Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection

 , ,
, ,  ,
, 
Abstract
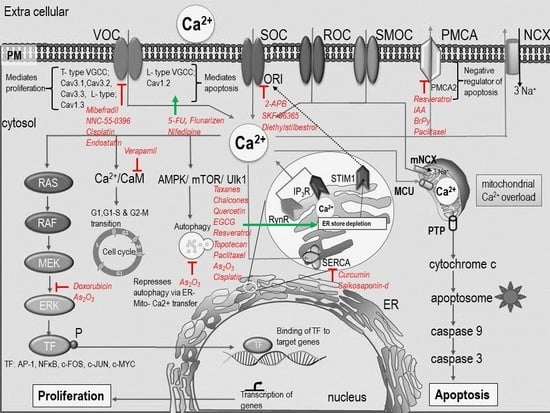
:
1. Intracellular Calcium Homeostasis and Calcium Signaling
2. [Ca2+]i -Signaling in Cell Proliferation and Apoptosis
2.1. [Ca2+]i -Signaling and Cell Proliferation
2.2. [Ca2+]o in Cell Proliferation
2.3. Role of Ca2+ Channels and Pumps in Proliferation
2.4. Store Operated Calcium Entry in Cell Proliferation
2.5. SERCA in Cell Proliferation
2.6. ER and Mitochondrial Axis in Proliferation
2.7. [Ca2+]i and Apoptosis
3. Targeting Calcium Signaling for Anti-Cancer Therapy
3.1. [Ca2+]o Influences Drug Efficiency
3.2. Anti-Cancer Drugs-Induced [Ca2+]i Modulation Triggers Apoptosis
3.2.1. Platinum Drugs
3.2.2. Anti-Metabolites
3.2.3. Inorganic Arsenic Compounds
3.2.4. Anthracyclines
3.2.5. Taxanes
3.2.6. Glucocorticoids
3.2.7. Natural Compounds
3.2.8. Hormonal Receptor Modulator
3.2.9. Epigenetic Modulators
3.3. Calcium Dependent Modulation of Aerobic Glycolysis by Anti-Cancer Agents
3.4. Calcium Modulators in Combination with Anti-Cancer Agents
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| [Ca2+]i | intracellular calcium |
| [Ca2+]o | extra cellular calcium |
| ALL | acute lymphoblastic leukemia |
| APAF-1 | apoptotic protease activating factor 1 |
| As2O3 | arsenic trioxide |
| ATP | adenosine triphosphate |
| BrPy | 3-bromopyruvate |
| Calreticulin | calcium buffering protein in the ER |
| CamK | Ca2+/calmodulin-dependent protein kinase |
| CaSR | calcium sensing receptor |
| CDDP | cis-diamminedichloridoplatinum(II) |
| CML | chronic myelogenous leukemia |
| CRAC | channel Calcium release-activated channels |
| CREB | cAMP response element-binding protein |
| DJM-1 | a human skin squamous carcinoma cell line |
| EC | endometrial cancer |
| EGF | epidermal growth factor |
| EGCG | Epigallocatechin gallate |
| EMT | epithelial–mesenchymal transition |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular signal–regulated kinases |
| GC | glucocorticoid |
| GPER | G protein-coupled estrogen receptor |
| HSC-1 | a human skin squamous carcinoma cell line |
| IAA | sodium iodoacetate |
| IP3R | inositol trisphosphate receptor |
| IAA | sodium iodoacetate |
| MAPK | mitogen-activated protein kinase |
| MCU | mitochondrial calcium uniporter |
| mTOR | mechanistic target of rapamycin |
| NB | neuroblastoma |
| NCS-1 | neuronal Ca2+ sensor 1 |
| NHBE | normal human bronchial epithelial |
| NSAID | non-steroidal anti-inflammatory drugs |
| Orai1 | calcium release-activated calcium channel protein 1 |
| PMCA | plasma membrane calcium ATPase |
| PML | promyelocytic leukemia protein |
| PTP | permeability transition pore |
| RyR | ryanodine receptors |
| SCID | severe combined immune deficiency syndrome |
| SERCA | sarco/endoplasmic reticulum Ca2+-ATPase |
| SK3 | small-conductance calcium-activated potassium channel |
| SOC | store operated channel |
| STIM1 | stromal interaction molecule 1 |
| TGFβ | transforming growth factor beta |
| TM | tamoxifen |
| TF | transcription factor |
| TSA | trichostatin A |
| TRPC1 | transient receptor potential channel 1 |
| TRPV1 | transient receptor potential vanilloid 1 |
| V-ATPase | vacuolar-type H+ -ATPase |
| VDAC1 | voltage-dependent anion channel |
| VGCC | voltage gated calcium channel |
| VGEF | vascular endothelial growth factor |
| VOC | voltage operated calcium channel |
| 2-APB | 2-Aminoethoxydiphenyl borate |
| 5-FU | 5-Fluorouracil |
References
