on selectivity (cross-dimerization vs. homodimerization or oligomerization) could be provided.
The most interesting and impressive impact of ligand structure arises from the feasibility for the development of efficient enantioselective processes, where subtle modifications can significantly affect the level of enantioselectivity of the reaction. These rare examples could also be found in the group of carbene ligands bearing an acenaphthylene backbone.
Cramer and co-workers have performed a detailed analysis of the effect of substituents at the 4,5-positions of the imidazole ring in the NHC ligand on the catalytic performance of nickel complexes catalyzing enantioselective pyridone C–H functionalization (Scheme 55) [52]. The application of the NHC-BIAN type ligand (20a) gave promising results (80% ee), but comparable to other complexes (124a–c). Further increase in steric shielding around the metal center by the introduction of sterically crowded 3,5-xylyl as the wingtip N-aryl substituent (structure 20c) resulted in higher enantioselectivity, namely 92% ee. This unambiguously proved the significance of the acenaphthylene motif in comparison to complex 124d.
The influence of substituent at positions 4 and 5 of the NHC core on the enantioselectivity was further rationalized by Cramer and co-workers [52] by careful analysis of steric properties of ligands used (Figure 4). The steric features of a given ligand could be easily quantified by the percent buried volume [23,24] (%Vbur) in a global sense or, more precisely, by topographic steric maps. It should be mentioned that topographic steric maps express steric hindrance around the metal center in quadrants, which can be useful for identification of the catalytic pocket or origin of enantioselectivity [100].
Direct comparison of %Vbur for Ni, Cu or Au complexes did not appear to be decisive for the correlation of enantioselectivity level with steric hindrance for the enantioselective pyridone C–H functionalization [52]. In particular, the value of %Vbur estimated by web application SambVca 2.1 [101] was higher for imidazolium NHC(Cp)NiCl 125a and NHCAuCl 126a complexes in comparison to the acenaphthylene congeners (125b and 126b, respectively), while the situation for copper complexes 127 and 111 was the opposite. More detailed analysis of steric congestion by steric maps revealed that the acenaphthene backbone affects the arrangement of aromatic substituents around the metallic center. In particular, the acenaphthene backbone forces the spatial arrangement of the flanking aryl groups creating C2-symmetric binding pocket, with accessible NW and SE quadrants. In contrast, C2-symmetry is less pronounced for the imidazolium derivatives, as was readily observed for the gold complexes (126a).
Similar observations have been made by Shi for copper-catalyzed borylation of terminal alkenes (Scheme 56) [53]. The substituents at the 4- and 5-position in the imidazole ring of copper NHC complexes significantly affected the catalytic properties. The use of acenaphthene-derived complex 111 increased the regioselectivity of the addition and enantioselectivity in product 109 formation in comparison to chiral IPr derivative 127.
4. Conclusions and Outlook
Since the first synthesis of an NHC-BIAN-type ligand almost 15 years ago, this family of ligands have enjoyed growing interest in catalysis as the ancillary ligands for transition-metal catalysis. The unique π-extended backbone and ease of modification of N-wingtip substituents have brought many excellent carbene ligands, in particular for palladium, nickel, and gold catalysis. The excellent stability of NHC-BIAN transition metal complexes allowed to perform many cross-coupling reactions of challenging low-reactivity compounds under aerobic conditions at very low catalyst loading, not accessible with classical imidazolium or imidazolinium salts. Furthermore, the unique reactivity allowed to develop new exciting transformations, such as gold-catalyzed sulfonylation.
The excellent catalytic performance has recently been translated into the area of enantioselective catalysis. Although many chiral NHC ligand precursors have been developed so far, there is still lack of privileged structures in this field. The chiral C2 symmetric NHC-BIAN type carbene precursor celebrated an enormous triumph in the last years, enabling the development of cutting-edge transformations, in particular Ni-catalyzed transformations.
Despite the significant step forward with NHC-BIAN-type ligands in catalysis, some challenges have remained. First, the remarkable activity of NHC-BIAN-type ligands have been ascribed the so-called “buttressing effect”, resulting in higher steric hindrance than the classical imidazolium ligands. In addition, many authors suggested that NHC-BIAN-type ligands are stronger σ-donors compared to imidazolium carbenes. As exemplified in this review, no clear evidence for higher steric hindrance or modulated electronic properties could be gained from IR spectroscopy, electrochemical studies or X-ray analyses. For these reasons, in-depth investigation of electronic and steric parameters of NHC-BIAN ligands and their metal complexes is required for better understanding and, hence, further development of the field. The second important feature to be resolved is the impractical and user-unfriendly synthetic approach to chiral NHC-BIAN type ligands via enantioselective reduction using a very expensive bisphosphine DuanPhos ligand. Although chiral NHC-BIAN type ligands have shown excellent activity, a user-friendly low-cost protocol must be developed to make NHC-BIAN ligands privileged ones in asymmetric catalysis.
Funding
Financial support for this work was provided by Polish National Science Centre; Grant SONATA BIS 2017/26/E/ST5/00510).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Artur Ulikowski for helpful discussion during manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, T.W.; Ugarte, R.A.; Perera, T.A. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools (2); The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 178–237. [Google Scholar]
- Doddi, A.; Peters, M.; Tamm, M. N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry. Chem. Rev. 2019, 119, 6994–7112. [Google Scholar] [CrossRef]
- Michalak, M.; Kośnik, W. Chiral N-heterocyclic Carbene Gold Complexes: Synthesis and Applications in Catalysis. Catalysts 2019, 9, 890. [Google Scholar] [CrossRef] [Green Version]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef] [PubMed]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-Heterocyclic Carbene Complexes of Copper, Nickel, and Cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef] [PubMed]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef]
- Janssen-Muller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Jin, Z.; Chi, Y.R. NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions. Chem. Sci. 2021, 12, 5037–5043. [Google Scholar] [CrossRef]
- Das, T.K.; Biju, A.T. Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 1–82. [Google Scholar]
- Chen, X.-Y.; Gao, Z.-H.; Ye, S. Bifunctional N-Heterocyclic Carbenes Derived from l-Pyroglutamic Acid and Their Applications in Enantioselective Organocatalysis. Acc. Chem. Res. 2020, 53, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Sau, S.C.; Hota, P.K.; Mandal, S.K.; Soleilhavoup, M.; Bertrand, G. Stable abnormal N-heterocyclic carbenes and their applications. Chem. Soc. Rev. 2020, 49, 1233–1252. [Google Scholar] [CrossRef] [PubMed]
- Valente, C.; Çalimsiz, S.; Hoi, K.H.; Mallik, D.; Sayah, M.; Organ, M.G. The Development of Bulky Palladium NHC Complexes for the Most-Challenging Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2012, 51, 3314–3332. [Google Scholar] [CrossRef]
- Froese, R.D.J.; Lombardi, C.; Pompeo, M.; Rucker, R.P.; Organ, M.G. Designing Pd–N-Heterocyclic Carbene Complexes for High Reactivity and Selectivity for Cross-Coupling Applications. Acc. Chem. Res. 2017, 50, 2244–2253. [Google Scholar] [CrossRef]
- Savka, R.; Plenio, H. Metal Complexes of Very Bulky N,N′-Diarylimidazolylidene N-Heterocyclic Carbene (NHC) Ligands with 2,4,6-Cycloalkyl Substituents. Eur. J. Inorg. Chem. 2014, 2014, 6246–6253. [Google Scholar] [CrossRef]
- Huynh, H.V. Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination. Chem. Rev. 2018, 118, 9457–9492. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, F.-S.; Szostak, M. BIAN-NHC Ligands in Transition-Metal-Catalysis: A Perfect Union of Sterically Encumbered, Electronically Tunable N-Heterocyclic Carbenes? Chem. Eur. J. 2021, 27, 4478–4499. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.V.; Butorac, R.R.; Abernethy, C.D.; Cowley, A.H. Synthesis and coordination compounds of a bis(imino)acenaphthene (BIAN)-supported N-heterocyclic carbene. Dalton Trans. 2010, 39, 7401–7408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Frémont, P.; Scott, N.M.; Stevens, E.D.; Ramnial, T.; Lightbody, O.C.; Macdonald, C.L.B.; Clyburne, J.A.C.; Abernethy, C.D.; Nolan, S.P. Synthesis of Well-Defined N-Heterocyclic Carbene Silver(I) Complexes. Organometallics 2005, 24, 6301–6309. [Google Scholar] [CrossRef]
- Fructos, M.R.; Belderrain, T.R.; de Frémont, P.; Scott, N.M.; Nolan, S.P.; Díaz-Requejo, M.M.; Pérez, P.J. A Gold Catalyst for Carbene-Transfer Reactions from Ethyl Diazoacetate. Angew. Chem. Int. Ed. 2005, 44, 5284–5288. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef] [Green Version]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Gottumukkala, A.L.; Teichert, J.F.; Heijnen, D.; Eisink, N.; van Dijk, S.; Ferrer, C.; van den Hoogenband, A.; Minnaard, A.J. Pd-diimine: A highly selective catalyst system for the base-free oxidative Heck reaction. J. Org. Chem. 2011, 76, 3498–3501. [Google Scholar] [CrossRef]
- Wang, J.; Ganguly, R.; Yongxin, L.; Díaz, J.; Soo, H.S.; García, F. Synthesis and the Optical and Electrochemical Properties of Indium(III) Bis(arylimino)acenaphthene Complexes. Inorg. Chem. 2017, 56, 7811–7820. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Zysman-Colman, E. Synthesis, UV–Vis and CV properties of a structurally related series of bis(Arylimino)acenaphthenes (Ar-BIANs). J. Phys. Org. Chem. 2013, 26, 274–279. [Google Scholar] [CrossRef]
- Evans, D.A.; Lee, L.M.; Vargas-Baca, I.; Cowley, A.H. Aggregation-Induced Emission of Bis(imino)acenaphthene Zinc Complexes: Photophysical Tuning via Methylation of the Flanking Aryl Substituents. Organometallics 2015, 34, 2422–2428. [Google Scholar] [CrossRef]
- Gasperini, M.; Ragaini, F.; Cenini, S. Synthesis of Ar-BIAN Ligands (Ar-BIAN = Bis(aryl)acenaphthenequinonediimine) Having Strong Electron-Withdrawing Substituents on the Aryl Rings and Their Relative Coordination Strength toward Palladium(0) and -(II) Complexes. Organometallics 2002, 21, 2950–2957. [Google Scholar] [CrossRef]
- Merino, E.; Poli, E.; Díaz, U.; Brunel, D. Synthesis and characterization of new ruthenium N-heterocyclic carbene Hoveyda II-type complexes. Study of reactivity in ring closing metathesis reactions. Dalton Trans. 2012, 41, 10913–10918. [Google Scholar] [CrossRef]
- Butorac, R.R.; Al-Deyab, S.S.; Cowley, A.H. Syntheses, structures and antimicrobial activities of bis(imino)acenaphthene (BIAN) imidazolium salts. Molecules 2011, 16, 3168–3178. [Google Scholar] [CrossRef]
- El-Ayaan, U.; Murata, F.; El-Derby, S.; Fukuda, Y. Synthesis, structural and solvent influence studies on solvatochromic mixed-ligand copper(II) complexes with the rigid nitrogen ligand: Bis[N-(2,4,6-trimethylphenyl)imino]acenaphthene. J. Mol. Struct. 2004, 692, 209–216. [Google Scholar] [CrossRef]
- Mak, C.S.K.; Wong, H.L.; Leung, Q.Y.; Tam, W.Y.; Chan, W.K.; Djurišić, A.B. The use of sublimable chlorotricarbonyl bis(phenylimino)acenaphthene rhenium(I) complexes as photosensitizers in bulk-heterojunction photovoltaic devices. J. Organomet. Chem. 2009, 694, 2770–2776. [Google Scholar] [CrossRef] [Green Version]
- Coventry, D.N.; Batsanov, A.S.; Goeta, A.E.; Howard, J.A.K.; Marder, T.B. Synthesis and molecular structures of α-diimines and their zinc and palladium dichloride complexes. Polyhedron 2004, 23, 2789–2795. [Google Scholar] [CrossRef]
- Tu, T.; Sun, Z.; Fang, W.; Xu, M.; Zhou, Y. Robust Acenaphthoimidazolylidene Palladium Complexes: Highly Efficient Catalysts for Suzuki–Miyaura Couplings with Sterically Hindered Substrates. Org. Lett. 2012, 14, 4250–4253. [Google Scholar] [CrossRef] [PubMed]
- Tronnier, A.; Pöthig, A.; Metz, S.; Wagenblast, G.; Münster, I.; Strassner, T. Enlarging the π System of Phosphorescent (C^C*) Cyclometalated Platinum(II) NHC Complexes. Inorg. Chem. 2014, 53, 6346–6356. [Google Scholar] [CrossRef] [PubMed]
- van Asselt, R.; Elsevier, C.J.; Smeets, W.J.J.; Spek, A.L.; Benedix, R. Synthesis and characterization of rigid bidentate nitrogen ligands and some examples of coordination to divalent palladium. X-ray crystal structures of bis (p-tolylimino) acenaphthene and methylchloro [bis(o,o′-diisopropylphenyl-imino) acenaphthene] palladium (II). Recl. Trav. Chim. Pays-Bas 1994, 113, 88–98. [Google Scholar] [CrossRef]
- Balogh, J.; Hlil, A.R.; El-Zoghbi, I.; Rafique, M.G.; Chouikhi, D.; Al-Hashimi, M.; Bazzi, H.S. Phase-Separable Polyisobutylene Palladium-PEPPSI Precatalysts: Synthesis and Application in Buchwald–Hartwig Amination. Macromol. Rapid. Comm. 2017, 38, 1700214. [Google Scholar] [CrossRef]
- Wang, J.; Ganguly, R.; Yongxin, L.; Díaz, J.; Soo, H.S.; García, F. A multi-step solvent-free mechanochemical route to indium(iii) complexes. Dalton Trans. 2016, 45, 7941–7946. [Google Scholar] [CrossRef] [Green Version]
- Rhinehart, J.L.; Mitchell, N.E.; Long, B.K. Enhancing α-Diimine Catalysts for High-Temperature Ethylene Polymerization. ACS Catal. 2014, 4, 2501–2504. [Google Scholar] [CrossRef]
- Guo, L.; Kong, W.; Xu, Y.; Yang, Y.; Ma, R.; Cong, L.; Dai, S.; Liu, Z. Large-scale synthesis of novel sterically hindered acenaphthene-based α-diimine ligands and their application in coordination chemistry. J. Organomet. Chem. 2018, 859, 58–67. [Google Scholar] [CrossRef]
- Ma, X.; Hu, X.; Zhang, Y.; Mu, H.; Cui, L.; Jian, Z. Preparation and in situ chain-end-functionalization of branched ethylene oligomers by monosubstituted α-diimine nickel catalysts. Polym. Chem. 2019, 10, 2596–2607. [Google Scholar] [CrossRef]
- Lu, D.-D.; He, X.-X.; Liu, F.-S. Bulky Yet Flexible Pd-PEPPSI-IPentAn for the Synthesis of Sterically Hindered Biaryls in Air. J. Org. Chem. 2017, 82, 10898–10911. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.; Gong, Q.; Zhang, S.; Liu, B.; Dai, S. Systematic Investigations of Ligand Steric Effects on α-Diimine Nickel Catalyzed Olefin Polymerization and Copolymerization. Organometallics 2019, 38, 2919–2926. [Google Scholar] [CrossRef]
- Lan, X.-B.; Li, Y.; Li, Y.-F.; Shen, D.-S.; Ke, Z.; Liu, F.-S. Flexible Steric Bulky Bis(Imino)acenaphthene (BIAN)-Supported N-Heterocyclic Carbene Palladium Precatalysts: Catalytic Application in Buchwald–Hartwig Amination in Air. J. Org. Chem. 2017, 82, 2914–2925. [Google Scholar] [CrossRef]
- Zhang, F.-Y.; Lan, X.-B.; Xu, C.; Yao, H.-G.; Li, T.; Liu, F.-S. Rigid hindered N-heterocyclic carbene palladium precatalysts: Synthesis, characterization and catalytic amination. Org. Chem. Front. 2019, 6, 3292–3299. [Google Scholar] [CrossRef]
- Savka, R.; Foro, S.; Plenio, H. Pentiptycene-based concave NHC-metal complexes. Dalton Trans. 2016, 45, 11015–11024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyatos, M.; Mata, J.A.; Peris, E. Complexes with Poly(N-heterocyclic carbene) Ligands: Structural Features and Catalytic Applications. Chem. Rev. 2009, 109, 3677–3707. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Hiller, J.; Peris, E. Pyracene-Linked Bis-Imidazolylidene Complexes of Palladium and Some Catalytic Benefits Produced by Bimetallic Catalysts. Chem. Eur. J. 2013, 19, 10405–10411. [Google Scholar] [CrossRef]
- Vasudevan, K.V.; Findlater, M.; Cowley, A.H. Synthesis and reactivity of tetrakis(imino)pyracene (TIP) ligands; bifunctional analogues of the BIAN ligand class. Chem. Commun. 2008, 1918–1919. [Google Scholar] [CrossRef] [PubMed]
- Prades, A.; Peris, E.; Alcarazo, M. Pyracenebis(imidazolylidene): A New Janus-Type Biscarbene and Its Coordination to Rhodium and Iridium. Organometallics 2012, 31, 4623–4626. [Google Scholar] [CrossRef]
- Diesel, J.; Finogenova, A.M.; Cramer, N. Nickel-Catalyzed Enantioselective Pyridone C–H Functionalizations Enabled by a Bulky N-Heterocyclic Carbene Ligand. J. Am. Chem. Soc. 2018, 140, 4489–4493. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, X.-T.; Zhang, S.-Q.; Li, F.; Li, Y.-Q.; Ruan, L.-X.; Hong, X.; Shi, S.-L. Copper-Catalyzed Enantioselective Markovnikov Protoboration of α-Olefins Enabled by a Buttressed N-Heterocyclic Carbene Ligand. Angew. Chem. Int. Ed. 2018, 57, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Diesel, J.; Grosheva, D.; Kodama, S.; Cramer, N. A Bulky Chiral N-Heterocyclic Carbene Nickel Catalyst Enables Enantioselective C−H Functionalizations of Indoles and Pyrroles. Angew. Chem. Int. Ed. 2019, 58, 11044–11048. [Google Scholar] [CrossRef]
- Albright, A.; Eddings, D.; Black, R.; Welch, C.J.; Gerasimchuk, N.N.; Gawley, R.E. Design and Synthesis of C2-Symmetric N-Heterocyclic Carbene Precursors and Metal Carbenoids. J. Org. Chem. 2011, 76, 7341–7351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spahn, E.; Albright, A.; Shevlin, M.; Pauli, L.; Pfaltz, A.; Gawley, R.E. Double-Asymmetric Hydrogenation Strategy for the Reduction of 1,1-Diaryl Olefins Applied to an Improved Synthesis of CuIPhEt, a C2-Symmetric N-Heterocyclic Carbenoid. J. Org. Chem. 2013, 78, 2731–2735. [Google Scholar] [CrossRef] [Green Version]
- Organ, M.G.; Avola, S.; Dubovyk, I.; Hadei, N.; Kantchev, E.A.B.; O’Brien, C.J.; Valente, C. A User-Friendly, All-Purpose Pd–NHC (NHC=N-Heterocyclic Carbene) Precatalyst for the Negishi Reaction: A Step Towards a Universal Cross-Coupling Catalyst. Chem. Eur. J. 2006, 12, 4749–4755. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem. Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Fang, W.; Jiang, J. A highly efficient precatalyst for amination of aryl chlorides: Synthesis, structure and application of a robust acenaphthoimidazolylidene palladium complex. Chem. Commun. 2011, 47, 12358–12360. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, N.; Xu, M.; Sun, Z.; Tu, T. Mild Negishi Cross-Coupling Reactions Catalyzed by Acenaphthoimidazolylidene Palladium Complexes at Low Catalyst Loadings. J. Org. Chem. 2013, 78, 7436–7444. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, H.; Shen, Y.; Tu, T. Acenaphthoimidazolium chloride-enabled nickel-catalyzed amination of bulky aryl tosylates. Org. Chem. Front. 2014, 1, 1172–1175. [Google Scholar] [CrossRef]
- Hu, L.-Q.; Deng, R.-L.; Li, Y.-F.; Zeng, C.-J.; Shen, D.-S.; Liu, F.-S. Developing Bis(imino)acenaphthene-Supported N-Heterocyclic Carbene Palladium Precatalysts for Direct Arylation of Azoles. Organometallics 2018, 37, 214–226. [Google Scholar] [CrossRef]
- Huang, F.-D.; Xu, C.; Lu, D.-D.; Shen, D.-S.; Li, T.; Liu, F.-S. Pd-PEPPSI-IPentAn Promoted Deactivated Amination of Aryl Chlorides with Amines under Aerobic Conditions. J. Org. Chem. 2018, 83, 9144–9155. [Google Scholar] [CrossRef]
- Ouyang, J.-S.; Li, Y.-F.; Huang, F.-D.; Lu, D.-D.; Liu, F.-S. The Highly Efficient Suzuki–Miyaura Cross-Coupling of (Hetero)aryl Chlorides and (Hetero)arylboronic Acids Catalyzed by “Bulky-yet-Flexible” Palladium–PEPPSI Complexes in Air. ChemCatChem 2018, 10, 371–375. [Google Scholar] [CrossRef]
- Leuthäußer, S.; Schmidts, V.; Thiele, C.M.; Plenio, H. π-Face Donor Properties of N-Heterocyclic Carbenes in Grubbs II Complexes. Chem. Eur. J. 2008, 14, 5465–5481. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.H.; Konstandaras, N.; Cole, M.L.; Harper, J.B. Targeted and Systematic Approach to the Study of pKa Values of Imidazolium Salts in Dimethyl Sulfoxide. J. Org. Chem. 2017, 82, 7324–7331. [Google Scholar] [CrossRef]
- Rayner, P.; Norcott, P.; Appleby, K.; Iali, W.; John, R.; Hart, S.; Whitwood, A.; Duckett, S. Fine-tuning the efficiency of para-hydrogen-induced hyperpolarization by rational N-heterocyclic carbene design. Nat. Commun. 2018, 9, 4251. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.-B.; Chen, F.-M.; Ma, B.-B.; Shen, D.-S.; Liu, F.-S. Pd-PEPPSI Complexes Bearing Bulky [(1,2-Di-(tert-butyl)acenaphthyl] (DtBu-An) on N-Heterocarbene Backbones: Highly Efficient for Suzuki–Miyaura Cross-Coupling under Aerobic Conditions. Organometallics 2016, 35, 3852–3860. [Google Scholar] [CrossRef]
- Sinha, N.; Heijnen, D.; Feringa, B.L.; Organ, M.G. Murahashi Cross-Coupling at −78 °C: A One-Pot Procedure for Sequential C−C/C−C, C−C/C−N, and C−C/C−S Cross-Coupling of Bromo-Chloro-Arenes. Chem. Eur. J. 2019, 25, 9180–9184. [Google Scholar] [CrossRef]
- Shen, A.; Wu, Z.; Fang, Y.; Yang, J.; Zhu, H.; Tu, T. A Concerted Catalytic System for Sonogashira Coupling Reactions: Combination of N-Heterocyclic Carbene Palladium and Copper Complexes. Asian J. Org. Chem. 2018, 7, 1113–1117. [Google Scholar] [CrossRef]
- Fang, W.; Deng, Q.; Xu, M.; Tu, T. Highly Efficient Aminocarbonylation of Iodoarenes at Atmospheric Pressure Catalyzed by a Robust Acenaphthoimidazolyidene Allylic Palladium Complex. Org. Lett. 2013, 15, 3678–3681. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shen, Y.; Deng, Q.; Chen, J.; Tu, T. Pd(NHC)-catalyzed alkylsulfonylation of boronic acids: A general and efficient approach for sulfone synthesis. Chem. Commun. 2017, 53, 12473–12476. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, Q.; Fang, W.; Gong, J.-F.; Song, M.-P.; Xu, M.; Tu, T. Efficient and scalable Pd-catalyzed double aminocarbonylations under atmospheric pressure at low catalyst loadings. Org. Chem. Front. 2014, 1, 1261–1265. [Google Scholar] [CrossRef]
- Deng, Q.; Zhang, Y.; Zhu, H.; Tu, T. Robust Acenaphthoimidazolylidene Palladacycles: Highly Efficient Catalysts for the Amination of N-Heteroaryl Chlorides. Chem. Asian. J. 2017, 12, 2364–2368. [Google Scholar] [CrossRef]
- Deng, Q.; Shen, Y.; Zhu, H.; Tu, T. A magnetic nanoparticle-supported N-heterocyclic carbene-palladacycle: An efficient and recyclable solid molecular catalyst for Suzuki–Miyaura cross-coupling of 9-chloroacridine. Chem. Commun. 2017, 53, 13063–13066. [Google Scholar] [CrossRef]
- Cao, Q.; Howard, J.L.; Wheatley, E.; Browne, D.L. Mechanochemical Activation of Zinc and Application to Negishi Cross-Coupling. Angew. Chem. Int. Ed. 2018, 57, 11339–11343. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Nicholson, W.; Jones, A.; Browne, D. Robust Buchwald–Hartwig amination enabled by ball-milling. Org. Biomol. Chem. 2018, 17, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Makida, Y.; Meiries, S.; Slawin, A.M.Z.; Nolan, S.P. Enhanced Activity of [Ni(NHC)CpCl] Complexes in Arylamination Catalysis. Organometallics 2013, 32, 6265–6270. [Google Scholar] [CrossRef]
- Ben Halima, T.; Masson-Makdissi, J.; Newman, S.G. Nickel-Catalyzed Amide Bond Formation from Methyl Esters. Angew. Chem. Int. Ed. 2018, 57, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.-W.; Li, R.; Li, J.-F.; Sun, J.; Ye, M. NHC ligand-enabled Ni-catalyzed reductive coupling of alkynes and imines using isopropanol as a reductant. Green Chem. 2019, 21, 2240–2244. [Google Scholar] [CrossRef]
- Huang, J.-Q.; Ho, C.-Y. NHC/Nickel(II)-Catalyzed [3+2] Cross-Dimerization of Unactivated Olefins and Methylenecyclopropanes. Angew. Chem. Int. Ed. 2020, 59, 5288–5292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-B.; Yang, X.-T.; Ma, J.-B.; Su, Z.-M.; Shi, S.-L. Regio- and Enantioselective C–H Cyclization of Pyridines with Alkenes Enabled by a Nickel/N-Heterocyclic Carbene Catalysis. J. Am. Chem. Soc. 2019, 141, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-C.; Xie, P.-P.; Xu, Y.; Hong, X.; Shi, S.-L. Low-Temperature Nickel-Catalyzed C−N Cross-Coupling via Kinetic Resolution Enabled by a Bulky and Flexible Chiral N-Heterocyclic Carbene Ligand. Angew. Chem. Int. Ed. 2021, 60, 16077–16084. [Google Scholar] [CrossRef] [PubMed]
- Grela, K.; Harutyunyan, S.; Michrowska, A. A Highly Efficient Ruthenium Catalyst for Metathesis Reactions. Angew. Chem. Int. Ed. 2002, 41, 4038–4040. [Google Scholar] [CrossRef] [Green Version]
- Hlil, A.R.; Moncho, S.; Tuba, R.; Elsaid, K.; Szarka, G.; Brothers, E.N.; Grubbs, R.H.; Al-Hashimi, M.; Bazzi, H.S. Synthesis and catalytic activity of supported acenaphthoimidazolylidene N-heterocyclic carbene ruthenium complex for ring closing metathesis (RCM) and ring opening metathesis polymerization (ROMP). J. Catal. 2016, 344, 100–107. [Google Scholar] [CrossRef]
- Butorac, R.; Al-Deyab, S.; Cowley, A. Antimicrobial Properties of Some Bis(Iminoacenaphthene (BIAN)-Supported N-Heterocyclic Carbene Complexes of Silver and Gold. Molecules 2011, 16, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Elzatahry, A.A.; Al-Enizi, A.M.; Elsayed, E.A.; Butorac, R.R.; Al-Deyab, S.S.; Wadaan, M.A.M.; Cowley, A.H. Nanofiber composites containing N-heterocyclic carbene complexes with antimicrobial activity. Int. J. Nanomed. 2012, 7, 2829–2832. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Taha, N.; Butorac, R.; Evans, D.; Elzatahry, A.; Elsayed, E.; Wadaan, M.; Al-Deyab, S.; Cowley, A. Biological Screening of Newly Synthesized BIAN N-Heterocyclic Gold Carbene Complexes in Zebrafish Embryos. Int. J. Mol. Sci. 2015, 16, 24718–24731. [Google Scholar] [CrossRef] [Green Version]
- Ciancaleoni, G.; Biasiolo, L.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D.; Belpassi, L. NHC-Gold-Alkyne Complexes: Influence of the Carbene Backbone on the Ion Pair Structure. Organometallics 2013, 32, 4444–4447. [Google Scholar] [CrossRef]
- Visbal, R.; Laguna, A.; Gimeno, M.C. Simple and efficient synthesis of [MCI(NHC)] (M = Au, Ag) complexes. Chem. Commun. 2013, 49, 5642–5644. [Google Scholar] [CrossRef]
- Collado, A.; Gomez-Suarez, A.; Martin, A.R.; Slawin, A.M.Z.; Nolan, S.P. Straightforward synthesis of [Au(NHC)X] (NHC = N-heterocyclic carbene, X = Cl, Br, I) complexes. Chem. Commun. 2013, 49, 5541–5543. [Google Scholar] [CrossRef] [PubMed]
- Plenio, H.; Heidrich, M.; Bergmann, M.; Müller-Borges, D. Bispentiptycenyl-N-Heterocyclic Carbene (NHC) Gold Complexes: Highly Active Catalysts for the Room Temperature Hydration of Alkynes. Adv. Synth. Catal. 2018, 360, 3572–3578. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Deng, Q.; Chen, J.; Tu, T. Acenaphthoimidazolylidene Gold Complex-Catalyzed Alkylsulfonylation of Boronic Acids by Potassium Metabisulfite and Alkyl Halides: A Direct and Robust Protocol to Access Sulfones. ACS Catal. 2017, 7, 4655–4659. [Google Scholar] [CrossRef]
- Gatto, M.; Del Zotto, A.; Segato, J.; Zuccaccia, D. Hydration of Alkynes Catalyzed by L–Au–X under Solvent- and Acid-Free Conditions: New Insights into an Efficient, General, and Green Methodology. Organometallics 2018, 37, 4685–4691. [Google Scholar] [CrossRef]
- Zhu, H.; Shen, Y.; Wen, D.; Le, Z.-G.; Tu, T. Selective Synthesis of ortho-Substituted Diarylsulfones by Using NHC-Au Catalysts under Mild Conditions. Org. Lett. 2019, 21, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, R.; Gong, X.; Li, Z.; Huang, Q.; Wang, H.; Song, G. Synthesis and Herbicidal Activity of N,N-Diethyl-3-(arylselenonyl)-1H-1,2,4-triazole-1-carboxamide. J. Agric. Food Chem. 2006, 54, 7724–7728. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Tobisu, M.; Chatani, N. C–H Borylation by Platinum Catalysis. Bull. Chem. Soc. Jpn. 2017, 90, 332–342. [Google Scholar] [CrossRef]
- Savka, R.; Plenio, H. Facile synthesis of [(NHC)MX(cod)] and [(NHC)MCl(CO)2] (M = Rh, Ir; X = Cl, I) complexes. Dalton Trans. 2015, 44, 891–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastgir, S.; Coleman, K.S.; Cowley, A.R.; Green, M.L.H. Stable crystalline annulated diaminocarbenes: Coordination with rhodium(i), iridium(i) and catalytic hydroformylation studies. Dalton Trans. 2009, 35, 7203–7214. [Google Scholar] [CrossRef]
- Gómez-Suárez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [Green Version]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The structure of commonly used NHC and BIAN-NHC ligands.

