A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Plasma and Liver Biochemical Analysis
2.3. Histology
2.4. Transcriptome Analysis
2.5. Statistical Analysis
3. Results
3.1. HFD and FFD Induce Features of the Metabolic Syndrome in Ldlr−/−.Leiden Mice
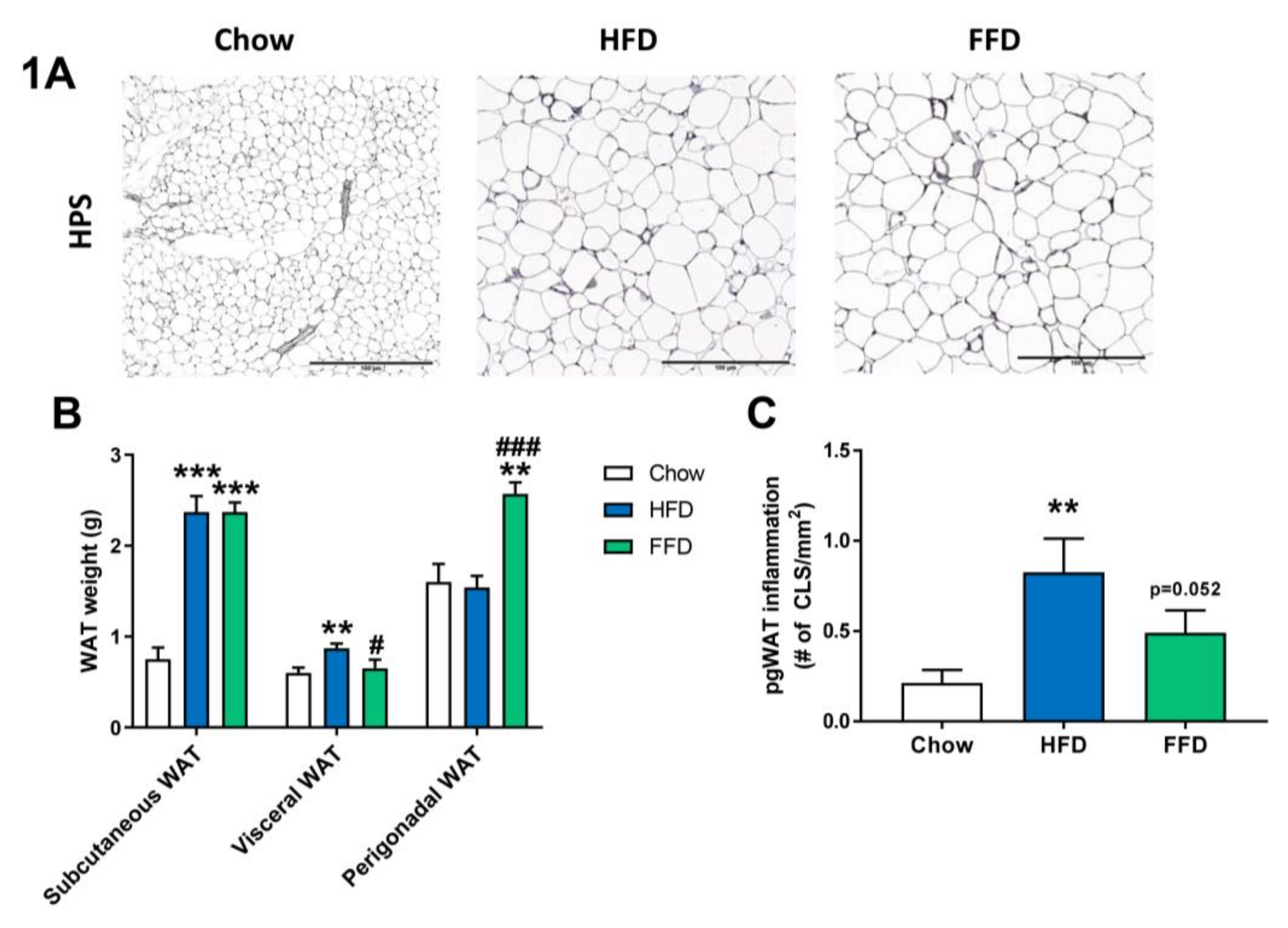
3.2. HFD and FFD Differentially Affect WAT
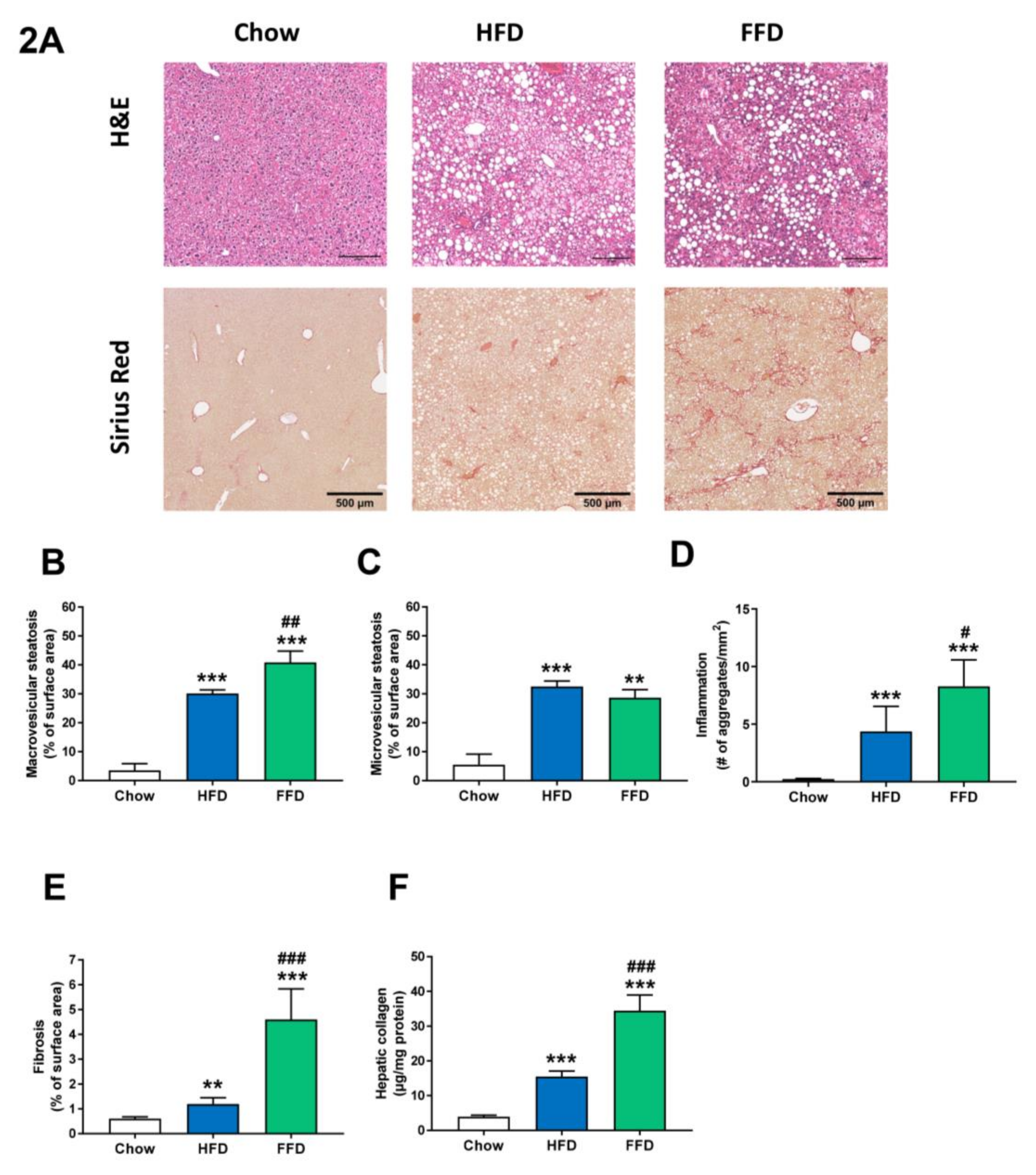
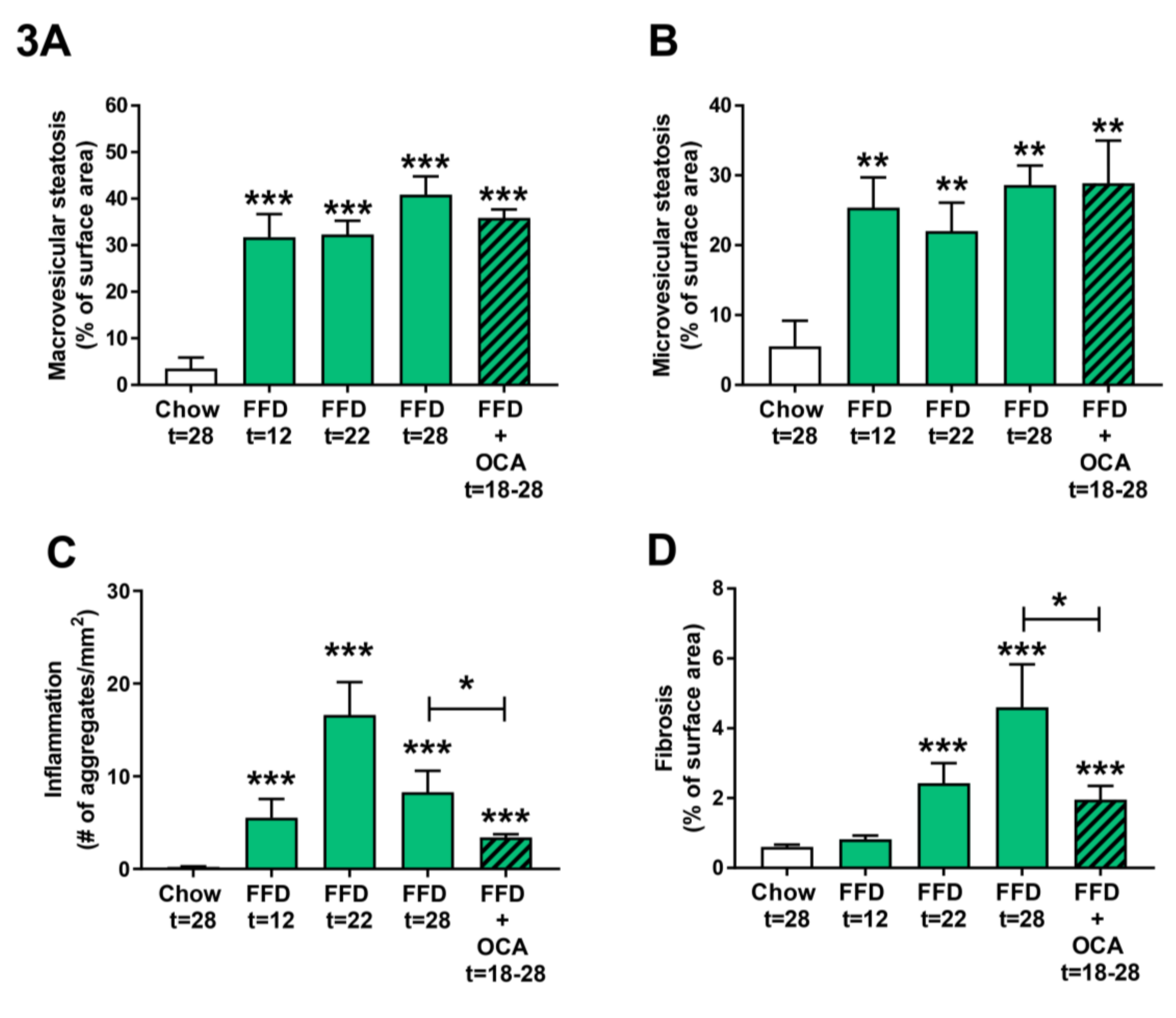
3.3. Ldlr−/−.Leiden Mice Develop Steatosis, Hepatic Inflammation, and Fibrosis.
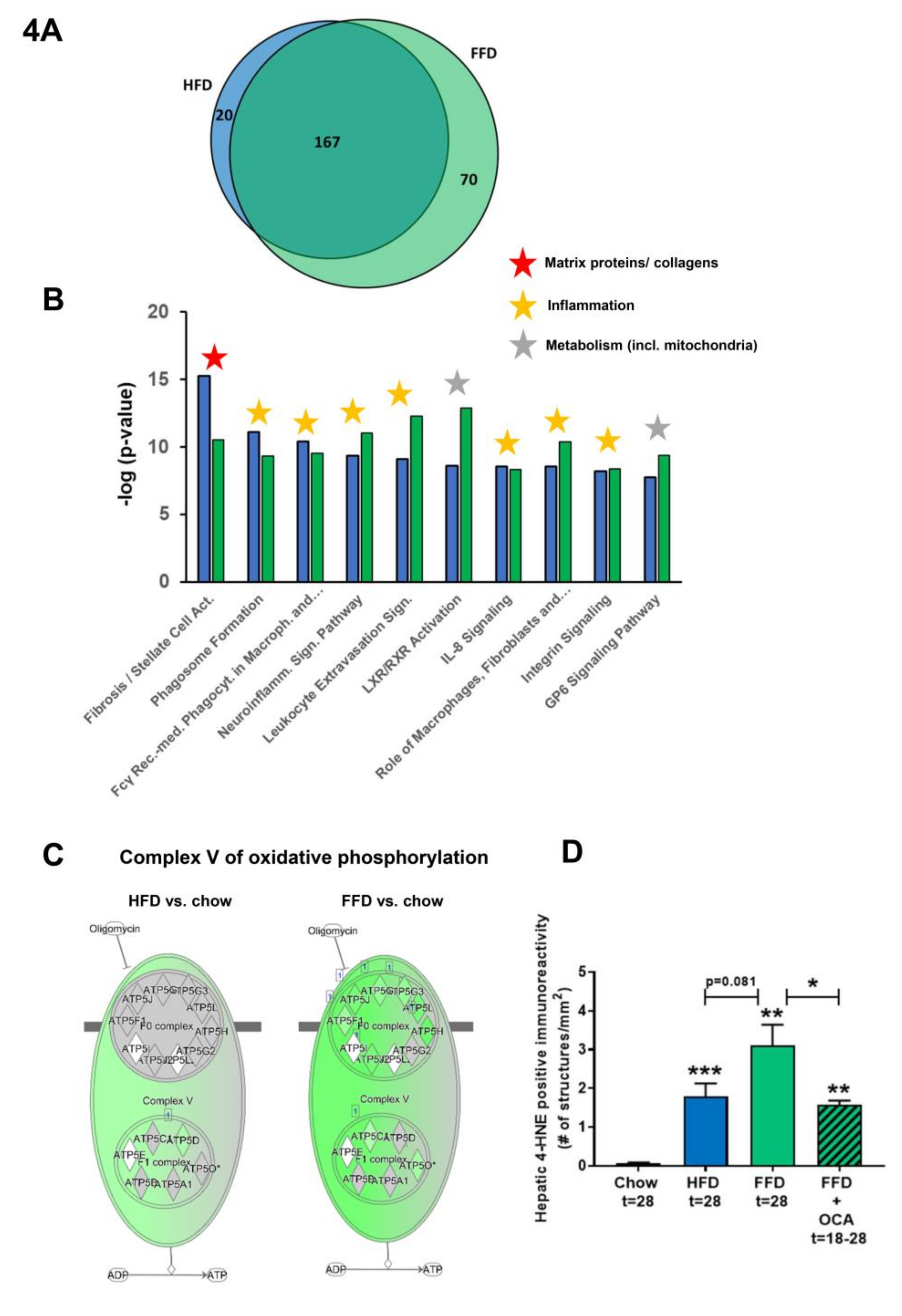
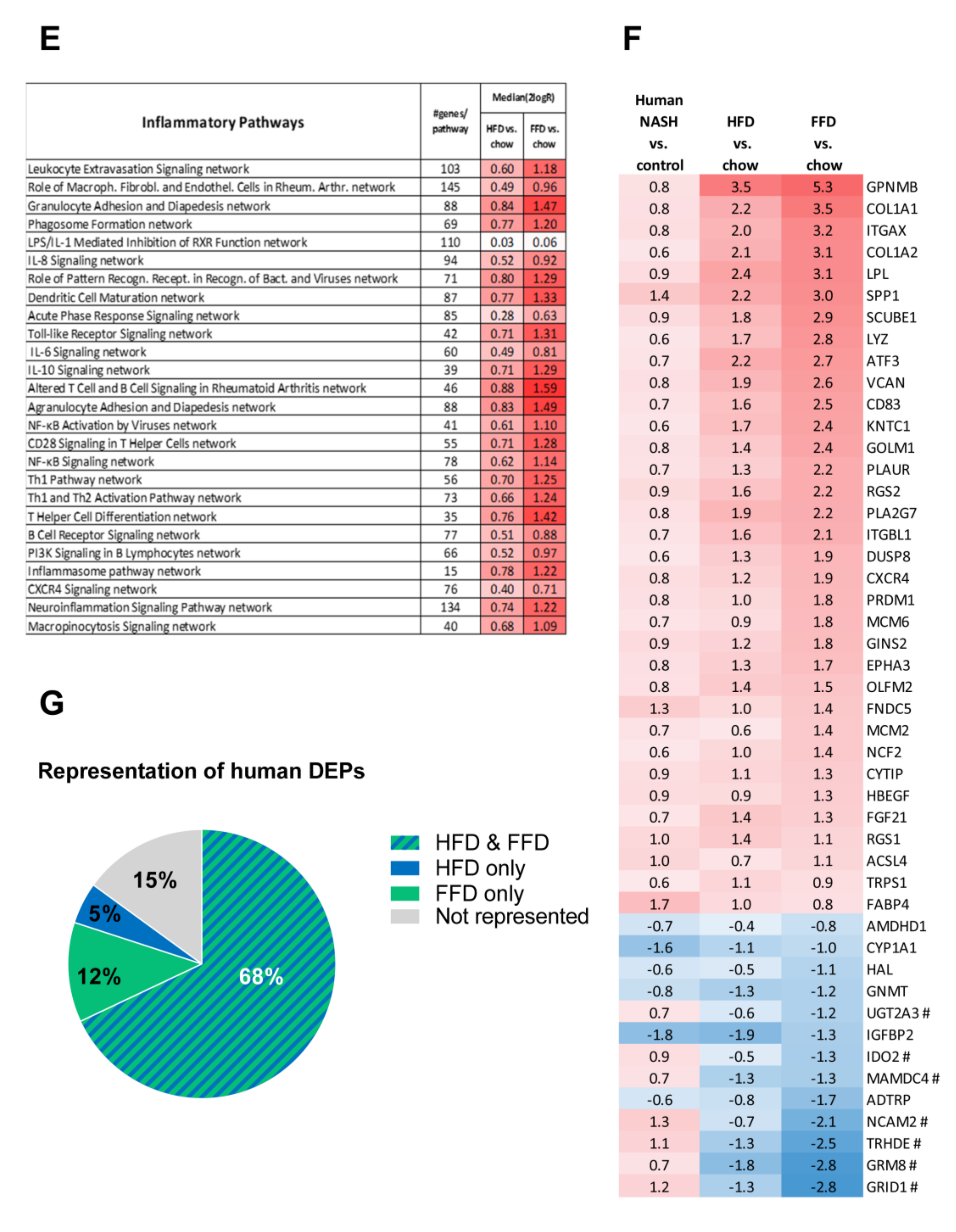
3.4. Ldlr−/−.Leiden Mice on HFD and FFD Recapitulate Human NASH and Fibrosis Pathways
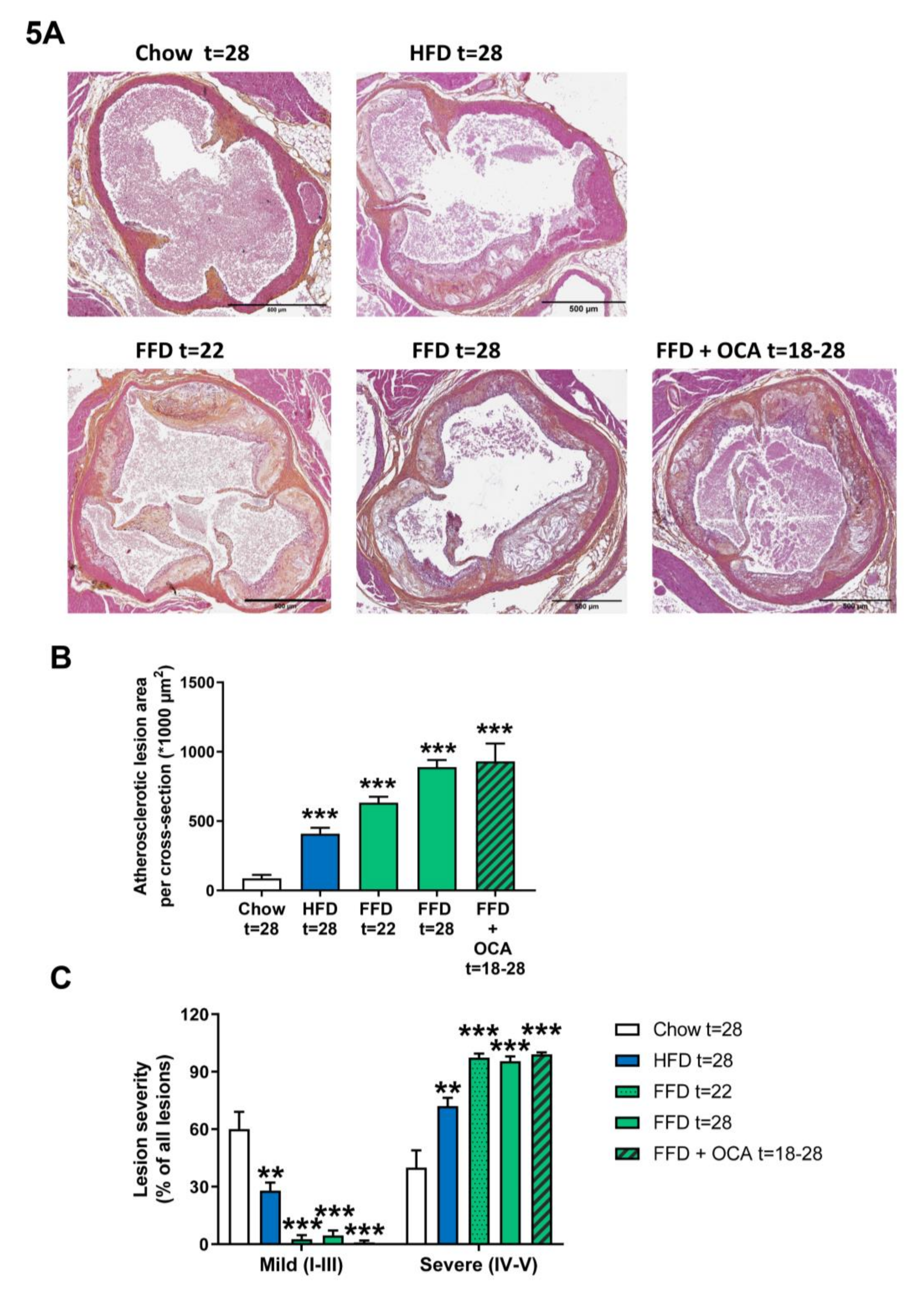
3.5. Ldlr−/−.Leiden Mice Develop Atherosclerosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but no Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.; Warda, A.S.; Verbeek, J.; Cassiman, D.; Spincemaille, P. An Overview of Mouse Models of Nonalcoholic Steatohepatitis: From Past to Present. Curr. Protoc. Mouse Biol. 2016, 6, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wu, N.; Chen, X.; Wang, W.; Chu, Y.; Liu, H.; Li, W.; Chen, D.; Li, X.; Xu, B. Pathogenesis of and major animal models used for nonalcoholic fatty liver disease. J. Int. Med. Res. 2019, 47, 1453–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef]