- Bootman, M.D.; Rietdorf, K.; Hardy, H.; Dautova, Y.; Corps, E.; Pierro, C.; Stapleton, E.; Kang, E.; Proudfoot, D. Calcium Signalling and Regulation of Cell Function. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Munaron, L.; Antoniotti, S.; Lovisolo, D. Intracellular calcium signals and control of cell proliferation: How many mechanisms? J. Cell. Mol. Med. 2004, 8, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S. Calcium signalling during mammalian fertilization. Ciba Found. Symp. 1995, 188, 235–247; discussion 247–251. [Google Scholar] [PubMed]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilon, P.; Chae, H.-Y.; Rutter, G.A.; Ravier, M.A. Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Anderson Mark, E. Mechanisms of Altered Ca2+ Handling in Heart Failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Wang, W.; Ren, Y.; Wang, L.; Zhao, W.; Dong, X.; Pan, J.; Gao, H.; Tian, Y. Orai1 and Stim1 Mediate the Majority of Store-Operated Calcium Entry in Multiple Myeloma and Have Strong Implications for Adverse Prognosis. Cell. Physiol. Biochem. 2018, 48, 2273–2285. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Reddish, F.N.; Miller, C.L.; Gorkhali, R.; Yang, J.J. Calcium Dynamics Mediated by the Endoplasmic/Sarcoplasmic Reticulum and Related Diseases. Int. J. Mol. Sci. 2017, 18, 1024. [Google Scholar] [CrossRef]
- Williams, J.A.; Hou, Y.; Ni, H.-M.; Ding, W.-X. Role of intracellular calcium in proteasome inhibitor-induced endoplasmic reticulum stress, autophagy, and cell death. Pharm. Res. 2013, 30, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca(2+) transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, M.; Gil-Longo, J.; Campos-Toimil, M. Calcium Binding Proteins. In Calcium Signaling; Islam, M.S., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 461–482. [Google Scholar]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Resende, R.R.; Andrade, L.M.; Oliveira, A.G.; Guimarães, E.S.; Guatimosim, S.; Leite, M.F. Nucleoplasmic calcium signaling and cell proliferation: Calcium signaling in the nucleus. Cell Commun. Signal. 2013, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.F.; Thrower, E.C.; Echevarria, W.; Koulen, P.; Hirata, K.; Bennett, A.M.; Ehrlich, B.E.; Nathanson, M.H. Nuclear and cytosolic calcium are regulated independently. Proc. Natl. Acad. Sci. USA 2003, 100, 2975–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allbritton, N.L.; Oancea, E.; Kuhn, M.A.; Meyer, T. Source of nuclear calcium signals. Proc. Natl. Acad. Sci. USA 1994, 91, 12458–12462. [Google Scholar] [CrossRef]
- Echevarria, W.; Leite, M.F.; Guerra, M.T.; Zipfel, W.R.; Nathanson, M.H. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003, 5, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Ikura, M.; Osawa, M.; Ames, J.B. The role of calcium-binding proteins in the control of transcription: Structure to function. BioEssays 2002, 24, 625–636. [Google Scholar] [CrossRef]
- Hiraoki, T.; Vogel, H.J. Structure and Function of Calcium-Binding Proteins. J. Cardiovasc. Pharmacol. 1987, 10, S14–S31. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Bidaux, G.; Flourakis, M.; Shuba, Y. Ion channels in death and differentiation of prostate cancer cells. Cell Death Differ. 2007, 14, 1295. [Google Scholar] [CrossRef]
- Florea, A.M.; Busselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.-M.; Splettstoesser, F.; Büsselberg, D. Arsenic trioxide (As2O3) induced calcium signals and cytotoxicity in two human cell lines: SY-5Y neuroblastoma and 293 embryonic kidney (HEK). Toxicol. Appl. Pharmacol. 2007, 220, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Busselberg, D. Auranofin, an anti-rheumatic gold compound, modulates apoptosis by elevating the intracellular calcium concentration ([Ca2+]i) in mcf-7 breast cancer cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef] [PubMed]
- Capiod, T.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Calcium signalling and cancer cell growth. In Calcium Signalling and Disease; Springer: Dordrecht, The The Netherlands, 2007; Volume 45, pp. 405–427. [Google Scholar]
