A Narrative Review of Cancer-Related Fatigue (CRF) and Its Possible Pathogenesis
by
 ,
,
Songwei Yang
1,2,3, 
Shifeng Chu
1,2,3,
Yan Gao
3,
Qidi Ai
1,2,3,
Yingjiao Liu
1,2,3,
Xun Li
1,2,3 and
Naihong Chen
1,2,3,* 1
College of Pharmacy, Hunan University of Chinese Medicine, Changsha 410208, Hunan, China
2
Hunan Engineering Technology Center of Standardization and Function of Chinese Herbal Decoction Pieces, Changsha 410208, Hunan, China
3
State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medical, Neuroscience Center, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
*
Author to whom correspondence should be addressed.
Cells 2019, 8(7), 738; https://doi.org/10.3390/cells8070738
Submission received: 23 May 2019
/
Revised: 10 July 2019
/
Accepted: 17 July 2019
/
Published: 18 July 2019
Abstract
:Many cancer patients suffer from severe fatigue when treated with chemotherapy or radiotherapy; however, the etiology and pathogenesis of this kind of fatigue remains unknown. Fatigue is associated with cancer itself, as well as adjuvant therapies and can persist for a long time. Cancer patients present a high degree of fatigue, which dramatically affects the quality of their everyday life. There are various clinical research studies and reviews that aimed to explore the mechanisms of cancer-related fatigue (CRF). However, there are certain limitations in these studies: For example, some studies have only blood biochemical texts without histopathological examination, and there has been insufficient systemic evaluation of the dynamic changes in relevant indexes. Thus, we present this narrative review to summarize previous studies on CRF and explore promising research directions. Plenty of evidence suggests a possible association between CRF and physiological dysfunction, including skeletal muscular and mitochondrial dysfunction, peripheral immune activation and inflammation dysfunction, as well as central nervous system (CNS) disorder. Mitochondrial DNA (mtDNA), mitochondrial structure, oxidative pressure, and some active factors such as ATP play significant roles that lead to the induction of CRF. Meanwhile, several pro-inflammatory and anti-inflammatory cytokines in the peripheral system, even in the CNS, significantly contribute to the occurrence of CRF. Moreover, CNS function disorders, such as neuropeptide, neurotransmitter, and hypothalamic-pituitary-adrenal (HPA) axis dysfunction, tend to amplify the sense of fatigue in cancer patients through various signaling pathways. There have been few accurate animal models established to further explore the molecular mechanisms of CRF due to different types of cancer, adjuvant therapy schedules, living environments, and physical status. It is imperative to develop appropriate animal models that can mimic human CRF and to explore additional mechanisms using histopathological and biochemical methods. Therefore, the main purpose of this review is to analyze the possible pathogenesis of CRF and recommend future research that will clarify CRF pathogenesis and facilitate the formulation of new treatment options.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Background
Fatigue is both a symptom and measurable sign of many diseases, but adequate criteria for the identification of fatigue are lacking. According to the CDC, fatigue is defined by several characteristics: (1) It happens gradually, (2) it can be mitigated by rest, (3) it lasts more than six months [1]. Moreover, fatigue ought to be distinguished from mental fatigue, which is associated with cognitive function disorder and muscle weakness [2]. Sleep studies have revealed that mental fatigue is different from somnolence and is interrelated with sleep disturbances and depression [3]. Muscular fatigue, which often occurs following exhaustive muscular exercise, is caused by the disordered electrophysiological rhythm of muscle relaxation and contraction [4]. Although they are distinct conditions, it is difficult to discriminate between mental and muscular fatigue since mental fatigue can occur with physical effects that are also present in muscular fatigue.
However, with increasing cancer morbidity as well as viable treatments, a different type of fatigue has become regularly recognized as a condition associated with cancer or cancer treatment, namely, cancer-related fatigue (CRF). CRF has been defined as: ”a distressing, persistent, subjective sense of tiredness or exhaustion related to cancer or cancer treatment which is not proportional to recent activity and interferes with usual functioning” [5]. Most cancer survivors are present with severe fatigue after the completion of cancer treatment, despite the lack of any signs of cancer recurrence, indicating that chemotherapy or radiotherapy promotes CRF [6,7]. A cross-sectional population-based study found that 39% of survivors of colorectal cancer (CRC) reported extreme fatigue compared with 22% of the healthy population, and a longitudinal study reported a CRF rate of 10% before therapy and 26% after therapy [8,9]. Thus, severe fatigue seems to occur at higher rates after adjuvant chemotherapy or radiotherapy. Putative mechanisms leading to CRF include cytokines, the hypothalamic-pituitary-adrenal (HPA) axis, autonomic nervous system function, insulin signaling, neurotransmitters, anemia, and psychosocial or medical factors [10]. To date, the underlying molecular mechanisms and treatment-related hazards that lead to CRF remain unknown [11]. To reveal the current knowledge of the molecular mechanisms that induce CRF, and explore directions of further research, we review the existing literature on CRF.
2. Prevalence of CRF and Its Association with Cancer and Cancer Therapy
CRF is a symptom of cancer cachexia, which is triggered by cancer or cancer therapies. Cancer cachexia significantly influences the patient’s quality of life and decreases survival rates [12,13,14]. In 2014, the prevalence of cancer cachexia in cancer patients was estimated to be 50%–80% and accounted for 20% of cancer-related deaths [15]. The main symptoms of cachexia include the decrease of body mass index (BMI) on the account of reduction of skeletal muscle mass, and can’t be alleviated by conventional nutritional support, which naturally induce extremely serious fatigue in cancer patients [16,17]. It was reported that cancer patients always experienced highest levels of fatigue at the end of cancer treatment, meanwhile some female patients felt equivalent severe fatigue throughout their treatment trajectory [18]. Previous studies indicated that CRF was a distressing symptom for female patients with stage I and II breast cancer, and cancer itself or cancer treatment can cause unremitting fatigue [19].
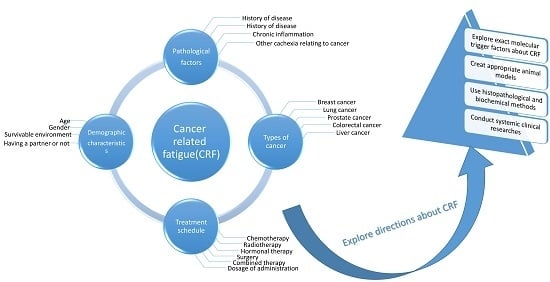
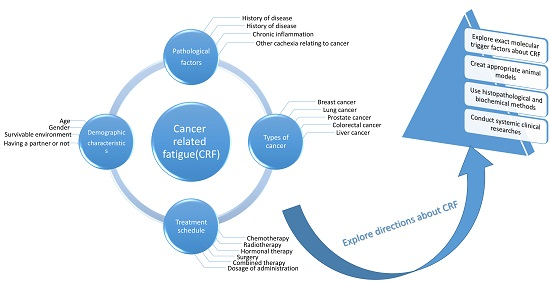
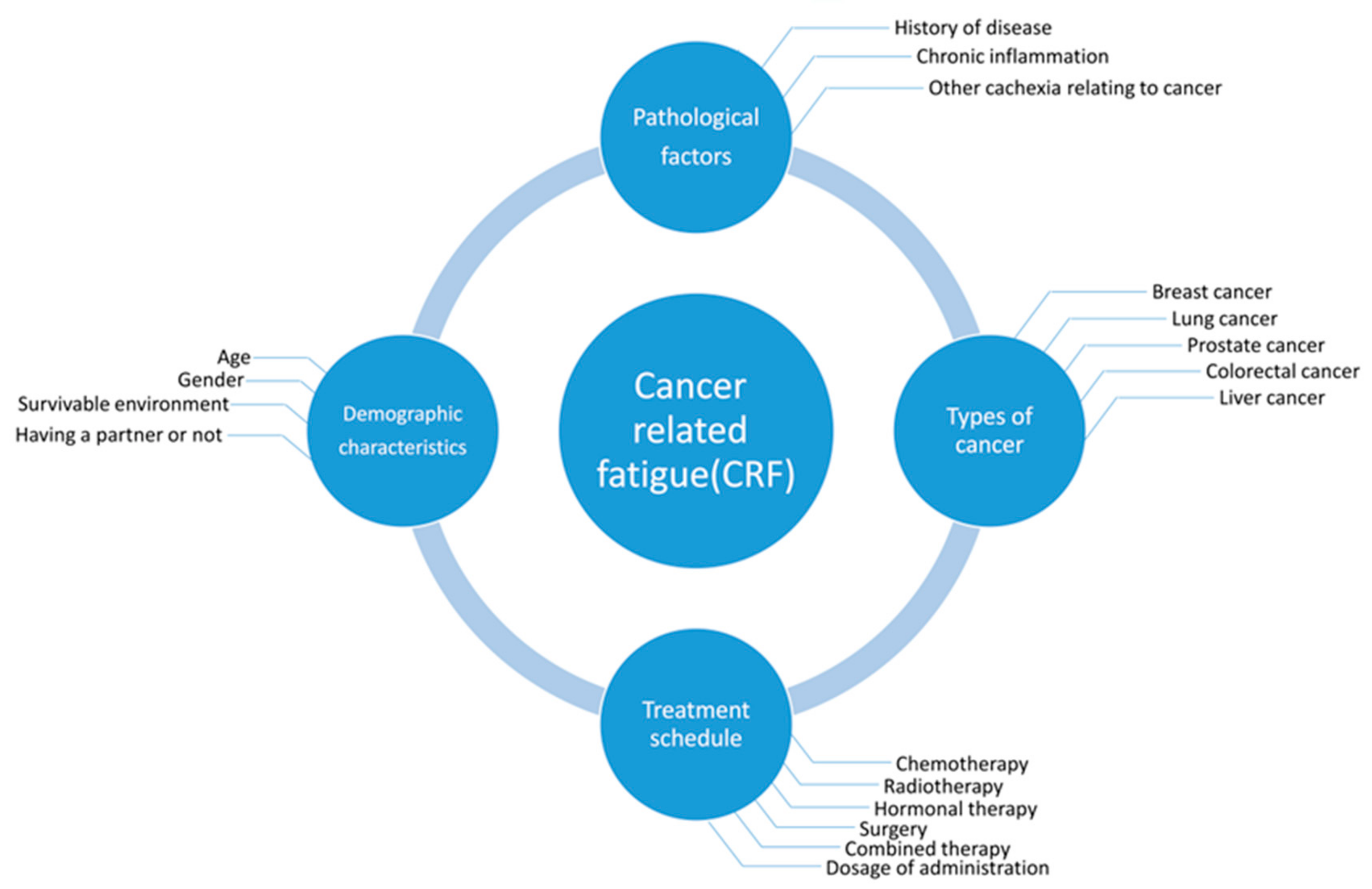
The pathogenic factors of severe fatigue include greater disease severity, radiotherapy, chemotherapy, surgery, hormone therapy, and a combination of these therapies, as well as demographic, lifestyle, and treatment characteristics. Breast cancer (BC) survivors with a partner were found to have a lower risk of developing severe fatigue, but the risk increased with advanced stages of BC; chemotherapy; a combination of surgery, chemotherapy, and radiotherapy; and hormone therapy combined with these treatments. The prevalence rate of severe fatigue ranges from 7% to 52%, and fatigue severity scores vary with different types of chemotherapy drugs or drug combinations [20] (Figure 1). It was reported that patients who received combinations of Cytoxan (cyclophosphamide), fluorouracil, Adriamycin (Doxorubicin) and/or Taxotere (docetaxel) experienced more severe fatigue than those who received only Taxol (paclitaxel) [21]. However, patients who received dose-dense or standard-dose of taxane treatments presented no significant difference in fatigue scores, indicating that fatigue severity changed significantly over time with different chemotherapy treatment types and a combination of strategies [22].
