Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study

Abstract
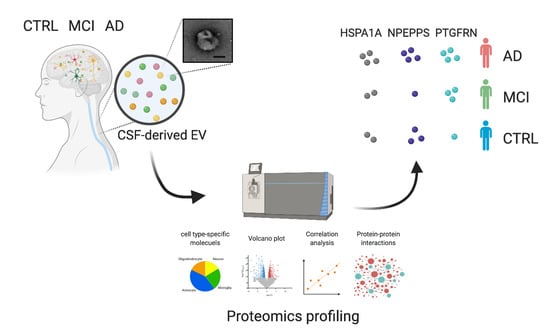
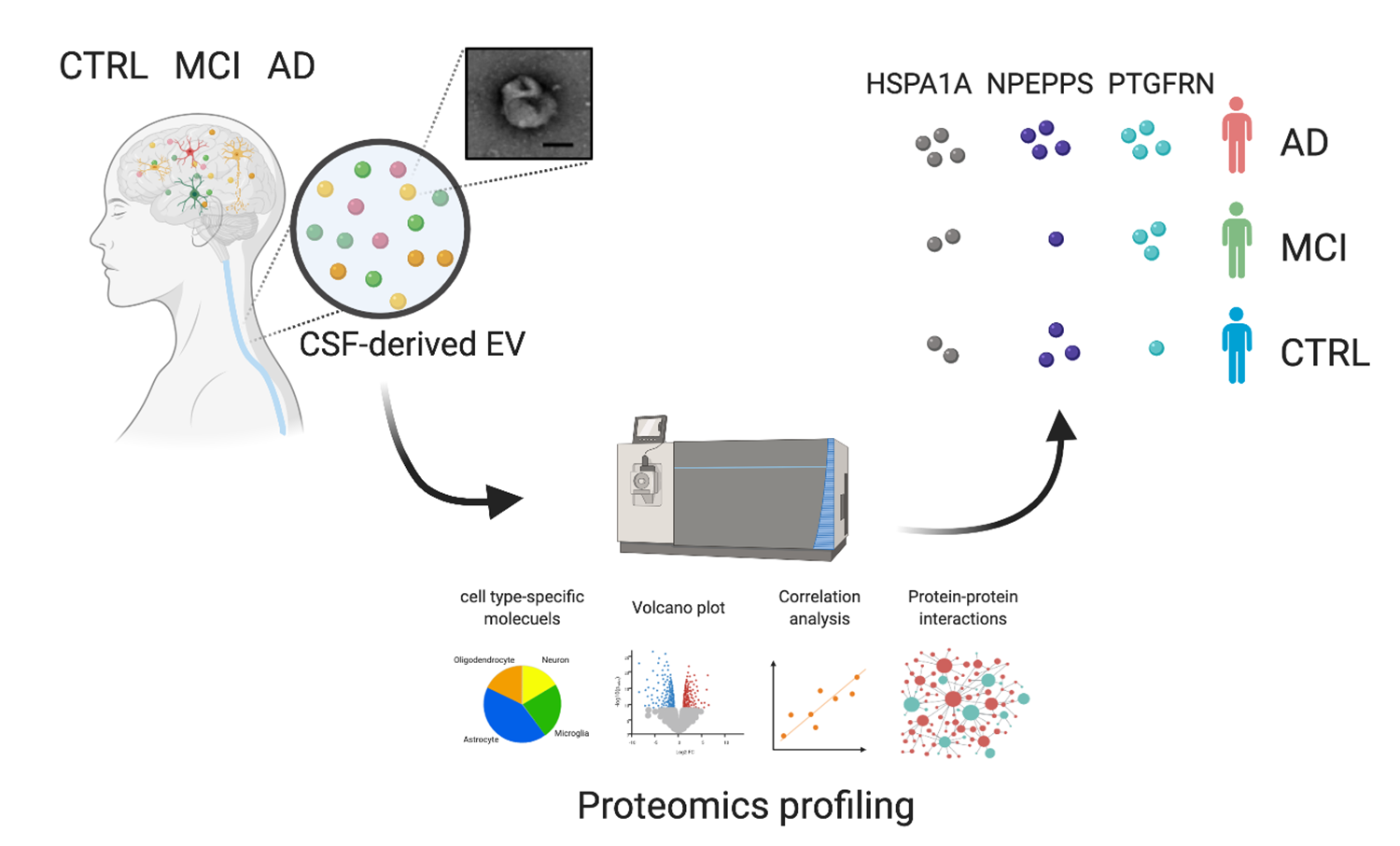
:
1. Introduction
2. Materials and Methods
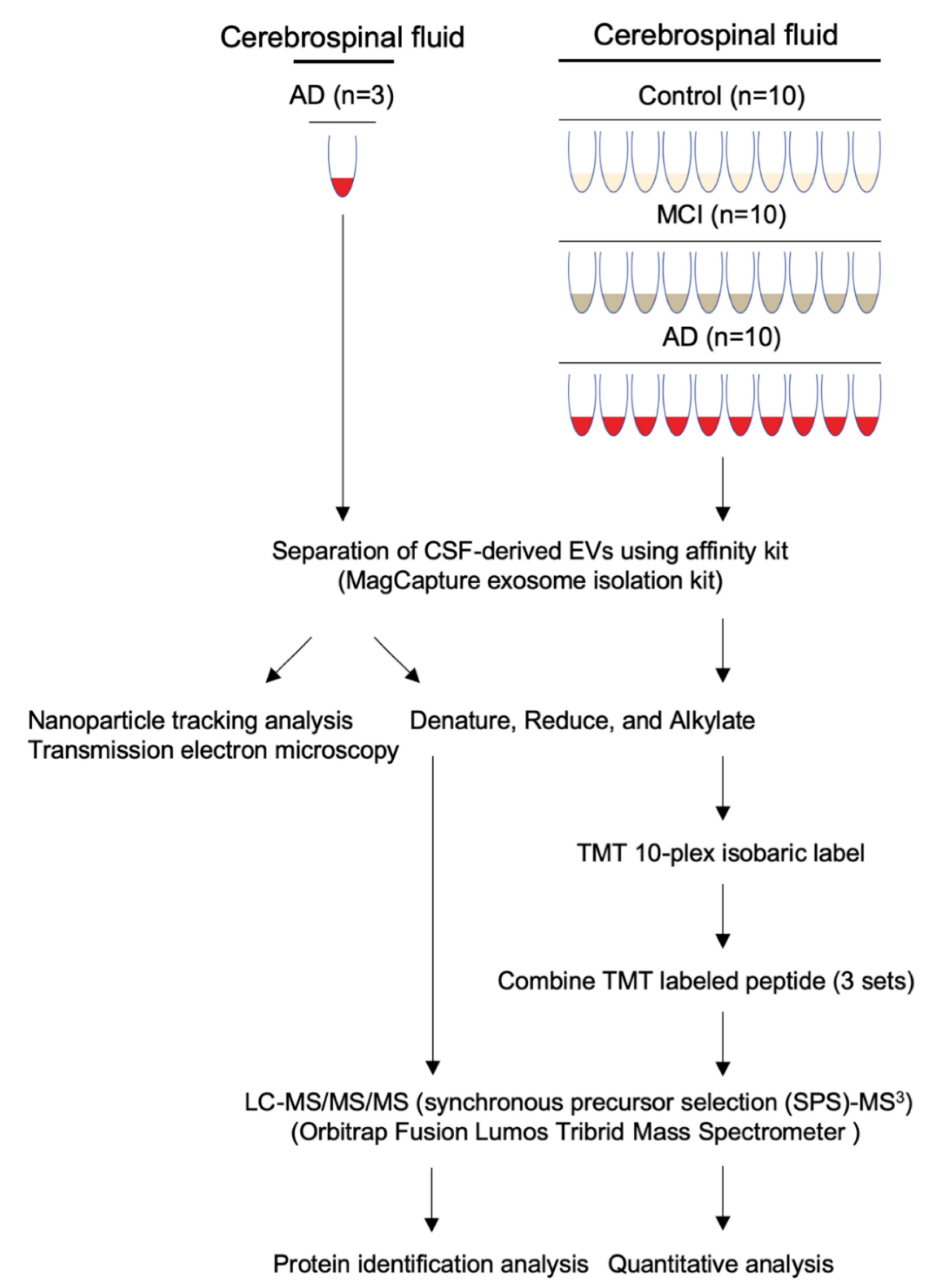
2.1. Sample Selection
2.2. Separation of EVs from Human CSF Samples
2.3. Nanoparticle Tracking Analysis (NTA)
2.4. Transmission Electron Microscopy (TEM)
2.5. Protein Concentrations
2.6. In-Solution Digestion
2.7. Mass Spectrometry
2.7.1. Peptide Labeling with TMT 10-Plex Isobaric Labeling Kit
2.7.2. Nano-Liquid Chromatography and Tandem Mass-Spectrometry
2.7.3. Mass-Spectrometry Data Analysis
2.8. Measurement of Total Tau (t-tau) and Tau Phosphorylated on Threonine 231 (p-tau231)
2.9. Statistical Analysis
3. Results
3.1. Workflow for Protein Profiling of Extracellular Vesicles Separated from Human CSF
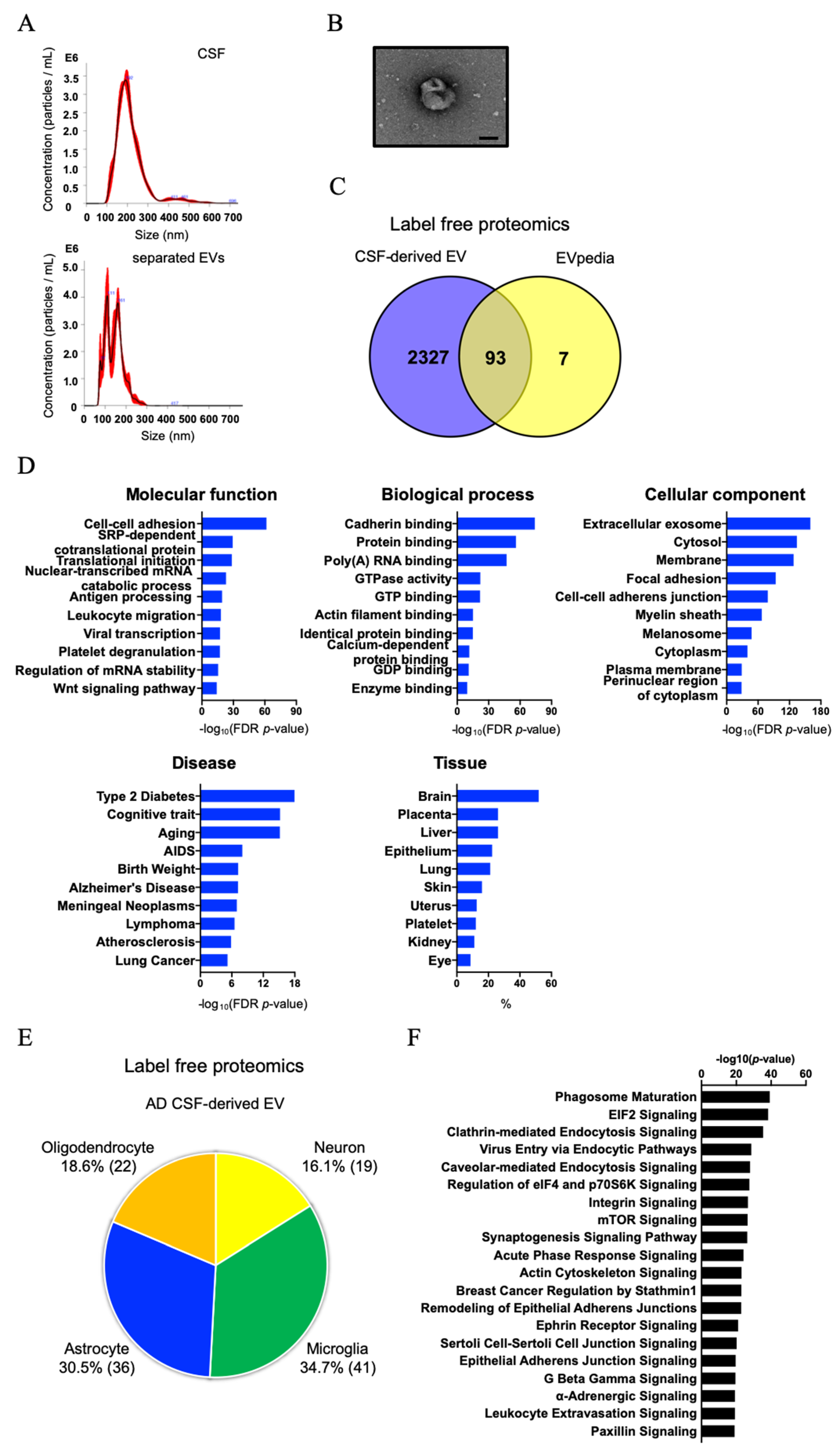
3.2. Biochemical and Morphological Characterization of EVs Separated from CSF
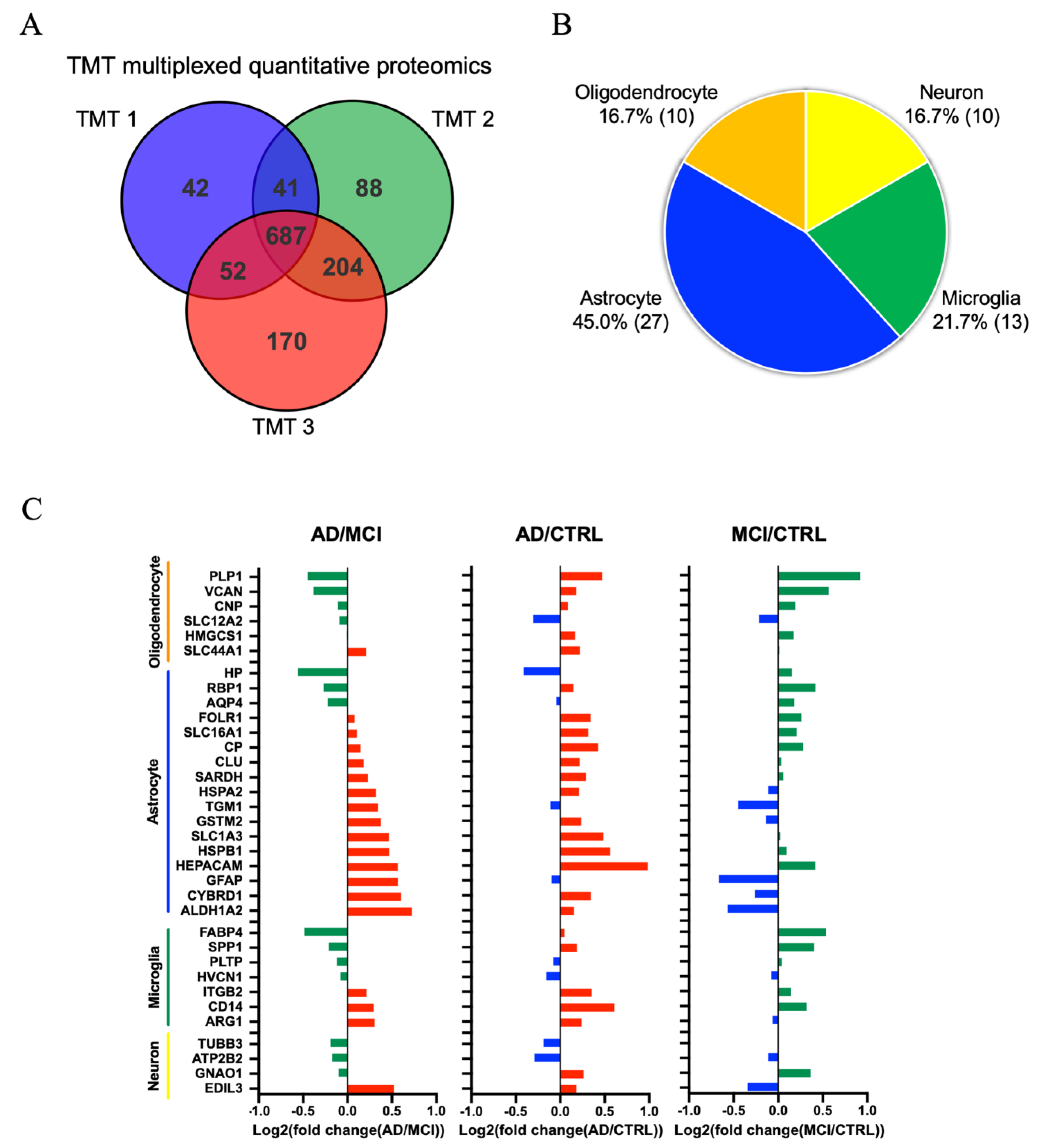
3.3. Comparison of AD, MCI, and CTRL CSF-Derived EV Proteins by TMT-Labeled Quantitative Proteomics Analysis
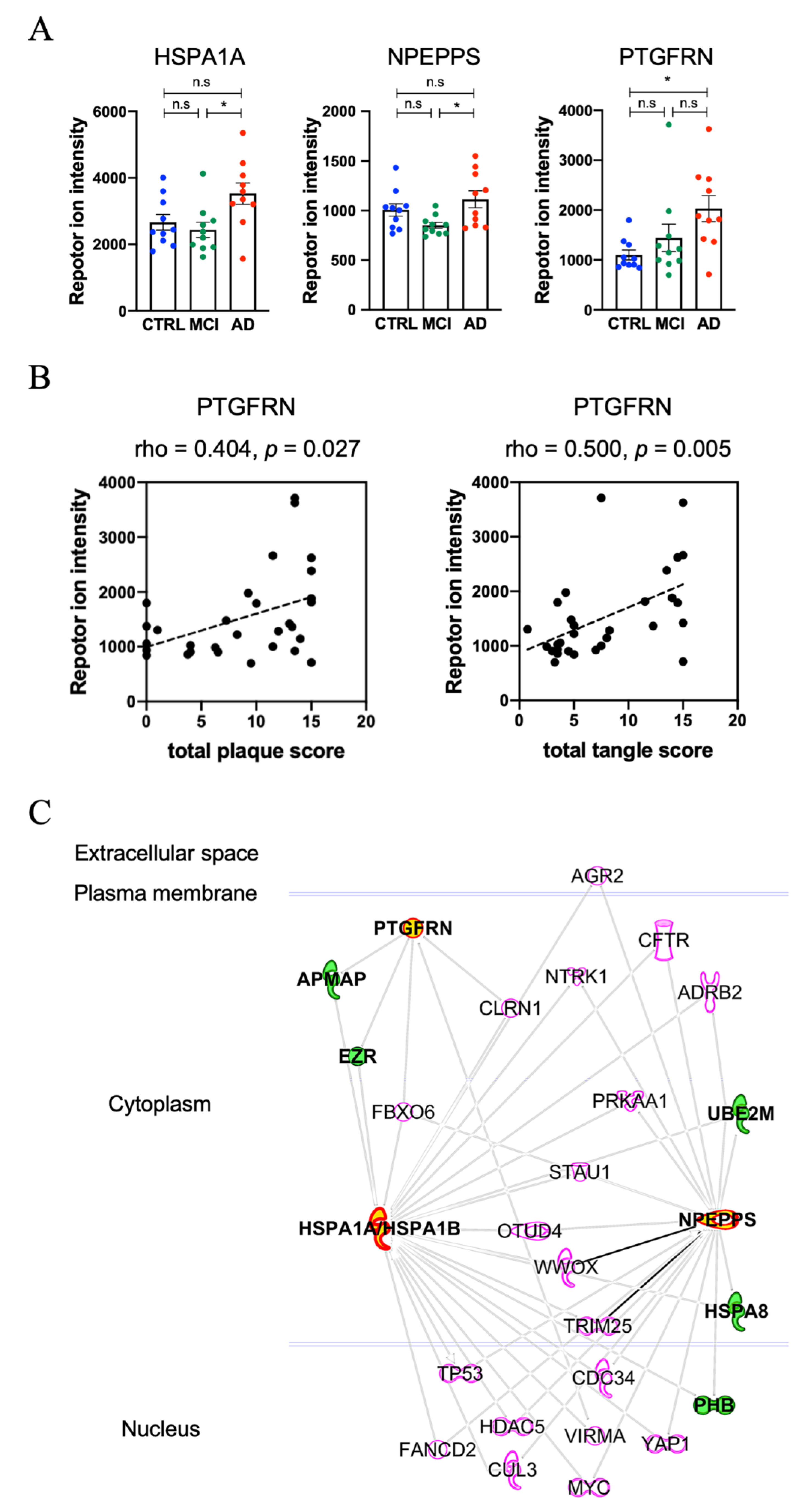
3.4. Molecules Altered during Progression to Alzheimer’s Disease from Mild Cognitive Impairment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef]
- Andreasen, N.; Hesse, C.; Davidsson, P.; Minthon, L.; Wallin, A.; Winblad, B.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Cerebrospinal fluid beta-amyloid(1-42) in Alzheimer disease: Differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch. Neurol. 1999, 56, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Spies, P.E.; Verbeek, M.M.; van Groen, T.; Claassen, J.A.H.R. Reviewing reasons for the decreased CSF Abeta42 concentration in Alzheimer disease. Front. Biosci. 