
From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization


Rappaport Faculty of Medicine, R-TICC, Technion-IIT, Efron St. Bat-Galim, Haifa 3109610, Israel
*
Author to whom correspondence should be addressed.
†
These authors contributed to this work equally.
Cells 2021, 10(9), 2374; https://doi.org/10.3390/cells10092374
Submission received: 11 August 2021
/
Revised: 30 August 2021
/
Accepted: 4 September 2021
/
Published: 9 September 2021
(This article belongs to the Special Issue Ubiquitin and Ubiquitin-Like Pathways in Development and Disease)
Abstract
:A hallmark of cancer is dysregulated protein turnover (proteostasis), which involves pathologic ubiquitin-dependent degradation of tumor suppressor proteins, as well as increased oncoprotein stabilization. The latter is due, in part, to mutation within sequences, termed degrons, which are required for oncoprotein recognition by the substrate-recognition enzyme, E3 ubiquitin ligase. Stabilization may also result from the inactivation of the enzymatic machinery that mediates the degradation of oncoproteins. Importantly, inactivation in cancer of E3 enzymes that regulates the physiological degradation of oncoproteins, results in tumor cells that accumulate multiple active oncoproteins with prolonged half-lives, leading to the development of “degradation-resistant” cancer cells. In addition, specific sequences may enable ubiquitinated proteins to evade degradation at the 26S proteasome. While the ubiquitin-proteasome pathway was originally discovered as central for protein degradation, in cancer cells a ubiquitin-dependent protein stabilization pathway actively translates transient mitogenic signals into long-lasting protein stabilization and enhances the activity of key oncoproteins. A central enzyme in this pathway is the ubiquitin ligase RNF4. An intimate link connects protein stabilization with tumorigenesis in experimental models as well as in the clinic, suggesting that pharmacological inhibition of protein stabilization has potential for personalized medicine in cancer. In this review, we highlight old observations and recent advances in our knowledge regarding protein stabilization.
1. Overview
The dynamic stability of proteins, which is a fundamental concept in biology, was discovered by Rudolph Schonheimer [1]. It is, however, well known that proteomes exhibit a wide range of protein half-lives [2]. While some intracellular proteins are extremely long-lived (LLPs; [3]), many regulatory proteins degrade rapidly, exhibiting a half-life of only minutes. In this study, we relate to protein abundance/stabilization and to changes in protein half-life that are determined by regulated degradation rather than by the biophysical structural properties of the protein. A central system that mediates ATP-dependent degradation of proteins is the ubiquitin-proteasome system (UPS; [4,5,6]). The degradation rate of a given protein is not, however, fixed; it is physiologically dynamic and dramatically affected by post-translational modifications such as phosphorylation, hydroxylation, acetylation, SUMOylation, and methylation, that may directly effects degrons [7]. For example, upon immune challenge, signal-induced phosphorylation of the inhibitory protein, IκBα, converts the extremely stable protein into a rapidly degraded phospho-protein, enabling immune response [8,9].
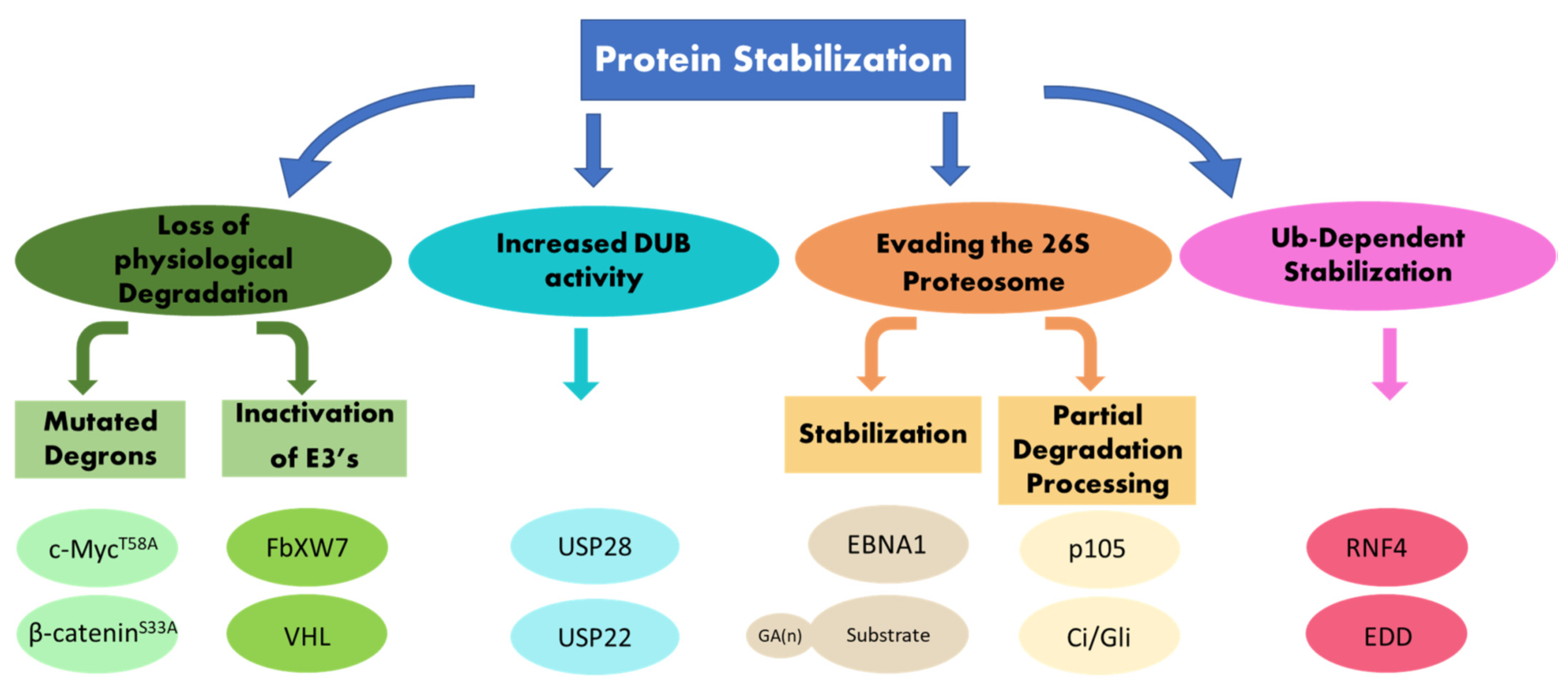
Protein stability depends on intrinsic structures within the targeted proteins, called degrons, as well as on enzymes that mediate the ubiquitination of the protein substrate. A recent systemic analysis of primary tumors established that stabilizing mutations in degrons and inactivating mutations in ubiquitin ligases (E3), which mediate the recognition of such degrons, are driving events in tumorigenesis [10]. Active stabilization, or limited proteolysis, also depends on the type of ubiquitin chains catalyzed on the protein substrates. In addition, a ubiquitin-dependent protein stabilization pathway actively stabilizes proteins in cancer. This pathway is situated upstream to and independent of the machinery involved in their degradation. A central enzyme in this pathway is the ubiquitin ligase RNF4 [11]. In this review, we address several aspects of protein stabilization. First, we discuss cases of protein stabilization that result from mutations in degrons or inactivation of the enzymes that mediate the degradation of these proteins. Second, we focus on cases of protein stabilization that stem from active ubiquitin-dependent protein stabilization, which is highly relevant to cancer (Figure 1).
2. Evading Recognition I: Protein Stabilization Due to Lacking or Mutated Degradation Signals
Degrons are short amino-acid sequences that determine the stability of proteins. They are divided according to their position on the protein substrate to N-terminal, C-terminal, and internal degrons. The biochemical nature of the N- and C-terminal degrons and the enzymatic pathways dedicated to the recognition thereof are discussed in detail in a recent review by Varshavsky [12], who also discovered the contribution of N-terminal degrons to protein stability in 1986 [13].
Internal degrons serve as recognition motifs for the diverse group of E3 ubiquitin ligases [14]. Internal degrons were initially thought to mediate only the docking and physical interaction between the substrate and the E3 ubiquitin ligases that catalyze their ubiquitination and subsequent degradation. However, it is now clear that these motifs also contain sequences that enable stabilizing signals [15,16,17]. Among the first degrons to be identified was the Delta motif, a stretch of 27 amino acids that is present in the c-Jun proto-oncogene and is required for its ubiquitin-dependent degradation. This motif is not present in v-Jun, the oncogenic viral protein, or in its closely related JunD, which exhibits a longer half-life [18,19].
