
Oncogenic FGFR Fusions Produce Centrosome and Cilia Defects by Ectopic Signaling


1
Department of Biology, Faculty of Medicine, Masaryk University, 62500 Brno, Czech Republic
2
Institute of Animal Physiology and Genetics of the CAS, 60200 Brno, Czech Republic
3
International Clinical Research Center, St. Anne’s University Hospital, 65691 Brno, Czech Republic
*
Author to whom correspondence should be addressed.
Cells 2021, 10(6), 1445; https://doi.org/10.3390/cells10061445
Submission received: 26 April 2021
/
Revised: 27 May 2021
/
Accepted: 7 June 2021
/
Published: 9 June 2021
(This article belongs to the Collection Fibroblast Growth Factor Receptor (FGFR) Signaling Pathway in Cancer and Inflammation)
Abstract
:A single primary cilium projects from most vertebrate cells to guide cell fate decisions. A growing list of signaling molecules is found to function through cilia and control ciliogenesis, including the fibroblast growth factor receptors (FGFR). Aberrant FGFR activity produces abnormal cilia with deregulated signaling, which contributes to pathogenesis of the FGFR-mediated genetic disorders. FGFR lesions are also found in cancer, raising a possibility of cilia involvement in the neoplastic transformation and tumor progression. Here, we focus on FGFR gene fusions, and discuss the possible mechanisms by which they function as oncogenic drivers. We show that a substantial portion of the FGFR fusion partners are proteins associated with the centrosome cycle, including organization of the mitotic spindle and ciliogenesis. The functions of centrosome proteins are often lost with the gene fusion, leading to haploinsufficiency that induces cilia loss and deregulated cell division. We speculate that this complements the ectopic FGFR activity and drives the FGFR fusion cancers.
1. Primary Cilium and Its Role in Cancer Development
A majority of the vertebrate cells are capable of forming a primary cilium, a microtubule-based organelle that projects from the centrosome to integrate signaling pathways and mediate cell-to-cell communication. Mutations in genes that control cilia structure or function produce a growing list of diseases called ciliopathies. To this day, at least 35 ciliopathies exist, and more than 400 candidate proteins have been identified [1]. Virtually all annotated ciliopathies are genetic developmental disorders; however, function of cilia in the tissue homeostasis is also beginning to emerge [2].
During cell division, the centrosomes need to function in the mitotic apparatus. Therefore, the cilium is typically disassembled during mitosis, even though cilia rudiments may be preserved [3,4]. The presence of a primary cilium is, therefore, tightly coupled with the cell cycle. In the majority of the cilia-competent cells, the primary cilium is formed during the G0/G1 phase of the cell cycle and resorbs before the S phase [5,6]. Several mitotic kinases, Aurora A [7,8], polo-like kinase 1 (PLK1) [9] and NIMA-related kinase 2 (NEK2) [10], were shown to block assembly and induce disassembly of the primary cilium, and upregulated activity of these kinases is frequently found in cancer [11,12,13,14,15,16,17,18,19]. Inhibition of the cilia disassembly signaling using small chemical inhibitors restored ciliogenesis and suppressed tumor growth in cholangiocarcinoma [20] or chondrosarcoma [21].
It is mainly the loss of primary cilia, as well as of their regulatory function in cellular signaling and cell division, that has been associated with neoplastic transformation and tumor progression [22,23,24,25]. In glioblastoma, disruption of ciliogenesis was observed at all stages, starting at early tumor lesions [26]. In a mouse model of Kirsten rat sarcoma virus protein (Kras)-driven pancreatic cancer, neoplastic lesions were coupled with cilia loss [27], and a similar observation was in precursor lesions of pancreatic cancer patients [27,28]. In breast cancer, inhibited ciliogenesis was reported within the tumor tissue [29,30,31]. Importantly, in a mouse model of breast cancer, genetic ablation of primary cilia led to earlier tumor formation, faster tumor growth rate, and increased metastasis [32]. Reduced ciliation has also been associated with the onset of prostate cancer [33], rhabdomyosarcoma [34] or chondrosarcoma [35], altogether supporting the role of primary cilia as tumor suppressors.
The Hedgehog (Hh) pathway plays fundamental roles in tissue morphogenesis and homeostasis [36,37,38,39,40,41,42,43,44], and is frequently activated in cancer [45,46]. In vertebrates, the canonical Hh signaling depends on primary cilium. Briefly, activation of the pathway allows for ciliary accumulation of Smoothened, which is accompanied by posttranslational activation of the effector transcription factors from the glioma family, Gli2 and Gli3, within the cilia, and induction of the target genes [47,48,49,50,51]. In Hh-addicted cancers such as medulloblastoma and basal cell carcinoma, the presence of a primary cilium can both promote and suppress tumorigenesis, depending on the oncogene identity. The following studies introduced this paradigm. In a mouse model of medulloblastoma, conditional expression of a constitutively active Smoothened variant SmoM2 leads to tumor formation. Genetic ablation of cilia in the SmoM2-expressing cells completely blocked medulloblastoma formation [52]. Mice harboring only one copy of the cilia-resident Hh pathway inhibitor Patched also develop medulloblastoma, which is abrogated by conditional deletion of cilia [53]. Another medulloblastoma mouse model depends on ectopic expression of the Hh effector Gli2. Tumor development in these mice, however, occurs only after conditional removal of primary cilia, as the cilia presence effectively reduced the Gli2 activity [52]. Notably, similar conclusions were obtained in the Hh-driven basal cell carcinoma. Abundant ciliogenesis was found in patient biopsies and primary lesions in a mouse model constitutively expressing SmoM2 in keratinocytes [54]. Removal of primary cilia abolished tumor development in SmoM2 animals, but accelerated cancerogenesis in mice with conditional expression of active Gli2.
Persistent or increased ciliation has also been associated with other types of cancer. In the choroid plexus, the ectopic presence of Hh-responsive cells harboring a primary cilium produced neoplasm in the mouse [55]. During epithelial–mesenchymal transition of the mammary cancer stem cells, the ciliation increases together with tumorigenic properties of the transplanted cells. Epigenetic or chemical ablation of cilia inhibited Hh signaling and tumorigenic ability of these cells [56]. Taken together, the initiation and progression of the Hh-driven cancers takes advantage of the primary cilium if that is needed to achieve the oncogene activity.
2. FGFR Regulates Cilia Motility and Signaling during Morphogenesis
The fibroblast growth factor receptors (FGFR) have a well-recognized function in the regulation of cilia. Four members of the FGFR family exist, denoted as FGFR1-4 [57,58,59,60], and respond to at least 18 secreted FGF ligands [61,62,63] by dimerization, transactivation and engagement of multiple intracellular signaling pathways [63,64,65,66]. The FGF–FGFR interaction is facilitated by the low affinity co-receptors, i.e., heparan sulfate proteoglycans for most FGF ligands that signal in a paracrine fashion, and Klotho proteins for endocrine FGF19, FGF21 and FGF23 [67,68,69,70,71,72,73,74,75,76,77,78,79]. FGFRs regulate a variety of physiological processes, including morphogenesis [80,81,82,83,84,85,86,87], metabolism [88,89,90,91,92,93] and regeneration [94,95,96,97,98]. Consequently, disrupted FGFR signaling manifests in a plethora of pathological conditions such as developmental ciliopathies [99,100,101] and cancer [70,102,103,104,105,106].
Mounting experimental evidence points towards a functional relationship between FGFR signaling and cilia. In the Xenopus organ of laterality, the gastrocoel roof plate, shorter cilia were obtained after expression of dominant-negative Fgfr1. In zebrafish, morpholino knockdown of fgfr1, expression of dominant-negative Fgfr1, treatment with FGFR kinase inhibitor or loss of fgf4, fgf8 or fgf24 all reduced cilia length in Kupffer’s vesicle and perturbed the cilia-mediated directional fluid flow that is required for left-right patterning of the zebrafish embryo [107,108,109]. In a follow-up study, the zebrafish fgfr2c morphants had shorter cilia in the Kupffer´s vesicle, and showed multiple developmental defects coupled with abnormal left-right polarization, including randomized positioning of the liver and pancreas, disrupted heart looping, and defective brain morphogenesis [110]. A similar phenotype was observed in Xenopus with depleted fgfr4 [111]. Morpholino knockdown of the zebrafish FGF target genes ier2 and fibp1, or of the proteoglycan sulfotransferase 3-OST-5 also shortened cilia in the Kupffer´s vesicle and induced laterality defects [112,113], and this was associated with lower expression of genes important for ciliogenesis [107,113,114,115,116]. Taken together, the cilia length and motility within the organ of laterality is regulated by FGFR signaling, which is critical for establishment of the left-right body asymmetry.
The FGFR signaling also regulates cilia during the later stages of development. Injection of a FGFR kinase inhibitor into neonatal mice produced cilia shortening in the biliary duct, proximal kidney tubules and lungs [99]. The zebrafish fgfr1 morphants had shorter tethering cilia in the otic vesicle and motile cilia in the pronephric ducts [107]. In the inner ear mechanosensory hair cells, FGFR1 localizes to kinocilia and regulates its length and stability [117]. In cultured mammalian cells, a ligand-mediated FGFR activation elongated primary cilia, via accelerated ciliary transport [99,118]. This was coupled with reduced ciliary Smoothened trafficking and inhibited Hh signaling. The molecular mechanism of the FGF-mediated cilia elongation involves ERK MAP (extracellular signal-regulated kinase mitogen-activated protein) kinase and mechanistic target of rapamycin complex 1/2 (mTORC1/2) pathways [99], and the phosphorylation-mediated inactivation of the conserved cilia regulator kinase CILK1 (ciliogenesis associated kinase 1) [101,119,120,121,122,123,124,125,126]. These data further connect the FGFR signaling with the cilia functions.
3. Aberrant FGFR Signaling Affects Primary Cilia
Pathological FGFR activity has been associated with shortening of primary cilia [99,100]. Gain-of-function missense mutations in FGFR3 produce human skeletal dysplasias, including achondroplasia and thanatophoric dysplasia [127,128,129,130,131], and frequently occur in cancer [132,133,134,135,136]. Several studies pointed towards a cross-talk of FGFR3 signaling with the cilia-associated Hh pathway that was found inhibited in mouse models of achondroplasia [137,138,139], due to the defective ciliogenesis [99,100]. Shorter cilia were also found in the cartilage of humans with thanatophoric dysplasia, and in cells overexpressing a pathological FGFR3 variant [99,100]. In cultured cells, pathological FGFR3 activity inhibited the Hh pathway, reduced ciliary Smoothened trafficking, and shortened cilia, possibly via reduced ciliary transport which limited the tubulin flux necessary for cilia maintenance [99,100,140]. FGFR kinase inhibitors normalized the cilia length in vitro [99,100], and Hh signaling in the cartilage in vivo [107,141]. Taken together the pathological FGFR activity interfered with ciliogenesis and cilia function. This was in part due to increased Aurora A and PLK1 activity [12,142,143], that are also found upregulated in the FGFR1-driven cancers [144,145,146]. Therefore, it is likely that the FGFR cancers are driven, at least partly, by cilia disassembly that alleviates the mitotic brakes and increases availability of centrosomes for the mitotic spindles [147,148].
4. FGFR Gene Fusions in Cancer
Deregulated FGFR signaling, mostly caused by increased FGFR activity, has been implicated mainly in tumor progression, through poorly understood mechanisms involving accelerated proliferation, resistance to apoptosis and enhanced angiogenesis [93,149,150,151,152]. Among the 4853 tumor samples analyzed by next generation sequencing, a FGFR aberration was found in 7.1% of all cases [153]. The most frequent lesion was gene amplification, accounting for 66% of FGFR aberrations [153], and typically resulting in FGFR overexpression and increased activity [154,155,156,157,158]. FGFR mutations were less frequent, covering 26% of the identified aberrations [153]. More than 200 distinct FGFR point mutations have been identified in cancer, targeting the extracellular, transmembrane and kinase domains of all four FGFRs [133,159,160,161]. The majority of the mutations lead to ligand-independent FGFR dimerization and increased pathway activity [162,163,164,165]. Interestingly, somatic mutations found in cancer frequently overlap with those causing developmental disorders (extensively reviewed in [133]); however, increased incidence of tumors has not been reported in these disorders. This can be exemplified by activating FGFR3-K650E/M mutation, causing thanatophoric dysplasia type II and SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans), respectively [128,129,166,167]. Although this mutation has been detected in aggressive cancers, it failed to induce neoplastic transformation in mice. Additional mutation, involving deletion of the tumor suppressor PTEN (phosphatase and tensin homolog) or activating KRAS mutation were required to induce the FGFR3 cancerogenesis [168,169]. These data suggest that FGFR missense mutations are not likely to initiate the neoplastic transformation, but rather occur later to promote tumor progression and metastasis.
A gene fusion originates from the chromosomal rearrangement involving two genes, and results in a fusion protein capable of neoplastic transformation and oncogene addiction [170,171]. FGFR fusions are relatively rare, accounting for 8% of all FGFR aberrations found in cancer [153,172]. Additional missense mutations are sporadic [172], suggesting that the FGFR fusion protein holds sufficient oncogenic properties. In type I fusions, typically driving the hematological malignancies [173], the FGFR extracellular and transmembrane domains are excluded, and the fusion occurs at the N-terminus of the FGFR kinase domain (Figure 1). In type II fusions that are mostly found in solid tumors [173], the breakpoint usually occurs between exons 17 and 19, affecting only a varying part of the C-terminal region of FGFR [133]. In both types of fusion, the partner typically contains domains that facilitate dimerization such as the coiled-coil domain, the sterile alpha motif, the leucine rich repeat or the leucine zipper, leading to ligand-independent FGFR dimerization and signaling activity. The FGFR fusion protein may also be sequestered to an alternate subcellular location, trough features gained via the fusion partner, which can result in misplaced and deregulated activity. Finally, a substantial part of the fusion partner is typically lost during chromosomal rearrangement, producing haploinsufficiency or gaining novel function that may contribute to neoplastic transformation.
A substantial portion of the FGFR fusion partners are proteins associated with the centrosome functions, including spindle organization and ciliogenesis (8 of 14 recurrent FGFR fusions with at least partially characterized signaling properties; based on PubMed search in April 2021). This led us to speculation that disruption of the centrosome cycle may drive pathogenesis of the FGFR fusion cancers. In the following sections, we review the current knowledge of such oncogenic FGFR fusions, and discuss the possible involvement of both fusion partners in cancerogenesis. For a complete reference, the recurrent and characterized, yet not included fusions comprise FGFR2-CCDC6 [149,174], FGFR2-AHCYL1 [175,176], FGFR2-PPHLN1 [177,178], FGFR3-BAIAP2L1 [136,179,180], ZMYM2-FGFR1 [181,182,183], and BCR-FGFR1 [182,183,184].