2012, 17, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Olsson, A.; Vanderstichele, H.; Andreasen, N.; De Meyer, G.; Wallin, A.; Holmberg, B.; Rosengren, L.; Vanmechelen, E.; Blennow, K. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 2005, 51, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.-K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buerger, K.; Zinkowski, R.; Teipel, S.J.; Tapiola, T.; Arai, H.; Blennow, K.; Andreasen, N.; Hofmann-Kiefer, K.; DeBernardis, J.; Kerkman, D.; et al. Differential diagnosis of Alzheimer disease with cerebrospinal fluid levels of tau protein phosphorylated at threonine 231. Arch. Neurol. 2002, 59, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Pirraglia, E.; Osorio, R.S.; Glodzik, L.; Li, Y.; Tsui, W.; Saint Louis, L.A.; Randall, C.; Butler, T.; Xu, J.; et al. Greater specificity for cerebrospinal fluid P-tau231 over P-tau181 in the differentiation of healthy controls from Alzheimer’s disease. J. Alzheimers Dis. 2016, 49, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. Alzheimer’s Disease Neuroimaging Initiative CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- Kvartsberg, H.; Portelius, E.; Andreasson, U.; Brinkmalm, G.; Hellwig, K.; Lelental, N.; Kornhuber, J.; Hansson, O.; Minthon, L.; Spitzer, P.; et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimers Res. Ther. 2015, 7, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Sutphen, C.L.; McCue, L.; Herries, E.M.; Xiong, C.; Ladenson, J.H.; Holtzman, D.M.; Fagan, A.M. ADNI Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimers Dement. 2018, 14, 869–879. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Brinkmalm, A.; Dumurgier, J.; Brinkmalm, G.; Hansson, O.; Zetterberg, H.; Bouaziz-Amar, E.; Hugon, J.; Paquet, C.; Blennow, K. The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sandelius, Å.; Portelius, E.; Källén, Å.; Zetterberg, H.; Rot, U.; Olsson, B.; Toledo, J.B.; Shaw, L.M.; Lee, V.M.Y.; Irwin, D.J.; et al. Elevated CSF GAP-43 is Alzheimer’s disease specific and associated with tau and amyloid pathology. Alzheimers Dement. 