Figure 2.
Comparison of the electronic properties of NHC ligands with NHC-BIAN-type one.

Figure 3.
Angle between N-wingtip substituents and the selected bond distance, based on crystallographic data. IPracnAgCl [20], IPrAgCl [21], IPracnAuCl [20], IPrAuCl [22].

Scheme 1.
Structure of bisimine 3 and NHC precursors 4 bearing an acenaphthylene subunit. a The yield of bisimine 3a formation was not provided.
Scheme 1.
Structure of bisimine 3 and NHC precursors 4 bearing an acenaphthylene subunit. a The yield of bisimine 3a formation was not provided.


Scheme 2.
Synthesis of bisimines 5 and NHC precursors 6 from anilines bearing benzhydryl subunits.

Scheme 3.
Synthesis of C1-symmetric NHC precursors bearing an acenaphthylene subunit.

Scheme 4.
The synthesis of NHC precursors bearing a pentiptycene subunit.

Scheme 5.
Synthetic pathways leading to bisacenaphthylene derivatives.

Scheme 6.
Synthetic pathway to chiral C2-symmetric NHC precursor 20.

Scheme 7.
Improved synthetic pathway to chiral C2-symmetric NHC precursor.

Scheme 8.
Synthetic pathway leading to chiral C2-symmetric anilines via enantioselective reduction.

Scheme 9.
Synthetic efforts towards C2-symmetric PEPPSI-type palladium complexes.

Scheme 10.
Synthetic efforts towards C1-symmetric PEPPSI-type palladium complexes.

Scheme 11.
Application of Pd-PEPPSI-type complexes for Suzuki coupling.

Scheme 12.
Application of Pd-PEPPSI-type complexes for Negishi coupling.

Scheme 13.
Application of Pd-PEPPSI type complexes for Murahashi coupling.

Scheme 14.
Buchwald-Hartwig coupling catalyzed by Pd-PEPPSI-type complexes.

Scheme 15.
Sonogashira coupling catalyzed by Pd-PEPPSI-type complexes.

Scheme 16.
Azole arylation catalyzed by Pd-PEPPSI-type complexes.

Scheme 17.
Synthesis of π-allyl palladium complexes.

Scheme 18.
Aminocarbonylation catalyzed by π-allyl palladium complexes.

Scheme 19.
Sulfonation of boronic acids catalyzed by π-allyl palladium complexes.

Scheme 20.
Synthesis of palladium complexes bearing a chelating arm.

Scheme 21.
Synthetic approach to NHC-BIAN-Pd complexes with modified backbone.

Scheme 22.
Application of polyfunctional NHC-BIAN-Pd complexes for the acylation of aldehydes and Sonogashira coupling.
Scheme 22.
Application of polyfunctional NHC-BIAN-Pd complexes for the acylation of aldehydes and Sonogashira coupling.

Scheme 23.
Synthesis of Pd-PEPPSI complexes bearing polymer subunits and application for the Buchwald-Hartwig reaction.
Scheme 23.
Synthesis of Pd-PEPPSI complexes bearing polymer subunits and application for the Buchwald-Hartwig reaction.

Scheme 24.
Immobilization of palladium complexes on magnetic nanoparticles and their application in the Suzuki-Miyaura coupling of chloroacridines.
Scheme 24.
Immobilization of palladium complexes on magnetic nanoparticles and their application in the Suzuki-Miyaura coupling of chloroacridines.

Scheme 25.
Application of Pd-PEPPSI complexes under mechanochemical conditions.

Scheme 26.
Synthetic approach to chiral nickelocenes bearing acenaphthylene backbone.

Scheme 27.
Application of NHC-BIAN-Ni complexes for the amidation of esters.

Scheme 28.
Nickel-catalyzed addition of imines to internal alkynes.

Scheme 29.
Nickel-catalyzed Buchwald-Hartwig amination of tosylate.

Scheme 30.
Nickel-catalyzed cross-dimerization of methylidene cyclopropanes with alkenes.

Scheme 31.
Enantioselective C-H activation of pyridinones and indoles.

Scheme 32.
Enantioselective cyclization of alkenylpyridines.

Scheme 33.
Enantioselective addition of imine to disubstituted alkynes.

Scheme 34.
Enantioselective synthesis of tertiary alcohols through the arylation of ketones and tetrahydroquinolines via kinetic Buchwald-Hartwig coupling.
Scheme 34.
Enantioselective synthesis of tertiary alcohols through the arylation of ketones and tetrahydroquinolines via kinetic Buchwald-Hartwig coupling.

Scheme 35.
Ring-closing metathesis catalyzed by NHC-BIAN Hoveyda-type complexes.

Scheme 36.
Application of NHC-BIAN-Ru complexes for alkene metathesis reactions.

Scheme 37.
Synthesis of gold(I) complexes bearing sterically hindered N-wingtip substituents.

Scheme 38.
Synthetic approach to NHC-BIAN-Au complex via transmetalation.

Scheme 39.
Synthesis of NHC-BIAN-Au complexes directly from NHC precursors.

Scheme 40.
Application of NHC-BIAN-Au complexes for hydration of alkynes.

Scheme 41.
Application of NHC-BIAN-Au complexes for sulfone synthesis.

Scheme 42.
NHC-BIAN-Au-catalyzed cycloisomerization of enynes.

Scheme 43.
Enantioselective NHC-BIAN-Cu-catalyzed hydroboration of terminal alkenes.

Scheme 44.
Sonogashira reaction mediated by the simultaneous use of NHC-BIAN-Pd and NHC-BIAN-Cu complexes.
Scheme 44.
Sonogashira reaction mediated by the simultaneous use of NHC-BIAN-Pd and NHC-BIAN-Cu complexes.

Scheme 45.
Synthesis of MHC-BIAN-Pt complexes and their application for C-H activation/borylation.

Scheme 46.
Synthetic approach to NHC-BIAN-Rh complexes.

Scheme 47.
Synthetic approach to NHC-BIAN-Ir complexes.

Scheme 48.
Synthetic approach to iridium complexes bearing a polyaromatic backbone.

Scheme 49.
Synthetic approach to NHC-BIAN-Ag complexes.

Scheme 50.
Influence of backbone modification of NHC ligands on catalytic activity in the Negishi coupling.
Scheme 50.
Influence of backbone modification of NHC ligands on catalytic activity in the Negishi coupling.

Scheme 51.
Influence of backbone modification of NHC ligands on catalytic activity in the Murahashi coupling.
Scheme 51.
Influence of backbone modification of NHC ligands on catalytic activity in the Murahashi coupling.

Scheme 52.
Influence of backbone modification of NHC ligands on catalytic activity in the Suzuki coupling.
Scheme 52.
Influence of backbone modification of NHC ligands on catalytic activity in the Suzuki coupling.

Scheme 53.
Influence of backbone modification of NHC ligands on the reductive coupling of alkynes with imines.
Scheme 53.
Influence of backbone modification of NHC ligands on the reductive coupling of alkynes with imines.

Scheme 54.
Influence of backbone modification of NHC ligands on cross-dimerization of methylene cyclopropanes with alkynes.
Scheme 54.
Influence of backbone modification of NHC ligands on cross-dimerization of methylene cyclopropanes with alkynes.

Scheme 55.
Influence of backbone modification of NHC ligands on enantioselective C-H activation of 2-pyridones.
Scheme 55.
Influence of backbone modification of NHC ligands on enantioselective C-H activation of 2-pyridones.

Figure 4.
Comparison of %Vbur and steric maps for Ni, Co and Au complexes. Adapted with permission from Ref. [52] Diesel, J.; Finogenova, A.M.; Cramer, N. J. Am. Chem. Soc. 2018, 140, 4489–4493. Copyright 2021 American Chemical Society.
Figure 4.
Comparison of %Vbur and steric maps for Ni, Co and Au complexes. Adapted with permission from Ref. [52] Diesel, J.; Finogenova, A.M.; Cramer, N. J. Am. Chem. Soc. 2018, 140, 4489–4493. Copyright 2021 American Chemical Society.

Scheme 56.
Influence of the NHC ligand structure on enantioselective hydroboration of alkynes.


