Degradation of the Tumor Suppressor PDCD4 Is Impaired by the Suppression of p62/SQSTM1 and Autophagy

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Development of ATG5 Mutant-16 Huh7 Cell Line
2.3. Reagents
2.4. Antibodies
2.5. Transfection of Plasmids
2.6. Western Blotting Analyses
2.7. siRNA-Mediated Knockdown of p62
2.8. Immunoprecipitation
2.9. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.10. Microscopy and Imaging Analyses
2.11. Statistical Analyses
3. Results
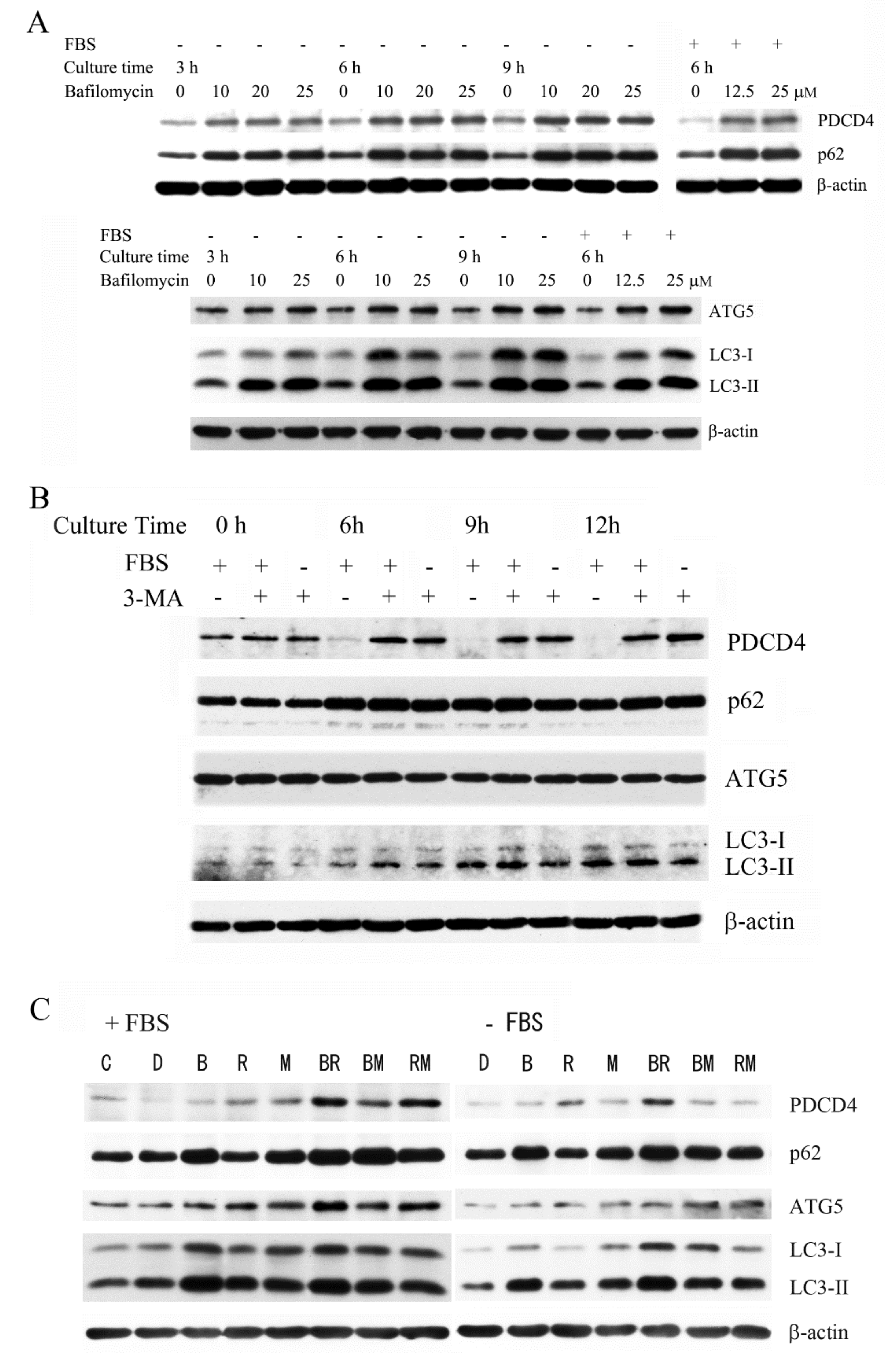
3.1. The Autophagy Inhibitors Bafilomycin A1 and 3-Methyladenine Upregulated PDCD4 in Huh7 Hepatoma Cells
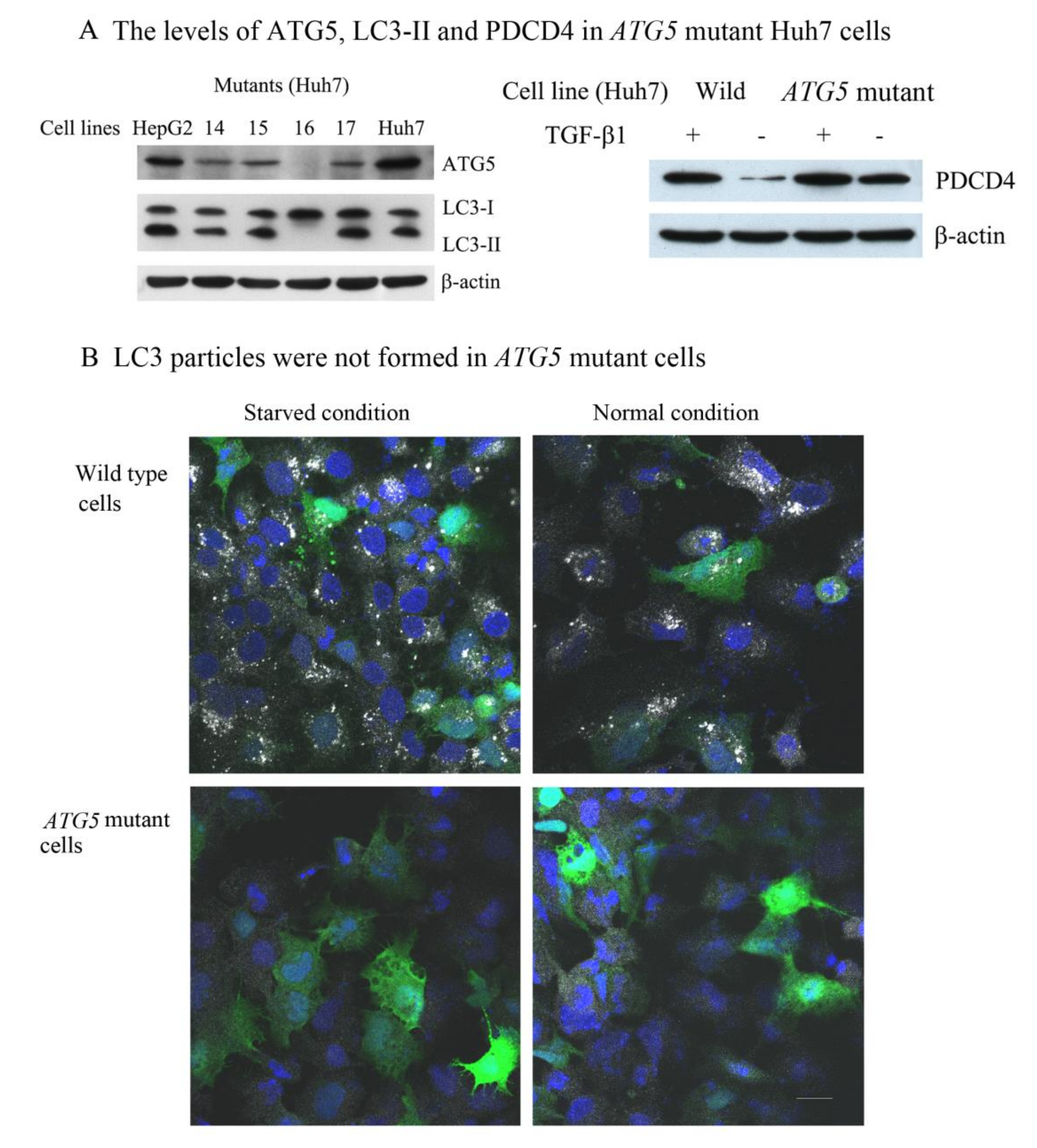
3.2. The Autophagy Inhibitors Upregulated the PDCD4 Levels in Huh7 ATG5 Mutant Cells
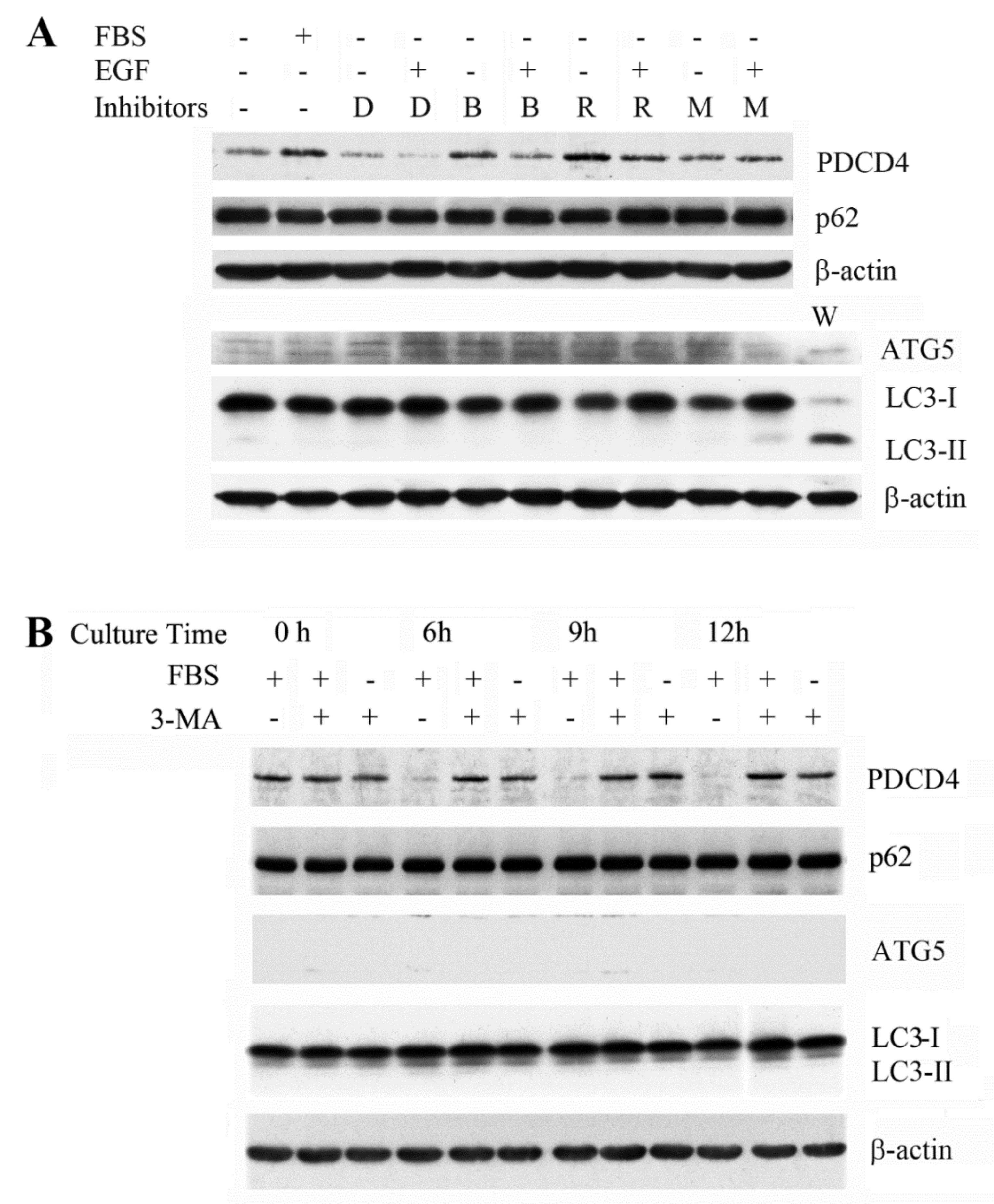
3.3. The PI3K Inhibitor 3-Methyladenine Upregulated the PDCD4 Level by Inhibiting Autophagy in Both Wild-Type and ATG5 Mutant Huh7 Cells
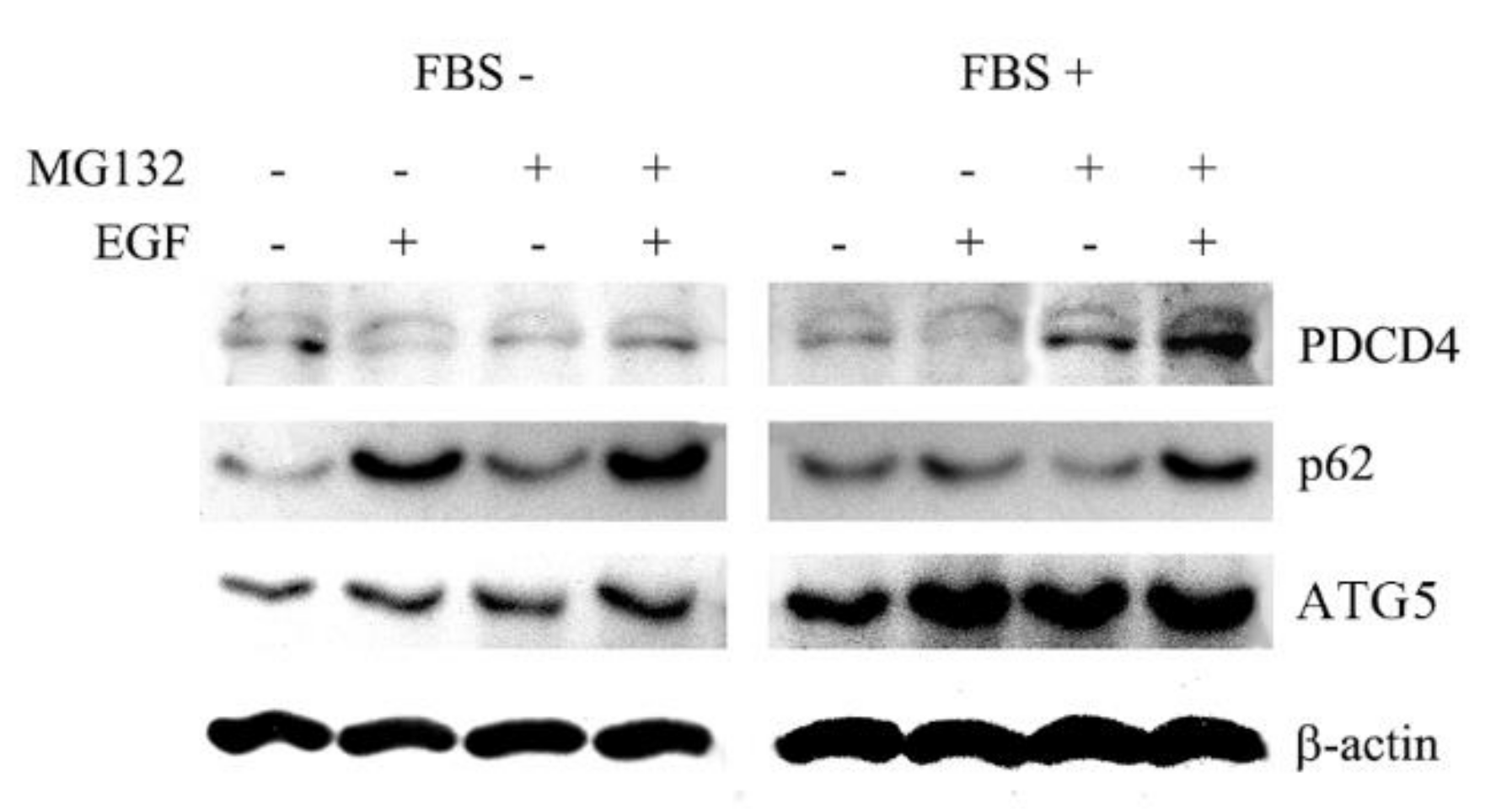
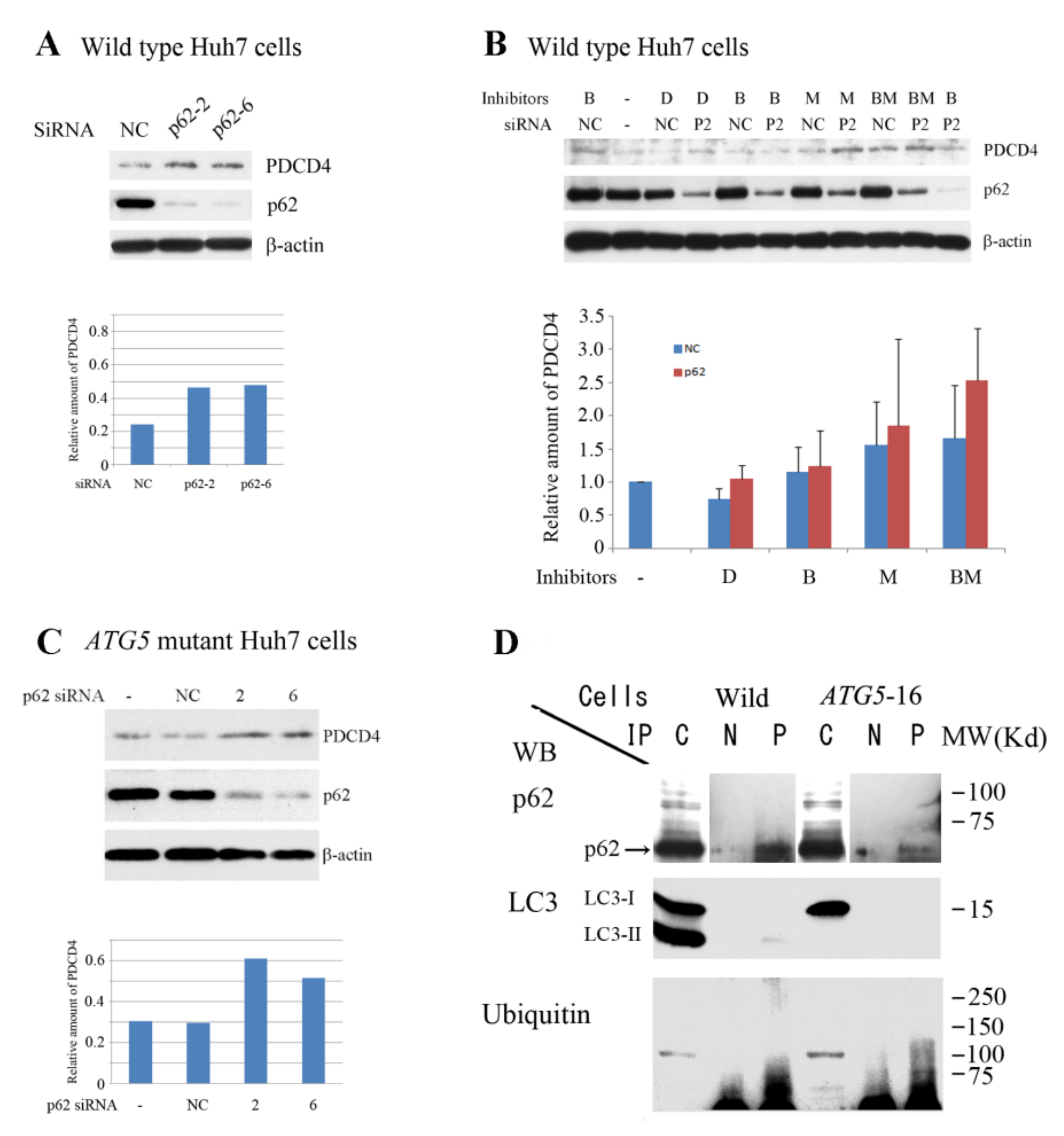
3.4. p62 is Involved in the Degradation of PDCD4
3.5. PDCD4 mRNA Levels Are Not Correlated with the Protein Levels
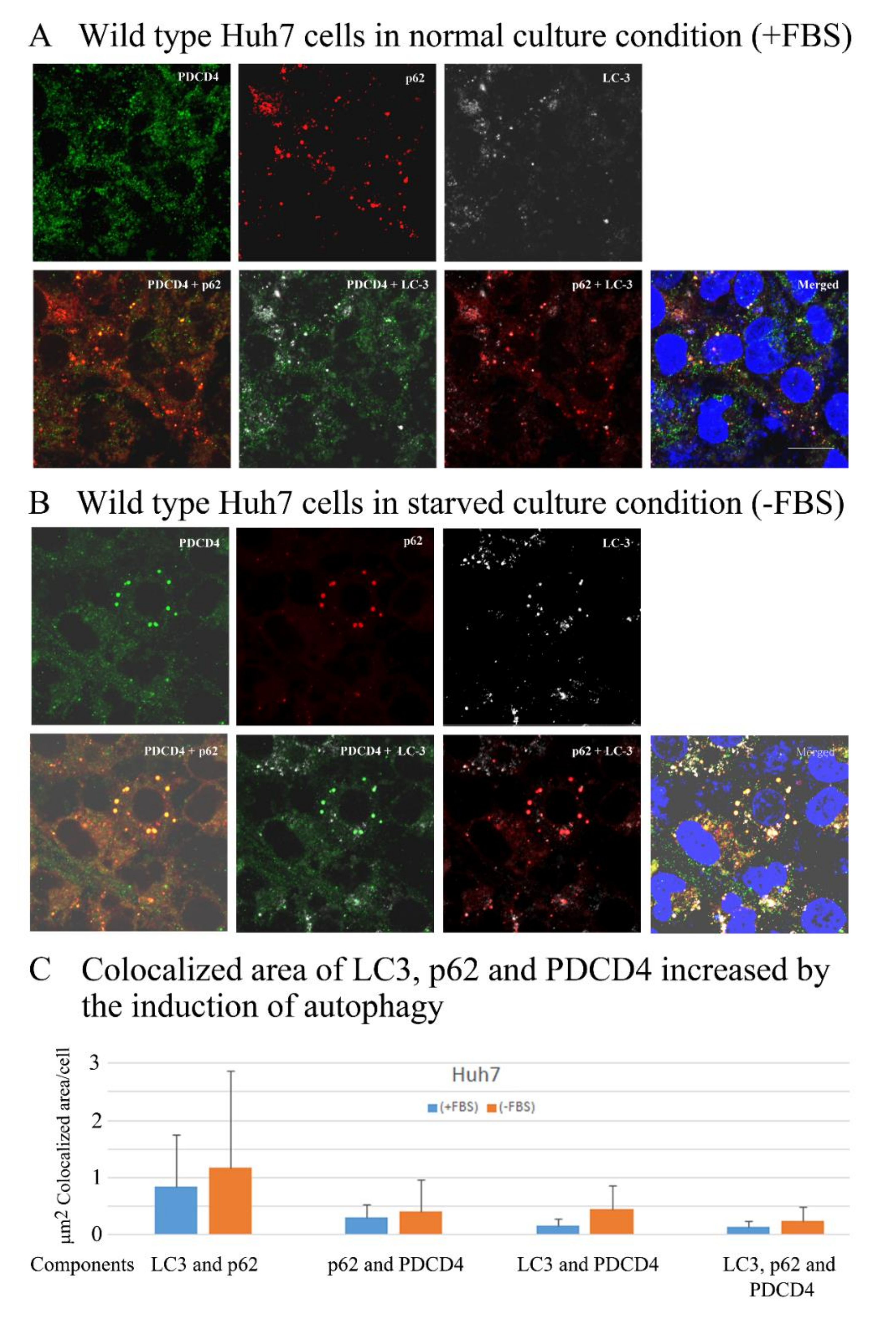
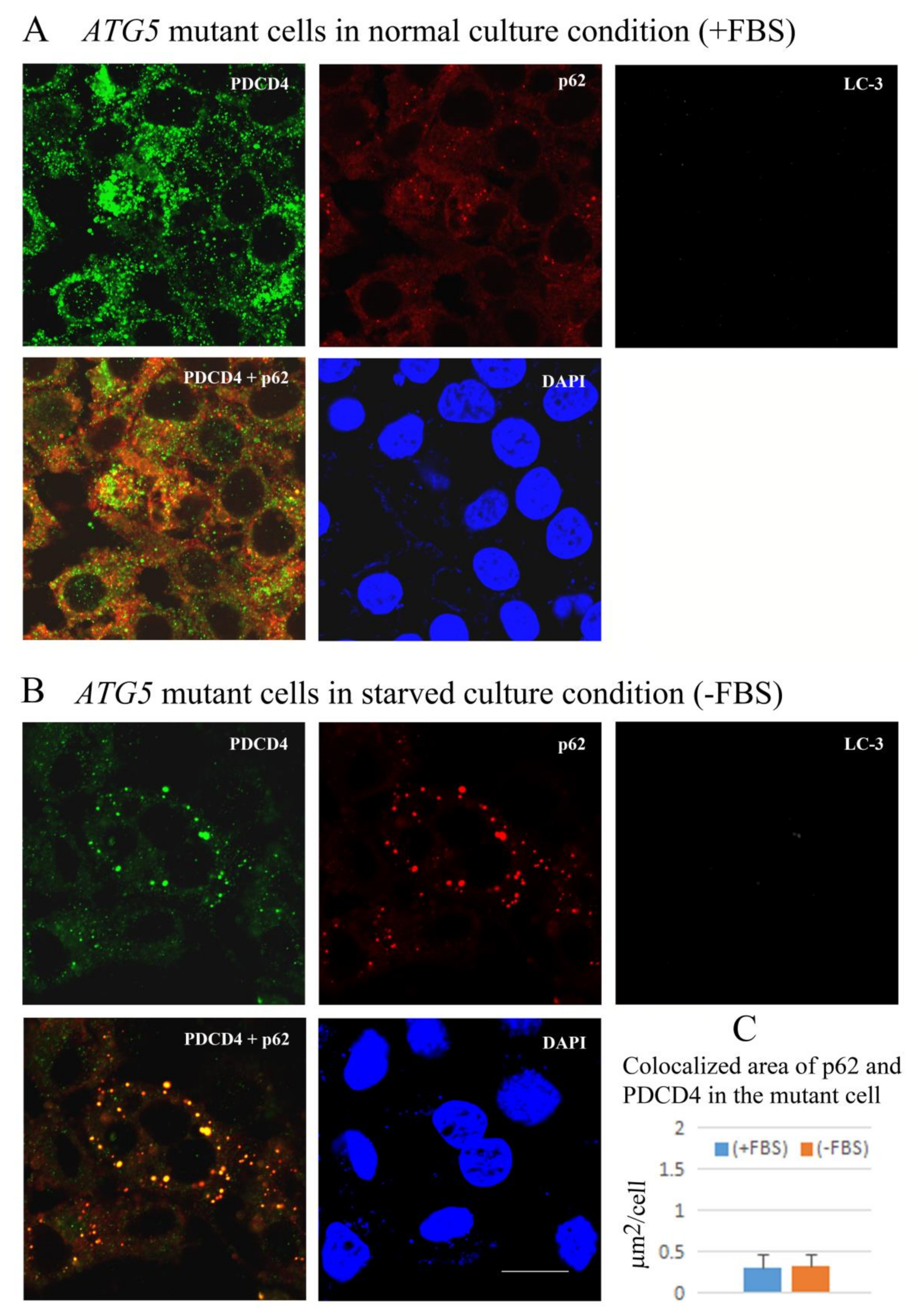
