
Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance

Centro Andaluz de Biología Molecular y Medicina Regenerativa (CABIMER), Consejo Superior de Investigaciones Científicas, Universidad de Sevilla, Universidad Pablo de Olavide, 41092 Seville, Spain
Genes 2021, 12(10), 1550; https://doi.org/10.3390/genes12101550
Submission received: 7 September 2021
/
Revised: 20 September 2021
/
Accepted: 23 September 2021
/
Published: 29 September 2021
(This article belongs to the Special Issue The Key of DNA Recombination and Replication—Recombination Mediator Proteins)
{kind=link}
{kind=link}
Abstract
:The DNA damage tolerance (DDT) response is aimed to timely and safely complete DNA replication by facilitating the advance of replication forks through blocking lesions. This process is associated with an accumulation of single-strand DNA (ssDNA), both at the fork and behind the fork. Lesion bypass and ssDNA filling can be performed by translation synthesis (TLS) and template switching mechanisms. TLS uses low-fidelity polymerases to incorporate a dNTP opposite the blocking lesion, whereas template switching uses a Rad51/ssDNA nucleofilament and the sister chromatid to bypass the lesion. Rad51 is loaded at this nucleofilament by two mediator proteins, BRCA2 and Rad52, and these three factors are critical for homologous recombination (HR). Here, we review recent advances showing that Rad51, BRCA2, and Rad52 perform some of these functions through mechanisms that do not require the strand exchange activity of Rad51: the formation and protection of reversed fork structures aimed to bypass blocking lesions, and the promotion of TLS. These findings point to the central HR proteins as potential molecular switches in the choice of the mechanism of DDT.
1. Introduction
Homologous recombination (HR) mechanisms repair DNA breaks using intact homologous DNA sequences as template (donor molecule). If the template contains some heterology, the recombination process might be associated with a transfer of genetic information, otherwise parental and recombinant products will be identical (e.g., sister chromatin recombination). The search for homology and strand exchange are central molecular steps for HR mechanisms associated with both double-strand break (DSB) repair and single-strand DNA (ssDNA) filling. These steps are carried out by a ssDNA molecule coated with the Rad51 protein (RecA in bacteria), which is the central protein in HR. Even though a number of additional factors participate in the dynamics of Rad51 on this nucleofilament, two are critical for Rad51 loading: BRCA2 in mammalian cells and Rad52 in yeast. The organisms where HR mechanisms have been studied in more details [1]. The analyses of stalled replication forks by different stress conditions revealed important non-recombinogenic functions (defined as those that do not require the strand exchange activity of Rad51) of these factors in the dynamics of reversed fork structures [2,3]. The number of non-recombinogenic functions of Rad51 and Rad52 have been recently extended to translesion synthesis (TLS) [4,5,6], an error-prone mechanism of DNA damage tolerance (DDT) in which specialized polymerases incorporate a dNTP opposite a lesion that blocks DNA replication [7].
2. DSB Repair by HR
Most of our knowledge about the mechanisms and factors involved in HR comes from the study of DSB repair both in mitosis and meiosis, where the recombinational repair of programmed DSBs is critical for the formation of viable meiotic products. Indeed, for a long time, the fact that DSBs induce HR led to the assumption that hyper-recombination in mutant backgrounds, or in response to genotoxic agents, was always mediated by a DSB. Genetic and molecular studies on DSB repair, together with the biochemical characterization of the factors required for HR, allowed to outline the main HR mechanisms [1,8]. They all start with the resection by specialized DNA nucleases of the 5′-ends of the DSB to generate 3′-ended ssDNA molecules (Figure 1). These nucleases include the MRX/N complex—formed by Mre11, Rad50 and Xrs2 (yeast), or Nbs1 (mammal), Sae2 (yeast)/CtIP (mammal), Exo1, and Dna2, which works together with the helicase Sgs1 (yeast)/BLM (mammal) [9]. The 3′-ended ssDNA molecule is covered and protected by the ssDNA binding heterotrimeric complex RPA. BRCA2 and Rad52 promote the loading of Rad51, thus displacing RPA and generating a ssDNA/Rad51 nucleofilament. BRCA2 and Rad52 are helped in this task by accessory factors: Rad55, Rad57, and the Shu complex in yeast; RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3 in mammals; and Rad54 in yeast and mammals.
Rad51 forms onto the ssDNA molecule a right-handed helical nucleoprotein filament where each Rad51 monomer binds three nucleotides. Rad51 polymerization is mediated by monomer–monomer interactions [10]. Importantly, Rad51 can also bind double-strand DNA (dsDNA) with an affinity similar to that displayed for ssDNA [11]; this binding impedes homologous pairing, and thereby HR [12]. The ssDNA/Rad51 nucleofilament (presynaptic filament) searches for a homologous template by sampling DNA in 8-nucleotides (nt) increments and capturing those tracts with 8-nt microhomology [13]. Once the ssDNA/Rad51 nucleofilament is paired with a homologous DNA sequence (synaptic complex), Rad51 promotes the strand exchange reaction in precise 3-nt steps [13,14]. The interaction of the ssDNA/Rad51 nucleofilament with the dsDNA template requires a low-affinity DNA binding site in Rad51 that is different from the high-affinity binding site that promotes the polymerization of Rad51 on ssDNA [15,16]. The strand exchange reaction generates a DNA joint molecule where Rad51 binds to the heteroduplex formed by the invading strand and its complementary strand in the donor molecule (postsynaptic complex). As a consequence of this exchange, a displacement loop (D-loop) is formed in which the invading 3′-end primes new DNA synthesis once Rad51 is disassembled by the translocase Rad54 [17]. Once this intermediate is formed, HR can proceed through different mechanisms (double-strand break repair, DSBR; synthesis-dependent strand annealing, SDSA; break-induced replication BIR) that may or may not involve gene conversion and/or crossover depending on whether there is transfer of information or reciprocal exchange of DNA between the recombining molecules, respectively (Figure 1) (for details about these mechanisms see [1,8]).
Despite its essential role in the search for homology and strand exchange, Rad51 is dispensable for some recombination events in yeast, and only a double mutant lacking Rad51 and Rad59 displays recombination defects as severe as a mutant lacking Rad52 [18]. This is due to the fact that Rad51 is not essential for BIR [19] and is not required for single-strand annealing (SSA) [20], a recombination process between direct repeats where the resected homologous sequences are directly annealed. Both Rad52 and Rad59 display strand annealing activity [21,22], which can promote strand invasion of a broken DNA end in BIR—though with low efficiency—and anneal the exposed homologous sequences in SSA [19,20]. In contrast to yeast, the lack of Rad52 in mammalian cells causes mild recombination defects. Instead, the breast and ovarian tumor suppressor protein BRCA2 maintains a central role in HR in mammals [23]. The relevance of Rad52 becomes evident in BRCA2-deficient cells [24]. The reason for these differences stems from the fact that yeast Rad52 contains both mediator (Rad51 loading onto DNA) and DNA annealing activities, which are separated in BRCA2 (mediator activity) and Rad52 (annealing activity) in mammals [25]. Yeast Rad52 mediator function is performed through physical interactions with ssDNA, Rad51, and RPA [26]. In mammalian cells, the loading of Rad51 onto the ssDNA molecule requires BRCA1, which interacts with PALB2 to recruit BRCA2. The physical interactions of BRC repeats in the middle of BRCA2 with Rad51 promoting the formation of the nucleofilament and prevent Rad51 binding to dsDNA [27,28,29]. In addition, BRCA2 stabilizes the nucleofilament through a second Rad51 interaction site in the carboxy-terminal domain (CTD) that is negatively regulated by CDK1/2-mediated phosphorylation of serine 3291 [30].