- Xu, M.; Seas, A.; Kiyani, M.; Ji, K.S.Y.; Bell, H.N. A temporal examination of calcium signaling in cancer- from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Boynton, A.L.; Whitfield, J.F.; Isaacs, R.J.; Morton, H.J. Control of 3T3 cell proliferation by calcium. In Vitro 1974, 10, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.X.; Kihara, A.H.; Goulart, V.A.M.; Tonelli, F.M.P.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Borowiec, A.-S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium channels, external calcium concentration and cell proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Splettstoesser, F.; Florea, A.M.; Busselberg, D. IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef]
- Capiod, T. Extracellular Calcium Has Multiple Targets to Control Cell Proliferation. In Calcium Entry Pathways in Non-Excitable Cells; Rosado, J.A., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 133–156. [Google Scholar]
- Flucher, B.E.; Tuluc, P. How and why are calcium currents curtailed in the skeletal muscle voltage-gated calcium channels? J. Physiol. 2017, 595, 1451–1463. [Google Scholar] [CrossRef]
- Phan, N.N.; Wang, C.-Y.; Chen, C.-F.; Sun, Z.; Lai, M.-D.; Lin, Y.-C. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef]
- Hao, J.; Bao, X.; Jin, B.; Wang, X.; Mao, Z.; Li, X.; Wei, L.; Shen, D.; Wang, J.-L. Ca2+ channel subunit α 1D promotes proliferation and migration of endometrial cancer cells mediated by 17β-estradiol via the G protein-coupled estrogen receptor. FASEB J. 2015, 29, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Han, Z.; Shao, L.; Zhao, Y. Ultrasound-targeted microbubble destruction of calcium channel subunit α 1D siRNA inhibits breast cancer via G protein-coupled receptor 30. Oncol. Rep. 2016, 36, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, X.; Zhang, R.; Huang, J.; Kuang, X.; Yang, J.; Liu, J.; Tawfik, O.; Brantley Thrasher, J.; Li, B. Cav1.3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Triggle, D.J. The Physiological and Pharmacological Significance of Cardiovascular T-Type, Voltage-gated Calcium Channels. Am. J. Hypertens. 1998, 11, 80S–87S. [Google Scholar] [CrossRef]
- Antal, L.; Martin-Caraballo, M. T-type Calcium Channels in Cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflügers Archiv 2014, 466, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, T.; Yamazaki, J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef]
- James, A.D.; Chan, A.; Erice, O.; Siriwardena, A.K.; Bruce, J.I.E. Glycolytic ATP fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 2013, 288, 36007–36019. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Giachini, F.R.; Lima, V.V.; Hannan, J.L.; Carneiro, F.S.; Webb, R.C.; Tostes, R.C. STIM1/Orai1-mediated store-operated Ca2+ entry: The tip of the iceberg. Braz. J. Med. Biol. Res. 2011, 44, 1080–1087. [Google Scholar] [CrossRef]
- Lipskaia, L.; Hulot, J.-S.; Lompré, A.-M. Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflügers Archiv 2009, 457, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Emeriau, N.; de Clippele, M.; Gailly, P.; Tajeddine, N. Store operated calcium entry is altered by the inhibition of receptors tyrosine kinase. Oncotarget 2018, 9, 16059–16073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca2+ entry and promote TGF-β-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-Mediated Calcium Influx in Lactation and in Breast Cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.; Amberger, A.; Widschwendter, M.; Margreiter, R.; Öfner, D.; Dietl, P. Inhibition of store-operated calcium entry contributes to the anti-proliferative effect of non-steroidal anti-inflammatory drugs in human colon cancer cells. Int. J. Cancer 2001, 92, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Mo, P.; Yang, S. The store-operated calcium channels in cancer metastasis: From cell migration, invasion to metastatic colonization. Front. Biosci. 2018, 23, 1241–1256. [Google Scholar]
- Yang, Z.; Pan, L.; Liu, S.; Li, F.; Lv, W.; Shu, Y.; Dong, P. Inhibition of stromal-interacting molecule 1-mediated store-operated Ca(2+) entry as a novel strategy for the treatment of acquired imatinib-resistant gastrointestinal stromal tumors. Cancer Sci. 2018, 109, 2792–2800. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef]