- Morrison, M.C.; Kleemann, R.; van Koppen, A.; Hanemaaijer, R.; Verschuren, L. Key Inflammatory Processes in Human NASH Are Reflected in Ldlr(-/-).Leiden Mice: A Translational Gene Profiling Study. Front. Physiol. 2018, 9, 132. [Google Scholar] [CrossRef]
- Oseini, A.M.; Cole, B.K.; Issa, D.; Feaver, R.E.; Sanyal, A.J. Translating scientific discovery: The need for preclinical models of nonalcoholic steatohepatitis. Hepatol. Int. 2018, 12, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR-/-. Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gole, L.; Yu, W.M.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-. Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelgrim, C.E.; Franx, B.A.A.; Snabel, J.; Kleemann, R.; Arnoldussen, I.A.C.; Kiliaan, A.J. Butyrate Reduces HFD-Induced Adipocyte Hypertrophy and Metabolic Risk Factors in Obese LDLr-/-. Leiden Mice. Nutrients 2017, 9, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoemaker, M.H.; Kleemann, R.; Morrison, M.C.; Verheij, J.; Salic, K.; van Tol, E.A.F.; Kooistra, T.; Wielinga, P.Y. A casein hydrolysate based formulation attenuates obesity and associated non-alcoholic fatty liver disease and atherosclerosis in LDLr-/-. Leiden mice. PLoS ONE 2017, 12, e0180648. [Google Scholar] [CrossRef] [PubMed]
- Gart, E.; Souto Lima, E.; Schuren, F.; de Ruiter, C.G.F.; Attema, J.; Verschuren, L.