3. Skeletal Muscular and Mitochondrial Dysfunction
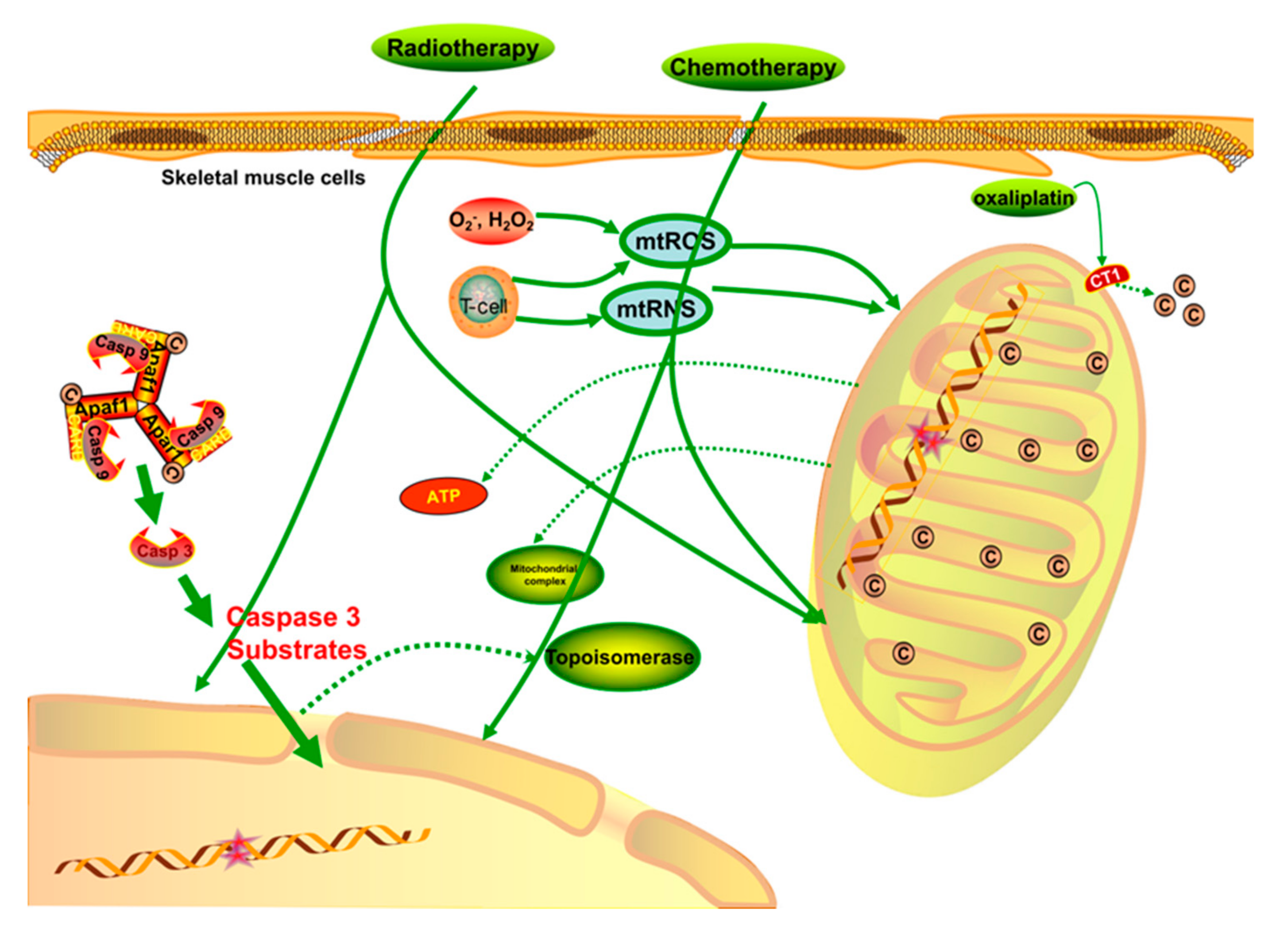
Once the structure and function of mitochondria are disrupted, the energy supply of cells decrease, leading to various unpleasant conditions, such as fatigue, muscle wasting, impaired regenerative tolerance, reduced exercise capacity, pain, and severe neurological diseases that are associated with anti-cancer chemotherapy [23,24]. Research on chemotherapy-receiving patients reported that chemotherapy always nonspecifically targets skeletal musculature, especially the mitochondria, inducing adverse side effects due to low energy supply and high oxidative stress [25]. Chemotherapy treatments cause long-term side effects in skeletal muscle that are different from those induced by the progression of cancer itself. Several mechanisms have been proposed to explain these effects: (1) DNA impairment that inhibits protein biosynthesis and transcription processes [26,27]; (2) generation of free radicals and oxidative products, which induce cellular damage, apoptosis, and necrosis [28,29]; (3) inhibited production of the topoisomerase II, which is important for nuclear DNA (nDNA) transcription [30]; and (4) activation and progression of intrinsic mitochondrial apoptosis. Moreover, mtDNA seems to be more vulnerable to the negative effects of chemotherapies through an increased rate of transcriptional errors and the consequent disruption of the circular and covalently closed mtDNA [31].
The mitochondrial function is modulated by various factors, such as mtDNA, nDNA, nuclear and cellular signaling molecules, mitochondrial reactive oxygen species (mtROS), and ATP formed in other organelles [32,33]. It was demonstrated that chemotherapy such as doxorubicin-an anthracycline used for leukemia and prostate, ovarian, lung, and breast cancer treatment-increases ROS production as an outcome of the redox cycling process [34,35]. Further, chemotherapy can increase the content of O2− and H2O2; these molecules constitute the mtROS, and increase oxidative pressure if not neutralized by endogenous antioxidants [36,37]. Single-stranded DNA damaged by chemotherapies stimulates the activation of enzymes to repair the damage, although this is harmful to ATP stores [38]. NAD+ is also a key mitochondrial substrate that impacts many metabolic pathways, such as glycolysis and the tricarboxylic acid cycle [39]. Skeletal muscle is a highly metabolic organ that needs adequate ATP generation; therefore, reduced ATP generation ultimately leads to skeletal muscle dysfunction [40].
Another type of chemotherapy, platinum-derived alkylating agents, causes similar symptoms of skeletal muscle fatigue but through different mechanisms. This is especially the case for oxaliplatin, which was demonstrated to cause dysregulated mitochondrial and energy homeostasis when used to treat colorectal cancer. Its potential mechanism is the induction of mutations in mtDNA or nuclear-encoded mitochondrial proteins, which are beneficial by stimulating the cell death pathways of cancer cells but also lead to mitochondrial dysfunction and the disruption of energy homeostasis in non-cancerous cells, especially in highly metabolic organs such as skeletal muscle [41]. It was proposed that chemotherapy drugs such as oxaliplatin competitively substitute copper (Cu2+) on copper transporter 1 (CT1), thus reducing the transportation of Cu2+ and leading to a decrease in the mitochondrial Cu2+ pool, which is critical for complex IV and COX17 function, as well as oxidative phosphorylation [42]. Moreover, chemotherapies such as the anti-metabolite and topoisomerase inhibitor families regularly induce an increase in mtROS generation and a decrease in mitochondrial pool viability [28]. Excessive ROS generation and impaired mitochondrial respiratory proteins can result in a destructive feedback cycle, leading to mtROS production, respiratory chain defects, mtDNA deletion, and macroautophagy in skeletal muscle (mitophagy) [43]. Therefore, reducing mtROS generation seems to be a potential intervention strategy and would be conducive to the attenuation of muscular atrophy, mitophagy, necrosis, and apoptosis signaling pathways [44,45].
Mitochondrial dysfunction plays a major role in the development of diseases associated with energy metabolism, and the impaired energy production or longitudinal depletion of ATP induces increased physical disability, such as that observed in CRF or chronic fatigue syndrome (CFS) [46,47]. The mitochondrial abnormalities associated with these disorders include the loss of mitochondrial membrane integrity, oxidative corruption of translocator proteins [48,49], abnormal muscle mitochondrial morphology, and defective aerobic metabolism, which are different from muscle disuse [50] and oxidative phosphorylation in the striated muscle [51,52]. Some cachexia diseases involve certain T cell disorders, including persistent mitochondrial membrane hyperpolarization and increased ROS and reactive nitrogen species (RNS) synthesis, along with decreased levels of glutathione and ATP [53,54]. The release of disease-associated molecular patterns (DAMPs) into circulation as a consequence of necrosis can lead to systemic inflammation, which in turn amplifies mitochondrial dysfunction in a vicious feedforward loop [47,55]. The possible mechanisms of mitochondrial dysfunction, together with chronic inflammation and oxidative stress or elevated levels of ROS and RNS, cause the destruction of proteins, DNA, and lipid membranes [47]. Nitric oxide (NO) and peroxynitrite can markedly reduce the generation of ATP by inhibiting or inactivating crucial enzymes; meanwhile, the inhibition of the electron transport chain (ETC) increase the formation of ROS, leading to more severe impairment of mitochondrial function [37,56]. Studies have shown that decreased energy production and increased lactate production, as well as the impairment of ATP synthesis, are connected with physiological fatigue, indicating that muscular disorders are likely to play a critical role in CRF [57,58,59]. It has been demonstrated that the origin of severe fatigue lies in mitochondrial respiratory dysfunction and impaired generation of ATP, but the deeper and more accurate picture of the molecular mechanisms that underlie mitochondrial dysfunction and CRF remains unclear [47] (Figure 2).
4. Peripheral Immune Activation and Inflammation Dysfunction
CRF is one of multiple concurrent symptoms associated with cancer itself and cancer treatment; however, the interrelations between these symptoms and the underlying mechanisms remain unknown [60,61]. As a symptom of cancer cachexia, CRF is induced by various mediating factors, such as cytokines (tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), IL-6, and interferon-γ (INF-γ)), hormones (insulin, glucagon, leptin, ghrelin, and adiponectin), and neuropeptide-Y [62]. It has been reported that hormones can regulate body weight by adjusting food intake and energy metabolism [63,64,65]. Similar to cachexia, CRF is characterized by systemic inflammation dysfunction, which disturbs the protein and energy balance. For certain neuro-inflammatory, autoimmune, and inflammatory disorders, severe fatigue is related to peripheral immune responses and inflammation due to elevated levels of pro-inflammatory cytokines [66,67]. A variety of cytokines are correlated with the prevalence of sickness behaviors, which are proposed to be the mechanisms of symptom clusters [68,69]. Cytokines are proteins that are generated and/or released by body cells (especially immune cells, such as T cells) that are not only peripheral but also throughout the whole body. Their role is to regulate immune responses through specific receptors [70]. The activation of NF-κB and the subsequent synthesis of pro-inflammatory cytokines are induced by the myeloid differentiation primary response gene (88) (MYD88), which is a general adapter protein used by almost all Toll-like receptors (TLRs) in diverse pathways [71,72,73]. Pro-inflammatory cytokines, such as IL-1, IL-6 and TNF-α act as cell-to-cell mediators in response to external immunologic stressors [74]. However, anti-inflammatory cytokines, such as IL-10, IL-19 and IL-20, are able to regulate the generation and activation of pro-inflammatory cytokines [75]. These pro/anti-inflammatory systems facilitate the body’s return to homeostasis through an inflammatory process by regulating autocrine and paracrine communications in immune cells, as well as immunocytes and other peripheral cells [76].