Another example of a short recognition motif is a motif present in P53 tumor suppressor, which binds to a cleft on the surface of the oncogenic RING ubiquitin ligases HDM2 and MDM2 [20]. Targeting this interaction surface and blocking it with a group of small molecules stabilizes P53. These small molecules are currently being tested in clinical trials in tumors harboring wild-type P53 [21,22]. Moreover, MDM2 itself is stabilized by Akt-phosphorylation [23], as well as by the HECT-ubiquitin ligases, WWP1 and NEDD-4 (see review by Chenghua Li in this volume and [24,25]).
A subgroup of internal degrons is recognized by Cullin-based RING ubiquitin ligases and includes Skp-Cullin-F-box ubiquitin ligase complexes (SCF), where the F-box protein serves as a receptor subunit that binds to a phosphorylated degron via its C-terminal WD40 domain. One example of such a degron is that recognized by the F-box protein β-TRCP, DpSGXX (X) pS (where X is any amino acid). Among the substrates that contain this degron are IκBα and β-catenin [26,27,28]. To date, more than 200 substrates containing this degron have been identified [29].
In cancer, oncoprotein stabilization is the direct result of mutations within degrons. Specifically, mutations in the phosphorylated residues abolish the binding of F-box proteins that recognize phosphorylated degrons. It was observed early on that colon cancer and melanoma genomes are characterized by mutations, substituting Ser33 and Ser37 with Ala within the β-catenin degron, resulting in a stable and active nuclear co-activator protein [30,31]. Later, it was shown that the binding and ubiquitination of β-catenin by the E3 ligase β-TRCP requires phosphorylation of the β-catenin degron at these sites by GSK-β [32,33]. Remarkably, the ubiquitin ligase EDD recognizes GSK-β-phosphorylated (Ser33) β-catenin, resulting in its stabilization, nuclear accumulation, and transcriptional hyper-activation [34].
The F-Box protein FbXW7, likewise, recognizes a short phospho-degron that is present in key G1/S regulators such as Myc proteins, CycE, c-Jun, Notch-intracellular domain (N-ICD) proteins, mTor, PGC-1α, and the simian virus-encoded oncoprotein Large-T antigen [35,36,37]. In Burkitt’s lymphoma, the oncogenic mutation Thr58Ala located in the c-Myc main degron abolishes its binding to FbXW7, stabilizes c-Myc, and is a hotspot for mutations [38,39,40]. A detailed analysis of degrons in cancers is discussed in [7] and the reader is referred to a detailed description of degrons therein (Table 1 in [7]). Beyond specific examples, leveraging machine learning and using cancer datasets, Takheim et al. found that mutations affecting degrons play an important role in driving tumorigenesis [41]. The authors developed “deepDegron”, a machine learning method that predicts the impact of mutations or degron loss on protein stability, and validated these predictions for mutations in the degrons of GATA3 and PPM1D that result in stable proteins. Interestingly, in many cases, degrons are localized to intrinsically disordered regions (IDRs), and recent studies suggest that recognition, ubiquitination, and degradation take place in a unique physical environment involving phase separation [42,43].
Transient protein stabilization may also be due to sequestration or inactivation of the kinase(s) phosphorylating the degron, a phosphorylation that is essential for E3 ligase recognition. During activation of Wnt/LRP6 signaling, the kinase GSK-β which phosphorylates β-catenin, and FbXW7 substrates, such as c-Myc, are sequestered, resulting in the stabilization of β-catenin, c-Myc, and other critical tumor-promoting regulators [44].
Thus, oncoprotein stabilization may stem from evolutionary forces and driving mutations within oncoprotein degrons, generating more stable and active proteins that evade recognition by the ubiquitination machinery that promotes tumorigenesis. It should be noted, however, that the real-life (“outside-the-textbook”) biology of degrons is more complex; the true degradation rate of a given protein is impacted by other weak degrons in the same protein, as well as additional factors such as the capacity of F-Box proteins to dimerize [45].
3. Evading Recognition II: Protein Stabilization Due to Impaired E3 Ubiquitin Ligase Activity, and the Emergences of Degradation-Resistant Tumors
Protein stabilization also results from genetic, epigenetic, or post-translational inactivation of the ubiquitin E3 ligases that mediate the ubiquitination and subsequent proteasomal degradation of short-lived oncoproteins. E3s ligases are, therefore, the enzymes that contribute most to the specificity of the ubiquitin system. The human genome encodes ~ 800 ubiquitin ligases that are divided into subfamilies based on central domains such as HECT, RING, RBR, and Ubox [14,46]. For example, enzymes such as Mdm2 or E6-AP, which ubiquitinate the tumor suppressor proteins P53, are oncogenic [47,48,49,50]. In contrast, ligases such as FbXW7 or β-TRCP, whose activity leads to the degradation of oncoproteins, are bona fide tumor suppressors. Furthermore, ubiquitination is linked to other protein destruction pathways such as lysosomal degradation of cell surface receptors. For example, inactivation of ubiquitin ligase Cbl results in stabilization of growth factors that drive receptors such as EGFR by increasing their half-life on the cell surface, resulting in sustained oncogenic signaling [51]. The reader is referred to excellent recent reviews of the biology of ubiquitin ligases in cancer [46,52]. Figure 2 in [46] presents an elegant scheme of ligases that are inactivated in cancer, as well as their substrates and relationship to the hallmark cancer.
One consequence of E3 inactivation is the development of degradation-resistant tumor cells that accumulate multiple, stabilized, and active oncoproteins. Loss of FbXW7, the receptor subunit within SCFFbXW7, results in stabilization of multiple oncoproteins that all drive G1/S progression (e.g., CycE, c-Myc, NICD, c-Jun). The collective stabilization of multiple oncogenic drivers is a frequent phenomenon that is also observed for other ligases; thus, inactivation of Cbl, results in stabilization of multiple cell surface receptors [53]. Another case is the stabilization of substrates of the VHL ligase complex beyond its classical substrate, the hydroxylated hypoxia-induced factor HIF [54]. Thus, the development of tumors due to loss of E3 ligase activity gives the transformed cell a dramatic evolutionary advantage over the surrounding non-transformed cells. It is a challenging entity, both clinically and for pharmacological targeting, unlike the case of cancer that arises from the stabilization of a single oncoprotein.
4. Evading the Proteasome: Stabilization and Limited Proteolysis Signals
Ubiquitinated proteins are recognized, unfolded, and degraded into short peptides by the 26S proteasome, a large ATP-dependent multi-subunit protease complex [55]. Nevertheless, short amino acid sequences or domains within proteins completely inhibit proteasomal degradation or serve as ”stop” signals, enforcing limited proteolysis/processing. One such inhibitory sequence is the Gly-Ala repeat (GAr), which is present in the Epstein--Barr virus (EBV) protein EBNA-1 as a 398-residue-long internal stretch [56]. EBNA-1 is expressed during latent EBV infection and is commonly detected in all chronic EBV carriers. It is the only viral protein regularly detected in tumors associated with EBV. EBNA1 does not stimulate a cytotoxic T-cell response as EBNA1-derived peptides are not generated nor presented by MHC-I antigen-presenting cells, a process that is mediated by the proteasome [57]. Wild-type EBNA1 is stable, and deletion mutants of EBNA-1 lacking the GAr degrade rapidly and are presented on the surface of MHC-Class I molecules, inducing T-cell activity. Moreover, addition of the GAr to the well-degraded and presented EBNA-4 results in a stable protein that is not presented by MHC Class I molecules [58]. Remarkably, the GAr is, in some cases, a transferable “stop” signal; it can stabilize and protect P53 from Mdm2- or E6AP-mediated degradation as well as chimeric proteins in yeast [59,60]. The exact mode of action of the GAr is unclear, and even short GA repeats are sufficient to halt degradation. Nevertheless, the presence of a strong degron was able to overcome even a long GAr (>200 a.a.) [61]. While these repeats do not interfere with the ubiquitination of the substrate, they likely reduce the residence time of the substrate on the proteasome and may impair substrate unfolding [62].