3. ssDNA Gap Filling by HR
A major source of DSBs occurs during S phase and is associated with the breakage of replication forks. These structures are especially vulnerable under stress conditions due to the dynamics of chromatin associated with DNA synthesis and the accumulation of ssDNA stretches and free ends [31]. Even though the mechanisms by which HR rescues broken forks remain poorly explored, BIR seems to play a major role by priming DNA synthesis via a bubble-like fork that results in Pol δ-mediated conservative replication [32,33]. Accordingly, situations that cause fork instability, such as mutations in replicative polymerases or histone depletion during S phase, activate Rad51-independent HR mechanisms [34,35].
The historical view of HR as a specialized DSB repair mechanism has been challenged by a large number of genetic and molecular evidence supporting an essential function of HR during S phase that is linked to the filling of ssDNA gaps. Genotoxic agents that stall replication forks either by generating DNA-blocking lesions (e.g., methyl methane sulfonate (MMS), UV light, 4-nitroquinoline oxide (NQO), H2O2) or reducing the pool of available dNTPs (e.g., hydroxyurea; HU, aphidicolin) uncouple the DNA unwinding and DNA synthesis activities of the replisome. This causes an accumulation of ssDNA at the fork that triggers different responses to stabilize, protect, and restart replication forks [36,37,38]. In the case of blocking lesions, the accumulation of ssDNA at the fork occurs preferentially at the leading strand, as the lagging strand can bypass the lesion by priming a new Okazaki fragment, leaving a gap behind the fork [39,40,41]. One specific consequence of the accumulation of ssDNA is the activation of the DDT response, aimed to timely complete DNA replication by promoting the bypass of the lesions and the filling of the stretches of ssDNA. This response is mediated by the ubiquitylation of the replication processivity factor PCNA (proliferating cell nuclear antigen) [42,43,44]. PCNA monoubiquitylation at lysine 164 by the heterodimer Rad6(E2)-Rad18(E3) promotes the recruitment of TLS polymerases, which provide a simple but mutagenic way to bypass the lesion and fill in the ssDNA gap at the fork (Figure 2A). Alternatively, extension of the K164 ubiquitylation with a K63-linked polyubiquitin chain by Mms2-Ubc13(E2)-Rad5 (yeast)/HLTF and SHPRH (mammal) (E3) promotes different template switching events mediated by HR proteins [7,45]. PCNA polyubiquitylation can be associated with the formation of reversed forks through a process in which the nascent strands are displaced and reannealed leading to a Holliday junction (HJ)-like structure (Figure 2B) [46]. These structures might facilitate the bypass of the blocking lesion without generating ssDNA behind the fork either by strand exchange ahead of the fork or DNA synthesis and fork restoration. Consistent with the former mechanism, yeast forks blocked by a replication fork barrier reinitiate DNA synthesis through Pol δ-mediated semiconservative replication [47]. This recombinational event might also occur without fork reversal by directly invading the intact, sister chromatid at the fork (Figure 2C), even though there are no physical evidence of this intermediate yet.
In some other cases, replication restart involves the formation of post-replicative ssDNA gaps in the daughter strand as a consequence of repriming DNA synthesis downstream of the blocking lesion (Figure 2D) [40,41]. Repriming in mammals is carried out by PrimPol, a DNA polymerase with primase activity required for progression in the presence of DNA blocking lesions [48,49]. Even though yeast cells lack a PrimPol homolog, the Pol α/primase complex seems to perform the repriming, as suggested by genetic evidence [50]. Thus, ssDNA gaps accumulate both at the leading and the lagging strand [40,41], and can be filled by TLS polymerases or processed by nucleases and helicases to generate the substrate for the HR machinery (Figure 2D) [41,51,52]. This template switching event uses most of HR central factors and generates sister chromatid junctions (SCJs) [53,54,55,56,57]. However, and in contrast to DSB-mediated HR, strand exchange during ssDNA gap filling is not initiated by the 3′-end but by the ssDNA gap through the reannealing of the parental strands, which exposes the intact newly synthesized chromatid as a template for the blocked nascent strand [58].
Both SCJs and reversed forks have been detected by electron microscopy and/or two-dimensional electrophoresis [56,58,59,60], but their abundance and relevance seem to depend on the organism and type of lesion. In mammalian cells, SCJs are hardly detected [61], whereas reversed forks are abundant structures preferentially confined to stalled forks [59,62]. In contrast, SCJs are detected in response to MMS-induced blocking lesions in yeast cells [56], whereas MMS- and UV-induced reversed forks are rare structures except in checkpoint or primase/Ctf4 mutants defective in fork stability and repriming, respectively [50,60], or in response to the topoisomerase I inhibitor camptothecin [63]. Thus, the scarcity of reversed forks in UV and MMS-treated yeast cells might reflect transient structures or a specific response to hard-to-bypass lesions. These results suggest that template switching events are predominant at the fork in mammals and behind the fork in yeast. At least for the later, this expectation is consistent with the formation of MMS- and UV-induced HR foci far away from sites of ongoing replication [64]. It must be stressed, though, that yeast Rad51 is also required at the fork as it facilitates replication fork advance in the presence of MMS-induced lesions [65,66,67] and protects nascent DNA at forks even under unperturbed conditions [41].
4. Non-Recombinogenic Roles of Rad51, BRCA2 and Rad52 in the Dynamics of Reversed Forks
The study of the cell response to different replication stress conditions established critical roles for Rad51 and its mediators, BRCA2 and Rad52, in the stability, protection, and restart of stalled forks. Unexpectedly, though, a number of studies along the last decade have revealed that reversed fork dynamics requires non-recombinogenic activities of these factors. The first evidence came from the study in mouse cells of stalled forks by HU, which showed that BRCA2 prevents the degradation of nascent strands by the nuclease Mre11 (Figure 2B) [68]. The analysis of brca2 mutants demonstrated that this protective role is not mediated by the binding of BRCA2 to DNA; however, it requires its Rad51 interaction site at the CTD (eliminated in a brca2-S3291A mutant), suggesting that BRCA2 protects stalled forks by stabilization of the Rad51 nucleofilament. Accordingly, over-expression of a Rad51-K133R mutant protein defective in Rad51 dissociation from DNA rendered brca2 mutant cells resistant to stalled fork degradation. Most importantly, the brca2-S3291A mutant was proficient in HR, indicating that stable Rad51 filaments protect stressed forks from Mre11 degradation through a non-recombinogenic function [68]. This line of research uncovered additional factors, such as BRCA1, FANCD2, RAD51C, and XRCC3, in protecting stressed forks by stabilizing Rad51 nucleofilaments [69,70]. Additional evidence for this non-recombinogenic role of Rad51 in protecting stalled forks came from the analysis of a rad51-T131P mutation in a patient with Fanconi anemia. The mutant protein destabilizes Rad51 nucleofilaments by constitutive activation of the Rad51 ATPase activity and renders cells sensitive to DNA interstrand crosslinks (ICLs) due to nucleolytic processing by the nuclease Dna2 and the helicase WRN; however, mutant cells remain HR proficient [71]. Importantly, a Rad51-II3A mutant protein proficient in DNA binding and nucleofilament formation but defective in strand exchange and HR protected forks arrested by a replication fork barrier in yeast or the replication inhibitor HU in human cells from nuclease degradation by Exo1 (yeast) and Mre11 (mammalian cells), further supporting a non-recombinogenic role for Rad51 in the protection of stressed forks [16,72].