4.1. FGFR3-TACC3
Gene fusion involving FGFR3 and the transforming acidic coiled-coil containing protein 3 (TACC3) is one of the recurrent gene fusions, found in glioblastoma (29 of 103), non-small-cell lung carcinoma (28 of 103), head and neck squamous cell carcinoma (11 of 103), bladder cancer (10 of 103), and other types of cancer (Table 1) [133,149,153,179,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200]. FGFR3-TACC3 transformed NIH3T3 and Rat1A fibroblasts [179,187,201,202], and the xenografted astrocytes or glioblastoma cells stably expressing FGFR3-TACC3 gave rise to gliomas [187,203]. Mice with hippocampal cells transduced with FGFR3-TACC3 developed invasive, rapidly growing high-grade gliomas [187], proposing FGFR3-TACC3 as an oncogenic driver.
The chromosomal rearrangement produces loss of the FGFR3 3′UTR containing miR-99a that normally regulates the FGFR3 levels; this leads to overexpression of FGFR3-TACC3 [203] and abundant transactivation of the FGFR3 residues [201]. Similar to the majority of the type II FGFR fusions, the FGFR3-TACC3 protein lacks the C-terminus of FGFR3 that is necessary for phospholipase C γ (PLCγ) binding (Figure 1), leading to silencing of this signaling branch [179,249]. Conversely, the ERK MAP kinase and STAT (signal transducer and activator of transcription proteins) signaling is increased in FGFR3-TACC3 expressing cells [201,203], and silencing of these pathways was partially successful in targeting the oncogene-driven growth of cell lines and xenografts [36,149,186,187,202,203,250,251,252].
TACC3 is an important component of the mitotic spindles, ensuring proper attachment of chromosomes to the microtubules [253,254]. During mitosis, FGFR3-TACC3 mislocalizes to the spindle poles while sequestering also the endogenous TACC3 from the mitotic spindle, through interaction of their coiled-coil domains [188,255,256]. This delays mitotic progression, and induces chromosome segregation defects and aneuploidy that increases by greater than 2.5 fold [187]. Interestingly, targeting TACC3 proved a viable strategy in TACC3-overexpressing cancers, likely by inducing abundant multipolar spindles, which led to mitotic arrest and apoptosis [257,258,259]. Elevated cellular levels of TACC3 were shown to induce loss of primary cilia through Aurora A induction and disruption of the transmembrane protein 67 (TMEM67)-filamin A complex [260,261], and promoted oncogenic transformation and shortened survival of the patients with prostate cancer [262]. Knockdown of TACC3 rescued ciliogenesis, reduced transformation and inhibited xenograft growth [262]. Taken together, FGFR3-TACC3 could lead to neoplastic transformation partly through induction of cilia disassembly and deregulated cell division, which are both druggable targets.
4.2. FGFR1-TACC1
The FGFR1 fusion with transforming acidic coiled-coil containing protein 1 (TACC1) was found in various types of tumors arising within the central nervous system (14 of 15; Table 1) [186,187,204,205,206,207,208,209,210,211]. FGFR1-TACC1 transformed C3H10T1/2 and Rat1A fibroblasts [187,263], and the xenografted astrocytes stably expressing FGFR1-TACC1 gave rise to gliomas [187].
The biological and oncogenic functions of FGFR1-TACC1 appear similar to those assigned to FGFR3-TACC3 [187]. TACC1 has a coiled-coil domain at the C-terminus, that is preserved in the fusion protein (Figure 1), and that mediates localization to the mitotic spindle [264,265,266]. FGFR1-TACC1 expression increased the rate of errors in chromosomal segregation about five times [187], likely through mislocalization and sequestration of endogenous TACC1, and similar spindle defects were observed in HeLa cells with depleted TACC1 [266]. TACC1 interacts with Aurora A, which appears critical for spindle formation, and the expression levels of the two proteins seem to correlate in cancers [266]. This suggests that TACC1 overexpression caused by FGFR1-TACC1 fusion could participate in neoplastic transformation through deciliation caused by increased Aurora A activity and deregulated cell division, similar to FGFR3-TACC3 cancers.
4.3. FGFR2-BICC1
About 45% of the intrahepatic cholangiocarcinoma cases are coupled with FGFR2 fusion, half of which are with bicaudal C1 (BICC1) [149,175,177,212,213,214,215]; identification of FGFR2-BICC1 in other types of cancer is rare [175] (Table 1). FGFR2-BICC1 transformed NIH3T3 cells that formed tumors in mice [175], and the xenografted FGFR2-BICC1 expressing liver organoids gave rise to tumors [267].
As a consequence of the chromosomal rearrangement, the FGFR2 3´UTR is truncated which results in upregulation of the FGFR2-BICC1 fusion protein [214]. FGFR2-BICC1 dimerizes likely via the sterile alpha motifs of BICC1 [268], leading to ligand-independent dimerization [149] and activation of the ERK MAP kinase, but not STAT3 or AKT signaling [175,212,267]. FGFR inhibitors were partially successful in targeting the oncogene-driven growth of cell lines, xenografts and patients’ tumors [175,215,269,270]; acquired resistance through gatekeeper FGFR2-V564F mutation was also reported [270]. The FGFR2V546F-BICC1 cells showed oncogene addiction that was fully inhibited by a synergistic effect of the FGFR and ERK MAP kinase pathway inhibitors [267].
BICC1 is a conserved RNA-binding protein that represses translation of selected mRNAs to control development [271,272,273,274,275]; the domains responsible for RNA binding are, however, partly lost during the chromosomal rearrangement, suggesting that this function is lost with the FGFR2-BICC1 fusion. Deletion of BICC1 leads to classical ciliopathy features, including randomization of the left-right asymmetry, and cystic development in the kidney, liver and pancreas [276,277,278,279,280,281,282,283]. Loss of BICC1 disrupted alignment of motile cilia and establishment of the cilia-driven fluid flow in the mouse embryonic node and Xenopus gastrocoel [279], producing laterality defects. This may be due to disrupted protein synthesis machinery at the centrosome that appears important for the adjacent cilia [284,285]. In humans, mutations in BICC1 were identified in patients with kidney dysplasia, likely caused by ectopic Wingless-related integration site (WNT)/β-catenin signaling [286]. Decreased levels of BICC1, or loss of some of the three RNA-binding domains which are also relevant for the FGFR2-BICC1 fusion, also upregulated WNT/β-catenin signaling [275,279,287,288,289]. Taken together, the FGFR2-BICC1 fusion is likely to produce a BICC1 haploinsufficiency that leads to disrupted ciliogenesis and cilia-associated signaling, which may contribute to cancerogenesis.
4.4. FGFR2-NDC80
A cholangiocarcinoma patient was described with a fusion comprising FGFR2 and NDC80 (or HEC1, highly expressed in cancer 1) [216]. FGFR2-NDC80 was overexpressed in the tumor cells, and activated the ERK MAP kinase, PLCγ, and STAT3 signaling [216]. Considering the PLCγ binding site is lost with the fusion (Figure 1), it is possible that FGFR2-NDC80 activates this pathway through heterodimerization with the endogenous FGFR. The fusion protein retains the kinetochore microtubule binding region of NDC80 [290], suggesting possible mislocalization that was, however, not experimentally addressed; within the tumor samples, FGFR2-NDC80 localized predominantly to the cell membrane [216].
NDC80 localizes to the centrosomes and mitotic spindles where it is necessary for assembly and stabilization of the kinetochore microtubules (reviewed in [290]). High NDC80 levels were found in cancers [291,292,293,294], and overexpression of NDC80 in mice led to abnormal spindle formation, hyperactivation of the mitotic checkpoint and initiation of the tumorigenic events [295]. Depletion or inhibition of NDC80 induced mitotic arrest, and suppressed xenograft tumor growth [294,296,297,298]. Taken together, these data suggest a possible involvement of mitotic defects in the FGFR2-NDC80 cancerogenesis, through ectopic FGFR and NDC80 activity.
4.5. FGFR2-CIT
Fusions of FGFR2 with the citron Rho-interacting kinase (CIT) were identified in non-small cell lung cancer and cholangiocarcinoma [215,217,218] (Table 1). FGFR2-CIT dimerized in cells, likely using the coiled-coil domain of CIT [149] (Figure 1), and induced oncogene addiction in Ba/F3 cells that was efficiently targeted by FGFR kinase inhibitors [267,299].
CIT functions in spindle orientation and during late cytokinesis [300,301,302,303]. CIT overexpression has been associated with cancers of various origin [304,305,306,307,308,309,310], likely through its kinase function that is, however, lost during chromosomal rearrangement in the FGFR2-CIT fusion (Figure 1). Transgenic mice expressing CIT variant lacking the kinase domain show defects in neurogenesis and spermatogenesis [311,312], due to aberrant cytokinesis that is followed by massive apoptosis. CIT also associates with primary cilia [313], and CIT downregulation inhibited ciliogenesis [314] and altered cilia length [315]. Therefore, it is possible that the FGFR2-CIT fusion produces CIT haploinsufficiency that may trigger cancerogenesis through cilia loss and mitotic defects.
4.6. FGFR2-OFD1
Fusions involving FGFR2 and the oral-facial-digital type 1 (OFD1) gene were reported in thyroid and endometrial cancer [149,219] (Table 1). FGFR2-OFD1 induced transformation of RK3E cells, that was abolished by FGFR kinase inhibitors [316]. Dimerization of the fusion protein likely occurs through the coiled-coil domains of OFD1 [149], which are preserved in the fusion protein (Figure 1), leading to transactivation of the FGFR2 kinase domain and activated ERK MAP kinase signaling [316].
OFD1 localizes to centrosome [317] where it is required for centriole maturation and primary ciliogenesis [318,319]. This localization requires the N-terminal part of OFD1 [320] that is, however, lost in the FGFR2-OFD1 fusion. Heterozygous loss-of-function mutations in OFD1 produce the OFD1 syndrome, an X-linked dominant disorder lethal in males that is characterized by systemic ciliopathy features [306,321,322,323,324]. The Ofd1+/− female mice reproduced the main patient phenotypes [318,325], suggesting haploinsufficiency in the heterozygous animals. The cilia were severely disrupted or lost, producing defects in laterality and Hh-dependent tissue patterning [318,326]. The zebrafish ofd1 morphants also displayed laterality defects, due to cilia abnormalities in the Kupffer´s vesicle, as well as additional ciliopathy features [327]. These data suggest that the decreased levels of endogenous and centrosome-competent OFD1 in the FGFR2-OFD1 cancers may lead to deregulated ciliogenesis and cilia signaling, potentially contributing to neoplastic transformation.
4.7. FOP-FGFR1
The type I fusion involving FGFR1 and the FGFR1 oncogene partner (FOP) is associated with a stem cell myeloproliferative disorder, acute myeloid leukemia (AML) [220,221,222,223,224,225,226,227] (Table 1). FOP-FGFR1 induced oncogene addiction in Ba/F3 cells [328,329,330], and transplanted FOP-FGFR1+ hematopoietic stem cells developed a fatal myeloproliferative disorder in mice [331].
FOP-FGFR1 comprises the leucine rich N-terminal region of FOP that facilitates dimerization and transactivation of the catalytic domain of FGFR1, and produces a constitutively active fusion protein [220,330,332] (Figure 1). Correspondingly, ERK MAP kinase and STAT signaling is increased in FOP-FGFR1 expressing cells [329,330]. Phosphoinositide 3-kinase (PI3K)/AKT pathway is also employed to sequester FOP-FGFR1 to the centrosome [328,330]. The mislocalization of FOP-FGFR1 [328,333,334,335,336] is also mediated by interaction with the centrosomal protein CAP350, through FOP [328,337]. The ectopic centrosomal FOP-FGFR1 activity then drives abundant cell division that was abolished by FGFR, PI3K and ERK pathway inhibitors [328,329,330,338]. The centrosomal localization appears critical for PLCγ phosphorylation [328,330,339,340] that is necessary for activation of the anti-apoptotic signaling in FOP-FGFR1 expressing cells [139,328,330,341]. Disruption of the PLCγ binding site delayed onset and prolonged survival of the mice transplanted with FOP-FGFR1 hematopoietic stem cells [331].
The FOP haploinsufficiency may contribute to FOP-FGFR1 cancerogenesis, as reduced FOP levels were shown to disrupt the centrosome structure and inhibit ciliogenesis [341,342,343], and similar defects were observed in FOP-FGFR1 expressing cells [227,340]. Although the hematopoietic cells do not produce cilia [344,345], the centrosome defects have also been associated with other myeloproliferative neoplasms [340,346], suggesting a common pathogenesis.
4.8. CEP110-FGFR1
The fusion of FGFR1 with the centrosomal protein 110 (CEP110) drives expansion of the hematopoietic stem cell population, and causes malignancies that frequently turn into AML [221,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248] (Table 1). When expressed in cells, CEP110-FGFR1 likely dimerizes through the leucine zippers in CEP110 (Figure 1) which drives constitutive autophosphorylation of the FGFR1 kinase domains [247]. CEP110-FGFR1 induced oncogene addiction in Ba/F3 cells [241,347,348], that could be targeted by tyrosine kinase inhibitors [241,348]. Transplantation of murine bone marrow or human CD34+ cord blood cells transduced with CEP110-FGFR1 produced AML in the recipient mice [347], further supporting the role of CEP110-FGFR1 as an oncogenic driver.
Pluripotent stem cells derived from the AMLCEP110-FGFR1 patient showed aberrant hematopoietic differentiation, which was restored by tyrosine kinase inhibitors; a growth inhibition was also achieved with isolated primary AMLCEP110-FGFR1 cells [240]. This is in a sharp contrast with the clinical observation, as patients with CEP110-FGFR1 disease do not respond to tyrosine kinase inhibitors and have particularly poor prognosis; allogeneic hematopoietic stem cell transplantation appears the only viable option [238,349]. These data suggest that inhibition of the ectopic FGFR1 kinase activity in CEP110-FGFR1 cancers [241,350] does not bring clinical benefits, and that perhaps additional mechanisms contribute to the disease pathogenesis.
CEP110 is a structural protein of the centrosome [351,352], for which it requires a 170-aa region in the C-terminus that is retained in the CEP110-FGFR1 fusion (Figure 1) [247]. The centrosome localization of the fusion may, therefore, interfere with centrosome maturation, likely due to combination of the steric effects of the fusion and its ectopic kinase activity, which in turn produces centrosomal and spindle abnormalities and drives the oncogenesis [351,353,354].
5. Conclusions and Perspectives
The FGFR fusion proteins are oncogenic drivers; therefore, patients typically show a good initial response to the targeted therapy using FGFR tyrosine kinase inhibitors [171,186,215,219,269,270,355]. However, secondary gatekeeper mutations occur during therapy [270,356], and inhibition of effectors downstream from the FGFR oncogene has not delivered strong clinical benefit; therefore, alternate approaches are being developed. One such strategy takes advantage of the general overexpression of type II FGFR fusion proteins [268], which makes them a good target for cytotoxic conjugates specifically binding FGFR. For example, FGF2 conjugated with auristatin induced endocytosis of the FGFR1-FGF2/auristatin complexes, which released auristatin and produced a strong cytotoxic effect on cancer cells overexpressing FGFR1 [357]. Similarly, the FGFR-specific antibodies or antibody fragments conjugated to a cytotoxic molecule enter the cells via endocytosis to induce cell death [358,359]. Clinical trials evaluating cytotoxic conjugates in FGFR fusion-driven cancers are yet to emerge.