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Diet-Independent Correlations between Bacteria and Dysfunction of Gut, Adipose Tissue, and Liver: A Comprehensive Microbiota Analysis in Feces and Mucosa of the Ileum and Colon in Obese Mice with NAFLD. Int. J. Mol. Sci. 2018, 20, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque-Sierra, A.; Alvarez-Amor, L.; Kleemann, R.; Martín, F.; Varela, L.M. Extra-Virgin Olive Oil with Natural Phenolic Content Exerts an Anti-Inflammatory Effect in Adipose Tissue and Attenuates the Severity of Atherosclerotic Lesions in Ldlr-/-. Leiden Mice. Mol. Nutr. Food Res. 2018, 62, e1800295. [Google Scholar] [CrossRef] [PubMed]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/-. Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef]
- Jacobs, S.A.H.; Gart, E.; Vreeken, D.; Franx, B.A.A.; Wekking, L.; Verweij, V.G.M.; Worms, N.; Schoemaker, M.H.; Gross, G.; Morrison, M.C.; et al. Sex-Specific Differences in Fat Storage, Development of Non-Alcoholic Fatty Liver Disease and Brain Structure in Juvenile HFD-Induced Obese Ldlr-/-. Leiden Mice. Nutrients 2019, 11, 1861. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferell, L.D.; Liu, Y.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [Green Version]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [Green Version]
- Salic, K.; Gart, E.; Seidel, F.; Verschuren, L.; Caspers, M.; van Duyvenvoorde, W.; Wong, K.E.; Keijer, J.; Bobeldijk-Pastorova, I.; Wielinga, P.Y.; et al. Combined treatment with L-carnitine and nicotinamide riboside improves hepatic