Fatigue is a common symptom for individuals living with chronic or acute diseases, such as rheumatologic diseases, autoimmune type 1 diabetes, inflammatory bowel diseases, systemic autoimmune diseases, cancers, and infections [67,77,78]. Therefore, in terms of peripheral immune activation or inflammatory dysfunction, CRF may have a similar pathogenesis to other types of pathogenic fatigue. There are variety of factors that contribute to fatigue, such as illness-related characteristics (pain, inflammation, joint damage), physical functions (sleep disturbance and disability), emotional impairment (depression and anxiety), and personal conditions (gender, work, social relationship, education, and whether the patient has a partner a partner) [67,79]. Some research has reported that the prevalence and severity of fatigue are associated with the serum levels of inflammatory cytokines, such as IL-6, TNF-α, IL-1 receptor antagonist (IL-1RA), and especially IL-8, a significant factor of pain and fatigue in lung cancer patients [80,81]. Upregulation of IL-6 and NF-κB has also been reported in cancer patients with sleep disturbances [82]. Furthermore, chemotherapy is one of the CRF-inducing factors because it triggers the generation and secretion of inflammatory cytokines by tumor or immune cells [83]. The increased levels of TNF-α and other cytokines in the periphery are significant predictors of the development of the active disease and fatigue severity that affect the majority of patients with cancer or those receiving cancer treatments [84,85]. The sense of fatigue can persist for over 10 years after chemotherapy treatment, meanwhile, pro-inflammatory cytokine levels remain high, and affect central nervous system (CNS) functions and other behavioral symptoms [86,87]. Moreover, high levels of fatigue have been reported among HIV/AIDS patients undergoing immunotherapies, and their plasma IFN-γ, IL-2, and TNF-α levels were determined to be lowered by antiretroviral therapy, suggesting that there is an intimate relationship between changes in inflammatory cytokines and the degree of fatigue [88].
It is generally accepted that fatigue is related to the loss of muscle mass or altered mood, but research has found that phenotypic fatigue and depressed mood behaviors, such as decreased voluntary wheel-running activity, resignation, and anhedonia, are not correlated with the contractile function of skeletal muscle, indicating that fatigue is likely associated with behavioral activity rather than muscle dysfunction [89]. After injecting murine models with IL-1, their social exploration ability and body weight decreased, and hypersomnia increased. These phenomena were improved by the administration of anti-inflammatory agents, such as IL-1RA or IL-10 [90,91]. Furthermore, in research on military personnel with insomnia, there was a higher degree of CRF in the restorative sleep group than the persistent insomnia group [92]. A therapeutic study showed that when pro-inflammatory cytokines were reduced by biological agents, fatigue severity was reduced accordingly, indicating that inflammatory disorders might be factors that induce or exacerbate CRF [93]. Additionally, fatigue has been found to co-exist with inflammation-associated anemia that was caused by a decrease in iron levels due to thyroid insufficiency or impaired HPA axis function mediated by IL-6 [94,95].
Indeed, early stimulators of chronic inflammation include pro-inflammatory cytokines and other molecules synthesized in response to pathogen invasion, as well as NF-κB activated by macrophages and other sentinel cells [96,97]. Cytokines, ROS, and RNS upregulated by NF-κB tend to maintain and amplify inflammatory and immune responses in a TLR-radical cycle in a feed-forward manner [37,98,99,100]. Moreover, pro-inflammatory cytokines can amplify the function of NF-κB through canonical pathways and lead to mutually elevated activity [101,102]. ROS and RNS are harmful to lipids, proteins, and DNA and lead to the formation of redox-derived DAMPs [103,104]. In return, these redox-derived DAMPs, along with TLRs, facilitate the genesis of NF-κB, cytokines, ROS, and RNS [105]. Briefly, the engagement of TLRs mediated by DAMPs can maintain and amplify chronic inflammation and immune activation [37,73]. Furthermore, elevated levels of NF-κB, pro-inflammatory cytokines, ROS and RNS tend to disrupt epithelial tight junctions in the intestine, resulting in the release of gram-negative bacteria containing lipopolysaccharides (LPS) into the circulation system. As a pathogen-associated molecular pattern (PAMP), this can amplify the TLR-radical cycle [66]. Thus, intestinal permeability is further altered by the translocation of LPS (from gut bacteria), which interacts with TLRs. This is a potential cause of chronic immune activation, which can lead to major depression, CRF, neuro-inflammatory disorders, and other autoimmune diseases [37,56,106]. In other words, the TLR-radical cycle, which leads to increased levels of pro-inflammatory cytokines and ROS or RNS, is likely to be a potential mechanism that drives the development of severe fatigue in cancer patients [73] (Figure 3).
5. Neuron and Central Nervous System (CNS) Disorder
The CNS is a significant factor in the induction of CRF by sensing inflammation, integrating signals from the peripheral nervous system, and stimulating downstream alterations in body mass and systemic metabolism [107]. Fatigue is also one of the characteristics of major depression induced by chronic systemic inflammation and the activation of neuroglia cells [108,109,110,111]. It is generally believed that CRF is related to peripheral mechanisms, such as skeletal muscular metabolism, and pro-/anti-inflammatory cytokines. However, a study investigating the mechanisms of neuromuscular fatigue in children and adults found that children experienced more severe central fatigue than adults, which could be instructive in developing a therapeutic strategy that limit the recruitment of motor units to avoid extensive peripheral fatigue [112]. Lower peripheral fatigue can be transmitted to the CNS via feedback mechanisms and be translated to reduced central fatigue, indicating that fatigue is highly related to CNS dysregulation [113]. In pro-inflammatory stress, signals integrated by the hypothalamus from peripheral systems are translated into neuroendocrine dysregulation, neural signaling alteration, and systematic metabolic derangements. Peripheral pro-inflammatory cytokines induced by systemic inflammatory responses enter the brain by various routes to activate microglia and astrocytes and trigger the production of cytokines and other neurotoxins, which lead to neuroinflammation in the CNS [56,114]. Research has demonstrated that the intracerebroventricular (ICV) injection of pro-inflammatory cytokines stimulates anorexia, lethargy, and catabolism at lower doses of peripheral injection. In addition, the attenuated dynamic regulation of the vascular access and blood-brain-barrier (BBB) function induce higher sensitivity of specifical brain areas to metabolic and inflammatory signaling molecules [115,116,117,118,119]. Inflammatory cytokines secreted by tumor cells can enter the brain through the damaged BBB and trigger neuroinflammatory responses, which leads to severe fatigue symptoms that are unlikely to be alleviated by regular physical movement.
The mediobasal hypothalamus (MBH) contains several neuronal peptides, including proopiomelanocortin (POMC), and cocaine-and-amphetamine-regulated transcript (CART) [120]. The POMC is a precursor polypeptide that can synthesize α-melanocyte-stimulating hormone (αMSH), adrenocorticotropic hormone (ACTH), enkephalin, and β-endorphine (β-EP) through several pathways [121]. The αMSH in the brain can decrease appetite and energy storage. CART is a neuropeptide that can increase locomotor activity and decrease food intake and act as an endogenous psychostimulant [122]. Agouti-related peptide (AgRP) is a significant orexigenic peptide that is characterized as an antagonist of melanocortin receptor 4 (MCR4) and an inhibitor of POMC neuron activity, thus inducing increased appetite and energy storage [123,124]. Neuropeptide Y (NPY) is another orexigenic peptide that has been proposed to be an effective orexigenic factor in the brain and autonomic nervous system, similar to intestinal peptide YY (PYY) [125,126]. These substances are considered to be closely related to the onset of CRF. It has been proposed that the MBH is closely related to CRF by receiving central and peripheral inflammatory signals and providing primary neuronal substrates that link inflammation to fatigue, anorexia, skeletal muscle catabolism dysfunction, and other diseases [127,128,129]. Furthermore, researches have shown that catabolic pathways in the muscle that are related to CNS regulation and muscle are the main targets of pathologic tissue impairment in the disease. This observation indicates that the catabolism of skeletal muscle is one of the factors of cachexia [130,131]. The cytokines entering the CNS stimulate the HPA axis function to promote catabolism of carbohydrates, proteins, and lipids in peripheral tissue, including skeletal muscle and adipose tissue [132].
Studies have indicated that peripheral fatigue results from the depletion of energy stores, synthesis of side-products, and decrease in muscle contractile capacity, which are related to immunological and genetic responses [133,134,135]. The locomotor system is regulated by neuronal networks of central pattern generators (CPGs) in the spinal cord. These are special groups of neurons that produce basic rhythmic patterns [136]. Some specific regions in the brain, such as the prefrontal cortex, the motor cortex, cerebellum, and basal ganglia, are essential for locomotor function and voluntary rhythmic motor patterns [137,138]. Moreover, nitric oxide (NO), the cyclic guanosine 3′, 5′- monophosphate pathway and soluble guanylyl cyclase regulate locomotor activity through NO-dependent or NO/cGMP/KATP pathways, which was demonstrated in anti-nociception in rats [139]. Increased levels of nitrite in both the plasma and brain of resistance-exercised rat models were found in an experimental study [140]. Intracerebral monoamines/neurotransmitters, such as serotonin (5-HT), dopamine (DA), and noradrenaline (NA), have been demonstrated to be associated with central fatigue [141]. 5-HT regulates neurotransmission and various physiological functions related to central fatigue and locomotor activities [142]. Higher 5-HT concentrations in the brain are stimulated by increased tryptophan levels, and can also facilitate 5-HT’s crossing of the BBB, which affects locomotor performance [138]. Meanwhile, increased DA in the brain induces blood calcium influx into the CNS through the proposed mechanism of calcium/calmodulin-dependent DA synthesis and upregulation of DA receptors [143]. Additionally, the noradrenergic neurons that synthesize NA by the hydroxylation of DA can regulate movement through the innervation of the cerebral cortex, subcortical areas, cerebellum, and the brain stem [144]. Nevertheless, the neurotransmitter regulation, synthesis, and secretion functions of the CNS can be influenced by muscle metabolism byproducts, such as ammonia, a metabolic end-product from gastrointestinal tracts [145].