- Primeau, J.O.; Armanious, G.P.; Fisher, M.L.E.; Young, H.S. The SarcoEndoplasmic Reticulum Calcium ATPase. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; pp. 229–258. [Google Scholar]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Legrand, G.; Humez, S.; Slomianny, C.; Dewailly, E.; Vanden Abeele, F.; Mariot, P.; Wuytack, F.; Prevarskaya, N. Ca2+ pools and cell growth. Evidence for sarcoendoplasmic Ca2+-ATPases 2B involvement in human prostate cancer cell growth control. J. Biol. Chem. 2001, 276, 47608–47614. [Google Scholar] [CrossRef] [PubMed]
- Bergner, A.; Kellner, J.; Tufman, A.; Huber, R.M. Endoplasmic reticulum Ca2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J. Exp. Clin. Cancer Res. 2009, 28, 25. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, H.; Kerkhofs, M.; La Rovere, R.M.; Bultynck, G. Endoplasmic Reticulum-Mitochondrial Ca(2+) Fluxes Underlying Cancer Cell Survival. Front. Oncol. 2017, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Marchi, S.; Vitto, V.A.M.; Danese, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle 2019, 18, 1068–1083. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisthal, S.; Keinan, N.; Ben-Hail, D.; Arif, T.; Shoshan-Barmatz, V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Dixit, V.M. Characterization of Calcium Release-activated Apoptosis of LNCaP Prostate Cancer Cells. J. Biol. Chem. 2000, 275, 11470–11477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca(2+)-ATPase by thapsigargin analogs induces cell death via ER Ca(2+) depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günes, D.A.; Florea, A.-M.; Splettstoesser, F.; Büsselberg, D. Co-application of arsenic trioxide (As2O3) and cisplatin (CDDP) on human SY-5Y neuroblastoma cells has differential effects on the intracellular calcium concentration ([Ca2+]i) and cytotoxicity. Neurotoxicology 2009, 30, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Al-Taweel, N.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium Ca2+ homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Shen, L.; Wen, N.; Xia, M.; Zhang, Y.U.; Liu, W.; Xu, Y.E.; Sun, L. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol. Lett. 2016, 11, 2411–2419. [Google Scholar] [CrossRef]
- Can, G.; Akpinar, B.; Baran, Y.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil signaling through a calcium–calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2012, 32, 4529. [Google Scholar] [CrossRef]
- Deveci, H.A.; Nazıroğlu, M.; Nur, G. 5-Fluorouracil-induced mitochondrial oxidative cytotoxicity and apoptosis are increased in MCF-7 human breast cancer cells by TRPV1 channel activation but not Hypericum perforatum treatment. Mol. Cell. Biochem. 2018, 439, 189–198. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca2+ signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 334. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafà, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwama, K.; Nakajo, S.; Aiuchi, T.; Nakaya, K. Apoptosis induced by arsenic trioxide in leukemia U937 cells is dependent on activation of p38, inactivation of ERK and the Ca2+-dependent production of superoxide. Int. J. Cancer 2001, 92, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Abdoul-Azize, S.; Buquet, C.; Li, H.; Picquenot, J.-M.; Vannier, J.-P. Integration of Ca2+ signaling regulates the breast tumor cell response to simvastatin and doxorubicin. Oncogene 2018, 37, 4979–4993. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.C.; Holton, M.; Baggott, R.R.; Roux-Soro, S.C.; Armesilla, A.L.; Oceandy, D.; Cartwright, E.J.; Neyses, L.; Mohamed, T.M.A.; Brown, S.; et al. Disruption of the interaction between PMCA2 and calcineurin triggers apoptosis and enhances paclitaxel-induced cytotoxicity in breast cancer cells. Carcinogenesis 2012, 33, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Avila, A.; Gollahon, L. Paclitaxel induces apoptosis in breast cancer cells through different calcium--regulating mechanisms depending on external calcium conditions. Int. J. Mol. Sci. 2014, 15, 2672–2694. [Google Scholar] [CrossRef] [PubMed]