{kind=link}
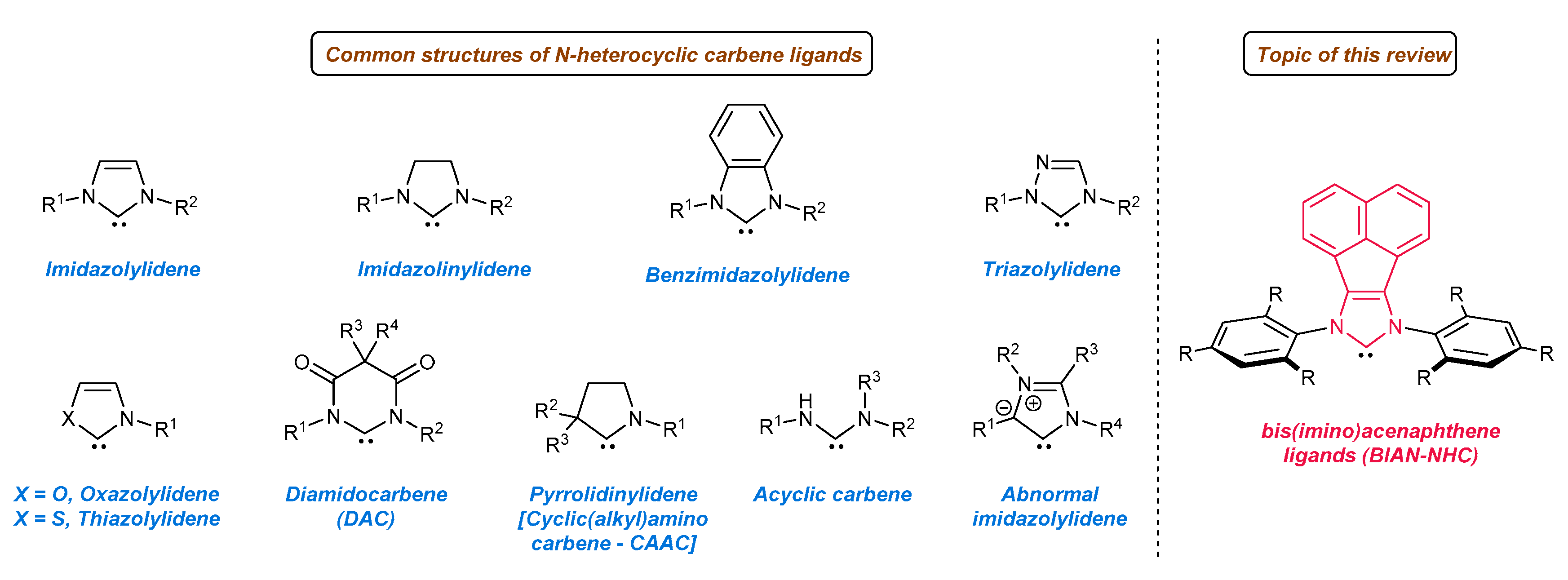{kind=link}
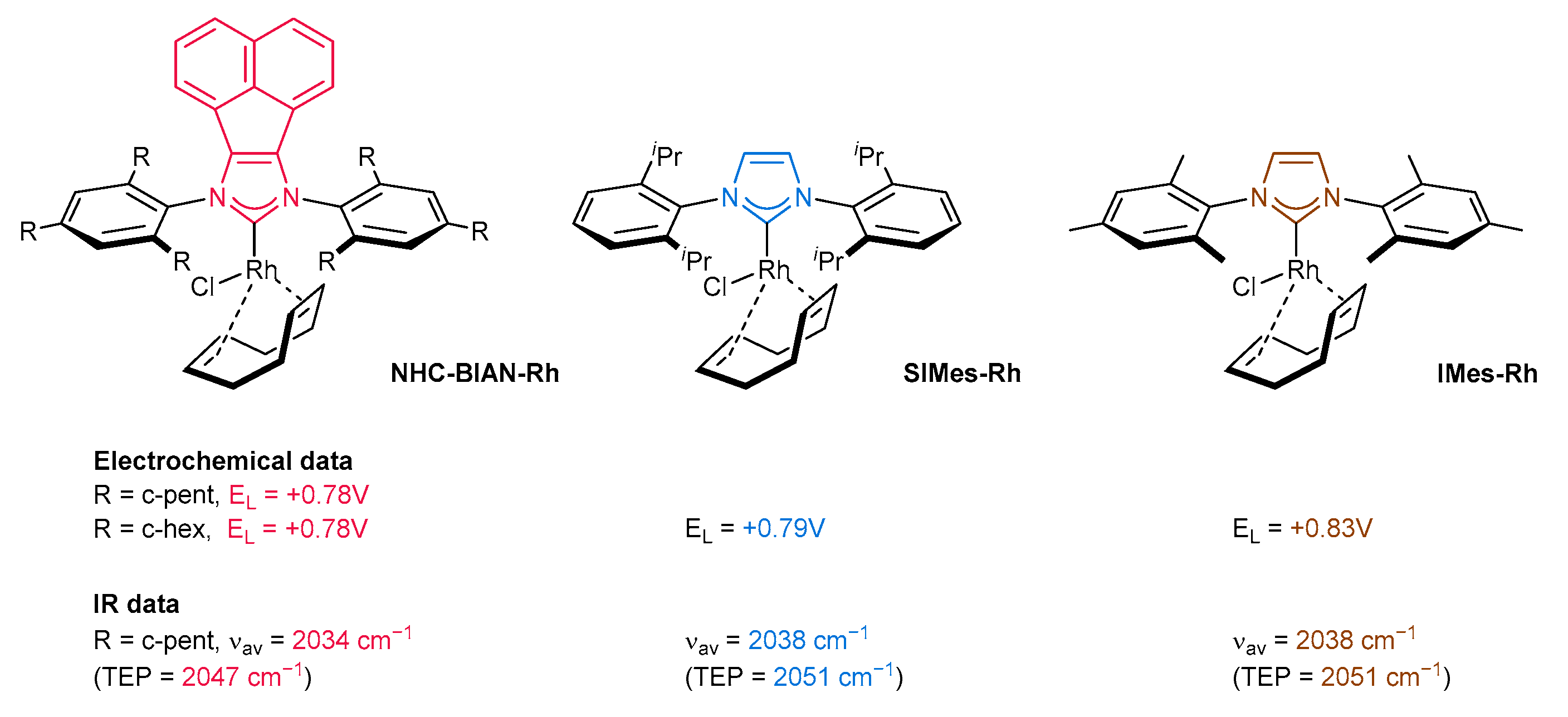{kind=link}
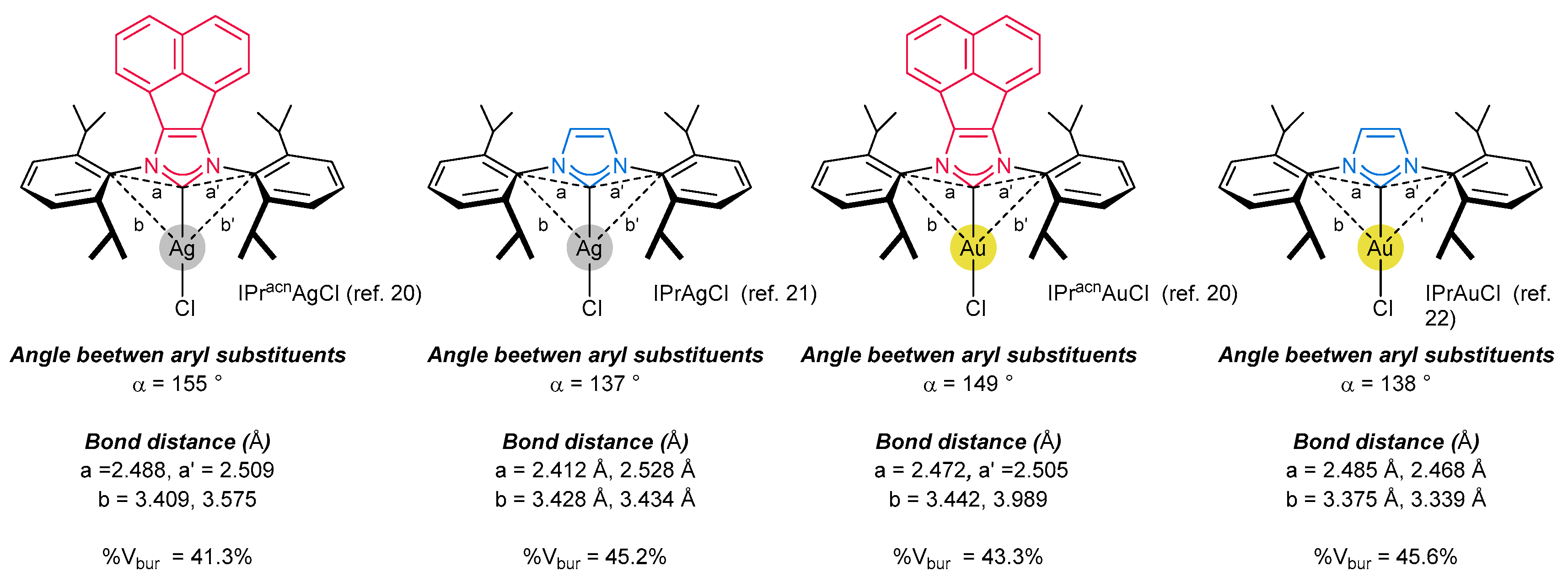{kind=link}
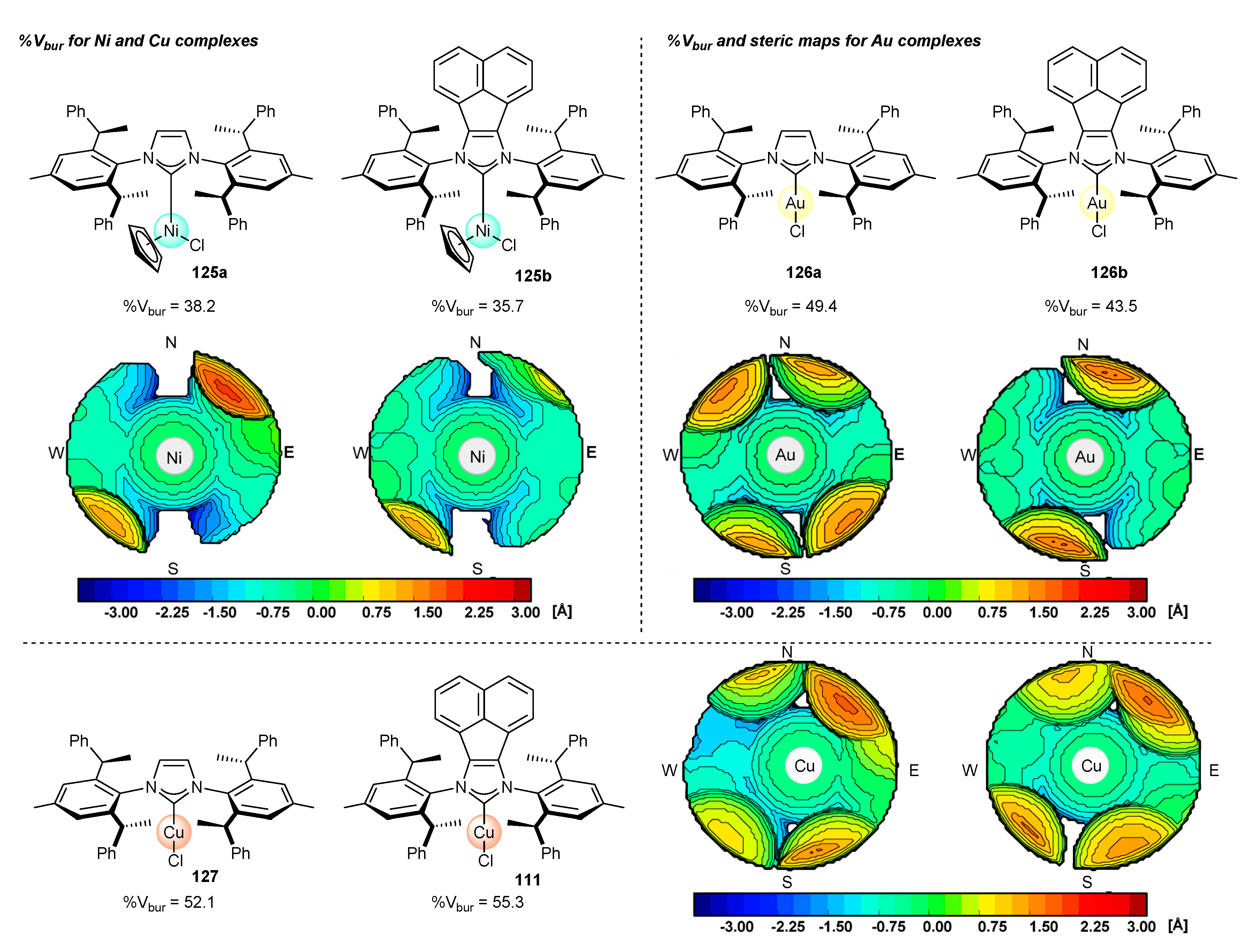{kind=link}
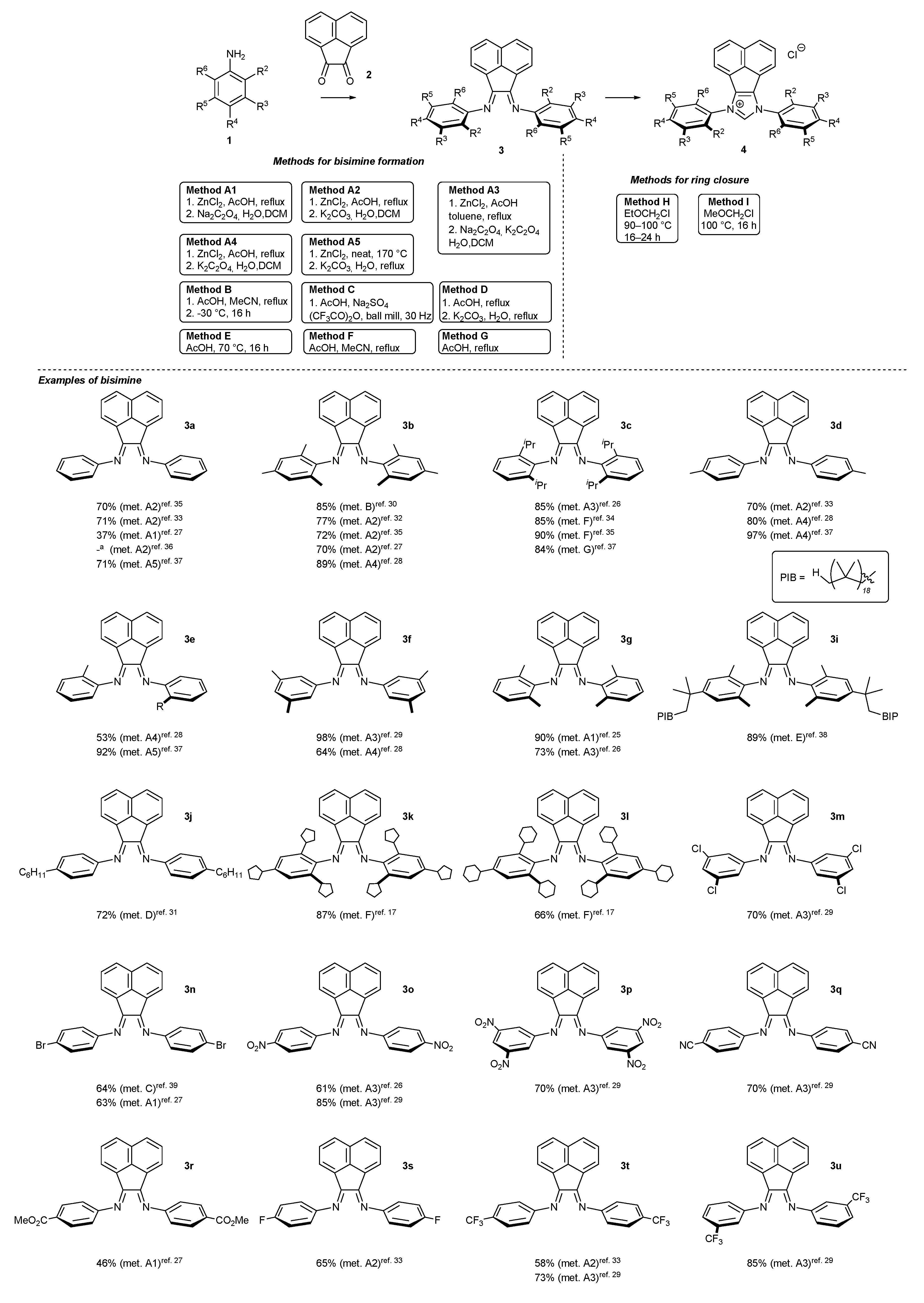{kind=link}
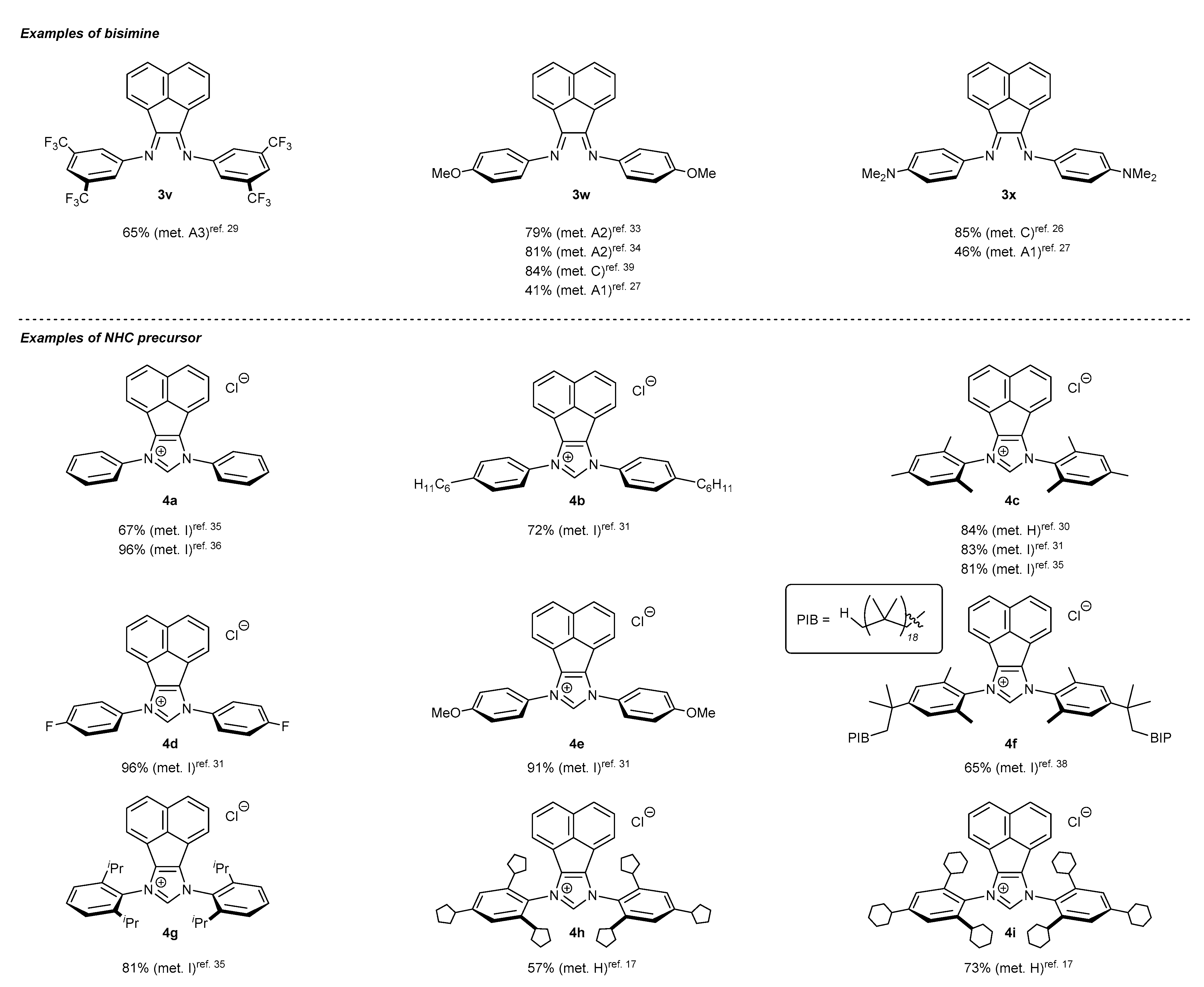{kind=link}
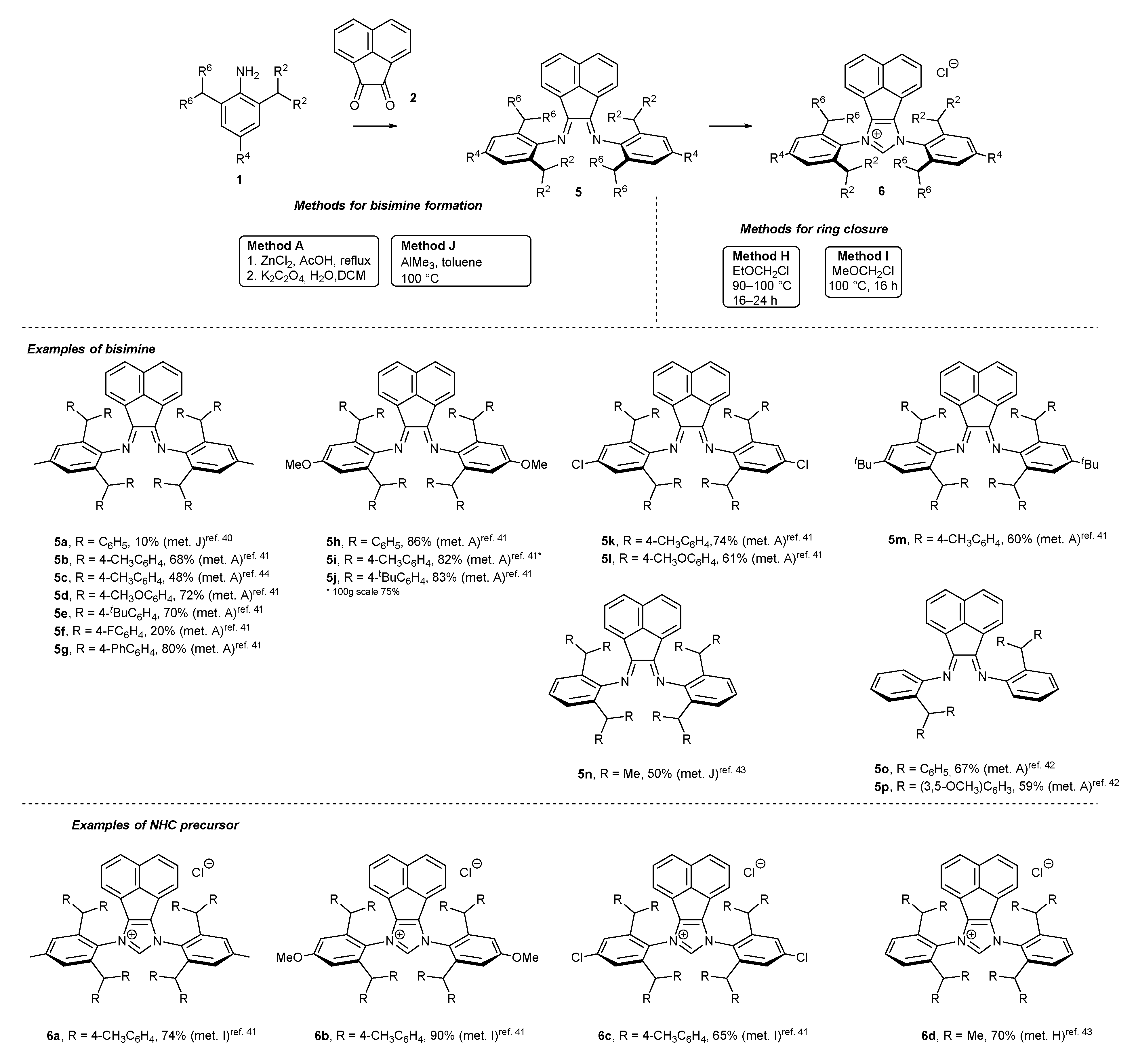{kind=link}
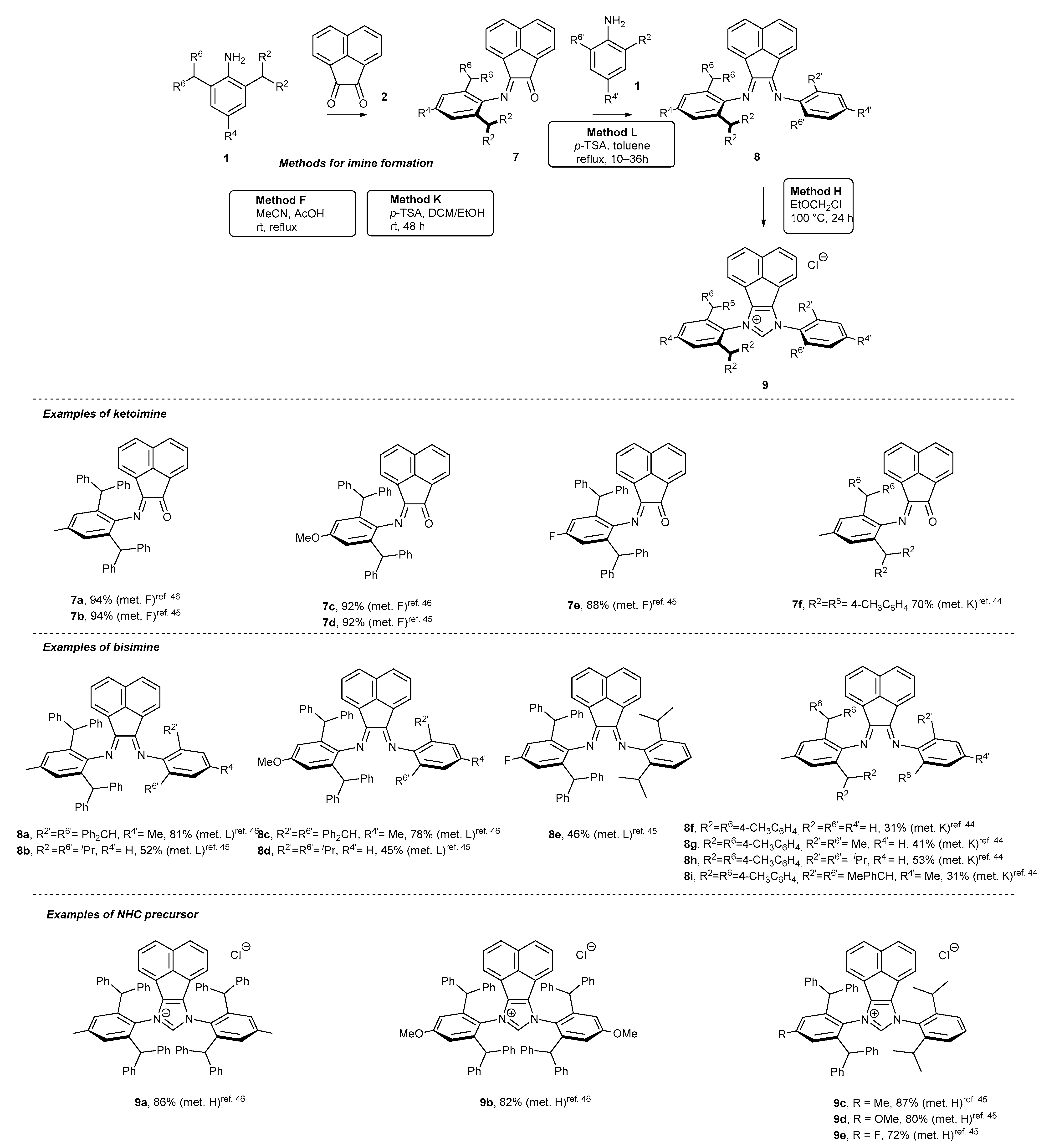{kind=link}
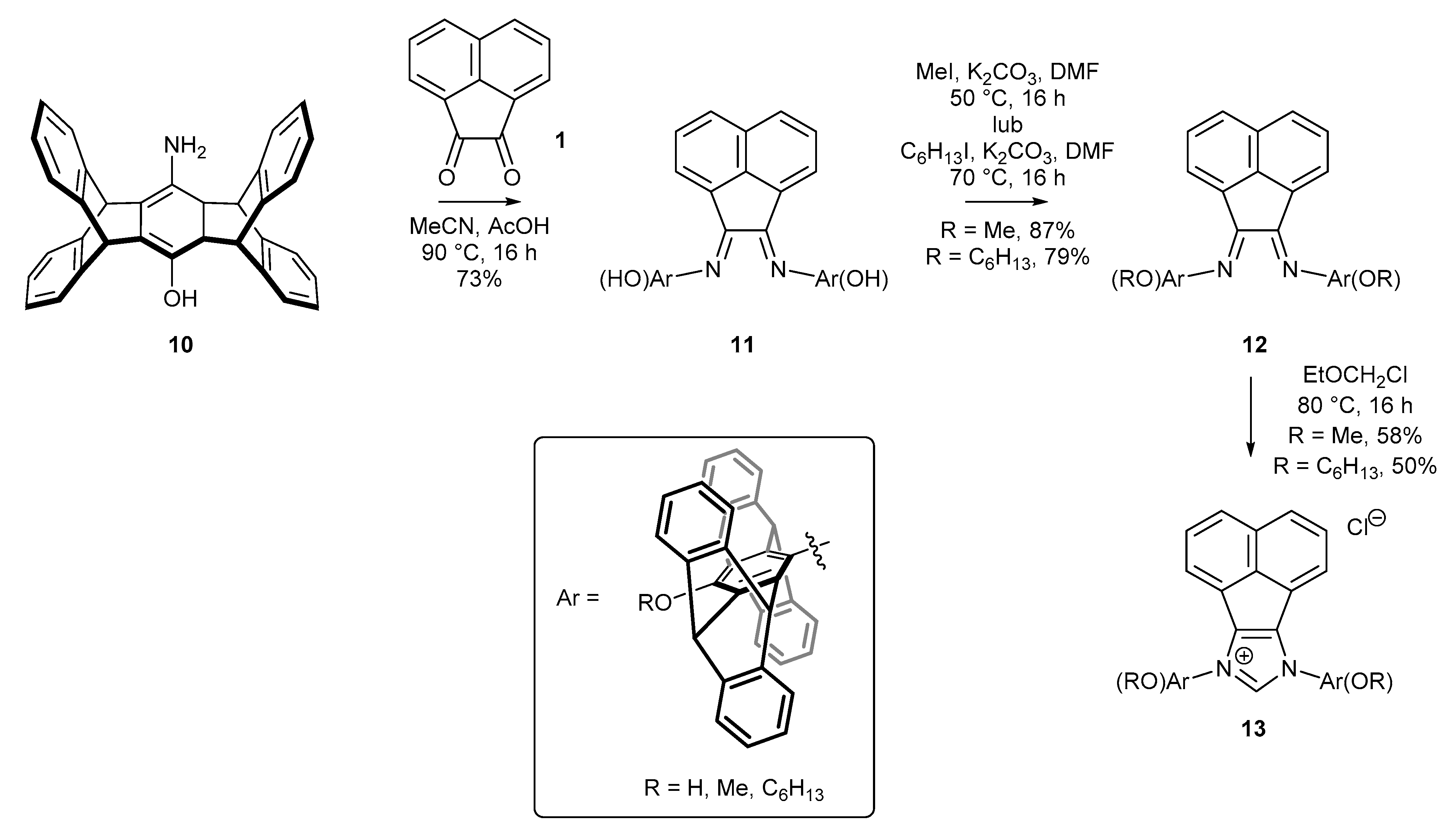{kind=link}
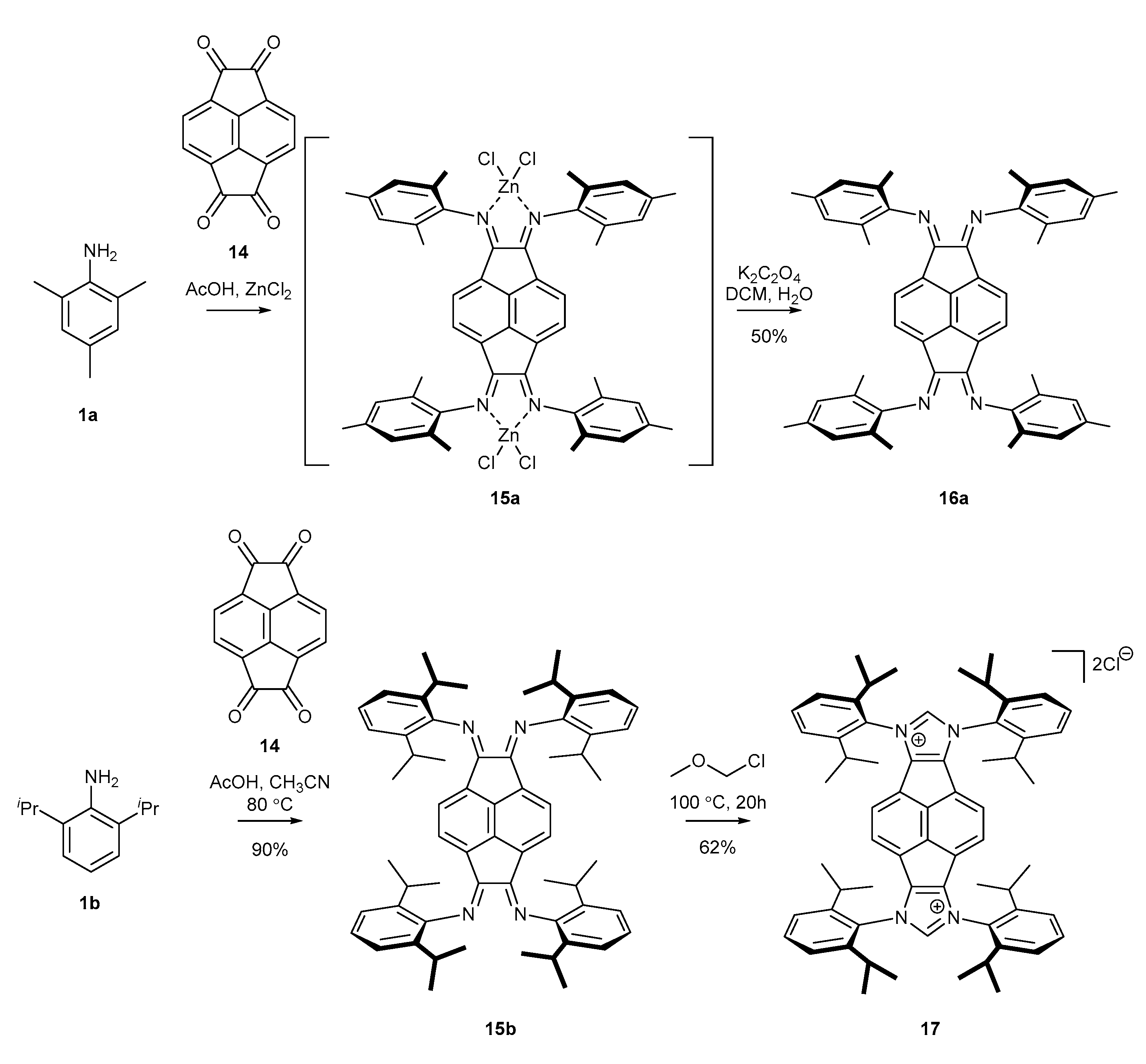{kind=link}
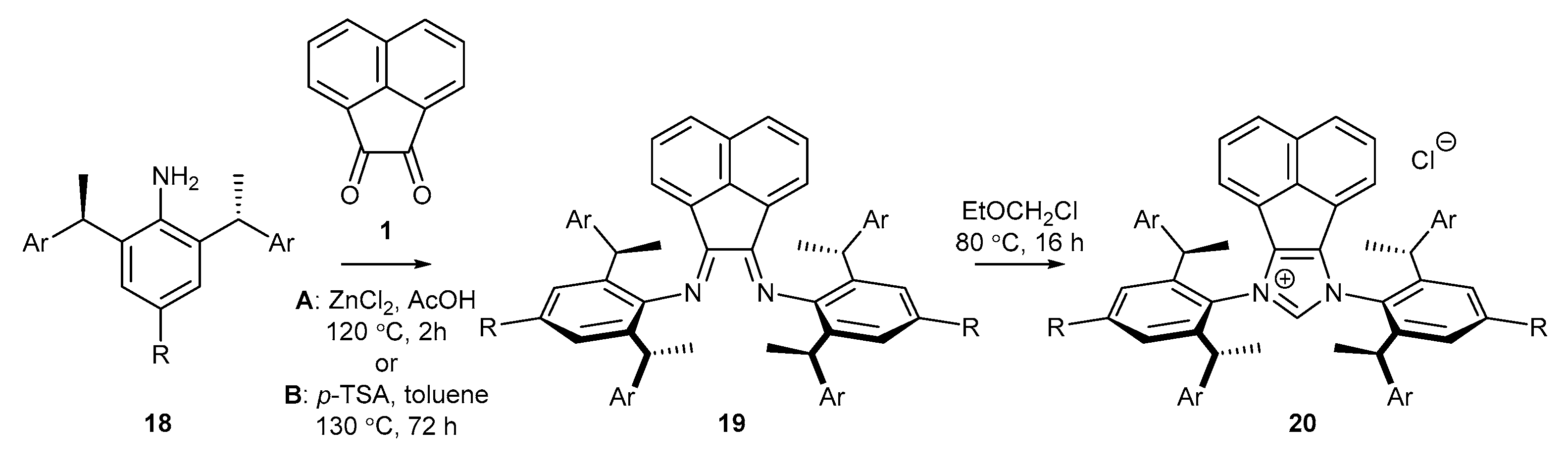{kind=link}
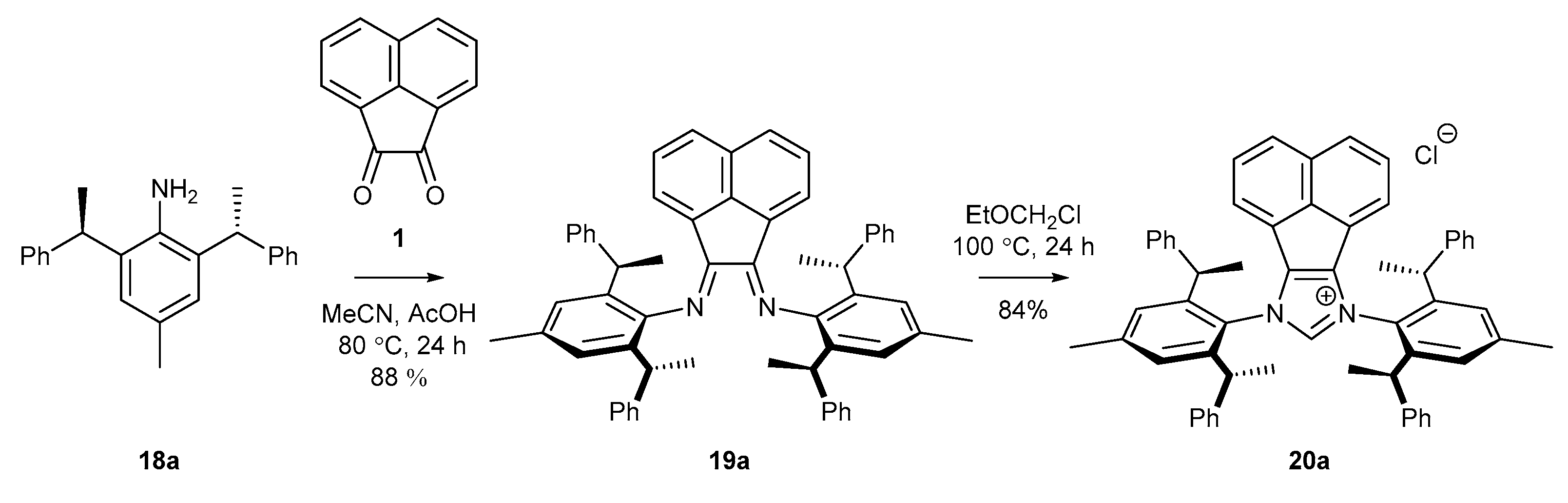{kind=link}
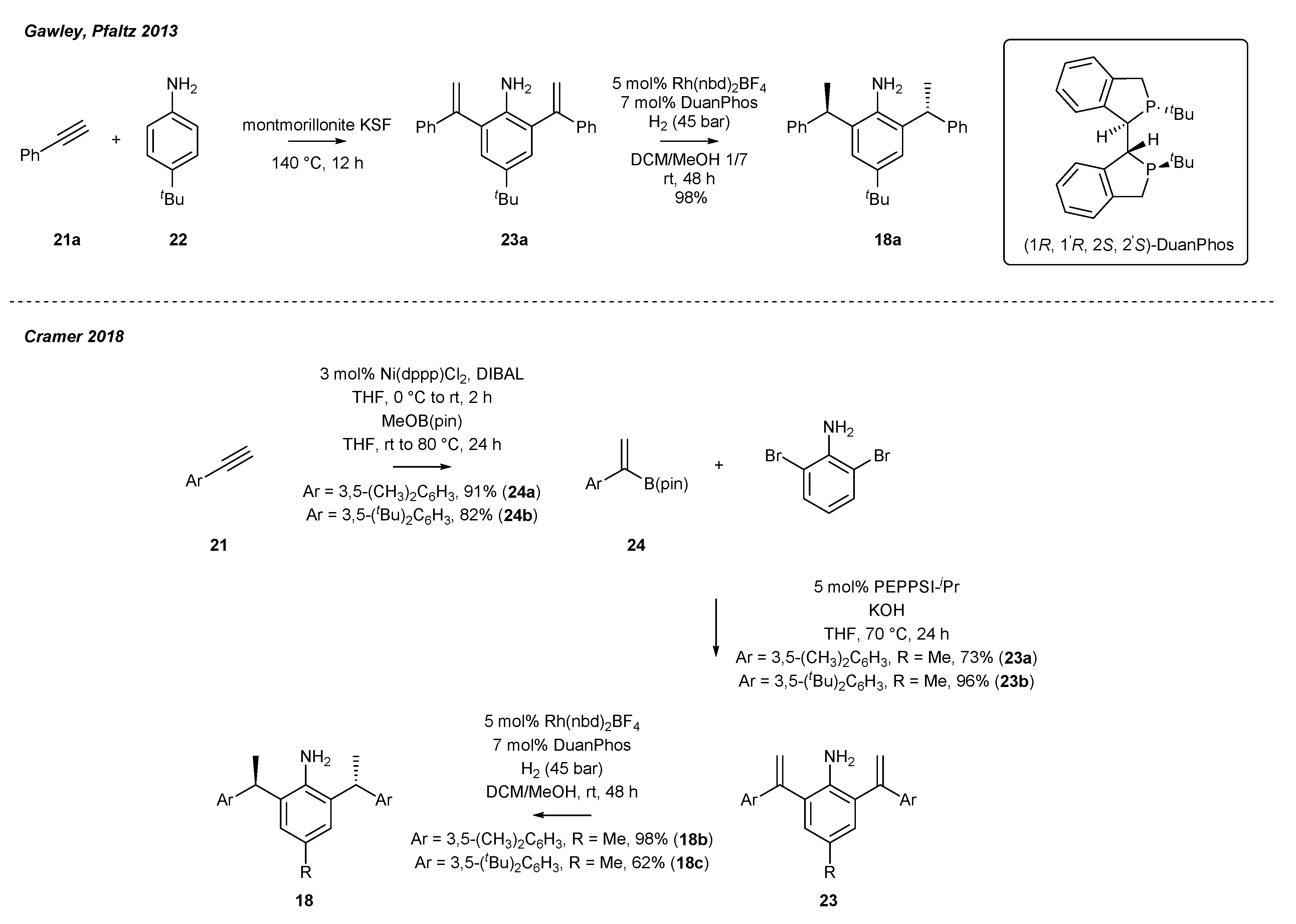{kind=link}
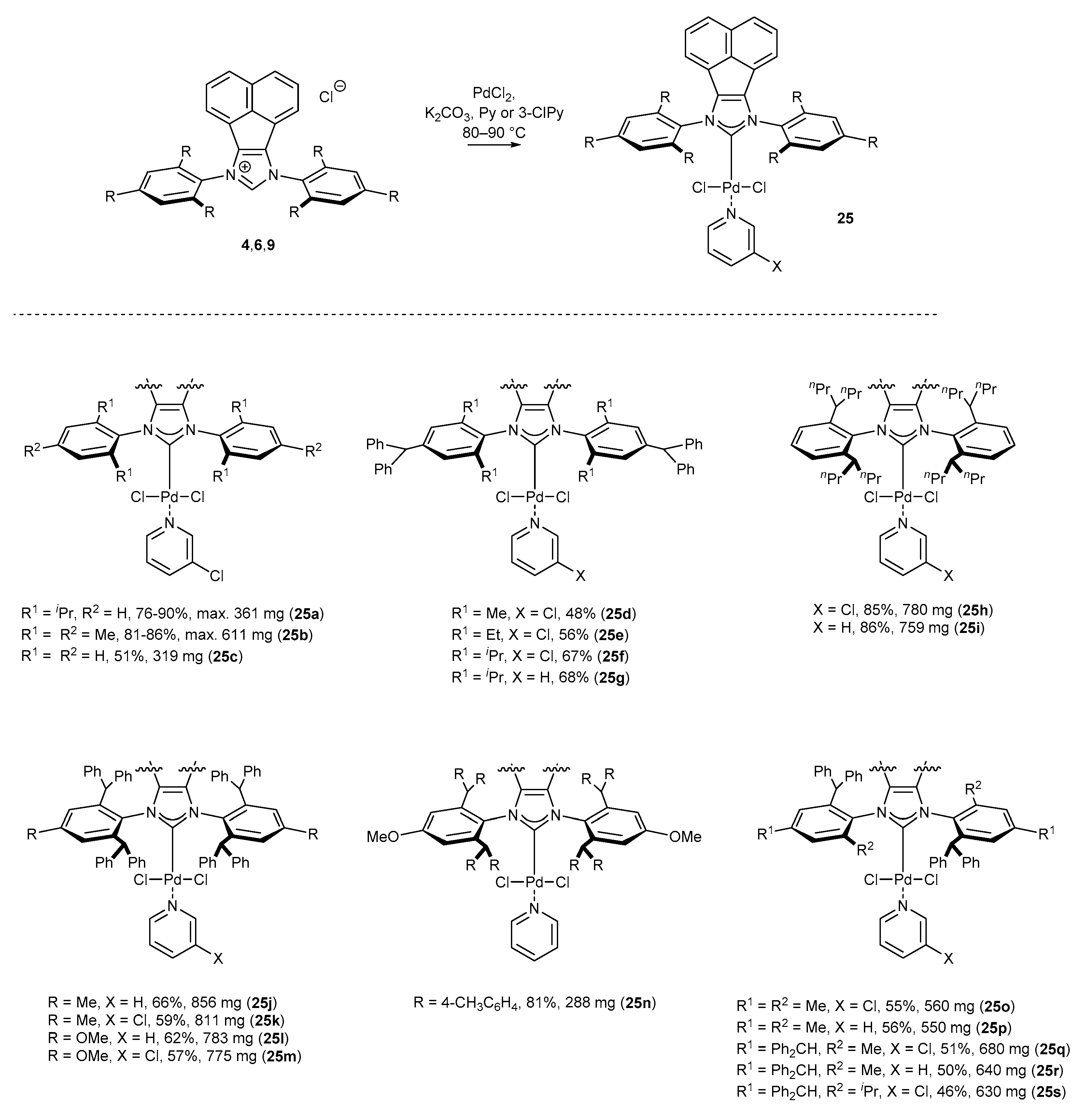{kind=link}