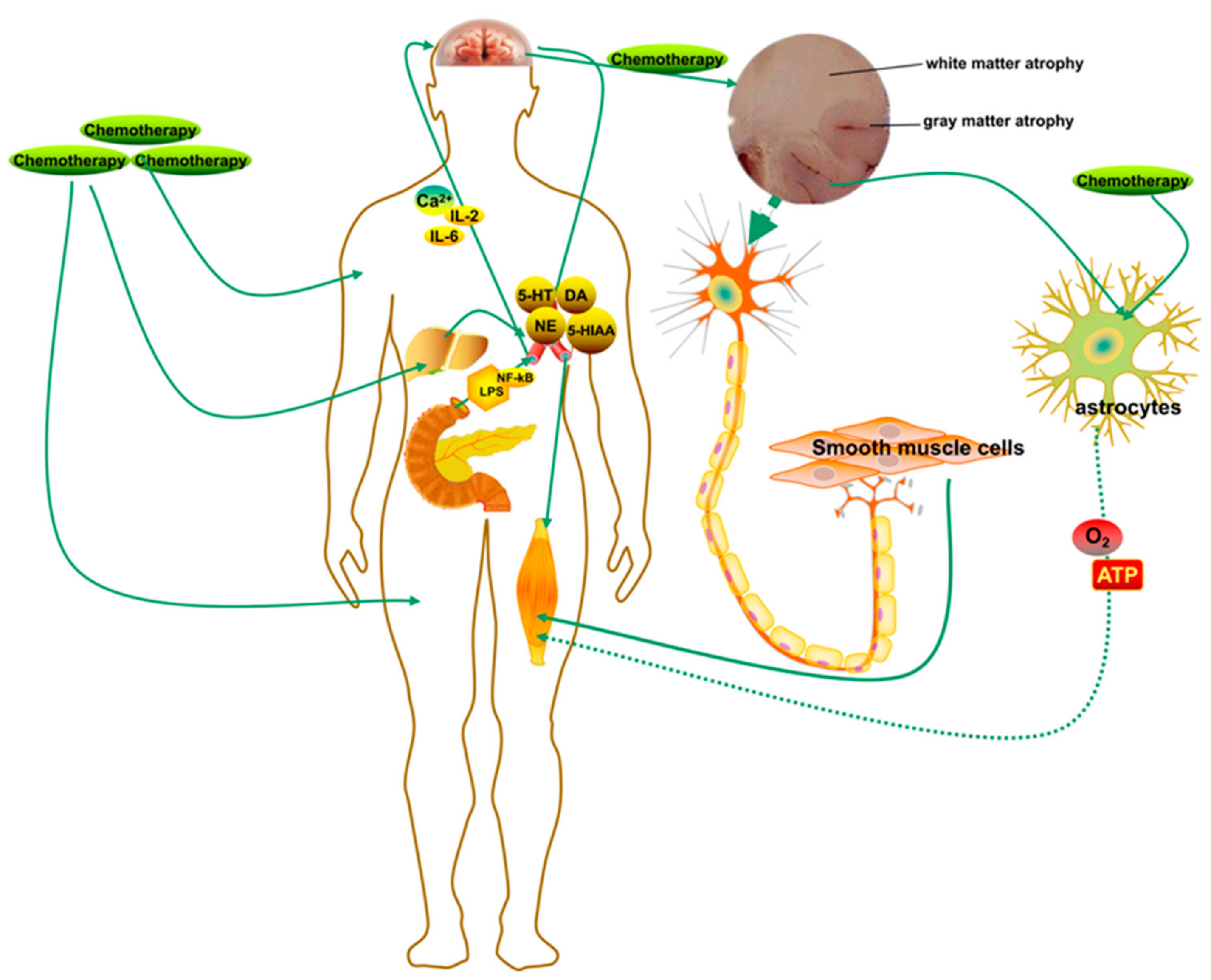
There is acceptance of the positive association between changes in brain activity and cognitive fatigue, and these connections typically occur in several areas of the brain, such as the basal ganglia and prefrontal cortex [146,147,148]. One hypothesis is that the stimulation of fatigue in neurological diseases is caused by the inordinate signal integration processed in the basal ganglia, which regulate inputs from the limbic system and outputs to the motor cortex, and, potentially, pathological states in the white and gray matter [149,150,151]. Gray matter atrophy occurs in some early diseases, such as clinically isolated syndrome (CIS), and the extent of gray matter atrophy correlates significantly with the degree of cognitive impairment and physical disability [152,153,154]. In fatigue, a clear relationship has been demonstrated to exit between global or localized gray matter atrophy and hypoperfusion [155,156]. Moreover, other research has found an association between fatigue and glucose hypometabolism in the frontal cortex and basal ganglia and a decreased N-acetyl asparagine/creatine ratio in basal ganglia [157]. Glial cells, such as astrocytes and protoplasmic astrocytes, play crucial roles in neurometabolic and neurovascular coupling and the delivery of oxygen and energy to central neurons [158,159,160]. It was reported that astrocytes form the vast bulk of gray matter, and the loss of gray matter in the early development of many diseases is due to the loss of astrocytes, which correlates significantly with the severity of inflammation [161]. These findings could explain the basal ganglion dysfunction, which leads to changes in brain activity and the progression of cognitive fatigue that correlates with CRF (Figure 4).
6. Conclusion and Discussion
CRF is frequently experienced by patients after receiving cancer treatment, including chemotherapy or radiotherapy, and the condition persists for a long time. It is often emphasized that the early natural history of CRF has not been systematically researched, so its symptoms, mechanisms, and prevention or treatment strategies are not entirely understood [6,162]. Various clinical studies have indicated that potential mechanisms of CRF lie in skeletal muscle metabolism, as well as pro- and anti- inflammatory functions. Moreover, neural or CNS dysfunction are the primary factors that induce the occurrence or persistence of CRF. However, there seem to be no treatment-related factors that are concordant with systematic reviews that could predict the incidence or treatment of CRF [163]. Recent research has found that, in the context of sickness such as cachexia, peripheral leukocytes concentrate in the brain and significantly affect the neuroinflammatory response. Meanwhile, research on cancer immunology has shown that leukocytes also significantly contribute to the inflammatory immune response in the brain [107,163]. Furthermore, a growing body of evidence indicates that the hypothalamus plays a driving role in the pathophysiology of the acute or chronic illness response. When stimulated by inflammatory cytokines, the hypothalamus stimulates skeletal muscle catabolism and lipolysis by regulating neurotransmitters, systemic circulation, and autocrine and paracrine signaling. However, the method by which cancer treatments such as chemotherapy and radiotherapy trigger the occurrence of CRF remains unknown, despite the increasing amount of research that indicates that skeletal metabolism, the inflammatory response, and CNS function are potential mechanisms of CRF [107]. Furthermore, rodent models are imperative to providing insight into the underlying stimulating molecular factors of CRF.
It is proven that the inflammation disorder drives the pathology in early stages of the disease, and a significant role in the progress of the disease is played by mitochondrial dysfunction, induced by oxidative damage, which leads to the reduced activity of mtDNA and mitochondrial complexes [164]. However, there is a great variability in patients’ cytokine levels and degree of fatigue during their treatment trajectories [165]. The hypothesis that genetic differences in pro-inflammatory cytokines could influence this phenomenon is currently accepted. Although a Genetic Risk Index was developed to predict a high risk of fatigue in women, there is no systemic explanation of the variability in CRF, and additional research is needed to identify CRF genetic variants [165,166].
CRF is characterized by a subjective feeling of severe fatigue, which is greater than general fatigue that would be expected under a special condition. However, the accurate pathophysiology of CRF has not been clearly explained, and whether CRF is peripheral or central or both has never been clarified [167,168]. Generally, fatigue can occur from brain cells to skeletal muscle basic contractile units. Central fatigue refers to the lack of motivation, central nervous system transmission or recruitment changes, and cognitive disorder or depression. However, peripheral fatigue includes decreased contact transfer, muscle point activity, and muscle contraction activity. To now, the paramount question then becomes to what extent can peripheral or central fatigue explain the pathogenesis of CRF. From a developmental perspective, a well understanding of peripheral or central fatigue can provide a pathological correlation that is lacking in the current cognition of CRF. Production of pro-inflammatory cytokines, oxidative stress, and muscle tissue injury occur from cancer or its treatment and promote a systemic effect some distance from the origin of cancer. On the other hand, many patients with breast cancer receiving chemotherapy present a chemo-brain phenomenon, which can be manifested as depression or impaired cognition [169]. This is an interesting phenomenon, suggesting that we ought to explore the pathogenesis of CRF from the perspective of the intrinsic relationship between peripheral and central fatigue, which is based on the sufficient understanding of peripheral and central fatigue. Inflammatory cytokine release, oxidative stress, and mitochondrial energy supply disorders can all occur in the peripheral and central system, which seem to be responsible for CRF, but the intrinsic relationship between muscle fatigue and neuromuscular fatigue in unknown.
In summary, systemic clinical and animal research on the CRF is essential to identify the triggering factors and mechanisms of CRF via histopathological, molecular biological, and biochemical methods. Clarifying the changes in the microstructure of the skeletal muscle, mitochondrial membrane, and brain tissue would be conducive to understanding the physiological mechanisms of CRF. Furthermore, a comparison between patients with different kinds of cancer, therapeutic schedules, and lifestyles would contribute to finding common factors of the mechanisms of CRF. Current research indicates that serum eotaxin, IL-1RA, monocyte chemoattractant protein 1 (MCP-1), and neurocognitive performance are inversely associated with fatigue before chemotherapy; however, serum MCP-1, macrophage inflammatory protein (MIP)-1a, and daily energy expenditure were significantly associated with fatigue after chemotherapy. Furthermore, only the serum IL-12 has been correlated with fatigue both before and after chemotherapy [170]. Hence, animal models should be developed to explore the exact neural and molecular pathways of these phenomena [171]. Creating accurate animal models that mimic the pathological features of CRF should be a priority for exploring the exact mechanisms of CRF before and after cancer treatment. Furthermore, exploring the intrinsic relationship between peripheral and central fatigue may also contribute to a better understanding of the pathogenesis of CRF. In summary, skeletal muscular and mitochondrial dysfunction, pro/anti-inflammatory cytokines (both peripheral and neural), and CNS disorders-especially hypothalamus function-are promising research directions to further the current understanding of the pathogenesis and clinical intervention strategies of CRF.
Author Contributions
Conceptualization, N.C., S.Y. and S.C., Resources, S.Y., Y.G., Y.L., X.L. and N.C.; Writing-Original Draft Preparation, S.Y., Y.G. and S.C.; Writing-Review and Editing, S.Y., Y.G., Q.A. and S.C.; Project Administration, Q.A., S.Y., S.C. and N.C.; Funding acquisition, N.C., S.C. and Q.A.
Funding
Financial support was from the Hunan University of Chinese Medicine First-class Disciple Construction Project (201803); the Hunan Engineering Technology Center of Standardization and Function of Chinese Herbal Decoction Pieces (2016TP2008)
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yancey, J.R.; Thomas, S.M. Chronic fatigue syndrome: Diagnosis and treatment. Am. Fam. Physician 2012, 86, 741–746. [Google Scholar] [PubMed]
- Hawley, J.A.; Reilly, T. Fatigue revisited. J. Sports Sci. 1997, 15, 245–246. [Google Scholar] [PubMed]
- Urrila, A.S.; Paunio, T.; Palomaki, E.; Marttunen, M. Sleep in adolescent depression: Physiological perspectives. Acta Physiol. 2015, 213, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Ce, E.; Rampichini, S.; Limonta, E.; Esposito, F. Fatigue effects on the electromechanical delay components during the relaxation phase after isometric contraction. Acta Physiol. 2014, 211, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Cancer-related fatigue. Clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 2003, 1, 308–331.