metabolism and attenuates obesity and liver steatosis. Int. J. Mol. Sci. 2019, 20, 4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsing, D.J.; Offerman, E.H.; van Duyvenvoorde, W.; van der Boom, H.; de Wit, E.C.; Gijbels, M.J.; van der Laarse, A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M. l-CoA:cholesterol acyltransferase inhibitor avasimibe reduces atherosclerosis in addition to its cholesterol-lowering effect in ApoE*3-Leiden mice. Circulation 2001, 103, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, S.; van der Tuin, S.J.; van der Hoorn, J.W.; van Klinken, J.B.; Simic, B.; Pieterman, E.; Havekes, L.M.; Landmesser, U.; Lüscher, T.F.; Willems van Dijk, K.; et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J. 2015, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, L.; Wielinga, P.Y.; Kelder, T.; Radonjic, M.; Salic, K.; Kleemann, R.; van Ommen, B.; Kooistra, T. A systems biology approach to understand the pathophysiological mechanisms of cardiac pathological hypertrophy associated with rosiglitazone. BMC Med. Genom. 2014, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef]
- Palmer, C.; Diehn, M.; Alizadeh, A.A.; Brown, P.O. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom. 2006, 7, 115. [Google Scholar] [CrossRef]
- Hu, F.B.; Manson, J.E.; Willett, W.C. Types of dietary fat and risk of coronary heart disease: A critical review. J. Am. Coll. Nutr. 2001, 20, 5–19. [Google Scholar] [CrossRef]