Consistent with the separation of the mediator and annealing activities between BRCA2 and Rad52 in mammalian cells, yeast but not human Rad52 is required for stalled fork protection [72,73]. Indeed, mammalian Rad52, together with the PTIP complex, facilitate Mre11 targeting to stalled forks through unknown mechanisms [73]. Interestingly, the absence of either PTIP, Rad52, or Mre11 not only prevents nascent DNA degradation under stress conditions but also rescues lethality in mouse stem cells and human tumor cells defective in BRCA2, suggesting that the essential role of BRCA2 is associated with fork protection and not with HR [73,74]. However, this essential role was not observed in non-transformed human mammary epithelial cells, indicating that the contribution of HR and fork protection by Rad51 and BRCA2 is modulated by the cell context [75].
The analysis of replication intermediates by EM revealed low levels of reversed forks in brca2 cells that were restored after eliminating PTIP, Rad52, or Mre11, indicating that the reversed fork is the entry point for the nuclease in brca2 cells [73]; indeed, genetic conditions that prevent reversed fork formation avoid fork degradation, even though lead to elevated levels of chromosomal breakage and genetic instability [72,73]. This is, in part, due to the fact that processing reversed forks by Mre11 is necessary for their recombinational restart [76]. In this scenario, BRCA2-mediated stable Rad51 nucleofilaments would display a critical role under replication stress conditions by protecting reversed forks from excessive degradation [73,74]. This protective role, together with its DNA repair functions, through HR is critical to prevent spontaneous replication-associated DNA damage—including under-replication—that lead to mitotic abnormalities, chromosome segregation defects, and G1 arrest [75,77,78].
Studies in human cells have shown that Rad51 is not only required for the protection but also for the formation of reversed forks [59], a function that is facilitated by the RAD51B/RAD51C/RAD51D/XRCC2 subcomplex of Rad51 paralogs [79]. As previously mentioned, reversed forks form both in BRCA2-deficient and Rad51-T131P-expressing cells [73], indicating that metastable Rad51 nucleofilaments would be sufficient to promote fork reversal. However, purified human Rad51 does not have fork remodelling activity in vitro [80], suggesting that fork reversal is not mediated by the strand exchange activity of Rad51. In accordance, reversed forks form in yeast and human cells expressing the strand exchange deficient rad51-II3A allele [16,72]. Recent analyses in human cells has connected DDT activation with reversed fork formation through the recruitment of the translocase ZRANB3 to stalled forks by physical interactions with polyubiquitylated PCNA [61,81]. In agreement with these results, fork reversal is impeded in the absence of HLTF, leading to PrimPol and Rev1-dependent discontinuous and mutagenic replication [82]. Nevertheless, it must be stressed that the role of HLTF in fork reversal might also be associated with its DNA translocase activity [83]. Actually, a number of different DNA translocases have been shown to promote fork reversal in vitro and in vivo [3], some of which (e.g., SMARCAL1) are directed by RPA to selectively reverse forks stalled at the leading strand and restore normal forks with a lagging-strand gap [84]. In this frame, Rad51 might stimulate the fork reversal activity of these translocases (e.g., Rad54) [80]. Alternatively, it might bind to the ssDNA end of the reversed fork to stabilize the structure [2].
An immediate consequence of these studies is that the function of the Rad51 nucleofilament is associated with its stability, and that both a defect and an excess of Rad51 can lead to genomic instability. It is thereby not surprising that the role of Rad51 at stalled forks is modulated by a dynamic interplay with RPA, which in turn can regulate the activity of additional factors, such as the human translocase SMARCAL1 [2]. Mammalian cells maintain a physiological level of Rad51 by the ssDNA binding protein RADX, which counteracts BRCA2 activity by interacting with and stimulating Rad51 ATPase hydrolysis to destabilize the nucleofilament [85,86,87,88]. Strikingly, RADX-dependent Rad51 nucleofilament destabilization can either inhibit or promote fork reversal depending on whether replication forks are unperturbed or stalled, respectively. The mechanistic of this regulation might be related to the amount of ssDNA at forks; in the short tracts that accumulate at unperturbed forks, Rad51 destabilization would prevent fork reversal. In contrast, Rad51 destabilization in the long ssDNA tracts that accumulate at stalled forks would lead to metastable and dynamic filaments that would facilitate its interplay with RPA without generating roadblocks that would impair strand reannealing during fork reversal [62].
Although the strand invasion activity of Rad51 is required for efficient fork restart as determined in yeast and human cells expressing the rad51-II3A allele, this requirement was greater upon persistence stalling in human cells [16,72]. This has led to propose a non-recombinogenic role for Rad51 at early times of fork stalling that would favour fork restoration, whereas at late times reversed fork processing would favour fork restart by HR (Figure 2B) [16,72]. Fork restart requires the RAD51C/XRCC3 subcomplex of Rad51 paralogs but, in contrast to DSB-mediated HR, is independent of BRCA2 [74,79]. Therefore, the stability and extent of the Rad51/ssDNA nucleofilament seem to be critical for their different recombinogenic and non-recombinogenic functions.
Finally, HR plays an additional role during mitosis by promoting mitotic DNA synthesis (MiDAS) of under-replicated regions through a BIR-like mechanism that deal with DNA ends generated by nuclease digestion of stalled forks [89,90]. Interestingly, Rad51 also facilitates MiDAS at DNA regions that are not associated with DNA repair foci or γ-H2A, two DSB markers. This process is enhanced in human cells expressing Rad51-K133 and is regulated by Polo-like kinase 1, which induces Rad51 recruitment to ssDNA upon phosphorylation. These results suggest a non-recombinogenic role for Rad51 in MiDAS by protecting stalled forks against nucleases [91].
5. Non-Recombinogenic Roles of Rad51 and Rad52 in TLS
In contrast to HR, which is mostly error free, TLS mechanisms can be mutagenic as most TLS polymerases display low fidelity. In Saccharomyces cerevisiae, there are three TLS polymerases that operate with different affinities depending on the dose and type of blocking lesion: Rev1, Pol ζ (formed by the catalytic subunit Rev3 and the regulatory subunits Rev7, Pol31, and Pol32), and Pol η (encoded by RAD30) [7]. Their activity—at least for Rev1/Pol ζ—is most dominant in G2/M [92], where they compete with a UbPCNA-independent salvage pathway of HR (inhibited during S phase) to deal with DNA gaps left unrepaired by the main, UbPCNA-dependent mechanism of HR [45]. This salvage pathway, in contrast to the latter, can lead to chromosomal rearrangements [93]. Therefore, the choice of the DDT mechanism is critical for the maintenance of genome integrity.
The analysis of DNA damage-induced repair foci in yeast revealed the requirement of the TLS machinery (Rad6/Rad18-mediated PCNA ubiquitylation and polymerases Rev1/Pol ζ) for the resolution of Rad52 but not Rad54-associated foci, indicating that this phenotype was not a consequence of HR being the only operative ssDNA filling process. Likewise, persistence of MMS-induced Rad52 foci in TLS mutants was not due to a demand for TLS polymerases during MMS or UV-induced HR, but a consequence of yeast Rad52 acting in concert with the TLS machinery to fill in the stretches of ssDNA generated during DDT in response to these genotoxic agents. In accordance, yeast Rad52 is partially required for MMS- and UV-induced mutagenesis [5]. This result was unexpected as inferred by epistatic analyses of DNA damage sensitivity and mutagenesis between HR and TLS mutants [94,95,96]. This synergism is explained by the fact that DNA damage-induced TLS is still partially operative in the rad52∆ mutant [5]. Yeast Rad52 is shown to facilitate Rad6/Rad18 chromatin binding and, in response to DNA damage, PCNA ubiquitylation [5]. Importantly, this function was not observed in the absence of Rad54, which is essential for HR during DDT [5,54,97,98]. This means that Rad52 facilitates Rad6/Rad18 recruitment to chromatin and subsequent PCNA ubiquitylation through non-recombinogenic activities. However, Rad51 and Rad57 cooperate with Rad52 in this mechanism but are dispensable for TLS-mediated ssDNA filling and mutagenesis, suggesting that Rad6/Rad18 recruitment to chromatin alone cannot explain the TLS defects observed in the rad52∆ mutant [5].