- Winter, E.; Chiaradia, L.D.; Silva, A.H.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: Advantages of multi-target action. Toxicol. In Vitro 2014, 28, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Ginkel, P.R.; Yan, M.B.; Bhattacharya, S.; Polans, A.S.; Kenealey, J.D. Natural products induce a G protein-mediated calcium pathway activating p53 in cancer cells. Toxicol. Appl. Pharmacol. 2015, 288, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Büsselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Parichatikanond, W.; Eroglu, E.; Klec, C.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell. Physiol. Biochem. 2016, 39, 1404–1420. [Google Scholar] [CrossRef]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Varghese, E.; McCallum, J.E.; Mahgoub, S.; Helmy, I.; Varghese, S.; Gopinath, N.; Sass, S.; Theis, F.J.; Reifenberger, G.; et al. Calcium-regulatory proteins as modulators of chemotherapy in human neuroblastoma. Oncotarget 2017, 8, 22876–22893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Couldwell, W.T.; Song, H.; Takano, T.; Lin, J.H.C.; Nedergaard, M. Tamoxifen-induced Enhancement of Calcium Signaling in Glioma and MCF-7 Breast Cancer Cells. Cancer Res. 2000, 60, 5395. [Google Scholar] [PubMed]
- Jan, C.-R.; Cheng, J.-S.; Chou, K.-J.; Wang, S.-P.; Lee, K.C.; Tang, K.-Y.; Tseng, L.-L.; Chiang, H.-T. Dual Effect of Tamoxifen, an Anti-Breast-Cancer Drug, on Intracellular Ca2+ and Cytotoxicity in Intact Cells. Toxicol. Appl. Pharmacol. 2000, 168, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Jiann, B.-P.; Chang, H.-T.; Huang, J.-K.; Chen, W.-C.; Su, W.; Jan, C.-R. Effect of the Anti-Breast Cancer Drug Tamoxifen on Ca2+ Movement in Human Osteosarcoma Cells. Pharmacol. Toxicol. 2002, 91, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-T.; Huang, J.-K.; Wang, J.-L.; Cheng, J.-S.; Lee, K.-C.; Lo, Y.-K.; Liu, C.-P.; Chou, K.-J.; Chen, W.-C.; Su, W.; et al. Tamoxifen-Induced Increases in Cytoplasmic Free Ca2+ Levels in Human Breast Cancer Cells. Breast Cancer Res. Treat. 2002, 71, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, G.; Akatsuka, K.; Nakashima, Y.; Yokoe, Y.; Higo, N.; Shimonaka, M. Tamoxifen inhibits the proliferation of non-melanoma skin cancer cells by increasing intracellular calcium concentration. Int. J. Oncol. 2018, 53, 2157–2166. [Google Scholar] [CrossRef]
- Meneses-Morales, I.; Izquierdo-Torres, E.; Flores-Peredo, L.; Rodríguez, G.; Hernández-Oliveras, A.; Zarain-Herzberg, Á. Epigenetic regulation of the human ATP2A3 gene promoter in gastric and colon cancer cell lines. Mol. Carcinog. 2019, 58, 887–897. [Google Scholar] [CrossRef]
- Raynal, N.J.M.; Lee, J.T.; Wang, Y.; Beaudry, A.; Madireddi, P.; Garriga, J.; Malouf, G.G.; Dumont, S.; Dettman, E.J.; Gharibyan, V.; et al. Targeting Calcium Signaling Induces Epigenetic Reactivation of Tumor Suppressor Genes in Cancer. Cancer Res. 2016, 76, 1494–1505. [Google Scholar] [CrossRef]
- Kim, J.-A.; Kang, Y.S.; Jung, M.-W.; Lee, S.H.; Lee, Y.S. Involvement of Ca2+ influx in the mechanism of tamoxifen-induced apoptosis in HepG2 human hepatoblastoma cells. Cancer Lett. 1999, 147, 115–123. [Google Scholar] [CrossRef]