2019, 15, 55–64. [Google Scholar] [CrossRef]
- Fang, Q.; Strand, A.; Law, W.; Faca, V.M.; Fitzgibbon, M.P.; Hamel, N.; Houle, B.; Liu, X.; May, D.H.; Poschmann, G.; et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol. Cell Proteom. 2009, 8, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraoka, S.; Lin, W.; Chen, M.; Hersh, S.W.; Emili, A.; Xia, W.; Ikezu, T. Assessment of separation methods for extracellular vesicles from human and mouse brain tissues and human cerebrospinal fluids. Methods 2020, 177, 35–49. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, A.M.; Ikezu, T. Extracellular Vesicle Biology in Alzheimer’s Disease and Related Tauopathy. J. Neuroimmune Pharmacol. 2018, 13, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.-J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Ikezu, T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiol. Dis. 2019, 130, 104512. [Google Scholar] [CrossRef]
- Karttunen, J.; Heiskanen, M.; Lipponen, A.; Poulsen, D.; Pitkänen, A. Extracellular Vesicles as Diagnostics and Therapeutics for Structural Epilepsies. Int. J. Mol. Sci. 2019, 20, 1259. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 32. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Tatebe, H.; DeLeo, A.M.; Ikezu, S.; Tokuda, T.; Gygi, S.P.; Stern, R.A.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Isolated From Cerebrospinal Fluid of Former National Football League Players at Risk for Chronic Traumatic Encephalopathy. Front. Neurosci. 2019, 13, 1059. [Google Scholar] [CrossRef] [Green Version]
