
Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NF-κB
2.1. NF-κB Subunits
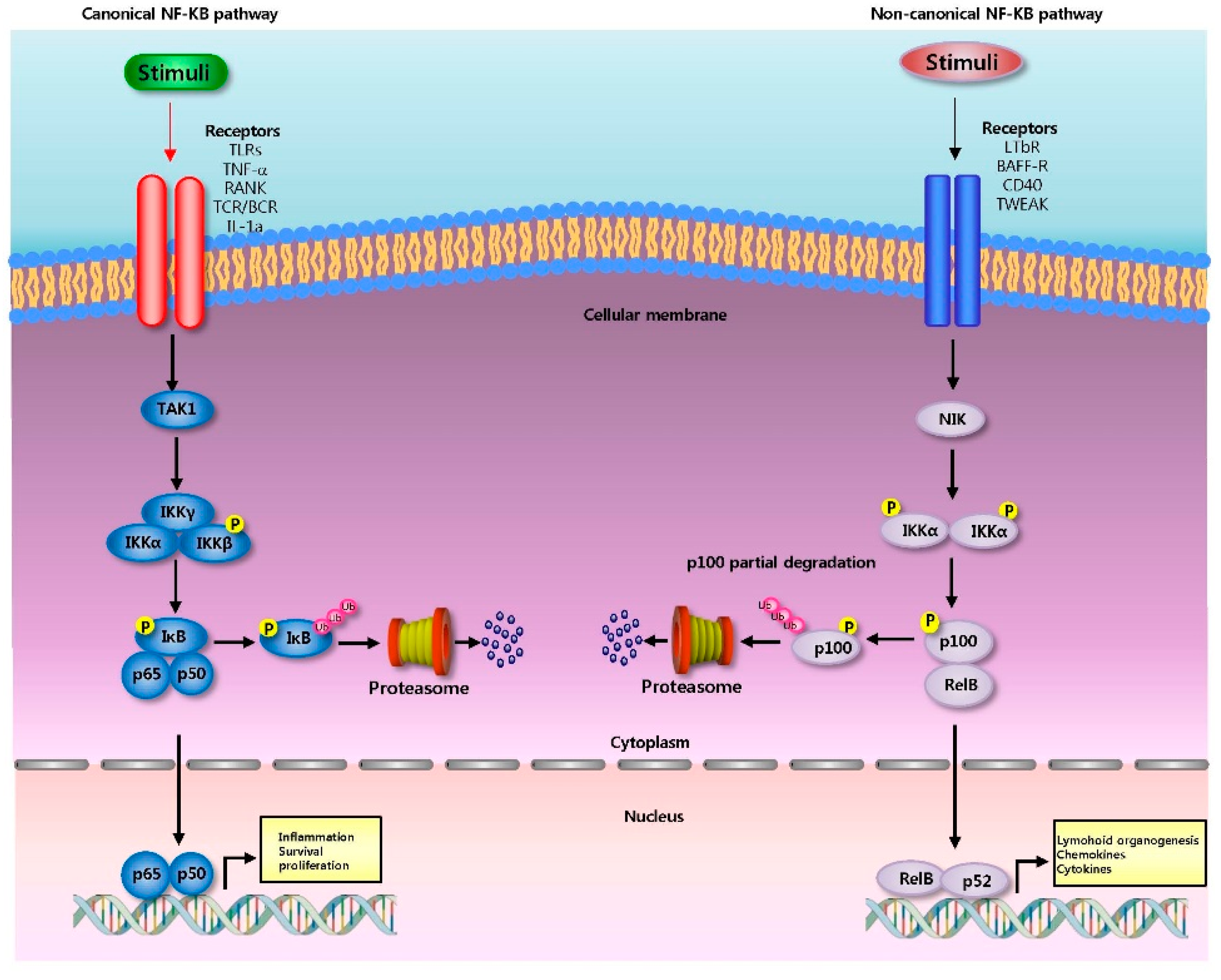
2.2. NF-κB Signaling Pathway
2.2.1. Canonical Pathway
2.2.2. Non-Canonical Pathway
3. Role of NF-κB in Diseases
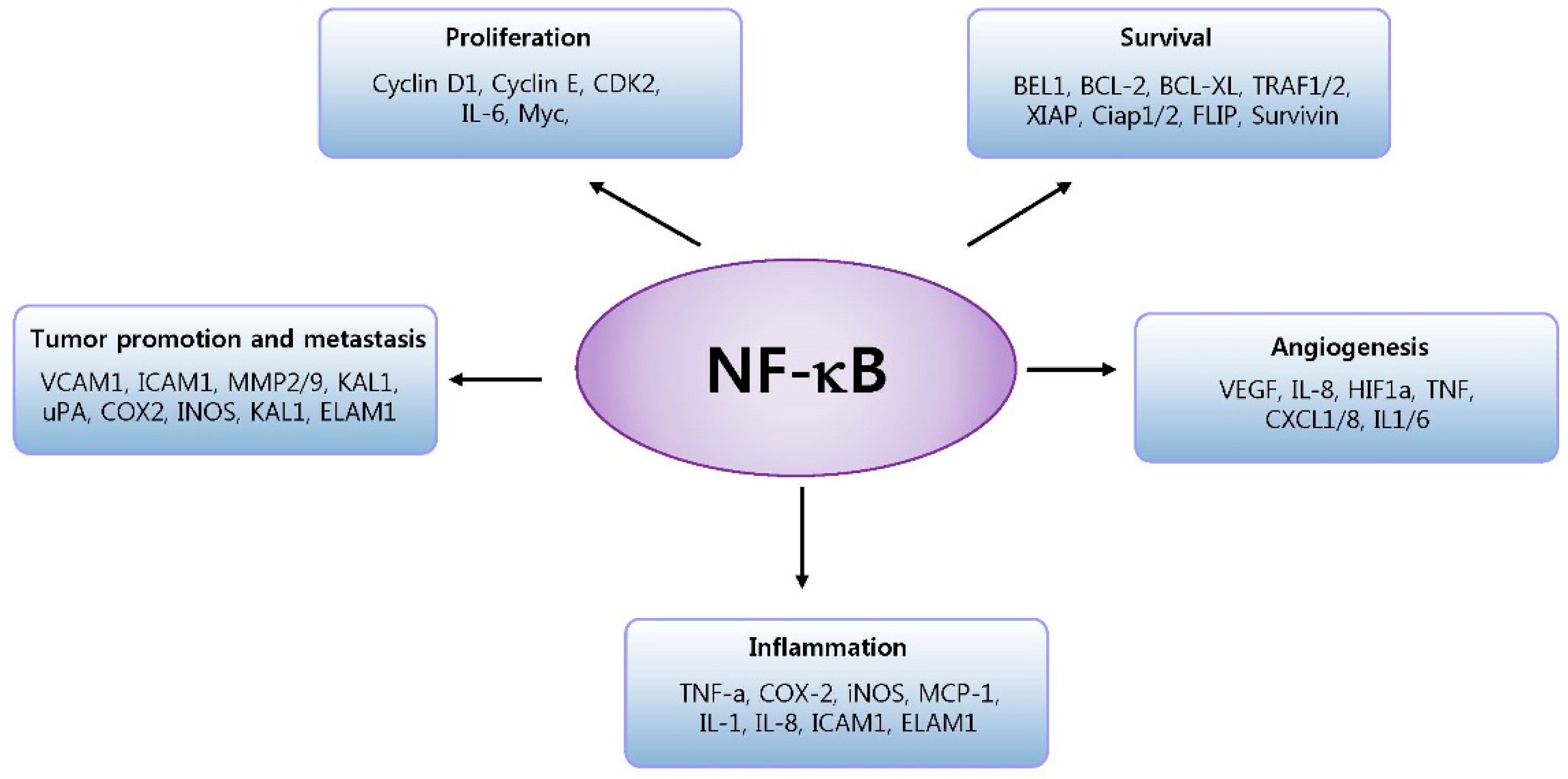
3.1. NF-κB and Cancer
3.2. NF-κB and Inflammatory Disease
4. Therapeutic Approaches for Targeting NF-κB
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Levchenko, A.; Scott, M.L.; Baltimore, D. The IκB-NF-κB signaling module: Temporal control and selective gene activation. Science 2002, 298, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Solan, N.J.; Miyoshi, H.; Carmona, E.M.; Bren, G.D.; Paya, C.V. Relb cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem. 2002, 277, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-κB in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, A.M.; Szakmary, A.; Schiestl, R.H. Mechanisms of intestinal inflammation and development of associated cancers: Lessons learned from mouse models. Mutat. Res. 2010, 705, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-κB in aging and disease. Aging Dis. 2011, 2, 449–465. [Google Scholar] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.-C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Hou, S.; Ricciardi, R.P. DNA binding of repressor nuclear factor-κB p50/p50 depends on phosphorylation of Ser337 by the protein kinase a catalytic subunit. J. Biol. Chem. 2005, 280, 9957–9962. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth IκB protein within the NF-κB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Basak, S.; James, E.S.; Quiambo, R.S.; Kinsella, M.C.; Alegre, M.-L.; Weih, F.; Franzoso, G.; Hoffmann, A.; Fu, Y.-X. Coordination between NF-κB family members p50 and p52 is essential for mediating ltβr signals in the development and organization of secondary lymphoid tissues. Blood 2006, 107, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glöckner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of nemo with the IκB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. Nf-[κ]b at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Delhase, M. The IκB kinase (IKK) and NF-κB: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Harhaj, E.W.; Sun, S.-C. NF-κB-inducing kinase regulates the processing of NF-kB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Coope, H.J.; Atkinson, P.G.P.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.J.; Klaus, G.G.B.; Johnston, L.H.; Ley, S.C. Cd40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef] [PubMed]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Körner, M.; Baud, V. Relb/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.-T.; Gong, X.-J.; Sun, H.-B.; Li, Y.-M.; Ji, H. Protective effects of oleanolic acid on cerebral ischemic damage in vivo and H2O2-induced injury in vitro. Pharm. Biol. 2011, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. Baff-induced nemo-independent processing of NF-κB2 in maturing b cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Novack, D.V.