- Mulder, P.; Morrison, M.C.; Wielinga, P.Y.; van Duyvenvoorde, W.; Kooistra, T.; Kleemann, R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non-alcoholic steatohepatitis in obesity. Int. J. Obes. 2016, 40, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, P.; van den Hoek, A.M.; Kleemann, R. The CCR2 Inhibitor Propagermanium Attenuates Diet-Induced Insulin Resistance, Adipose Tissue Inflammation and Non-Alcoholic Steatohepatitis. PLoS ONE 2017, 12, e0169740. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.; Liang, W.; Wielinga, P.Y.; Verschuren, L.; Toet, K.; Havekes, L.M.; van den Hoek, A.M.; Kleemann, R. Macrovesicular steatosis is associated with development of lobular inflammation and fibrosis in diet-induced non-alcoholic steatohepatitis (NASH). Inflamm. Cell Signal. 2015, 2, e804. [Google Scholar]
- Krishnan, A.; Abdullah, T.S.; Mounajjed, T.; Hartono, S.; McConico, A.; White, T.; LeBrasseur, N.; Lanza, I.; Nair, S.; Gores, G.; et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G666–G680. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Poekes, L.; Wang, H.; Mridha, A.R.; Barn, V.; Haigh, W.G.; Ioannou, G.N.; Yeh, M.M.; Leclercq, I.A.; Teoh, N.; et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity 2017, 25, 155–165. [Google Scholar] [CrossRef]
