
Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine

1
Merck & Co., Inc., Kenilworth, NJ 07033, USA
2
Sanofi, 94400 Vitry-sur-Seine, France
3
Sanofi, 91380 Chilly-Mazarin, France
*
Authors to whom correspondence should be addressed.
†
These authors contribute equally to this work.
Cells 2021, 10(10), 2715; https://doi.org/10.3390/cells10102715
Submission received: 13 September 2021
/
Revised: 1 October 2021
/
Accepted: 1 October 2021
/
Published: 11 October 2021
(This article belongs to the Special Issue Hippo Pathway in Cancer: Toward Anticancer Drugs or Regenerative Medicines for Tissue Repair)
Abstract
:The Hippo pathway is an evolutionary conserved signaling network that regulates essential processes such as organ size, cell proliferation, migration, stemness and apoptosis. Alterations in this pathway are commonly found in solid tumors and can lead to hyperproliferation, resistance to chemotherapy, compensation for mKRAS and tumor immune evasion. As the terminal effectors of the Hippo pathway, the transcriptional coactivators YAP1/TAZ and the transcription factors TEAD1–4 present exciting opportunities to pharmacologically modulate the Hippo biology in cancer settings, inflammation and regenerative medicine. This review will provide an overview of the progress and current strategies to directly and indirectly target the YAP1/TAZ protein–protein interaction (PPI) with TEAD1–4 across multiple modalities, with focus on recent small molecules able to selectively bind to TEAD, block its autopalmitoylation and inhibit YAP1/TAZ–TEAD-dependent transcription in cancer.
1. Introduction
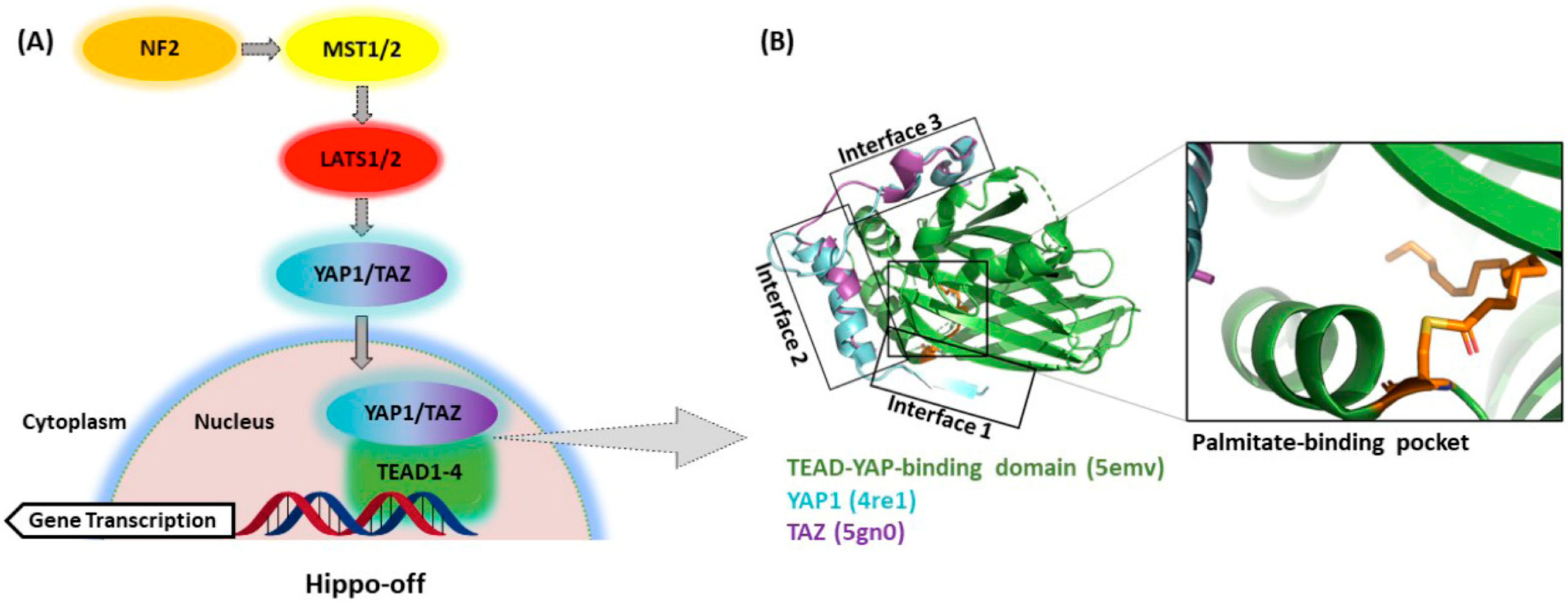
Originally described as an organ size and tissue growth control mechanism [1], the Hippo pathway has gained a tremendous amount of recognition in the fields of regenerative medicine and oncology in the past 16 years. Stunning organ growth phenotypes observed in the first mammalian liver models closely resembled those initially found in Drosophila studies, highlighting the conserved nature of this pathway, and its probable importance to human biology [2,3]. At a high level, Hippo functions as a tissue growth and cell proliferation inhibitory pathway. Extracellular stimuli activate the core kinase cascade that results in the phosphorylation of the transcriptional coactivators yes-associated protein 1 (YAP1) and transcriptional coactivator with PDZ-binding motif (TAZ, gene name WWTR1), resulting in their sequestration in the cytoplasm (Hippo-on state). When in the nucleus, YAP1 and TAZ interact with the TEAD transcription factors (TEAD1–4), the most well-known transcriptional mediators of the YAP1 and TAZ function (Hippo-off state) [4]. In the Hippo-off state (Figure 1A), the YAP1/TAZ–TEAD complex drives the induction of genes involved in proliferation and cell survival [5]. This interaction with TEADs is critical for the growth-promoting properties of YAP1 and TAZ as many of the effects of the YAP1 transcriptional activity can be blunted by eliminating the YAP1–TEAD interaction [6,7,8,9]. Many of the growth-promoting phenotypes observed after YAP1 activation eventually lead to tumor formation, and thus TEADs likely play an integral role in this process [3,8,10,11,12]. Whether the four TEAD protein family members, TEAD1–4, have the same or slightly different functions in this process is not yet understood and still a matter of research (reviewed in [13]. From the structure and sequence points of view, all TEAD proteins share the same domain structures and are highly homologous in their YAP1-binding domain [14].
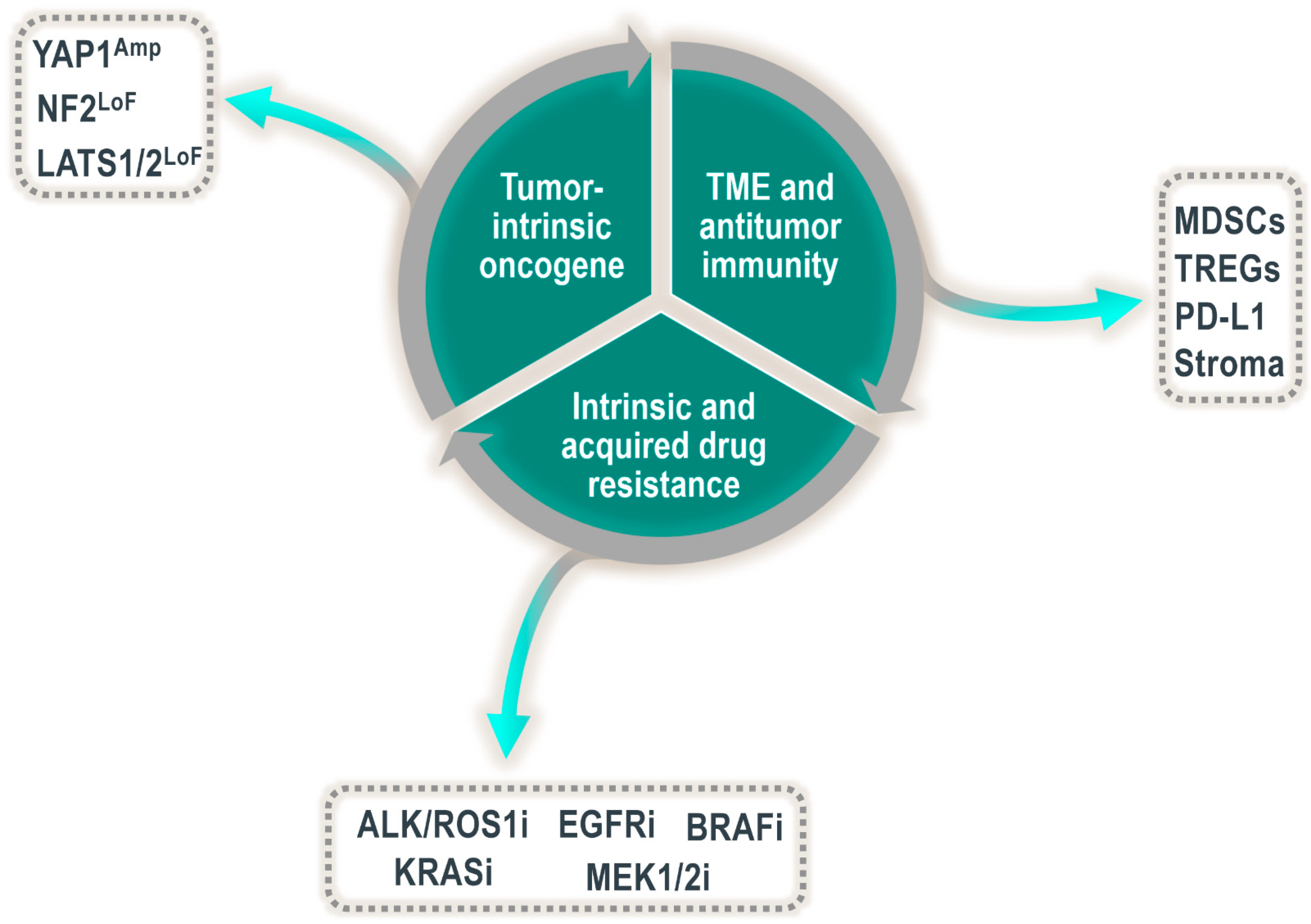
Based on the strong growth-promoting and tumor-initiating phenotypes in mice and Drosophila, it is unsurprising that the Hippo pathway would play an integral role in human neoplasms. Indeed, overexpression of, or increased levels of nuclear localized YAP1 have been found in numerous solid tumor indications. In addition to general overexpression or accumulation of YAP1 staining in tumor cells (often termed “YAP1 activation”), several members of the Hippo pathway are known to be altered in human cancer at the DNA level, including amplification of YAP1 and TAZ, as well as genomic deletions or truncating mutations in NF2, LATS1 and LATS2 [15,16]. Although much of the early research related to the oncogenic potential of YAP1 and TAZ centered on their role as cell-autonomous drivers, there is growing evidence that these transcriptional cofactors might play a much wider function than simply stimulating proliferation or inducing antiapoptotic factors. It is becoming clear that YAP1 and TAZ are multifaceted regulators of the processes driving tumor growth. Some of these attributes include, but are not limited to, (1) broadly regulating the tumor microenvironment (TME), including impacting antitumor immunity, (2) driving resistance to a wide array of drugs (including cytotoxic and targeted agents) and (3) acting as a tumor-intrinsic oncogenic driver [4,17,18]. Until very recently, it has been difficult to systematically probe the tumor growth requirements for the YAP1/TAZ–TEAD interaction on a broad scale. Additionally, many published studies looking at the role of YAP1/TAZ and TEAD in regulating tumor growth rely on analyzing tumor initiation. Whether YAP1/TAZ and TEADs are critical for the continued growth of already established tumors is still a largely unanswered question critical to successfully targeting YAP1/TAZ–TEAD in cancer.