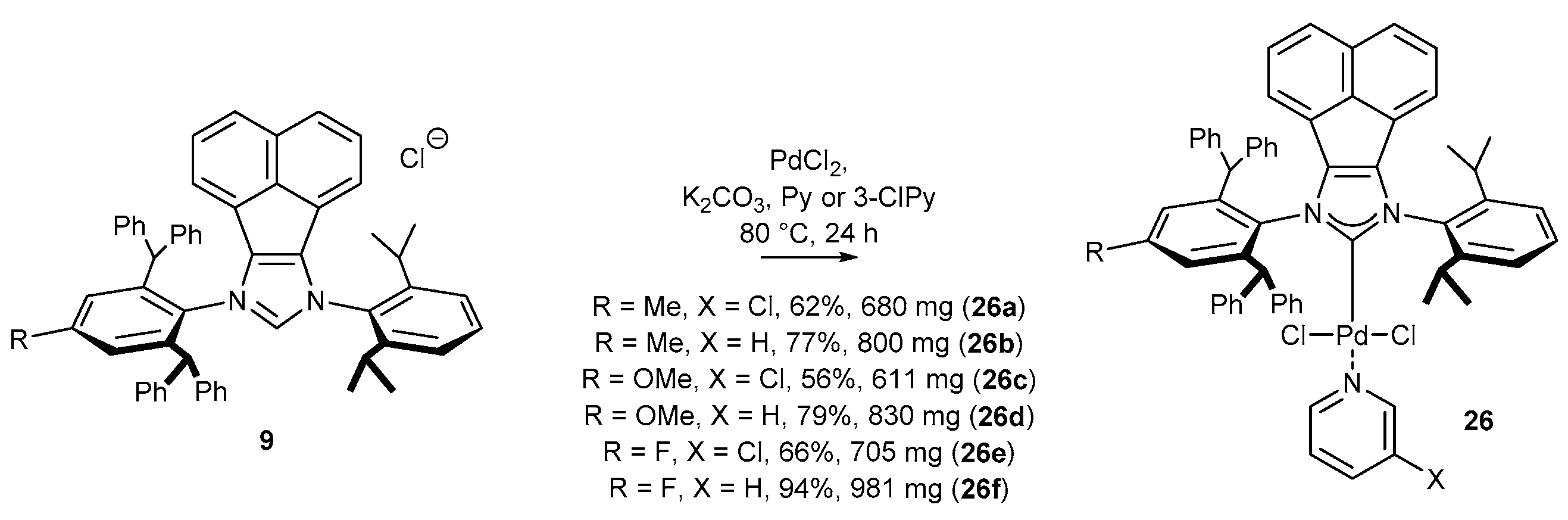{kind=link}
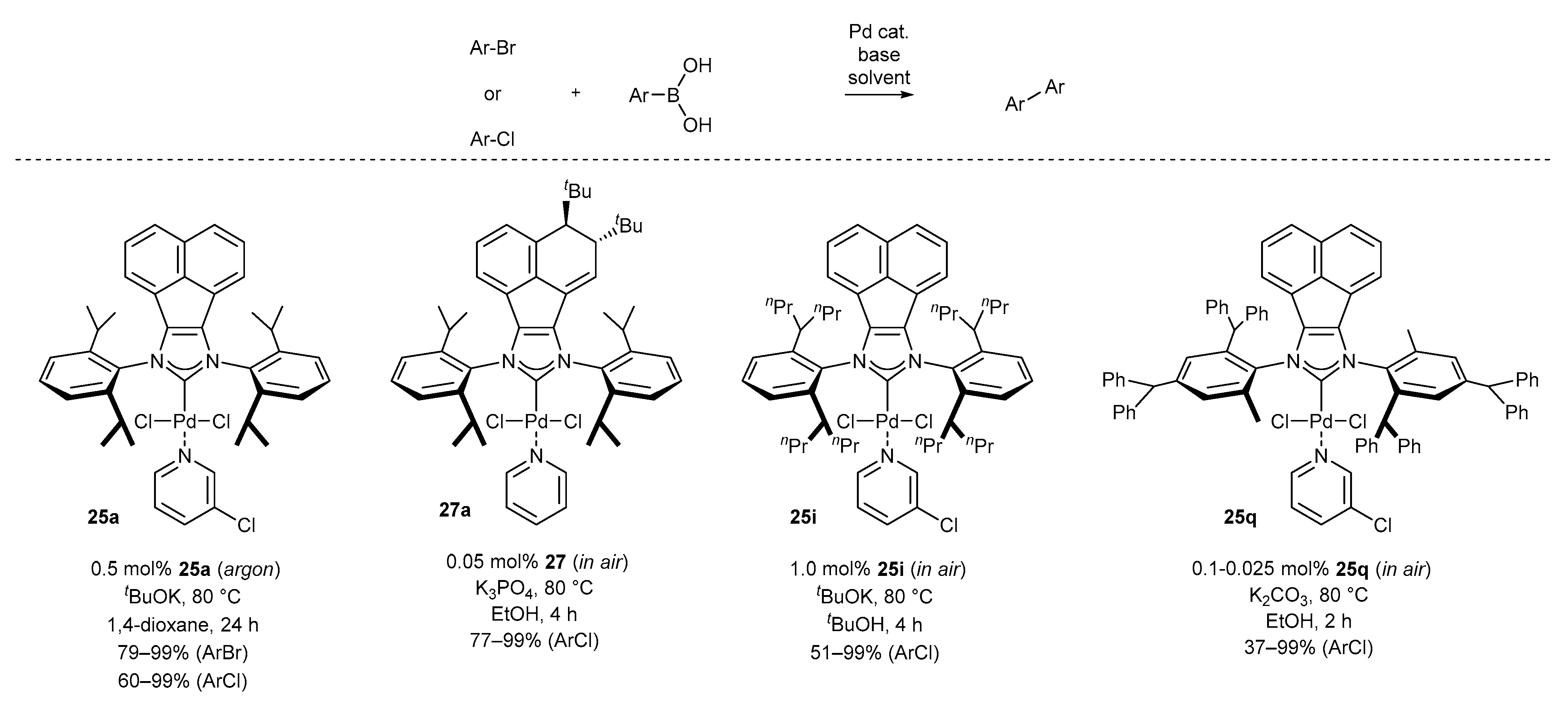{kind=link}
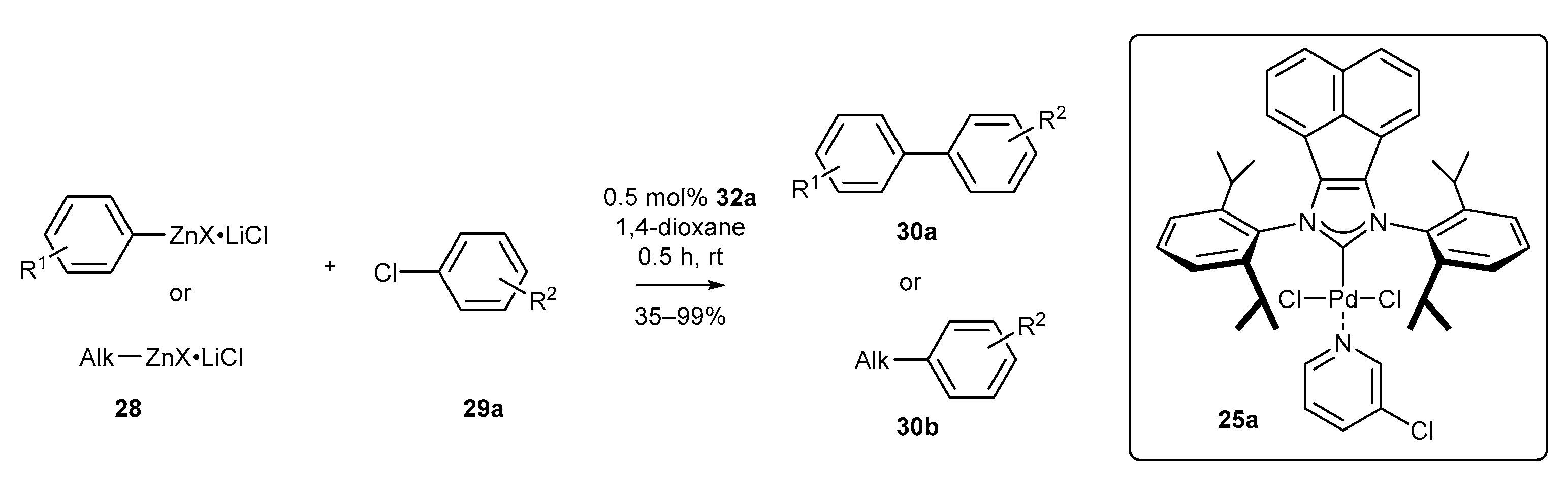{kind=link}
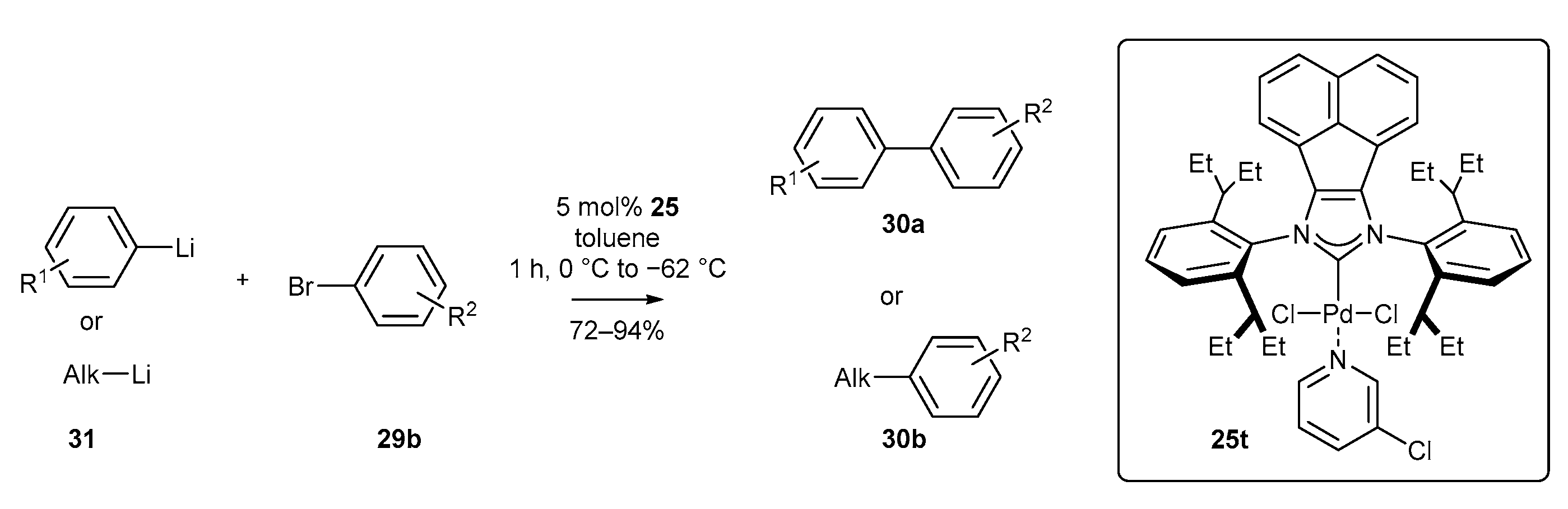{kind=link}
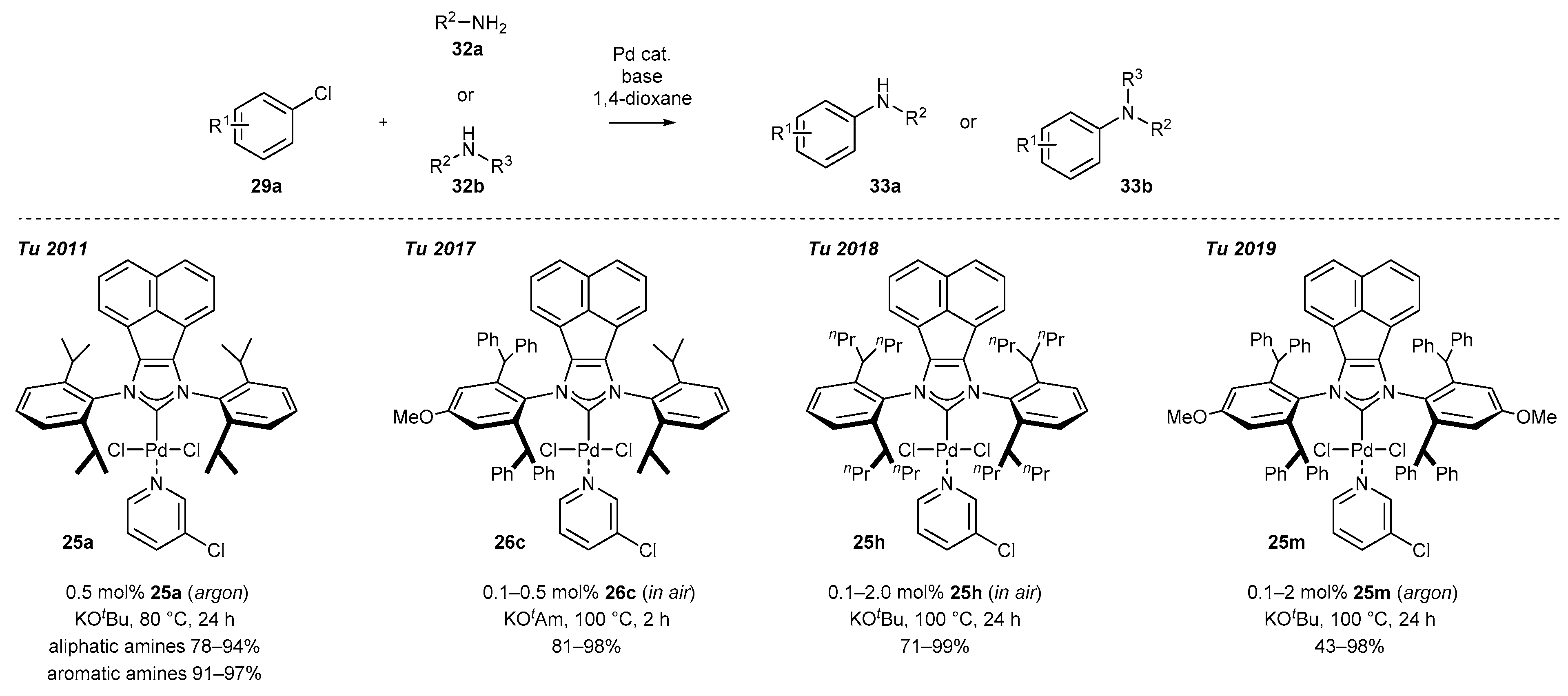{kind=link}
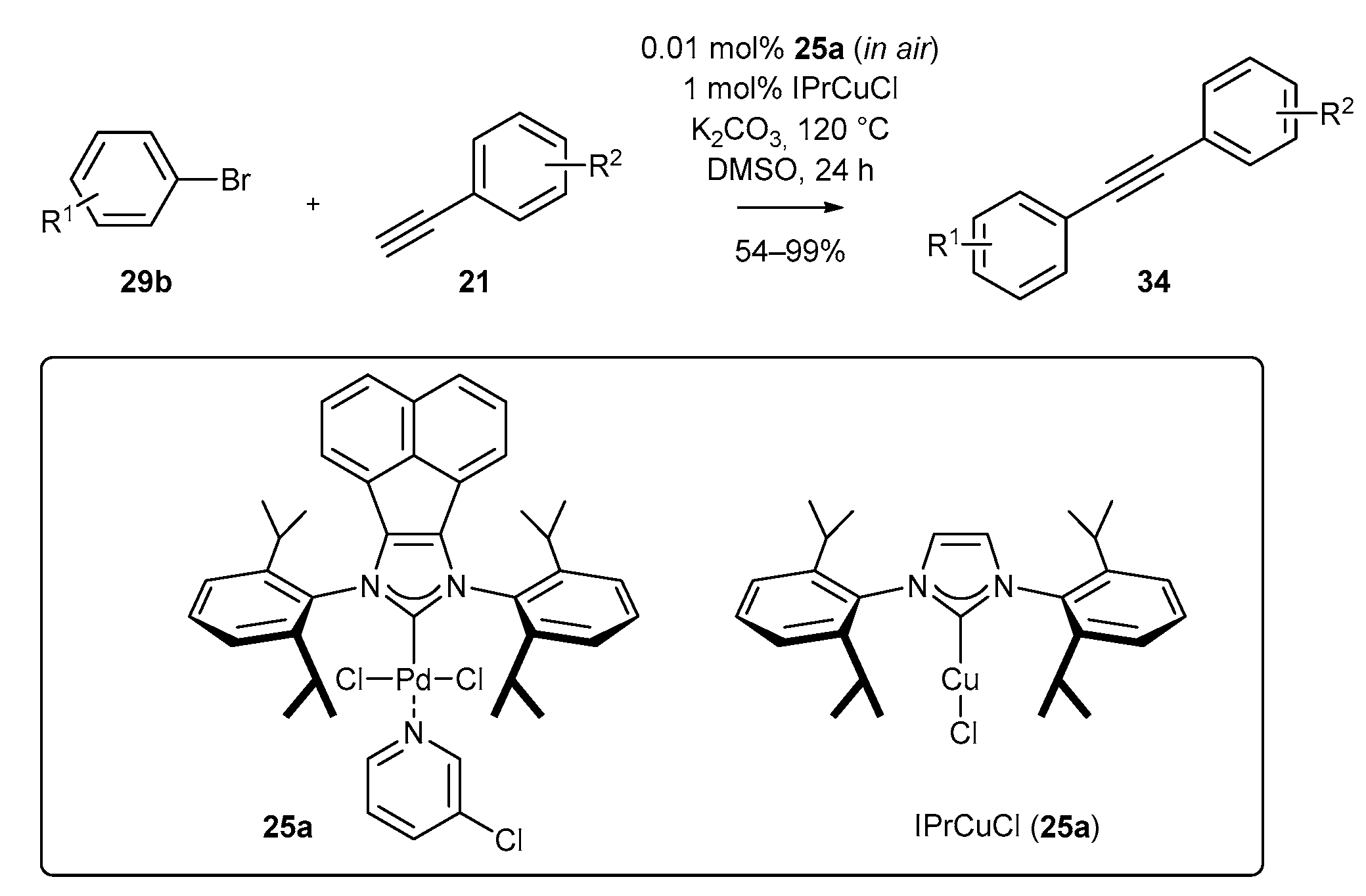{kind=link}
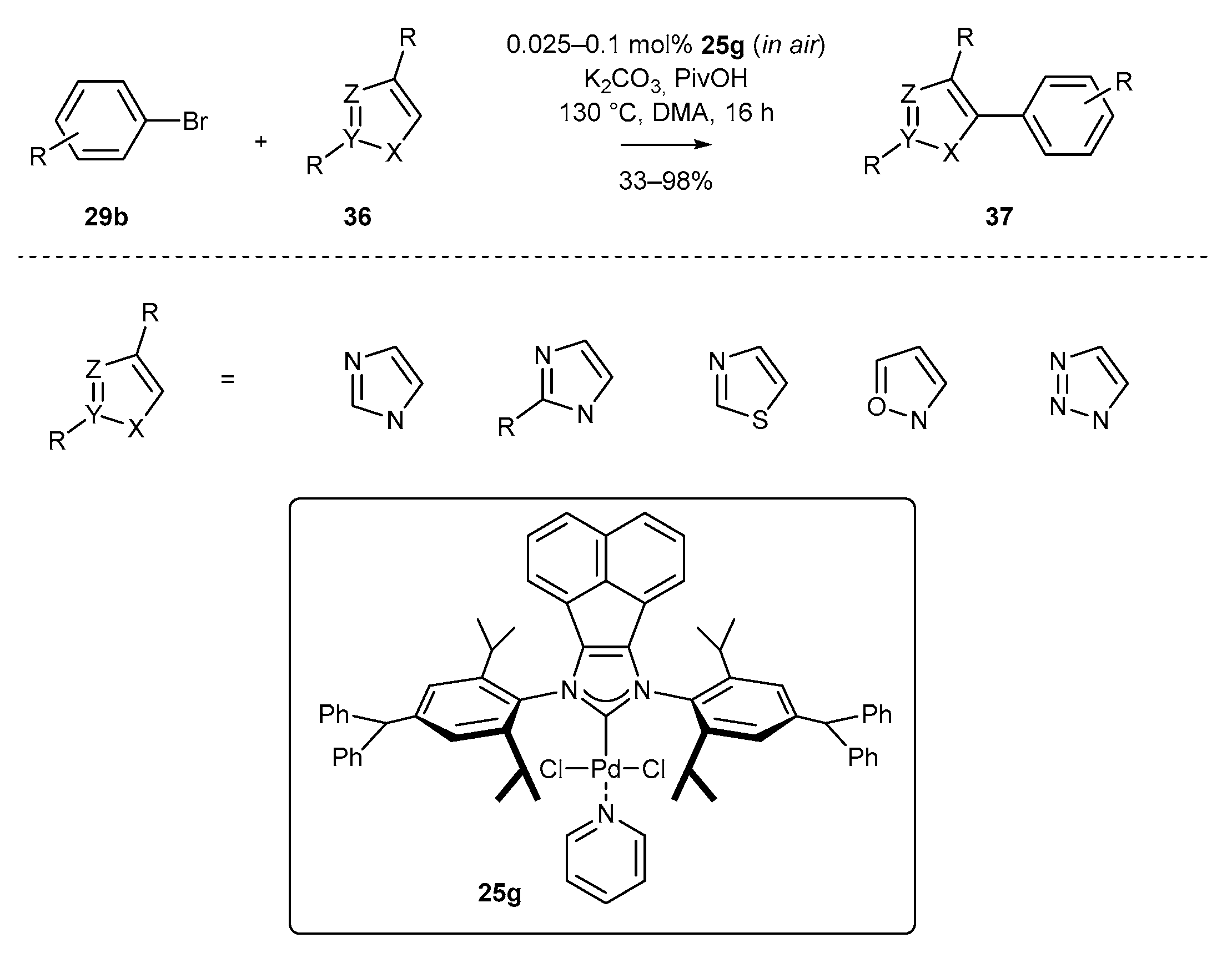{kind=link}
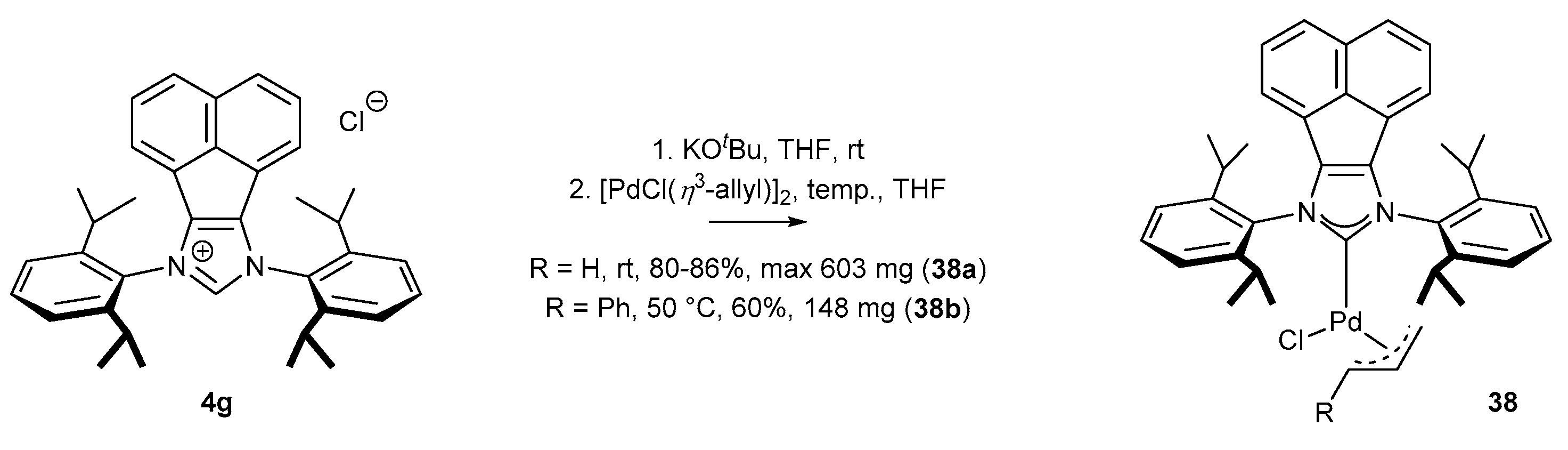{kind=link}
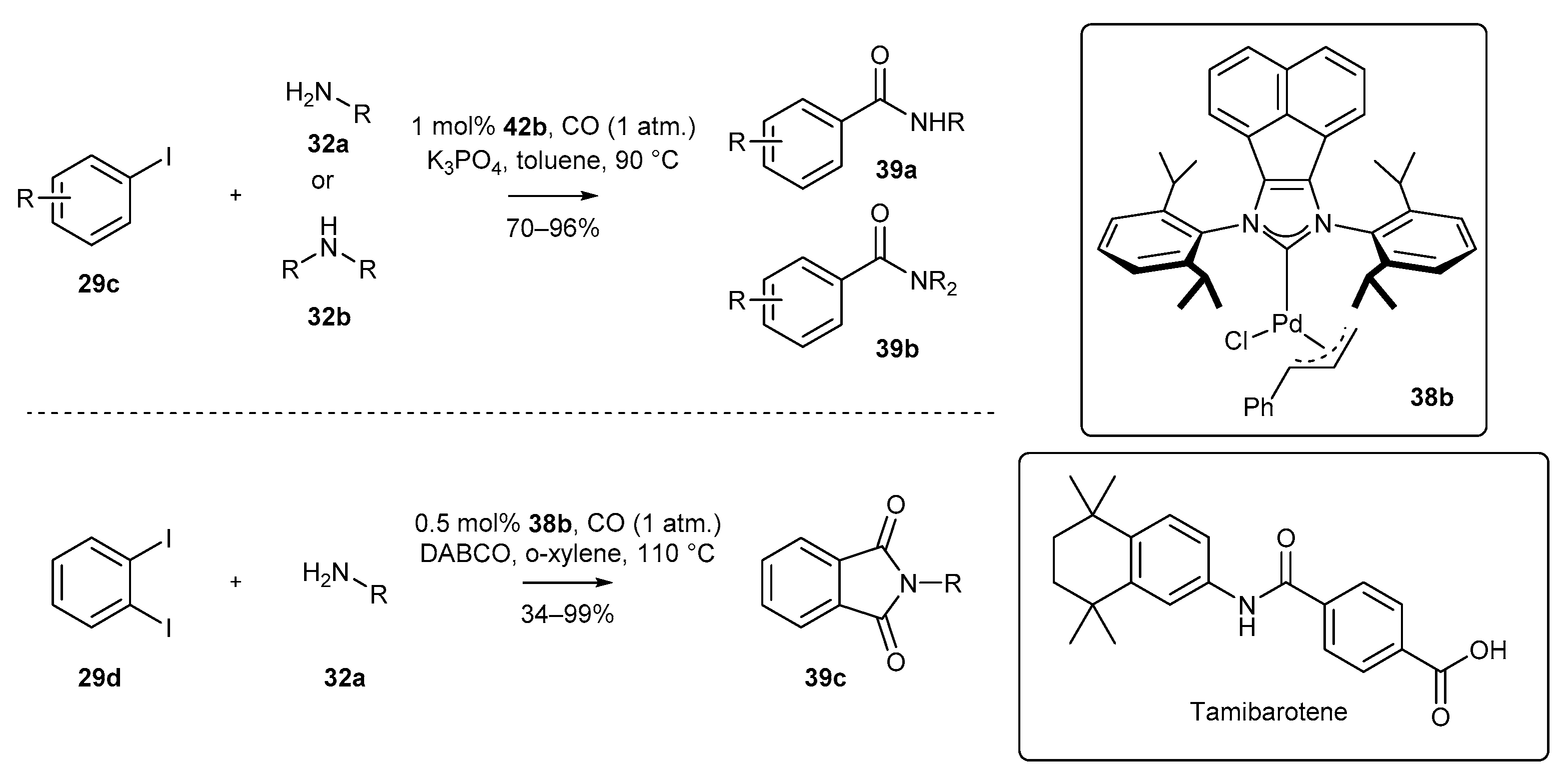{kind=link}
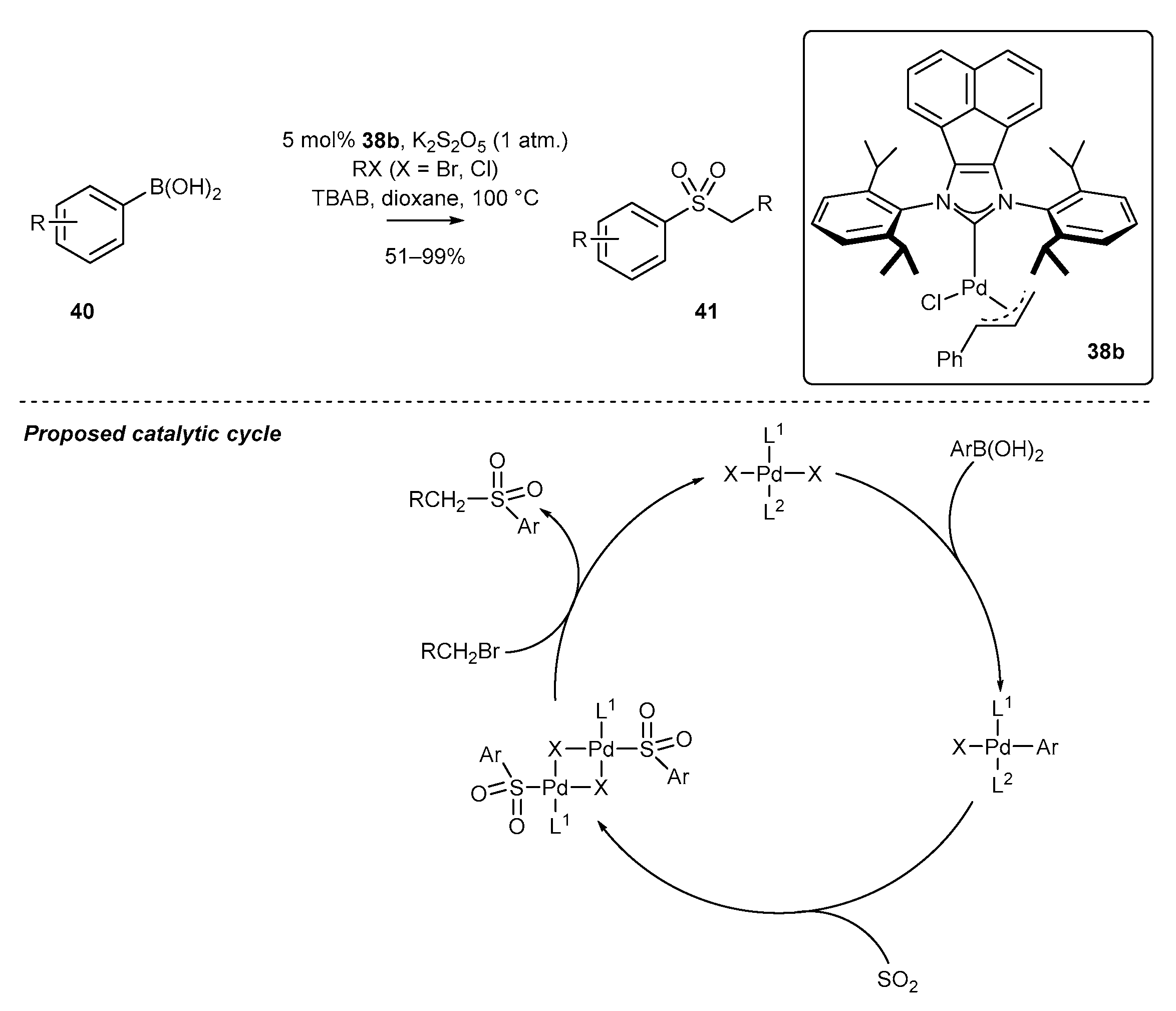{kind=link}
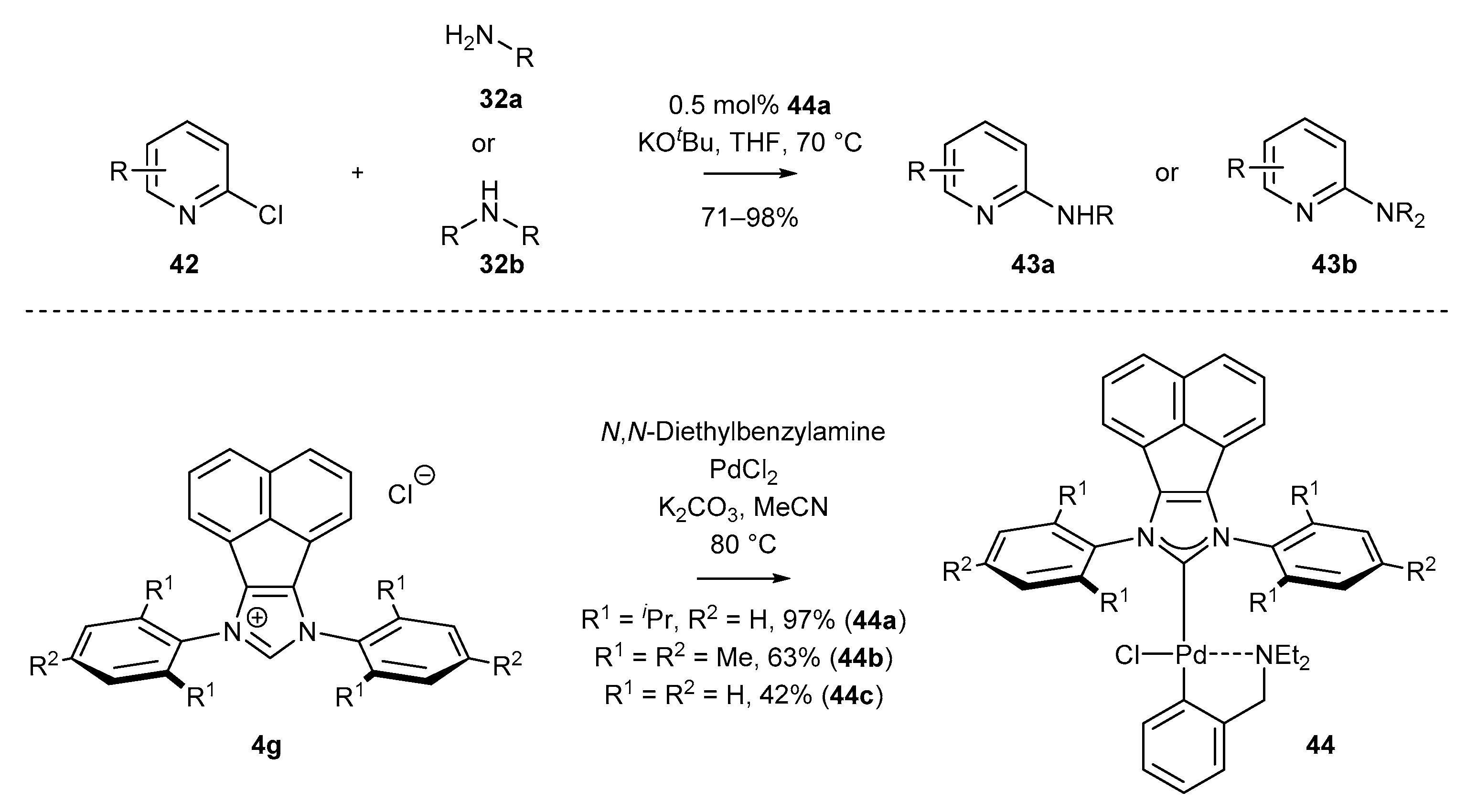{kind=link}
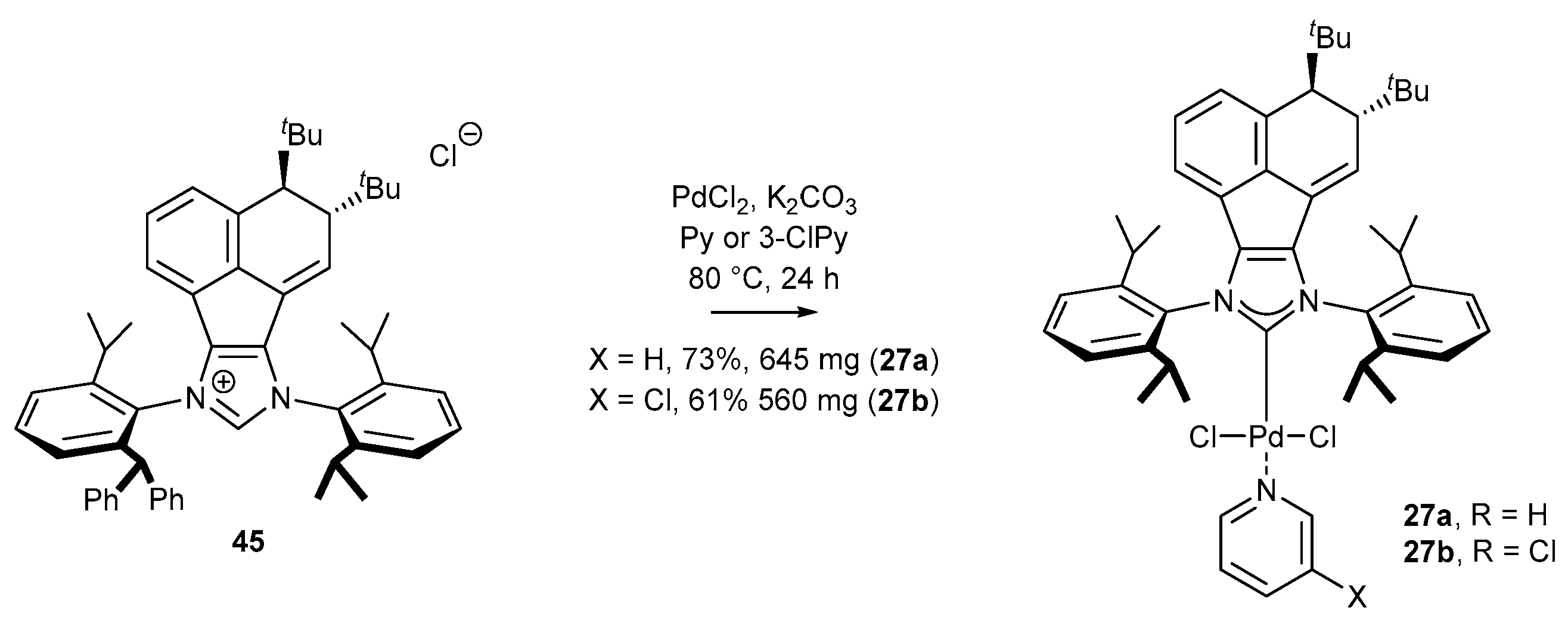{kind=link}
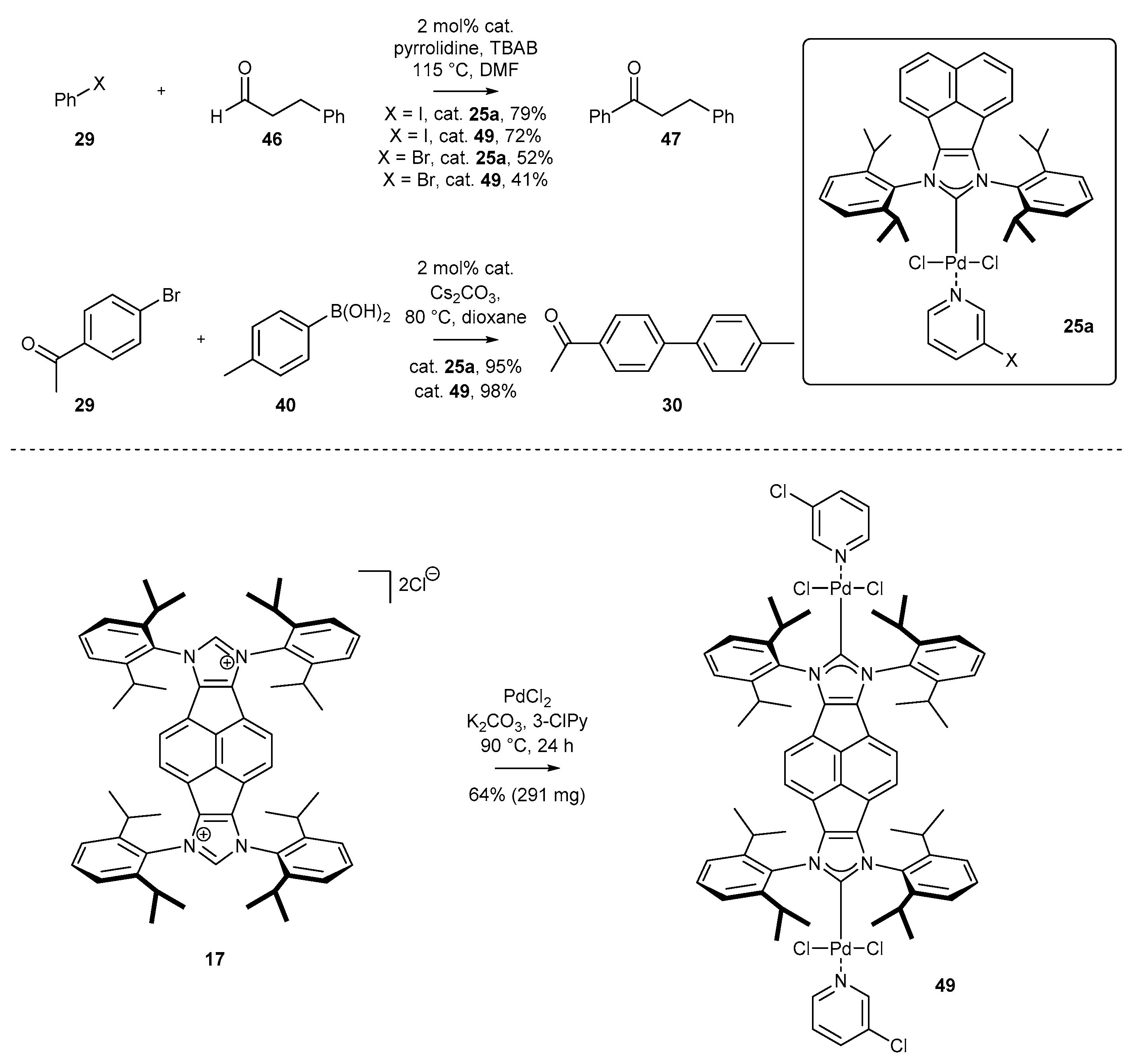{kind=link}
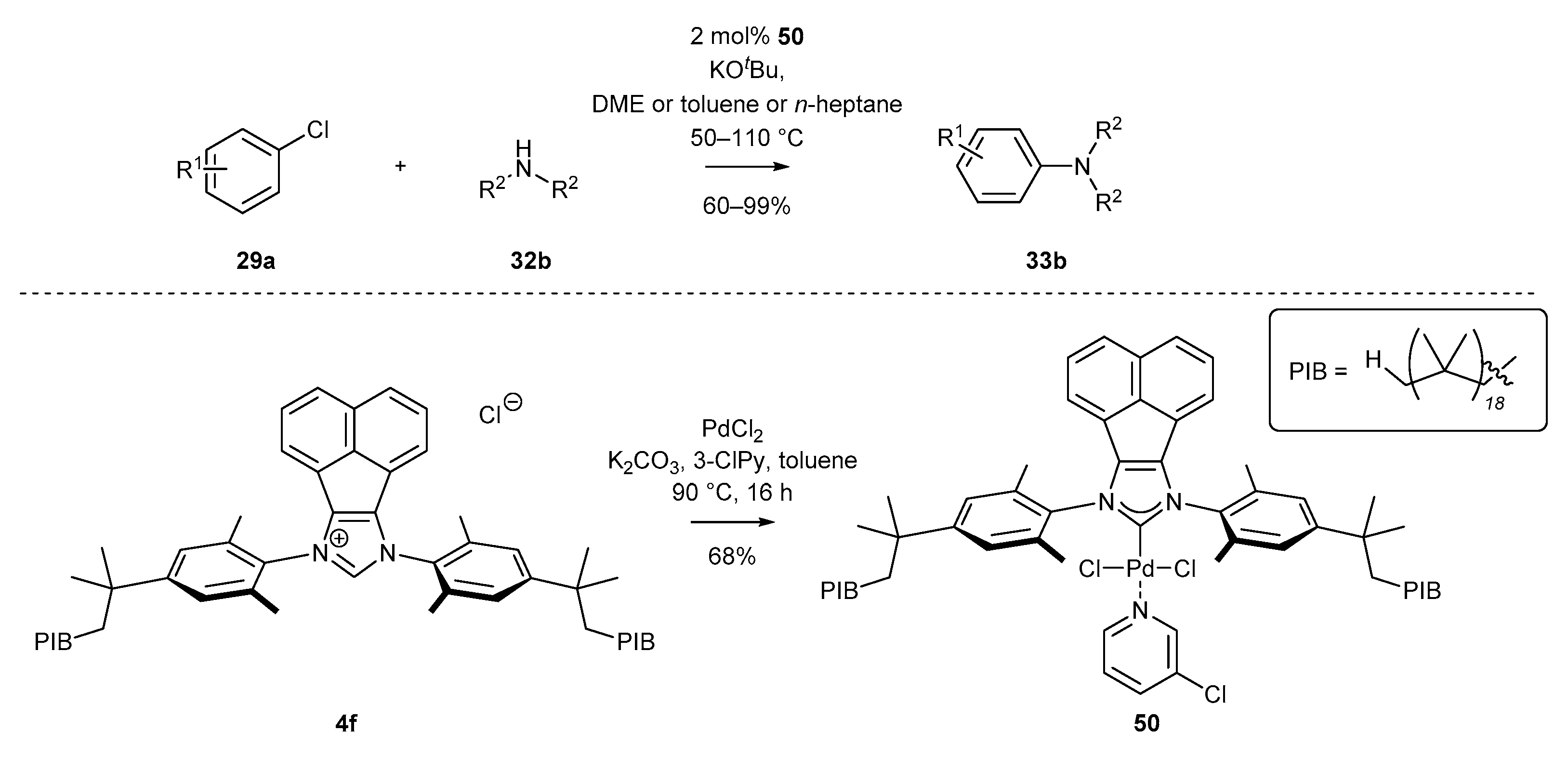{kind=link}