- Goldstein, D.; Bennett, B.K.; Webber, K.; Boyle, F.; de Souza, P.L.; Wilcken, N.R.; Scott, E.M.; Toppler, R.; Murie, P.; O’Malley, L.; et al. Cancer-related fatigue in women with breast cancer: Outcomes of a 5-year prospective cohort study. J. Clin. Oncol. 2012, 30, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Servaes, P.; Gielissen, M.F.; Verhagen, S.; Bleijenberg, G. The course of severe fatigue in disease-free breast cancer patients: A longitudinal study. Psychooncology 2007, 16, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Andrykowski, M.A.; Schmidt, J.E.; Salsman, J.M.; Beacham, A.O.; Jacobsen, P.B. Use of a case definition approach to identify cancer-related fatigue in women undergoing adjuvant therapy for breast cancer. J. Clin. Oncol. 2005, 23, 6613–6622. [Google Scholar] [CrossRef]
- Thong, M.S.; Mols, F.; Wang, X.S.; Lemmens, V.E.; Smilde, T.J.; van de Poll-Franse, L.V. Quantifying fatigue in (long-term) colorectal cancer survivors: A study from the population-based patient reported outcomes following initial treatment and long term evaluation of survivorship registry. Eur. J. Cancer 2013, 49, 1957–1966. [Google Scholar] [CrossRef]
- Bower, J.E.; Lamkin, D.M. Inflammation and cancer-related fatigue: Mechanisms, contributing factors, and treatment implications. Brain Behav. Immun. 2013, 30, S48–S57. [Google Scholar] [CrossRef]
- Prue, G.; Rankin, J.; Allen, J.; Gracey, J.; Cramp, F. Cancer-related fatigue: A critical appraisal. Eur. J. Cancer 2006, 42, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Morley, J.E.; Argiles, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Vanhoutte, G.; van de Wiel, M.; Wouters, K.; Sels, M.; Bartolomeeussen, L.; de Keersmaecker, S.; Verschueren, E.; de Vroey, V.; de Wilde, A.; Smits, E.; et al. Cachexia in cancer: What is in the definition? BMJ. Open Gastroenterol. 2016, 3, e000097. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Grimble, R.F. Nutritional therapy for cancer cachexia. Gut 2003, 52, 1391–1392. [Google Scholar] [CrossRef] [PubMed]
- Gullett, N.P.; Mazurak, V.C.; Hebbar, G.; Ziegler, T.R. Nutritional interventions for cancer-induced cachexia. Curr. Probl. Cancer 2011, 35, 58–90. [Google Scholar] [CrossRef]
- Miller, M.; Maguire, R.; Kearney, N. Patterns of fatigue during a course of chemotherapy: Results from a multi-centre study. Eur. J. Oncol. Nurs. 2007, 11, 126–132. [Google Scholar] [CrossRef]
- Cleeland, C.S.; Bennett, G.J.; Dantzer, R.; Dougherty, P.M.; Dunn, A.J.; Meyers, C.A.; Miller, A.H.; Payne, R.; Reuben, J.M.; Wang, X.S. Are the symptoms of cancer and cancer treatment due to a shared biologic mechanism? Cancer 2003, 97, 2919–2925. [Google Scholar] [CrossRef]
- Abrahams, H.J.; Gielissen, M.F.; Schmits, I.C.; Verhagen, C.A.; Rovers, M.M.; Knoop, H. Risk factors, prevalence, and course of severe fatigue after breast cancer treatment: A meta-analysis involving 12,327 breast cancer survivors. Ann. Oncol. 2016, 27, 965–974. [Google Scholar] [CrossRef]
- Prigozin, A.; Uziely, B.; Musgrave, C.F. The relationship between symptom severity and symptom interference, education, age, marital status, and type of chemotherapy treatment in Israeli women with early-stage breast cancer. Oncol. Nurs. Forum 2010, 37, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.M.; Lockhart, K.; Agrawal, S. Variability of patterns of fatigue and quality of life over time based on different breast cancer adjuvant chemotherapy regimens. Oncol. Nurs. Forum 2009, 36, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-induced weakness and fatigue in skeletal muscle: The role of oxidative stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Muscle wasting in cancer: The role of mitochondria. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, D.; Bourassa, P.; Berube, G.; Tajmir-Riahi, H.A. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: Structural features and biological implications. Int. J. Biol. Macromol. 2014, 66, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Quach, B.; Birk, A.; Szeto, H. Mechanism of preventing doxorubicin-induced mitochondrial toxicity with cardiolipin-targeted peptide, SS-31. FASEB J. 2014, 28, 966. [Google Scholar]
- Cheregi, B.; Timpani, C.; Nurgali, K.; Hayes, A.; Rybalka, E. Chemotherapy-induced mitochondrial respiratory dysfunction, oxidant production and death in healthy skeletal muscle C2C12 myoblast and myotube models. Neuromuscul. Disord. 2015, 25, S202. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef]
- Sawyer, D.B.; Peng, X.; Chen, B.; Pentassuglia, L.; Lim, C.C. Mechanisms of anthracycline cardiac injury: Can we identify strategies for cardioprotection? Prog. Cardiovasc. Dis. 2010, 53, 105–113. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Dirks-Naylor, A.J.; Tran, N.T.; Yang, S.; Mabolo, R.; Kouzi, S.A. The effects of acute doxorubicin treatment on proteome lysine acetylation status and apical caspases in skeletal muscle of fasted animals. J. Cachexia Sarcopenia Muscle 2013, 4, 239–243. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Callahan, L.A.; Sumandea, M.P.; Reid, M.B. Doxorubicin causes diaphragm weakness in murine models of cancer chemotherapy. Muscle Nerve 2011, 43, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Dorchies, O.M.; Perozzo, R.; Strosova, M.K.; Scapozza, L.; Ruegg, U.T. Inhibition of iPLA2 beta and of stretch-activated channels by doxorubicin alters dystrophic muscle function. Br. J. Pharmacol. 2013, 169, 1537–1550. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Walder, K.; Maes, M. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med. 2015, 13, 28. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004, 18, 1272–1282. [Google Scholar] [CrossRef] [Green Version]
- Niere, M.; Kernstock, S.; Koch-Nolte, F.; Ziegler, M. Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix. Mol. Cell. Biol. 2008, 28, 814–824. [Google Scholar] [CrossRef]
- Gourdier, I.; Crabbe, L.; Andreau, K.; Pau, B.; Kroemer, G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene 2004, 23, 7449–7457. [Google Scholar] [CrossRef] [Green Version]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends Endocrinol. Metab. 2013, 24, 635–643. [Google Scholar] [CrossRef]
- Lind, M.J. Principles of cytotoxic chemotherapy. Medicine 2004, 32, 20–25. [Google Scholar] [CrossRef]
- Singh, R.; Teel, C.; Sabus, C.; McGinnis, P.; Kluding, P. Fatigue in Type 2 Diabetes: Impact on Quality of Life and Predictors. PLoS ONE 2016, 11, e0165652. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Eikelenboom, M.J.; Killestein, J.; Belli, A.; di Pietro, V.; Tavazzi, B.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.; et al. Cerebrospinal fluid ATP metabolites in multiple sclerosis. Mult. Scler. J. 2010, 16, 549–554. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef]
- Booth, N.E.; Myhill, S.; McLaren-Howard, J. Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int. J. Clin. Exp. Med. 2012, 5, 208–220. [Google Scholar]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Targeting mitochondrial dysfunction in the treatment of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)—A clinical audit. Int. J. Clin. Exp. Med. 2013, 6, 1. [Google Scholar]
- Behan, W.M.; Mcdonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef]
- Hollingsworth, K.G.; Jones, D.E.; Taylor, R.; Blamire, A.M.; Newton, J.L. Impaired cardiovascular response to standing in chronic fatigue syndrome. Eur. J. Clin. Investig. 2010, 40, 608–615. [Google Scholar] [CrossRef]
- Jones, D.E.; Hollingsworth, K.G.; Taylor, R.; Blamire, A.M.; Newton, J.L. Abnormalities in pH handling by peripheral muscle and potential regulation by the autonomic nervous system in chronic fatigue syndrome. J. Intern. Med. 2010, 267, 394–401. [Google Scholar] [CrossRef]
- Perl, A.; Hanczko, R.; Doherty, E. Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol. Biol. 2012, 900, 61–89. [Google Scholar]
- Perl, A.; Nagy, G.; Gergely, P.; Puskas, F.; Qian, Y.; Banki, K. Apoptosis and mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol. Med. 2004, 102, 87–114. [Google Scholar]
- Nagy, G.; Koncz, A.; Fernandez, D.; Perl, A. Nitric oxide, mitochondrial hyperpolarization, and T cell activation. Free Radic. Biol. Med. 2007, 42, 1625–1631. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Maes, M. Oxidative and Nitrosative Stress and Immune-Inflammatory Pathways in Patients with Myalgic Encephalomyelitis (ME)/Chronic Fatigue Syndrome (CFS). Curr. Neuropharmacol. 2014, 12, 168–185. [Google Scholar] [CrossRef]
- Lutz, N.W.; Cozzone, P.J. Metabolic profiling in multiple sclerosis and other disorders by quantitative analysis of cerebrospinal fluid using nuclear magnetic resonance spectroscopy. Curr. Pharm. Biotechnol. 2011, 12, 1016–1025. [Google Scholar] [CrossRef]
- Lutz, N.W.; Viola, A.; Malikova, I.; Confort-Gouny, S.; Audoin, B.; Ranjeva, J.P.; Pelletier, J.; Cozzone, P.J. Inflammatory multiple-sclerosis plaques generate characteristic metabolic profiles in cerebrospinal fluid. PLoS ONE 2007, 2, e595. [Google Scholar] [CrossRef]
- Reinke, S.N.; Broadhurst, D.L.; Sykes, B.D.; Baker, G.B.; Catz, I.; Warren, K.G.; Power, C. Metabolomic profiling in multiple sclerosis: Insights into biomarkers and pathogenesis. Mult. Scler. J. 2014, 20, 1396–1400. [Google Scholar] [CrossRef]
- Miaskowski, C.; Aouizerat, B.E.; Dodd, M.; Cooper, B. Conceptual issues in symptom clusters research and their implications for quality-of-life assessment in patients with cancer. J. Natl. Cancer Inst. Monogr. 2007, 2007, 39–46. [Google Scholar] [CrossRef]
- Teunissen, S.C.; Wesker, W.; Kruitwagen, C.; de Haes, H.C.; Voest, E.E.; de Graeff, A. Symptom prevalence in patients with incurable cancer: A systematic review. J. Pain Symptom Manag. 2007, 34, 94–104. [Google Scholar] [CrossRef]
- Molfino, A.; Formiconi, A.; Rossi Fanelli, F.; Muscaritoli, M. Ghrelin: From discovery to cancer cachexia therapy. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 471–476. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.J.; Yun, J.; Kim, K.H.; Kim, S.H.; Lee, S.C.; Bae, S.B.; Kim, C.K.; Lee, N.S.; Lee, K.T.; et al. Pathophysiological role of hormones and cytokines in cancer cachexia. J. Korean Med. Sci. 2012, 27, 128–134. [Google Scholar] [CrossRef]
- Mak, R.H.; Cheung, W.W.; Gertler, A. Exploiting the therapeutic potential of leptin signaling in cachexia. Curr. Opin. Support. Palliat. Care 2014, 8, 352–357. [Google Scholar] [CrossRef]