3.6. PDCD4, p62, and LC3 Are Colocalized in Particles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chang, J.H.; Cho, Y.H.; Sohn, S.Y.; Choi, J.M.; Kim, A.; Kim, Y.C.; Jang, S.K.; Cho, Y. Crystal structure of the eIF4A–PDCD4 complex. Proc. Natl. Acad. Sci. USA 2009, 106, 3148–3153. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, C.; Garces, R.G.; Edmonds, K.A.; Hiller, S.; Hyberts, S.G.; Marintchev, A.; Wagner, G. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc. Natl. Acad. Sci. USA 2008, 105, 327–3279. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-S.; Matthews, C.P.; Clair, T.; Wang, Q.; Baker, A.R.; Li, C.-C.H.; Tan, T.-H.; Colburn, N.H. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol. Cell. Biol. 2006, 26, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor Suppressor PDCD4 Represses Internal Ribosome Entry Site-Mediated Translation of Antiapoptotic Proteins and Is Regulated by S6 Kinase 2. Mol. Cell. Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Wedeken, L.; Waters, L.C.; Carr, M.D.; Klempnauer, K.H. Pdcd4 directly binds the coding region of c-myb mRNA and suppresses its translation. Oncogene 2011, 30, 4864–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.-K.; Baker, A.R.; Young, M.R.; Colburn, N.H. Tumor suppressor PDCD4 inhibits NF-κB-dependent transcription in human glioblastoma cells by direct interaction with p65. Carcinogenesis 2014, 35, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Izumi, H.; Tanimoto, A.; Takahashi, M.