Another possibility is to specifically target the fusion protein. For example, no therapy protocol is available for FOP-FGFR1-driven cancers, which are very aggressive [221,222,328,331]. FOP-FGFR1 saturates at the centrosome, which appears critical for oncogenic transformation [329,331]. An adeno-associated virus-mediated delivery of interfering RNA, peptide or a coding sequence, specifically targeting the FOP-FGFR1 fusion or its interaction interface with the centrosome, therefore represents an attractive therapeutic possibility [360,361,362].
Finally, the ectopic activity of the FGFR fusion protein, together with decreased levels of the endogenous fusion partner, may contribute to neoplastic transformation through loss of primary cilia and deregulated cell division. Restoration of ciliogenesis and/or cilia function is, therefore, an attractive and so far unappreciated strategy to attenuate tumor growth. NSC12, an orally available analog of the naturally occurring FGF ligand trap pentraxin 3 (PTX3), was developed to target the FGF-driven pathologies [363]. NSC12 rescued ciliogenesis defects in three FGFR-driven cancer cell lines and a xenograft, and inhibited tumor growth [363]. The clinical studies evaluating cilia targeting as a cancer therapy are however yet to emerge.
Author Contributions
A.N., S.P.A. and M.B.; writing—original draft preparation, P.K. and M.B.; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Education, Youth and Sports of the Czech Republic, grant number LTAUSA19030. S.P.A. and A.N. were supported by IGA MU project No. CZ.02.2.69/0.0/0.0/19_073/0016943. A.N. is a Brno Ph.D. Talent Scholarship Holder—Funded by the Brno City Municipality.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Kopinke, D.; Norris, A.M.; Mukhopadhyay, S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin. Cell Dev. Biol. 2021, 110, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.J.; Yeyati, P.L.; Mali, G.R.; Keighren, M.A.; Waddell, S.H.; Mjoseng, H.K.; Douglas, A.T.; Hall, E.A.; Sakaue-Sawano, A.; Miyawaki, A.; et al. A Cell/Cilia Cycle Biosensor for Single-Cell Kinetics Reveals Persistence of Cilia after G1/S Transition Is a General Property in Cells and Mice. Dev. Cell 2018, 47, 509–523.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paridaen, J.T.M.L.; Wilsch-Bräuninger, M.; Huttner, W.B. Asymmetric Inheritance of Centrosome-Associated Primary Cilium Membrane Directs Ciliogenesis after Cell Division. Cell 2013, 155, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Primary Cilia and the Cell Cycle, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 94, ISBN 9780123750242. [Google Scholar]
- Ke, Y.-N.; Yang, W.-X. Primary cilium: An elaborate structure that blocks cell division? Gene 2014, 547, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-Dependent Aurora A Activation Induces Disassembly of the Primary Cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J. Cell Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Chen, Q.; Zhang, X.; Zhang, B.; Zhuo, X.; Liu, J.; Jiang, Q.; Zhang, C. PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. J. Cell Sci. 2013, 126, 1355–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappello, P.; Blaser, H.; Gorrini, C.; Lin, D.C.C.; Elia, A.J.; Wakeham, A.; Haider, S.; Boutros, P.C.; Mason, J.M.; Miller, N.A.; et al. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 2014, 33, 2375–2384. [Google Scholar] [CrossRef] [Green Version]
- Dere, R.; Perkins, A.L.; Bawa-Khalfe, T.; Jonasch, D.; Walker, C.L. β-Catenin links von Hippel-Lindau to Aurora kinase A and loss of primary cilia in renal cell carcinoma. J. Am. Soc. Nephrol. 2015, 26, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Egeberg, D.L.; Lethan, M.; Manguso, R.; Schneider, L.; Awan, A.; Jørgensen, T.S.; Byskov, A.G.; Pedersen, L.B.; Christensen, S.T. Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia 2012, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Sarkisian, M.R.; Li, W.; Di Cunto, F.; D’Mello, S.R.; LoTurco, J.J. Citron-kinase, a protein essential to cytokinesis in neuronal progenitors, is deleted in the flathead mutant rat. J. Neurosci. 2002, 22, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Hosoba, K.; Ochiai, H.; Royba, E.; Izumi, H.; Sakuma, T.; Yamamoto, T.; Dynlacht, B.D.; Matsuura, S. The Microtubule-Depolymerizing activity of a mitotic kinesin protein KIF2A drives primary cilia disassembly coupled with cell proliferation. Cell Rep. 2015, 10, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Michaud, E.J.; Yoder, B.K. The primary cilium in cell signaling and cancer. Cancer Res. 2006, 66, 6463–6467. [Google Scholar] [CrossRef] [Green Version]
- Frett, B.; Brown, R.V.; Ma, M.; Hu, W.; Han, H.; Li, H.Y. Therapeutic melting pot of never in mitosis gene a related kinase 2 (Nek2): A perspective on Nek2 as an oncology target and recent advancements in Nek2 small molecule inhibition. J. Med. Chem. 2014, 57, 5835–5844. [Google Scholar] [CrossRef]
- Gradilone, S.A.; Habringer, S.; Masyuk, T.V.; Howard, B.N.; Masyuk, A.I.; Larusso, N.F. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am. J. Pathol. 2014, 184, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo Pisarello, M.; Masyuk, T.V.; Gradilone, S.A.; Masyuk, A.I.; Ding, J.F.; Lee, P.Y.; LaRusso, N.F. Combination of a Histone Deacetylase 6 Inhibitor and a Somatostatin Receptor Agonist Synergistically Reduces Hepatorenal Cystogenesis in an Animal Model of Polycystic Liver Disease. Am. J. Pathol. 2018, 188, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Sarkisian, M.R.; Siebzehnrubl, D.; Hoang-Minh, L.; Deleyrolle, L.; Silver, D.J.; Siebzehnrubl, F.A.; Guadiana, S.M.; Srivinasan, G.; Semple-Rowland, S.; Harrison, J.K.; et al. Detection of primary cilia in human glioblastoma. J. Neurooncol. 2014, 117, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; LaRusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Guo, F.; Cheng, W.; Zhang, J.; Huang, J.; Wang, R.; Ma, Z.; Xu, K. HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia. Oncol. Rep. 2017, 38, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer (Review). Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef]
- Peixoto, E.; Richard, S.; Pant, K.; Biswas, A.; Gradilone, S.A. The primary cilium: Its role as a tumor suppressor organelle. Biochem. Pharmacol. 2020, 175, 113906. [Google Scholar] [CrossRef]
- Kiseleva, A.A.; Nikonova, A.S.; Golemis, E.A. Patterns of Ciliation and Ciliary Signaling in Cancer. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Sabanovic, B.; Giulietti, M.; Piva, F. Role of primary cilium in pancreatic ductal adenocarcinoma (Review). Int. J. Oncol. 2020, 57, 1095–1102. [Google Scholar] [CrossRef]
- Moser, J.J.; Fritzler, M.J.; Rattner, J.B. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 2009, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Seeley, E.S.; Carrière, C.; Goetze, T.; Longnecker, D.S.; Korc, M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009, 69, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Callahan, C.A.; Dupree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; De Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [Green Version]
- Yuan, K.; Frolova, N.; Xie, Y.; Wang, D.; Cook, L.; Kwon, Y.J.; Steg, A.D.; Serra, R.; Frost, A.R. Primary cilia are decreased in breast cancer: Analysis of a collection of human breast cancer cell lines and tissues. J. Histochem. Cytochem. 2010, 58, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Nobutani, K.; Shimono, Y.; Yoshida, M.; Mizutani, K.; Minami, A.; Kono, S.; Mukohara, T.; Yamasaki, T.; Itoh, T.; Takao, S.; et al. Absence of primary cilia in cell cycle-arrested human breast cancer cells. Genes Cells 2014, 19, 141–152. [Google Scholar] [CrossRef]
- Menzl, I.; Lebeau, L.; Pandey, R.; Hassounah, N.B.; Li, F.W.; Nagle, R.; Weihs, K.; McDermott, K.M. Loss of primary cilia occurs early in breast cancer development. Cilia 2014, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Hassounah, N.B.; Nunez, M.; Fordyce, C.; Roe, D.; Nagle, R.; Bunch, T.; McDermott, K.M. Inhibition of ciliogenesis promotes Hedgehog signaling, tumorigenesis, and metastasis in breast cancer. Mol. Cancer Res. 2017, 15, 1421–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassounah, N.B.; Nagle, R.; Saboda, K.; Roe, D.J.; Dalkin, B.L.; McDermott, K.M. Primary Cilia Are Lost in Preinvasive and Invasive Prostate Cancer. PLoS ONE 2013, 8, e68521. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Asp, P.; Canter, B.; Dynlacht, B.D. Primary cilia control hedgehog signaling during muscle differentiation and are deregulated in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 9151–9156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, L.; Ali, S.A.; Al-Jazrawe, M.; Kandel, R.; Wunder, J.S.; Alman, B.A. Primary cilia attenuate hedgehog signalling in neoplastic chondrocytes. Oncogene 2013, 32, 5388–5396. [Google Scholar] [CrossRef] [Green Version]
- Jackman, W.R.; Yoo, J.J.; Stock, D.W. Hedgehog signaling is required at multiple stages of zebrafish tooth development. BMC Dev. Biol. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Cobourne, M.T.; Sharpe, P.T. Sonic Hedgehog Signaling and the Developing Tooth. Curr. Top. Dev. Biol. 2004, 65, 255–287. [Google Scholar] [CrossRef]
- Rallis, A.; Navarro, J.A.; Rass, M.; Hu, A.; Birman, S.; Schneuwly, S.; Thérond, P.P. Hedgehog Signaling Modulates Glial Proteostasis and Lifespan. Cell Rep. 2020, 30, 2627–2643.e5. [Google Scholar] [CrossRef] [Green Version]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Büller, N.V.J.A.; Rosekrans, S.L.; Westerlund, J.; van den Brink, G.R. Hedgehog signaling and maintenance of homeostasis in the intestinal epithelium. Physiology 2012, 27, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.; DiNardo, S. Drosophila hedgehog acts as a morphogen in cellular patterning. Cell 1994, 76, 449–460. [Google Scholar] [CrossRef]
- Chapouly, C.; Guimbal, S.; Hollier, P.L.; Renault, M.A. Role of hedgehog signaling in vasculature development, differentiation, and maintenance. Int. J. Mol. Sci. 2019, 20, 3076. [Google Scholar] [CrossRef] [Green Version]
- Ehlen, H.W.A.; Buelens, L.A.; Vortkamp, A. Hedgehog signaling in skeletal develoment. Birth Defects Res. Part C Embryo Today Rev. 2006, 78, 267–279. [Google Scholar] [CrossRef]
- Hebrok, M. Hedgehog signaling in pancreas development. Mech. Dev. 2003, 120, 45–57. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic hedgehog signaling in organogenesis, tumors, and tumor microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [Green Version]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e10053. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Ospina, J.K.; Zhang, J.; Michelson, A.P.; Schoen, A.M.; Zhu, A.J. Sequential phosphorylation of smoothened transduces graded hedgehog signaling. Sci. Signal. 2011, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, M.T.; Humke, E.W.; Scott, M.P. Kif3a is necessary for initiation and maintenance of medulloblastoma. Carcinogenesis 2013, 34, 1382–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Y.; Seol, A.D.; So, P.-L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway–dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Grausam, K.B.; Wang, J.; Lun, M.P.; Ohli, J.; Lidov, H.G.W.; Calicchio, M.L.; Zeng, E.; Salisbury, J.L.; Wechsler-Reya, R.J.; et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nat. Cell Biol. 2016, 18, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Guen, V.J.; Chavarria, T.E.; Kröger, C.; Ye, X.; Weinberg, R.A.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.L.; Johnson, D.E.; Cousens, L.S.; Fried, V.A.; Williams, L.T. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science 1989, 245, 57–60. [Google Scholar] [CrossRef]
- Kornbluth, S.; Paulson, K.E.; Hanafusa, H. Novel tyrosine kinase identified by phosphotyrosine antibody screening of cDNA libraries. Mol. Cell. Biol. 1988, 8, 5541–5544. [Google Scholar] [CrossRef] [Green Version]
- Keegan, K.; Johnson, D.E.; Williams, L.T.; Hayman, M.J. Isolation of an additional member of the fibroblast growth factor receptor family, FGFR-3. Proc. Natl. Acad. Sci. USA 1991, 88, 1095–1099. [Google Scholar] [CrossRef] [Green Version]
- Partanen, J.; Makela, T.P.; Eerola, E.; Korhonen, J.; Hirvonen, H.; Claesson-Welsh, L.; Alitalo, K. FGFR-4, a novel acidic fibroblast growth factor receptor with a distinct expression pattern. EMBO J. 1991, 10, 1347–1354. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Xu, J.; Colvin, J.S.; McEwen, D.G.; MacArthur, C.A.; Coulier, F.; Gao, G.; Goldfarb, M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996, 271, 15292–15297. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, reviews3005.1. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Schlessinger, J.; Hubbard, S.R.; Mohammadi, M. Structural Basis for FGF Receptor Dimerization and Activation et al Dimerization of the extracellular domains leads to juxtaposition of the cytoplasmic domains and. Cell 1999, 98, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal Structure of a Ternary FGF-FGFR-Heparin Complex Reveals a Dual Role for Heparin in FGFR Binding and Dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Luo, Y.; Ye, S.; Kan, M.; McKeehan, W.L. Control of Fibroblast Growth Factor (FGF) 7- and FGF1-induced mitogenesis and downstream signaling by distinct heparin octasaccharide motifs. J. Biol. Chem. 2006, 281, 21052–21061. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Ohnishi, M.; Ding, X.; Kurosu, H.; Wang, L.; Akiyoshi, J.; Ma, J.; Gai, W.; Sidis, Y.; Pitteloud, N.; et al. Klotho Coreceptors Inhibit Signaling by Paracrine Fibroblast Growth Factor 8 Subfamily Ligands. Mol. Cell. Biol. 2012, 32, 1944–1954. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [Green Version]