- Wolf, I.; Sadetzki, S.; Kanety, H.; Kundel, Y.; Pariente, C.; Epstein, N.; Oberman, B.; Catane, R.; Kaufman, B.; Shimon, I. Adiponectin, ghrelin, and leptin in cancer cachexia in breast and colon cancer patients. Cancer 2006, 106, 966–973. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Galecki, P.; Berk, M.; Maes, M. A narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med. 2013, 11, 64. [Google Scholar] [CrossRef]
- Norheim, K.B.; Jonsson, G.; Omdal, R. Biological mechanisms of chronic fatigue. Rheumatology 2011, 50, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.N.; Dantzer, R.; Langley, K.E.; Bennett, G.J.; Dougherty, P.M.; Dunn, A.J.; Meyers, C.A.; Miller, A.H.; Payne, R.; Reuben, J.M.; et al. A cytokine-based neuroimmunologic mechanism of cancer-related symptoms. Neuroimmunomodulation 2004, 11, 279–292. [Google Scholar] [CrossRef]
- Miaskowski, C.; Aouizerat, B.E. Is there a biological basis for the clustering of symptoms? Semin. Oncol. Nurs. 2007, 23, 99–105. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine, sickness behavior, and depression. Neurol. Clin. 2006, 24, 441–460. [Google Scholar] [CrossRef]
- Fernandez-Gonzalo, R.; de Paz, J.A.; Rodriguez-Miguelez, P.; Cuevas, M.J.; Gonzalez-Gallego, J. Effects of eccentric exercise on toll-like receptor 4 signaling pathway in peripheral blood mononuclear cells. J. Appl. Physiol. 2012, 112, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Jialal, I.; Kaur, H.; Devaraj, S. Toll-like receptor status in obesity and metabolic syndrome: A translational perspective. J. Clin. Endocrinol. Metab. 2014, 99, 39–48. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Ader, R. Psychoneuroimmunology. ILAR J. 1998, 39, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Li, M.C.; He, S.H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J. Gastroenterol. 2004, 10, 620–625. [Google Scholar] [CrossRef]
- Gilbertson-White, S.; Aouizerat, B.E.; Miaskowski, C. Methodologic issues in the measurement of cytokines to elucidate the biological basis for cancer symptoms. Biol. Res. Nurs. 2011, 13, 15–24. [Google Scholar] [CrossRef]
- Goedendorp, M.M.; Tack, C.J.; Steggink, E.; Bloot, L.; Bazelmans, E.; Knoop, H. Chronic fatigue in type 1 diabetes: Highly prevalent but not explained by hyperglycemia or glucose variability. Diabetes Care 2014, 37, 73–80. [Google Scholar] [CrossRef]
- Willems, L.M.; Kwakkenbos, L.; Leite, C.C.; Thombs, B.D.; van den Hoogen, F.H.; Maia, A.C.; Vliet Vlieland, T.P.; van den Ende, C.H. Frequency and impact of disease symptoms experienced by patients with systemic sclerosis from five European countries. Clin. Exp. Rheumatol. 2014, 32, 88–93. [Google Scholar]
- Hewlett, S.; Chalder, T.; Choy, E.; Cramp, F.; Davis, B.; Dures, E.; Nicholls, C. Kirwan, J. Fatigue in rheumatoid arthritis: Time for a conceptual model. Rheumatology 2011, 50, 1004–1006. [Google Scholar] [CrossRef]
- Meyers, C.A.; Albitar, M.; Estey, E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005, 104, 788–793. [Google Scholar] [CrossRef]
- Reyes-Gibby, C.C.; Wang, J.; Spitz, M.; Wu, X.; Yennurajalingam, S.; Shete, S. Genetic variations in interleukin-8 and interleukin-10 are associated with pain, depressed mood, and fatigue in lung cancer patients. J. Pain Symptom Manag. 2013, 46, 161–172. [Google Scholar] [CrossRef]
- Miaskowski, C.; Cooper, B.A.; Dhruva, A.; Dunn, L.B.; Langford, D.J.; Cataldo, J.K.; Baggott, C.R.; Merriman, J.D.; Dodd, M.; Lee, K.; et al. Evidence of Associations between Cytokine Genes and Subjective Reports of Sleep Disturbance in Oncology Patients and Their Family Caregivers. PLoS ONE 2012, 7, e40560. [Google Scholar] [CrossRef]
- Irwin, M.R. Inflammation at the intersection of behavior and somatic symptoms. Psychiatr. Clin. N. Am. 2011, 34, 605–620. [Google Scholar] [CrossRef]
- Flachenecker, P.; Bihler, I.; Weber, F.; Gottschalk, M.; Toyka, K.V.; Rieckmann, P. Cytokine mRNA expression in patients with multiple sclerosis and fatigue. Mult. Scler. J. 2004, 10, 165–169. [Google Scholar] [CrossRef]
- Heesen, C.; Nawrath, L.; Reich, C.; Bauer, N.; Schulz, K.H.; Gold, S.M. Fatigue in multiple sclerosis: An example of cytokine mediated sickness behaviour? J. Neurol. Neurosurg. Psychiatry 2006, 77, 34–39. [Google Scholar] [CrossRef]
- Bower, J.E. Cancer-related fatigue: Links with inflammation in cancer patients and survivors. Brain Behav. Immun. 2007, 21, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Bower, J.E. Cancer-related fatigue—Mechanisms, risk factors, and treatments. Nat. Rev. Clin. Oncol. 2014, 11, 597–609. [Google Scholar] [CrossRef]
- Lee, Y.C.; Frits, M.L.; Iannaccone, C.K.; Weinblatt, M.E.; Shadick, N.A.; Williams, D.A.; Cui, J. Subgrouping of patients with rheumatoid arthritis based on pain, fatigue, inflammation, and psychosocial factors. Arthritis Rheumatol. 2014, 66, 2006–2014. [Google Scholar] [CrossRef]
- Norden, D.M.; Bicer, S.; Clark, Y.; Jing, R.F.; Henry, C.J.; Wold, L.E.; Reiser, P.J.; Godbout, J.P.; McCarthy, D.O. Tumor growth increases neuroinflammation, fatigue and depressive-like behavior prior to alterations in muscle function. Brain Behav. Immun. 2015, 43, 76–85. [Google Scholar] [CrossRef]
- Arnett, S.V.; Clark, I.A. Inflammatory fatigue and sickness behavior—Lessons for the diagnosis and management of chronic fatigue syndrome. J. Affect Disord. 2012, 141, 130–142. [Google Scholar] [CrossRef]
- Bluthe, R.M.; Beaudu, C.; Kelley, K.W.; Dantzer, R. Differential effects of IL-1ra on sickness behavior and weight loss induced by IL-1 in rats. Brain Res. 1995, 677, 171–176. [Google Scholar] [CrossRef]
- Heinzelmann, M.; Lee, H.; Rak, H.; Livingston, W.; Barr, T.; Baxter, T.; Scattergood-Keepper, L.; Mysliwiec, V.; Gill, J. Sleep restoration is associated with reduced plasma C-reactive protein and depression symptoms in military personnel with sleep disturbance after deployment. Sleep Med. 2014, 15, 1565–1570. [Google Scholar] [CrossRef]
- Chauffier, K.; Salliot, C.; Berenbaum, F.; Sellam, J. Effect of biotherapies on fatigue in rheumatoid arthritis: A systematic review of the literature and meta-analysis. Rheumatology 2012, 51, 60–68. [Google Scholar] [CrossRef]
- Chrousos, G.P. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef]
- Masson, C. Rheumatoid anemia. Jt. Bone Spine 2011, 78, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Peng, B.; Li, Z.; Sclabas, G.M.; Fujioka, S.; Niu, J.; Schmidt-Supprian, M.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. Mechanisms of proinflammatory cytokine-induced biphasic NF-kappaB activation. Mol. Cell 2003, 12, 1287–1300. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Memet, S.; Ave, P.; Verhaeghe, C.; Mayo, K.H.; Struman, I.; Martial, J.A.; Griffioen, A.W. NF-kappaB activation in endothelial cells is critical for the activity of angiostatic agents. Mol. Cancer Ther. 2009, 8, 2645–2654. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef]
- Nakata, S.; Tsutsui, M.; Shimokawa, H.; Yamashita, T.; Tanimoto, A.; Tasaki, H.; Ozumi, K.; Sabanai, K.; Morishita, T.; Suda, O.; et al. Statin treatment upregulates vascular neuronal nitric oxide synthase through Akt/NF-kappaB pathway. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 92–98. [Google Scholar] [CrossRef]
- Sultani, M.; Stringer, A.M.; Bowen, J.M.; Gibson, R.J. Anti-inflammatory cytokines: Important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother. Res. Pract. 2012, 2012, 490804. [Google Scholar] [CrossRef]
- Sonis, S.T. A biological approach to mucositis. J. Support. Oncol. 2004, 2, 21–32. [Google Scholar]
- Sonis, S.T. Pathobiology of oral mucositis: Novel insights and opportunities. J. Support. Oncol. 2007, 5, 3–11. [Google Scholar]
- Maes, M.; Kubera, M.; Obuchowiczwa, E.; Goehler, L.; Brzeszcz, J. Depression’s multiple comorbidities explained by (neuro)inflammatory and oxidative & nitrosative stress pathways. Neuroendocr. Endocrinol. Lett. 2011, 32, 7–24. [Google Scholar]
- Maes, M.; Mihaylova, I.; Leunis, J.C. Chronic fatigue syndrome is accompanied by an IgM-related immune response directed against neopitopes formed by oxidative or nitrosative damage to lipids and proteins. Neuroendocr. Endocrinol. Lett. 2006, 27, 615–621. [Google Scholar]
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2001, 249, 61–74. [Google Scholar] [CrossRef]
- Morris, G.; Maes, M. A neuro-immune model of Myalgic Encephalomyelitis/Chronic fatigue syndrome. Metab. Brain Dis. 2013, 28, 523–540. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2016, 54, 42–52. [Google Scholar] [CrossRef]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef]
- Kreisel, T.; Frank, M.G.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.V.; Maier, S.F.; Yirmiya, R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar] [CrossRef]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Galecki, P.; Leonard, B. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 66. [Google Scholar] [CrossRef]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; Schwabedissen, L.M.Z.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 2013, 10, 34. [Google Scholar] [CrossRef]
- Ratel, S.; Kluka, V.; Vicencio, S.G.; Jegu, A.G.; Cardenoux, C.; Morio, C.; Coudeyre, E.; Martin, V. Insights into the Mechanisms of Neuromuscular Fatigue in Boys and Men. Med. Sci. Sport Exerc. 2015, 47, 2319–2328. [Google Scholar] [CrossRef]
- Amann, M.; Blain, G.M.; Proctor, L.T.; Sebranek, J.J.; Pegelow, D.F.; Dempsey, J.A. Implications of group III and IV muscle afferents for high-intensity endurance exercise performance in humans. J. Physiol. 2011, 589, 5299–5309. [Google Scholar] [CrossRef]
- Perry, V.H.; Cunningham, C.; Boche, D. Atypical inflammation in the central nervous system in prion disease. Curr. Opin. Neurol. 2002, 15, 349–354. [Google Scholar] [CrossRef]
- Bodnar, R.J.; Pasternak, G.W.; Mann, P.E.; Paul, D.; Warren, R.; Donner, D.B. Mediation of anorexia by human recombinant tumor necrosis factor through a peripheral action in the rat. Cancer Res. 1989, 49, 6280–6284. [Google Scholar]
- Cone, R.D.; Cowley, M.A.; Butler, A.A.; Fan, W.; Marks, D.L.; Low, M.J. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. 2002, 25, S63. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Scarlett, J.M.; Zhu, X.; Bowe, D.D.; Batra, A.K.; Braun, T.P.; Marks, D.L. Arcuate nucleus proopiomelanocortin neurons mediate the acute anorectic actions of leukemia inhibitory factor via gp130. Endocrinology 2010, 151, 606–616. [Google Scholar] [CrossRef]