- Izadpanah, M.; Seddigh, A.; Ebrahimi Barough, S.; Fazeli, S.A.S.; Ai, J. Potential of Extracellular Vesicles in Neurodegenerative Diseases: Diagnostic and Therapeutic Indications. J. Mol. Neurosci. 2018, 66, 172–179. [Google Scholar] [CrossRef]
- Delpech, J.-C.; Herron, S.; Botros, M.B.; Ikezu, T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019, 42, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Sharples, R.A.; Nisbet, R.M.; Cappai, R.; Hill, A.F. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 2008, 37, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [Green Version]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 12093. [Google Scholar] [CrossRef]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, S.; DeLeo, A.M.; Sethi, M.K.; Yukawa-Takamatsu, K.; Yang, Z.; Ko, J.; Hogan, J.D.; Ruan, Z.; You, Y.; Wang, Y.; et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimers Dement. 2020, 16, 896–907. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, M.L.; Ilmer, M.; Silva, L.P.; Hawke, D.H.; Recio, A.; Vorontsova, M.A.; Alt, E.; Vykoukal, J. Benchtop isolation and characterization of functional exosomes by sequential filtration. J. Chromatogr. A 2014, 1371, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, S.S.; Dunlay, C.J.; Simeone, D.M.; Nagrath, S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab. Chip. 2014, 14, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Krumeich, S.; Raposo, G.; Thery, C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Onódi, Z.; Pelyhe, C.; Terézia Nagy, C.; Brenner, G.B.; Almási, L.; Kittel, Á.; Manček-Keber, M.; Ferdinandy, P.; Buzás, E.I.; Giricz, Z. Isolation of High-Purity Extracellular Vesicles by the Combination of Iodixanol Density Gradient Ultracentrifugation and Bind-Elute Chromatography From Blood Plasma. Front. Physiol. 