GArs serve not only as protein stabilizers but also as internal barriers leading to limited proteolysis/processing and protein aggregation. A well-studied case is the generation of the NF-κB subunit p50 by proteolytic processing from its NFκB1 p105 precursor protein. A 30 a.a.-long glycine-rich region (GRR) is required for the limited processing of p105 and deletion of the GRR results in complete degradation of the p105 precursor. In this case, a short GAGAGA sequence is sufficient for generating the p50 subunit, but with lower yield, likely inducing the full degradation of the precursor. The GRR also confers stability to the newly formed p50 subunits [63,64]. A GRR is also present and required for the increased stability and aggregation of the 43 kDa TAR DNA-binding protein (TDP-43), and the short peptide related to the GRR is capable of forming aggregate-prone fibrils [65,66].
Another case of ubiquitin-dependent processing is the generation of the transcriptional repressors Cubitus interruptus (Ci)/Gli proteins that function downstream hedgehog signaling. The Ci75 repressor protein is generated by processing its precursor Ci155 [67], but the structural “stop” signal limiting Ci proteolysis is not a GAr/GRR but rather part of a zinc-finger domain [68]. In the case of Ci, β-TRCP is the ubiquitin ligase involved in processing, while ubiquitination by HIB, a BTB E3 ligase, leads to the complete degradation of Ci155 [69,70].
Interestingly, in the case of p105, limited proteolysis is mediated by the E3 KPC, while the ubiquitin ligase β-TRCP mediates the complete degradation of p105 [71,72,73,74]. In both cases, it is still not clear mechanistically how ubiquitination by distinct ligases has differential effects on the same protein. It is possible that in addition to the structural barrier, the type of ubiquitin chains generated by the E2/E3, potentially together with ancillary factors at the proteasome level, may determine the exact outcome: complete destruction or limited proteolysis.
Another domain that confers protein stabilization in the vicinity of the proteasome is the ubiquitin-associated 2 domain (UBA2) of Rad23, which is a bona fide LLP. Rad23 serves as a ubiquitin receptor that delivers ubiquitinated proteins to the proteasome and the UBA2 domain protects Rad23 from proteasomal degradation [75,76]. It is possible that UBA2 functions as a tightly folded domain that prevents the unfolding of Rad23 by the proteasomal ATPases, protecting Rad23 from degradation.
Increased stabilization may also stem from reduced proteasomal degradation. During aging, a unique frame shift mechanism generates a mutant ubiquitin molecule termed UBB (+1). UBB (+1) caps existing substrate-free ubiquitin chains and is a natural inhibitor of the 26S proteasome. Thus, by inhibiting the proteasome, endogenous substrates exhibit increased stabilization. Ubb (+1) chains may also interfere with the activity of protein kinases that are regulated by ubiquitination. Ubb (+1) was shown to be intimately related to the development of Alzheimer’s disease, where it is linked to protein aggregation [77,78,79,80,81].
While the aberrant activity of the proteasome affecting proteostasis is discussed in depth by Staller et al. in this volume, it should be noted that protein stabilization may be due to mutations in genes coding to proteasome subunits. Collectively, these mutations are the molecular basis for proteasome-associated auto-inflammatory syndromes (PRAAS). One example of such syndrome, which results from a mutation in the proteasome β subunit type 8, is CANDLE syndrome (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature) [82,83]. Moreover, reduced global protein clearance due to impaired proteasome activity is observed upon aging and results in the global accumulation of damaged proteins [84,85,86].
5. Ubiquitin-Dependent Oncoprotein Stabilization
Over the lifetime of a cell, the stability of a given protein is actively and transiently increased by post-translational modifications, of which phosphorylation is one of the more prominent. Stabilizing phosphorylations convey signals from both the extracellular and intracellular environments, granting a given protein or group of proteins enhanced activity for a limited time. P53 is stabilized and its transcriptional activity is enhanced by cellular stress signals such as UV or ionizing radiation. Such stress signals lead to P53 phosphorylation and stabilization by diverse kinases such as ATM (mutated in ataxia-telangiectasia), ATR (A-T and Rad3-related), the checkpoint kinases (Chk1 and Chk2), as well as Jun NH2-terminal kinase (JNK), p38, and others. Among the most phosphorylated sites that lead to stabilization of human P53 is Ser 15 [87,88,89,90,91,92].
In the case of multiple oncoproteins, a priming phosphorylation, which is mediated by growth-promoting signaling and mitogenic kinases such as Akt and Ras/MAPK, first stabilizes and enhances their activity. These priming phosphorylations subsequently enable the recruitment of glycogen synthase kinase-3β (GSK3-β) that catalyzes the second destabilizing phosphorylation. One well-studied case is the phosphorylation of Ser62 of c-Myc by the Ras/MAPK pathway that primes for the secondary destabilizing phosphorylation by GSK3β at Thr58, enabling the binding of the E3 ligase receptor subunit FbXW7. Phosphorylation of Ser45 of β-catenin by casein-kinase-I (CKI) is likewise stabilizing [93]. Similar stabilizing and degradation-priming phosphorylations have been observed in all FbXW7 substrates including CycE, c-Jun, Notch intracellular domain (NICD), and PGC1α [40,94,95,96].
How is the transient phosphorylation translated into protein stabilization? Recent studies from our laboratory suggest that once such oncoproteins are initially phosphorylated, they are recognized by a ubiquitin-dependent pathway that catalyzes a-typical polyubiquitin chains leading to their stabilization (see below). The ubiquitin ligase RNF4 is the central E3 enzyme in this pathway [11,97,98]. RNF4 is a RING-type ubiquitin ligase that belongs to a small group of SUMO-targeted ubiquitin ligases (STUbL). RNF4 binds to SUMOylated proteins via its four SUMO-interacting motifs (SIMs) and targets them for ubiquitination via its RING domain, connecting the ubiquitin with the SUMO pathways [99,100,101]. STUbLs are conserved from yeast to humans. The Drosophila genome codes for a single STUbL gene, degringolade (dgrn) [102,103], while the human genome encodes for two STUbL proteins, RNF4 and RNF111.
RNF4 and Dgrn are present in both the nucleus and the cytoplasm. Remarkably, during early Drosophila embryogenesis, Dgrn localization dramatically alternates between the cytoplasm to the nucleus in an embryonic cycle-regulated manner [103]. Likewise, RNF4 is localized to the cytoplasm, along the secretory pathway and the nucleus. Indeed, RNF4 substrates include the predominantly cytoplasmic translation initiation factor eIF2α, the transmembrane protein CFTR, and nuclear oncoproteins.
RNF4 and Dgrn, but not RNF111, harbor an arginine-rich region (ARM) that is the docking site for the phosphorylated oncoproteins. STUbL also contains a nucleosome-targeting motif that is required for binding to chromatin [100,104]. RNF4 regulates diverse cellular processes and serves as a transcriptional co-activator, a function that may involve CpG demetylation [105,106,107,108]. RNF4 is also required for the DNA damage response and nuclear protein quality control, both of which are topics that are beyond the scope of this review (for example see [109,110,111]; and reviewed in [112,113]).
In cancer, RNF4 has both tumor-suppressive as well as tumor-promoting activities. The cancer suppressive activities of RNF4 are SUMOylation dependent, while its cancer- promoting activities involving protein stabilization are independent of de-novo SUMOylation. Stabilization takes place in cells in which the SUMO E1 subunits SAE2 was inactivated, as was well observed upon expression of RNF4 that lacks the SIM domains.
Table 1 presents examples of SUMO-dependent and independent substrates of RNF4. A tumor-suppressive role for RNF4 was discovered in acute promyelocytic leukemia (APL), in which the oncogenic driver is a fusion protein that combines the promyelocytic leukemia protein (PML) with RARα (PML-RARα). Arsenic or retinoic acid treatment induces the SUMOylation of PML-RARα. The SUMOylated oncoprotein is bound and ubiquitinated by RNF4 and is subsequently degraded, resulting in differentiation of the tumor cells [114,115]. Remarkably, a short course of treatment with arsenic forms the basis of the current therapy and cure for APL [116].