Generally considered to be extremely difficult or impossible to drug by traditional small-molecule approaches, transcription factor/cofactor complexes are of critical importance to cellular differentiation, as well as diseases, including cancer, and thus represent highly attractive drug targets [19]. Although YAP1 and TAZ lack a defined druggable pocket, this is not the case for TEAD proteins, which harbor a recently uncovered hydrophobic pocket (Figure 1B) containing a cysteine residue conserved in each of the four TEAD family members residing in the YAP1-binding domain [20,21,22]. This conserved cysteine is the site of S-palmitoylation and is thought to modulate TEAD stability and, thus, transcriptional output [18]. This newly discovered foothold for medicinal chemistry efforts could pave the way to selective and on-target TEAD allosteric inhibitors that inhibit palmitoylation and thus disrupt YAP1/TAZ–TEAD transcriptional activity. In addition to the allosteric palmitate pocket, TEAD interfaces 2 and 3 have also been identified as druggable pockets within the YAP1-binding domain (YBD), albeit more amenable to the binding of larger molecules (e.g., peptides, peptidomimetics, etc.) able to directly disrupt the YAP1/TAZ–TEAD protein–protein interaction (PPI). Many questions surrounding the utility and feasibility of targeting this transcription factor complex still persist, however. In this review, we discuss some of the potential oncology settings where a drug-targeting YAP1/TAZ–TEAD would be of interest and what progress has been made towards the discovery of novel, selective and high-quality inhibitors of this critical transcription factor/cofactor complex. We highlight several molecules that have now either entered phase I clinical trials or are slated to do so in the near future.
2. Opportunities to Modulate the Hippo Pathway
2.1. YAP1 in Embryogenesis and Developed Tissues
In a healthy organism, YAP1 activation is apparent during embryonic development where cell proliferation and tissue growth are essential. Homozygous Yap1-knockout mouse embryos arrest development around embryonic day 8.5 and have defects in yolk sac vasculogenesis and embryonic axis elongation [23]. Similarly, conditional deletion of Yap1 in the embryonic heart or lung impair cardiomyocyte proliferation and the development of basal airway stem cells in the lungs, respectively [24]. Conversely, in developed adult tissues, YAP1 activity is kept at a low steady-state level through the continuous and active signaling control from the upstream Hippo pathway. Induction of YAP1 in adults is restricted to the temporary activation in response to injury, where YAP1 initiates short-term transcriptional programs required for wound healing and repair processes (reviewed in [25]). Thus, the conditional knockout of Yap1 in adult mouse intestines had no consequences for the normal intestinal function but did impair tissue repair post-dextran sodium sulfate (DSS) injury [26]. Indeed, the therapeutic activation of YAP1 has been considered for the regeneration of injured organs, a subject covered in more depth in other reviews [25,27,28].
2.2. YAP1 as a Tumor-Intrinsic Oncogenic Driver
In contrast to the temporary activation of YAP1 observed in wound healing, the continuous and persistent activation of YAP1 in adult tissues is associated with cancer and resistance to therapy (Figure 2). Many different cancer types harbor aberrant YAP1 activation, and examples range from tumors of the lungs, liver, skin and pancreas to tumors of the breasts, uterus, prostate, head and neck cancers and gliomas (reviewed in [17,29]). However, in which cases YAP1 is a functional driver in these malignancies still requires further investigation. In some of the abovementioned indications, the origin of the YAP1 activation can be directly traced to genetic alterations in the Hippo pathway or in YAP1 itself. For example, analysis of the TCGA database of patient tumors demonstrates that a subset of roughly ten percent of patients with cervical cancer shows focal amplification of YAP1 [30], which in turn is associated with an increase in YAP1 transcriptional activity. Similarly, a large set of more than sixty percent of patients with malignant pleural mesothelioma, an asbestos-induced tumor of the lungs, harbor genetic alterations in the Hippo signaling pathway or its direct upstream regulators. These alterations predominantly comprise deletions or loss-of-function mutations in LATS1 or NF2 and are, once again, linked to an increase in the transcriptional activity of YAP1 [30].
For malignant mesothelioma tumors, it has been shown that the aberrant activation of YAP1 is an actual driver of tumor maintenance. The inhibition of YAP1 in preclinical models of malignant mesothelioma with Hippo pathway alterations decreased YAP1-dependent gene transcription and inhibited tumor cell growth. This effect was initially demonstrated by two small-molecule inhibitors of TEAD palmitoylation, as well as by genetic studies. These observations were found both in vitro as well as in vivo [31,32,33]. Specifically, the inhibition of YAP1 activation and the subsequent reduction of already established tumors in vivo clearly demonstrate that YAP1 is a driver of tumor maintenance in malignant mesothelioma with the Hippo pathway or NF2 deletions. Furthermore, NF2 deletions are also found in other tumor types such as renal cell carcinoma, cervical squamous cell carcinoma, schwannomas and meningiomas, which may hence respond equally well to the inhibition of YAP1 activation [33,34].
2.3. YAP1 as a Mechanism of Intrinsic and Acquired Drug Resistance
Not all cancers with YAP1 activation are responsive to the inhibition of YAP1 alone. In many contexts, YAP1 is not the driver of tumor growth but rather an acquired mechanism that adds to a set of already present driver alterations and increases tumor aggressiveness or resistance to therapy (Figure 2). In lung cancer, for example, YAP1 activation confers resistance to therapies targeted against common lung cancer oncogenes, such as anaplastic lymphoma kinase (ALK), BRAF or mutated epidermal growth factor receptor (EGFR) tyrosine kinase. The mechanism of action by which YAP1 confers resistance to EGFR-targeted therapies is best understood and seems to be linked to the concept of persister cells, a small set of cells that resist treatment to therapy. In EGFR mutant non-small-cell lung cancer (NSCLC), it has been shown that the population of persister cells remaining viable after the combined treatment with the EGFR and MEK inhibitors osimertinib and trametinib, respectively, is in fact YAP1-activated and permits tumor outgrowth upon treatment stop. In turn, genetic downregulation of YAP1 resensitizes these tumors to osimertinib and trametinib treatment [35,36]. Mechanistically, Kurppa et al. attribute the role of YAP1 in this context to the evasion of apoptosis by YAP1–TEAD-mediated repression of the proapoptotic protein BMF via the EMT transcription factor SLUG. YAP1 activation itself in response to osimertinib or trametinib treatment is thought to be the result of epigenetic changes. Along the same lines, YAP1 activation was associated with poor response to treatment with the ALK/ROS1 inhibitor crizotinib in ALK-rearranged lung cancers. YAP1 silencing suppressed tumor growth in resistant cells, patient-derived xenografts, and EML4–ALK transgenic mice, whereas YAP1 overexpression decreased the responsiveness of parental cells to the ALK inhibitor [37]. Similarly, YAP1 activation is a mechanism of resistance to crizotinib therapy for ROS1-rearranged lung cancers [38].
YAP1 activation is also a major resistance mechanism in other tumor indications with increased MAPK pathway signaling. Treatment of BRAF mutant skin tumors with BRAF or MEK inhibitors vemurafenib and trametinib leads to resistance to therapy through YAP1 activation via actin cytoskeleton remodeling and RhoA-mediated inhibition of Hippo kinase LATS1 [39,40]. A recent cellular screen identified YAP1 activation as a novel mechanism of resistance to the triple combination of EGFR, BRAF and MEK inhibitors cetuximab, dabrafenib and trametinib in BRAF mutant colorectal cancer cell lines [41], which is in line with studies showing that the knockdown of YAP1 increases sensitivity to mitogen-activated protein kinase (MAPK) pathway inhibitors [42].
Finally, YAP1 activation is also described as a bypass mechanism to KRAS inactivation in cancers driven by oncogenic mutant KRAS signaling. Most of these studies are based on genetic knockdown or knockout of KRAS since specific inhibitors of mutant KRAS proteins are not yet available for most KRAS mutant forms. Thus, Yap1 gene amplification appears to occur in mice under KRAS ablation in a KRAS G12D-dependent mouse pancreatic tumor model [7], while a genome-scale cDNA screen identified activation of YAP1 as a factor for survival upon KRAS knockdown in KRAS G13D mutant colorectal cancer cell lines [43]. YAP1 upregulation was further identified as a mediator of resistance to combined TBK1 and MEK inhibition in KRAS mutant lung cancer cells and in a KRAS G12D mutant genetically engineered lung cancer mouse models [44]. The mechanisms by which YAP1 activation bypass KRAS inactivation span from (1) the induction of the epithelial mesenchymal transition (EMT) through (2) the RAS–YAP1 mediated activation of transcription factor FOS to (3) the RAS-mediated protein stabilization of YAP1 via the downregulation of SOCS-box proteins which serve as substrate recognition modules of elongin B/C ubiquitin ligase complexes [45] or (4) the stabilization of the YAP1-binding partners TEADs [46]. Studies in Drosophila suggest that Hippo–YAP1 signaling controls the transcriptional response to RAS via the transcriptional regulators Pointed and Capicua [47].
While YAP1 activation is a major bypass mechanism to EGFR and MAPK pathway inhibition, the role of YAP1 as a mechanism of resistance to therapy seems to be general in many cancer types and independent of the type of treatment used. Numerous reports describe the activation of YAP1 in response to chemotherapy treatment covering resistance to anti-microtubule, antimetabolite and DNA-damaging agents [48]. In this context, YAP1-mediated regulation of ferroptosis is starting to be discussed as one of the potential mechanistic links between chemoresistance and high YAP1 activity [49,50,51,52,53]. In addition, an increasing number of studies have shown that the Hippo pathway is closely linked to endocrine therapy resistance in breast and prostate cancer [54,55].
In summary, YAP1-mediated resistance to therapy typically includes an initial upregulation of YAP1 activity. This upregulation may result from the inactivation of the NF2–Hippo axis via mechanisms such as genetic alterations (for example, of the NF2 and LATS1/2 genes), inhibition of LATS kinases through RhoA signaling or downregulation of LATS / upregulation of YAP1 mRNA via micro RNAs [56,57]. Additionally, YAP1 activation may come from signals increasing YAP1 gene expression, protein half-life or nuclear translocation. The consequence of YAP1 activation is always an increased transcriptional output of YAP1 target genes leading to resistance to therapy by evasion of apoptosis (e.g., through upregulation of apoptosis inhibitors CYR61, CTGF and antiapoptotic BCLXL), increase of cell proliferation (e.g., through upregulation of growth factors such as EGFR, HER3 and AXL) or EMT transition (e.g., through interaction with transcription factors SLUG and FOS). Whether all these mechanisms for YAP1 activation and subsequent resistance to therapy exist in parallel and within the same tumor type or tumor cell requires further investigation. It is also important to better understand under which of the circumstances YAP1 requires partnering with TEAD, as this will impact the potential to pharmacologically target resistance to therapy.