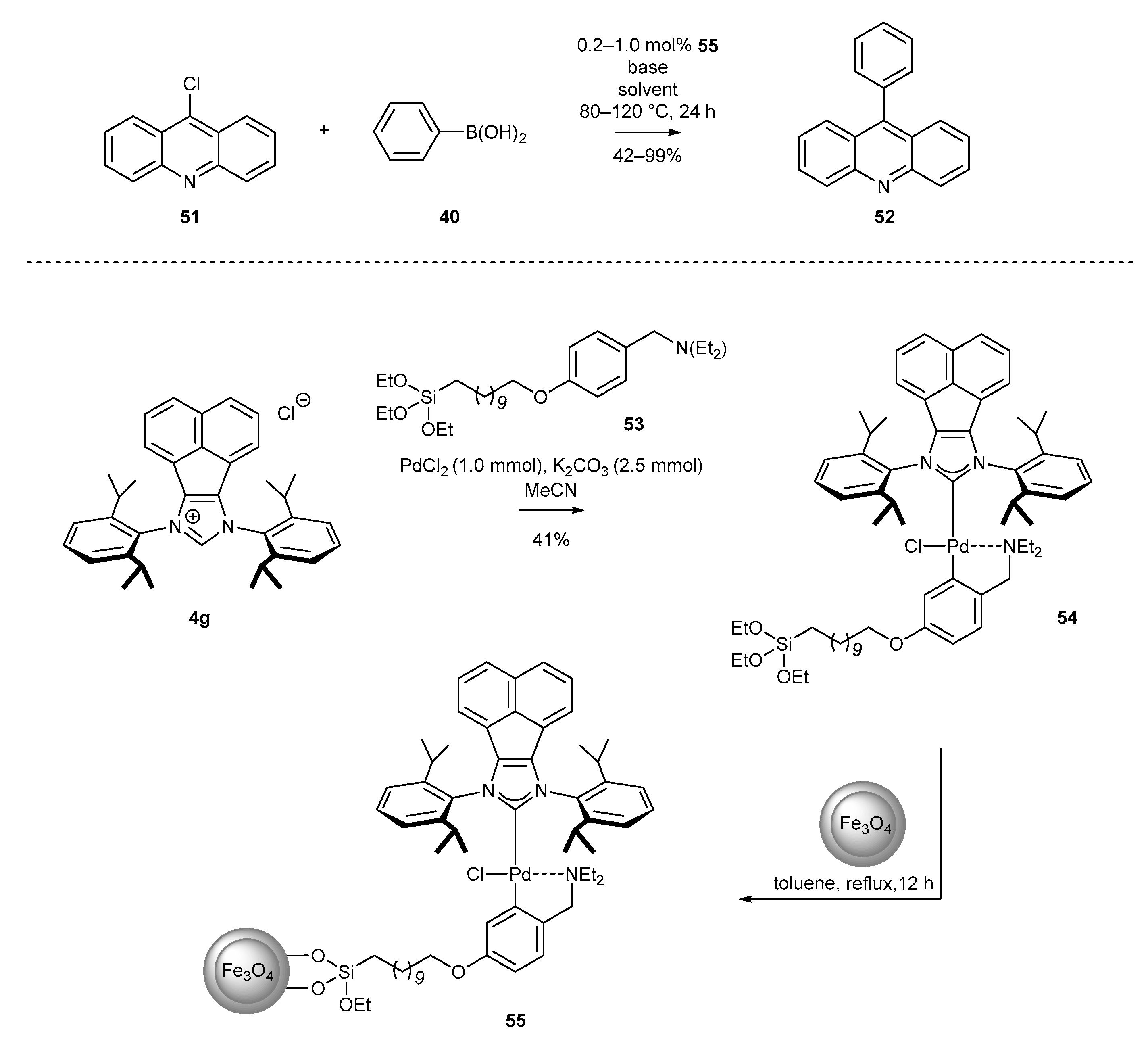{kind=link}
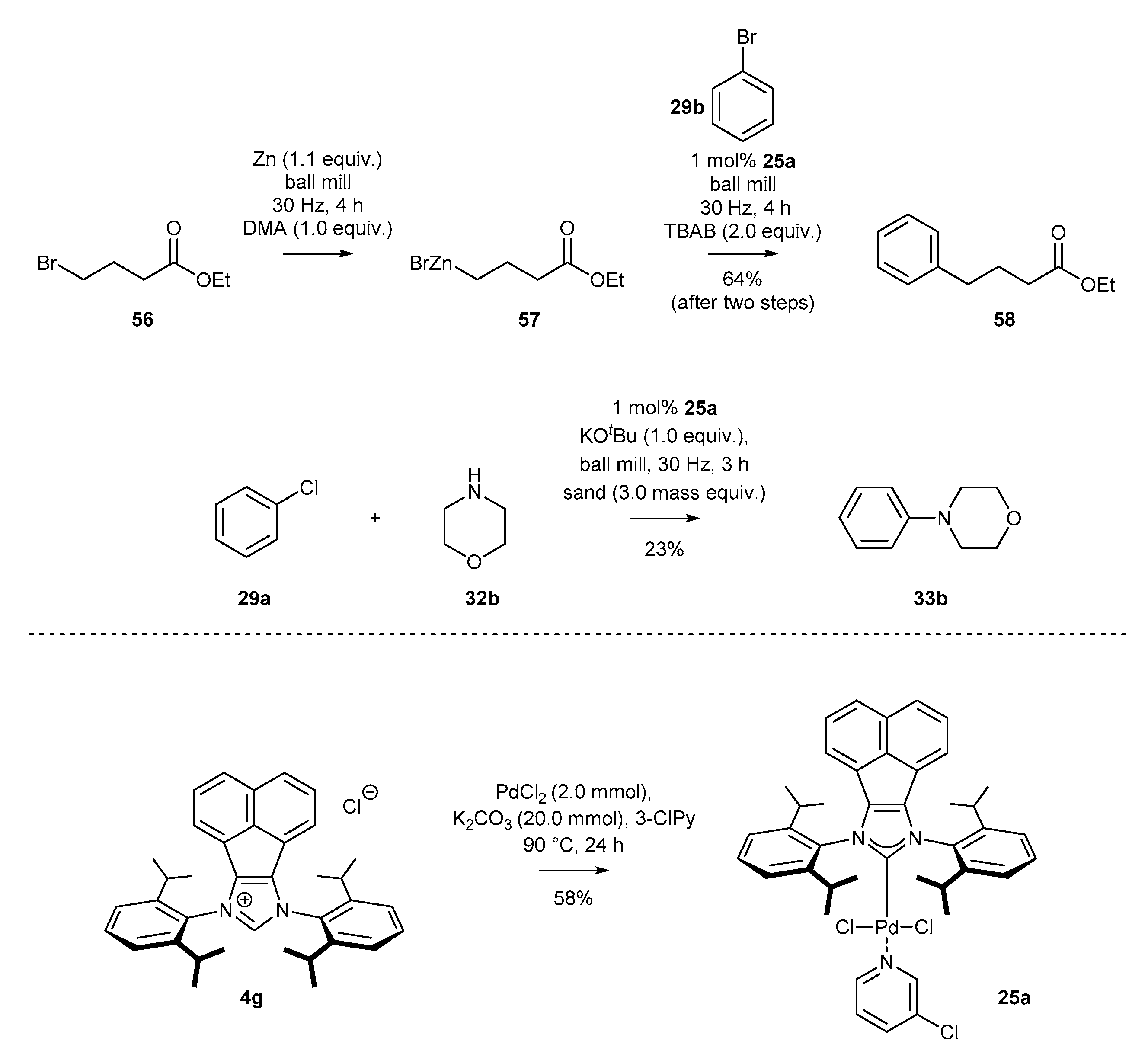{kind=link}
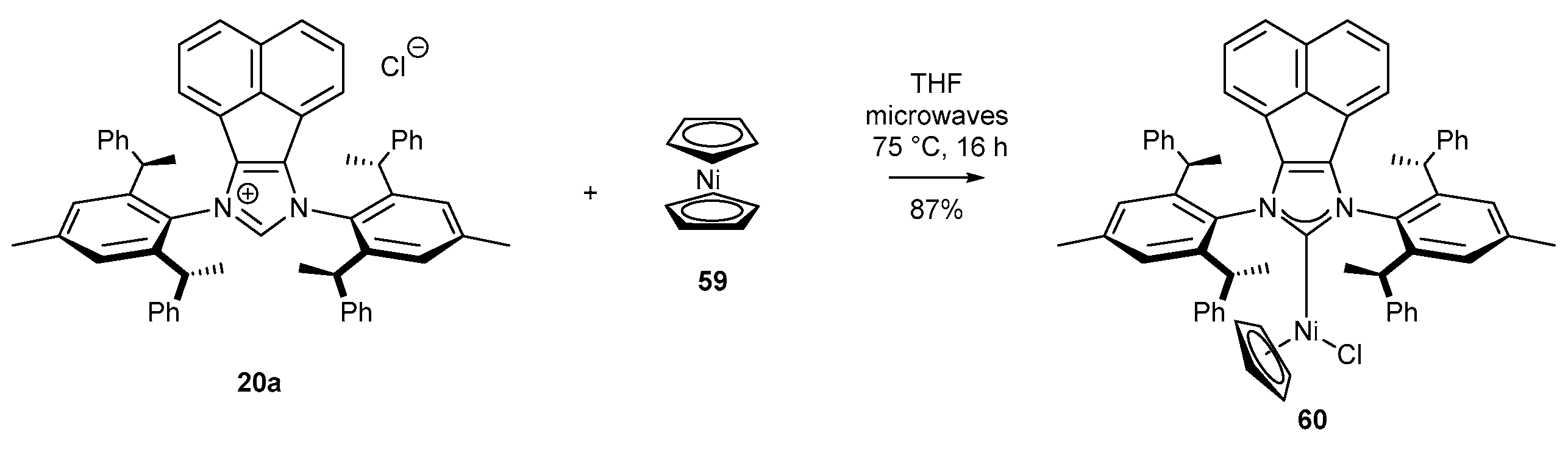{kind=link}
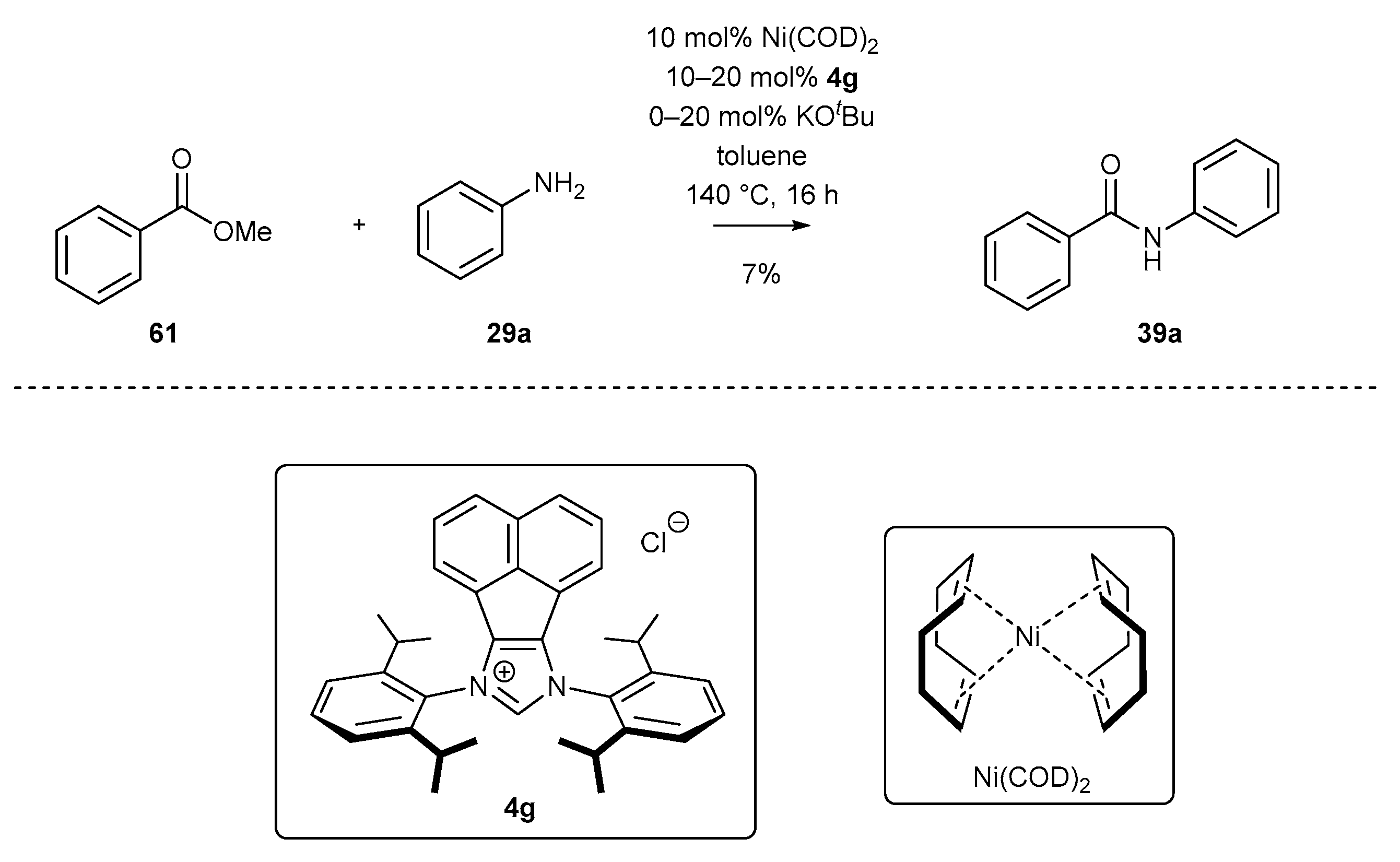{kind=link}
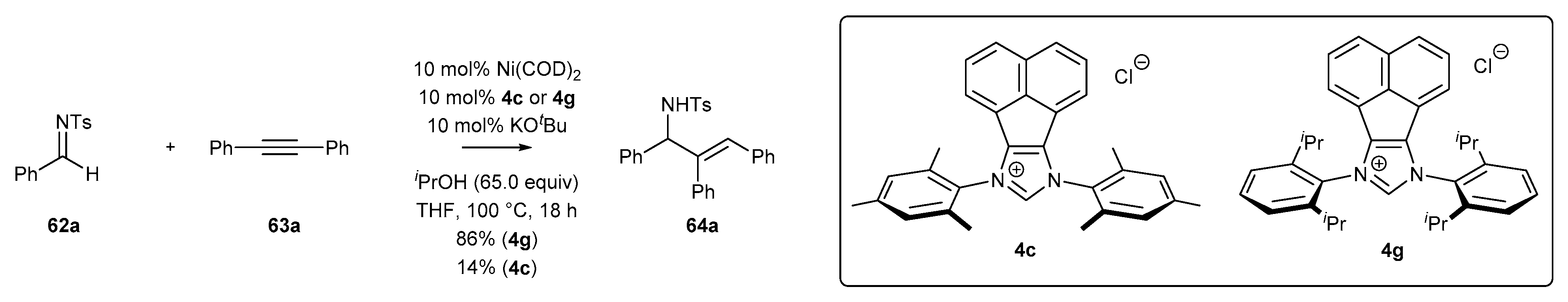{kind=link}
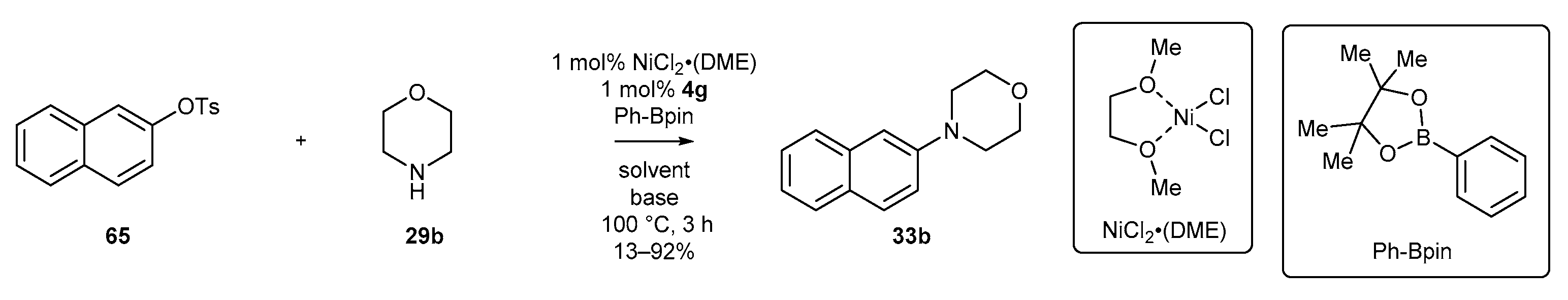{kind=link}
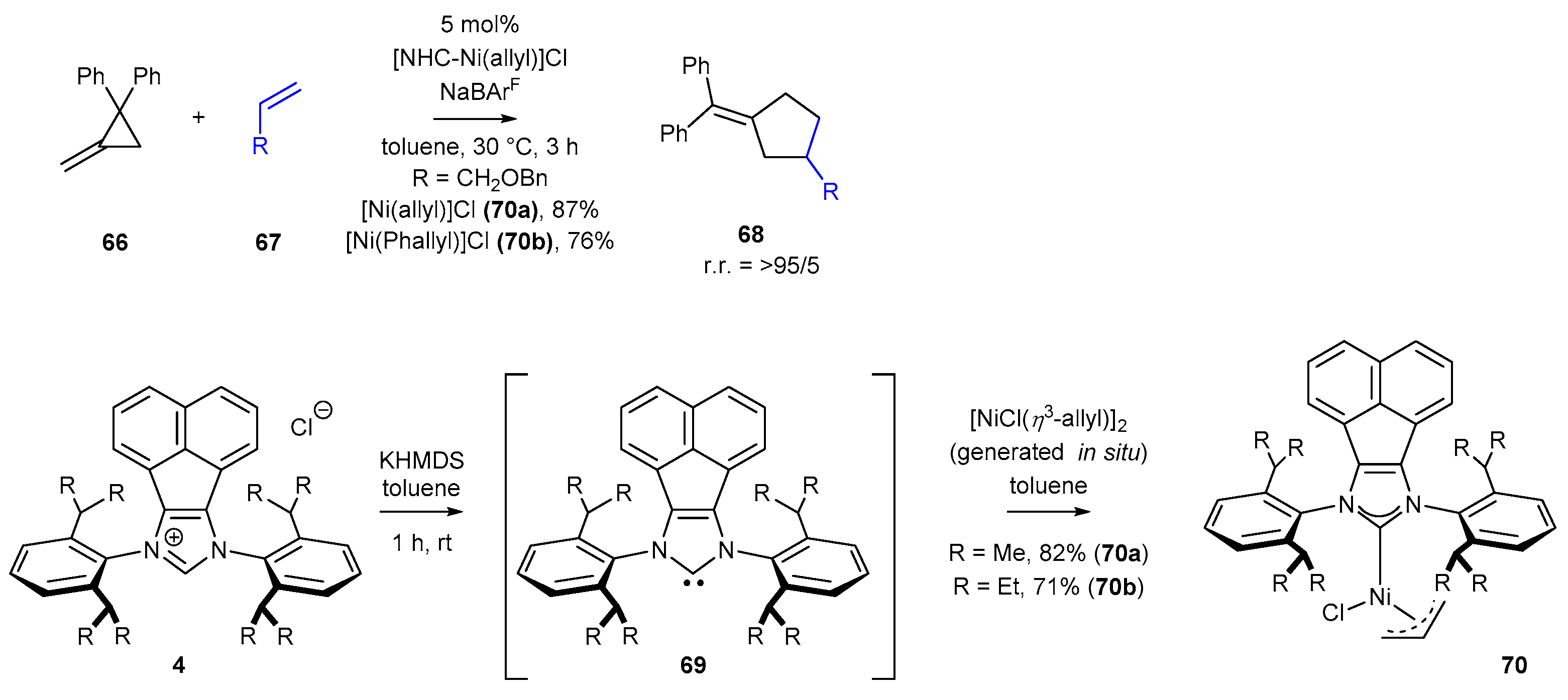{kind=link}
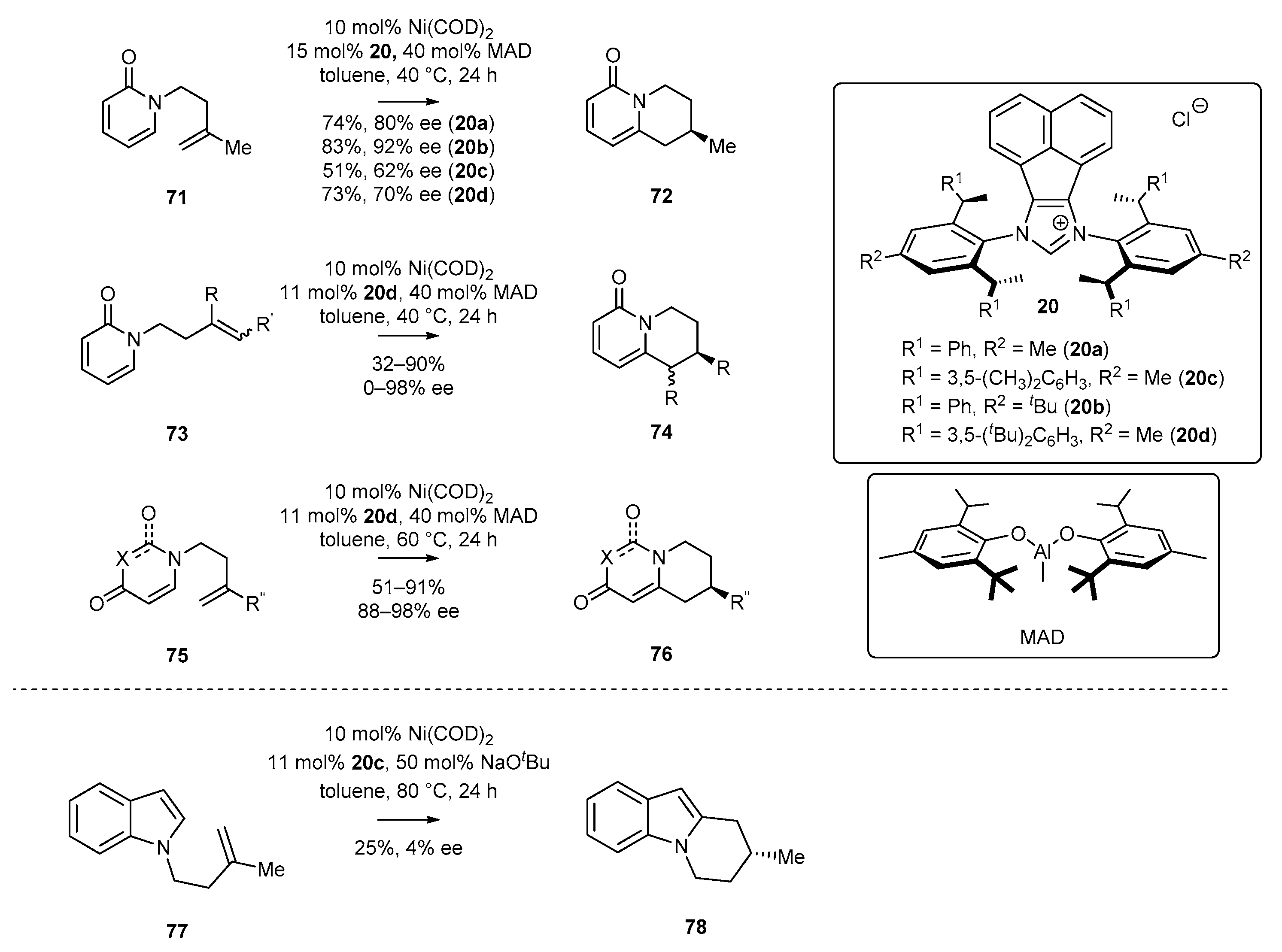{kind=link}
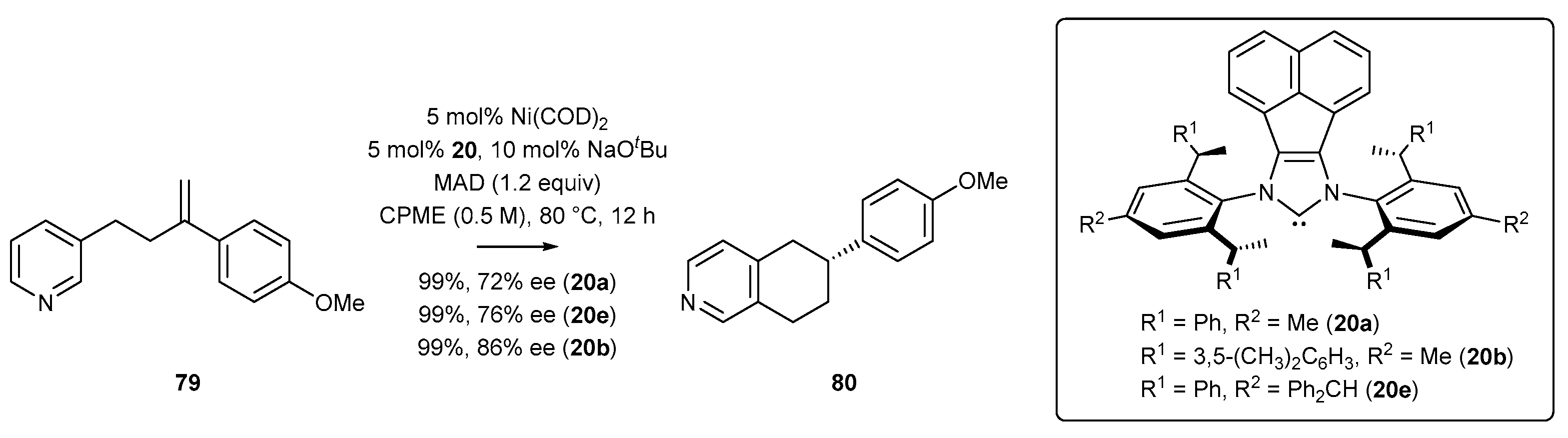{kind=link}
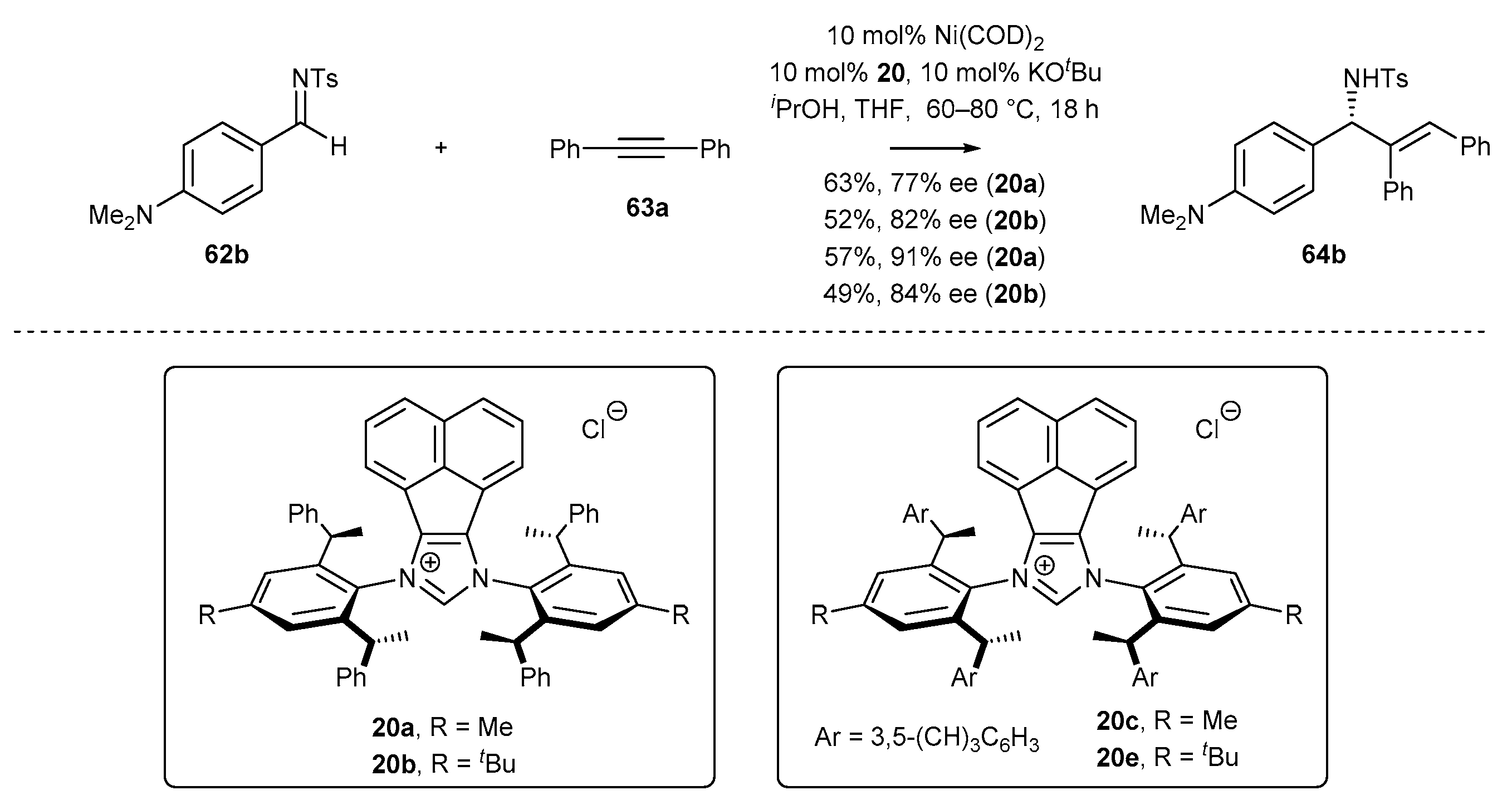{kind=link}
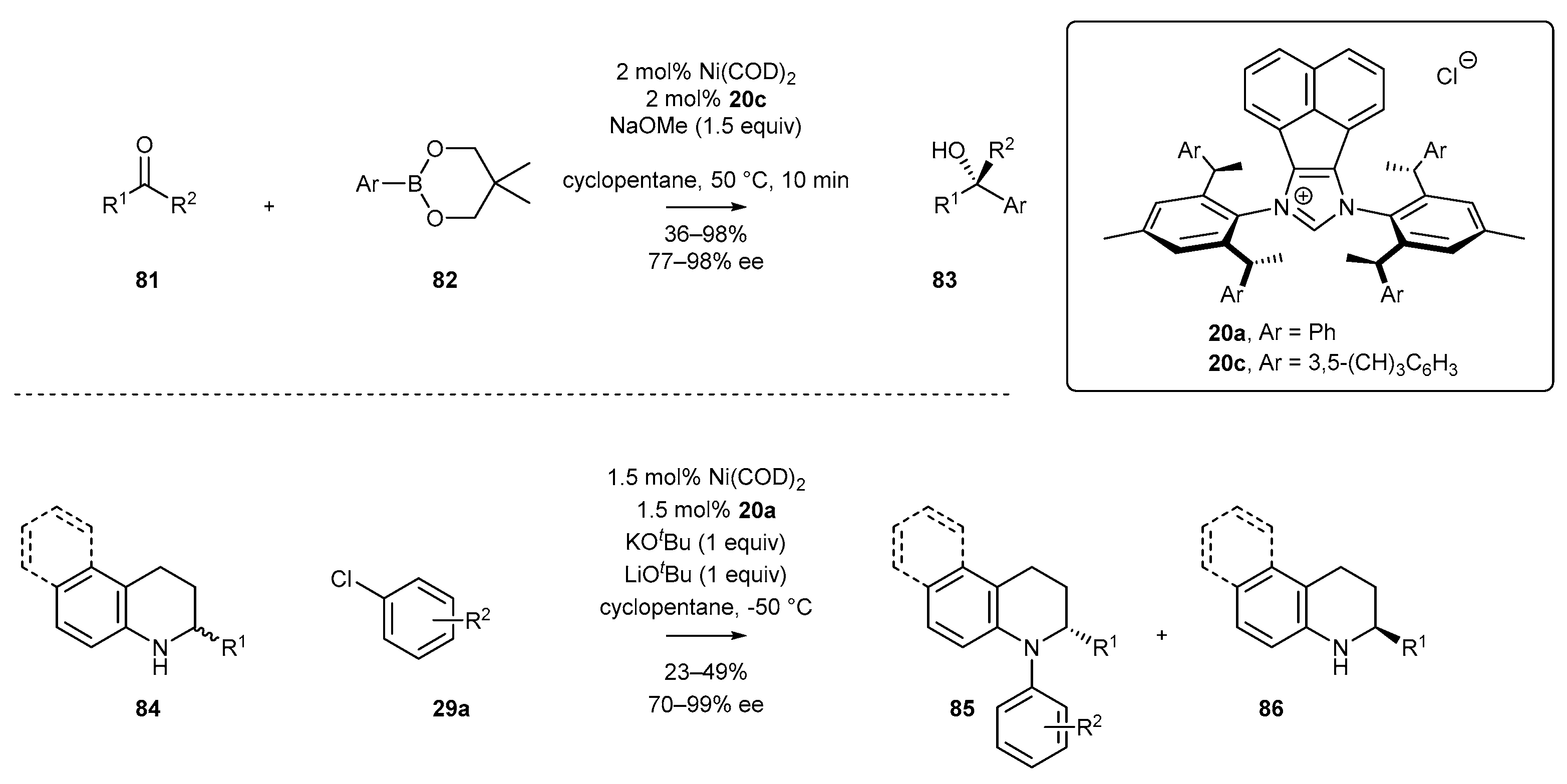{kind=link}
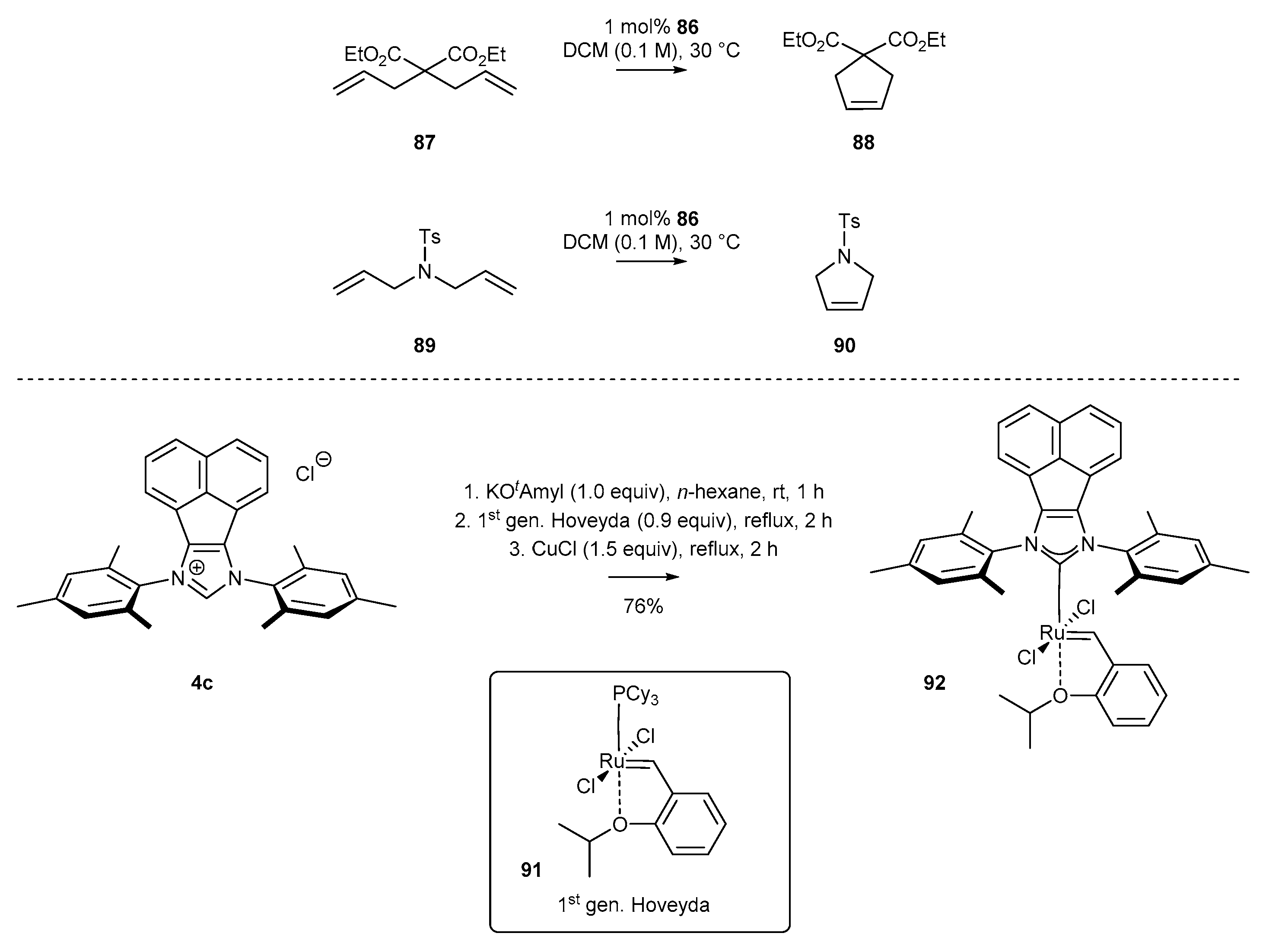{kind=link}
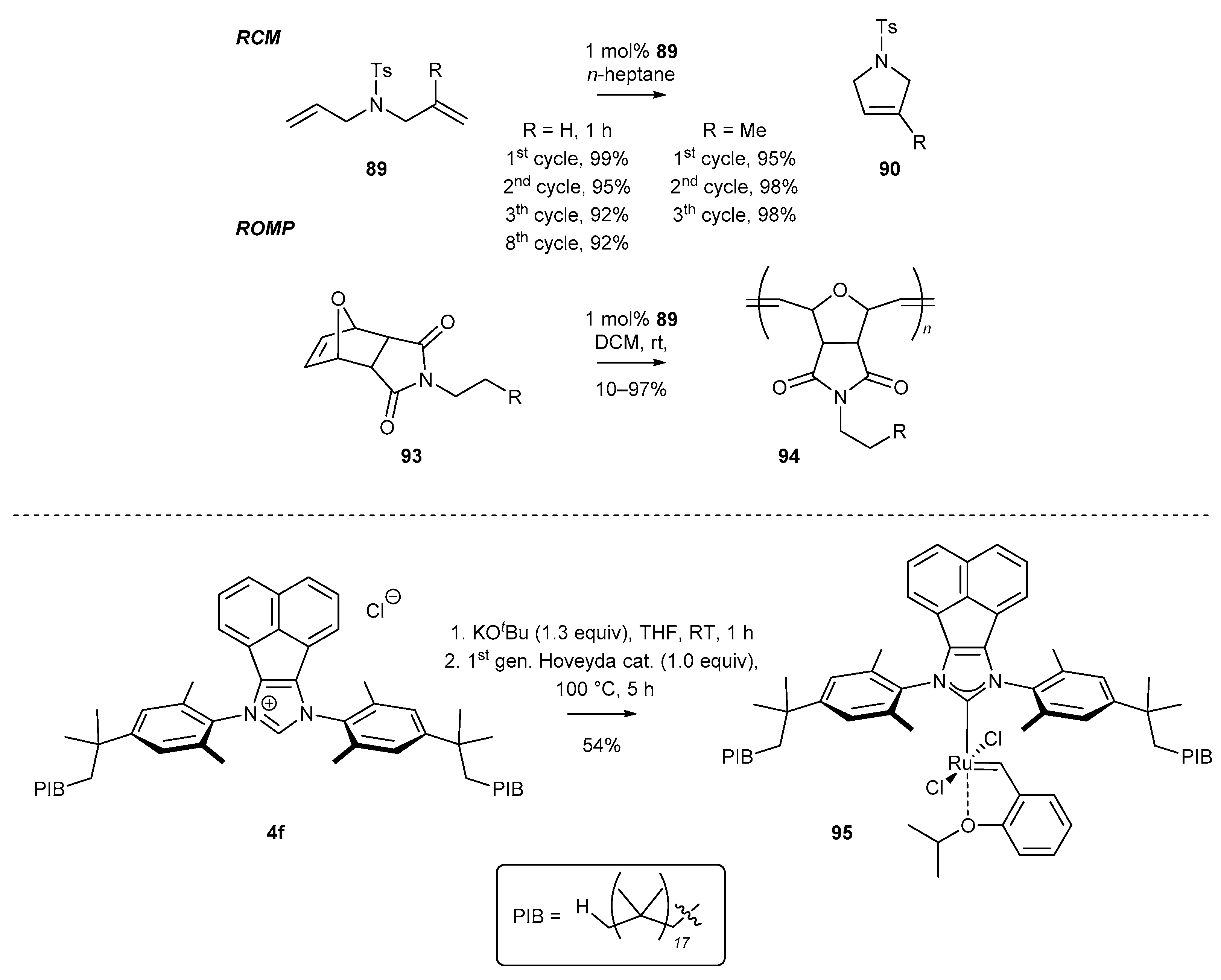{kind=link}
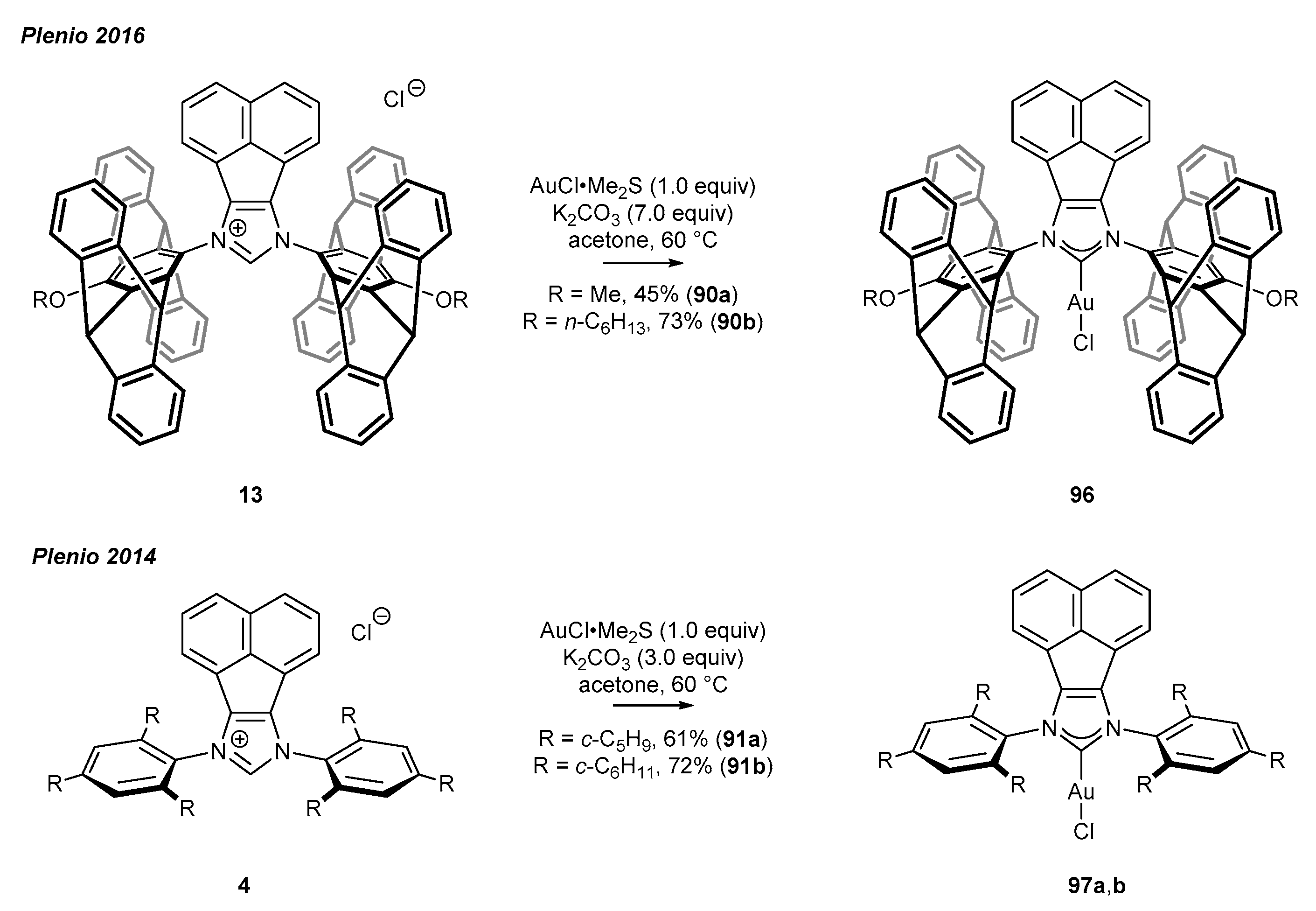{kind=link}
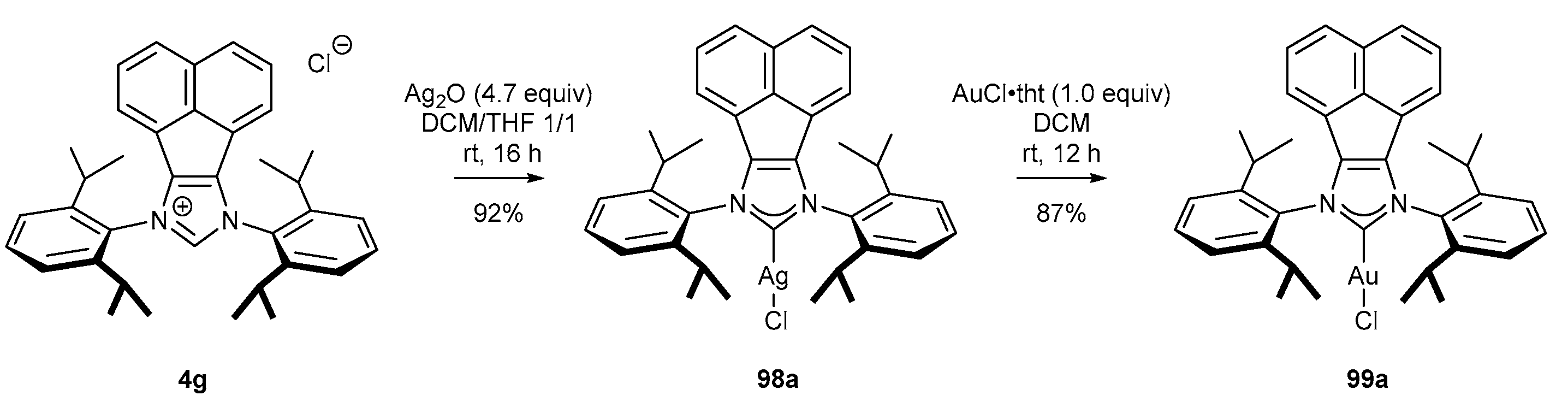{kind=link}