- Quarto, N.; Amalric, F. Heparan sulfate proteoglycans as transducers of FGF-2 signalling. J. Cell Sci. 1994, 107, 3201–3212. [Google Scholar] [CrossRef]
- Zhang, Z.; Coomans, C.; David, G. Membrane heparan sulfate proteoglycan-supported FGF2-FGFR1 signaling: Evidence in support of the “cooperative end structures” model. J. Biol. Chem. 2001, 276, 41921–41929. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Yayon, A.; Flanagan, J.G.; Svahn, C.M.; Levi, E.; Leder, P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 1992, 12, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Spivak-Kroizman, T.; Lemmon, M.A.; Dikic, I.; Ladbury, J.E.; Pinchasi, D.; Huang, J.; Jaye, M.; Crumley, G.; Schlessinger, J.; Lax, I. Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell 1994, 79, 1015–1024. [Google Scholar] [CrossRef]
- Rapraeger, A.C.; Krufka, A.; Olwin, B.B. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science 1991, 252, 1705–1708. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Hu, M.C.; Shiizaki, K.; Kuro-O, M.; Moe, O.W. Fibroblast growth factor 23 and klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [Green Version]
- Tacer, K.F.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [Green Version]
- Arman, E.; Haffner-Krausz, R.; Gorivodsky, M.; Lonai, P. Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 11895–11899. [Google Scholar] [CrossRef] [Green Version]
- Danopoulos, S.; Thornton, M.E.; Grubbs, B.H.; Frey, M.R.; Warburton, D.; Bellusci, S.; Al Alam, D. Discordant roles for FGF ligands in lung branching morphogenesis between human and mouse. J. Pathol. 2019, 247, 254–265. [Google Scholar] [CrossRef] [PubMed]
- McDougall, K.; Kubu, C.; Verdi, J.M.; Meakin, S.O. Developmental expression patterns of the signaling adapters FRS-2 and FRS-3 during early embryogenesis. Mech. Dev. 2001, 103, 145–148. [Google Scholar] [CrossRef]
- Weinstein, M.; Xu, X.; Ohyama, K.; Deng, C.X. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 1998, 125, 3615–3623. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.T.; Godin, R.E.; Robertson, E.J. Interaction between FGF and BMP signaling pathways regulates development of metanephric mesenchyme. Genes Dev. 1999, 13, 1601–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastner, S.; Elias, M.C.; Rivera, A.J.; Yablonka-Reuveni, Z. Gene expression patterns of the fibroblast growth factors and their receptors during myogenesis of rat satellite cells. J. Histochem. Cytochem. 2000, 48, 1079–1096. [Google Scholar] [CrossRef] [Green Version]
- Walker, K.A.; Sims-Lucas, S.; Bates, C.M. Fibroblast growth factor receptor signaling in kidney and lower urinary tract development. Pediatr. Nephrol. 2016, 31, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Colvin, J.S.; Bohne, B.A.; Harding, G.W.; McEwen, D.G.; Ornitz, D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 1996, 12, 390–397. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, J.; Chen, M.; Yang, H.; Learned, R.M.; DePaoli, A.M.; Tian, H.; Ling, L. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J. Hepatol. 2017, 66, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Floss, T.; Arnold, H.H.; Braun, T. A role for FGF-6 in skeletal muscle regeneration. Genes Dev. 1997, 11, 2040–2051. [Google Scholar] [CrossRef] [Green Version]
- Schmid, G.J.; Kobayashi, C.; Sandell, L.J.; Ornitz, D.M. Fibroblast growth factor expression during skeletal fracture healing in mice. Dev. Dyn. 2009, 238, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, A.; Nakajima, F.; Shimizu, S.; Ogasawara, A.; Wanaka, A.; Moriya, H.; Einhorn, T.A.; Yamazaki, M. Spatial and temporal gene expression for fibroblast growth factor type I receptor (FGFR1) during fracture healing in the rat. Bone 2001, 29, 458–466. [Google Scholar] [CrossRef]
- Goebel, S.; Lienau, J.; Rammoser, U.; Seefried, L.; Wintgens, K.F.; Seufert, J.; Duda, G.; Jakob, F.; Ebert, R. FGF23 is a putative marker for bone healing and regeneration. J. Orthop. Res. 2009, 27, 1141–1146. [Google Scholar] [CrossRef]
- Hurley, M.M.; Adams, D.J.; Wang, L.; Jiang, X.; Burt, P.M.; Du, E.; Xiao, L. Accelerated fracture healing in transgenic mice overexpressing an anabolic isoform of fibroblast growth factor 2. J. Cell. Biochem. 2016, 117, 599–611. [Google Scholar] [CrossRef]
- Kunova Bosakova, M.; Varecha, M.; Hampl, M.; Duran, I.; Nita, A.; Buchtova, M.; Dosedelova, H.; Machat, R.; Xie, Y.; Ni, Z.; et al. Regulation of ciliary function by fibroblast growth factor signaling identifies FGFR3-related disorders achondroplasia and thanatophoric dysplasia as ciliopathies. Hum. Mol. Genet. 2018, 27, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Kaci, N.; Estibals, V.; Goudin, N.; Garfa-Traore, M.; Benoist-Lasselin, C.; Dambroise, E.; Legeai-Mallet, L. Constitutively-active FGFR3 disrupts primary cilium length and IFT20 trafficking in various chondrocyte models of achondroplasia. Hum. Mol. Genet. 2018, 27, 1–13. [Google Scholar] [CrossRef]
- Kunova Bosakova, M.; Nita, A.; Gregor, T.; Varecha, M.; Gudernova, I.; Fafilek, B.; Barta, T.; Basheer, N.; Abraham, S.P.; Balek, L.; et al. Fibroblast growth factor receptor influences primary cilium length through an interaction with intestinal cell kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 4316–4325. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Nakagama, H. FGF Receptors: Cancer Biology and Therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Salvesen, H.B.; Chen, T.H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Lee, J.H.; Abdullaev, Z.; Park, K.S.; Pineda, M.; Saidkhodjaeva, L.; Miettinen, M.; Wang, Y.; Pack, S.D.; Giaccone, G. Characterization of fibroblast growth factor receptor 1 in small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Rosty, C.; Aubriot, M.H.; Cappellen, D.; Bourdin, J.; Cartier, I.; Thiery, J.P.; Sastre-Garau, X.; Radvanyi, F. Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Mol. Cancer 2005, 4, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Neugebauer, J.M.; Amack, J.D.; Peterson, A.G.; Bisgrove, B.W.; Yost, H.J. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature 2009, 458, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Essner, J.J.; Amack, J.D.; Nyholm, M.K.; Harris, E.B.; Yost, H.J. Kupffer’s vesicle is a ciliated organ of asymmetry in the zebrafish embryo that initiates left-right development of the brain, heart and gut. Development 2005, 132, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, H.; Miyakawa, N.; Miyake, A.; Itoh, N. Fgf4 is required for left-right patterning of visceral organs in zebrafish. Dev. Biol. 2009, 332, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-W.W.; Hsu, C.-H.H.; Tsai, S.-M.M.; Hsiao, C.-D.; Wang, W.-P.P. A Variant of Fibroblast Growth Factor Receptor 2 (Fgfr2) Regulates Left-Right Asymmetry in Zebrafish. PLoS ONE 2011, 6, e21793. [Google Scholar] [CrossRef] [Green Version]
- Sempou, E.; Lakhani, O.A.; Amalraj, S.; Khokha, M.K. Candidate Heterotaxy Gene FGFR4 Is Essential for Patterning of the Left-Right Organizer in Xenopus. Front. Physiol. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Hong, S.K.; Dawid, I.B. FGF-dependent left-right asymmetry patterning in zebrafish is mediated by Ier2 and Fibp1. Proc. Natl. Acad. Sci. USA 2009, 106, 2230–2235. [Google Scholar] [CrossRef] [Green Version]
- Neugebauer, J.M.; Cadwallader, A.B.; Amack, J.D.; Bisgrove, B.W.; Joseph Yost, H. Differential roles for 3-OSTs in the regulation of cilia length and motility. Development 2013, 140, 3892–3902. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.; Xu, X.; Lin, X. Wnt/β-catenin signaling directly regulates Foxj1 expression and ciliogenesis in zebrafish Kupffer’s vesicle. Development 2012, 139, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Bonnafe, E.; Touka, M.; AitLounis, A.; Baas, D.; Barras, E.; Ucla, C.; Moreau, A.; Flamant, F.; Dubruille, R.; Couble, P.; et al. The Transcription Factor RFX3 Directs Nodal Cilium Development and Left-Right Asymmetry Specification. Mol. Cell. Biol. 2004, 24, 4417–4427. [Google Scholar] [CrossRef] [Green Version]
- Bisgrove, B.W.; Snarr, B.S.; Emrazian, A.; Yost, H.J. Polaris and Polycystin-2 in dorsal forerunner cells and Kupffer’s vesicle are required for specification of the zebrafish left-right axis. Dev. Biol. 2005, 287, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Honda, A.; Kita, T.; Seshadri, S.V.; Misaki, K.; Ahmed, Z.; Ladbury, J.E.; Richardson, G.P.; Yonemura, S.; Ladher, R.K. FGFR1-mediated protocadherin-15 loading mediates cargo specificity during intraflagellar transport in inner ear hair-cell kinocilia. Proc. Natl. Acad. Sci. USA 2018, 115, 8388–8393. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Liu, M.; Cao, X.; Yang, S. Ciliary IFT80 regulates dental pulp stem cells differentiation by FGF/FGFR1 and Hh/BMP2 signaling. Int. J. Biol. Sci. 2019, 15, 2087–2099. [Google Scholar] [CrossRef]
- Taylor, S.P.; Bosakova, M.K.; Varecha, M.; Balek, L.; Barta, T.; Trantirek, L.; Jelinkova, I.; Duran, I.; Vesela, I.; Forlenza, K.N.; et al. An inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome. Hum. Mol. Genet. 2016, 25, 3998–4011. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Song, J.; Shin, J.O.; Lee, H.; Kim, H.K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, aprotein associated with endocrine-cerebro-osteodysplasia syndrome is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546. [Google Scholar] [CrossRef] [Green Version]
- Chaya, T.; Omori, Y.; Kuwahara, R.; Furukawa, T. ICK is essential for cell type-specific ciliogenesis and the regulation of ciliary transport. EMBO J. 2014, 33, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Park, S.H.; Wu, D.; Xu, W.; Guillot, S.J.; Jin, L.; Li, X.; Wang, Y.; Lin, C.-S.; Fu, Z. An essential role of intestinal cell kinase in lung development is linked to the perinatal lethality of human ECO syndrome. FEBS Lett. 2017, 591, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Jin, L.; Xie, L.; Park, S.H.; Tong, Y.; Wu, D.; Chhabra, A.B.; Fu, Z.; Li, X. A Murine Model for Human ECO Syndrome Reveals a Critical Role of Intestinal Cell Kinase in Skeletal Development. Calcif. Tissue Int. 2018, 102, 348–357. [Google Scholar] [CrossRef]
- Okamoto, S.; Chaya, T.; Omori, Y.; Kuwahara, R.; Kubo, S.; Sakaguchi, H.; Furukawa, T. Ick ciliary kinase is essential for planar cell polarity formation in inner ear hair cells and hearing function. J. Neurosci. 2017, 37, 2073–2085. [Google Scholar] [CrossRef] [Green Version]
- Berman, S.A.; Wilson, N.F.; Haas, N.A.; Lefebvre, P.A. A Novel MAP Kinase Regulates Flagellar Length in Chlamydomonas. Curr. Biol. 2003, 13, 1145–1149. [Google Scholar] [CrossRef] [Green Version]
- Burghoorn, J.; Dekkers, M.P.J.; Rademakers, S.; De Jong, T.; Willemsen, R.; Jansen, G. Mutation of the MAP kinase DYF-5 affects docking and undocking of kinesin-2 motors and reduces their speed in the cilia of Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2007, 104, 7157–7162. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, F.; El Ghouzzi, V.; Delezoide, A.L.; Legeai-Mallet, L.; Le Merrer, M.; Munnich, A.; Bonaventure, J. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1). Hum. Mol. Genet. 1996, 5, 509–512. [Google Scholar] [CrossRef] [Green Version]
- Tavormina, P.L.; Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Wilkin, D.J.; Lachman, R.S.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Wasmuth, J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995, 9, 321–328. [Google Scholar] [CrossRef]
- Tavormina, P.L.; Rimoin, D.L.; Cohn, D.H.; Zhu, Y.Z.; Shiang, R.; Wasmuth, J.J. Another mutation that results in the substitution of an unpaired cysteine residue in the extracellular domain of FGFR3 in thanatophoric dysplasia type I. Hum. Mol. Genet. 1995, 4, 2175–2177. [Google Scholar] [CrossRef]
- Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Church, D.M.; Fielder, T.J.; Bocian, M.; Winokur, S.T.; Wasmuth, J.J. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 1994, 78, 335–342. [Google Scholar] [CrossRef]
- Bellus, G.A.; Hefferon, T.W.; De Luna, R.I.O.; Hecht, J.T.; Horton, W.A.; Machado, M.; Kaitila, I.; McIntosh, I.; Francomano, C.A. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am. J. Hum. Genet. 1995, 56, 368–373. [Google Scholar]