Rad6/Rad18 is targeted to chromatin by Rad18 binding to ssDNA, RPA (yeast and mammal), and sumoylated PCNA (only yeast) [44,99,100,101,102]. The involvement of Rad52 and Rad57 in the recruitment of Rad6/Rad18 suggests that this function is mediated by the Rad51 nucleofilament. Therefore, RPA and Rad51 are likely cooperating to target Rad6/Rad18 and promote PCNA ubiquitylation in response to DNA damage (Figure 2D; note that this event might also occur at the fork). The competition between RPA and Rad51 for ssDNA binding would explain the mild defects in TLS observed in the HR mutants [5]. Remarkably, the Rad6/Rad18 recruitment to chromatin is observed even in the absence of DNA damage [5]. Therefore, these interactions might occur at not-yet defined regions and, in response to DNA damage, mobilize Rad6/Rad18 to ssDNA gaps to ubiquitylate PCNA. This ubiquitylation would facilitate template switching events in S phase and TLS in G2/M. Some aspects of this mechanism seem to be conserved in human cells, where Rad51 physically interact with Rad18 and FANCD2 in a complex that is stimulated specifically by HU. In response to this agent, these factors promote PCNA monoubiquitylation and chromatin recruitment of the TLS polymerase Pol H; importantly, these events are not affected by the absence of BRCA2 or the pharmacological inhibition of Rad51, indicating that they are independent of HR [4].
Another piece of evidence supporting the non-recombinogenic role of Rad51 and Rad52 in the DDT response comes from the analysis in S. cerevisiae of the physical interactions between these proteins and the MCM complex, an essential component of the CMG (Cdc45/MCM/GINS) replicative helicase [6]. A rad51 mutation that disrupts the interaction between Rad51 and MCM was proficient in MMS and DSB-induced HR but partially defective in MMS-induced ssDNA gap filling, supporting a role for this interaction in TLS. Actually, this mutant was also partially defective in replication fork progression through damaged DNA and this defect could be bypassed by forcing the interaction through the simultaneous expression of Mcm4-GFP and Rad51-GBP (GFP-binding protein) chimeras. Thus, the MCM–Rad51 interaction facilitates DNA damage-induced ssDNA gap filling and fork progression through DNA blocking lesions by non-recombinogenic functions, presumably TLS (Figure 2A,D) [6].
How does the MCM–Rad51 interaction promote replicative and repair functions during yeast DDT? The biochemical characterization of these interactions revealed interesting aspects about its regulation. First, MCM also interacts with Rad52, but Rad52 does not bridge the MCM–Rad51 interaction. Second, these interactions occur in G1 and are lost in S phase, unless cells are released in the presence of replicative stress. Third, these interactions are prevented at origins and replication forks; instead, they occur at a cellular fraction that, in contrast to most chromatin, is insoluble after DNA digestion with nucleases. Rad51 accumulates in a MCM- and DNA binding-independent manner, whereas MCM seems to bind DNA. In this nucleoprotein scaffold, MCM and Rad51 (and also Rad52) display dynamic interactions that are regulated by the kinase activity of Cdc7, which prevents the release of Rad51 and Rad52 from the insoluble scaffold in response to replicative stress [6]. One possibility is that these physical interactions provide a platform to facilitate the chromatin recruitment of Rad6/Rad18 by Rad51 and Rad52. PCNA ubiquitylation by Rad6/Rad18 would promote the targeting of TLS polymerases to fill in ssDNA gaps [103] and would facilitate replication fork advance in the presence of damaged DNA [104], mechanistically connecting the two functions reported for the MCM–Rad51 interaction. In this regard, it is worth noting that Rad51-dependent PCNA monoubiquitylation in human cells is not affected in the absence of BRCA2, which prevents Rad51 binding to chromatin [4]. However, yeast Rad51 and Rad52 promote Rad6/Rad18 binding to DNA also under unperturbed replication conditions [5] that do not sustain the MCM–Rad51–Rad52 interactions [6], suggesting that they could also operate through alternative mechanisms.
Remarkably, this nuclease insoluble fraction also accumulates a number of factors involved in replication fork stability: the topoisomerase Top2, the checkpoint effector Rad53, the helicase Sgs1, the origin recognition complex (ORC) and the Cdc7 regulator Dbf4 [105,106]. We have hypothesized that these factors, together with MCM, Rad51, and Rad52, aggregate in G1 for replication assistance [6]. Under unperturbed replication conditions, Rad51 and Rad52 (and may be others) have to be removed to prevent toxic protein–DNA interactions [107,108]; however, in response to replicative DNA damage, Cdc7 would maintain these physical interactions to assist stressed forks by facilitating their advance and the repair of ssDNA gaps generated during lesion bypass [6]. This function might be conserved in mammalian cells, where the MCM–Rad51–Rad52 interactions have also been observed [109,110]. In this regard, it worth noting that mammalian replication origins and components of the pre-replication complex, including MCM, are associated during G1 and early S phase with a nuclease-insoluble scaffold, where DNA would be replicated at static replisomes [111,112].
6. Concluding Remarks
Thus far, the role of Rad51, BRCA2, and Rad52 in DDT was restricted to HR mechanisms. A new scenario has emerged in which they additionally operate in DDT through non-recombinogenic functions. A major conclusion of these results is that Rad51 and its mediators take part in most DDT mechanisms, and therefore they become a potential molecular switch to decide how to tolerate the lesion at different stages. At the fork, the Rad51 nucleofilament is critical for the formation, protection, and restart of reversed forks [2]. The involvement of Rad51 and Rad52 in TLS and fork advance through damaged DNA opens up the possibility that they could also facilitate fork restart by this mutagenic pathway [4,5,6]. Indeed, RecA can promote the switch from the replicative to a TLS polymerase in the bacterial replisome [113]. Interestingly, the analysis of stressed forks in Xenopus laevis showed that Rad51, apart from promoting fork reversion in a BRCA2-independent manner, interacts and stabilizes Pol α, and therefore might play a role in DNA synthesis resumption and/or repriming [114]. Behind the fork, yeast Rad51 and Rad52 may channel the filling of unrepaired ssDNA gaps in G2/M, either by TLS (via Rad6/Rad18 recruitment and not-yet defined Rad52 functions) or HR (via DNA strand exchange) [5]. Since the consequences for genome integrity are different depending on the mechanism of fork restart and ssDNA filling [115], it will be important to gain a deeper insight into these novel non-recombinogenic functions to understand how Rad51 and its mediators are regulated in response to the type and extent of DNA lesions and the cell cycle phase.