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S.L. The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by relb:p52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Shih, V.F.-S.; Hoffmann, A. Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system. Mol. Cell. Biol. 2008, 28, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-κB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. Nf-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46, 705–716. [CrossRef]
- Nelson, D.E.; Ihekwaba, A.E.C.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; et al. Oscillations in NF-κB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Ravindran, J.; Aggarwal, B.B. NF-κB and cancer: How intimate is this relationship. Mol. Cell. Biochem. 2010, 336, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Guttridge, D.C.; Albanese, C.; Reuther, J.Y.; Pestell, R.G.; Baldwin, A.S. NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin d1. Mol. Cell. Biol. 1999, 19, 5785–5799. [Google Scholar] [CrossRef] [PubMed]
- Escárcega, R.O.; Fuentes-Alexandro, S.; García-Carrasco, M.; Gatica, A.; Zamora, A. The transcription factor nuclear factor-κB and cancer. Clin. Oncol. 2007, 19, 154–161. [Google Scholar] [CrossRef]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-κB function in growth control: Regulation of cyclin D1 expression and G(0)/G(1)-to-S-phase transition. Mol. Cell. Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Bonizzi, G.; Seagroves, T.N.; Greten, F.R.; Johnson, R.; Schmidt, E.V.; Karin, M. IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 2001, 107, 763–775. [Google Scholar] [CrossRef]
- Beg, A.A.; Baltimore, D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-α-induced apoptosis by NF-κB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.R.; Liu, J.; Bock, P.J.; Schols, D.; Coffey, M.J.; Strieter, R.M.; Polverini, P.J.; Markovitz, D.M. Interleukin-8 and growth-regulated oncogene alpha mediate angiogenesis in Kaposi’s sarcoma. J. Virol. 2002, 76, 11570–11583. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Lopez-Otin, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Cohen, J.; Arun, P.; Chen, Z.; Van Waes, C. NF-κB in carcinoma therapy and prevention. Expert Opin. Ther. Targets 2008, 12, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Sovak, M.A.; Bellas, R.E.; Kim, D.W.; Zanieski, G.J.; Rogers, A.E.; Traish, A.M.; Sonenshein, G.E. Aberrant nuclear factor-κB/rel expression and the pathogenesis of breast cancer. J. Clin. Investig. 1997, 100, 2952–2960. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J.; Sledge, G.W. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, S.; Vyas, D.; Hollis, M.; Aekka, A.; Vyas, A. Nuclear factor κB role in inflammation associated gastrointestinal malignancies. World J. Gastroenterol. 2015, 21, 3174–3183. [Google Scholar] [PubMed]