2018, 9, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, W.; Yoshida, T.; Diez, D.; Miyatake, Y.; Nishibu, T.; Imawaka, N.; Naruse, K.; Sadamura, Y.; Hanayama, R. A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci. Rep. 2016, 6, 33935. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Serrano, G.; Shill, H.A.; Walker, D.G.; Lue, L.; Roher, A.E.; Dugger, B.N.; Maarouf, C.; et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015, 35, 354–389. [Google Scholar] [CrossRef]
- Morris, J.C.; Heyman, A.; Mohs, R.C.; Hughes, J.P.; van Belle, G.; Fillenbaum, G.; Mellits, E.D.; Clark, C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989, 39, 1159–1165. [Google Scholar] [CrossRef]
- Gearing, M.; Mirra, S.S.; Hedreen, J.C.; Sumi, S.M.; Hansen, L.A.; Heyman, A. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s disease. Neurology 1995, 45, 461–466. [Google Scholar] [CrossRef]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wühr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, L.; Rad, R.; Gygi, S.P.; Haas, W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Matsumura, S.; Minamisawa, T.; Suga, K.; Kishita, H.; Akagi, T.; Ichiki, T.; Ichikawa, Y.; Shiba, K. Subtypes of tumour cell-derived small extracellular vesicles having differently externalized phosphatidylserine. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-K.; Lee, J.; Kim, S.R.; Choi, D.-S.; Yoon, Y.J.; Kim, J.H.; Go, G.; Nhung, D.; Hong, K.; Jang, S.C.; et al. EVpedia: A community web portal for extracellular vesicles research. Bioinformatics 2015, 31, 933–939. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Schmitt, S.; Bergner, C.G.; Tyanova, S.; Kannaiyan, N.; Manrique-Hoyos, N.; Kongi, K.; Cantuti, L.; Hanisch, U.-K.; Philips, M.-A.; et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015, 18, 1819–1831. [Google Scholar] [CrossRef]