- Lawrence, C.B.; Rothwell, N.J. Anorexic but Not Pyrogenic Actions of Interleukin-1 are Modulated by Central Melanocortin-3/4 Receptors in the Rat. J. Neuroendocrinol. 2001, 13, 490–495. [Google Scholar] [CrossRef]
- Sonti, G.; Ilyin, S.E.; Plata-Salamán, C.R. Anorexia induced by cytokine interactions at pathophysiological concentrations. Am. J. Physiol. 1996, 270, 1394–1402. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Millington, G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 2007, 4, 18. [Google Scholar] [CrossRef]
- Murphy, K.G. Dissecting the role of cocaine- and amphetamine-regulated transcript (CART) in the control of appetite. Brief. Funct. Genom. Proteom. 2005, 4, 95–111. [Google Scholar] [CrossRef]
- Cowley, M.A.; Dinulescu, D.M.; Pronchuk, N.; Fan, W.; Colmers, W.F.; Cone, R.D. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: Evidence of a cellular basis for the adipostat. Neuron 1999, 24, 155–163. [Google Scholar] [CrossRef]
- Pritchard, L.E.; Armstrong, D.; Davies, N.; Oliver, R.L.; Schmitz, C.A.; Brennand, J.C.; Wikinson, G.F.; White, A. Agouti-related protein (83-132) is a competitive antagonist at the human melanocortin-4 receptor: No evidence for differential interactions with pro-opiomelanocortin-derived ligands. J. Endocrinol. 2004, 180, 183–191. [Google Scholar] [CrossRef]
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.W.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 2007, 13, 803–811. [Google Scholar] [CrossRef]
- Tatemoto, K.; Carlquist, M.; Mutt, V. Neuropeptide Y—A novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature 1982, 296, 659–660. [Google Scholar] [CrossRef]
- Scarlett, J.M.; Jobst, E.E.; Enriori, P.J.; Bowe, D.D.; Batra, A.K.; Grant, W.F.; Cowley, M.A.; Marks, D.L. Regulation of central melanocortin signaling by interleukin-1 beta. Endocrinology 2007, 148, 4217–4225. [Google Scholar] [CrossRef]
- Scarlett, J.M.; Zhu, X.; Enriori, P.J.; Bowe, D.D.; Batra, A.K.; Levasseur, P.R.; Grant, W.F.; Meguid, M.M.; Cowley, M.A.; Marks, D.L. Regulation of agouti-related protein messenger ribonucleic acid transcription and peptide secretion by acute and chronic inflammation. Endocrinology 2008, 149, 4837–4845. [Google Scholar] [CrossRef]
- Wisse, B.E.; Ogimoto, K.; Tang, J.; Harris, M.K., Jr.; Raines, E.W.; Schwartz, M.W. Evidence that lipopolysaccharide-induced anorexia depends upon central, rather than peripheral, inflammatory signals. Endocrinology 2007, 148, 5230–5237. [Google Scholar] [CrossRef]
- Braun, T.P.; Marks, D.L. The regulation of muscle mass by endogenous glucocorticoids. Front. Physiol. 2015, 6, 12. [Google Scholar] [CrossRef]
- Braun, T.P.; Szumowski, M.; Levasseur, P.R.; Grossberg, A.J.; Zhu, X.; Agarwal, A.; Marks, D.L. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014, 9, e106489. [Google Scholar] [CrossRef]
- Johns, N.; Stephens, N.A.; Fearon, K.C. Muscle wasting in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 2215–2229. [Google Scholar] [CrossRef]
- Finsterer, J. Biomarkers of peripheral muscle fatigue during exercise. BMC Musculoskelet. Disord. 2011, 13, 218. [Google Scholar] [CrossRef]
- Keyser, R.E. Peripheral fatigue: High-energy phosphates and hydrogen ions. PM & R 2010, 2, 347–358. [Google Scholar]
- Vanhatalo, A.; Fulford, J.; DiMenna, F.J.; Jones, A.M. Influence of hyperoxia on muscle metabolic responses and the power-duration relationship during severe-intensity exercise in humans: A 31P magnetic resonance spectroscopy study. Exp. Physiol. 2010, 95, 528–540. [Google Scholar] [CrossRef]
- Guertin, P.A. Central pattern generator for locomotion: Anatomical, physiological, and pathophysiological considerations. Front. Neurol. 2013, 3, 183. [Google Scholar] [CrossRef]
- Green, H.J. Mechanisms of muscle fatigue in intense exercise. J. Sports Sci. 1997, 15, 247–256. [Google Scholar] [CrossRef]
- Zajac, A.; Chalimoniuk, M.; Maszczyk, A.; Golas, A.; Lngfort, J. Central and Peripheral Fatigue During Resistance Exercise—A Critical Review. J. Hum. Kinet. 2015, 49, 159–169. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Chrapusta, S.J.; Lukacova, N.; Langfort, J. Endurance training upregulates the nitric oxide/soluble guanylyl cyclase/cyclic guanosine 3’,5’-monophosphate pathway in the striatum, midbrain and cerebellum of male rats. Brain Res. 2015, 1618, 29–40. [Google Scholar] [CrossRef]
- Galdino, G.S.; Xavier, C.H.; Almeida, R.; Silva, G.; Fontes, M.A.; Menezes, G.; Duarte, I.D.; Perez, A.C. The Nitric oxide/CGMP/KATP pathway mediates systemic and central antinociception induced by resistance exercise in rats. Int. J. Neurosci. 2015, 125, 765–773. [Google Scholar] [CrossRef]
- Meeusen, R.; Watson, P. Amino acids and the brain: Do they play a role in “central fatigue”? Int. J. Sport Nutr. Exerc. Metab. 2007, 17, 37–46. [Google Scholar] [CrossRef]
- Meeusen, R.; Watson, P.; Hasegawa, H.; Roelands, B.; Piacentini, M.F. Central fatigue: The serotonin hypothesis and beyond. Sports Med. 2006, 36, 881–909. [Google Scholar] [CrossRef]
- Sutoo, D.; Akiyama, K. Regulation of brain function by exercise. Neurobiol. Dis. 2003, 13, 1–14. [Google Scholar] [CrossRef]
- Bouret, S.; Sara, S.J. Network reset: A simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 2005, 28, 574–582. [Google Scholar] [CrossRef]
- Romero-Gomez, M.; Jover, M.; Galan, J.J.; Ruiz, A. Gut ammonia production and its modulation. Metab. Brain Dis. 2009, 24, 147–157. [Google Scholar] [CrossRef]
- DeLuca, J.; Genova, H.M.; Capili, E.J.; Wylie, G.R. Functional neuroimaging of fatigue. Phys. Med. Rehabil. Clin. N. Am. 2009, 20, 325–337. [Google Scholar] [CrossRef]
- Genova, H.M.; Rajagopalan, V.; Deluca, J.; Das, A.; Binder, A.; Arjunan, A.; Chiaravalloti, N.; Wylie, G. Examination of cognitive fatigue in multiple sclerosis using functional magnetic resonance imaging and diffusion tensor imaging. PLoS ONE 2013, 8, e78811. [Google Scholar] [CrossRef]
- Kohl, A.D.; Wylie, G.R.; Genova, H.M.; Hillary, F.G.; Deluca, J. The neural correlates of cognitive fatigue in traumatic brain injury using functional MRI. Brain Inj. 2009, 23, 420–432. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Behan, P.O. Fatigue in neurological disorders. Lancet 2004, 363, 978–988. [Google Scholar] [CrossRef]
- Filippi, M.; Rocca, M.A. MR imaging of gray matter involvement in multiple sclerosis: Implications for understanding disease pathophysiology and monitoring treatment efficacy. Am. J. Neuroradiol. 2010, 31, 1171–1177. [Google Scholar] [CrossRef]
- Messina, S.; Patti, F. Gray matters in multiple sclerosis: Cognitive impairment and structural MRI. Mult. Scler. Int. 2014, 2014, 609694. [Google Scholar] [CrossRef]
- Ceccarelli, A.; Rocca, M.A.; Pagani, E.; Colombo, B.; Martinelli, V.; Comi, G.; Filippi, M. A voxel-based morphometry study of grey matter loss in MS patients with different clinical phenotypes. Neuroimage 2008, 42, 315–322. [Google Scholar] [CrossRef]
- Henry, R.G.; Shieh, M.; Okuda, D.T.; Evangelista, A.; Gorno-Tempini, M.L.; Pelletier, D. Regional grey matter atrophy in clinically isolated syndromes at presentation. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1236–1244. [Google Scholar] [CrossRef] [Green Version]
- Inglese, M.; Oesingmann, N.; Casaccia, P.; Fleysher, L. Progressive multiple sclerosis and gray matter pathology: An MRI perspective. Mt. Sinai J. Med. 2011, 78, 258–267. [Google Scholar] [CrossRef]
- Inglese, M.; Park, S.J.; Johnson, G.; Babb, J.S.; Miles, L.; Jaggi, H.; Herbert, J.; Grossman, R.I. Deep gray matter perfusion in multiple sclerosis: Dynamic susceptibility contrast perfusion magnetic resonance imaging at 3 T. Arch. Neurol. 2007, 64, 196–202. [Google Scholar] [CrossRef]
- Pellicano, C.; Gallo, A.; Li, X.; Ikonomidou, V.N.; Evangelou, I.E.; Ohayon, J.M.; Stern, S.K.; Ehrmantraut, M.; Cantor, F.; McFarland, H.F.; et al. Relationship of cortical atrophy to fatigue in patients with multiple sclerosis. Arch. Neurol. 2010, 67, 447–453. [Google Scholar] [CrossRef]
- Téllez, N.; Alonso, J.; Río, J.; Tintoré, M.; Nos, C.; Montalban, X.; Rovira, A. The basal ganglia: A substrate for fatigue in multiple sclerosis. Neuroradiology 2008, 50, 17–23. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods Mol. Biol. 2012, 814, 23–45. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef]
- Cella, D.; Peterman, A.; Passik, S.; Jacobsen, P.; Breitbart, W. Progress toward guidelines for the management of fatigue. Oncology 1998, 12, 369–377. [Google Scholar]
- Sugihara, A.Q.; Rolle, C.E.; Lesniak, M.S. Regulatory T cells actively infiltrate metastatic brain tumors. Int. J. Oncol. 2009, 34, 1533–1540. [Google Scholar] [Green Version]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef]
- Raudonis, B.M.; Kelley, I.H.; Rowe, N.; Ellis, J. A Pilot Study of Proinflammatory Cytokines and Fatigue in Women with Breast Cancer During Chemotherapy. Cancer Nurs. 2017, 40, 323–331. [Google Scholar] [CrossRef]
- Bower, J.E.; Ganz, P.A.; Irwin, M.R.; Castellon, S.; Arevalo, J.; Cole, S.W. Cytokine Genetic Variations and Fatigue Among Patients with Breast Cancer. J. Clin. Oncol. 2013, 31, 1656–1661. [Google Scholar] [CrossRef]
- Cai, B.; Allexandre, D.; Rajagopalan, V.; Jiang, Z.; Siemionow, V.; Ranganathan, V.K.; Davis, M.P.; Walsh, D.; Dai, K.; Yue, G.H. Evidence of Significant Central Fatigue in Patients with Cancer-Related Fatigue during Repetitive Elbow Flexions till Perceived Exhaustion. PLoS ONE 2014, 9, e115370. [Google Scholar] [CrossRef]
- Janda, M.; Gerstner, N.; Obermair, A.; Fuerst, A.; Wachter, S.; Dieckmann, K.; Pötter, R. Quality of life changes during conformal radiation therapy for prostate carcinoma. J. Cancer 2000, 89, 1322–1328. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.V. The underrecognized role of impaired muscle function in cancer-related fatigue. J. Support. Oncol. 2010, 8, 177. [Google Scholar]
- Mortimer, J.E.; Waliany, S.; Dieli-Conwright, C.M.; Patel, S.K.; Hurria, A.; Chao, J.; Tiep, B.; Behrendt, C.E. Objective physical and mental markers of self-reported fatigue in women undergoing (neo)adjuvant chemotherapy for early-stage breast cancer. Cancer 2017, 123, 1810–1816. [Google Scholar] [CrossRef]
- Kanzaki, A.; Okauchi, T.; Hu, D.; Shingaki, T.; Katayama, Y.; Koyama, H.; Watanabe, Y.; Cui, Y. Extension of recovery time from fatigue by repeated rest with short-term sleep during continuous fatigue load: Development of chronic fatigue model. J. Neurosci. Res. 2016, 94, 424–429. [Google Scholar] [CrossRef]
Figure 1.