In epithelial cancers, melanoma, and osteosarcoma, RNF4 has a pro-tumorigenic function, which is directly linked to its protein-stabilizing activity [97,98]. RNF4 binds via its ARM domain to selected phospho-oncoproteins that all share a stabilizing phosphorylation mediated by mitogenic/stress kinases. Examples are the binding of RNF4 to c-Myc upon phosphorylation of Ser62 or to β-catenin upon phosphorylation of Ser45.
Importantly, RNF4 acts upstream to the secondary destabilizing phosphorylations and the ubiquitin enzymes involved in the physiological degradation of these oncogenic substrates (e.g., E2 and E3). For example, it stabilizes cancer-mutated MycT58A and β-cateininS33A and its stabilizing activity is, accordingly, independent of FbXW7 and the E2s involved in c-Myc degradation.
6. Heterotypic Ubiquitin Chains Mediate Protein Stabilization
The exact molecular details involved in RNF4-dependent protein stabilization are still not fully clear. Nevertheless, molecular analysis suggests that it requires the catalysis of unique polyubiquitin chains on RNF4-stabilized substrates. Ubiquitin is a 76 amino-acid long protein that contains seven internal lysine residues (K6, K11, K27, K29, K33, K48, K63). These lysines enable the covalent linking of additional ubiquitin molecules to yield polyubiquitin chains [117,118,119]. Linkage-specific ubiquitin chains have distinct physiological outcomes; polyubiquitin chains in which all ubiquitin molecules are connected through the same internal lysine are called homotypic chains. K48-linked homotypic ubiquitin chains are involved in targeting proteins for proteasomal degradation, while K63-chains are involved in immune signaling cascades and serve as an inoculation/docking platform [120,121]. Polyubiquitin chains in which the expanding ubiquitin polymer is linked internally through diverse linkages are referred to as heterotypic or mixed chains. While the full physiological function of heterotypic ubiquitin chains (UbHet) is only starting to unfold, several reports indicate that UbHet chains promote protein stabilization [122].
In the case of RNF4, the catalysis of poly-UbHet chains by RNF4 is critical for substrate stabilization. These chains are characterized by K11 and K33 internal linkages on these substrates and enhance their stability. Interestingly, it was shown that in the case of c-Myc, phosphorylation of Ser62 is required not only for RNF4-dependent stabilization but also for its association with lamins, an association that may be linked to the formation of Ubhet chains and to stabilization [16].
The formation of poly-UbHet chains is a shared property of other stabilized proteins. As mentioned above, EDD-dependent stabilization of β-catenin requires the catalysis of K11- and K29-internally linked UbHet chains [34]. Another example is the case of UbHet chains that are catalyzed by the virus-encoded E3 ligase mLena, which stabilizes c-Myc independently of the phosphorylations that are associated with its turnover [123]. Self-ubiquitination of UbHet chains is likewise catalyzed by RING1B, protecting them from degradation, a self-ubiquitination that is required for H2B ubiquitination and gene activation [124]. UbHet chains are also catalyzed by the E3 TRAF6, stabilizing the mutant DJ-1 and α-synuclein proteins, which are localized to Lewy bodies in sporadic Parkinson’s disease brains [125]. UbHet chains were also shown to regulate cell cycle progression, in part by stabilizing p27Kip1/CDKN1B, a ubiquitination that is mediated by UBCH7/UBE2L3 enzymes [126].
While our focus in this review is on stabilizing E3 ubiquitin ligases, it is likely that the identity of the ubiquitin-conjugating enzyme (E2, UbC), which pairs with the stabilizing E3, also determines the nature of the internal linkage and the type of chain formed, in turn, affecting protein stability. Moreover, it is not yet clear whether UbHet chains confer stability passively, as they are poor substrates for the proteasome, or whether they are recognized by a yet-to-be-identified active stabilization machinery. Furthermore, while the activity of the ubiquitination machinery may be intact, increased stabilization of oncoproteins may result from increased protein deubiquitination by deubiquitinase (DUBs). The role of DUBs in protein stabilization, development, and in cancer, are the focus of reviews by Diefenbacher and Mohan in this volume.
7. Ubiquitin-Dependent Stabilization Is Evolutionarily Conserved
Much of what we have learned regarding RNF4 and ubiquitin-dependent protein stabilization was first observed in the fly. The Drosophila genome codes for a single Sumo-targeted ubiquitin ligase (STUbL) enzyme ortholog to RNF4 that we termed degringolade (dgrn CG10981). As with RNF4, Dgrn is required for Wnt signaling in the developing embryo and for Notch pathway activation in the fly wing [102,103]. Likewise, in the adult Drosophila, Dgrn is essential for Notch pathway activation during gut regeneration upon infection, similar to its enhancement of N-ICD stability and Notch pathway activation in mouse cells [97,127]. Moreover, dgrn null and heterozygous mutants are immunocompromised, as Dgrn is required for the transcriptional activation of anti-microbial peptide genes downstream of the NFκB pathway, by enhancing the stability of Dif, a central immunity transcription factor [127]. Moreover, expression of Dgrn in the ovary restored fertility to dMycdm1 hypomorphic sterile females, likely by increasing the reduced dMyc protein levels in the hypomorphic mutant [Orian, unpublished]. Collectively, these data suggest that Dgrn has similar effects on protein stabilization and oncogenic pathways as RNF4, opening the door to use Dgrn as a handle, together with the power of Drosophila genetics, to identify novel genes within the ubiquitin-dependent stabilization pathway.
8. The Tumor-Promoting Activity of RNF4
In cancer, the stabilizing effect of RNF4 results in increased transcriptional activity of oncogenic transcription factors and enhances tumorigenic properties of cancer cells both in culture and in vivo. RNF4 is required for, and enhances, the transcriptional activity of Myc and β-catenin, and promotes Wnt target gene expression as well as stabilization of Notch intracellular domain protein (NICD) and Delta-dependent Notch activation [97,98].
In melanoma, the stabilizing activity of RNF4 is not limited to oncogenic transcription factors but also requires the translation initiation factor eIF2α. In non-transformed cells, phosphorylation of eIF2α on Ser51 inhibits the formation of the initiation complex and attenuates general translation. Remarkably, in cancer cells, p-eIF2α promotes oncogenic translation; phosphorylation of eIF2α in cancer cells “hijacks” the translation machinery and results in enhanced activation of tumor-promoting translation via the use of non-conventional internal ribosome entry site (IRES)-containing oncogenic mRNAs and involves unique translation machinery including proteins such as eIF5B, eIF2D, and MCT-1 that together activate tumor promoting pathways [128,129,130].
eIF2α was identified as an upstream positive regulator of RNF4-dependent gene signature (but not of the RNF4 gene itself) in human melanoma xenograft tumors conditionally expressing RNF4. In melanoma patient-derived biopsies, the levels of p-eIF2α correlate with high levels of RNF4 protein. Indeed, RNF4 binds and stabilizes p-eIF2α catalyzing Ubhet chains (K11, K33) similar to its stabilizing activity towards c-Myc and other oncoproteins. The phosphorylation of eIF2α is mandatory for binding, ubiquitination, and stabilization by RNF4. eIF2α is phosphorylated on Ser51 by four distinct kinases (Heme-regulated kinase HRI, PERK, GCN2, and PKR) [131], yet the identity of the kinase(s) that act(s) in the context of RNF4-dependent stabilization is the focus of current studies. Subsequently, stabilized p-eIF2α enhances the translation of mRNAs of oncogenes such as c-myc and vegf. However, and unlike in the classical stress response in non-transformed cells, RNF4 activity in cancer cells enhances oncogenic translation without inhibiting general translation [98].
A hallmark of BRAF-mutated melanoma is the rapid development of resistance to receptor kinase (RTK) inhibitors such as Vemurafenib and its analog PLX4032 in experimental models and patients. The expression of RNF4 and its stabilized proteins, including p-eIF2α, is higher in Vemurafenib-resistant cell lines. Along this line, we found that eIF2α is critical for the resistance to therapy induced by RNF4 in melanoma. For example, expression of RNF4 confers resistance to PLX4032 in human melanoma cells, and its conditional elimination from PLX4032-resistant tumors in vivo resulted in collapse of the tumors. The inability of RNF4-deficient PLX4032-resistant melanoma cells to form colonies was restored upon co-expression of eIF2α a function that is dependent on eIF2α phosphorylation. Thus, establishing the critical role of this phosphorylation to and RNF4 tumorigenesis in culture [98].