- Bryk, A.H.; Wiśniewski, J.R. Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef] [Green Version]
- Zaccaria, A.; Roux-Dalvai, F.; Bouamrani, A.; Mombrun, A.; Mossuz, P.; Monsarrat, B.; Berger, F. Accessing to the minor proteome of red blood cells through the influence of the nanoparticle surface properties on the corona composition. Int. J. Nanomed. 2015, 10, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, S.B.; Hampton, M.B.; Winterbourn, C.C. Accumulation of oxidized peroxiredoxin 2 in red blood cells and its prevention. Transfusion 2015, 55, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Ravenhill, B.J.; Kanjee, U.; Ahouidi, A.; Nobre, L.; Williamson, J.; Goldberg, J.M.; Antrobus, R.; Dieye, T.; Duraisingh, M.T.; Weekes, M.P. Quantitative comparative analysis of human erythrocyte surface proteins between individuals from two genetically distinct populations. Commun. Biol. 2019, 2, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, P.; Mulzer, L.-M.; Oberstein, T.J.; Munoz, L.E.; Lewczuk, P.; Kornhuber, J.; Herrmann, M.; Maler, J.M. Microvesicles from cerebrospinal fluid of patients with Alzheimer’s disease display reduced concentrations of tau and APP protein. Sci. Rep. 2019, 9, 7089. [Google Scholar] [CrossRef] [Green Version]
- Scieglinska, D.; Krawczyk, Z. Expression, function, and regulation of the testis-enriched heat shock HSPA2 gene in rodents and humans. Cell Stress Chaperones 2015, 20, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Voss, K.; Combs, B.; Patterson, K.R.; Binder, L.I.; Gamblin, T.C. Hsp70 alters tau function and aggregation in an isoform specific manner. Biochemistry 2012, 51, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Yamashima, T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J. Neurochem. 2012, 120, 477–494. [Google Scholar] [CrossRef]