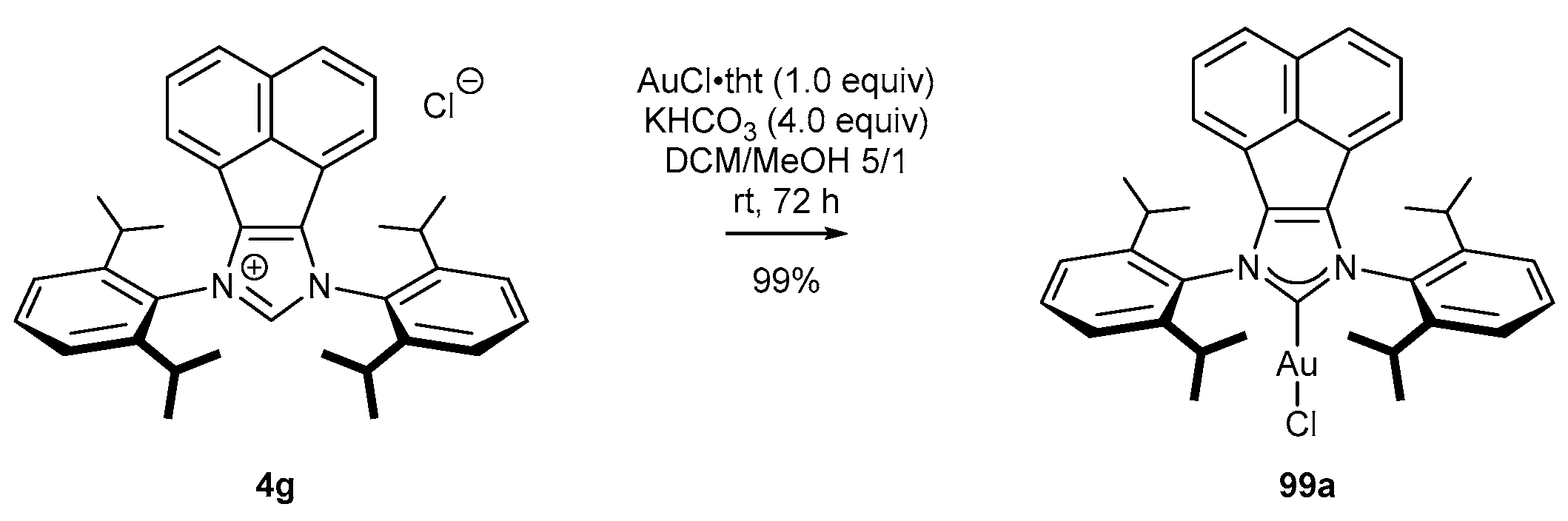{kind=link}
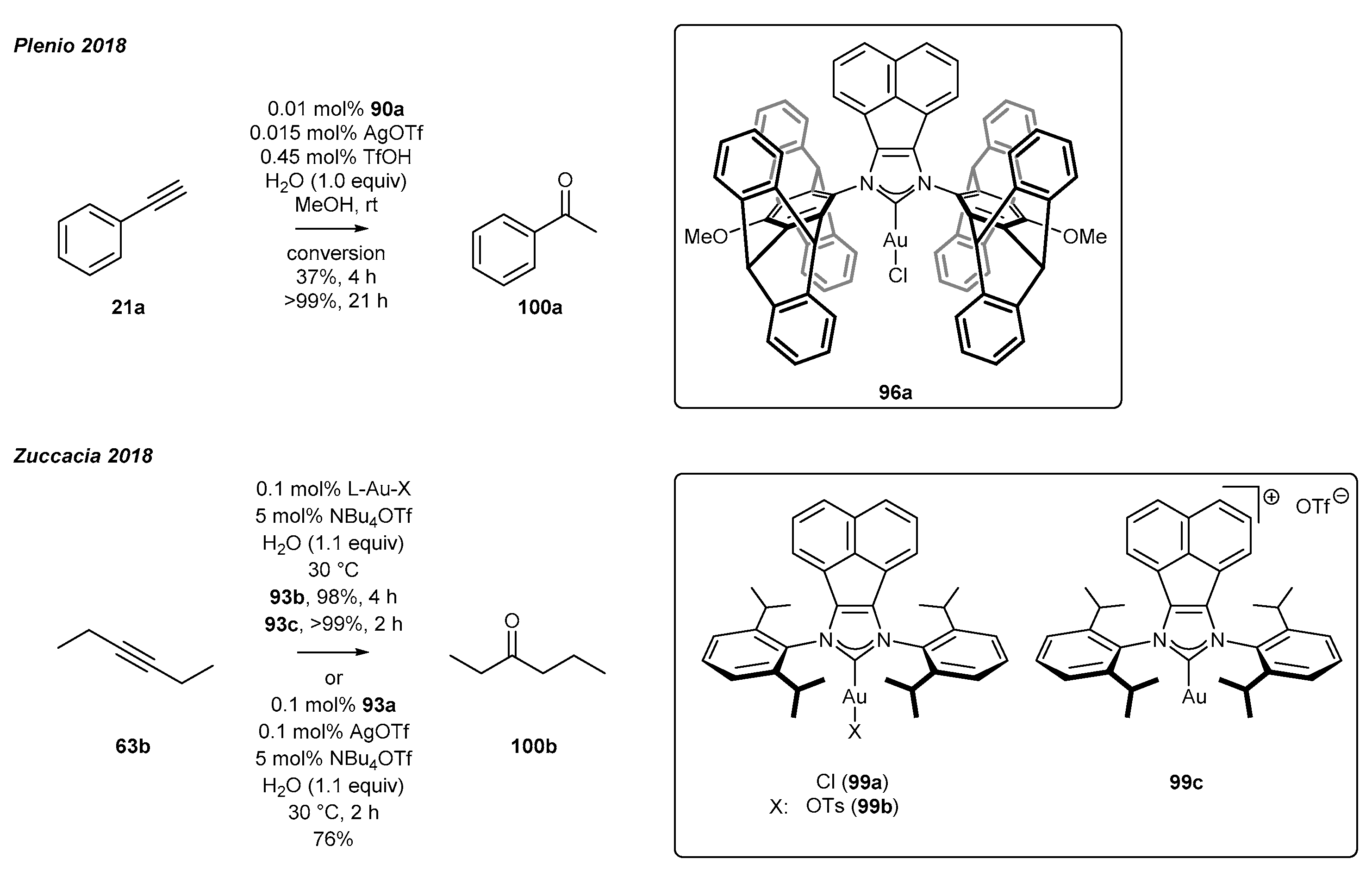{kind=link}
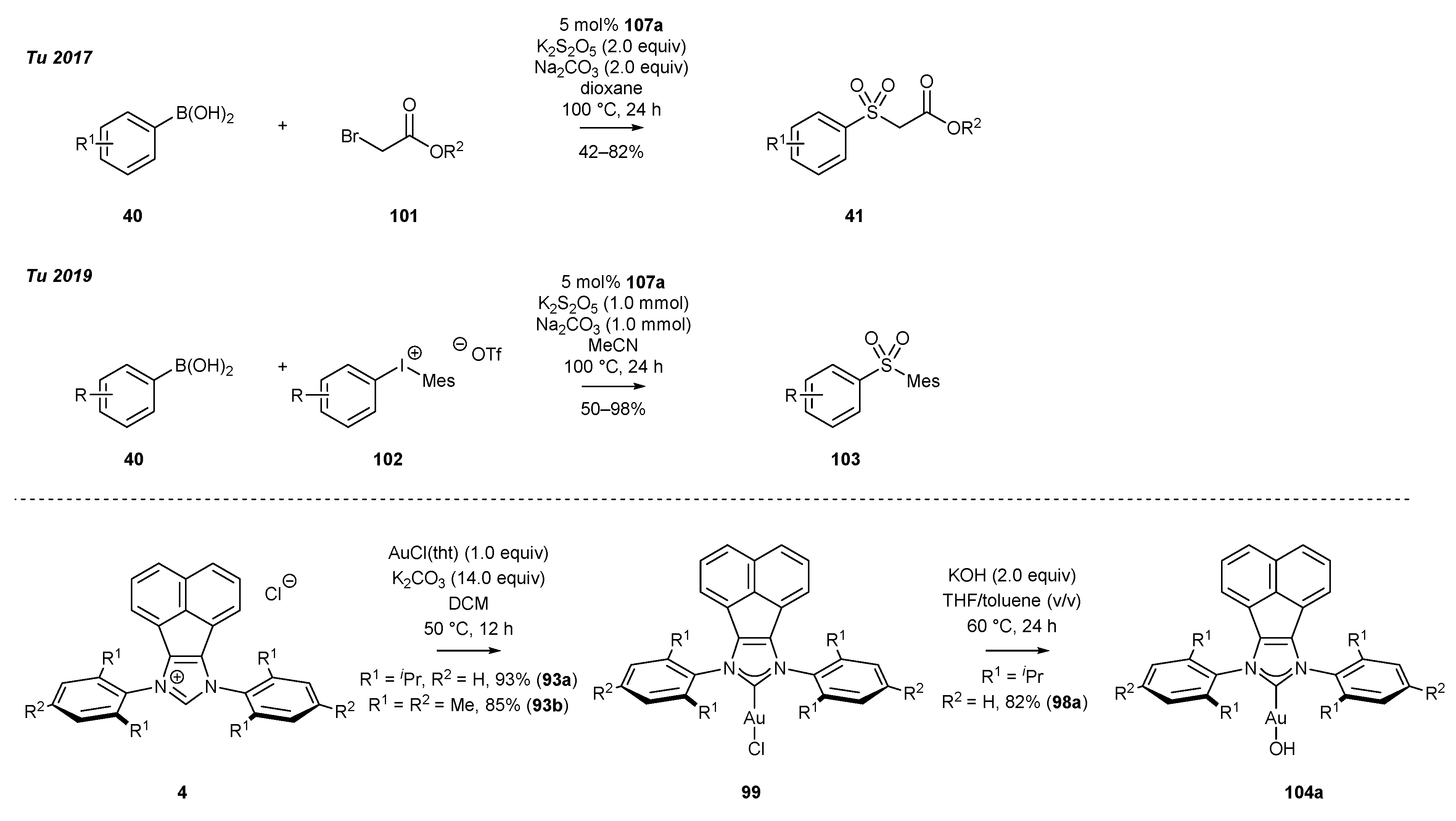{kind=link}
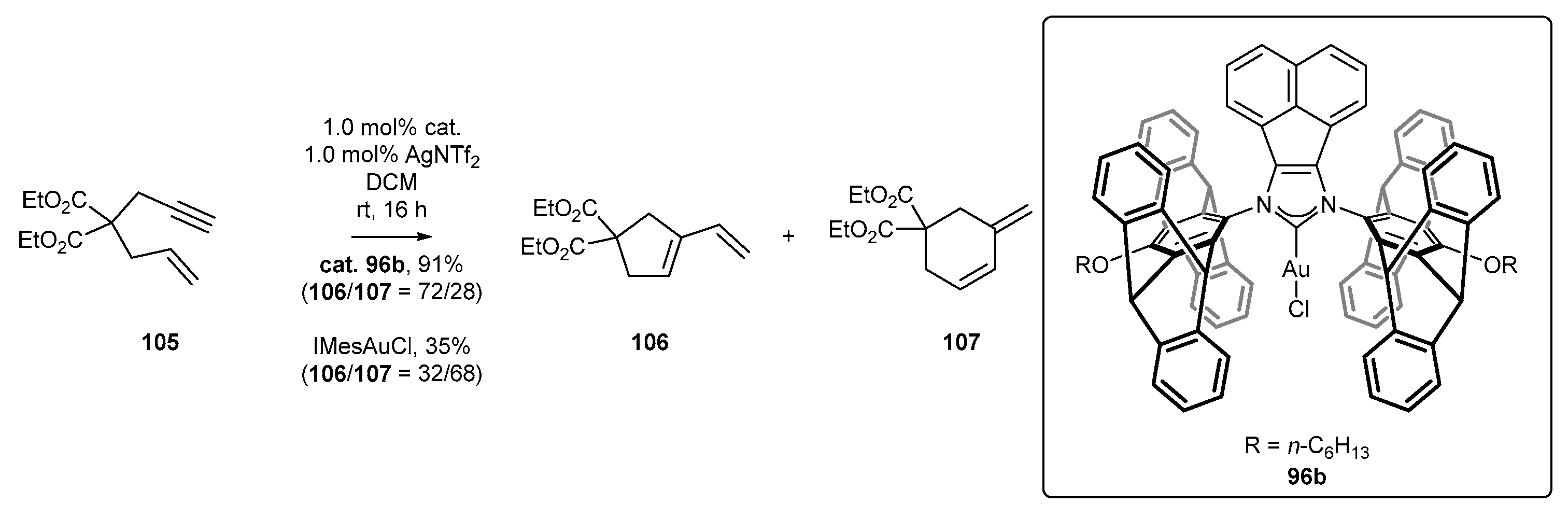{kind=link}
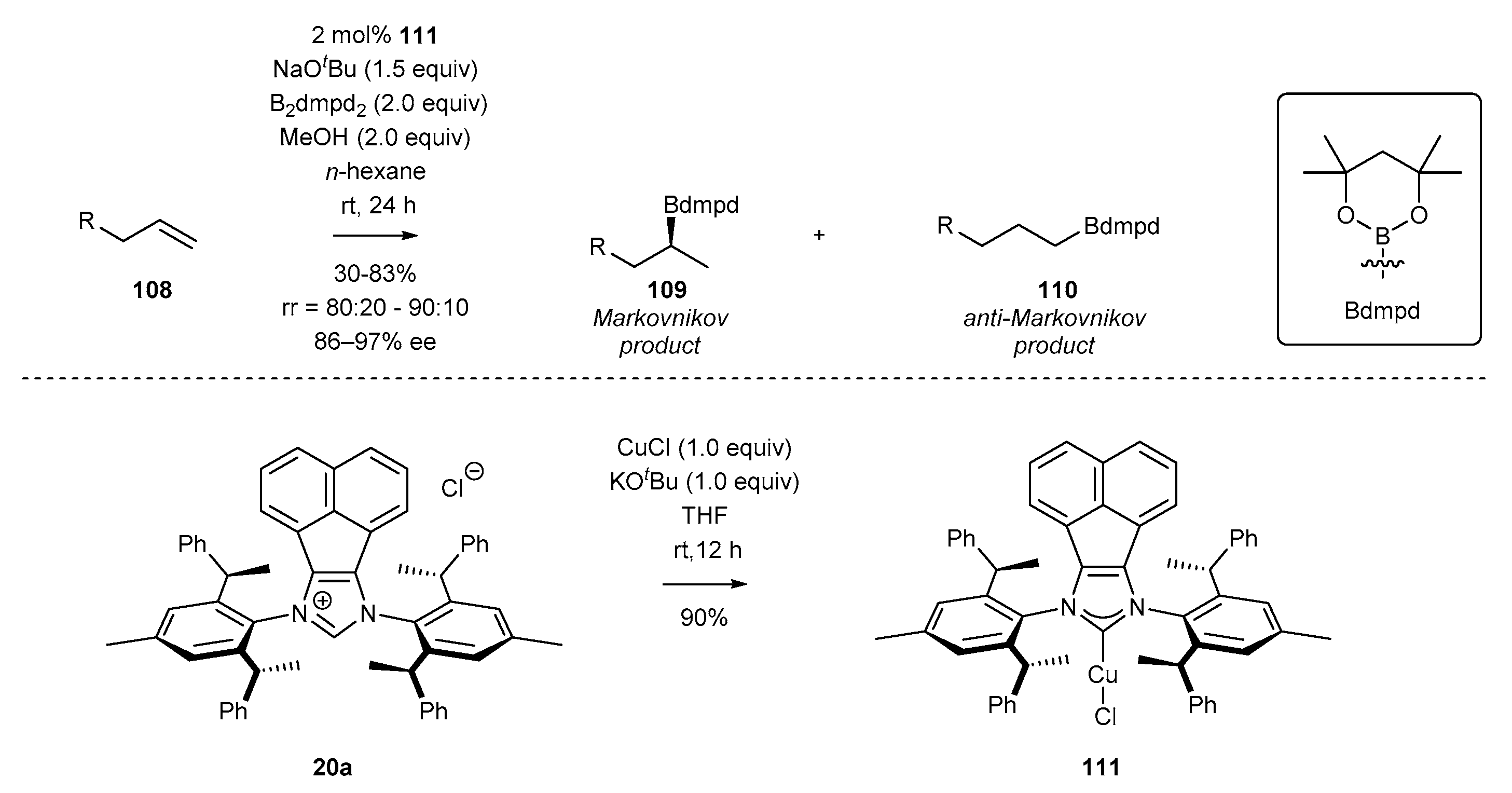{kind=link}
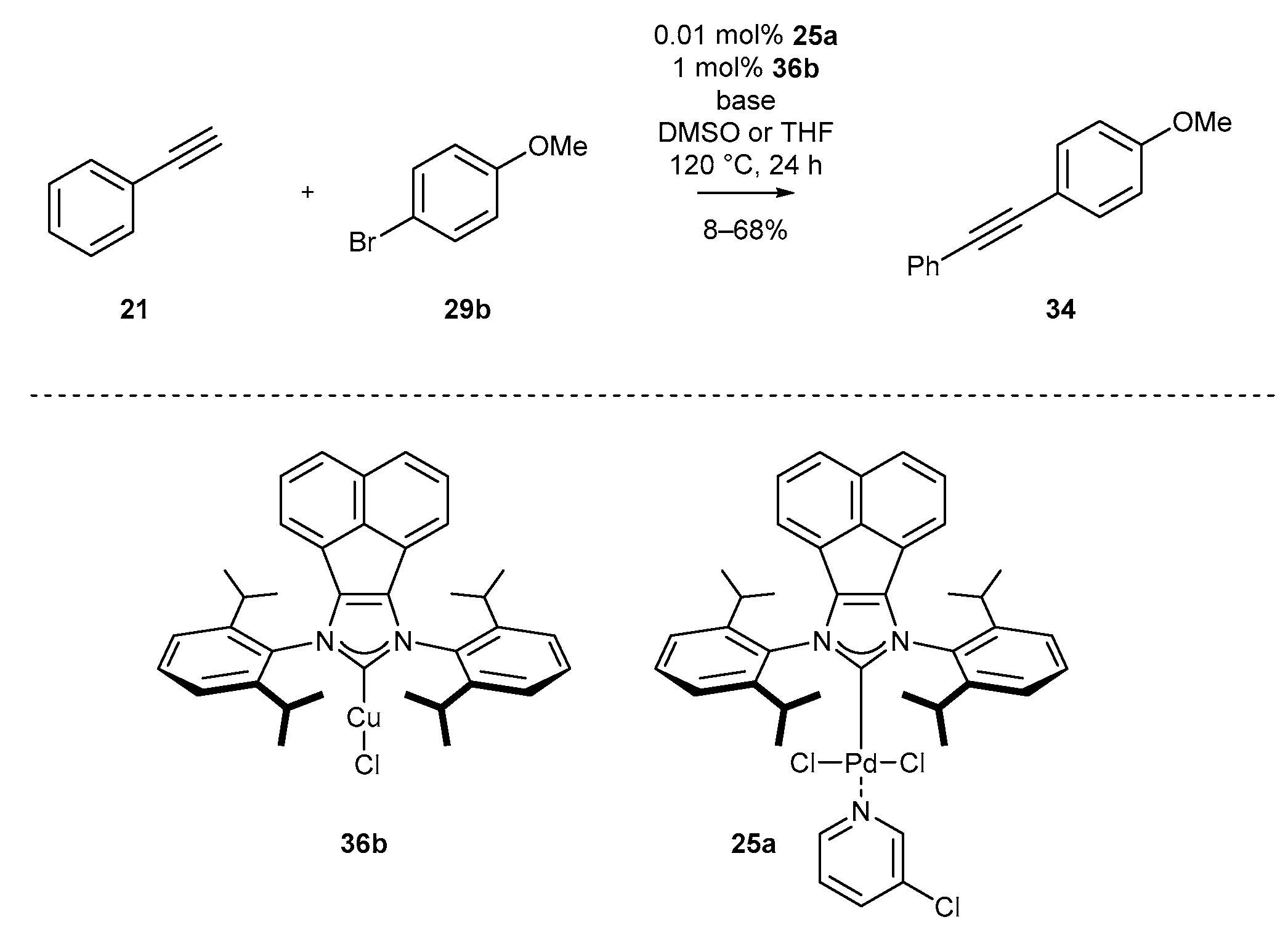{kind=link}
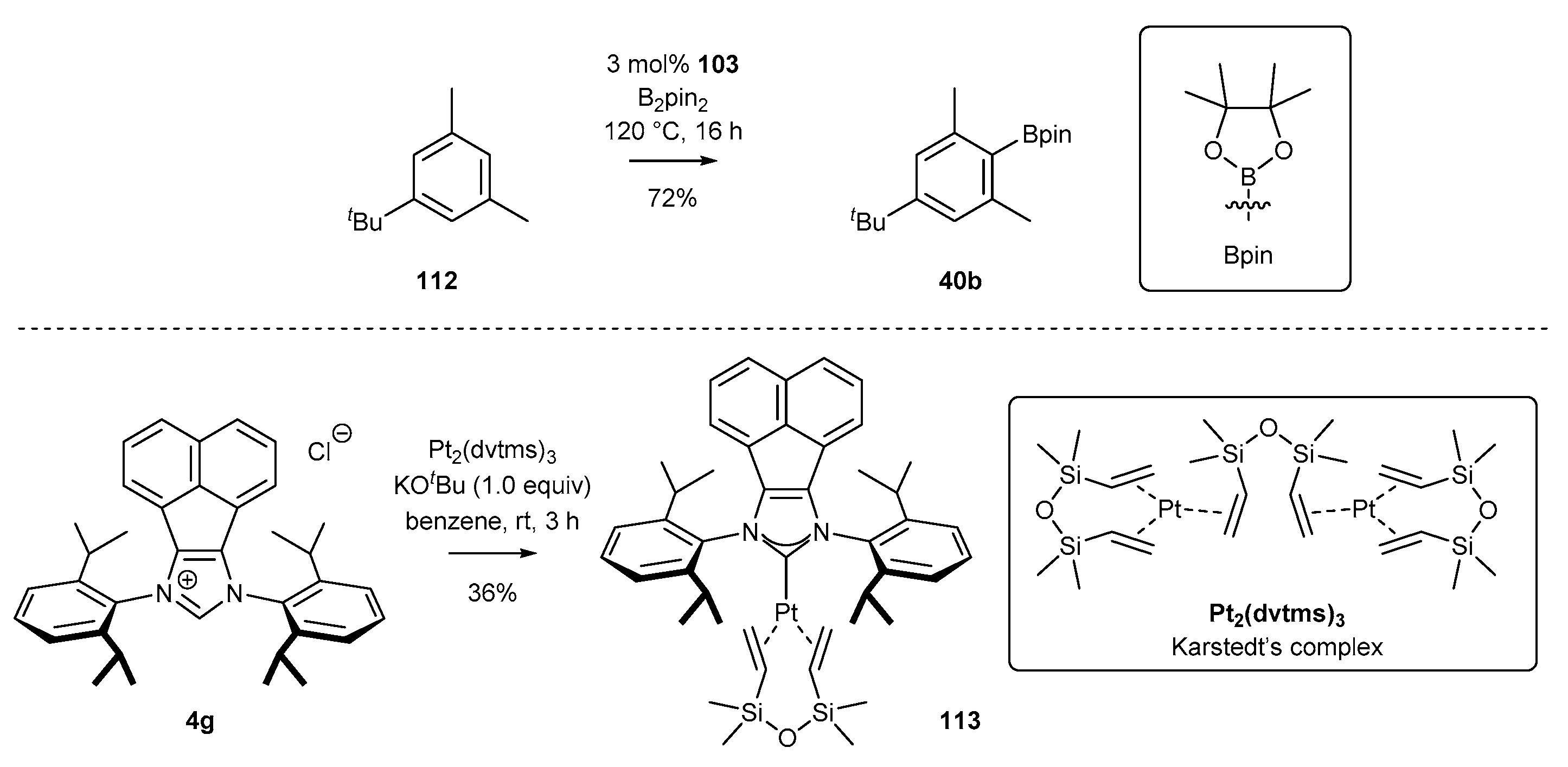{kind=link}
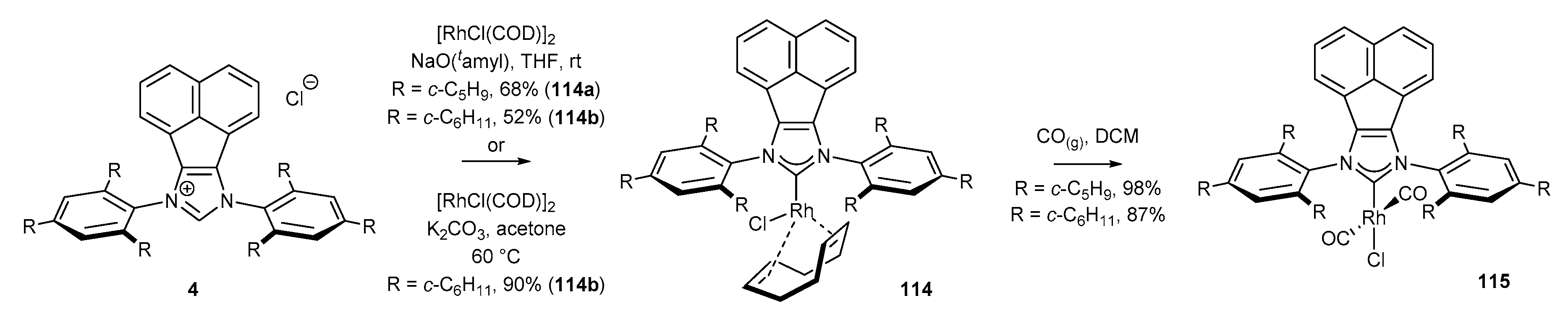{kind=link}
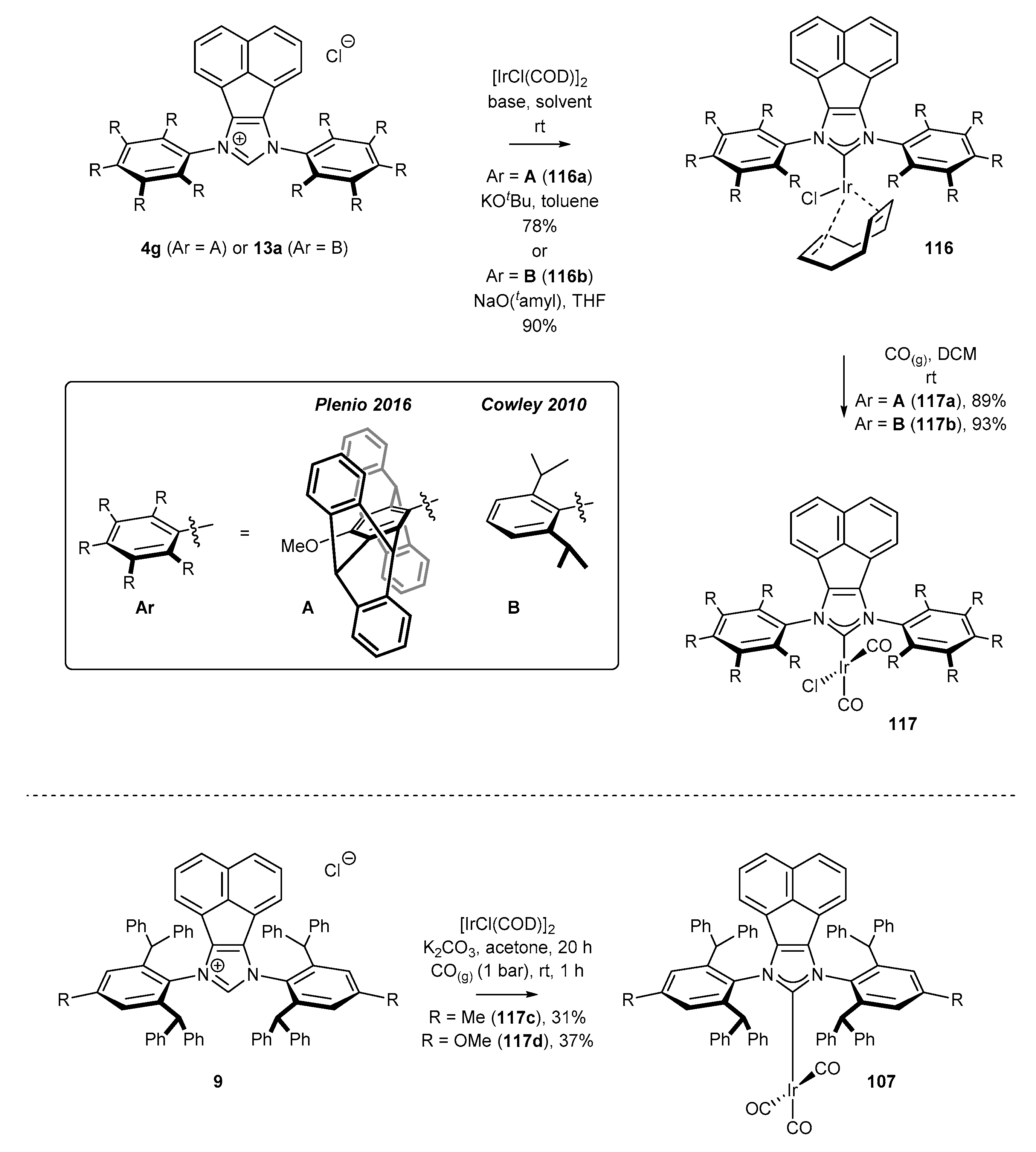{kind=link}
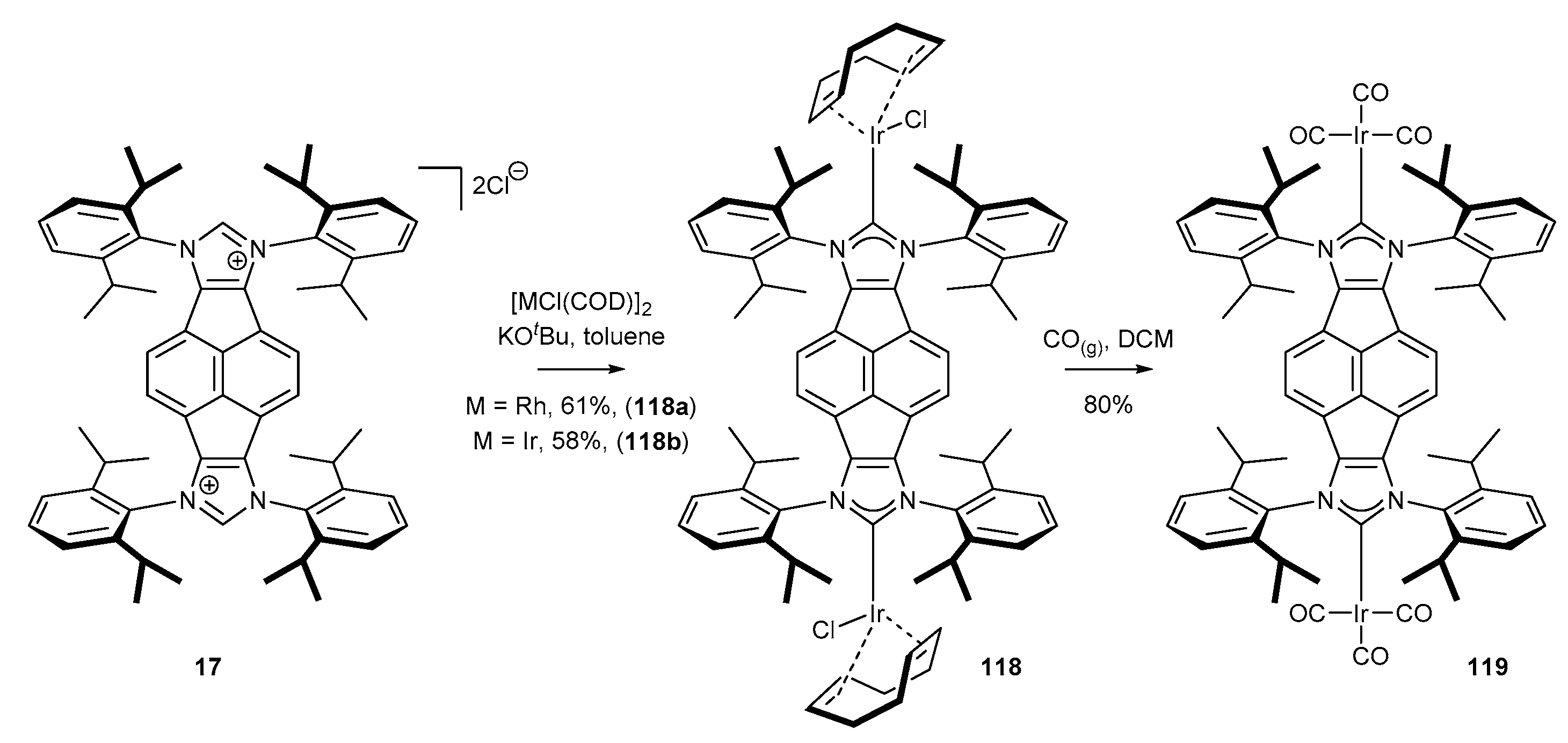{kind=link}
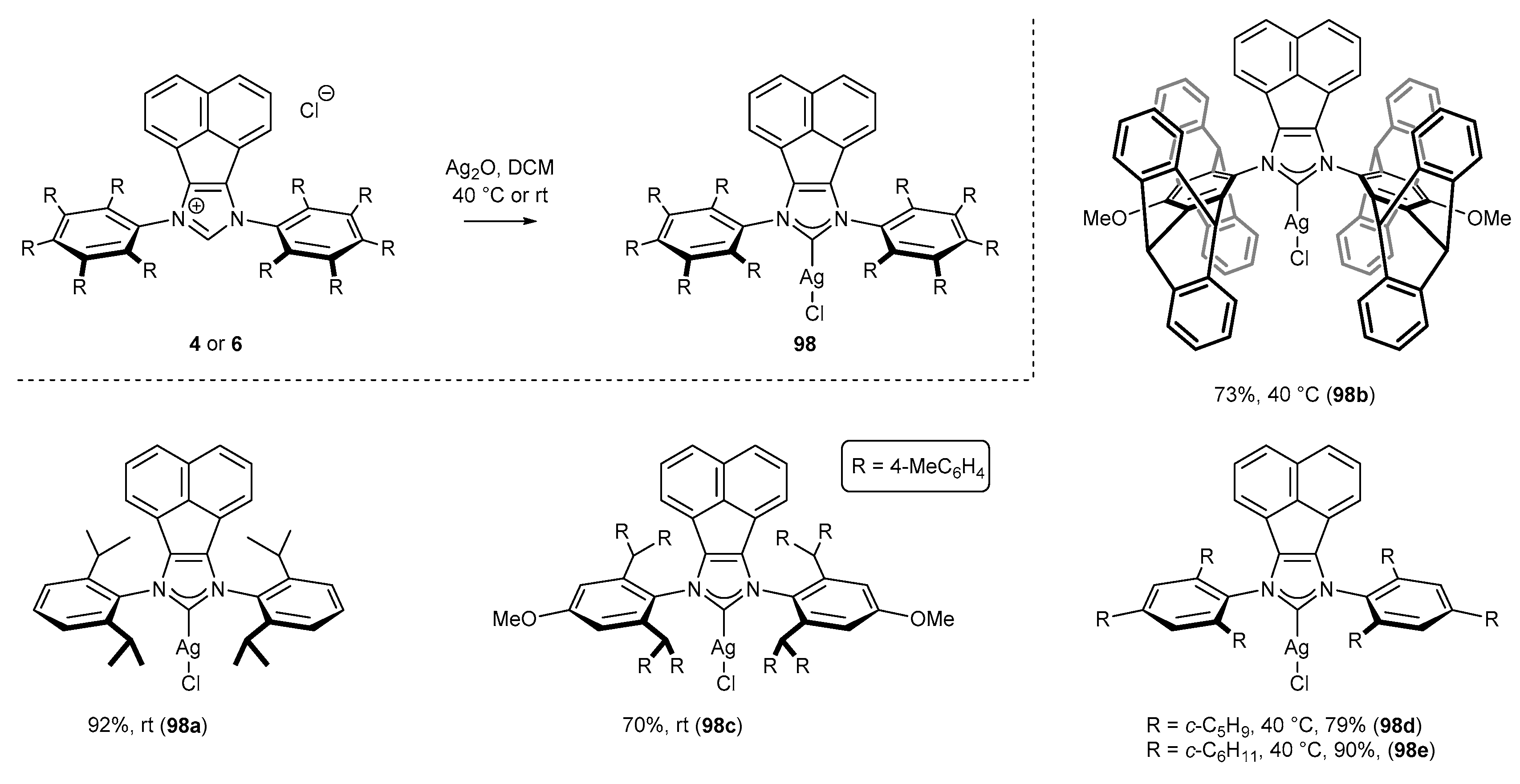{kind=link}
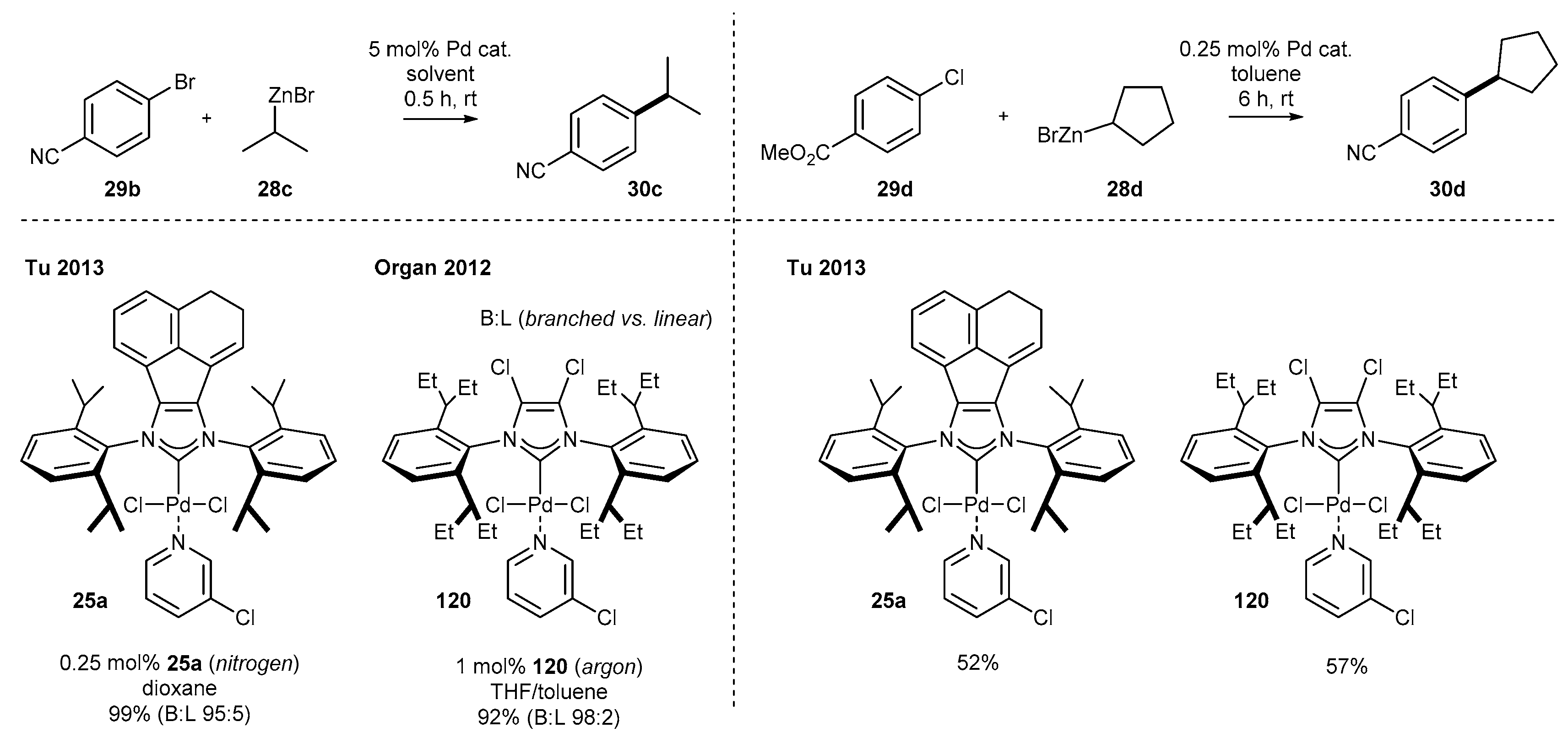{kind=link}
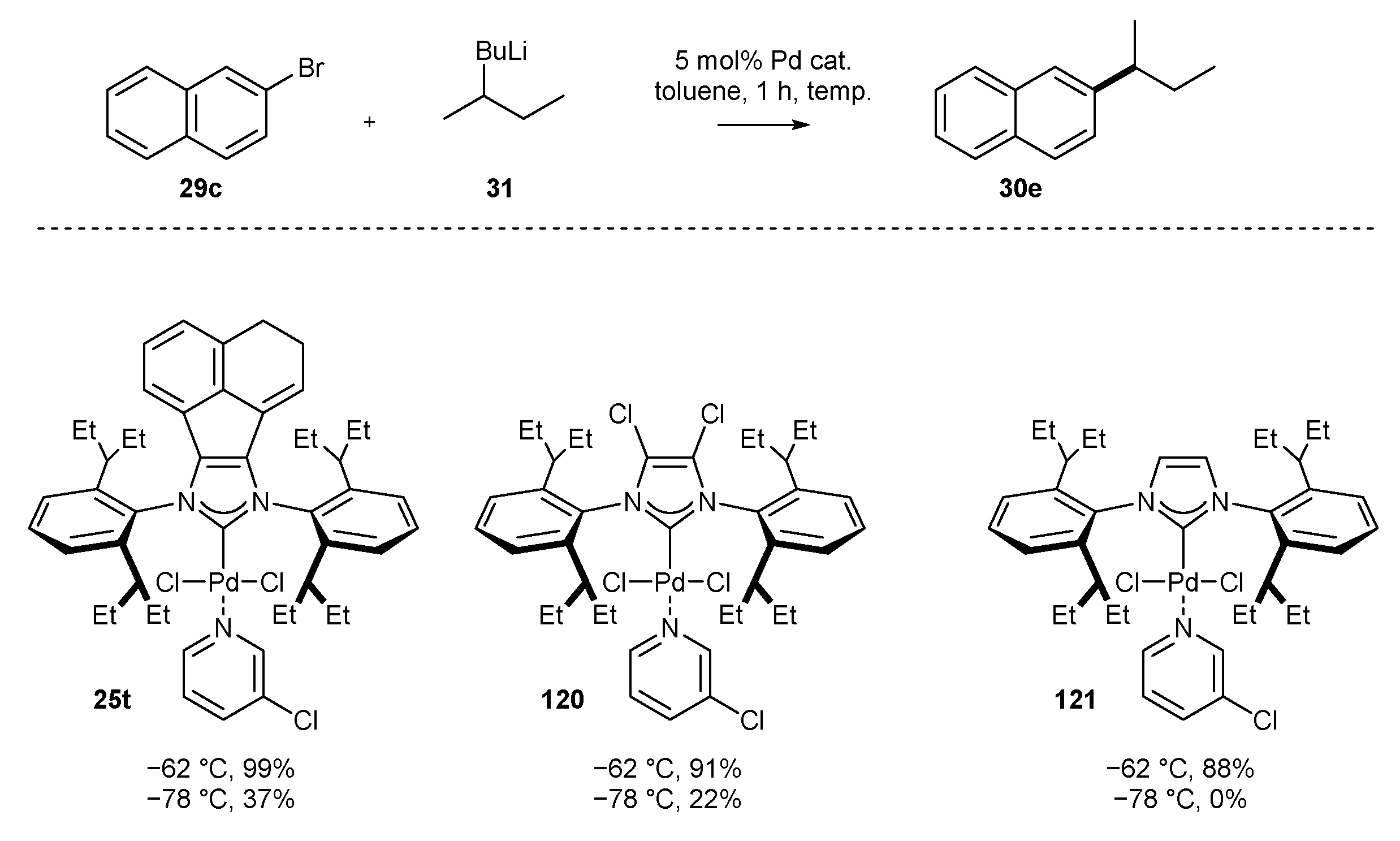{kind=link}
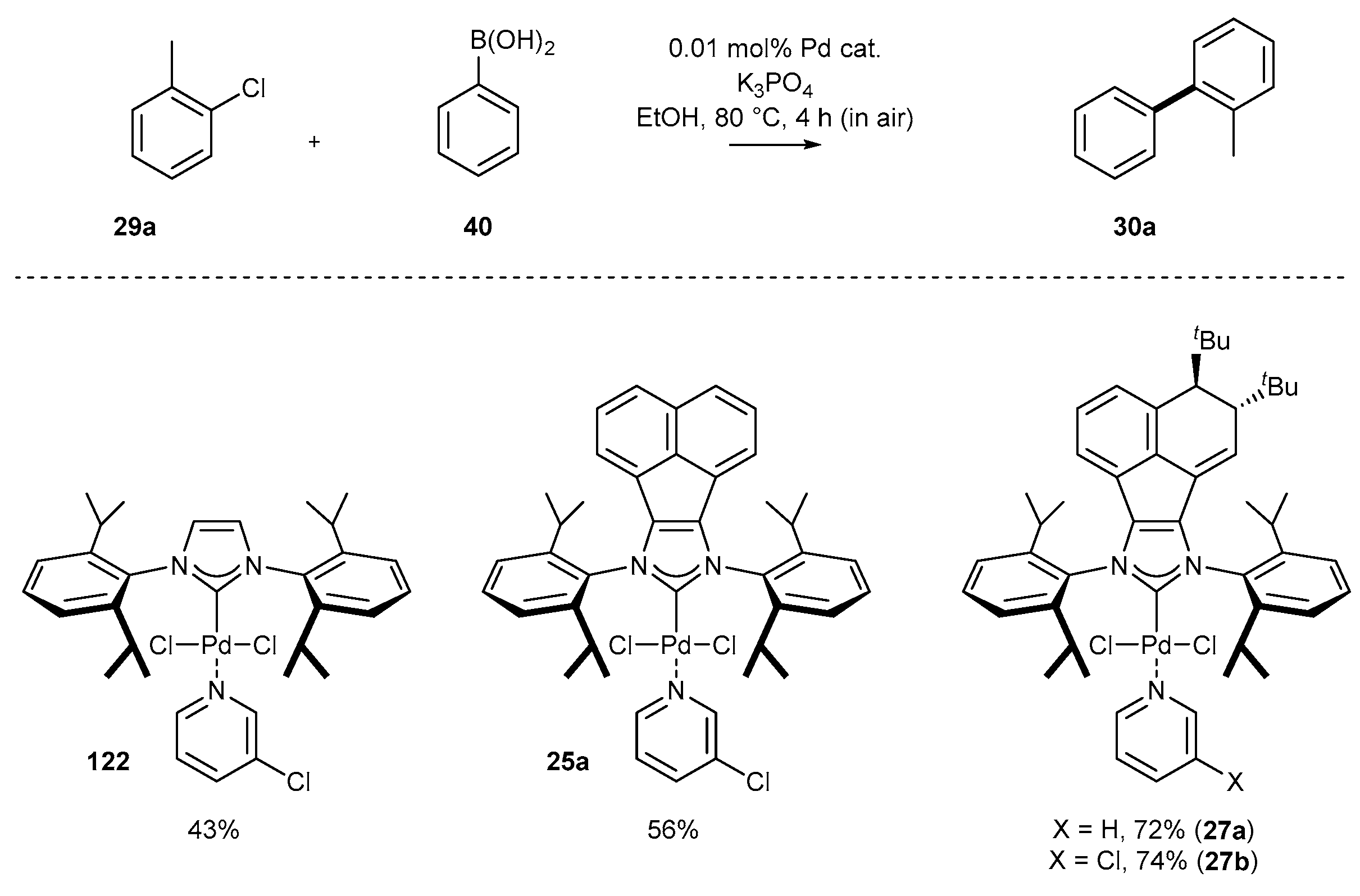{kind=link}