9. RNF4 Is Associated with Resistance to Therapy and Poor Prognosis in Human Cancer
High levels of RNF4 are observed in about 40% of melanoma patients and in 30% of colon cancer biopsies. Analysis of colon cancer biopsies suggests that an elevated level of RNF4 protein is observed specifically at the transition from adenoma to carcinoma. In melanoma, high levels of RNF4 are correlated with high levels of p-eIF2α and are in correlation with unresponsiveness to RTK therapy. In both melanoma and luminal type-A breast cancer, high levels of RNF4 are associated with poorer prognosis [97,98]. The importance of RNF4 in cancer is a new concept and the mechanisms by which RNF4 confers resistance to molecular therapy are still unclear. It is also unknown whether these resistance mechanisms are general, are relevant also to immune checkpoint inhibitors therapy, or are specific to RTK therapy. Additonally, the full spectrum of tumors that collapse upon loss of RNF4 is still unknown. For example, high levels of RNF4 correlate with longer survival times in lung cancer but are associated with poor prognosis in gastric cancer [132].
10. Implications of RNF4 and Ubiquitin-Dependent Protein Stabilization in Cancer
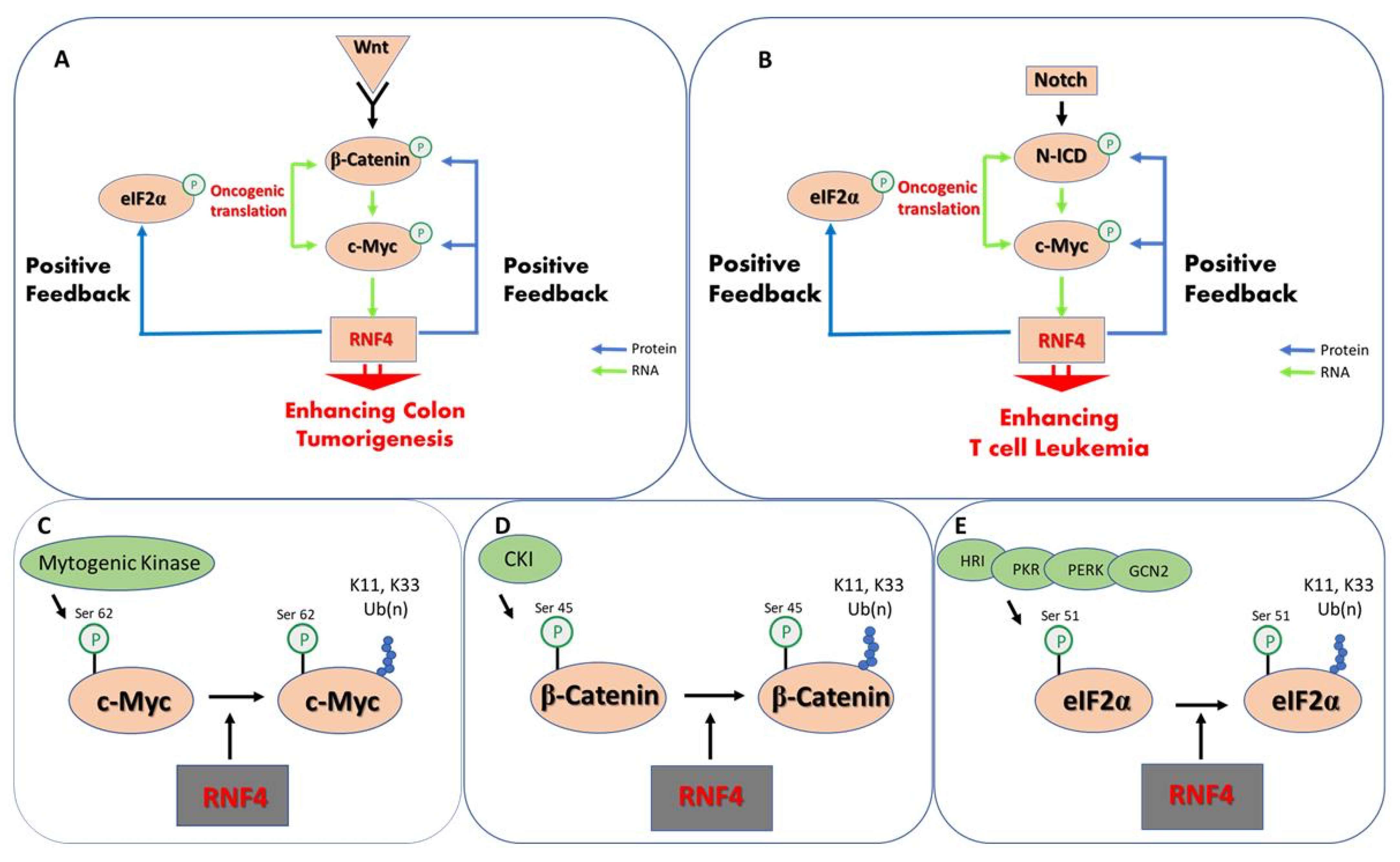
Ubiquitin-dependent protein stabilization emerges as a positive feed-forward loop that enhances the activity of key oncoproteins and promotes oncogenic translation (Figure 2). Several of the oncogenic signaling pathways, such as Notch and Wnt, activate the transcription of c-Myc that binds to the rnf4 gene and induces its expression [133,134]. Indeed, RNF4 expression is elevated in experimental models of Myc-driven tumors [135]. In turn, and once expressed, RNF4 stabilizes and enhances the activity of c-Myc, β-catenin, and N-ICD, enhancing the expression of pathway target genes [97]. In parallel, RNF4 enhances oncogenic translation by stabilizing p-eIF2α, including the translation of c-Myc and angiogenic factors such as VEGF [128,129].
Taken together, the accumulating data suggest that the activity of RNF4 and the ubiquitin-dependent protein stabilization pathway constitute a fundamental basis for tumorigenesis, are essential for cancer cell survival, and act as an Achilles’ heel for cancer cells. Inhibition of enzymes within this pathway will, therefore, will result in the eradication of degradation-resistant tumors, which will be highly relevant to a large yet specific group of cancer patients.

{kind=link}
{kind=link}
Table 1.
Examples of RNF4 substrates. Upper table: Substrates that are recognized once SUMOylated.
| SUMO-Dependent Substrates of RNF4 | |
| Protein | References |
| PML | [114,115,116,136] |
| JARID1B | [137] |
| SP100 * | [138] |
| IFI16 | [139] |
| CFTR | [140] |
| Pea3 | [141] |
| BLM | [109] |
| FANCI, FANCD2 (ID complex) | [142] |
| PIAS1, PIAS2, PIAS3 | [143] |
| ZNF451 | [143] |
| NSMCE2 | [143] |
| Ubc9 | [143] |
| BRCA1–BARD1 complex | [143] |
| MDC1 | [144] |
| PARIS/ZNF746 | [145] |
| KAP1 | [104] |
| Rta | [146] |
| NDRG2 | [147] |
| Tax | [148] |
| Ataxin-7 | [149] |
| FXR | [150] |
| SUMO-independent substrates of RNF4 | |
| Protein | Reference |
| c-Myc | [97] |
| c-Jun | [97] |
| β-catenin | [97] |
| NICD | [97] |
| PGC1 α | [97] |
| eIF2α | [98] |
| HIF1α | [98] |
“*” indicates partial proof. Lower table: Substrates that are recognized by RNF4 independent of SUMOylation.