2.4. Regulation of Immunity and the Tumor Microenvironment by YAP1/TAZ
The tumor microenvironment (TME) is a multifaceted ecosystem composed of a myriad of cell types and resources, each critical to the growth and survival of the tumor as a whole. Indeed, tumors undergo evolutionary processes during development driven by the tumor cells and the influence of the surrounding microenvironment [58]. The preponderance of published literature suggests that YAP1 and TAZ are critical in the evolution of some tumors, in part via their influence on the TME (Figure 2). The tumor immune microenvironment is a key component of the TME and is a critical regulator of neoplastic growth. YAP1 and TAZ are likely involved in this facet of tumor biology as intrinsic regulators of certain immune cell subsets as well as extrinsic regulators of the immune response via their oncogenic driver role in tumor cells [17,59,60].
Although YAP1 and TAZ are not required for normal hematopoiesis [61], they may be important for the function of some subsets of immune cells, as several groups have shown that YAP1 and TAZ are critical regulators of specialized T cells in a context-specific manner [62,63,64,65,66]. The function of TAZ in T cells has mainly been described as a regulator of the balance between Th17 and Treg differentiation, likely with implications for regulating autoimmunity [65]. Loss of TAZ results in CD4+ T cell differentiation to skew towards immunosuppressive Tregs at the expense of the proinflammatory Th17 subset. Knockout (KO) of YAP1 in naïve T cells results in hypersensitivity to in vitro stimulation and enhanced antitumor activity in vivo [62,63,64,66]. Interestingly, YAP1 KO CD4 and CD8+ T cells demonstrated superior tumor infiltration compared to wild-type counterparts, and tumor growth was either greatly delayed or nearly absent in tumor-bearing CD4–Cre or Foxp3–Cre mice [62,63,64]. These observations suggest that inhibition of YAP1 activity could help drive more profound antitumor T cell responses. YAP1 appears to overall play a functionally suppressive role in T cells; however, it is currently unclear if the interaction with TEAD is critical in this process [66], which has important implications for directly drugging YAP1-driven processes with small-molecule approaches. It is possible that YAP1 plays a role in the cytoplasm of T cells, modulating the activity of NFAT1 [66], suggesting that treatment modalities that force cytoplasmic localization of YAP1 could be of interest, for example, statins or SRC inhibitors (e.g., dasatinib) [67]. In preclinical mouse models, dasatinib has been shown to improve the antitumor activity of anti-PD1 antibodies, and prior statin use may be associated with improved patient outcomes in non-small-cell lung cancer as well as in mesotheliomas, although further studies are required to confirm this observation [68,69,70]. It will be interesting to see clinically how YAP1 and TEAD factor into these observations.
Both YAP1 and TAZ have also been shown to regulate the tumor immune microenvironment via numerous avenues, including tumor cell-intrinsic properties via directly regulating the expression of PD-L1, as well as recruitment of immunosuppressive cell types via induction of secreted factors [71,72,73,74,75,76,77]. YAP1 has also been shown to promote adaptive resistance to anti-PD1 therapy [78], and the YAP1/TAZ target gene signature has been shown to correlate with tumor immune cell infiltration in patient samples [16]. Direct upregulation of PD-L1 transcripts in tumor cells by both YAP1 and TAZ has been shown in multiple contexts and involves their interaction with TEAD transcription factors [72,73,74,75]. These observations are potentially of interest since tumor cells upregulate PD-L1 to avoid immune surveillance. Therefore, inhibitors of YAP1/TAZ could presumably alleviate some of this immune escape; however, testing this mechanistically in an immunocompetent setting may be challenging if YAP1/TAZ do not directly regulate PD-L1 in mouse cells [73]. Other mechanisms of immunosuppression have been linked to secreted factors induced by YAP1. These include (1) tumor stromal cell expansion via the expression of CTGF, CYR61, IL6 and MMP7 [76], (2) macrophage polarization [71] in pancreatic cancer models and (3) CXCL5-mediated expansion of myeloid-derived suppressor cells in prostate cancer [77]. It is clear from the current literature that the transcriptional effectors of the Hippo pathway likely drive the immunosuppressive tumor microenvironment on numerous fronts and it appears to be an attractive target from the antitumor immunity perspective.
3. Targeting the YAP1/TAZ–TEAD Interaction
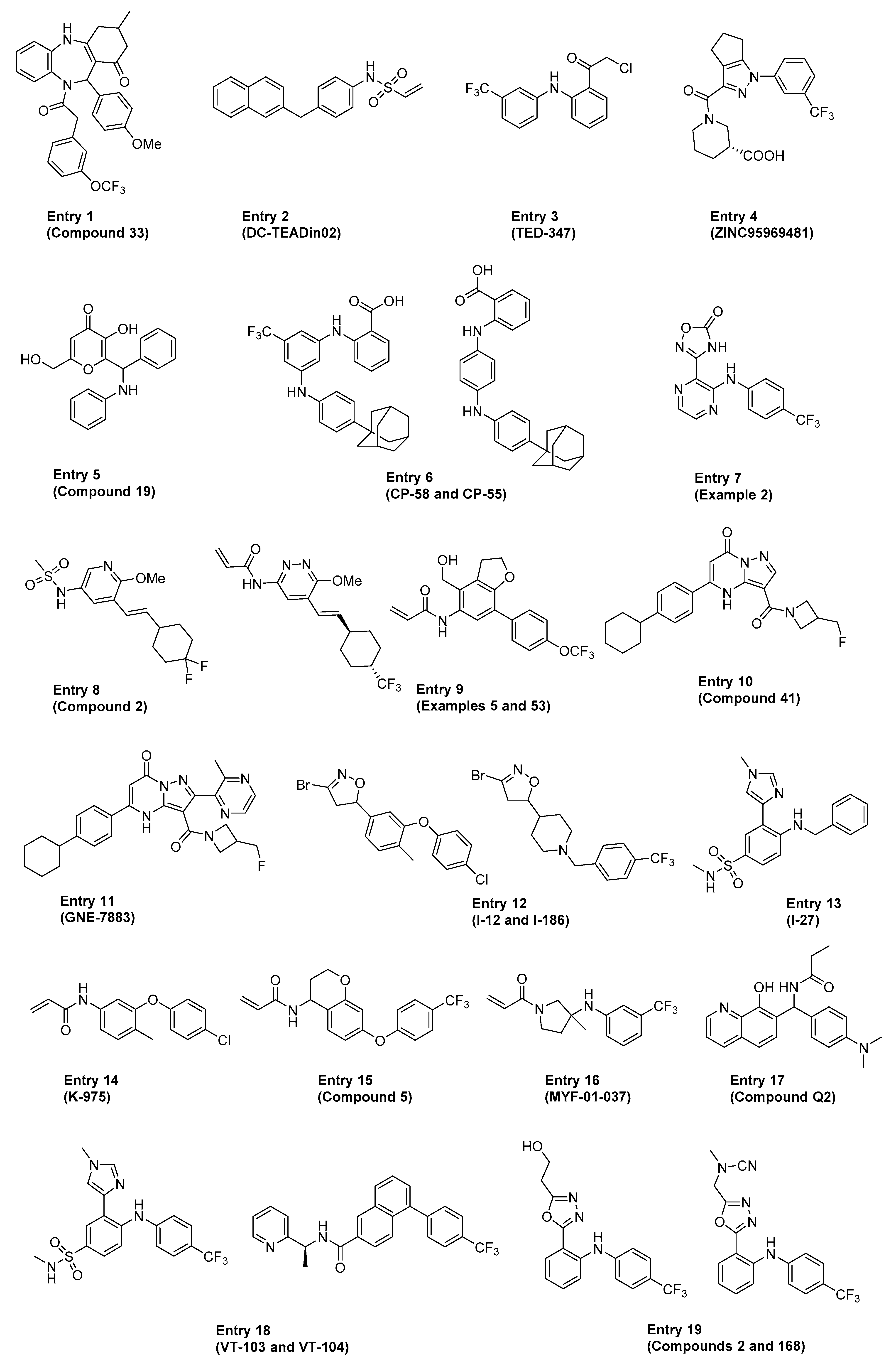
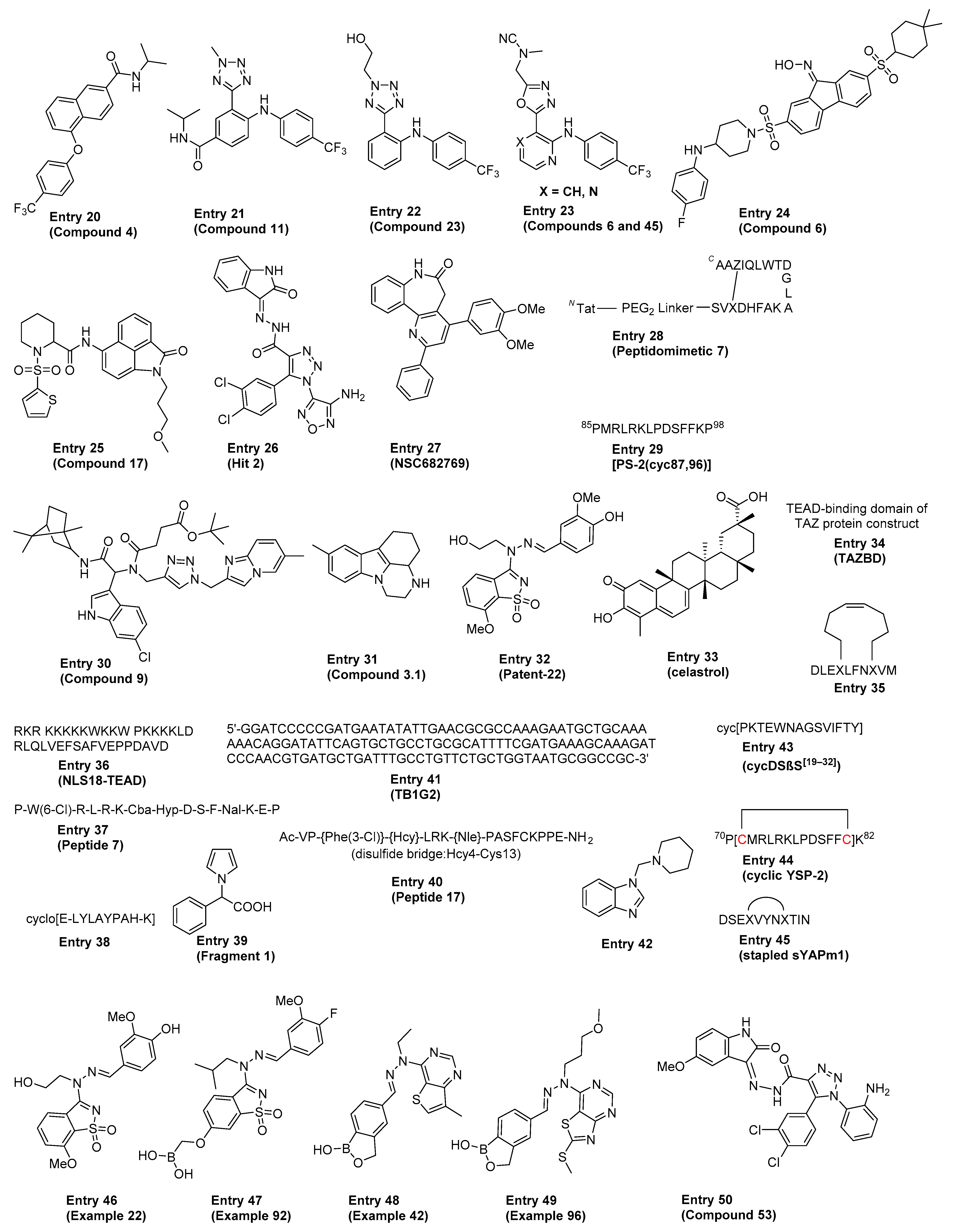
Several excellent reviews covered the various direct and indirect approaches to targeting the Hippo pathway in cancers and regenerative medicine, including a detailed discussion on the historical proof-of-concept tool molecules such as verteporfin, flufenamic acid, Peptide 17 and Super-TDU [79,80,81,82,83]. The ensuing section highlights some of the more recent advancements in the field, focusing on compounds and therapeutic approaches directed at disrupting the YAP1/TAZ–TEAD interaction either directly (i.e., classical PPI inhibition, also referred to as just “PPI” in Table 1) or allosterically by blocking TEAD palmitoylation and stabilization (referred to as “Allosteric, palmitate” in Table 1). The comprehensive summary in Table 1 is meant to provide the reader with an overview of the existing chemical matter shown in Figure 3 that spans across multiple modalities to disrupt the YAP1/TAZ–TEAD interaction. In cases where patent publications are discussed, we decided to highlight representative examples based on the data disclosed.