- Lind, D.S.; Hochwald, S.N.; Malaty, J.; Rekkas, S.; Hebig, P.; Mishra, G.; Moldawer, L.L.; Copeland, E.M., III; MacKay, S. Nuclear factor-κB is upregulated in colorectal cancer. Surgery 2001, 130, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Bardhan, K.; Yang, D.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Liles, G.B.; Lee, J.R.; Liu, K. NF-κB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J. Biol. Chem. 2012, 287, 25530–25540. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, J.; Hunter, C.A. NF-κB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.D.; Clark, M.R. B-cell antigen-receptor signalling in lymphocyte development. Immunology 2003, 110, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Kortum, R.L.; Rouquette-Jazdanian, A.K.; Miyaji, M.; Merrill, R.K.; Markegard, E.; Pinski, J.M.; Wesselink, A.; Nath, N.N.; Alexander, C.P.; Li, W.; et al. A PLC-γ1-independent, RasGRP1-ERK dependent pathway drives lymphoproliferative disease in LAT-Y136F mutant mice. J. Immunol. 2013, 190, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Diaz-Meco, M.T.; Rennert, P. NF-κB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003, 4, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor κB activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Gona, P.; Larson, M.G.; Massaro, J.M.; Lipinska, I.; Keaney, J.F.; Rong, J.; Corey, D.; Hoffmann, U.; Fox, C.S.; et al. Biomarkers of the osteoprotegerin pathway: Clinical correlates, subclinical disease, incident cardiovascular disease and mortality. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-Q.; Lv, L.-X.; Li, W.-Q.; Hao, Y.-H.; Zhao, J.-P. The interaction of nuclear factor-κB and cytokines is associated with schizophrenia. Biol. Psychiatry 2009, 65, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vidal, P.M.; Lemmens, E.; Dooley, D.; Hendrix, S. The role of “anti-inflammatory” cytokines in axon regeneration. Cytokine Growth Factor Rev. 2013, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A.; Cen, O.; Hengen, N.; Agan, J.; Moschovi, M.; Critselis, E.; Adamaki, M.; Bacopoulou, F.; Copland, J.A.; Boldogh, I.; et al. Dynamic aberrant NF-κB spurs tumorigenesis: A new model encompassing the microenvironment. Cytokine Growth Factor Rev. 2015, 26, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bonavita, E.; Galdiero, M.R.; Jaillon, S.; Mantovani, A. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation. Adv. Cancer Res. 2015, 128, 141–171. [Google Scholar] [PubMed]
- Mecollari, V.; Nieuwenhuis, B.; Verhaagen, J. A perspective on the role of class III semaphorin signaling in central nervous system trauma. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Matsushima-Nishiwaki, R.; Yamaguchi, S.; Iida, H.; Dohi, S.; Kozawa, O. Mechanisms of tumor necrosis factor-α-induced interleukin-6 synthesis in glioma cells. J. Neuroinflamm. 2010, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Rice, S.E.; Bhat-Nakshatri, P. Antitumor agent parthenolide reverses resistance of breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through sustained activation of C-jun N-terminal kinase. Oncogene 2004, 23, 7330–7344. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Arbiser, J.L.; Weitzmann, M.N. Honokiol stimulates osteoblastogenesis by suppressing NF-κB activation. Int. J. Mol. Med. 2011, 28, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; Di Paola, R.; Suzuki, H.; Paterniti, I.; Ahmad, A.; Mariotto, S.; Cuzzocrea, S. Costunolide and dehydrocostuslactone, two natural sesquiterpene lactones, ameliorate the inflammatory process associated to experimental pleurisy in mice. Eur. J. Pharmacol. 2014, 730, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Whipple, R.A.; Vitolo, M.I.; Boggs, A.E.; Charpentier, M.S.; Thompson, K.; Martin, S.S. Parthenolide and costunolide reduce microtentacles and tumor cell attachment by selectively targeting detyrosinated tubulin independent from NF-κB inhibition. Breast Cancer Res. 2013, 15, R83. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Ho, O.-M.; Kim, D.-W. Magnolol attenuates neointima formation by inducing cell cycle arrest via inhibition of ERK1/2 and NF-κB activation in vascular smooth muscle cells. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, S.; Wang, M.-W. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/AkT/caspase and Fas-L/NF-κB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol. In Vitro 2010, 24, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; He, H.; Ye, Y.; Liu, W.; Fan, S.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Nitric oxide augments oridonin-induced efferocytosis by human histocytic lymphoma U937 cells via autophagy and the NF-κB-COX-2-IL-1β pathway. Free Radic. Res. 2012, 46, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Huang, H.; Zhao, S.; Liu, W.; Liu, C.-X.; Chen, L.; Li, J.-M.; Wu, Y.-L.; Yan, H. Alantolactone induces apoptosis in chronic myelogenous leukemia sensitive or resistant to imatinib through NF-κB inhibition and Bcr/Abl protein deletion. Apoptosis 2013, 18, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.-Q.; Wang, J.-C.; Long, J.; Zhao, Y.; Chen, S.-J.; Zhai, J.-D.; Wei, L.-B.; Zhang, Q.; Chen, Y.; Long, H.-B. Sesquiterpene lactones and their derivatives inhibit high glucose-induced NF-κB activation and MCP-1 and TGF-β1 expression in rat mesangial cells. Molecules 2013, 18, 13061–13077. [Google Scholar] [CrossRef] [PubMed]
- Koh, D.-J.; Ahn, H.-S.; Chung, H.-S.; Lee, H.; Kim, Y.; Lee, J.-Y.; Kim, D.-G.; Hong, M.; Shin, M.; Bae, H. Inhibitory effects of casticin on migration of eosinophil and expression of chemokines and adhesion molecules in A549 lung epithelial cells via NF-κB inactivation. J. Ethnopharmacol. 2011, 136, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wong, V.K.W.; Yi, X.Q.; Wong, Y.F.; Zhou, H.; Liu, L. Pseudolaric acid b suppresses T lymphocyte activation through inhibition of NF-κB signaling pathway and p38 phosphorylation. J. Cell. Biochem. 2009, 108, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Bae, E.-A.; Park, E.-K.; Shin, Y.-W.; Baek, N.-I.; Han, E.-J.; Chung, H.