Funding
This work was funded by FEDER/Ministerio de Ciencia e Innovación—Agencia Estatal de Investigación (PGC2018-099182-B-100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I thank Pablo Huertas for critical reading of the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Jiang, G.; Cao, L.; Huang, J. Replication Fork Reversal and Protection. Front. Cell. Dev. Biol. 2021, 9, 670392. [Google Scholar] [CrossRef]
- Chen, X.; Bosques, L.; Sung, P.; Kupfer, G.M. A Novel Role for Non-Ubiquitinated FANCD2 in Response to Hydroxyurea-Induced DNA Damage. Oncogene 2016, 35, 22–34. [Google Scholar] [CrossRef]
- Cano-Linares, M.I.; Yáñez-Vilches, A.; García-Rodríguez, N.; Barrientos-Moreno, M.; González-Prieto, R.; San-Segundo, P.; Ulrich, H.D.; Prado, F. Non-recombinogenic Roles for Rad52 in Translesion Synthesis during DNA Damage Tolerance. EMBO Rep. 2021, 22, e50410. [Google Scholar] [CrossRef]
- Cabello-Lobato, M.J.; González-Garrido, C.; Cano-Linares, M.I.; Wong, R.P.; Yáñez-Vílchez, A.; Morillo-Huesca, M.; Roldán-Romero, J.M.; Vicioso, M.; González-Prieto, R.; Ulrich, H.D.; et al. Physical Interactions between MCM and Rad51 Facilitate Replication Fork Lesion Bypass and ssDNA Gap Filling by Non-Recombinogenic Functions. Cell Rep. 2021, 36, 109440. [Google Scholar] [CrossRef]
- Zhao, L.; Washington, M. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhao, L.; Xu, Y.; Zhao, W.; Sung, P.; Wang, H.-W. Cryo-EM Structures of Human RAD51 Recombinase Filaments during Catalysis of DNA-Strand Exchange. Nat. Struct. Mol. Biol. 2017, 24, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, F.E.; Stasiak, A.; West, S.C. Purification and Characterization of the Human Rad51 Protein, an Analogue of E. Coli RecA. EMBO J. 1994, 13, 5764–5771. [Google Scholar] [CrossRef]
- Sung, P.; Robberson, D.L. DNA Strand Exchange Mediated by a RAD51-SsDNA Nucleoprotein Filament with Polarity Opposite to That of RecA. Cell 1995, 82, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell 2015, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Terakawa, T.; Qi, Z.; Steinfeld, J.B.; Redding, S.; Kwon, Y.; Gaines, W.A.; Zhao, W.; Sung, P.; Greene, E.C. Base Triplet Stepping by the Rad51/RecA Family of Recombinases. Science 2015, 349, 977–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloud, V.; Chan, Y.-L.; Grubb, J.; Budke, B.; Bishop, D.K. Rad51 Is an Accessory Factor for Dmc1-Mediated Joint Molecule Formation during Meiosis. Science 2012, 337, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.M.; Chan, Y.-L.; Weichselbaum, R.W.; Bishop, D.K. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat. Commun. 2019, 10, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solinger, J.A.; Kiianitsa, K.; Heyer, W.-D. Rad54, a Swi2/Snf2-like Recombinational Repair Protein, Disassembles Rad51:DsDNA Filaments. Mol. Cell 2002, 10, 1175–1188. [Google Scholar] [CrossRef]
- Prado, F.; Cortés-Ledesma, F.; Huertas, P.; Aguilera, A. Mitotic Recombination in Saccharomyces Cerevisiae. Curr. Genet. 2003, 42, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-Strand Break Repair in the Absence of RAD51 in Yeast: A Possible Role for Break-Induced DNA Replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef] [Green Version]
- Prado, F.; Aguilera, A. Role of Reciprocal Exchange, One-Ended Invasion Crossover and Single-Strand Annealing on Inverted and Direct Repeat Recombination in Yeast: Different Requirements for the RAD1, RAD10, and RAD52 Genes. Genetics 1995, 139, 109–123. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA Strand Annealing Is Promoted by the Yeast Rad52 Protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [Green Version]
- Petukhova, G.; Stratton, S.A.; Sung, P. Single Strand DNA Binding and Annealing Activities in the Yeast Recombination Factor Rad59*. J. Biol. Chem. 1999, 274, 33839–33842. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2017, 2, 313–336. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 Inactivation Is Synthetically Lethal with BRCA2 Deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Heyer, W.D. Who’s Who in Human Recombination: BRCA2 and RAD52. Proc. Natl. Acad. Sci. USA 2011, 108, 441–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, U.H.; Lisby, M.; Rothstein, R. Rad52. Curr. Biol. 2009, 19, R676–R677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC Repeats of BRCA2 Modulate the DNA-Binding Selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA Recombination from the Structure of a RAD51–BRCA2 Complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The BRCA2 Homologue Brh2 Nucleates RAD51 Filament Formation at a DsDNA–SsDNA Junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 Nucleoprotein Filaments by the C-Terminal Region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef]
- Prado, F.; Maya, D. Regulation of Replication Fork Advance and Stability by Nucleosome Assembly. Genes 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Ramakrishnan, S.; Elango, R.; Ayyar, S.; Zhang, Y.; Deem, A.; Ira, G.; Haber, J.E.; Lobachev, K.S.; Malkova, A. Migrating Bubble during Break-Induced Replication Drives Conservative DNA Synthesis. Nature 2013, 502, 389–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.A.; Kwon, Y.; Xu, Y.; Chung, W.-H.; Chi, P.; Niu, H.; Mayle, R.; Chen, X.; Malkova, A.; Sung, P.; et al. Pif1 Helicase and Polδ Promote Recombination-Coupled DNA Synthesis via Bubble Migration. Nature 2013, 502, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Rothstein, R. Holliday Junctions Accumulate in Replication Mutants via a RecA Homolog-Independent Mechanism. Cell 1997, 90, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Ruiz, M.; Prado, F. Chromatin Assembly Controls Replication Fork Stability. EMBO Rep. 2009, 10, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Cortez, D.; Lopes, M. The Plasticity of DNA Replication Forks in Response to Clinically Relevant Genotoxic Stress. Nat. Rev. Mol. Cell. Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA Damage Checkpoint Response to Replication Stress: A Game of Forks. Front. Genet. 2013, 4, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, F. Homologous Recombination: To Fork and Beyond. Genes 2018, 9, 603. [Google Scholar] [CrossRef] [Green Version]
- Pagès, V.; Fuchs, R.P. Uncoupling of Leading- and Lagging-Strand DNA Replication during Lesion Bypass in Vivo. Science 2003, 300, 1300–1303. [Google Scholar] [CrossRef]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple Mechanisms Control Chromosome Integrity after Replication Fork Uncoupling and Restart at Irreparable UV Lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Chaudhuri, A.R.; Lopes, M.; Costanzo, V. Rad51 Protects Nascent DNA from Mre11-Dependent Degradation and Promotes Continuous DNA Synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [Green Version]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-Dependent DNA Repair Is Linked to Modification of PCNA by Ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Niimi, A.; Brown, S.; Sabbioneda, S.; Kannouche, P.L.; Scott, A.; Yasui, A.; Green, C.M.; Lehmann, A.R. Regulation of Proliferating Cell Nuclear Antigen Ubiquitination in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16125–16130. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of Ubiquitin-Dependent DNA Damage Bypass Is Mediated by Replication Protein A. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef]