- Van Rhijn, B.W.G.; van Tilborg, A.A.G.; Lurkin, I.; Bonaventure, J.; de Vries, A.; Thiery, J.P.; van der Kwast, T.H.; Zwarthoff, E.C. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur. J. Hum. Genet. 2002, 10, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard-Pierrot, I.; Brams, A.; Dunois-Lardé, C.; Caillault, A.; Diez de Medina, S.G.; Cappellen, D.; Graff, G.; Thiery, J.P.; Chopin, D.; Ricol, D.; et al. Oncogenic properties of the mutated forms of fibroblast growth factor receptor 3b. Carcinogenesis 2006, 27, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greulich, H.; Pollock, P.M. Targeting mutant fibroblast growth factor receptors in cancer. Trends Mol. Med. 2011, 17, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Kato, H.; Matsuura, S.; Inoue, Y.; Igarashi, H.; Nagura, K.; Nakamura, S.; Maruyama, K.; Tajima, M.; Funai, K.; et al. A novel somatic FGFR3 mutation in primary lung cancer. Oncol. Rep. 2014, 31, 1219–1224. [Google Scholar] [CrossRef]
- Naski, M.C.; Colvin, J.S.; Coffin, J.D.; Ornitz, D.M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 1998, 125, 4977–4988. [Google Scholar] [CrossRef]
- Chen, J.; Chien, K.R. Complexity in simplicity: Monogenic disorders and c complex cardiomyopathies. J. Clin. Invest. 1999, 103, 1483–1485. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, C.; Qiao, W.; Xu, X.; Deng, C. A Ser365→Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates lhh/PTHrP signals and causes severe achondroplasia. Hum. Mol. Genet. 2001, 10, 457–465. [Google Scholar] [CrossRef]
- Wren, K.N.; Craft, J.M.; Tritschler, D.; Schauer, A.; Patel, D.K.; Smith, E.F.; Porter, M.E.; Kner, P.; Lechtreck, K.F. A Differential Cargo-Loading Model of Ciliary Length Regulation by IFT. Curr. Biol. 2013, 23, 2463–2471. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Xie, Y.; Tang, J.; Huang, J.; Huang, Q.; Xu, W.; Wang, Z.; Luo, F.; Wang, Q.; Chen, H.; et al. FGFR3 Deficiency Causes Multiple Chondroma-like Lesions by Upregulating Hedgehog Signaling. PLoS Genet. 2015, 11, e1005214. [Google Scholar] [CrossRef]
- Du, E.; Lu, C.; Sheng, F.; Li, C.; Li, H.; Ding, N.; Chen, Y.; Zhang, T.; Yang, K.; Xu, Y. Analysis of potential genes associated with primary cilia in bladder cancer. Cancer Manag. Res. 2018, 10, 3047–3056. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.S.; Johmura, Y.; Yu, L.R.; Park, J.E.; Gao, Y.; Bang, J.K.; Zhou, M.; Veenstra, T.D.; Yeon Kim, B.; Lee, K.H.S. Identification of a novel Wnt5a-CK1ε-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J. 2012, 31, 3104–3117. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Kao, C.Y.; Chung, Y.F.; Lee, D.C.; Liu, J.W.; Chiu, I.M. Activation of Aurora A kinase through the FGF1/FGFR signaling axis sustains the stem cell characteristics of glioblastoma cells. Exp. Cell Res. 2016, 344, 153–166. [Google Scholar] [CrossRef]
- Li, X.; Martinez-Ledesma, E.; Zhang, C.; Gao, F.; Zheng, S.; Ding, J.; Wu, S.; Nguyen, N.; Clifford, S.C.; Wen, P.Y.; et al. TIE2–FGFR1 interaction induces adaptive PI3K inhibitor resistance by upregulating Aurora A/PlK1/CDK1 signaling in glioblastoma. Cancer Res. 2019, 79, 5088–5101. [Google Scholar] [CrossRef] [Green Version]
- Montaudon, E.; Nikitorowicz-Buniak, J.; Sourd, L.; Morisset, L.; El Botty, R.; Huguet, L.; Dahmani, A.; Painsec, P.; Nemati, F.; Vacher, S.; et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Sánchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Pimenta-Marques, A.; Bettencourt-Dias, M. Maintaining centrosomes and cilia. J. Cell Sci. 2017, 130, 3789–3800. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [Green Version]
- L’Hôte, C.G.M.; Knowles, M.A. Cell responses to FGFR3 signalling: Growth, differentiation and apoptosis. Exp. Cell Res. 2005, 304, 417–431. [Google Scholar] [CrossRef]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem. Biophys. Res. Commun. 1992, 189, 824–831. [Google Scholar] [CrossRef]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR landscape in cancer: Analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortes, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563. [Google Scholar] [CrossRef]
- Kim, S.; Dubrovska, A.; Salamone, R.J.; Walker, J.R.; Grandinetti, K.B.; Bonamy, G.M.C.; Orth, A.P.; Elliott, J.; Porta, D.G.; Garcia-Echeverria, C.; et al. FGFR2 Promotes Breast Tumorigenicity through Maintenance of Breast Tumor-Initiating Cells. PLoS ONE 2013, 8, e51671. [Google Scholar] [CrossRef] [Green Version]
- Tsimafeyeu, I.; Demidov, L.; Stepanova, E.; Wynn, N.; Ta, H. Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma. Scand. J. Urol. Nephrol. 2011, 45, 190–195. [Google Scholar] [CrossRef]
- Giri, D.; Ropiquet, F.; Ittmann, M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin. Cancer Res. 1999, 5, 1063–1071. [Google Scholar]
- Di Martino, E.; Tomlinson, D.C.; Knowles, M.A. A decade of FGF receptor research in bladder cancer: Past, present, and future challenges. Adv. Urol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Byron, S.A.; Gartside, M.; Powell, M.A.; Wellens, C.L.; Gao, F.; Mutch, D.G.; Goodfellow, P.J.; Pollock, P.M. Fgfr2 point mutations in 466 endometrioid endometrial tumors: Relationship with msi, kras, pik3ca, ctnnb1 mutations and clinicopathological features. PLoS ONE 2012, 7, e30801. [Google Scholar] [CrossRef]
- Cappellen, D.; De Oliveira, C.; Ricol, D.; de Medina, S.; Bourdin, J.; Sastre-Garau, X.; Chopin, D.; Thiery, J.P.; Radvanyi, F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 1999, 23, 18–20. [Google Scholar] [CrossRef]
- Ahmed, Z.; Schüller, A.C.; Suhling, K.; Tregidgo, C.; Ladbury, J.E. Extracellular point mutations in FGFR2 elicit unexpected changes in intracellular signalling. Biochem. J. 2008, 413, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Neilson, K.M.; Friesel, R. Ligand-independent activation of fibroblast growth factor receptors by point mutations in the extracellular, transmembrane, and kinase domains. J. Biol. Chem. 1996, 271, 25049–25057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, O.A.; Yeh, B.K.; Eliseenkova, A.V.; Zhang, F.; Olsen, S.K.; Igarashi, M.; Aaronson, S.A.; Linhardt, R.J.; Mohammadi, M. Analysis of Mutations in Fibroblast Growth Factor (FGF) and a Pathogenic Mutation in FGF Receptor (FGFR) Provides Direct Evidence for the Symmetric Two-End Model for FGFR Dimerization. Mol. Cell. Biol. 2005, 25, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, M.K.; D’Avis, P.Y.; Robertson, S.C.; Donoghue, D.J. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol. Cell. Biol. 1996, 16, 4081–4087. [Google Scholar] [CrossRef] [Green Version]
- Naski, M.C.; Wang, Q.; Xu, J.; Ornitz, D.M. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 1996, 13, 233–237. [Google Scholar] [CrossRef]
- Bellus, G.A.; Bamshad, M.J.; Przylepa, K.A.; Dorst, J.; Lee, R.R.; Hurko, O.; Jabs, E.W.; Curry, C.J.R.; Wilcox, W.R.; Lachman, R.S.; et al. Severe achondroplasia with developmental delay and Acanthosis nigricans (SADDAN): Phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. Am. J. Med. Genet. 1999, 85, 53–65. [Google Scholar] [CrossRef]
- Foth, M.; Ahmad, I.; Van Rhijn, B.W.G.; Van Der Kwast, T.; Bergman, A.M.; King, L.; Ridgway, R.; Leung, H.Y.; Fraser, S.; Sansom, O.J.; et al. Fibroblast growth factor receptor 3 activation plays a causative role in urothelial cancer pathogenesis in cooperation with Pten loss in mice. J. Pathol. 2014, 233, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Singh, L.B.; Foth, M.; Morris, C.A.; Taketo, M.M.; Wu, X.R.; Leung, H.Y.; Sansom, O.J.; Iwata, T. K-Ras and β-catenin mutations cooperate with Fgfr3 mutations in mice to promote tumorigenesis in the skin and lung, but not in the bladder. DMM Dis. Model. Mech. 2011, 4, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- De Luca, A.; Abate, R.E.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR fusions in cancer: From diagnostic approaches to therapeutic intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, X.; Wang, S.; Moser, C.D.; Shaleh, H.M.; Mohamed, E.A.; Chaiteerakij, R.; Allotey, L.K.; Chen, G.; Miyabe, K.; et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016, 380, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Ochiai, M.; Yoshihara, Y.; Maru, Y.; Tetsuya, M.; Izumiya, M.; Imai, T.; Hippo, Y. Kras-driven heterotopic tumor development from hepatobiliary organoids. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Meyer, A.N.; Peiris, M.N.; Nelson, K.N.; Donoghue, D.J. Oncogenic fusion protein FGFR2-PPHLN1: Requirements for biological activation, and efficacy of inhibitors. Transl. Oncol. 2020, 13, 100853. [Google Scholar] [CrossRef]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Akiyama, N.; Tsukaguchi, T.; Fujii, T.; Satoh, Y.; Ishii, N.; Aoki, M. Mechanism of Oncogenic Signal Activation by the Novel Fusion Kinase FGFR3-BAIAP2L1. Mol. Cancer Ther. 2015, 14, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Cowell, J.K. Constitutive Notch pathway activation in murine ZMYM2-FGFR1-induced T-cell lymphomas associated with atypical myeloproliferative disease. Blood 2011, 117, 6837–6847. [Google Scholar] [CrossRef] [Green Version]
- Agerstam, H.; Järås, M.; Andersson, A.; Johnels, P.; Hansen, N.; Lassen, C.; Rissler, M.; Gisselsson, D.; Olofsson, T.; Richter, J.; et al. Modeling the human 8p11-myeloproliferative syndrome in immunodeficient mice. Blood 2010, 116, 2103–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, A.; Grand, F.H.; Cross, N.C.P.P. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood 2007, 110, 3729–3734. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Bisom-Rapp, E.W.; Donoghue, D.J. Oncogenic fusion protein BCR-FGFR1 requires the breakpoint cluster region-mediated oligomerization and chaperonin Hsp90 for activation. Haematologica 2020, 105, 1262–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, D.M.; Mirante, T.; Mignogna, C.; Scrima, M.; Migliozzi, S.; Rocco, G.; Franco, R.; Corcione, F.; Viglietto, G.; Malanga, D.; et al. Simultaneous identification of clinically relevant single nucleotide variants, copy number alterations and gene fusions in solid tumors by targeted next-generation sequencing. Oncotarget 2018, 9, 22749–22768. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, L.; Li, Y.; Hu, H.; Shen, L.; Shen, X.; Pan, Y.; Ye, T.; Zhang, Y.; Luo, X.; et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4107–4114. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Hammerman, P.S.; Kim, J.; Yoon, J.A.; Lee, Y.; Sun, J.M.; Wilkerson, M.D.; Pedamallu, C.S.; Cibulskis, K.; Yoo, Y.K.; et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J. Clin. Oncol. 2014, 32, 121–128. [Google Scholar] [CrossRef]
- Javle, M.; Rashid, A.; Churi, C.; Kar, S.; Zuo, M.; Eterovic, A.K.; Nogueras-Gonzalez, G.M.; Janku, F.; Shroff, R.T.; Aloia, T.A.; et al. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum. Pathol. 2014, 45, 701–708. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 2014, 15, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.A.; Elvin, J.A.; Kamath, S.D.; Ali, S.M.; Paintal, A.S.; Restrepo, A.; Berry, E.; Giles, F.J.; Johnson, M.L. FGFR3-TACC3: A novel gene fusion in cervical cancer. Gynecol. Oncol. Rep. 2015, 13, 53–56. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Majewski, I.J.; Mittempergher, L.; Davidson, N.M.; Bosma, A.; Willems, S.M.; Horlings, H.M.; De Rink, I.; Greger, L.; Hooijer, G.K.J.; Peters, D.; et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred RNA sequencing. J. Pathol. 2013, 230, 270–276. [Google Scholar] [CrossRef]
- Mizukami, T.; Sakai, K.; Naruki, S.; Taniyama, T.; Horie, Y.; Izawa, N.; Tsuda, T.; Fujino, T.; Boku, N.; Yasuda, H.; et al. Identification of a FGFR3-TACC3 fusion in esophageal cancer. Ann. Oncol. 2017, 28, 437–438. [Google Scholar] [CrossRef]
- Capelletti, M.; Dodge, M.E.; Ercan, D.; Hammerman, P.S.; Park, S.-I.; Kim, J.; Sasaki, H.; Jablons, D.M.; Lipson, D.; Young, L.; et al. Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 6551–6558. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.N.; Meyer, A.N.; Siari, A.; Campos, A.R.; Motamedchaboki, K.; Donoghue, D.J. Oncogenic gene fusion FGFR3-TACC3 Is regulated by tyrosine phosphorylation. Mol. Cancer Res. 2016, 14, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.N.; Meyer, A.N.; Wang, C.G.; Donoghue, D.J. Oncogenic driver FGFR3-TACC3 is dependent on membrane trafficking and ERK signaling. Oncotarget 2018, 9, 34306–34319. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.; et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Invest. 2013, 123, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, P.; Stichel, D.; Schrimpf, D.; Sahm, F.; Koelsche, C.; Reuss, D.E.; Wefers, A.K.; Reinhardt, A.; Huang, K.; Ebrahimi, A.; et al. FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol. 2018, 136, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.A. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol. Commun. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.H.G.; Gupta, R.; Doo, P.; Lee, J.C.; Cadwell, C.R.; Ramani, B.; Hofmann, J.W.; Sloan, E.A.; Kleinschmidt-Demasters, B.K.; Lee, H.S.; et al. Comprehensive analysis of diverse low-grade neuroepithelial tumors with FGFR1 alterations reveals a distinct molecular signature of rosette-forming glioneuronal tumor. Acta Neuropathol. Commun. 2020, 8, 1–17. [Google Scholar] [CrossRef]