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Masubuchi, D.; Fukunaka, Y.; et al. Programmed Cell Death Protein 4 Down-regulates Y-Box Binding Protein-1 Expression via a Direct Interaction with Twist1 to Suppress Cancer Cell Growth. Cancer Res. 2009, 69, 3148–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leupold, J.H.; Yang, H.S.; Colburn, N.H.; Asangani, I.; Post, S.; Allgayer, H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 2007, 26, 4550–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Y.; Liu, Z.; Yang, H.; Yu, X.; Wu, Q.; Hua, S.; Long, X.; Jiang, Q.; Song, Y.; Cheng, C.; et al. Tumor suppressor PDCD4 modulates miR-184-mediated direct suppression of C-MYC and BCL2 blocking cell growth and survival in nasopharyngeal carcinoma. Cell Death Amp. Dis. 2013, 4, e872. [Google Scholar] [CrossRef]
- Afonja, O.; Juste, D.; Das, S.; Matsuhashi, S.; Samuels, H.H. Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 2004, 23, 8135. [Google Scholar] [CrossRef] [Green Version]
- Lankat-Buttgereit, B.; Gregel, C.; Knolle, A.; Hasilik, A.; Arnold, R.; Göke, R. Pdcd4 inhibits growth of tumor cells by suppression of carbonic anhydrase type II. Mol. Cell. Endocrinol. 