Funding
This research was funded by grants from the Israeli Science Foundation (Grant no. 318/20) and the Flinkman-Marandy Family cancer research grant to AO.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Susan Spira for the excellent linguistic editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schoenheimer, R. The Dynamic State of Body Constituents. Cancer Res. 1942, 2, 810. [Google Scholar] [CrossRef]
- Cambridge, S.B.; Gnad, F.; Nguyen, C.; Bermejo, J.L.; Krüger, M.; Mann, M. Systems-wide proteomic analysis in mammalian cells reveals conserved, functional protein turnover. J. Proteome Res. 2011, 10, 5275–5284. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; DiGiuseppe, J.A.; Bercovich, B.; Orian, A.; Richter, J.D.; Schwartz, A.L.; Brodeur, G.M. Degradation of nuclear oncoproteins by the ubiquitin system in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Akopian, D.; Rape, M. Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef]
- Mészáros, B.; Kumar, M.; Gibson, T.J.; Uyar, B.; Dosztányi, Z. Degrons in cancer. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent IκBα phosphorylation marks the NF-κb inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; López-Arribillaga, E.; Lopez-Bigas, N.; Gonzalez-Perez, A. Systematic analysis of alterations in the ubiquitin proteolysis system reveals its contribution to driver mutations in cancer. Nat. Cancer 2020, 1, 122–135. [Google Scholar] [CrossRef] [Green Version]
- Diefenbacher, M.; Orian, A. Stabilization of nuclear oncoproteins by RNF4 and the ubiquitin system in cancer. Mol. Cell. Oncol. 2017, 4, e1260671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Myant, K.; Qiao, X.; Halonen, T.; Come, C.; Laine, A.; Janghorban, M.; Partanen, J.I.; Cassidy, J.; Ogg, E.L.; Cammareri, P.; et al. Serine 62-phosphorylated MYC Associates with nuclear lamins and its regulation by CIP2A is essential for regenerative proliferation. Cell Rep. 2015, 12, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wong, C.C.; Gong, B.; Yu, J. Functional significance and therapeutic implication of ring-type E3 ligases in colorectal cancer. Oncogene 2018, 37, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treier, M.; Staszewski, L.M.; Bohmann, D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 1994, 78, 787–798. [Google Scholar] [CrossRef]
- Musti, A.M.; Treier, M.; Peverali, F.A.; Bohmann, D. Differential regulation of c-Jun and JunD by ubiquitin-dependent protein degradation. Biol. Chem. 1996, 377, 619–624. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Yang, Y.; Ludwig, R.L.; Jensen, J.P.; Pierre, S.A.; Medaglia, M.V.; Davydov, I.V.; Safiran, Y.J.; Oberoi, P.; Kenten, J.H.; Phillips, A.C.; et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005, 7, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.K.; Khatik, G.L.; Narang, R.; Monga, V.; Chopra, H.K. p53-Mdm2 Interaction Inhibitors as Novel Nongenotoxic Anticancer Agents. Curr. Cancer Drug Targets 2018, 18, 749–772. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tamaskovic, R.; Yang, Z.; Brazil, D.P.; Merlo, A.; Hess, D.; Hemmings, B.A. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J. Biol. Chem. 2004, 279, 35510–35517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007, 26, 1477–1483. [Google Scholar] [CrossRef]
- Xu, C.; Fan, C.D.; Wang, X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene 2015, 34, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the IκBα–ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [CrossRef]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Concordet, J.P.; Lassot, I.; Albert, I.; Del Los Santos, R.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Low, T.Y.; Peng, M.; Magliozzi, R.; Mohammed, S.; Guardavaccaro, D.; Heck, A.J.R. A systems-wide screen identifies substrates of the SCFβTrCP ubiquitin ligase. Sci. Signal. 2014, 7, rs8. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Rubinfeld, B.; Tice, D.A.; Polakis, P. Axin-dependent Phosphorylation of the Adenomatous Polyposis Coli Protein Mediated by Casein Kinase 1ε. J. Biol. Chem. 2001, 276, 39037–39045. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kato, Y.; Zhang, Z.; Do, V.M.; Yankner, B.A.; He, X. β-Trcp couples β-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc. Natl. Acad. Sci. USA 1999, 96, 6273–6278. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.-H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of β-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Hay-Koren, A.; Caspi, M.; Zilberberg, A.; Rosin-Arbesfeld, R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates β-catenin. Mol. Biol. Cell 2011, 22, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Hio, C.K.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [Green Version]
- Yumimoto, K.; Nakayama, K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin. Cancer Biol. 2020, 67, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Westermarck, J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle 2008, 7, 592–596. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Tokheim, C.; Wang, X.; Timms, R.T.; Zhang, B.; Mena, E.L.; Wang, B.; Chen, C.; Ge, J.; Chu, J.; Zhang, W.; et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol. Cell 2021, 81, 1292–1308. [Google Scholar] [CrossRef]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic Wnt Signaling Promotes Protein Stabilization and Regulates Cell Size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Fagman, J.B.; Chen, C.; Alberti, S.; Liu, B. Protein phase separation and its role in tumorigenesis. eLife 2020, 9, e60264. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Hajdu-soltész, B.; Zeke, A.; Dosztányi, Z. Mutations of intrinsically disordered protein regions can drive cancer but lack therapeutic strategies. Biomolecules 2021, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. Oncoprotein ubiquitylation: Dimers, degrons, and degradation. Cell Cycle 2014, 13, 1829–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Pagano, M. Ubiquitin ligases in cancer: Functions and clinical potentials. Cell Chem. Biol. 2021, 28, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Hao, Q.; Chen, Y.; Zhou, X. The Janus Face of p53-Targeting Ubiquitin Ligases. Cells 2020, 9, 1656. [Google Scholar] [CrossRef]
- Levkowitz, G.; Waterman, H.; Ettenberg, S.A.; Katz, M.; Tsygankov, A.Y.; Alroy, I.; Lavi, S.; Iwai, K.; Reiss, Y.; Ciechanover, A.; et al. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 1999, 4, 1029–1040. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.F.; Langdon, W.Y. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q. VHL and hypoxia signaling: Beyond HIF in cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999, 17, 739–779. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Dantuma, N.P.; Tessarz, P.; Jellne, M.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent proteolysis in Saccharomyces cerevisiae by a Gly-Ala repeat. FEBS Lett. 2003, 555, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Leonchiks, A.; Issaeva, N.; Sharipo, A.; Selivanova, G.; Masucci, M.G.; Dantuma, N.P. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA 2002, 99, 1532–1537. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Heessen, S.; Lindsten, K.; Jellne, M.; Masucci, M.G. Inhibition of proteasomal degradation by the Gly-Ala repeat of Epstein-Barr virus is influenced by the length of the repeat and the strength of the degradation signal. Proc. Natl. Acad. Sci. USA 2000, 97, 8381–8385. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, M.A.; Zich, J.; Takeuchi, J.; Zhang, M.; Govaerts, C.; Coffino, P. Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 2006, 25, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Ghosh, S. A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol. Cell. Biol. 1996, 16, 2248–2254. [Google Scholar] [CrossRef] [Green Version]