- Prado, F. Homologous Recombination Maintenance of Genome Integrity during DNA Damage Tolerance. Mol. Cell. Oncol. 2014, 1, e957039. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication Fork Reversal in Eukaryotes: From Dead End to Dynamic Response. Nat. Rev. Mol. Cell. Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Miyabe, I.; Mizuno, K.; Keszthelyi, A.; Daigaku, Y.; Skouteri, M.; Mohebi, S.; Kunkel, T.A.; Murray, J.M.; Carr, A.M. Polymerase δ Replicates Both Strands after Homologous Recombination–Dependent Fork Restart. Nat. Struct. Mol. Biol. 2015, 22, 932–938. [Google Scholar] [CrossRef]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA Synthesis at Stalled Replication Forks by Human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, N.G.; Wong, R.P.; Ulrich, H.D. The Helicase Pif1 Functions in the Template Switching Pathway of DNA Damage Bypass. Nucleic Acids Res. 2018, 46, 8347–8356. [Google Scholar] [CrossRef]
- Rodríguez, N.G.; Morawska, M.; Wong, R.P.; Daigaku, Y.; Ulrich, H.D. Spatial Separation between Replisome- and Template-Induced Replication Stress Signaling. EMBO J. 2018, 37, 779. [Google Scholar] [CrossRef]
- Vanoli, F.; Fumasoni, M.; Szakal, B.; Maloisel, L.; Branzei, D. Replication and Recombination Factors Contributing to Recombination-Dependent Bypass of DNA Lesions by Template Switch. PLoS Genet. 2010, 6, e1001205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankouri, H.W.; Ngo, H.-P.; Hickson, I.D. Shu Proteins Promote the Formation of Homologous Recombination Intermediates That Are Processed by Sgs1-Rmi1-Top3. Mol. Biol. Cell 2007, 18, 4062–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation Regulates Rad18-Mediated Template Switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Liberi, G.; Maffioletti, G.; Lucca, C.; Chiolo, I.; Baryshnikova, A.; Cotta-Ramusino, C.; Lopes, M.; Pellicioli, A.; Haber, J.E.; Foiani, M. Rad51-Dependent DNA Structures Accumulate at Damaged Replication Forks in Sgs1 Mutants Defective in the Yeast Ortholog of BLM RecQ Helicase. Genes Dev. 2005, 19, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Shor, E.; Weinstein, J.; Rothstein, R. A Genetic Screen for Top3 Suppressors in Saccharomyces Cerevisiae Identifies SHU1, SHU2, PSY3 and CSM2: Four Genes Involved in Error-Free DNA Repair. Genetics 2005, 169, 1275–1289. [Google Scholar] [CrossRef] [Green Version]
- Giannattasio, M.; Zwicky, K.; Lopes, M.; Branzei, D. Visualization of Recombination-Mediated Damage Bypass by Template Switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-Mediated Replication Fork Reversal Is a Global Response to Genotoxic Treatments in Human Cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef]
- Sogo, J.M.; Lopes, M.; Foiani, M. Fork Reversal and SsDNA Accumulation at Stalled Replication Forks Owing to Checkpoint Defects. Science 2002, 297, 599–602. [Google Scholar] [CrossRef]
- Vujanovic, M.; Krietsch, J.; Raso, M.C.; Terraneo, N.; Zellweger, R.; Schmid, J.A.; Taglialatela, A.; Huang, J.-W.; Holland, C.L.; Zwicky, K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67, 882–890.e5. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, A.; Jackson, J.; Mohamed, T.; Adolph, M.; Vindigni, A.; Cortez, D. RADX Prevents Genome Instability by Confining Replication Fork Reversal to Stalled Forks. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I Poisoning Results in PARP-Mediated Replication Fork Reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Wong, R.P.; Rodríguez, N.G.; Zilio, N.; Hanulová, M.; Ulrich, H.D. Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 2020, 77, 3–16.e4. [Google Scholar] [CrossRef]
- Alabert, C.; Bianco, J.N.; Pasero, P. Differential Regulation of Homologous Recombination at DNA Breaks and Replication Forks by the Mrc1 Branch of the S-Phase Checkpoint. EMBO J. 2009, 28, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, M.V.; Rojas, V.; Tercero, J.A. Multiple Pathways Cooperate to Facilitate DNA Replication Fork Progression through Alkylated DNA. DNA Repair 2008, 7, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- González-Prieto, R.; Muñoz-Cabello, A.M.; Cabello-Lobato, M.J.; Prado, F. Rad51 Replication Fork Recruitment Is Required for DNA Damage Tolerance. EMBO J. 2013, 32, 1307–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somyajit, K.; Saxena, S.; Babu, S.; Mishra, A.; Nagaraju, G. Mammalian RAD51 Paralogs Protect Nascent DNA at Stalled Forks and Mediate Replication Restart. Nucleic Acids Res. 2015, 43, gkv880-21. [Google Scholar] [CrossRef]
- Wang, A.T.; Kim, T.; Wagner, J.E.; Conti, B.A.; Lach, F.P.; Huang, A.L.; Molina, H.; Sanborn, E.M.; Zierhut, H.; Cornes, B.K.; et al. A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol. Cell 2015, 59, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Ait Saada, A.A.; Teixeira-Silva, A.; Iraqui, I.; Costes, A.; Hardy, J.; Paoletti, G.; Fréon, K.; Lambert, S.A.E. Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol. Cell 2017, 66, 398–410.e4. [Google Scholar] [CrossRef] [Green Version]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Chaudhuri, A.R.; Nussenzweig, A.; Janscak, P.; et al. Replication Fork Reversal Triggers Fork Degradation in BRCA2-Defective Cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Jasin, M. BRCA2 Suppresses Replication Stress-Induced Mitotic and G1 Abnormalities through Homologous Recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP Is Activated at Stalled Forks to Mediate Mre11-dependent Replication Restart and Recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous Slow Replication Fork Progression Elicits Mitosis Alterations in Homologous Recombination-Deficient Mammalian Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.-S.; Tarsounas, M. MUS81 Nuclease Activity Is Essential for Replication Stress Tolerance and Chromosome Segregation in BRCA2-Deficient Cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Teloni, F.; Mijic, S.; Ursich, S.; Fuchs, J.; Palumbieri, M.D.; Krietsch, J.; Schmid, J.A.; Garcin, E.B.; Gon, S.; et al. Sequential Role of RAD51 Paralog Complexes in Replication Fork Remodeling and Restart. Nat. Commun. 2020, 11, 3531. [Google Scholar] [CrossRef] [PubMed]
- Bugreev, D.V.; Rossi, M.J.; Mazin, A.V. Cooperation of RAD51 and RAD54 in Regression of a Model Replication Fork. Nucleic Acids Res. 2011, 39, 2153–2164. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, G.; Kermi, C.; Stoy, H.; Schiltz, C.J.; Bacal, J.; Zaino, A.M.; Hadden, M.K.; Eichman, B.F.; Lopes, M.; Cimprich, K.A. HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol. Cell 2020, 78, 1237–1251.e7. [Google Scholar] [CrossRef] [PubMed]