- Farrell, G.C.; Mridha, A.R.; Yeh, M.M.; Arsov, T.; van Rooyen, D.M.; Brooling, J.; Nguyen, T.; Heydet, D.; Delghingaro-Augusto, V.; Nolan, C.J.; et al. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int. 2014, 34, 1084–1093. [Google Scholar] [CrossRef]
- Tolbol, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Daniels, S.J.; Leeming, D.J.; Detlefsen, S.; Bruun, M.F.; Hjuler, S.T.; Henriksen, K.; Hein, P.; Karsdal, M.A.; Brockbank, S.; Cruwys, S. Biochemical and histological characterisation of an experimental rodent model of non-alcoholic steatohepatitis—Effects of a peroxisome proliferator-activated receptor gamma (PPAR-gamma) agonist and a glucagon-like peptide-1 analogue. Biomed. Pharmacother. 2019, 111, 926–933. [Google Scholar] [CrossRef]
- Abe, N.; Kato, S.; Tsuchida, T.; Sugimoto, K.; Saito, R.; Verschuren, L.; Kleemann, R.; Oka, K. Longitudinal characterization of diet-induced genetic murine models of non-alcoholic steatohepatitis with metabolic, histological, and transcriptomic hallmarks of human patients. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Van Koppen, A.; Verschuren, L.; van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuru, H.; Costessi, A.; Caspers, M.M.; van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 83–98. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Invest. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Papazyan, R.; Liu, X.; Liu, J.; Dong, B.; Plummer, E.M.; Lewis, R.D.; Roth, J.D.; Young, M.A. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J. Lipid Res. 2018, 59, 982–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salic, K.; Morrison, M.; Brosnan, M.J.; Gart, E.; Bence, K.; Kleemann, R.; Tesz, G. Inhibition of DGAT2 improves hepatic steatosis, inflammation and cardiovascular risk factors in the LDLr-/-. Leiden mouse. In Proceedings of the EASL NAFLD Summit, Geneva, Switzerland, 20–22 September 2018. Abstract Number P06-09, 95. [Google Scholar]
- Arnoldussen, I.A.C.; Wiesmann, M.; Pelgrim, C.E.; Wielemaker, E.M.; van Duyvenvoorde, W.; Amaral-Santos, P.L.; Verschuren, L.; Keijser, B.J.F.; Heerschap, A.; Kleemann, R.; et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int. J. Obes. 2017, 41, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef]
- Krishnasamy, Y.; Gooz, M.; Li, L.; Lemasters, J.J.; Zhong, Z. Role of mitochondrial depolarization and disrupted mitochondrial homeostasis in non-alcoholic steatohepatitis and fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 190–204. [Google Scholar]
- Leveille, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chow | HFD | FFD | |
|---|---|---|---|
| Body weight (g) | 38.3 ± 1.5 | 52.3 ± 1.1 *** | 48.7 ± 1.5 *** |
| Food intake (kCal/mouse/day) | 13.5 ± 0.8 | 12.5 ± 0.6 | 14.9 ± 1.9 |
| Liver weight (g) | 1.8 ± 0.1 | 3.1 ± 0.2 *** | 5.9 ± 0.4 * |
| Liver weight (% of body weight) | 4.8 ± 0.2 | 3.5 ± 0.2 *** | 7.1 ± 0.3 *** |
| Blood glucose (mM) | 7.7 ± 0.4 | 7.7 ± 0.2 | 6.5 ± 0.3 ** |