- Orian, A.; Schwartz, A.L.; Israël, A.; Whiteside, S.; Kahana, C.; Ciechanover, A. Structural Motifs Involved in Ubiquitin-Mediated Processing of the NF-κB Precursor p105: Roles of the Glycine-Rich Region and a Downstream Ubiquitination Domain. Mol. Cell. Biol. 1999, 19, 3664–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 Is Directed to Stress Granules by Sorbitol, a Novel Physiological Osmotic and Oxidative Stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimonaka, S.; Nonaka, T.; Suzuki, G.; Hisanaga, S.I.; Hasegawa, M. Templated aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J. Biol. Chem. 2016, 291, 8896–8907. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Struhl, G. Regulation of the Hedgehog and Wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature 1998, 391, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Price, M.A. A Unique Protection Signal in Cubitus interruptus Prevents Its Complete Proteasomal Degradation. Mol. Cell. Biol. 2008, 28, 5555–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, L.; Wang, B.; Ou, C.Y.; Chien, C.T.; Jiang, J. A Hedgehog-Induced BTB Protein Modulates Hedgehog Signaling by Degrading Ci/Gli Transcription Factor. Dev. Cell 2006, 10, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravtsova-Ivantsiv, Y.; Shomer, I.; Cohen-Kaplan, V.; Snijder, B.; Superti-Furga, G.; Gonen, H.; Sommer, T.; Ziv, T.; Admon, A.; Naroditsky, I.; et al. KPC1-mediated ubiquitination and proteasomal processing of nf-κb1 p105 to p50 restricts tumor growth. Cell 2015, 161, 333–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belich, M.P.; Salmerón, A.; Johnston, L.H.; Ley, S.C. TPL-2 kinase regulates the proteolysis of the NF-κB-inhibitory protein NF-κB1 p105. Nature 1999, 397, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israël, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, Z.B.; Kofahl, B.; Beaudette, P.; Baum, K.; Ipenberg, I.; Weih, F.; Wolf, J.; Dittmar, G.; Scheidereit, C. Quantitative Dissection and Modeling of the NF-κB p100-p105 Module Reveals Interdependent Precursor Proteolysis. Cell Rep. 2014, 9, 1756–1769. [Google Scholar] [CrossRef] [Green Version]
- Heessen, S.; Masucci, M.G.; Dantuma, N.P. The UBA2 domain functions as an intrinsic stabilization signal that protects rad23 from proteasomal degradation. Mol. Cell 2005, 18, 225–235. [Google Scholar] [CrossRef]
- Heinen, C.; Ács, K.; Hoogstraten, D.; Dantuma, N.P. C-terminal UBA domains protect ubiquitin receptors by preventing initiation of protein degradation. Nat. Commun. 2011, 2, 191. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, F.W.; de Kleijn, D.P.V.; van den Hurk, H.H.; Neubauer, A.; Sonnemans, M.A.F.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.J.; Salehi, A.; Martens, G.J.M.; et al. Frameshift Mutants of β Amyloid Precursor Protein and Ubiquitin-B in Alzheimer’s and Down Patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef]
- Lindsten, K.; de Vrij, F.M.S.; Verhoef, L.G.G.C.; Fischer, D.F.; van Leeuwen, F.W.; Hol, E.M.; Masucci, M.G.; Dantuma, N.P. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002, 157, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leeuwen, F.W.; Hol, E.M.; Fischer, D.F. Frameshift proteins in Alzheimer’s disease and in other conformational disorders: Time for the ubiquitin-proteasome system. J. Alzheimers Dis. 2006, 9, 319–325. [Google Scholar] [CrossRef]
- Tank, E.M.H.; True, H.L. Disease-Associated Mutant Ubiquitin Causes Proteasomal Impairment and Enhances the Toxicity of Protein Aggregates. PLoS Genet. 2009, 5, e1000382. [Google Scholar] [CrossRef] [Green Version]
- Krutauz, D.; Reis, N.; Nakasone, M.A.; Siman, P.; Zhang, D.; Kirkpatrick, D.S.; Gygi, S.P.; Brik, A.; Fushman, D.; Glickman, M.H. Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 2014, 10, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ramot, Y.; Torrelo, A.; Paller, A.S.; Si, N.; Babay, S.; Kim, P.W.; Sheikh, A.; Lee, C.-C.R.; Chen, Y.; et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012, 64, 895–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, A.; Jacks, J.; Kessler, M.; Emanuel, P.D.; Gao, L. Proteasome-associated autoinflammatory syndromes: Advances in pathogeneses, clinical presentations, diagnosis, and management. Int. J. Dermatol. 2015, 54, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, B.; Haung Yu, W.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Publ. Gr. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [Green Version]
- Banin, S.; Moyal, L.; Shieh, S.Y.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Buschmann, T.; Potapova, O.; Bar-Shira, A.; Ivanov, V.N.; Fuchs, S.Y.; Henderson, S.; Fried, V.A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M.R.; et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 2001, 21, 2743–2754. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-mediated CKI phosphorylation of β-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.J.; Abed, M.; Heuberger, J.; Novak, R.; Zohar, Y.; Beltran Lopez, A.P.; Trausch-Azar, J.S.; Ilagan, M.X.G.; Benhamou, D.; Dittmar, G.; et al. RNF4-Dependent Oncogene Activation by Protein Stabilization. Cell Rep. 2016, 16, 3388–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitan-Hersh, E.; Feng, Y.; Oknin Vaisman, A.; Abu Ahmad, Y.; Zohar, Y.; Zhang, T.; Lee, J.S.; Lazar, I.; Sheikh Khalil, S.; Feiler, Y.; et al. Regulation of eIF2α by RNF4 Promotes Melanoma Tumorigenesis and Therapy Resistance. J. Investig. Dermatol. 2020, 140, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Abed, M.; Bitman-Lotan, E.; Orian, A. The biology of SUMO-targeted ubiquitin ligases in Drosophila development, immunity, and cancer. J. Dev. Biol. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Sabapathy, K. RNF4—A Paradigm for SUMOylation-Mediated Ubiquitination. Proteomics 2019, 19, 1900185. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Barry, K.C.; Kenyagin, D.; Koltun, B.; Phippen, T.M.; Delrow, J.J.; Parkhurst, S.M.; Orian, A. Degringolade, a SUMO-targeted ubiquitin ligase, inhibits Hairy/Groucho-mediated repression. EMBO J. 2011, 30, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, K.C.; Abed, M.; Kenyagin, D.; Werwie, T.R.; Boico, O.; Orian, A.; Parkhurst, S.M. The Drosophila STUbL protein Degringolade limits HES functions during embryogenesis. Development 2011, 138, 1759–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.Y.; Li, X.; Kong, X.Q.; Luo, C.; Chang, C.C.; Chung, Y.; Shih, H.M.; Li, K.K.; Ann, D.K. An arginine-rich motif of ring finger protein 4 (RNF4) oversees the recruitment and degradation of the phosphorylated and SUMOylated Krüppel-associated box domain-associated protein 1 (KAP1)/TRIM28 protein during genotoxic stress. J. Biol. Chem. 2014, 289, 20757–20772. [Google Scholar] [CrossRef] [Green Version]
- Moilanen, A.-M.; Poukka, H.; Karvonen, U.; Häkli, M.; Jänne, O.A.; Palvimo, J.J. Identification of a Novel RING Finger Protein as a Coregulator in Steroid Receptor-Mediated Gene Transcription. Mol. Cell. Biol. 1998, 18, 5128–5139. [Google Scholar] [CrossRef] [Green Version]
- Saville, B.; Poukka, H.; Wormke, M.; Jänne, O.; Palvimo, J.; Stoner, M.; Samudio, I.; Safe, S. Cooperative Coactivation of Estrogen Receptor α in ZR-75 Human Breast Cancer Cells by SNURF and TATA-binding Protein. J. Biol. Chem. 2002, 277, 2485–2497. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.V.; Rodrigues, T.M.A.; Tao, H.; Baker, R.K.; Miraglia, L.; Orth, A.P.; Lyons, G.E.; Schultz, P.G.; Wu, X. Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc. Natl. Acad. Sci. USA 2010, 107, 15087–15092. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y. RING finger protein 4 (RNF4) derepresses gene expression from DNA methylation. J. Biol. Chem. 2014, 289, 33808–33813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, C.M.; Berndsen, C.E.; Zhu, J.; Gupta, V.; Datta, A.; Greenberg, R.A.; Wolberger, C.; Matunis, M.J. RNF4-Dependent Hybrid SUMO-Ubiquitin Chains Are Signals for RAP80 and Thereby Mediate the Recruitment of BRCA1 to Sites of DNA Damage. Sci. Signal. 2012, 5, ra88. [Google Scholar] [CrossRef] [Green Version]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67. [Google Scholar] [CrossRef]
- Nagai, S.; Davoodi, N.; Gasser, S.M. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011, 21, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Oram, M.K.; Bielinsky, A.K. Sumo-targeted ubiquitin ligases and their functions in maintaining genome stability. Int. J. Mol. Sci. 2021, 22, 5391. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML–RARα through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Chen, Z.; De Thé, H. Curing APL through PML/RARA degradation by As2O3. Trends Mol. Med. 2012, 18, 36–42. [Google Scholar] [CrossRef]
- Behrends, C.; Harper, J.W. Constructing and decoding unconventional ubiquitin chains. Nat. Struct. Mol. Biol. 2011, 18, 520–528. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Karin, M. Emerging roles of Lys63-linked polyubiquitylation in immune responses. Immunol. Rev. 2015, 266, 161–174. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Rodrigues, L.; Popov, N.; Kaye, K.M.; Simas, J.P. Stabilization of Myc through Heterotypic Poly-Ubiquitination by mLANA Is Critical for γ-Herpesvirus Lymphoproliferation. PLoS Pathog. 2013, 9, e1003554. [Google Scholar] [CrossRef] [PubMed]