- Blastyak, A.; Hajdu, I.; Unk, I.; Haracska, L. Role of Double-Stranded DNA Translocase Activity of Human HLTF in Replication of Damaged DNA. Mol. Cell. Biol. 2010, 30, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bétous, R.; Couch, F.B.; Mason, A.C.; Eichman, B.F.; Manosas, M.; Cortez, D. Substrate-Selective Repair and Restart of Replication Forks by DNA Translocases. Cell Rep. 2013, 3, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Schubert, L.; Ho, T.; Hoffmann, S.; Haahr, P.; Guérillon, C.; Mailand, N. RADX Interacts with Single-stranded DNA to Promote Replication Fork Stability. EMBO Rep. 2017, 18, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Bhat, K.P.; Meur, R.L.; Chazin, W.J.; Ding, X.; Sharan, S.K.; Wessel, S.R.; Sathe, A.A.; Zhao, R.; Cortez, D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol. Cell 2017, 67, 374–386.e5. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolph, M.B.; Mohamed, T.M.; Balakrishnan, S.; Xue, C.; Morati, F.; Modesti, M.; Greene, E.C.; Chazin, W.J.; Cortez, D. RADX Controls RAD51 Filament Dynamics to Regulate Replication Fork Stability. Mol. Cell 2021. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication Stress Activates DNA Repair Synthesis in Mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Wassing, I.E.; Graham, E.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 Recombinase Protects Mitotic Chromatin in Human Cells. Nat. Commun. 2021, 12, 5380. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Walker, G.C. The Critical Mutagenic Translesion DNA Polymerase Rev1 Is Highly Expressed during G(2)/M Phase Rather than S Phase. Proc. Natl. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, A.; Kuntz, K.; Majeed, A.; Smith, S.; Myung, K. Regulation of Gross Chromosomal Rearrangements by Ubiquitin and SUMO Ligases in Saccharomyces Cerevisiae. Mol. Cell. Biol. 2006, 26, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zgaga, Z. Transformation of Saccharomyces Cerevisiae with UV-Irradiated Single-Stranded Plasmid. Mutat. Res. 1991, 263, 211–215. [Google Scholar] [CrossRef]
- Ball, L.G.; Zhang, K.; Cobb, J.A.; Boone, C.; Xiao, W. The Yeast Shu Complex Couples Error-Free Post-Replication Repair to Homologous Recombination. Mol. Microbiol. 2009, 73, 89–102. [Google Scholar] [CrossRef]
- Paulovich, A.G.; Armour, C.D.; Hartwell, L.H. The Saccharomyces Cerevisiae RAD9, RAD17, RAD24 and MEC3 Genes Are Required for Tolerating Irreparable, Ultraviolet-Induced DNA Damage. Genetics 1998, 150, 75–93. [Google Scholar] [CrossRef]
- Schiestl, R.H.; Prakash, S.; Prakash, L. The SRS2 Suppressor of Rad6 Mutations of Saccharomyces Cerevisiae Acts by Channeling DNA Lesions into the RAD52 DNA Repair Pathway. Genetics 1990, 124, 817–831. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.-L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-Modified PCNA Recruits Srs2 to Prevent Recombination during S Phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef]
- Parker, J.L.; Ulrich, H.D. A SUMO-Interacting Motif Activates Budding Yeast Ubiquitin Ligase Rad18 towards SUMO-Modified PCNA. Nucleic Acids Res. 2012, 40, 11380–11388. [Google Scholar] [CrossRef]
- Tsuji, Y.; Watanabe, K.; Araki, K.; Shinohara, M.; Yamagata, Y.; Tsurimoto, T.; Hanaoka, F.; Yamamura, K.; Yamaizumi, M.; Tateishi, S. Recognition of Forked and Single-stranded DNA Structures by Human RAD18 Complexed with RAD6B Protein Triggers Its Recruitment to Stalled Replication Forks. Genes Cells 2008, 13, 343–354. [Google Scholar] [CrossRef]
- Bailly, V.; Lamb, J.; Sung, P.; Prakash, S.; Prakash, L. Specific Complex Formation between Yeast RAD6 and RAD18 Proteins: A Potential Mechanism for Targeting RAD6 Ubiquitin-Conjugating Activity to DNA Damage Sites. Genes Dev. 1994, 8, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, V.; Lauder, S.; Prakash, S.; Prakash, L. Yeast DNA Repair Proteins Rad6 and Rad18 Form a Heterodimer That Has Ubiquitin Conjugating, DNA Binding, and ATP Hydrolytic Activities. J. Biol. Chem. 1997, 272, 23360–23365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-Binding Domains in Y-Family Polymerases Regulate Translesion Synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 Plays a Major Role in the Cellular Response to DNA Damage during Chromosome Replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Frei, C.; Gasser, S.M. The Yeast Sgs1p Helicase Acts Upstream of Rad53p in the DNA Replication Checkpoint and Colocalizes with Rad53p in S-Phase-Specific Foci. Genes Dev. 2000, 14, 81–96. [Google Scholar] [CrossRef]
- Pasero, P.; Duncker, B.P.; Schwob, E.; Gasser, S.M. A Role for the Cdc7 Kinase Regulatory Subunit Dbf4p in the Formation of Initiation-Competent Origins of Replication. Genes Dev. 1999, 13, 2159–2176. [Google Scholar] [CrossRef]
- Shah, P.P.; Zheng, X.; Epshtein, A.; Carey, J.N.; Bishop, D.K.; Klein, H.L. Swi2/Snf2-Related Translocases Prevent Accumulation of Toxic Rad51 Complexes during Mitotic Growth. Mol. Cell 2010, 39, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Muraszko, J.; Kramarz, K.; Argunhan, B.; Ito, K.; Baranowska, G.; Kurokawa, Y.; Murayama, Y.; Tsubouchi, H.; Lambert, S.; Iwasaki, H.; et al. Rrp1 Translocase and Ubiquitin Ligase Activities Restrict the Genome Destabilising Effects of Rad51 in Fission Yeast. Nucleic Acids Res. 2021, 49, 6832–6848. [Google Scholar] [CrossRef]
- Bailis, J.M.; Luche, D.D.; Hunter, T.; Forsburg, S.L. Minichromosome Maintenance Proteins Interact with Checkpoint and Recombination Proteins To Promote S-Phase Genome Stability. Mol. Cell. Biol. 2008, 28, 1724–1738. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Navadgi, V.M.; Mallikarjuna, K.; Rao, B.J. Interaction of HRad51 and HRad52 with MCM Complex: A Cross-Talk between Recombination and Replication Proteins. Biochem. Biophys. Res. Commu. 2005, 329, 1240–1245. [Google Scholar] [CrossRef]
- Hesketh, E.L.; Knight, J.R.P.; Wilson, R.H.C.; Chong, J.P.J.; Coverley, D. Transient Association of MCM Complex Proteins with the Nuclear Matrix during Initiation of Mammalian DNA Replication. Cell Cycle 2015, 14, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.H.C.; Coverley, D. Relationship between DNA Replication and the Nuclear Matrix. Genes Cells 2012, 18, 17–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indiani, C.; Patel, M.; Goodman, M.F.; O’Donnell, M.E. RecA Acts as a Switch to Regulate Polymerase Occupancy in a Moving Replication Fork. Proc. Natl. Acad. Sci. USA 2013, 110, 5410–5415. [Google Scholar] [CrossRef] [Green Version]
- Kolinjivadi, A.M.; Sannino, V.; Antoni, A.D.; Zadorozhny, K.; Kilkenny, M.; Técher, H.; Baldi, G.; Shen, R.; Ciccia, A.; Pellegrini, L.; et al. Smarcal1-Mediated Fork Reversal Triggers Mre11- Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol. Cell 2017, 67, 867–881.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To Skip or Not to Skip: Choosing Repriming to Tolerate DNA Damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Canonical functions of Rad51, BRCA2, and Rad52 during DSB repair. The central steps in DSB-induced HR are located for homology and strand exchange. They are carried out by a Rad51 nucleofilament formed upon resection of the 5′-ends of the DSB and subsequent replacement of the ssDNA binding complex RPA with Rad51. This replacement is mediated by BRCA2 in mammalian cells and Rad52 in yeast, with the help of accessory factors. DNA strand exchange leads to the formation of a D-loop structure that is enlarged by DNA synthesis once Rad54 removes Rad51. The final output of the repair will depend on the chosen mechanism: DSBR, SDSA, BIR, or SSA [1,8]. See text for more details.
Figure 1.