2004, 214, 149–153. [Google Scholar] [CrossRef]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Hamajima, H.; Yasutake, T.; Eguchi, Y.; Ideguchi, H.; Yamamoto, K.; Matsuhashi, S. Involvement of programmed cell death 4 in transforming growth factor-β1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006, 25, 6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhu, J.; Wang, Y.W.; Dai, Y.; Wang, Y.L.; Wang, C.; Liu, J.; Baker, A.; Colburn, N.H.; Yang, H.S. Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 2017, 36, 6225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zennami, K.; Choi, S.M.; Liao, R.; Li, Y.; Dinalankara, W.; Marchionni, L.; Rafiqi, F.H.; Kurozumi, A.; Hatano, K.; Lupold, S.E. PDCD4 is an androgen-repressed tumor suppressor that regulates prostate cancer growth and castration resistance. Mol. Cancer Res. 2018, 17, 618–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Yu, Y.; Li, H.; Zhang, D.; Gong, A.; Li, S.; Liu, W.; Cheng, L.; Qiu, Y.; Yao, W.; et al. TGF-â1-induced miR-503 controls cell growth and apoptosis by targeting PDCD4 in glioblastoma cells. Sci. Rep. 2017, 7, 11569. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Knösel, T.; Kristiansen, G.; Pietas, A.; Garber, M.E.; Matsuhashi, S.; Ozaki, I.; Petersen, I. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J. Pathol. 2003, 200, 640–646. [Google Scholar] [CrossRef]
- Gao, F.; Wang, X.; Zhu, F.; Wang, Q.; Zhang, X.; Guo, C.; Zhou, C.; Ma, C.; Sun, W.; Zhang, Y.; et al. PDCD4 gene silencing in gliomas is associated with 5′CpG island methylation and unfavourable prognosis. J. Cell. Mol. Med. 2009, 13, 4257–4267. [Google Scholar] [CrossRef] [Green Version]