- Sievers, P.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Hasselblatt, M.; Hagel, C.; Staszewski, O.; Hench, J.; Frank, S.; Brandner, S.; et al. Posterior fossa pilocytic astrocytomas with oligodendroglial features show frequent FGFR1 activation via fusion or mutation. Acta Neuropathol. 2020, 139, 403–406. [Google Scholar] [CrossRef]
- Daoud, E.V.; Patel, A.; Gagan, J.; Raisanen, J.M.; Snipes, G.J.; Mantilla, E.; Krothapally, R.; Hatanpaa, K.J.; Pan, E. Spinal Cord Pilocytic Astrocytoma With FGFR1-TACC1 Fusion and Anaplastic Transformation. J. Neuropathol. Exp. Neurol. 2021, 80, 283–285. [Google Scholar] [CrossRef]
- Devereaux, K.A.; Weiel, J.J.; Mills, A.M.; Kunder, C.A.; Longacre, T.A. Neurofibrosarcoma Revisited: An Institutional Case Series of Uterine Sarcomas Harboring Kinase-related Fusions With Report of a Novel FGFR1-TACC1 Fusion. Am. J. Surg. Pathol. 2021, 45, 638–652. [Google Scholar] [CrossRef]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.D.; et al. Integrated Genomic Characterization Reveals Novel, Therapeutically Relevant Drug Targets in FGFR and EGFR Pathways in Sporadic Intrahepatic Cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Ying, X.; Tu, J.; Wang, W.; Li, X.; Xu, C.; Ji, J. FGFR2-BICC1: A subtype of FGFR2 oncogenic fusion variant in cholangiocarcinoma and the response to sorafenib. Onco. Targets. Ther. 2019, 12, 9303–9307. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New Routes to Targeted Therapy of Intrahepatic Cholangiocarcinomas Revealed by Next-Generation Sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, V.; El-Rayes, B.F.; Droz dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Scheiter, A.; Keil, F.; Lüke, F.; Grosse, J.; Verloh, N.; Opitz, S.; Schlosser, S.; Kandulski, A.; Pukrop, T.; Dietmaier, W.; et al. Identification and In-Depth Analysis of the Novel FGFR2-NDC80 Fusion in a Cholangiocarcinoma Patient: Implication for Therapy. Curr. Oncol. 2021, 28, 112. [Google Scholar] [CrossRef]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non–Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Popovici, C.; Zhang, B.; Grégoire, M.J.; Jonveaux, P.; Lafage-Pochitaloff, M.; Birnbaum, D.; Pébusque, M.J. The t(6;8)(q27;p11) translocation in a stem cell myeloproliferative disorder fuses a novel gene, FOP, to Fibroblast Growth Factor Receptor 1. Blood 1999, 93, 1381–1389. [Google Scholar] [CrossRef]
- Sohal, J.; Chase, A.; Mould, S.; Corcoran, M.; Oscier, D.; Iqbal, S.; Parker, S.; Welborn, J.; Harris, R.I.; Martinelli, G.; et al. Identification of four new translocations involvingFGFR1 in myeloid disorders. Genes, Chromosom. Cancer 2001, 32, 155–163. [Google Scholar] [CrossRef]
- Vannier, J.P.; Bizet, M.; Bastard, C.; Bernard, A.; Ducastelle, T.; Tron, P. Simultaneous occurrence of a T-cell lymphoma and a chronic myelogenous leukemia with an unusual karyotype. Leuk. Res. 1984, 8, 647–657. [Google Scholar] [CrossRef]
- Vizmanos, J.L.; Hernández, R.; Vidal, M.J.; Larráyoz, M.J.; Odero, M.D.; Marín, J.; Ardanaz, M.T.; Calasanz, M.J.; Cross, N.C.P. Clinical variability of patients with the t(6;8)(q27;p12) and FGFR1OP-FGFR1 fusion: Two further cases. Hematol. J. 2004, 5, 534–537. [Google Scholar] [CrossRef]
- Chaffanet, M.; Popovici, C.; Leroux, D.; Jacrot, M.; Adélaïde, J.; Dastugue, N.; Grégoire, M.J.; Hagemeijer, A.; Lafage-Pochitaloff, M.; Birnbaum, D.; et al. t(6;8), t(8;9) and t(8;13) translocations associated with stem cell myeloproliferative disorders have close or identical breakpoints in chromosome region 8p11-12. Oncogene 1998, 16, 945–949. [Google Scholar] [CrossRef] [Green Version]
- Onozawa, M.; Ohmura, K.; Ibata, M.; Iwasaki, J.; Okada, K.; Kasahara, I.; Yamaguchi, K.; Kubota, K.; Fujisawa, S.; Shigematsu, A.; et al. The 8p11 myeloproliferative syndrome owing to rare FGFR1OP2-FGFR1 fusion. Eur. J. Haematol. 2011, 86, 347–349. [Google Scholar] [CrossRef]
- Macdonald, D.; Aguiar, R.C.; Mason, P.J.; Goldman, J.M.; Cross, N.C. A new myeloproliferative disorder associated with chromosomal translocations involving 8p11: A review. Leukemia 1995, 9, 1628–1630. [Google Scholar]
- Macdonald, D.; Reiter, A.; Cross, N.C.P. The 8p11 myeloproliferative syndrome: A distinct clinical entity caused by constitutive activation of FGFR1. Acta Haematol. 2002, 107, 101–107. [Google Scholar] [CrossRef]
- Friedhoff, F.; Rajendra, B.; Moody, R.; Alapatt, T. Novel reciprocal translocation between chromosomes 8 and 9 found in a patient with myeloproliferative disorder. Cancer Genet. Cytogenet. 1983, 9, 391–394. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kawano, H.; Nishikawa, S.; Yakushijin, K.; Okamura, A.; Matsui, T. A biphenotypic transformation of 8p11 myeloproliferative syndrome with CEP1/FGFR1 fusion gene. Eur. J. Haematol. 2006, 77, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Song, J.; Kim, J.S.; Yang, W.I.; Song, S.; Kim, S.J.; Suh, B.; Choi, J.R. 8p11 myeloproliferative syndrome preceded by t(8;9)(p11;q33), CEP110/FGFR1 fusion transcript: Morphologic, molecular, and cytogenetic characterization of myeloid neoplasms associated with eosinophilia and FGFR1 abnormality. Cancer Genet. Cytogenet. 2008, 181, 93–99. [Google Scholar] [CrossRef]
- Mozziconacci, M.J.; Carbuccia, N.; Prebet, T.; Charbonnier, A.; Murati, A.; Vey, N.; Chaffanet, M.; Birnbaum, D. Common features of myeloproliferative disorders with t(8;9)(p12;q33) and CEP110-FGFR1 fusion: Report of a new case and review of the literature. Leuk. Res. 2008, 32, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, W.; Yuan, Z.; Hou, J. Complete molecular remission after interferon alpha treatment in a case of 8p11 myeloproliferative syndrome. Leuk. Res. 2010, 34, e306–e307. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; He, Y.; Zhu, X.; Li, J.; He, H. Myeloproliferative disorders with t(8;9)(p12;q33): A case report and review of the literature. Pediatr. Hematol. Oncol. 2011, 28, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ebihara, Y.; Mochizuki, S.; Kawakita, T.; Kato, S.; Ooi, J.; Takahashi, S.; Tojo, A.; Yusa, N.; Furukawa, Y.; et al. Quantitative polymerase chain reaction detection of CEP110-FGFR1 fusion gene in a patient with 8p11 myeloproliferative syndrome. Leuk. Lymphoma 2013, 54, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Sarah, O.-O.; Anthony, A.; Titilope, A.; Alani, S. The 8p12 myeloproliferative syndrome. Niger. Med. J. 2014, 55, 176. [Google Scholar] [CrossRef]
- Wehrli, M.; Oppliger Leibundgut, E.; Gattiker, H.H.; Manz, M.G.; Müller, A.M.S.; Goede, J.S. Response to Tyrosine Kinase Inhibitors in Myeloproliferative Neoplasia with 8p11 Translocation and CEP110-FGFR1 Rearrangement. Oncologist 2017, 22, 480–483. [Google Scholar] [CrossRef] [Green Version]
- Sarthy, J.F.; Reddivalla, N.; Radhi, M.; Chastain, K. Pediatric 8p11 eosinophilic myeloproliferative syndrome (EMS): A case report and review of the literature. Pediatr. Blood Cancer 2017, 64, 1–8. [Google Scholar] [CrossRef]
- Chen, M.; Wang, K.; Cai, X.; Zhang, X.; Chao, H.; Chen, S.; Shen, H.; Wang, Q.; Zhang, R. Myeloid/lymphoid neoplasm with CEP110-FGFR1 fusion: An analysis of 16 cases show common features and poor prognosis. Hematology 2021, 26, 153–159. [Google Scholar] [CrossRef]
- Oscier, D.G.; Mufti, G.J.; Gardiner, A.; Hamblin, T.J. Reciprocal translocation between chromosomes 8 and 9 in atypical chronic myeloid leukaemia. J. Med. Genet. 1985, 22, 398–401. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Otsu, M.; Matsuzaka, E.; Konishi, C.; Takagi, H.; Hanada, S.; Mochizuki, S.; Nakauchi, H.; Imai, K.; Tsuji, K.; et al. Screening of drugs to treat 8p11 myeloproliferative syndrome using patient-derived induced pluripotent stem cells with fusion gene CEP110-FGFR1. PLoS ONE 2015, 10, e0120841. [Google Scholar] [CrossRef]
- Brown, L.M.; Bartolo, R.C.; Davidson, N.M.; Schmidt, B.; Brooks, I.; Challis, J.; Petrovic, V.; Khuong-Quang, D.A.; Mechinaud, F.; Khaw, S.L.; et al. Targeted therapy and disease monitoring in CNTRL-FGFR1-driven leukaemia. Pediatr. Blood Cancer 2019, 66, 1–5. [Google Scholar] [CrossRef]
- Lewis, J.P.; Welborn, J.L.; Meyers, F.J.; Levy, N.B.; Roschak, T. Mast cell disease followed by leukemia with clonal evolution. Leuk. Res. 1987, 11, 769–773. [Google Scholar] [CrossRef]
- Jotterand Bellomo, M.; Mühlematter, D.; Wicht, M.; Delacrétaz, F.; Schmidt, P.M. t(8;9)(p11;q32) in atypical chronic myeloid leukaemia: A new cytogenetic-clinicopathologic association? Br. J. Haematol. 1992, 81, 307–308. [Google Scholar] [CrossRef]
- Van den Berg, H.; Kroes, W.; van der Schoot, C.E.; Dee, R.; Pals, S.T.; Bouts, T.H.; Slater, R.M. A young child with acquired t(8;9)(p11;q34): Additional proof that 8p11 is involved in mixed myeloid/T lymphoid malignancies. Leukemia 1996, 10, 1252–1253. [Google Scholar]
- Nakayama, H.; Inamitsu, T.; Ohga, S.; Kai, T.; Suda, M.; Matsuzaki, A.; Ueda, K. Chronic myelomonocytic leukaemia with t(8;9)(p11;q34) in childhood: An example of the 8p11 myeloproliferative disorder? Br. J. Haematol. 1996, 92, 692–695. [Google Scholar] [CrossRef]
- Vandergoten, P.; Janssen, M.; Madoe, V.; Vanstraelen, D. A myeloproliferative disorder with eosinophilia, a translocation t(8; 9)(p22;23) and a cerebellar degeneration in regression with interferon alpha therapy. Acta Haematol. 1998, 100, 8. [Google Scholar]
- Guasch, G.; Mack, G.J.; Popovici, C.; Dastugue, N.; Birnbaum, D.; Rattner, J.B.; Pébusque, M.J. FGFR1 is fused to the centrosome-associated protein CEP110 in the 8p12 stem cell myeloproliferative disorder with t(8;9)(p12;q33). Blood 2000, 95, 1788–1796. [Google Scholar] [CrossRef]
- Heiss, S.; Erdel, M.; Gunsilius, E.; Nachbaur, D.; Tzankov, A. Myelodysplastic/myeloproliferative disease with erythropoietic hyperplasia (erythroid preleukemia) and the unique translocation (8;9)(p23;p24): First description of a case. Hum. Pathol. 2005, 36, 1148–1151. [Google Scholar] [CrossRef]
- Chen, J.; Williams, I.R.; Lee, B.H.; Duclos, N.; Huntly, B.J.P.; Donoghue, D.J.; Gilliland, D.G. Constitutively activated FGFR3 mutants signal through PLCγ-dependent and -independent pathways for hematopoietic transformation. Blood 2005, 106, 328–337. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl- piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Liu, P.C.C.; Koblish, H.; Wu, L.; Bowman, K.; Diamond, S.; DiMatteo, D.; Zhang, Y.; Hansbury, M.; Rupar, M.; Wen, X.; et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 2020, 15, e231877. [Google Scholar] [CrossRef]
- Hood, F.E.; Williams, S.J.; Burgess, S.G.; Richards, M.W.; Roth, D.; Straube, A.; Pfuhl, M.; Bayliss, R.; Royle, S.J. Coordination of adjacent domains mediates TACC3-ch-TOG-clathrin assembly and mitotic spindle binding. J. Cell Biol. 2013, 202, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Hood, F.E.; Royle, S.J. Pulling it together: The mitotic function of TACC3. Bioarchitecture 2011, 1, 105–109. [Google Scholar] [CrossRef]
- Nixon, F.M.; Gutiérrez-Caballero, C.; Hood, F.E.; Booth, D.G.; Prior, I.A.; Royle, S.J. The mesh is a network of microtubule connectors that stabilizes individual kinetochore fibers of the mitotic spindle. Elife 2015, 4, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Ryan, E.L.; Royle, S.J. FGFR3-TACC3 cancer gene fusions cause mitotic defects by removal of endogenous TACC3 from the mitotic spindle. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Natsume, Y.; Saiki, Y.; Shioya, H.; Takeuchi, K.; Yamori, T.; Toki, H.; Aoki, I.; Saga, T.; Noda, T. Disruption of Tacc3 function leads to in vivo tumor regression. Oncogene 2012, 31, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Oyanagi, J.; Natsume, Y.; Kusama, D.; Kato, Y.; Nagayama, S.; Noda, T. Suppression of intestinal tumors by targeting the mitotic spindle of intestinal stem cells. Oncogene 2016, 35, 6109–6119. [Google Scholar] [CrossRef]
- Akbulut, O.; Lengerli, D.; Saatci, O.; Duman, E.; Seker, U.O.S.; Isik, A.; Akyol, A.; Caliskan, B.; Banoglu, E.; Sahin, O. A highly potent TACC3 inhibitor as a novel anticancer drug candidate. Mol. Cancer Ther. 2020, 19, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K.; Noetzel, T.L.; Pelletier, L.; Mechtler, K.; Drechsel, D.N.; Schwager, A.; Lee, M.; Raff, J.W.; Hyman, A.A. Aurora A phosphorylation of TACC3/maskin is required for centrosome-dependent microtubule assembly in mitosis. J. Cell Biol. 2005, 170, 1047–1055. [Google Scholar] [CrossRef]
- Adams, M.; Simms, R.J.; Abdelhamed, Z.; Dawe, H.R.; Szymanska, K.; Logan, C.V.; Wheway, G.; Pitt, E.; Gull, K.; Knowles, M.A.; et al. A meckelin-filamin a interaction mediates ciliogenesis. Hum. Mol. Genet. 2012, 21, 1272–1286. [Google Scholar] [CrossRef]
- Qie, Y.; Wang, L.; Du, E.; Chen, S.; Lu, C.; Ding, N.; Yang, K.; Xu, Y. TACC3 promotes prostate cancer cell proliferation and restrains primary cilium formation. Exp. Cell Res. 2020, 390. [Google Scholar] [CrossRef] [PubMed]
- Still, I.H.; Hamilton, M.; Vince, P.; Wolfman, A.; Cowell, J.K. Cloning of TACC1, an embryonically expressed, potentially transforming coiled coil containing gene, from the 8p11 breast cancer amplicon. Oncogene 1999, 18, 4032–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peset, I.; Vernos, I. The TACC proteins: TACC-ling microtubule dynamics and centrosome function. Trends Cell Biol. 2008, 18, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Gergely, F.; Karlsson, C.; Still, I.; Cowell, J.; Kilmartin, J.; Raff, J.W. The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc. Natl. Acad. Sci. USA 2000, 97, 14352–14357. [Google Scholar] [CrossRef] [Green Version]