Cancer-related fatigue (CRF) is associated with various risk factors. The predominant factors are demographic characteristics, pathological factors, the types of cancer, and anti-cancer treatment schedules.
Figure 1.
Cancer-related fatigue (CRF) is associated with various risk factors. The predominant factors are demographic characteristics, pathological factors, the types of cancer, and anti-cancer treatment schedules.

Figure 2.
When treated with chemotherapy or radiotherapy, the regular structure and function of mitochondria are damaged through different signaling pathways. The processes involved in the transcription of nDNA and mtDNA are significantly destroyed, and the levels of mtROS and mtRNS are upregulated when skeletal muscle is nonspecifically targeted by chemotherapies. Meanwhile, the respiratory function of mitochondria is weakened by the impaired mitochondrial membrane. The Cu2+ capacity is critical for mitochondrial complexes and ATP generation, so when Cu2+ is competitively inhibited by some chemotherapies, such as oxaliplatin, the outflow of Cu2+ increases, which is harmful to the mitochondrial energy generation. In general, direct chemo/radio-therapy injuries, hyperoxidative stress, and a low energy supply are likely to cause physical fatigue via apoptosis or other detrimental signaling pathways.
Figure 2.
When treated with chemotherapy or radiotherapy, the regular structure and function of mitochondria are damaged through different signaling pathways. The processes involved in the transcription of nDNA and mtDNA are significantly destroyed, and the levels of mtROS and mtRNS are upregulated when skeletal muscle is nonspecifically targeted by chemotherapies. Meanwhile, the respiratory function of mitochondria is weakened by the impaired mitochondrial membrane. The Cu2+ capacity is critical for mitochondrial complexes and ATP generation, so when Cu2+ is competitively inhibited by some chemotherapies, such as oxaliplatin, the outflow of Cu2+ increases, which is harmful to the mitochondrial energy generation. In general, direct chemo/radio-therapy injuries, hyperoxidative stress, and a low energy supply are likely to cause physical fatigue via apoptosis or other detrimental signaling pathways.

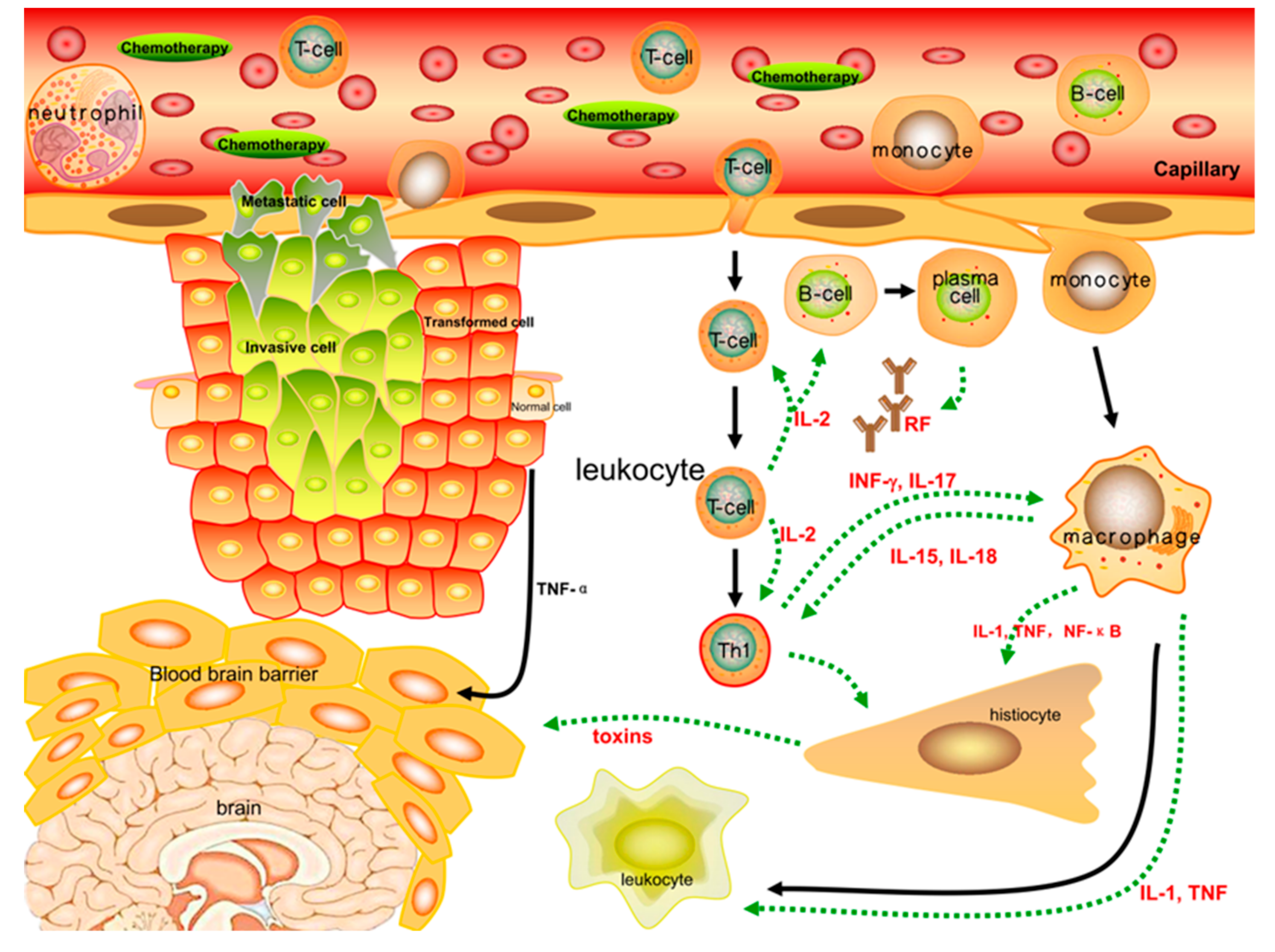
Figure 3.
Inflammatory cytokines such as TNF-α, IL-1β, IL-2, IL-6, and INF-γ, which are correlated with the severity of fatigue, play a significant role in peripheral immune activation. Cytokines are primarily generated by immune cells and regulate inflammatory responses through peripheral, neural, and even systematic circulations. The pro/anti-inflammatory function encourages the body to maintain relative homeostasis by autocrine and paracrine communication between immune cells. When stimulated by chemotherapies, the inflammatory responses of immune cells are further strengthened, and the secretion of cytokines such as NF-κB, IL-1β, and TNF-α into the peripheral or neural circulation is increased. These changes lead to more severe fatigue symptoms that are closely linked to the incidence of CRF.
Figure 3.
Inflammatory cytokines such as TNF-α, IL-1β, IL-2, IL-6, and INF-γ, which are correlated with the severity of fatigue, play a significant role in peripheral immune activation. Cytokines are primarily generated by immune cells and regulate inflammatory responses through peripheral, neural, and even systematic circulations. The pro/anti-inflammatory function encourages the body to maintain relative homeostasis by autocrine and paracrine communication between immune cells. When stimulated by chemotherapies, the inflammatory responses of immune cells are further strengthened, and the secretion of cytokines such as NF-κB, IL-1β, and TNF-α into the peripheral or neural circulation is increased. These changes lead to more severe fatigue symptoms that are closely linked to the incidence of CRF.

Figure 4.
The central nervous system (CNS) integrates the signals from peripheral circulations to inhibit or amplify immune signals through neural regulation mechanisms. On the account of inflammatory stress, peripheral inflammatory cytokines enter the brain through various routes to activate microglia and astrocytes and to generate neurotoxins that cause neuroinflammation in the CNS. Furthermore, inflammation of the nervous system leads to severe disorders of systemic circulations, such as decreased blood-brain barrier (BBB) strength, atrophy of spinal gray matter, or decreased muscular innervation. A nervous system affected by peripheral or central inflammatory responses is inclined to destroy muscle cells and inhibit the generation of energy and nutrition, leading to motor unit decrease and severe fatigue, both physical and mental.
Figure 4.
The central nervous system (CNS) integrates the signals from peripheral circulations to inhibit or amplify immune signals through neural regulation mechanisms. On the account of inflammatory stress, peripheral inflammatory cytokines enter the brain through various routes to activate microglia and astrocytes and to generate neurotoxins that cause neuroinflammation in the CNS. Furthermore, inflammation of the nervous system leads to severe disorders of systemic circulations, such as decreased blood-brain barrier (BBB) strength, atrophy of spinal gray matter, or decreased muscular innervation. A nervous system affected by peripheral or central inflammatory responses is inclined to destroy muscle cells and inhibit the generation of energy and nutrition, leading to motor unit decrease and severe fatigue, both physical and mental.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, S.; Chu, S.; Gao, Y.; Ai, Q.; Liu, Y.; Li, X.; Chen, N. A Narrative Review of Cancer-Related Fatigue (CRF) and Its Possible Pathogenesis. Cells 2019, 8, 738. https://doi.org/10.3390/cells8070738
AMA Style
Yang S, Chu S, Gao Y, Ai Q, Liu Y, Li X, Chen N. A Narrative Review of Cancer-Related Fatigue (CRF) and Its Possible Pathogenesis. Cells. 2019; 8(7):738. https://doi.org/10.3390/cells8070738
Chicago/Turabian StyleYang, Songwei, Shifeng Chu, Yan Gao, Qidi Ai, Yingjiao Liu, Xun Li, and Naihong Chen. 2019. "A Narrative Review of Cancer-Related Fatigue (CRF) and Its Possible Pathogenesis" Cells 8, no. 7: 738. https://doi.org/10.3390/cells8070738
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.