As previously discussed, YAP1/TAZ binding to TEAD regulates the transcriptional output of the Hippo pathway and therefore represents a promising strategy to target diseases with dysregulated Hippo signaling. YAP1 and TAZ are natively disordered proteins and therefore difficult to selectively target with traditional small-molecule or peptide modalities [84]. Instead, recent therapeutic approaches have focused on targeting the TEAD proteins, which consist of two well-defined DNA-binding and YAP1-binding domains. Targeting the DNA-binding domain of TEAD (DNA-TEAD) represents a viable and attractive strategy to regulate TEAD-mediated gene expression; however, this approach has yet to gain momentum and warrants additional attention [85]. Alternatively, recent efforts within the Hippo field have focused on targeting the TEAD coactivator domain (YAP1/TAZ–TEAD, also referred to as YAP1–TEAD below). The domain architecture and structure of YAP1/TAZ and TEAD have been extensively reviewed before, with the availability of several crystal structures of TEADs complexed with either a coactivator or small-molecule partner significantly enhancing our understanding of how to disrupt the YAP1–TEAD interaction [85,86]. These reviews discuss progress towards targeting the most druggable sites within the YAP1–TEAD binding domain (YBD), initially focusing on interfaces 2 and 3 shown in Figure 1B [4,85,86,87,88,89,90]. These early approaches to directly targeting the YAP1–TEAD PPI have primarily relied on larger molecules and peptidomimetics designed to bind to the YBD of TEAD and therefore directly inhibit formation of the transcriptionally active YAP1–TEAD complex. However, a recent development in targeting the Hippo pathway has been the discovery of a central lipophilic pocket in TEAD amenable to small-molecule binding that is the site of autopalmitoylation [20,22,91]. Within this lipophilic (palmitate) pocket, posttranslational S-palmitoylation of TEAD at a conserved catalytic cysteine (Cys) residue (e.g., C380 in TEAD2) leads to TEAD stabilization [22] and is believed to be critical for maintaining appropriate protein folding to enable formation of the transcriptionally active YAP1–TEAD complex. Therefore, targeting the palmitate pocket with small molecules to allosterically inhibit and disrupt formation of the YAP1–TEAD complex represents an attractive strategy to modulate TEAD-driven gene transcription. Within the palmitate pocket, it is important to note that the sequence similarity is 80–90% across the four TEAD isoforms, suggesting that the discovery of pan-TEAD ligands is feasible [22]. However, key questions in targeting TEAD palmitoylation remain with respect to the impact of isoform selectivity on both efficacy and depth of response, as well as safety.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reported key in vitro and in vivo data for each representative example in Figure 3 (MOA = mechanism of action; NR = not reported).
Table 1.
Reported key in vitro and in vivo data for each representative example in Figure 3 (MOA = mechanism of action; NR = not reported).
| Entry (Ref.) | MOA | In Vitro Data | In Vivo Data |
|---|---|---|---|
| 1 [92] | NR |
| NR |
| |||
| 2 [93] | Allosteric, palmitate (covalent) |
| |
| NR | ||
| 3 [94,95] | Allosteric, palmitate (covalent) |
| NR |
| |||
| 4 [96] | Allosteric, palmitate |
| NR |
| 5 [97] | Allosteric, palmitate (covalent) |
| NR |
| 6 [98] | Allosteric, palmitate |
| NR |
| 7 [99] | Allosteric, palmitate |
|
|
| 8 [100,101] | Allosteric, palmitate |
|
|
| 9 [102] | Allosteric, palmitate (covalent) |
| NR |
| 10 [103] | Allosteric, palmitate |
| NR |
| 11 [104] | Allosteric, palmitate |
|
|
| 12 [105] | Allosteric, palmitate (covalent) |
|
|
| 13 [106] | Allosteric, palmitate |
|
|
| 14 [32] | Allosteric, palmitate (covalent) |
|
|
| 15 [107] | Allosteric, palmitate (covalent) |
| NR |
| 16 [35,108] | Allosteric, palmitate (covalent) |
| NR |
| 17 [109] | Allosteric, palmitate |
|
|
| 18 [33,110,111] | Allosteric, palmitate |
|
|
| 19 [112] | Allosteric, palmitate |
| NR |
| 20 [113] | Allosteric, palmitate |
| NR |
| 21 [114] | Allosteric, palmitate |
| NR |
| 22 [115] | Allosteric, palmitate |
| NR |
| 23 [116] | Allosteric, palmitate |
| NR |
| 24 [117] | NR |
| NR |
| 25 [118] | NR |
|
|
| 26 [119] | PPI |
| NR |
| 27 [120] | PPI |
|
|
| 28 [121] | PPI |
| NR |
| 29 [122] | PPI |
| NR |
| 30 [123] | Allosteric, palmitate |
| NR |
| 31 [124] | PPI |
| NR |
| 32 [125] | PPI |
| NR |
| 33 [126] | PPI |
| NR |
| 34 [127] | PPI |
|
|
| 35 [128] | PPI |
| NR |
| 36 [129] | PPI |
|
|
| 37 [130] | PPI |
| NR |
| 38 [131] | PPI |
| NR |
| 39 [132] | PPI |
| NR |
| 40 [133,134] | PPI |
|
|
| 41 [135,136] | PPI |
| NR |
| 42 [137] | PPI |
| NR |
| 43 [138] | PPI |
| NR |
| 44 [139] | PPI |
| NR |
| 45 [140] | PPI |
| NR |
| 46 [141] | PPI |
| NR |
| 47 [142] | PPI |
| NR |
| 48 [143] | PPI |
| NR |
| 49 [144] | PPI |
| NR |
| 50 [145] | PPI |
| NR |
The section below highlights several recent examples from Table 1 that have not been reviewed before, as well as emphasizes the molecules that have either entered the clinic or are positioned to do so in the near future.
3.1. MGH (Entry 6)
Wu et al. have previously disclosed the discovery and characterization of adamantyl-containing MGH–CP1 inhibitor of autopalmitoylation of endogenous TEAD in cells, with the allosteric mechanism and palmitate pocket binding subsequently confirmed by co-IP and the X-ray structure, respectively [146]. In a more recent patent application, the authors merged the adamantyl aniline portion of MGH–CP1, which extends deep into the lipophilic pocket, with the flufenamic acid structure to arrive at two key analogs: CP-58 and CP-55. In a cell proliferation assay, CP-55 is quite potent with IC50 values of 38 nM in 92.1 cells and 330 nM in Huh7 cells [98].
3.2. Basilea Pharmaceutica (Entry 7)
Basilea Pharmaceutica is a Swiss biopharmaceutical company that recently described in a patent application a series of 1.2,4-oxadiazol-5-one derivatives [99]. Structurally, these molecules show similarity with flufenamic acid [91], where the carboxylic acid group has been replaced with an oxadiazol-5-one warhead. These molecules show TEAD2 binding by thermal shift assay with a range of stabilizations from 0.4–18 °C and inhibit luciferase activity in the MCF7 TEAD reporter gene assay with a range of activities between 39 nM and 6 µM. Notably, Example 2 (MCF7 TEAD RGA IC50 = 41 nM, thermal shift assay hTEAD2 ΔTm = 10.6 °C, no PK data reported) is active in vivo in a 62-day murine NCI-H226 xenograft efficacy model by showing dose-dependent activity and tumor stasis at the highest dose of 250 mg/kg PO QD.
3.3. Genentech (Entries 8–11)
Researchers at Genentech have recently disclosed the full characterization of Compound 2 (GNE-9886) as a potent TEAD binder that blocks S-palmitoylation, as measured by Lipid FP assay [100]. Despite inhibiting palmitoylation, mechanistically, this molecule does not disrupt the YAP1–TEAD interaction, but rather is hypothesized to drive its reported in vivo efficacy (e.g., 78% TGI in the Detroit X1 562 xenograft model) by transforming TEAD into the dominant negative transcriptional repressor which blocks the TEAD interaction with chromatin. More recently, related analogs of GNE-9886 were patented where the noncovalent Cys interaction with the sulfonamide in Compound 2 was replaced with a covalent acrylamide warhead to yield representative Examples 5 and 53 [102]. While the affinity of these molecules for the TEAD palmitate pocket was retained, it is unclear from the presented data whether the switch to covalent binding has any impact on YAP1–TEAD complex disruption.
A novel, structurally distinct class of pan-TEAD binders was recently disclosed, with GNE-7883 highlighted as the lead reversible allosteric inhibitor of the YAP1–TEAD interaction [104]. The potent inhibition of pan-TEAD lipidation with GNE-7883 translated well to the disruption of YAP1 binding to each of the four TEAD isoforms by the YAP1 HTRF assay. GNE-7883 potently inhibited proliferation of the OVCAR-8 and NCI-H226 cells, with EC50 values of 115 nM and 333 nM, respectively. At 250 mg/kg (subcutaneous dosing), this molecule showed strong in vivo tumor growth inhibition of 102% in the mesothelioma NCIH226 xenograft mouse model, with a favorable tolerability profile based on the reported body weight change data relative to placebo. Overall, GNE-7883 represents an attractive molecule to further probe the Hippo pathway in cancer and the impact of allosteric inhibition of the YAP1–TEAD complex on efficacy across different indications and genetic backgrounds.
3.4. Ikena Oncology (Entries 12–13)
Ikena Oncology, a Boston-based biotechnological company, has recently published two patent applications disclosing their TEAD palmitoylation inhibitors. In the first patent application [106], a series of noncovalent aryl sulfonamide derivatives bearing a heterocycle (e.g., imidazole) and an amine (e.g., aniline or benzylamine) are described. These compounds show potent inhibition of TEAD reporter activity in MCF7 cells (EC50 range from <0.1 µM to >0.5 µM) and proliferation of H226 cells while maintaining selectivity against the Hippo WT H28 cell line (H226 EC50 range from <0.1 µM to >0.5 µM). Example I-27 in Entry 13 (MCF7 TEAD cell reporter assay EC50 < 100 nM, H226 cell proliferation assay EC50 < 100 nM and selectivity in the H28 cell proliferation assay with EC50 > 500 nM) is further exemplified with both pharmacokinetic and pharmacodynamic data. When administered at 10 mg/kg PO in BALB/c mice, I-27 reaches Cmax = 1037 ng/mL and AUC0-last = 2252 ng·h/mL. After 3-day treatment at 30 mg/kg PO in immunodeficient mice bearing NCI-H226 tumors, I-27 led to >50% downregulation of CTGF mRNA in the tumor, a known transcriptional target of YAP1–TEAD.