-G.; Kim, D.-H. Inhibitory effect of eupatilin and jaceosidin isolated from artemisia princeps in IgE-induced hypersensitivity. Int. Immunopharmacol. 2007, 7, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Roy, S.; Packer, L. Involvement of intracellular Ca2+ in oxidant-induced NF-κB activation. FEBS Lett. 1996, 385, 58–62. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Beyaert, R.; Vandevoorde, V.; Haegeman, G.; Fiers, W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993, 12, 3095–3104. [Google Scholar] [PubMed]
- Manna, S.K.; Zhang, H.J.; Yan, T.; Oberley, L.W.; Aggarwal, B.B. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-κB and activated protein-1. J. Biol. Chem. 1998, 273, 13245–13254. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Aggarwal, B.B. Protein-tyrosine phosphatase inhibitors block tumor necrosis factor-dependent activation of the nuclear transcription factor NF-κB. J. Biol. Chem. 1995, 270, 10631–10639. [Google Scholar] [PubMed]
- Kopp, E.; Ghosh, S. Inhibition of NF-κB by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Bhardwaj, A.; Potdar, P.; Aggarwal, B.B. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-κB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 2004, 23, 9247–9258. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Pizzi, M.; Memo, M.; Spano, P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science 1996, 274, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Palayoor, S.T.; Bump, E.A.; Calderwood, S.K.; Bartol, S.; Coleman, C.N. Combined antitumor effect of radiation and ibuprofen in human prostate carcinoma cells. Clin. Cancer Res. 1998, 4, 763–771. [Google Scholar] [PubMed]
- Ettarh, R.; Cullen, A.; Calamai, A. Nsaids and cell proliferation in colorectal cancer. Pharmaceuticals 2010, 3, 2007–2021. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar] [PubMed]
- Paikin, J.S.; Eikelboom, J.W. Aspirin. Circulation 2012, 125, e439–e442. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Ittaman, S.V.; VanWormer, J.J.; Rezkalla, S.H. The role of aspirin in the prevention of cardiovascular disease. Clin. Med. Res. 2014, 12, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.-J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-B. Nature 1998, 396, 77–80. [Google Scholar] [PubMed]
- Dikshit, P.; Chatterjee, M.; Goswami, A.; Mishra, A.; Jana, N.R. Aspirin induces apoptosis through the inhibition of proteasome function. J. Biol. Chem. 2006, 281, 29228–29235. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.-W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IKKβ. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Fang, C.H. C-reactive protein is not only an inflammatory marker but also a direct cause of cardiovascular diseases. Med. Hypotheses 2004, 62, 499–506. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. Cross-talk between nuclear receptors and NF-κB. Oncogene 2006, 25, 6868–6886. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzidis, D.; Gilmore, T.D. Transcription factor cross-talk: The estrogen receptor and NF-κB. Trends Endocrinol. Metab. 2005, 16, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Olivier, S.; Close, P.; Castermans, E.; de Leval, L.; Tabruyn, S.; Chariot, A.; Malaise, M.; Merville, M.-P.; Bours, V.; Franchimont, N. Raloxifene-induced myeloma cell apoptosis: A study of NF-κB inhibition and gene expression signature. Mol. Pharmacol. 2006, 69, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Frantz, B.; Nordby, E.C.; Bren, G.; Steffan, N.; Paya, C.V.; Kincaid, R.L.; Tocci, M.J.; O’Keefe, S.J.; O’Neill, E.A. Calcineurin acts in synergy with PMA to inactivate IκB/MAD3, an inhibitor of NF-κB. EMBO J. 1994, 13, 861–870. [Google Scholar] [PubMed]
- Meyer, S.; Kohler, N.G.; Joly, A. Cyclosporine A is an uncompetitive inhibitor of proteasome activity and prevents NF-κB activation. FEBS Lett. 1997, 413, 354–358. [Google Scholar] [CrossRef]
- McCaffrey, P.G.; Kim, P.K.; Valge-Archer, V.E.; Sen, R.; Rao, A. Cyclosporin A sensitivity of the NF-κB site of the IL2R alpha promoter in untransformed murine T cells. Nucleic Acids Res. 1994, 22, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Venkataraman, L.; Shinkai, Y.; Pierce, J.W.; Alt, F.W.; Burakoff, S.J.; Sen, R. Expression and induction of nuclear factor-κB-related proteins in thymocytes. J. Immunol. 1995, 154, 3213–3221. [Google Scholar] [PubMed]
- Hirano, F.; Kobayashi, A.; Makino, I. Inhibition of TNF-α-induced rantes expression in human hepatocyte-derived cells by fibrates, the hypolipidemic drugs. Int. Immunopharmacol. 2003, 3, 225–232. [Google Scholar] [CrossRef]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Ludwiczek, S.; Enrich, B.; Gastl, G.; Weiss, G.; Tilg, H. The kinase inhibitor imatinib mesylate inhibits TNF-α production in vitro and prevents TNF-dependent acute hepatic inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 13622–13627. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.M.; Singh, R.S.; Franklin, D.P.; Carey, D.J.; Elmore, J.R. Rapamycin suppresses experimental aortic aneurysm growth. J. Vasc. Surg. 2004, 40, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Acarin, L.; González, B.; Castellano, B. Oral administration of the anti-inflammatory substance triflusal results in the downregulation of constitutive transcription factor NF-κB in the postnatal rat brain. Neurosci. Lett. 2000, 288, 41–44. [Google Scholar] [CrossRef]
- Ruan, H.; Pownall, H.J.; Lodish, H.F. Troglitazone antagonizes tumor necrosis factor-α-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-κB. J. Biol. Chem. 2003, 278, 28181–28192. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. https://doi.org/10.3390/cells5020015
Park MH, Hong JT. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells. 2016; 5(2):15. https://doi.org/10.3390/cells5020015
Chicago/Turabian StylePark, Mi Hee, and Jin Tae Hong. 2016. "Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches" Cells 5, no. 2: 15. https://doi.org/10.3390/cells5020015