- Conte, N.; Delaval, B.; Ginestier, C.; Ferrand, A.; Isnardon, D.; Larroque, C.; Prigent, C.; Séraphin, B.; Jacquemier, J.; Birnbaum, D. TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene 2003, 22, 8102–8116. [Google Scholar] [CrossRef] [Green Version]
- Cristinziano, G.; Porru, M.; Lamberti, D.; Buglioni, S.; Rollo, F.; Amoreo, C.A.; Manni, I.; Giannarelli, D.; Cristofoletti, C.; Russo, G.; et al. FGFR2 fusion proteins drive oncogenic transformation of mouse liver organoids towards cholangiocarcinoma. J. Hepatol. 2021, 11, 3–5. [Google Scholar] [CrossRef]
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol. 2014, 232, 4–15. [Google Scholar] [CrossRef]
- Bahleda, R.; Meric-Bernstam, F.; Goyal, L.; Tran, B.; He, Y.; Yamamiya, I.; Benhadji, K.A.; Matos, I.; Arkenau, H.-T. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 2020, 31, 1405–1412. [Google Scholar] [CrossRef]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Mahone, M.; Saffman, E.E.; Lasko, P.F. Localized Bicaudal-C RNA encodes a protein containing a KH domain, the RNA binding motif of FMR1. EMBO J. 1995, 14, 2043–2055. [Google Scholar] [CrossRef]
- Park, S.; Blaser, S.; Marchal, M.A.; Houston, D.W.; Sheets, M.D. A gradient of maternal Bicaudal-C controls vertebrate embryogenesis via translational repression of mRNAs encoding cell fate regulators. Development 2016, 143, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Wessely, O.; De Robertis, E.M. The Xenopus homologue of Bicaudal-C is a localized maternal mRNA that can induce endoderm formation. Development 2000, 127, 2053–2062. [Google Scholar] [CrossRef]
- Dowdle, M.E.; Park, S.; Blaser Imboden, S.; Fox, C.A.; Houston, D.W.; Sheets, M.D. A single KH domain in Bicaudal-C links mRNA binding and translational repression functions to maternal development. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Saffman, E.E.; Styhler, S.; Rother, K.; Li, W.; Richard, S.; Lasko, P. Premature translation of oskar in oocytes lacking the RNA-binding protein bicaudal-C. Mol. Cell. Biol. 1998, 18, 4855–4862. [Google Scholar] [CrossRef] [Green Version]
- Tran, U.; Zakin, L.; Schweickert, A.; Agrawal, R.; Döger, R.; Blum, M.; De Robertis, E.M.; Wessely, O. The RNA-binding protein bicaudal C regulates polycystin 2 in the kidney by antagonizing miR-17 activity. Development 2010, 137, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Cogswell, C.; Price, S.J.; Hou, X.; Guay-Woodford, L.M.; Flaherty, L.; Bryda, E.C. Positional cloning of jcpk/bpk locus of the mouse. Mamm. Genome 2003, 14, 242–249. [Google Scholar] [CrossRef]
- Lemaire, L.A.; Goulley, J.; Kim, Y.H.; Carat, S.; Jacquemin, P.; Rougemont, J.; Constam, D.B.; Grapin-Botton, A. Bicaudal C1 promotes pancreatic NEUROG3+ endocrine progenitor differentiation and ductal morphogenesis. Development 2015, 142, 858–870. [Google Scholar] [CrossRef] [Green Version]
- Maisonneuve, C.; Guilleret, I.; Vick, P.; Weber, T.; Andre, P.; Beyer, T.; Blum, M.; Constam, D.B. Bicaudal C, a novel regulator of Dvl signaling abutting RNA-processing bodies, controls cilia orientation and leftward flow. Development 2009, 136, 3019–3030. [Google Scholar] [CrossRef] [Green Version]
- Guay-Woodford, L.M.; Bryda, E.C.; Christine, B.; Lindsey, J.R.; Collier, W.R.; Avner, E.D.; D’Eustachio, P.; Flaherty, L. Evidence that two phenotypically distinct mouse PKD mutations, bpk and jcpk, are allelic. Kidney Int. 1996, 50, 1158–1165. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, L.; Bryda, E.C.; Collins, D.; Rudofsky, U.; Montogomery, J.C. New mouse model for polycystic kidney disease with both recessive and dominant gene effects. Kidney Int. 1995, 47, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Bouvrette, D.J.; Sittaramane, V.; Heidel, J.R.; Chandrasekhar, A.; Bryda, E.C. Knockdown of bicaudal C in zebrafish (Danio rerio) causes cystic kidneys: A nonmammalian model of polycystic kidney disease. Comp. Med. 2010, 60, 96–106. [Google Scholar]
- Gamberi, C.; Hipfner, D.R.; Trudel, M.; Lubell, W.D. Bicaudal C mutation causes myc and TOR pathway up-regulation and polycystic kidney disease-like phenotypes in Drosophila. PLoS Genet. 2017, 13, e1006694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaconis, D.; Monti, M.; Renda, M.; Van Koppen, A.; Tammaro, R.; Chiaravalli, M.; Cozzolino, F.; Pignata, P.; Crina, C.; Pucci, P.; et al. The centrosomal OFD1 protein interacts with the translation machinery and regulates the synthesis of specific targets. Sci. Rep. 2017, 7, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothé, B.; Gagnieux, C.; Leal-Esteban, L.C.; Constam, D.B. Role of the RNA-binding protein Bicaudal-C1 and interacting factors in cystic kidney diseases. Cell. Signal. 2020, 68, 109499. [Google Scholar] [CrossRef]
- Kraus, M.R.C.; Clauin, S.; Pfister, Y.; Di Maïo, M.; Ulinski, T.; Constam, D.; Bellanné-Chantelot, C.; Grapin-Botton, A. Two mutations in human BICC1 resulting in wnt pathway hyperactivity associated with cystic renal dysplasia. Hum. Mutat. 2012, 33, 86–90. [Google Scholar] [CrossRef]
- Mesner, L.D.; Ray, B.; Hsu, Y.H.; Manichaikul, A.; Lum, E.; Bryda, E.C.; Rich, S.S.; Rosen, C.J.; Criqui, M.H.; Allison, M.; et al. Bicc1 is a genetic determinant of osteoblastogenesis and bone mineral density. J. Clin. Invest. 2014, 124, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Rothé, B.; Leal-Esteban, L.; Bernet, F.; Urfer, S.; Doerr, N.; Weimbs, T.; Iwaszkiewicz, J.; Constam, D.B. Bicc1 Polymerization Regulates the Localization and Silencing of Bound mRNA. Mol. Cell. Biol. 2015, 35, 3339–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvrette, D.J.; Price, S.J.; Bryda, E.C. K homology domains of the mouse polycystic kidney disease-related protein, Bicaudal-C (Bicc1), mediate RNA binding in vitro. Nephron. Exp. Nephrol. 2008, 108, e27–e34. [Google Scholar] [CrossRef]
- Wimbish, R.T.; DeLuca, J.G. Hec1/Ndc80 Tail Domain Function at the Kinetochore-Microtubule Interface. Front. Cell Dev. Biol. 2020, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Li, J.; Cai, Q.; Liu, B. Hec1/Ndc80 is overexpressed in human gastric cancer and regulates cell growth. J. Gastroenterol. 2014, 49, 408–418. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, Q.; Zhang, C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J. Cell Sci. 2014, 127, 4111–4122. [Google Scholar] [CrossRef] [Green Version]
- Bièche, I.; Vacher, S.; Lallemand, F.; Tozlu-Kara, S.; Bennani, H.; Beuzelin, M.; Driouch, K.; Rouleau, E.; Lerebours, F.; Ripoche, H.; et al. Expression analysis of mitotic spindle checkpoint genes in breast carcinoma: Role of NDC80/HEC1 in early breast tumorigenicity, and a two-gene signature for aneuploidy. Mol. Cancer 2011, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.M.; Zhu, J.; Guo, X.E.; Chen, W.; Qiu, X.L.; Ngo, B.; Chien, R.; Wang, Y.V.; Tsai, C.Y.; Wu, G.; et al. Novel small molecules disrupting Hec1/Nek2 interaction ablate tumor progression by triggering Nek2 degradation through a death-trap mechanism. Oncogene 2015, 34, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rodríguez, E.; Sotillo, R.; Schvartzman, J.M.; Benezra, R. Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 16719–16724. [Google Scholar] [CrossRef] [Green Version]
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Wu, G.; Qiu, X.L.; Zhou, L.; Zhu, J.; Chamberlin, R.; Lau, J.; Chen, P.L.; Lee, W.H. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008, 68, 8393–8399. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Y.L.; Chang, C.C.; Lee, Y.S.; Huang, J.J.; Chuang, S.H.; Chang, J.M.; Kao, K.J.; Lau, G.M.G.; Tsai, P.Y.; Liu, C.W.; et al. Inhibition of Hec1 as a novel approach for treatment of primary liver cancer. Cancer Chemother. Pharmacol. 2014, 74, 511–520. [Google Scholar] [CrossRef]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.; Savage, R.E.; Lapierre, J.-M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef] [Green Version]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.A.; Annaratone, L.; et al. ASPM and CITK regulate spindle orientation by affecting the dynamics of astral microtubules. EMBO Rep. 2017, 18, 1870. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; De Zan, T.; Ishizaki, T.; Narumiya, S. Citron kinase mediates transition from constriction to abscission through its coiled-coil domain. J. Cell Sci. 2013, 126, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Bassi, Z.I.; Audusseau, M.; Riparbelli, M.G.; Callaini, G.; D’Avino, P.P. Citron kinase controls a molecular network required for midbody formation in cytokinesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9782–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruneberg, U.; Neef, R.; Li, X.; Chan, E.H.Y.; Chalamalasetty, R.B.; Nigg, E.A.; Barr, F.A. KIF14 and citron kinase act together to promote efficient cytokinesis. J. Cell Biol. 2006, 172, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Zhu, X.; Xu, W.; Zhang, Y.; Chen, L.; Qiu, F.; Zhang, B.; Wu, L.; Peng, Z.; Tang, H. Up-regulation of CIT promotes the growth of colon cancer cells. Oncotarget 2017, 8, 71954–71964. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Huang, J.; Wang, K.S.; Zhang, X.; Han, Z.G. RNA interference targeting CITRON can significantly inhibit the proliferation of hepatocellular carcinoma cells. Mol. Biol. Rep. 2011, 38, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Ehrlichova, M.; Mohelnikova-Duchonova, B.; Hrdy, J.; Brynychova, V.; Mrhalova, M.; Kodet, R.; Rob, L.; Pluta, M.; Gut, I.; Soucek, P.; et al. The association of taxane resistance genes with the clinical course of ovarian carcinoma. Genomics 2013, 102, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Yu, Q.; Feng, L.; Luo, M.; Shao, S.; Huang, S.; Wang, G.; Jing, X.; Tong, Z.; Zhao, X.; et al. Citron kinase (CIT-K) promotes aggressiveness and tumorigenesis of breast cancer cells in vitro and in vivo: Preliminary study of the underlying mechanism. Clin. Transl. Oncol. 2019, 21, 910–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yan, H.; Yang, Y.; Wei, L.; Xia, S.; Xiu, Y. Down-regulation of CIT can inhibit the growth of human bladder cancer cells. Biomed. Pharmacother. 2020, 124, 109830. [Google Scholar] [CrossRef]
- Liu, J.; Dou, J.; Wang, W.; Liu, H.; Qin, Y.; Yang, Q.; Jiang, W.; Liang, Y.; Liu, Y.; He, J.; et al. High expression of citron kinase predicts poor prognosis of prostate cancer. Oncol. Lett. 2020, 19, 1815–1823. [Google Scholar] [CrossRef] [Green Version]
- Pallavicini, G.; Iegiani, G.; Berto, G.E.; Calamia, E.; Trevisiol, E.; Veltri, A.; Allis, S.; Di Cunto, F. CITK loss inhibits growth of group 3 and group 4 medulloblastoma cells and sensitizes them to DNA-damaging agents. Cancers 2020, 12, 542. [Google Scholar] [CrossRef] [Green Version]
- Di Cunto, F.; Imarisio, S.; Hirsch, E.; Broccoli, V.; Bulfone, A.; Migheli, A.; Atzori, C.; Turco, E.; Triolo, R.; Dotto, G.P.; et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 2000, 28, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Di Cunto, F.; Imarisio, S.; Camera, P.; Boitani, C.; Altruda, F.; Silengo, L. Essential role of citron kinase in cytokinesis of spermatogenic precursors. J. Cell Sci. 2002, 115, 4819–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M. V Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhns, S.; Schmidt, K.N.; Reymann, J.; Gilbert, D.F.; Neuner, A.; Hub, B.; Carvalho, R.; Wiedemann, P.; Zentgraf, H.; Erfle, H.; et al. The microtubule affinity regulating kinase MARK4 promotes axoneme extension during early ciliogenesis. J. Cell Biol. 2013, 200, 505–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastas, S.B.; Mueller, D.; Semple-Rowland, S.L.; Breunig, J.J.; Sarkisian, M.R. Failed cytokinesis of neural progenitors in citron kinase-deficient rats leads to multiciliated neurons. Cereb. Cortex 2011, 21, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Karkera, J.D.; Cardona, G.M.; Bell, K.; Gaffney, D.; Portale, J.C.; Santiago-Walker, A.; Moy, C.H.; King, P.; Sharp, M.; Bahleda, R.; et al. Oncogenic characterization and pharmacologic sensitivity of activating Fibroblast Growth Factor Receptor (FGFR) genetic alterations to the selective FGFR inhibitor erdafitinib. Mol. Cancer Ther. 2017, 16, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Romio, L.; Fry, A.M.; Winyard, P.J.D.; Malcolm, S.; Woolf, A.S.; Feather, S.A. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J. Am. Soc. Nephrol. 2004, 15, 2556–2568. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dollé, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Singla, V.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Reiter, J.F. Ofd1, a Human Disease Gene, Regulates the Length and Distal Structure of Centrioles. Dev. Cell 2010, 18, 410–424. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.A.M.; Prosser, S.L.; Romio, L.; Hirst, R.A.; O’Callaghan, C.; Woolf, A.S.; Fry, A.M. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 2011, 124, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Chetty-John, S.; Piwnica-Worms, K.; Bryant, J.; Bernardini, I.; Fischer, R.E.; Heller, T.; Gahl, W.A.; Gunay-Aygun, M. Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD I). Am. J. Med. Genet. 2010, 152, 2640–2645. [Google Scholar] [CrossRef] [Green Version]