In the second patent application [105], a series of covalent derivatives with a variety of warheads is described (e.g., α-chloroketone, acrylamides, 3-bromodihydroisoxazole, 3-substituted dihydroisoxazole). Similar to the first patent application, compounds show potent inhibition of the TEAD reporter assay in MCF7 (EC50 range from <0.1 µM to >0.5 µM), with a select few also evaluated for inhibition of H226 proliferation and selectivity against the Hippo WT H28 cell line (H226 EC50 range from <0.1 µM up to >0.5 µM). Example I-186 is active in the MCF7 TEAD cell reporter assay with EC50 < 100 nM and inhibits H226 cell proliferation with EC50 < 100 nM while maintaining selectivity against H28 (EC50 >500 nM). PK/PD and efficacy data are reported for examples I-12 (Isomer 2) and I-186 (Isomer 1). Example I-12 (Isomer 2) administered at 10 mg/kg PO in BALB/c mice shows Cmax = 23 ng/mL and AUC0-last = 48 ng·h/mL, as well as > 50% downregulation of tumor CTGF mRNA upon 3 days of treatment at 50 mg/kg IP of mice bearing NCI-H226 tumors. Example I-186 (Isomer 1) administered at 10 mg/kg PO in BALB/c mice shows Cmax = 110 ng/mL and AUC0-last = 435 ng·h/mL, as well as >50% downregulation of tumor CTGF mRNA upon 3 days of treatment at 30 mg/kg PO of mice bearing NCI-H226 tumors. Both compounds were evaluated in the NCI-H226 tumor xenograft mouse model and showed promising antitumor activity after 28 days of treatment (e.g., 80% TGI for I-12 (Isomer 2) at 200 mg/kg PO QD dosing and 67% TGI for I-186 (Isomer 1) at 75 mg/kg PO QD dosing).
3.5. A*STAR (Entry 17)
While most reported palmitate pocket binders show inhibitory activity on TEAD palmitoylation and transcription, Hong and Probatti et al. recently reported quinolinol Q2 with palmitate-like TEAD stimulating activity [109]. Importantly Q2 has been shown to occupy the TEAD central pocket by SPR with a Kd = 2.6 µM and increase endogenous expression levels of TEAD target genes CTGF, CYR61 and ANKRD1 by RT-PCR. In vivo, quinolinol Q2 accelerates cutaneous wound healing in mice to as soon as day 7, which is consistent with the previously reported genetic experiments. This molecule further highlights the potential of modulating the Hippo pathway for wound healing and regenerative medicine that warrants further attention.
3.6. Vivace Therapeutics (Entries 18–24)
Vivace Therapeutics, a San Francisco-based biotechnological company, is one of the most active companies in the Hippo field with seven recent patent applications [110,111,112,113,114,115,116]; they also entered the clinic with their lead molecule VT3989 in early 2021. The Vivace patents can be characterized by two distinct chemical series. The first series derives from flufenamic acid [111,112,114,115,116] and consists of an aryl amine central core substituted with a five-membered heterocycle (e.g., imidazole, oxadiazole, tetrazole). The second series consists of a substituted bicyclic aromatic ring (e.g., naphthyl, quinoline), which contains an amide substitute and an aryl or an O-linked aryl group [110,113].
Within the first chemical series, compound VT-103 (Entry 18) and Compound 11 (Entry 21) [111,114] are derivatives of the representative Compound 23 (Entry 22) [115] where a carbonyl or a sulfone/sulfonamide group are introduced in the para-position of the aniline. Compounds 6 and 45 (Entry 23) [112] lack this third substitution, but the five-membered heterocycle is always a 1,3,4-oxadiazole. The reported cellular activities of these molecules in the reporter assay feature IC50 < 100 nM. In the second chemical series, the main difference between biaryl Compound 4 (Entry 20) [113] and VT-104 (Entry 18) [110] is the introduction of a flexible linker, mostly oxygen-based, between the aryl group and the bicyclic central core. As with prior compounds, the best reported activities in the cellular reporter assay feature IC50 < 100nM.
A series of key molecules included in the two abovementioned patent families are detailed in a recent paper by Vivace, which highlights the properties and characteristics of these TEAD inhibitors [33]. Among the compounds highlighted in the paper, VT-103 and VT-104 (Entry 18) are the most advanced examples, with both in vitro and in vivo data included. Based on the thermal shift and TEAD palmitoylation assay data, VT-103 is considered to be a TEAD1-selective binder, with low nanomolar IC50 in the proliferation assay using a panel of YAP1/TEAD-dependent cell lines. VT-103 has a favorable pharmacokinetics profile in mice with sufficient exposure for constant target engagement with daily oral dosing. VT-103 was evaluated in two malignant mesothelioma tumor xenograft mouse models with impressive outcomes, including (1) an NCI-H226 model with TGI = 106% at 3 mg/kg PO and (2) an NCI-H2373 model with TGI = 126% at 10 mg/kg PO.
VT-104 is part of the second family of bicyclic aromatics substituted with an amide functional group. Based on the thermal shift and TEAD palmitoylation assay data, VT-104 is a pan-TEAD binder, with low nanomolar IC50 values in proliferation assays using YAP1/TEAD-dependent cell lines. Crystal structure (PDB: 7CNL) of a close analog of VT-104 in the TEAD3 palmitoylation pocket confirms the noncovalent binding mode and palmitate pocket MOA of this derivative. VT-104 also exhibits a favorable pharmacokinetics profile with exposure sufficient for daily dosing in mice. In the NCI-H226 xenograft efficacy model, VT-104 is highly active with a TGI = 102% at 3 mg/kg PO.
3.7. Astra Zeneca (Entry 28)
Astra Zeneca reported the design and characterization of Proteomimetic 7 (Tat–PEG2–4E), a stabilized ternary protein structure that disrupts the PPI between TEAD and its corepressor VGL4 [121]. Tat–PEG2–4E was derived from the VGL4 233-252 amino acid sequence and crosslinked via lactamization of residues E235 and K250. By SPR, 4E shows binding to hTEAD1 with a Kd = 0.7 µM. As a disruptor of the suppressive VGL4–TEAD complex, Tat–PEG2–4E significantly stimulates the expression of TEAD target genes CYR61, CTGF, ANKRD1 and SEPINE1 in human cardiomyocytes. In addition, it showed increased levels of YAP1 nuclear translocation and cell cycle activity in primary juvenile rat heart cells which is assumed to be required for cardiomyocyte proliferation. The cell cycle activity was stimulated by Tat–PEG2–4E to the same extent as positive controls SB203580 (p38MAPK inhibitor) and CHIR99021 (GSK3β inhibitor), thus highlighting the importance of peptidomimetic PPI inhibition of repressor complexes towards the activation of transcription factors.
3.8. University of Lille (Entry 50)
Compound 53 [145] is an optimized analog of the previously reported Hit 2 (Entry 26) [119] with an improvement in activity in the TEAD reporter assay; IC50 = 1.7 µM. Compound 53 inhibits the proliferation of MDA-MB-231 cells with CC50 = 6.9 µM and in the same model decreases TEAD target genes CYR61, ANKRD1 and CTGF by 50%. On the basis of thermal shift and molecular modeling tests, it has been proposed that Compound 53 is a classical and direct protein–protein interaction inhibitor of YAP1–TEAD.
4. Clinical Trials Overview
The exciting progress in targeting the Hippo pathway described above has recently culminated in three molecules shown in Table 2 reaching the clinic in 2021. Amongst the various cancer indications where the Hippo pathway is deregulated, malignant mesothelioma is the primary indication selected in these clinical studies due to the high unmet medical need and the highest reported frequency of somatic mutations in NF2–Hippo, although other solid tumor indications will also be evaluated as discussed below.
Vivace Therapeutics is the sponsor of a phase I clinical study (NCT04665206) to evaluate the safety, tolerability, PK and biological activity of VT3989, a reported TEAD inhibitor in patients with refractory metastatic solid tumors, including refractory pleural malignant mesotheliomas. The chemical structure of VT3989 has not been disclosed, but several patent applications have been published and reviewed above [110,111,112,113,114,115,116].
Ionis Pharmaceuticals is developing ION537, an antisense oligonucleotide (ASO)-targeting YAP1 mRNA which is currently undergoing a phase I trial in patients with molecularly selected advanced solid tumors [147,148]. ION537 is administered by subcutaneous (SC) or intravenous (IV) injection, with the chemical structure recently presented at the AACR 2021 meeting as AksAdsGdsTdsGdsTdsAdsTdsGdsTdsmGdsAksGesAksAesGk. ION537 also corresponds to compound number 1198440 in a recent Ionis patent application [147]. Structurally, the backbone is modified with a phosphorothioate linker between each ribose for added stability, while a 2′–5′ constrained ethyl modification is introduced at positions 1, 12, 14 and 16 of the ASO for improved affinity, stability and tolerability. Additional modification of the ribose with a 2′ methoxyethyl (MOE) group is also introduced at positions 13 and 15.
ION537 inhibits cell proliferation in YAP1-activated head and neck tumors (SCC25 GI50 = 80 nM; BIC56 GI50 = 80 nM). In addition, ION537 inhibits YAP1 mRNA expression in several tumor xenografts models: (1) in the human hepatocellular carcinoma SNU449 xenograft tumor model, Yap1 mRNA is decreased by 51% after five days of treatment with 50 mg/kg ION537 twice weekly, (2) in the human epidermoid carcinoma A-431 xenograft tumor model, Yap1 mRNA is decreased in tumors by 32% after five days of treatment with 25 mg/kg ION537 daily, (3) in the human squamous cell carcinoma CAL27 xenograft tumor model, Yap1 mRNA is decreased in tumors by 67% after five days of treatment with 15 mg/kg ION537 daily, and (4) in the CAL33 human squamous cell carcinoma xenograft tumor model, Yap1 mRNA is decreased by 88% in tumors after five days of treatment with 50 mg/kg ION537 twice weekly. In the same experiment, the weight of the tumor decreased by 74% compared to the vehicle after 34 days of treatment.
The latest arrival in the clinic, Novartis, has initiated a phase I clinical study with a molecule IAG933. The purpose of that study is to characterize the safety and tolerability of IAG933 in patients with mesothelioma, NF2/LATS1/LATS2-mutated tumors and tumors with functional YAP1/TAZ fusions, as well as to identify the maximum tolerated dose. The structure, the binding partner, and the mechanism of action of IAG933 have not been disclosed yet, although a new patent application has recently been disclosed by Novartis, at the time of publication of this review [149].
5. Concluding Remarks
The past decade and a half of Hippo/YAP1 research has uncovered countless connections to human diseases ranging from Ebola [150] to oncology and even applications in laboratory-grown meat [151]. This wide array of biological functions has sparked a plethora of research targeting the pathway, and in particular the YAP1–TEAD interaction. We have now entered a new phase of investigation, where the safety and relevance of YAP1–TEAD in driving human disease will be tested clinically, in particular for oncology and tumors such as malignant mesotheliomas. In addition, the disclosure of structures of selective and potent compounds that disrupt the YAP1–TEAD association could allow for the probing of more biological questions that cannot be easily answered with genetic tools, particularly, which tumor indications may respond to the YAP1–TEAD dissociation. This line of investigation could lead to an expansion of potential indications outside of malignant mesothelioma tumors.