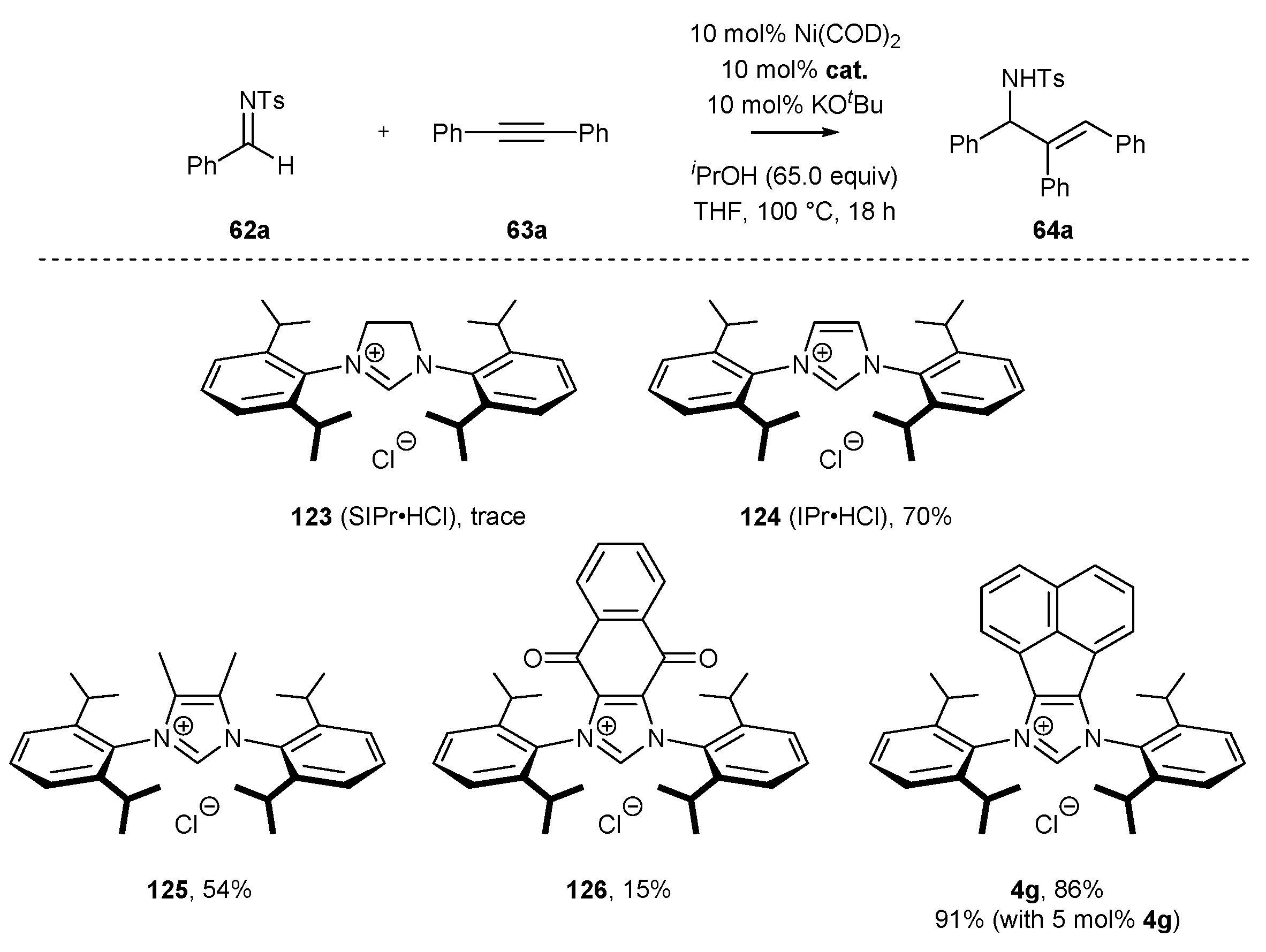{kind=link}
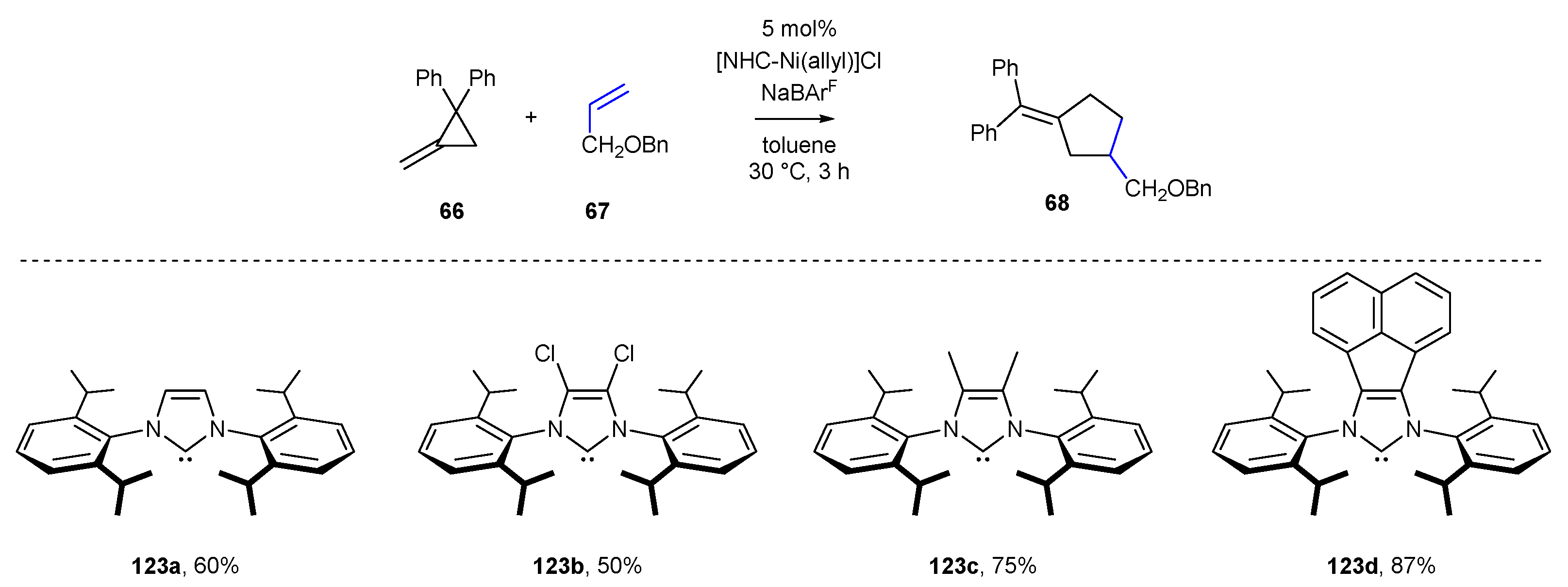{kind=link}
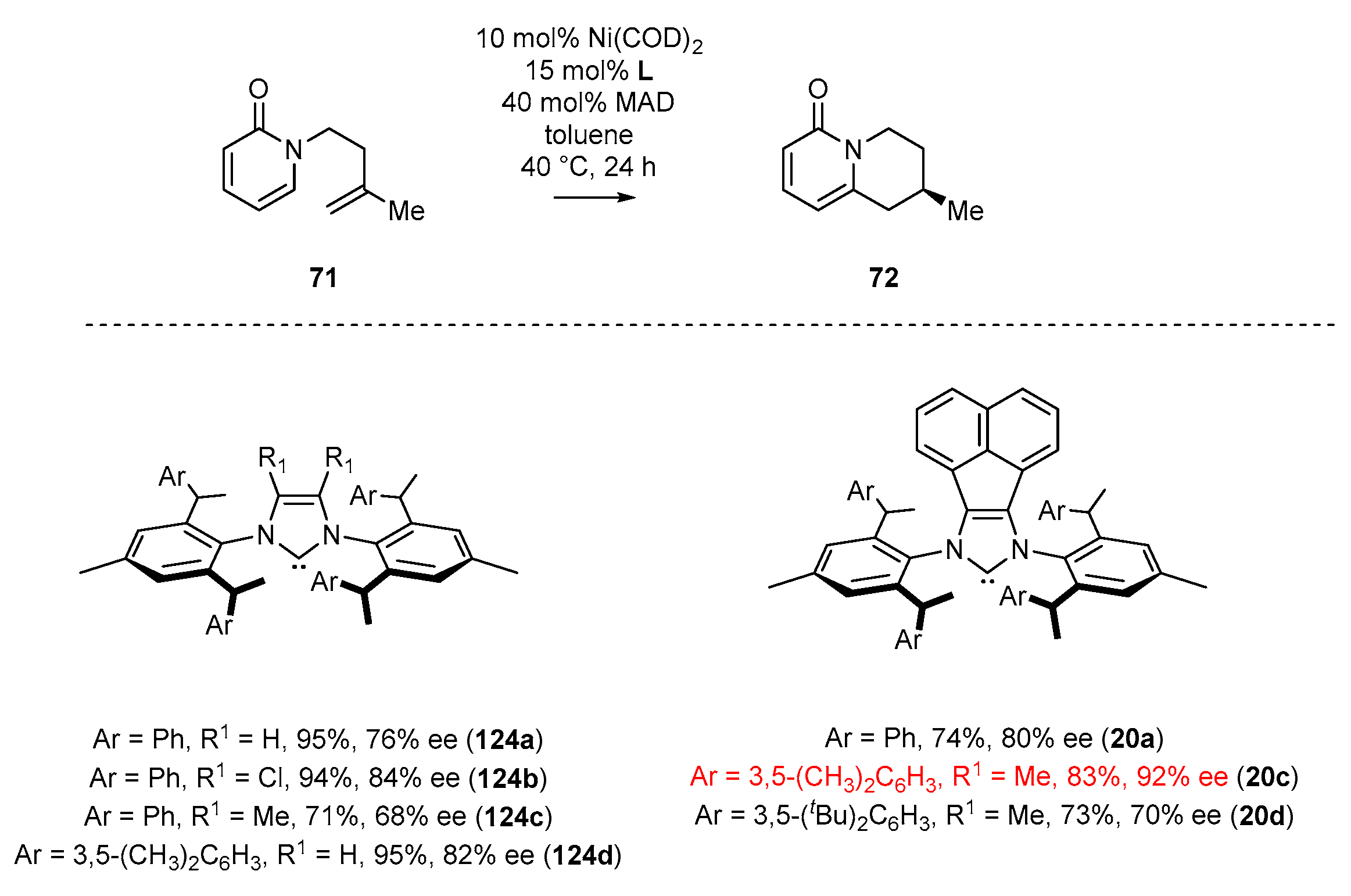{kind=link}
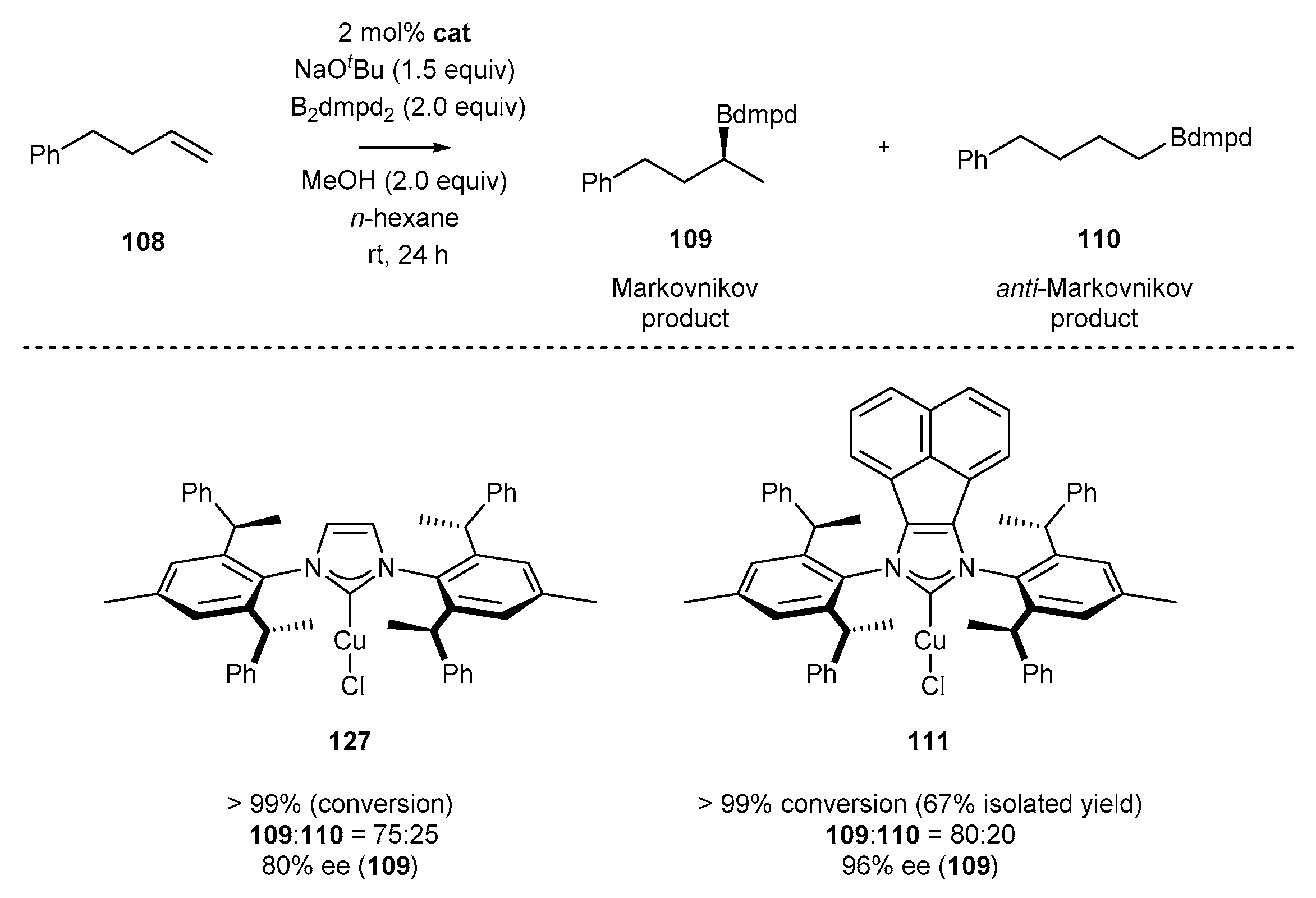{kind=link}
Table 1.
Preparation of salts 20.
| Entry | Ar | R | Conditions | 19 (%) | 20 (%) |
|---|---|---|---|---|---|
| 1 | Ph | Me | A | 70 (19a) | 40 (20a) |
| 2 | Ph | tBu | A | 60 (19b) | 17 (20b) |
| 3 | 3,5−(CH3)2C6H3 | Me | A | 54 (19c) | 52 (20c) |
| 4 | 3,5−(tBu)2C6H3 | Me | B | 23 (19d) | 10 (20d) |
Table 2.
Reaction conditions for Sonogashira coupling.
| Entry | Solvent | Base | 34 (%) |
|---|---|---|---|
| 1 | DMSO | K3PO4 | 65 |
| 2 | DMSO | CsF | 26 |
| 3 | DMSO | Na2CO3 | 42 |
| 4 | DMSO | K2CO3 | 68 |
| 5 | DMSO | Cs2CO3 | 20 |
| 6 | THF | K2CO3 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Baczewska, P.; Śniady, K.; Kośnik, W.; Michalak, M. Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts 2021, 11, 972. https://doi.org/10.3390/catal11080972
AMA Style
Baczewska P, Śniady K, Kośnik W, Michalak M. Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis. Catalysts. 2021; 11(8):972. https://doi.org/10.3390/catal11080972
Chicago/Turabian StyleBaczewska, Paulina, Katarzyna Śniady, Wioletta Kośnik, and Michał Michalak. 2021. "Acenaphthene-Based N-Heterocyclic Carbene Metal Complexes: Synthesis and Application in Catalysis" Catalysts 11, no. 8: 972. https://doi.org/10.3390/catal11080972
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.