- Ben-Saadon, R.; Zaaroor, D.; Ziv, T.; Ciechanover, A. The Polycomb Protein Ring1B Generates Self Atypical Mixed Ubiquitin Chains Required for Its In Vitro Histone H2A Ligase Activity. Mol. Cell 2006, 24, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Codrich, M.; Marcuzzi, F.; Pinto, M.; Vilotti, S.; Biagioli, M.; Ferrer, I.; Gustincich, S. TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 2010, 19, 3759–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcomb, E.A.; Tsai, Y.C.; Basappa, J.; Liu, K.; Le Feuvre, A.K.; Weissman, A.M.; Taylor, A. Stabilization of p27Kip1/CDKN1B by UBCH7/UBE2L3 catalyzed ubiquitinylation: A new paradigm in cell-cycle control. FASEB J. 2019, 33, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Koltun, B.; Shackelford, E.; Bonnay, F.; Matt, N.; Reichhart, J.M.; Orian, A. The SUMO-targeted ubiquitin ligase, Dgrn, is essential for Drosophila innate immunity. Int. J. Dev. Biol. 2017, 61, 319–327. [Google Scholar] [CrossRef]
- Sendoel, A.; Dunn, J.G.; Rodriguez, E.H.; Naik, S.; Gomez, N.C.; Hurwitz, B.; Levorse, J.; Dill, B.D.; Schramek, D.; Molina, H.; et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef]
- Holcik, M. Could the eIF2α-independent translation be the achilles heel of cancer? Front. Oncol. 2015, 5, 264. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.L.; Kanellos, G.; Vlahov, N.; Alexandrou, C.; Willis, A.E.; Knight, J.R.P.; Sansom, O.J. Translation initiation in cancer at a glance. J. Cell Sci. 2021, 134, jcs248476. [Google Scholar] [CrossRef]
- Koromilas, A.E. Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Palomero, T.; Wei, K.L.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [Green Version]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; Da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Reymann, S.; Borlak, J. Transcription profiling of lung adenocarcinomas of c-myc-transgenic mice: Identification of the c-myc regulatory gene network. BMC Syst. Biol. 2008, 2, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koidl, S.; Eisenhardt, N.; Fatouros, C.; Droescher, M.; Chaugule, V.K.; Pichler, A. The SUMO2/3 specific E3 ligase ZNF451-1 regulates PML stability. Int. J. Biochem. Cell Biol. 2016, 79, 478–487. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Treffers, L.W.; Verlaan-de Vries, M.; Olsen, J.V.; Vertegaal, A.C.O. SUMO-2 Orchestrates Chromatin Modifiers in Response to DNA Damage. Cell Rep. 2015, 10, 1778–1791. [Google Scholar] [CrossRef] [Green Version]
- Maarifi, G.; Maroui, M.A.; Dutrieux, J.; Dianoux, L.; Nisole, S.; Chelbi-Alix, M.K. Small Ubiquitin-like Modifier Alters IFN Response. J. Immunol. 2015, 195, 2312–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Asmi, F.; McManus, F.P.; Brantis-de-Carvalho, C.E.; Valle-Casuso, J.C.; Thibault, P.; Chelbi-Alix, M.K. Cross-talk between SUMOylation and ISGylation in response to interferon. Cytokine 2020, 129, 155025. [Google Scholar] [CrossRef]
- Ahner, A.; Gong, X.; Schmidt, B.Z.; Peters, K.W.; Rabeh, W.M.; Thibodeau, P.H.; Lukacs, G.L.; Frizzell, R.A. Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 2013, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Sharrocks, A.D. Extracellular signal-regulated kinase mitogen-activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol. Cell. Biol. 2009, 29, 3204–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs-Seymour, I.; Oka, Y.; Rajendra, E.; Weinert, B.T.; Passmore, L.A.; Patel, K.J.; Olsen, J.V.; Choudhary, C.; Bekker-Jensen, S.; Mailand, N. Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Mol. Cell 2015, 57, 150–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; González-Prieto, R.; Xiao, Z.; Verlaan-De Vries, M.; Vertegaal, A.C.O. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 2017, 8, 1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012, 31, 3008–3019. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Yamada, Y. RNF4-mediated SUMO-targeted ubiquitination relieves PARIS/ZNF746-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 2020, 526, 110–116. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Yoshikai, Y.; Hsu, S.-W.; Saitoh, H.; Chang, L.-K. Role of RNF4 in the ubiquitination of Rta of Epstein-Barr virus. J. Biol. Chem. 2013, 288, 12866–12879. [Google Scholar] [CrossRef] [Green Version]
- Tantai, J.; Pan, X.; Hu, D. RNF4-mediated SUMOylation is essential for NDRG2 suppression of lung adenocarcinoma. Oncotarget 2016, 7, 26837–26843. [Google Scholar] [CrossRef] [Green Version]
- Fryrear, K.A.; Guo, X.; Kerscher, O.; Semmes, O.J. The Sumo-targeted ubiquitin ligase RNF4 regulates the localization and function of the HTLV-1 oncoprotein Tax. Blood 2012, 119, 1173–1181. [Google Scholar] [CrossRef]
- Marinello, M.; Werner, A.; Giannone, M.; Tahiri, K.; Alves, S.; Tesson, C.; den Dunnen, W.; Seeler, J.-S.; Brice, A.; Sittler, A. SUMOylation by SUMO2 is implicated in the degradation of misfolded ataxin-7 via RNF4 in SCA7 models. Dis. Model. Mech. 2019, 12, dmm036145. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, S.; Caron, V.; Gagnon, J.; Kuftedjian, A.; Tremblay, A. A CK2-RNF4 interplay coordinates non-canonical SUMOylation and degradation of nuclear receptor FXR. J. Mol. Cell Biol. 2017, 9, 195–208. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of protein stabilization. Schematic diagram for mechanisms resulting in protein stabilization that are discussed in this review, see text for details.
Figure 1.
Mechanisms of protein stabilization. Schematic diagram for mechanisms resulting in protein stabilization that are discussed in this review, see text for details.

Figure 2.
Protein stabilization is a feed-forward mechanism enhancing tumorigenesis. (A) In colon cancer activation of Wnt signaling results in the nuclear translocation of the β-catenin co-activator that together with TCF activates the transcription of c-Myc. c-Myc binds to the rnf4 gene and induces its expression. RNF4 protein in turn stabilizes both p(S45)-β-catenin and p(S62)-c-Myc and enhances their stability and transcriptional activity. In parallel, RNF4 stabilizes p(S51)eIF2α that is central for the activation of oncogenic translation including c-Myc and VEGF. (B) A similar mechanism operates in acute T-cell leukemia, where Notch pathway activation induces the nuclear accumulation of Notch intracellular domain protein (N-ICD) that binds to a c-Myc enhancer inducing c-Myc expression, that subsequently increases RNF4 expression. (C–E) Molecular details of kinases and site of phosphorylation of the indicated oncoproteins that prime for RNF4-dependent K11, K33 polyubiquitination and protein stabilization.
Figure 2.
Protein stabilization is a feed-forward mechanism enhancing tumorigenesis. (A) In colon cancer activation of Wnt signaling results in the nuclear translocation of the β-catenin co-activator that together with TCF activates the transcription of c-Myc. c-Myc binds to the rnf4 gene and induces its expression. RNF4 protein in turn stabilizes both p(S45)-β-catenin and p(S62)-c-Myc and enhances their stability and transcriptional activity. In parallel, RNF4 stabilizes p(S51)eIF2α that is central for the activation of oncogenic translation including c-Myc and VEGF. (B) A similar mechanism operates in acute T-cell leukemia, where Notch pathway activation induces the nuclear accumulation of Notch intracellular domain protein (N-ICD) that binds to a c-Myc enhancer inducing c-Myc expression, that subsequently increases RNF4 expression. (C–E) Molecular details of kinases and site of phosphorylation of the indicated oncoproteins that prime for RNF4-dependent K11, K33 polyubiquitination and protein stabilization.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Abu Ahmad, Y.; Oknin-Vaisman, A.; Bitman-Lotan, E.; Orian, A. From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells 2021, 10, 2374. https://doi.org/10.3390/cells10092374
AMA Style
Abu Ahmad Y, Oknin-Vaisman A, Bitman-Lotan E, Orian A. From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization. Cells. 2021; 10(9):2374. https://doi.org/10.3390/cells10092374
Chicago/Turabian StyleAbu Ahmad, Yamen, Avital Oknin-Vaisman, Eliya Bitman-Lotan, and Amir Orian. 2021. "From the Evasion of Degradation to Ubiquitin-Dependent Protein Stabilization" Cells 10, no. 9: 2374. https://doi.org/10.3390/cells10092374
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.