Author Contributions
Writing—review and editing, E.R.B., V.S., I.V. and O.V. All authors contribute equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We wish to thank Sebastian Eckehart Schneider (Merck & Co., Inc., Kenilworth, 07033, NJ, USA) for conducting the druggability assessment shown in Figure 1B. We also wish to acknowledge Livia Valtingojer for the Hippo art included in the graphical abstract.
Conflicts of Interest
Iris Valtingojer and Olivier Venier are Sanofi employees and may hold shares and/or stock options in the company.
Abbreviations
| AACR | American Association for Cancer Research |
| ABT | 1-Aminobenzotriazole |
| ALK | Anaplastic lymphoma kinase |
| ANKRD1 | Ankyrin repeat domain 1 |
| AXL | AXL receptor tyrosine kinase |
| BCLXL | B cell lymphoma-extra large |
| BMF | Bcl2-modifying factor |
| CETSA | Cellular thermal shift assay |
| Co-IP | Complex immunoprecipitation |
| CTGF | Connective tissue growth factor |
| CXCL5 | C–X–C motif chemokine 5 |
| CYR61 | Cysteine-rich angiogenic inducer 61 |
| DSS | Dextran sodium sulfate |
| EdU | Ethynyl deoxyuridine |
| EGFR | Epidermal growth factor receptor |
| EML4 | Echinoderm microtubule-associated protein-like 4 |
| EMT | Epithelial mesenchymal transition |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit |
| FP | Fluorescence polarization |
| HER3 | Human epidermal growth factor receptor 3 |
| HTRF | Homogeneous time-resolved fluorescence |
| IP | Intraperitoneal |
| ITC | Isothermal titration calorimetry |
| KRAS | Kirsten rat sarcoma virus |
| LATS | Large tumor suppressor kinase |
| MAPK | Mitogen-activated protein kinase |
| MDSC | Myeloid-derived suppressor cell |
| MEK | MAPK/ERK kinase |
| mKRAS | Mutant KRAS |
| MMP7 | Matrix metalloproteinase-7 |
| MOA | Mechanism of action |
| MOE | 2′-O-methoxyethyl |
| MST | Mammalian sterile 20-like kinase |
| NF2 | Neurofibromin 2 |
| NFAT1 | Nuclear factor of activated T cells, cytoplasmic 2 |
| NR | Not reported |
| NSCLC | Non-small-cell lung carcinoma |
| PDB | Protein Data Bank |
| PD-L1 | Programmed death-ligand 1 |
| PO | Per os (oral dosing) |
| PPI | Protein–protein interaction |
| QD | Quaque die (once a day dosing) |
| RGA | Reporter gene assay |
| ROS1 | Proto-oncogene tyrosine-protein kinase |
| SLUG | Snail family transcriptional repressor 2 |
| SPR | Surface plasmon resonance |
| SRC | Proto-oncogene tyrosine-protein kinase Src |
| TAZ | WW domain-containing transcription regulator protein 1 |
| TBK1 | TANK-binding kinase 1 |
| TCGA | The Cancer Genome Atlas |
| TEAD | TEA domain family member |
| TGI | Tumor growth inhibition |
| TME | Tumor microenvironment |
| TREG | Regulatory T cells |
| VGL4 | Vestigial like family member 4 |
| YAP1 | Yes-associated protein 1 |
| YBD | YAP-binding domain |
References
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [Green Version]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, Y.; Zheng, Y.; Dong, J.; Pan, D. The TEAD/TEF Family Protein Scalloped Mediates Transcriptional Output of the Hippo Growth-Regulatory Pathway. Dev. Cell 2008, 14, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 Acts Downstream of α-Catenin to Control Epidermal Proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; Lai, Z.-C.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Bai, H.; David, K.K.; Dong, J.; Zheng, Y.; Cai, J.; Giovannini, M.; Liu, P.; Anders, R.A.; Pan, D. The Merlin/NF2 Tumor Suppressor Functions through the YAP Oncoprotein to Regulate Tissue Homeostasis in Mammals. Dev. Cell 2010, 19, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.-G.; Hynes, R.O.; Lamar, J. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, C.-Y.; Zha, Z.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.-Y.; Guan, K.-L. TEAD Transcription Factors Mediate the Function of TAZ in Cell Growth and Epithelial-Mesenchymal Transition. J. Biol. Chem. 2009, 284, 13355–13362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currey, L.; Thor, S.; Piper, M. TEAD family transcription factors in development and disease. Development 2021, 148, 196675. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, T.; Cheng, A.S.L.; Yu, J.; Kang, W.; To, K.F. The TEAD Family and Its Oncogenic Role in Promoting Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, X.; Maglic, D.; Dill, M.; Mojumdar, K.; Ng, P.K.-S.; Jeong, K.J.; Tsang, Y.H.; Moreno, D.; Bhavana, V.H.; et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018, 25, 1304–1317. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [Green Version]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug. Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Mesrouze, Y.; Meyerhofer, M.; Bokhovchuk, F.; Fontana, P.; Zimmermann, C.; Martin, T.; Delaunay, C.; Izaac, A.; Kallen, J.; Schmelzle, T.; et al. Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ. Protein Sci. 2017, 26, 2399–2409. [Google Scholar] [CrossRef] [Green Version]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin-Kensicki, E.M.; Boone, B.N.; Howell, M.; Stonebraker, J.R.; Teed, J.; Alb, J.G.; Magnuson, T.R.; O’Neal, W.; Milgram, S.L. Defects in Yolk Sac Vasculogenesis, Chorioallantoic Fusion, and Embryonic Axis Elongation in Mice with Targeted Disruption of Yap. Mol. Cell. Biol. 2006, 26, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, I.M.; Halder, G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, N.; Zheng, Y.; De Wilde, R.F.; Maitra, A.; Pan, D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010, 24, 2383–2388. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Li, Y.; Liu, Q.; Chen, Q.; Chen, L.; Zhou, D. The Hippo Signaling Pathway in Regenerative Medicine. In Embryonic Stem Cell Protocols; Humana Press: New York, NY, USA, 2019, 1893; pp. 353–370. [Google Scholar]
- Wang, Y.; Yu, A.; Yu, F.-X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell 2017, 8, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Spanakis, E.; Calvet, L.; Dos Santos, O.; Dib, C.; Sidhu, S.; Moll, J.; Debussche, L.; Pollard, J.; Valtingojer, I. Abstract 2161: A transcriptomic signature for measuring YAP1 activity in patient samples and tumor models. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021; Volume 81, p. 2161. [Google Scholar]
- Calvet, L.; Dos Santos, O.; Jean-Baptiste, V.; Spanakis, E.; Ruffin, Y.; Sanchez, I.; Mestadier, J.; Soubigou, S.; Feteanu, S.; Picard, P.; et al. Abstract 4858: Oncogenic HIPPO-YAP1:in vivotarget validation of YAP1 in malignant mesothelioma. In Proceedings of the AACR Annual Meeting 2020, Virtual, 24–29 April 2020; Volume 80, p. 4858. [Google Scholar]
- Kaneda, A.; Seike, T.; Danjo, T.; Nakajima, T.; Otsubo, N.; Yamaguchi, D.; Tsuji, Y.; Hamaguchi, K.; Yasunaga, M.; Nishiya, Y.; et al. The novel potent TEAD inhibitor, K-975, inhibits YAP1/TAZ-TEAD protein-protein interactions and exerts an anti-tumor effect on malignant pleural mesothelioma. Am. J. Cancer Res. 2020, 10, 4399–4415. [Google Scholar] [PubMed]
- Tang, T.T.; Konradi, A.W.; Feng, Y.; Peng, X.; Ma, M.; Li, J.; Yu, F.-X.; Guan, K.-L.; Post, L. Small Molecule Inhibitors of TEAD Auto-palmitoylation Selectively Inhibit Proliferation and Tumor Growth of NF2-deficient Mesothelioma. Mol. Cancer Ther. 2021, 20, 986–998. [Google Scholar] [CrossRef]
- Ammoun, S.; Maze, E.A.; Agit, B.; Belshaw, R.; Hanemann, C.O. Abstract 1164: Human endogenous retrovirus type K promotes proliferation of Merlin negative schwannoma and meningioma which can be inhibited by anti-retroviral and anti-TEAD drugs. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021; Volume 81, p. 1164. [Google Scholar]
- Kurppa, K.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122. [Google Scholar] [CrossRef]
- Pfeifer, M.; Brammeld, J.S.; Price, S.; Martin, M.; Thorpe, H.; Bornot, A.; Banks, E.; Guan, N.; Dunn, S.; Guerriero, M.L.; et al. Abstract 1100: Gain and loss of function genome-wide CRISPR screens identify Hippo signaling as an important driver of resistance in EGFR mutant lung cancer. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021; Volume 81, p. 1100. [Google Scholar]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Na Kang, H.; Pyo, K.-H.; Shin, E.J.; et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK -rearranged lung cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef]
- Yamazoe, M.; Ozasa, H.; Ohgimoto, T.; Hosoya, K.; Ajimizu, H.; Funazo, T.; Yasuda, Y.; Tsuji, T.; Yoshida, H.; Itotani, R.; et al. Abstract 1098: Activation of YAP1 confers ROS1 inhibitor resistance in ROS1-rearranged lung cancer. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021; Volume 81, p. 1098. [Google Scholar]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.; Lee, J.-K.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.S.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Brammeld, J.S.; Thorpe, H.; Garcia, M.A.; Price, S.; Young, J.; Pfeifer, M.; Lupo, B.; Yusa, K.; Trusolino, L.; Garnett, M.; et al. Abstract 1081: Genome-wide CRISPR screens reveal Hippo pathway activation as a resistance mechanism in BRAF mutant colon cancer. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021; Volume 81, p. 1081. [Google Scholar]
- Su, M.; Zhan, L.; Zhang, Y.; Zhang, J. Yes-activated protein promotes primary resistance of BRAF V600E mutant metastatic colorectal cancer cells to mitogen-activated protein kinase pathway inhibitors. J. Gastrointest. Oncol. 2021, 12, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Asahina, H.; Chen, T.; Guo, S.; Quiceno, L.G.; Cavanaugh, J.D.; Merlino, A.A.; Tange, S.; Terai, H.; Kim, J.W.; et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell 2018, 34, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Hagenbeek, T.J.; Lee, H.-J.; Li, J.; Rose, C.M.; Lin, E.; Yu, M.; Martin, S.E.; Piskol, R.; Lacap, J.A.; et al. Machine-Learning and Chemicogenomics Approach Defines and Predicts Cross-Talk of Hippo and MAPK Pathways. Cancer Discov. 2021, 11, 778–793. [Google Scholar] [CrossRef]