Canonical functions of Rad51, BRCA2, and Rad52 during DSB repair. The central steps in DSB-induced HR are located for homology and strand exchange. They are carried out by a Rad51 nucleofilament formed upon resection of the 5′-ends of the DSB and subsequent replacement of the ssDNA binding complex RPA with Rad51. This replacement is mediated by BRCA2 in mammalian cells and Rad52 in yeast, with the help of accessory factors. DNA strand exchange leads to the formation of a D-loop structure that is enlarged by DNA synthesis once Rad54 removes Rad51. The final output of the repair will depend on the chosen mechanism: DSBR, SDSA, BIR, or SSA [1,8]. See text for more details.

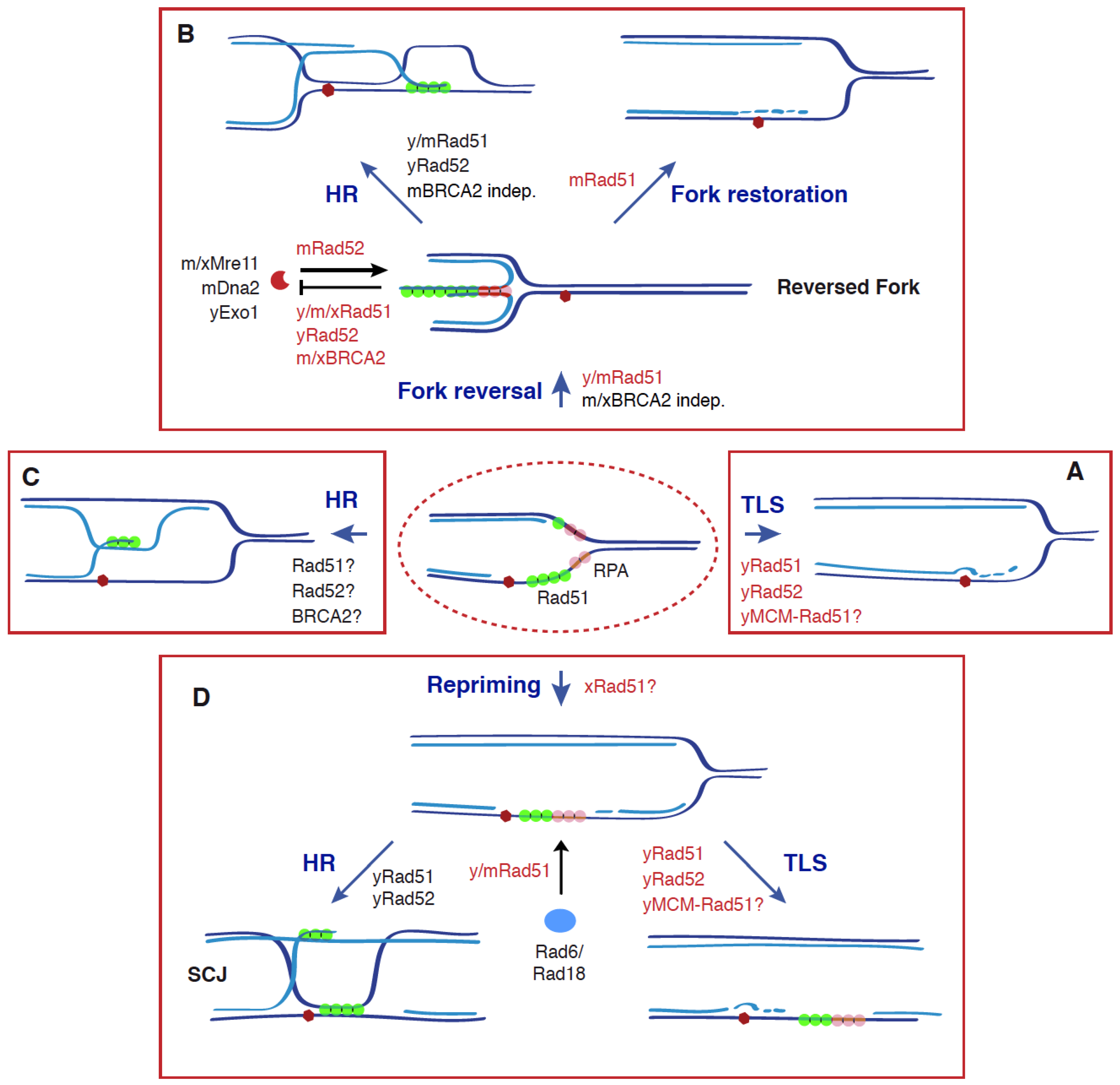
Figure 2.
Recombinogenic and non-recombinogenic functions of Rad51, BRCA2, and Rad52 in DDT. The DDT response is triggered by an accumulation of ssDNA at the fork as a consequence of stress conditions (e.g., blocking lesions, dNTP depletion) that uncouple DNA unwinding and DNA synthesis activities. This response is characterized by ubiquitylation of PCNA, which facilitates the bypass of the blocking lesion and the fill in of the ssDNA gaps through different mechanisms: TLS (A), fork reversal, followed by HR or fork restoration (B), HR (C), and repriming, followed by HR or TLS (D). Apart from its canonical role in HR by promoting strand exchange, Rad51 plays additional, non-recombinogenic roles together with BRCA2 and Rad52 in (1) the formation, protection, and restart of reversed forks through Rad51 nucleofilaments; (2) TLS, by facilitating Rad6/Rad18 recruitment to chromatin and subsequent PCNA ubiquitylation, and by physically interacting with the helicase MCM at nucleoprotein scaffolds. All these functions make Rad51 a potential molecular switch to choose the DDT mechanism. See text for more details. Dark and light blue indicate parental and nascent strands; a red hexagon indicates a blocking DNA lesion; non-recombinogenic functions of Rad51, Rad52, and BRCA2 are highlighted in red; y (yeast), m (mammal), and x (Xenopus) indicate the species in which those molecular functions have been shown for each protein; a question mark indicates a putative function.
Figure 2.
Recombinogenic and non-recombinogenic functions of Rad51, BRCA2, and Rad52 in DDT. The DDT response is triggered by an accumulation of ssDNA at the fork as a consequence of stress conditions (e.g., blocking lesions, dNTP depletion) that uncouple DNA unwinding and DNA synthesis activities. This response is characterized by ubiquitylation of PCNA, which facilitates the bypass of the blocking lesion and the fill in of the ssDNA gaps through different mechanisms: TLS (A), fork reversal, followed by HR or fork restoration (B), HR (C), and repriming, followed by HR or TLS (D). Apart from its canonical role in HR by promoting strand exchange, Rad51 plays additional, non-recombinogenic roles together with BRCA2 and Rad52 in (1) the formation, protection, and restart of reversed forks through Rad51 nucleofilaments; (2) TLS, by facilitating Rad6/Rad18 recruitment to chromatin and subsequent PCNA ubiquitylation, and by physically interacting with the helicase MCM at nucleoprotein scaffolds. All these functions make Rad51 a potential molecular switch to choose the DDT mechanism. See text for more details. Dark and light blue indicate parental and nascent strands; a red hexagon indicates a blocking DNA lesion; non-recombinogenic functions of Rad51, Rad52, and BRCA2 are highlighted in red; y (yeast), m (mammal), and x (Xenopus) indicate the species in which those molecular functions have been shown for each protein; a question mark indicates a putative function.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Prado, F. Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance. Genes 2021, 12, 1550. https://doi.org/10.3390/genes12101550
AMA Style
Prado F. Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance. Genes. 2021; 12(10):1550. https://doi.org/10.3390/genes12101550
Chicago/Turabian StylePrado, Félix. 2021. "Non-Recombinogenic Functions of Rad51, BRCA2, and Rad52 in DNA Damage Tolerance" Genes 12, no. 10: 1550. https://doi.org/10.3390/genes12101550
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.