- Matsuhashi, S.; Manirujjaman, M.; Hamajima, H.; Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int. J. Mol. Sci. 2019, 20, 2304. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, Y.; Liang, H.; Fan, Q.; Zhu, R.; Cui, J.; Zhang, W.; Zen, K.; Zhang, C.-Y.; Hou, D.; et al. miR-23a/b promote tumor growth and suppress apoptosis by targeting PDCD4 in gastric cancer. Cell Death Dis. 2017, 8, e3059. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Guo, R.-D.; Guo, R.-M.; Sheng, W.; Yin, L.-R. MicroRNA-182 promotes cell growth, invasion, and chemoresistance by targeting programmed cell death 4 (PDCD4) in human ovarian carcinomas. J. Cell. Biochem. 2013, 114, 1464–1473. [Google Scholar] [CrossRef]
- Yao, Q.; Xu, H.; Zhang, Q.-Q.; Zhou, H.; Qu, L.-H. MicroRNA-21 promotes cell proliferation and down-regulates the expression of programmed cell death 4 (PDCD4) in HeLa cervical carcinoma cells. Biochem. Biophys. Res. Commun. 2009, 388, 539–542. [Google Scholar] [CrossRef]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed Cell Death 4 (PDCD4) Is an Important Functional Target of the MicroRNA miR-21 in Breast Cancer Cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Rasheed, S.A.K.