- Pascual, J.; Jacobs, J.; Sansores-Garcia, L.; Natarajan, M.; Zeitlinger, J.; Aerts, S.; Halder, G.; Hamaratoglu, F. Hippo Reprograms the Transcriptional Response to Ras Signaling. Dev. Cell 2017, 42, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-H.; Lin, C.-C.; Wu, J.; Chao, P.-Y.; Chen, K.; Chen, P.-H.; Chi, J.-T. The Hippo Pathway Effector YAP Promotes Ferroptosis via the E3 Ligase SKP2. Mol. Cancer Res. 2021, 19, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Chi, J.-T. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis. 2021, 8, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Yang, W.H.; Ding, C.K.C.; Sun, T.; Rupprecht, G.; Lin, C.C.; Hsu, D.; Chi, J.T. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019, 28, 2501–2508. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.-N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Chen, J.; Wan, R.; Li, Q.; Rao, Z.; Wang, Y.; Zhang, L.; Teichmann, A.T. Utilizing the Hippo pathway as a therapeutic target for combating endocrine-resistant breast cancer. Cancer Cell Int. 2021, 21, 306. [Google Scholar] [CrossRef]
- Lee, H.-C.; Ou, C.-H.; Huang, Y.-C.; Hou, P.-C.; Creighton, C.J.; Lin, Y.-S.; Hu, C.-Y.; Lin, S.-C. YAP1 overexpression contributes to the development of enzalutamide resistance by induction of cancer stemness and lipid metabolism in prostate cancer. Oncogene 2021, 40, 2407–2421. [Google Scholar] [CrossRef]
- Lee, N.-H.; Kim, S.; Hyun, J. MicroRNAs Regulating Hippo-YAP Signaling in Liver Cancer. Biomedicines 2021, 9, 347. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, Z.; Hassan, M.K.; Okasha, S.; Mitamura, T.; Keshk, S.; Konno, Y.; Kato, T.; El-Khamisy, S.F.; Ohba, Y.; Watari, H. miR-363 confers taxane resistance in ovarian cancer by targeting the Hippo pathway member, LATS2. Oncotarget 2018, 9, 30053–30065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Morciano, G.; Vezzani, B.; Missiroli, S.; Boncompagni, C.; Pinton, P.; Giorgi, C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers 2021, 13, 3100. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Donato, E.; Biagioni, F.; Bisso, A.; Caganova, M.; Amati, B.; Campaner, S. YAP and TAZ are dispensable for physiological and malignant haematopoiesis. Leukemia 2018, 32, 2037–2040. [Google Scholar] [CrossRef] [Green Version]
- Lebid, A.; Chung, L.; Pardoll, D.M.; Pan, F. YAP Attenuates CD8 T Cell-Mediated Anti-tumor Response. Front. Immunol. 2020, 11, 580. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yu, S.; Zhao, H.; Sun, X.; Li, X.; Wang, P.; Xiong, X.; Hong, L.; Xie, C.; Gao, J.; et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 2017, 18, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.P.; Majedi, F.S.; Thauland, T.J.; Butte, M.J. Mechanosensing through YAP controls T cell activation and metabolism. J. Exp. Med. 2020, 217, 20200053. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Sorrentino, G.; Zannini, A.; Caroli, J.; Beneventano, D.; Anderlucci, L.; Lolli, M.L.; Bicciato, S.; Del Sal, G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget 2015, 6, 38854–38865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, L.; Pecci, F.; Hurkmans, D.P.; Belderbos, R.A.; Lanese, A.; Copparoni, C.; Aerts, S.; Cornelissen, R.; Dumoulin, D.W.; Fiordoliva, I.; et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur. J. Cancer 2021, 144, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Okuma, Y.; Hakozaki, T.; Hosomi, Y. Statins improve survival in patients previously treated with nivolumab for advanced non-small cell lung cancer: An observational study. Mol. Clin. Oncol. 2018, 10, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.M.; Lee, F.Y.F.; Jones, R.T.; Kimball, A.K.; Saravia, E.; Graziano, R.F.; Coleman, B.; Menard, K.; Yan, J.; Michaud, E.; et al. Targeting DDR2 enhances tumor response to anti–PD-1 immunotherapy. Sci. Adv. 2019, 5, eaav2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Van Rensburg, H.J.; Rensburg, H.J.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; et al. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.-A.; Park, S.-H.; Shin, E.-C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Miao, J.; Hsu, P.C.; Yang, Y.-L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP regulates PD-L1 expression in human NSCLC cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Peng, Z.; Qin, M.; Liu, Y.; Wang, J.; Zhang, C.; Lin, J.; Dong, T.; Wang, L.; Li, S.; et al. Interferon-gamma induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol. Cell 2021, 81, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Maehama, T.; Nishio, M.; Goto, H.; Kato, W.; Omori, H.; Miyachi, Y.; Togashi, H.; Shimono, Y.; Suzuki, A. Targeting the Hippo signalling pathway for cancer treatment. J. Biochem. 2017, 161, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.S.; Kim, S.M.; Lee, H. The Hippo signaling pathway provides novel anti-cancer drug targets. Oncotarget 2017, 8, 16084–16098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, R.; Yu, F.-X. Targeting the Hippo Pathway for Anti-cancer Therapies. Curr. Med. Chem. 2015, 22, 4104–4117. [Google Scholar] [CrossRef]
- Holden, J.; Cunningham, C. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Yu, J.; Tomchick, D.R.; Pan, D.; Luo, X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. USA 2010, 107, 7293–7298. [Google Scholar] [CrossRef] [Green Version]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (TEADs). J. Med. Chem. 2018, 61, 5057–5072. [Google Scholar] [CrossRef]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ–TEAD Protein–Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.J.; Bronner, S.M.; Zbieg, J.R. Hippo pathway inhibition by blocking the YAP/TAZ–TEAD interface: A patent review. Expert Opin. Ther. Patents 2018, 28, 867–873. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.-Y.; Kang, C.; Chia, C.S.B.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, L. Benzodiazepines Derivative and its preparation method and application. CN109734676A, 29 January 2019. [Google Scholar]
- Lu, W.; Wang, J.; Li, Y.; Tao, H.; Xiong, H.; Lian, F.; Gao, J.; Ma, H.; Lu, T.; Zhang, D.; et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur. J. Med. Chem. 2019, 184, 111767. [Google Scholar] [CrossRef]
- Bum-Erdene, K.; Zhou, D.; Gonzalez-Gutierrez, G.; Ghozayel, M.K.; Si, Y.; Xu, D.; Shannon, H.E.; Bailey, B.J.; Corson, T.W.; Pollok, K.E.; et al. Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEAD⋅Yap Protein-Protein Interaction. Cell Chem. Biol. 2019, 26, 378–389.e13. [Google Scholar] [CrossRef]
- Meroueh, S.; Bum-Erdene, K. Compounds and Methods to Attenuate Tumor Progression and Metastasis. WO2020087063A1, 30 April 2020. [Google Scholar]
- Pal, R.; Kumar, A.; Misra, G. Exploring TEAD2 as a drug target for therapeutic intervention of cancer: A multi-computational case study. Brief. Bioinform. 2021, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Akbarzadeh, M.; Adihou, H.; Hahne, G.; Pobbati, A.V.; Ng, E.Y.; Guéret, S.M.; Sievers, S.; Pahl, A.; Metz, M.; et al. Discovery of Covalent Inhibitors Targeting the Transcriptional Enhanced Associate Domain Central Pocket. J. Med. Chem. 2020, 63, 11972–11989. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Abbineni, C.; Talluri, K.C.; Panigrahi, S.K.; Wu, X.; Jarugumilli, K.G.; Sun, Y. Novel small molecule inhibitors of tead transcription factors. WO2020190774A1, 24 September 2020. [Google Scholar]
- Richalet, F.; Weiler, S.; Reinelt, S.; Groner, A.; Lane, H.; Nuoffer, C. 1,2,4-Oxadiazol-5-one Derivatives for the Treatment of Cancer. WO2021018869A1, 4 February 2021. [Google Scholar]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Beroza, P.P.; Crawford, J.J.; Lee, W.; Rene, O.; Zbeig, J.R.; Liao, J.; Wang, T.; Yu, C. Carboxamide and Sulfonamide Derivatives Useful as TEAD Modulators. WO2020051099A1, 12 March 2020. [Google Scholar]
- Zbieg, J.R.; Crawford, J.J.; Cunningham, C.N. Therapeutic compounds and methods of use. WO2021097110A1, 20 May 2021. [Google Scholar]
- Zbieg, J.R.; Beroza, P.P.; Crawford, J.J. Therapeutic compounds. WO2019232216A1, 5 December 2019. [Google Scholar]
- Zbieg, J.R. Discovery of GNE-7883, a novel reversable pan-TEAD binder which functions as an allosteric inhibitor against YAP/TAZ: Hit Identification, rational design and in vivo PK/PD results. In Proceedings of the American Chemical Society National Meeting (Spring 2021), Virtual, 5–16 April 2021. [Google Scholar]
- Castro, A.C. TEAD Inhibitors and Uses Thereof. WO2020243423A1, 3 December 2020. [Google Scholar]
- Castro, A.C. TEAD Inhibitors and Uses Thereof. WO2020243415A2, 3 December 2020. [Google Scholar]
- Danjo, T.; Yamada, H.; Nakajima, T. Preparation of α,β-Unsaturated Amide Compounds Having Anti-Cancer Activity; Kyowa Hakko Kirin Co., Ltd.: Tokyo, Japan, 2018; WO2018235926A1. [Google Scholar]
- Gray, N.S.; Zhang, T.; Liu, Y.; Fan, M.; Gao, Y. Transcriptional Enhanced Associate Domain (TEAD) Transcription Factor Inhibitors and Uses Thereof; Dana-Farber Cancer Institute, Inc.: Boston, MA, USA, 2020; WO2020081572A1. [Google Scholar]
- Pobbati, A.V.; Mejuch, T.; Chakraborty, S.; Karatas, H.; Bharath, S.R.; Guéret, S.M.; Goy, P.-A.; Hahne, G.; Pahl, A.; Sievers, S.; et al. Identification of Quinolinols as Activators of TEAD-Dependent Transcription. ACS Chem. Biol. 2019, 14, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Konradi, A.W.; Lin, T.T.-L.T. Bicyclic compounds. WO2020097389A1, 14 May 2020. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Benzosulfonyl Compounds. WO2019040380A1, 28 February 2019. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Oxadiazole Compounds. WO2019222431A1, 21 November 2019. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Bicyclic Compounds. WO2020214734A1, 22 October 2020. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Benzocarbonyl Compounds. WO2019113236A1, 13 June 2019. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Non-Fused Tricyclic Compounds. WO2018204532A1, 8 November 2018. [Google Scholar]
- Konradi, A.W.; Lin, T.T.-L.T. Heteroaryl compounds. WO2021102204A1, 27 May 2021. [Google Scholar]
- Lin, T.T.-L.T.; Konradi, A.W.; Vacca, J.; Shen, W.; Coburn, C. Preparation of Tricyclic Heterocyclic Compounds that are Useful for Treating Cancers or Congenital Diseases. WO2017058716A1, 6 April 2017. [Google Scholar]
- Lim, H.J.; Park, S.J.; Lee, C.H.; No, K.T.; Choi, J.; Jeung, H.-C.; Shin, Y.; Kim, J.W.; Jin, X. Compound inhibiting yap-tead binding, and pharmaceutical composition for preventing or treating cancer, comprising compound as active ingredient. WO2020096416A1, 14 May 2020. [Google Scholar]