- Gerber, H.; Mosser, S.; Boury-Jamot, B.; Stumpe, M.; Piersigilli, A.; Goepfert, C.; Dengjel, J.; Albrecht, U.; Magara, F.; Fraering, P.C. The APMAP interactome reveals new modulators of APP processing and beta-amyloid production that are altered in Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 13–18. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Ren, G.; Ma, Z.; Hui, M.; Kudo, L.C.; Hui, K.-S.; Karsten, S.L. Cu, Zn-superoxide dismutase 1 (SOD1) is a novel target of Puromycin-sensitive aminopeptidase (PSA/NPEPPS): PSA/NPEPPS is a possible modifier of amyotrophic lateral sclerosis. Mol. Neurodegener. 2011, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Karsten, S.L.; Sang, T.-K.; Gehman, L.T.; Chatterjee, S.; Liu, J.; Lawless, G.M.; Sengupta, S.; Berry, R.W.; Pomakian, J.; Oh, H.S.; et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006, 51, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Horowitz, P.M.; Karsten, S.L.; Jackson, G.R.; Geschwind, D.H.; Fu, Y.; Berry, R.W.; Binder, L.I. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry 2006, 45, 15111–15119. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, L.; Ping, L.; Dammer, E.B.; Duong, D.M.; Zhou, M.; Gearing, M.; Johnson, E.C.B.; Hajjar, I.; Lah, J.J.; Levey, A.I.; et al. Integrated Proteomics Reveals Brain-Based Cerebrospinal Fluid Biomarkers in Asymptomatic and Symptomatic Alzheimer’s Disease. bioRxiv 2019, 14, 806752. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, T.; Craessaerts, K.; Bammens, L.; Bentahir, M.; Borgions, F.; Herdewijn, P.; Staes, A.; Timmerman, E.; Vandekerckhove, J.; Rubinstein, E.; et al. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 2009, 11, 1340–1346. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| For Label-Free Proteomics | AD | |||||
| (n = 3) | ||||||