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and ßTRCP-Mediated Degradation of PDCD4 Promotes Protein Translation and Cell Growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Hamajima, H.; Xia, J.; Iwane, S.; Kwaguchi, Y.; Eguchi, Y.; Mizuta, T.; Fujimoto, K.; Ozaki, I.; Matsuhashi, S. Regulation of tumor suppressor PDCD4 by novel protein kinase C isoforms. Biochim. Biophys. Acta 2010, 1803, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuhashi, S.; Hamajima, H.; Xia, J.; Zhang, H.; Mizuta, T.; Anzai, K.; Ozaki, I. Control of a tumor suppressor PDCD4: Degradation mechanisms of the protein in hepatocellular carcinoma cells. Cell. Signal. 2014, 26, 603–610. [Google Scholar] [CrossRef]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Schläfli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 2481. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-s.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-i.; Ezaki, J.; Murata, S.; et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [PubMed]
- Komatsu, M.; Ichimura, Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010, 584, 1374–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolewska, A.; Gajewska, M.; Zarzyńska, J.; Gajkowska, B.; Motyl, T. IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur. J. Cell Biol. 2009, 88, 117–130. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Grasso, D.; Renna, F.J.; Vaccaro, M.I. Initial Steps in Mammalian Autophagosome Biogenesis. Front. Cell Dev. Biol. 2018, 6, 146. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.-i.; Natsume, T.; Guan, J.-L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, N.; Codogno, P. Non-canonical autophagy: Facts and prospects. Curr. Pathobiol. Rep. 2013, 1, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Invest. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2016, 11, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar]
- Mauvezin, C.; Nagy, P.; Juhász, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.C.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-Independent Essential Function for the Autophagy Protein Atg5 in Cellular Immunity to Intracellular Pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050. [Google Scholar] [CrossRef] [Green Version]
- Schmid, T.; Jansen, A.P.; Baker, A.R.; Hegamyer, G.; Hagan, J.P.; Colburn, N.H. Translation Inhibitor Pdcd4 Is Targeted for Degradation during Tumor Promotion. Cancer Res. 2008, 68, 1254–1260. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, S.; Honda, S.; Yamaguchi, H.; Shimizu, S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Knævelsrud, H.; Simonsen, A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010, 584, 2635–2645. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Wang, X. p62 Stages an Interplay Between the Ubiquitin-Proteasome System and Autophagy in the Heart of Defense Against Proteotoxic Stress. Trends Cardiovasc. Med. 2011, 21, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.-L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, F.; Altucci, L.; Cobellis, G. Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers 2019, 11, 1465. [Google Scholar] [CrossRef] [Green Version]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manirujjaman, M.; Ozaki, I.; Murata, Y.; Guo, J.; Xia, J.; Nishioka, K.; Perveen, R.; Takahashi, H.; Anzai, K.; Matsuhashi, S. Degradation of the Tumor Suppressor PDCD4 Is Impaired by the Suppression of p62/SQSTM1 and Autophagy. Cells 2020, 9, 218. https://doi.org/10.3390/cells9010218
Manirujjaman M, Ozaki I, Murata Y, Guo J, Xia J, Nishioka K, Perveen R, Takahashi H, Anzai K, Matsuhashi S. Degradation of the Tumor Suppressor PDCD4 Is Impaired by the Suppression of p62/SQSTM1 and Autophagy. Cells. 2020; 9(1):218. https://doi.org/10.3390/cells9010218
Chicago/Turabian StyleManirujjaman, M., Iwata Ozaki, Yuzo Murata, Jing Guo, Jinghe Xia, Kenichi Nishioka, Rasheda Perveen, Hirokazu Takahashi, Keizo Anzai, and Sachiko Matsuhashi. 2020. "Degradation of the Tumor Suppressor PDCD4 Is Impaired by the Suppression of p62/SQSTM1 and Autophagy" Cells 9, no. 1: 218. https://doi.org/10.3390/cells9010218