- Gibault, F.; Coevoet, M.; Sturbaut, M.; Farce, A.; Renault, N.; Allemand, F.; Guichou, J.F.; Drucbert, A.S.; Foulon, C.; Magnez, R.; et al. Toward the Discovery of a Novel Class of YAP(-)TEAD Interaction Inhibitors by Virtual Screening Approach Targeting YAP(-)TEAD Protein(-)Protein Interface. Cancers 2018, 10, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.T.; Holmes, B.; Benavides-Serrato, A.; Kumar, S.; Nishimura, R.N.; Gera, J. Targeting the YAP-TEAD interaction interface for therapeutic intervention in glioblastoma. J. Neuro-Oncol. 2021, 152, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Adihou, H.; Gopalakrishnan, R.; Förster, T.; Guéret, S.M.; Gasper, R.; Geschwindner, S.; García, C.C.; Karatas, H.; Pobbati, A.V.; Vazquez-Chantada, M.; et al. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun. 2020, 11, 5425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, D.; Pan, X.; Liu, L. Rational Design and Intramolecular Cyclization of Hotspot Peptide Segments at YAP-TEAD4 Complex Interface. Protein Pept. Lett. 2020, 27, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Kunig, V.B.K.; Potowski, M.; Akbarzadeh, M.; Klika Škopić, M.; dos Santos Smith, D.; Arendt, L.; Dormuth, I.; Adihou, H.; Andlovic, B.; Karatas, H.; et al. TEAD-YAP Interaction Inhibitors and MDM2 Binders from DNA-Encoded Indole-Focused Ugi Peptidomimetics. Angew. Chem. Int. Ed. Engl. 2020, 59, 20338–20342. [Google Scholar] [CrossRef]
- Smith, S.A.; Sessions, R.B.; Shoemark, D.K.; Williams, C.; Ebrahimighaei, R.; McNeill, M.C.; Crump, M.P.; McKay, T.R.; Harris, G.; Newby, A.C.; et al. Antiproliferative and Antimigratory Effects of a Novel YAP-TEAD Interaction Inhibitor Identified Using in Silico Molecular Docking. J. Med. Chem. 2019, 62, 1291–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Li, Y.; Song, J.; Li, C. Fluorescence polarization assay for the identification and evaluation of inhibitors at YAP-TEAD protein-protein interface. Anal. Biochem. 2019, 586, 113413. [Google Scholar]
- Nouri, K.; Azad, T.; Ling, M.; Van Rensburg, H.J.J.; Pipchuk, A.; Shen, H.; Hao, Y.; Zhang, J.; Yang, X. Identification of Celastrol as a Novel YAP-TEAD Inhibitor for Cancer Therapy by High Throughput Screening with Ultrasensitive YAP/TAZ-TEAD Biosensors. Cancers 2019, 11, 1596. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, L.; Tian, R.; Dong, Q.; Li, P.; Yan, Z.; Yang, X.; Huo, J.; Fei, Z.; Zhen, H. Truncated TEAD-binding protein of TAZ inhibits glioma survival through the induction of apoptosis and repression of epithelial-mesenchymal transition. J. Cell Biochem. 2019, 120, 17337–17344. [Google Scholar] [CrossRef]
- He, B.; Wu, T.; He, P.; Lv, F.; Liu, H. Structure-based derivation and optimization of YAP-like coactivator-derived peptides to selectively target TEAD family transcription factors by hydrocarbon stapling and cyclization. Chem. Biol. Drug Des. 2021, 97, 1129–1136. [Google Scholar] [CrossRef]
- Dominguez-Berrocal, L.; Cirri, E.; Zhang, X.; Andrini, L.; Marin, G.H.; Lebel-Binay, S.; Rebollo, A. New Therapeutic Approach for Targeting Hippo Signalling Pathway. Sci. Rep. 2019, 9, 4771. [Google Scholar] [CrossRef]
- Furet, P.; Salem, B.; Mesrouze, Y.; Schmelzle, T.; Lewis, I.; Kallen, J.; Chène, P. Structure-based design of potent linear peptide inhibitors of the YAP-TEAD protein-protein interaction derived from the YAP omega-loop sequence. Bioorg. Med. Chem. Lett. 2019, 29, 2316–2319. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.; Schneible, J.; Bacon, K.; Labar, C.; Menegatti, S.; Rao, B. Screening of Yeast Display Libraries of Enzymatically Treated Peptides to Discover Macrocyclic Peptide Ligands. Int. J. Mol. Sci. 2021, 22, 1634. [Google Scholar] [CrossRef] [PubMed]
- Kaan, H.Y.K.; Sim, A.Y.L.; Tan, S.K.J.; Verma, C.; Song, H. Targeting YAP/TAZ-TEAD protein-protein interactions using fragment-based and computational modeling approaches. PLoS ONE 2017, 12, e0178381. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Jia, Y.; Lou, H.; Ma, J.; Huang, Q.; Meng, Y.; Sun, C.; Yang, Z.; Li, X.; Xu, S.; et al. Targeting YAP suppresses ovarian cancer progression through regulation of the PI3K/Akt/mTOR pathway. Oncol. Rep. 2019, 42, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.M.; Crook, Z.; Bradley, P.H. Peptide Compositions and Methods of Use Thereof for Disrupting TEAD Interactions. WO2018136614A1, 26 July 2018. [Google Scholar]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.-Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Ng, E.Y.; Li, R.; Poulsen, A.; Hill, J.; Pobbati, A.V.; Hung, A.W.; Hong, W.; Keller, T.H.; et al. Structural and ligand-binding analysis of the YAP-binding domain of transcription factor TEAD4. Biochem. J. 2018, 475, 2043–2055. [Google Scholar] [CrossRef]
- Zheng, W.; Lan, J.; Feng, L.; Chen, Z.; Feng, S.; Gao, Y.; Ren, F.; Chen, Y. Structure-Based Optimization of Conformationally Constrained Peptides to Target Esophageal Cancer TEAD Transcription Factor. Int. J. Pept. Res.Ther. 2020, 27, 923–930. [Google Scholar] [CrossRef]
- Wu, D.; Luo, L.; Yang, Z.; Chen, Y.; Quan, Y.; Min, Z. Targeting Human Hippo TEAD Binding Interface with YAP/TAZ-Derived, Flexibility-Reduced Peptides in Gastric Cancer. Int. J. Pept. Res.Ther. 2020, 27, 119–128. [Google Scholar] [CrossRef]
- Gao, S.; Wang, Y.; Ji, L. Rational design and chemical modification of TEAD coactivator peptides to target hippo signaling pathway against gastrointestinal cancers. J. Recept. Signal Transduct. 2021, 41, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Barth, M.; Contal, S.; Montalbetti, C.; Spitzer, L. New compounds inhibitors of the yap/taz-tead interaction and their use in the treatment of malignant mesothelioma. WO2017064277A1, 20 April 2017. [Google Scholar]
- Barth, M.; Contal, S. New compounds inhibitors of the yap/taz-tead interaction and their use in the treatment of malignant mesothelioma. WO2018185266A1, 11 October 2018. [Google Scholar]
- Barth, M.; Contal, S.; Junien, J.-L.; Massardier, C.; Montalbetti, C.; Soude, A. Inhibitors of the yap/taz-tead interaction and their use in the treatment of cancer. EP3632908A1, 8 April 2020. [Google Scholar]
- Barth, M.; Contal, S.; Junien, J.-L.; Massardier, C.; Montalbetti, C.; Soude, A. Inhibitors of the yap/taz-tead interaction and their use in the treatment of cancer. WO2020070181A1, 9 April 2020. [Google Scholar]
- Bailly, F.; Gibault, F.; Sturbaut, M.; Coevoet, M.; Pugnière, M.; Burtscher, A.; Allemand, F.; Melnyk, P.; Hong, W.; Rubin, B.P. Design, Synthesis and Evaluation of a Series of 1,5-Diaryl-1,2,3-triazole-4-carbohydrazones as Inhibitors of the YAP-TAZ/TEAD Complex. ChemMedChem 2021, 16, 1–23. [Google Scholar]
- Wu, X. TEAD Transcription Factor Autopalmitoylation Inhibitors. WO2017053706A1, 30 March 2017. [Google Scholar]
- Kim, Y.; Luo, X.; MacLeod, R.; Freier, S.M.; Bui, H.H. Modulators of YAP1 Expression. WO2020160453A1, 6 August 2020. [Google Scholar]
- Macleod, R. The discovery and characterization of ION-537: A next generation antisense oligonucleotide inhibitor of YAP1 in preclinical cancer models. In Proceedings of the AACR Annual Meeting 2021, Virtual, 10–15 April and 17–21 May 2021. [Google Scholar]
- Bordas, V. Biaryl Derivatives as YAP/TAZ-TEAD Protein-Protein Interaction Inhibitors. WO2021186324A1, 23 September 2021. [Google Scholar]
- Han, Z.; Ruthel, G.; Dash, S.; Berry, C.T.; Freedman, B.D.; Harty, R.N.; Shtanko, O. Angiomotin regulates budding and spread of Ebola virus. J. Biol. Chem. 2020, 295, 8596–8601. [Google Scholar] [CrossRef] [PubMed]
- Genovese, N.J.; Firpo, M.T.; Dambournet, D. Compositions and Methods for Increasing the Culture Density of a Cellular Biomass within a Cultivation Infrastructure. WO2018208628A1, 15 November 2018. [Google Scholar]
Figure 1.
(A) In the Hippo-off state, unphosphorylated YAP1/TAZ accumulates in the nucleus and interacts with TEADs to drive the induction of genes involved in proliferation and cell survival. (B) Druggability assessment of the YAP1/TAZ–TEAD protein–protein interaction (PPI) has identified several druggable pockets amenable to small- and large-molecule inhibition, including interfaces 2 and 3, as well as the central lipophilic palmitate pocket (PDB: 5emv, 4re1 and 5gn0).
Figure 1.
(A) In the Hippo-off state, unphosphorylated YAP1/TAZ accumulates in the nucleus and interacts with TEADs to drive the induction of genes involved in proliferation and cell survival. (B) Druggability assessment of the YAP1/TAZ–TEAD protein–protein interaction (PPI) has identified several druggable pockets amenable to small- and large-molecule inhibition, including interfaces 2 and 3, as well as the central lipophilic palmitate pocket (PDB: 5emv, 4re1 and 5gn0).

Figure 2.
Multifaceted role of YAP1/TEAD in driving tumor growth and/or progression. Amp = amplification; LoF = loss of function; ALK/ROS1i, EGFRi, BRAFi, KRASi, MEK1/2i = inhibitors of ALK/ROS, EGFR, BRAF, KRAS and MEK.
Figure 2.
Multifaceted role of YAP1/TEAD in driving tumor growth and/or progression. Amp = amplification; LoF = loss of function; ALK/ROS1i, EGFRi, BRAFi, KRASi, MEK1/2i = inhibitors of ALK/ROS, EGFR, BRAF, KRAS and MEK.

Figure 3.
Structures of representative compounds with claims to disrupt the YAP1–TEAD interaction.


Table 2.
Recently initiated clinical trials to inhibit TEAD-driven transcription in cancers.
| Compound (Ref.) | Structure | Phase | Disease Indication | Sponsor (Trial Number) |
|---|---|---|---|---|
| VT3989 | Not disclosed | I |
| Vivace Therapeurics (NCT04665206) |
| ION537 [147,148] | AksAdsGdsTdsGdsTdsAdsTdsGdsTdsmGdsAksGesAksAesGk | I | ||
| Ionis Pharmaceuticals (NCT04659096) | |||
| IAG933 | Not disclosed | I |
| Novartis (NCT04857372) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10, 2715. https://doi.org/10.3390/cells10102715
AMA Style
Barry ER, Simov V, Valtingojer I, Venier O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells. 2021; 10(10):2715. https://doi.org/10.3390/cells10102715
Chicago/Turabian StyleBarry, Evan R., Vladimir Simov, Iris Valtingojer, and Olivier Venier. 2021. "Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine" Cells 10, no. 10: 2715. https://doi.org/10.3390/cells10102715
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.