| Plasma insulin (ng/mL) | 2.9 ± 0.6 | 14.7 ± 4.2 *** | 3.9 ± 0.4 |
| HOMA-IR | 1.0 ± 0.2 | 4.9 ± 1.4 ** | 1.1 ± 0.1 |
| Plasma cholesterol (mM) | 8.0 ± 0.7 | 32.2 ± 3.7 *** | 41.3 ± 4.7 *** |
| Plasma triglycerides (mM) | 1.5 ± 0.3 | 6.5 ± 1.3 ** | 8.3 ± 1.2 *** |
| Plasma ALT (U/L) | 53.3 ± 7.1 | 363.7 ± 64.8 ** | 348.6 ± 33.0 *** |
| Plasma AST (U/L) | 88.2 ± 8.2 | 449.1 ± 69.1 *** | 342.9 ± 58.4 *** |
| Plasma SAA (µg/mL) | 39.6 ± 4.2 | 244.6 ± 54.6 *** | 318.6 ± 73.3 *** |
| Plasma E-selectin (ng/mL) | 61.3 ± 3.7 | 83.0 ± 10.1 * | 83.7 ± 3.4 *** |
| Plasma MCP-1 (pg/mL) | 33.2 ± 6.5 | 75.4 ± 11.2 ** | 70.7 ± 6.3 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Hoek, A.M.; Verschuren, L.; Worms, N.; van Nieuwkoop, A.; de Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. https://doi.org/10.3390/cells9092014
van den Hoek AM, Verschuren L, Worms N, van Nieuwkoop A, de Ruiter C, Attema J, Menke AL, Caspers MPM, Radhakrishnan S, Salic K, et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells. 2020; 9(9):2014. https://doi.org/10.3390/cells9092014
Chicago/Turabian Stylevan den Hoek, Anita M., Lars Verschuren, Nicole Worms, Anita van Nieuwkoop, Christa de Ruiter, Joline Attema, Aswin L. Menke, Martien P. M. Caspers, Sridhar Radhakrishnan, Kanita Salic, and et al. 2020. "A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology" Cells 9, no. 9: 2014. https://doi.org/10.3390/cells9092014