- Thauvin-Robinet, C.; Cossée, M.; Cormier-Daire, V.; Van Maldergem, L.; Toutain, A.; Alembik, Y.; Bieth, E.; Layet, V.; Parent, P.; David, A.; et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: A French and Belgian collaborative study. J. Med. Genet. 2006, 43, 54–61. [Google Scholar] [CrossRef]
- Saal, S.; Faivre, L.; Aral, B.; Gigot, N.; Toutain, A.; Van Maldergem, L.; Destree, A.; Maystadt, I.; Cosyns, J.-P.P.; Jouk, P.-S.S.; et al. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin. Genet. 2010, 77, 258–265. [Google Scholar] [CrossRef]
- Marina, M.; Franco, B. The molecular basis of oral-facial-digital syndrome, type 1. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151, 318–325. [Google Scholar] [CrossRef]
- Zullo, A.; Iaconis, D.; Barra, A.; Cantone, A.; Messaddeq, N.; Capasso, G.; Dollé, P.; Igarashi, P.; Franco, B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum. Mol. Genet. 2010, 19, 2792–2803. [Google Scholar] [CrossRef] [Green Version]
- Bimonte, S.; De Angelis, A.; Quagliata, L.; Giusti, F.; Tammaro, R.; Dallai, R.; Ascenzi, M.-G.G.; Diez-Roux, G.; Franco, B. Ofd1 is required in limb bud patterning and endochondral bone development. Dev. Biol. 2011, 349, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, M.I.; Romio, L.; Castro, S.; Collins, J.E.; Goulding, D.A.; Stemple, D.L.; Woolf, A.S.; Wilson, S.W. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 2009, 18, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Lelièvre, H.; Chevrier, V.; Tassin, A.-M.; Birnbaum, D. Myeloproliferative disorder FOP-FGFR1 fusion kinase recruits phosphoinositide-3 kinase and phospholipase Cgamma at the centrosome. Mol. Cancer 2008, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Delaval, B.; Létard, S.; Lelièvre, H.; Chevrier, V.; Daviet, L.; Dubreuil, P.; Birnbaum, D. Oncogenic tyrosine kinase of malignant hemopathy targets the centrosome. Cancer Res. 2005, 65, 7231–7240. [Google Scholar] [CrossRef] [Green Version]
- Guasch, G.; Ollendorff, V.; Borg, J.-P.; Birnbaum, D.; Pébusque, M.-J. 8p12 Stem Cell Myeloproliferative Disorder: The FOP-Fibroblast Growth Factor Receptor 1 Fusion Protein of the t(6;8) Translocation Induces Cell Survival Mediated by Mitogen-Activated Protein Kinase and Phosphatidylinositol 3-Kinase/Akt/mTOR Pathways. Mol. Cell. Biol. 2001, 21, 8129–8142. [Google Scholar] [CrossRef] [Green Version]
- Guasch, G.; Delaval, B.; Arnoulet, C.; Xie, M.J.; Xerri, L.; Sainty, D.; Birnbaum, D.; Pébusque, M.J. FOP-FGFR1 tyrosine kinase, the product of a t(6;8) translocation, induces a fatal myeloproliferative disease in mice. Blood 2004, 103, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Mikolajka, A.; Yan, X.; Popowicz, G.M.; Smialowski, P.; Nigg, E.A.; Holak, T.A. Structure of the N-terminal Domain of the FOP (FGFR1OP) Protein and Implications for its Dimerization and Centrosomal Localization. J. Mol. Biol. 2006, 359, 863–875. [Google Scholar] [CrossRef]
- Hori, A.; Toda, T. Regulation of centriolar satellite integrity and its physiology. Cell. Mol. Life Sci. 2016, 74, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Odabasi, E.; Gul, S.; Kavakli, I.H.; Firat-Karalar, E.N. Centriolar satellites are required for efficient ciliogenesis and ciliary content regulation. EMBO Rep. 2019, 20, 1–20. [Google Scholar] [CrossRef]
- Tollenaere, M.A.X.; Mailand, N.; Bekker-Jensen, S. Centriolar satellites: Key mediators of centrosome functions. Cell. Mol. Life Sci. 2015, 72, 11–23. [Google Scholar] [CrossRef]
- Bärenz, F.; Mayilo, D.; Gruss, O.J. Centriolar satellites: Busy orbits around the centrosome. Eur. J. Cell Biol. 2011, 90, 983–989. [Google Scholar] [CrossRef]
- Yan, X.; Habedanck, R.; Nigg, E.A. A Complex of Two Centrosomal Proteins, CAP350 and FOP, Cooperates with EB1 in Microtubule Anchoring. Mol. Biol. Cell 2006, 17, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; McMahon, G.; Sun, L.; Tang, C.; Hirth, P.; Yeh, B.K.; Hubbard, S.R.; Schlessinger, J. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997, 276, 955–960. [Google Scholar] [CrossRef]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol. Cell. Biol. 1991, 11, 5068–5078. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Hong, W.J.; Majeti, R.; Stearns, T. Centrosome-kinase fusions promote oncogenic signaling and disrupt centrosome function in myeloproliferative neoplasms. PLoS ONE 2014, 9, e92641. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Stearns, T. FOP Is a Centriolar Satellite Protein Involved in Ciliogenesis. PLoS ONE 2013, 8, e58589. [Google Scholar] [CrossRef]
- Mojarad, B.A.; Gupta, G.D.; Hasegan, M.; Goudiam, O.; Basto, R.; Gingras, A.C.; Pelletier, L. CEP19 cooperates with FOP and CEP350 to drive early steps in the ciliogenesis programme. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabaud, O.; Roubin, R.; Comte, A.; Bascunana, V.; Sergé, A.; Sedjaï, F.; Birnbaum, D.; Rosnet, O.; Acquaviva, C. Mutation of FOP/FGFR1OP in mice recapitulates human short rib-polydactyly ciliopathy. Hum. Mol. Genet. 2018, 27, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [Green Version]
- Giehl, M.; Fabarius, A.; Frank, O.; Hochhaus, A.; Hafner, M.; Hehlmann, R.; Seifarth, W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005, 19, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Qin, H.; Kitamura, E.; Cowell, J.K. Dysregulated signaling pathways in the development of CNTRL-FGFR1-induced myeloid and lymphoid malignancies associated with FGFR1 in human and mouse models. Blood 2013, 122, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Qin, H.; Ren, R.; Tidwell, J.; Cowell, J.K. Src activation plays an important key role in lymphomagenesis induced by FGFR1 fusion kinases. Cancer Res. 2011, 71, 7312–7322. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Ren, M.; Qin, H.; Ren, R.; Cowell, J.K. Ponatinib suppresses the development of myeloid and lymphoid malignancies associated with FGFR1 abnormalities. Leukemia 2013, 27, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.Y.; Mack, G.J.; Zhang, M.; Rattner, J.B. CEP110 and ninein are located in a specific domain of the centrosome associated with centrosome maturation. J. Cell Sci. 2002, 115, 1825–1835. [Google Scholar] [CrossRef]
- Kashihara, H.; Chiba, S.; Kanno, S.-I.; Suzuki, K.; Yano, T.; Tsukita, S. Cep128 associates with Odf2 to form the subdistal appendage of the centriole. Genes Cells 2019, 24, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.-Y.; Wang, H.-Y.; Kwon, J.-W.; Yuan, B.; Lee, I.-W.; Cui, X.-S.; Kim, N.-H. Centriolin, a centriole-appendage protein, regulates peripheral spindle migration and asymmetric division in mouse meiotic oocytes. Cell Cycle 2017, 16, 1774–1780. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-T.; Hehnly, H.; Yu, Q.; Farkas, D.; Zheng, G.; Redick, S.D.; Hung, H.-F.; Samtani, R.; Jurczyk, A.; Akbarian, S.; et al. A unique set of centrosome proteins requires pericentrin for spindle-pole localization and spindle orientation. Curr. Biol. 2014, 24, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.; Gil-Gil, J.M.; Puduvalli, V.K.; Raizer, J.; De Vos, F.Y.; Wen, P.Y.; Butowski, N.; Clement, P.; et al. OS10.6 Infigratinib (BGJ398) in patients with recurrent gliomas with fibroblast growth factor receptor (FGFR) alterations: A multicenter phase II study. Neuro. Oncol. 2019, 21, iii21–iii22. [Google Scholar] [CrossRef]
- Goyal, L.; Shi, L.; Liu, L.Y.; de la Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 overcomes resistance to atp-competitive FGFR inhibitors in patients with FGFR2 fusion–positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Krzyscik, M.A.; Zakrzewska, M.; Otlewski, J. Site-Specific, Stoichiometric-Controlled, PEGylated Conjugates of Fibroblast Growth Factor 2 (FGF2) with Hydrophilic Auristatin y for Highly Selective Killing of Cancer Cells Overproducing Fibroblast Growth Factor Receptor 1 (FGFR1). Mol. Pharm. 2020, 17, 2734–2748. [Google Scholar] [CrossRef]
- Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. J. Clin. Med. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canning, P.; Park, K.; Gonçalves, J.; Li, C.; Howard, C.J.; Sharpe, T.D.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Harrington, K.J.; Hingorani, M.; Tanay, M.A.; Hickey, J.; Bhide, S.A.; Clarke, P.M.; Renouf, L.C.; Thway, K.; Sibtain, A.; McNeish, I.A.; et al. Phase I/II study of oncolytic HSVGM-CSFin combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. 2010, 16, 4005–4015. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Lu, M.; Elshaikh, M.; Aref, I.; Pradhan, D.; Levin, K.; Kim, J.H.; Peabody, J.; Siddiqui, F.; et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014, 89, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torrella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of the FGFR fusion proteins. The wild-type FGFR comprises the extracellular immunoglobulin-like domain, responsible for ligand binding, the transmembrane region, and the intracellular part that is responsible for binding and activation of the signal transducers including PLCγ (binding site indicated in green). The type II fusions lose a variable part of the C-terminal region of FGFR, frequently involving the PLCγ binding site, and attach a truncated C-terminal part of the fusion partner. In type I fusions, the FGFR extracellular and transmembrane parts are excluded, and the truncated fusion partner joins in just before the FGFR kinase domain. In both types of FGFR fusion, the partner possesses domains that facilitate dimerization—the coiled-coil domain, the sterile alpha motif, the leucine rich repeat or the leucine zipper. The positions of the fusion breakpoints are indicated.
Figure 1.
Schematic representation of the FGFR fusion proteins. The wild-type FGFR comprises the extracellular immunoglobulin-like domain, responsible for ligand binding, the transmembrane region, and the intracellular part that is responsible for binding and activation of the signal transducers including PLCγ (binding site indicated in green). The type II fusions lose a variable part of the C-terminal region of FGFR, frequently involving the PLCγ binding site, and attach a truncated C-terminal part of the fusion partner. In type I fusions, the FGFR extracellular and transmembrane parts are excluded, and the truncated fusion partner joins in just before the FGFR kinase domain. In both types of FGFR fusion, the partner possesses domains that facilitate dimerization—the coiled-coil domain, the sterile alpha motif, the leucine rich repeat or the leucine zipper. The positions of the fusion breakpoints are indicated.


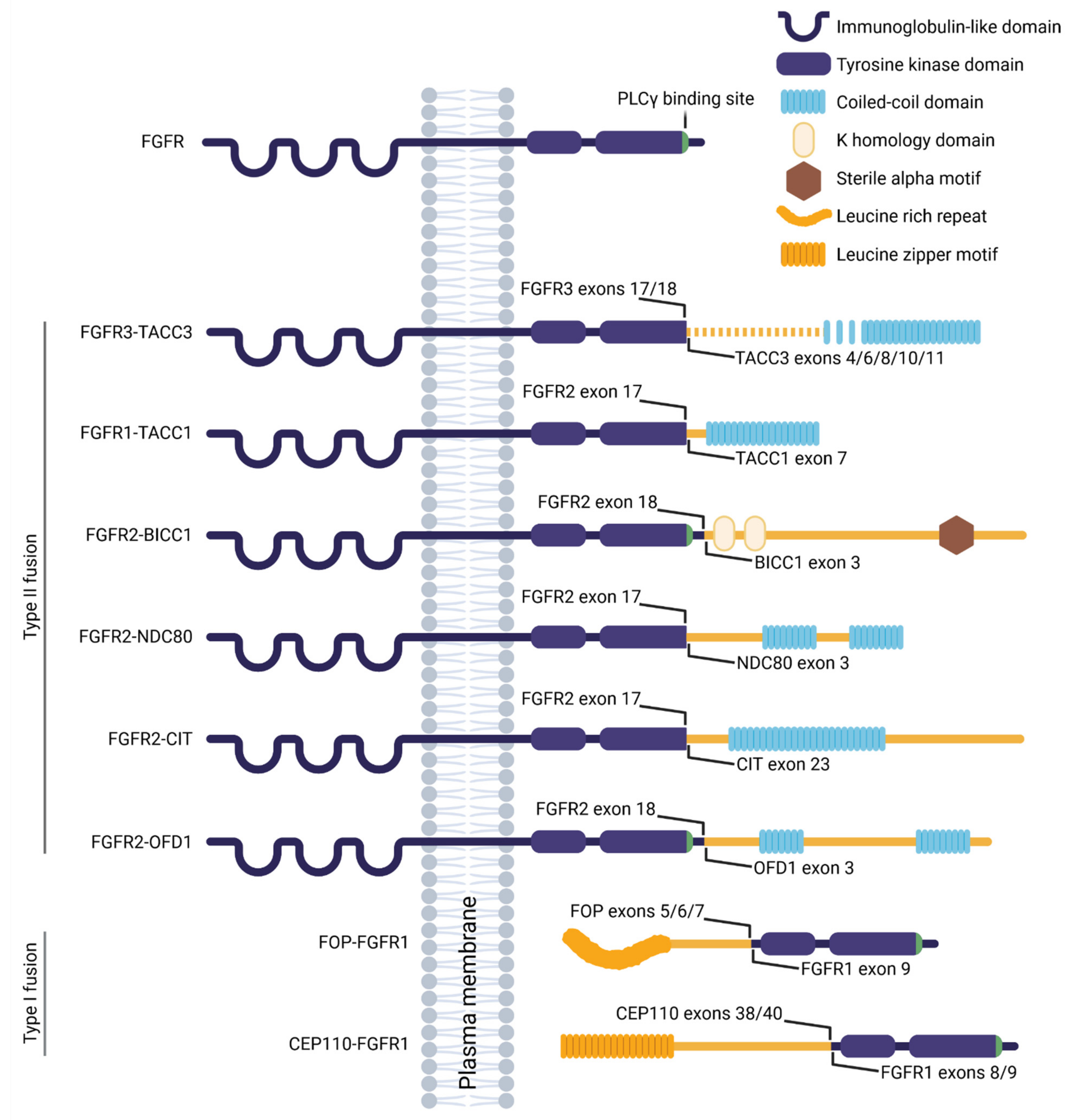{kind=link}
Table 1.
FGFR fusion proteins in cancer.
| Gene Fusion | Cancer Type (Cases) | Reference |
|---|---|---|
| FGFR3-TACC3 | glioblastoma (29); NSCLC (28); HNSCC (11); bladder cancer (10); urothelial carcinoma (4); nasopharyngeal carcinoma (4); LUSC (4); glioma (3); ESCC (2); cervical cancer (2); gallbladder cancer (2); oral cancer (1); renal cell cancer (1); endometrial adenocarcinoma (1) | [133,149,153,179,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200] |
| FGFR1-TACC1 | low grade glioma (7); extraventricular neurocytoma (3); glioblastoma (2); spinal cord pilocytic astrocytoma (2); GIST (1) | [186,187,204,205,206,207,208,209,210,211] |
| FGFR2-BICC1 | cholangiocarcinoma (47); hepatocellular cancer (1); colorectal cancer (1) | [149,175,177,212,213,214,215] |
| FGFR2-NDC80 | cholangiocarcinoma (1) | [216] |
| FGFR2-CIT | NSCLC (3); cholangiocarcinoma (1) | [215,217,218] |
| FGFR2-OFD1 | thyroid cancer (1); endometrial cancer (1) | [149,219] |
| FOP-FGFR1 | AML (9); EMS (1) | [220,221,222,223,224,225,226,227] |
| CEP110-FGFR1 | EMS (9); MPD (5); aCML (3); AML (2); CMML (1); AMML (1) | [221,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248] |
AML—acute myeloid leukemia; AMML—acute myelomonocytic leukemia; aCML—atypical chronic myeloid leukemia; CMML—chronic myelomonocytic leukemia; EMS—8p11 myeloproliferative syndrome; ESCC—esophageal squamous-cell carcinoma; GIST—gastrointestinal stromal tumor; HNSCC—head and neck squamous cell carcinoma; LUSC—lung squamous cell carcinoma; MPD—myeloproliferative disorder; NSCLC—non-small cell lung cancer.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nita, A.; Abraham, S.P.; Krejci, P.; Bosakova, M. Oncogenic FGFR Fusions Produce Centrosome and Cilia Defects by Ectopic Signaling. Cells 2021, 10, 1445. https://doi.org/10.3390/cells10061445
AMA Style
Nita A, Abraham SP, Krejci P, Bosakova M. Oncogenic FGFR Fusions Produce Centrosome and Cilia Defects by Ectopic Signaling. Cells. 2021; 10(6):1445. https://doi.org/10.3390/cells10061445
Chicago/Turabian StyleNita, Alexandru, Sara P. Abraham, Pavel Krejci, and Michaela Bosakova. 2021. "Oncogenic FGFR Fusions Produce Centrosome and Cilia Defects by Ectopic Signaling" Cells 10, no. 6: 1445. https://doi.org/10.3390/cells10061445
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.