| Age, mean | 82.7 ± 7.57 | |||||
| Gender | 2M, 1F | |||||
| For TMT-Label Proteomics | CTRL | MCI | AD | t-test a (p-value b) | t-test a (p-value b) | t-test a (p-value b) |
| (n = 10) | (n = 10) | (n = 10) | (CTRL_MCI) | (CTRL_AD) | (MCI_AD) | |
| Age, mean | 89.8 ± 6.12 | 89.0 ± 5.27 | 87.1 ± 4.41 | 0.313 (0.758) | 1.131 (0.273) | 0.874 (0.393) |
| Gender c | 5M, 5F | 5M, 5F | 5M, 5F | |||
| PMI, mean | 2.90 ± 0.85 | 2.94 ± 0.87 | 2.84 ± 0.49 | −0.106 (0.917) | 0.173 (0.865) | 0.299 (0.768) |
| Braak stage | 2.8 ± 0.42 | 3.3 ± 0.67 | 5.4 ± 0.52 | −1.987 (0.062) | −12.333 (<0.001) | −7.814 (<0.001) |
| Human CSF | EV Fraction | |
|---|---|---|
| Mode size (nm) a | 191 | 110 |
| Particle number a,b | 4.6 × 109/500 μL CSF | 2.36 × 109/100 μL |
| EV protein (μg) b,c | 1187.48 | 0.78 |
| Particles/proteins (μg) | 3.87 × 106 | 3.03 × 109 |
| Protein ID | Gene Symbol | log2 | log2 | log2 | One-Way ANOVA a | −log10 (p-Value | −log10 (p-Value | −log10 (p-Value |
|---|---|---|---|---|---|---|---|---|
| (AD/CTRL) | (MCI/CTRL) | (AD/MCI) | (AD_CTRL)) a | (MCI_CTRL)) | (AD_MCI)) | |||
| P08107 | HSPA1A | 0.405 | −0.128 | 0.533 | 0.017 | 1.149 | 0.087 | 1.721 |
| Q9P2B2 | PTGFRN | 0.882 | 0.391 | 0.491 | 0.024 | 1.721 | 0.268 | 0.75 |
| P55786 | NPEPPS | 0.145 | −0.243 | 0.388 | 0.025 | 0.319 | 0.666 | 1.699 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. https://doi.org/10.3390/cells9091959
Muraoka S, Jedrychowski MP, Yanamandra K, Ikezu S, Gygi SP, Ikezu T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells. 2020; 9(9):1959. https://doi.org/10.3390/cells9091959
Chicago/Turabian StyleMuraoka, Satoshi, Mark P. Jedrychowski, Kiran Yanamandra, Seiko Ikezu, Steven P. Gygi, and Tsuneya Ikezu. 2020. "Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study" Cells 9, no. 9: 1959. https://doi.org/10.3390/cells9091959