- Tomaszewski, A.; Büsselberg, D. Cisplatin modulates voltage gated channel currents of dorsal root ganglion neurons of rats. Neurotoxicology 2007, 28, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Gualdani, R.; de Clippele, M.; Ratbi, I.; Gailly, P.; Tajeddine, N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers 2019, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Satheesh, N.J.; Büsselberg, D. The role of intracellular calcium for the development and treatment of neuroblastoma. Cancers 2015, 7, 823–848. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lu, E.; Hu, X.; Cheng, H.; Zhang, J.-A.; Zhu, X. S100A9 regulates cisplatin chemosensitivity of squamous cervical cancer cells and related mechanism. Cancer Manag. Res. 2018, 10, 3753–3764. [Google Scholar] [CrossRef] [PubMed]
- Pelzl, L.; Hosseinzadeh, Z.; Alzoubi, K.; Al-Maghout, T.; Schmidt, S.; Stournaras, C.; Lang, F. Impact of Na+/Ca2+ Exchangers on Therapy Resistance of Ovary Carcinoma Cells. Cell. Physiol. Biochem. 2015, 37, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Schulze, C.; McGowan, M.; Jordt, S.-E.; Ehrlich, B.E. Prolonged oxaliplatin exposure alters intracellular calcium signaling: A new mechanism to explain oxaliplatin-associated peripheral neuropathy. Clin. Colorectal Cancer 2011, 10, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Muscella, A.; Vetrugno, C.; Fanizzi, F.P.; Manca, C.; De Pascali, S.A.; Marsigliante, S. A new platinum(II) compound anticancer drug candidate with selective cytotoxicity for breast cancer cells. Cell Death Dis. 2013, 4, e796. [Google Scholar] [CrossRef]
- Antman, K.H. Introduction: The history of arsenic trioxide in cancer therapy. Oncologist 2001, 6 (Suppl. 2), 1–2. [Google Scholar] [CrossRef]
- Liu, L.Z.; Jiang, Y.; Carpenter, R.L.; Jing, Y.; Peiper, S.C.; Jiang, B.H. Role and mechanism of arsenic in regulating angiogenesis. PLoS ONE 2011, 6, e20858. [Google Scholar] [CrossRef]
- Park, W.H.; Seol, J.G.; Kim, E.S.; Hyun, J.M.; Jung, C.W.; Lee, C.C.; Kim, B.K.; Lee, Y.Y. Arsenic Trioxide-mediated Growth Inhibition in MC/CAR Myeloma Cells via Cell Cycle Arrest in Association with Induction of Cyclin-dependent Kinase Inhibitor, p21, and Apoptosis. Cancer Res. 2000, 60, 3065–3071. [Google Scholar]
- Jing, Y.; Dai, J.; Chalmers-Redman, R.M.E.; Tatton, W.G.; Waxman, S. Arsenic Trioxide Selectively Induces Acute Promyelocytic Leukemia Cell Apoptosis Via a Hydrogen Peroxide-Dependent Pathway. Blood 1999, 94, 2102–2111. [Google Scholar] [PubMed]
- Nguyen, T.T.T.; Lim, Y.J.; Fan, M.H.M.; Jackson, R.A.; Lim, K.K.; Ang, W.H.; Ban, K.H.K.; Chen, E.S. Calcium modulation of doxorubicin cytotoxicity in yeast and human cells. Genes Cells 2016, 21, 226–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehmerle, W.; Splittgerber, U.; Lazarus, M.B.; McKenzie, K.M.; Johnston, D.G.; Austin, D.J.; Ehrlich, B.E. Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 18356–18361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenstein, S.; Ghias, K.; Krett, N.L.; Rosen, S.T. Mechanisms of Glucocorticoid-mediated Apoptosis in Hematological Malignancies. Clin. Cancer Res. 2002, 8, 1681–1694. [Google Scholar] [PubMed]
- Abdoul-Azize, S.; Dubus, I.; Vannier, J.-P. Improvement of dexamethasone sensitivity by chelation of intracellular Ca2+ in pediatric acute lymphoblastic leukemia cells through the prosurvival kinase ERK1/2 deactivation. Oncotarget 2017, 8, 27339–27352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareen, D.; Darjatmoko, S.R.; Albert, D.M.; Polans, A.S. Mitochondria, calcium, and calpain are key mediators of resveratrol-induced apoptosis in breast cancer. Mol. Pharmacol. 2007, 72, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Obakan, P.; Barrero, C.; Coker-Gurkan, A.; Arisan, E.D.; Merali, S.; Palavan-Unsal, N. SILAC-Based Mass Spectrometry Analysis Reveals That Epibrassinolide Induces Apoptosis via Activating Endoplasmic Reticulum Stress in Prostate Cancer Cells. PLoS ONE 2015, 10, e0135788. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, W.; You, S.; Song, G. Ameliorative effects of luteolin against endometriosis progression in vitro and in vivo. J. Nutr. Biochem. 2019, 67, 161–172. [Google Scholar] [CrossRef]
- Park, S.; Lim, W.; Song, G. Delphinidin induces antiproliferation and apoptosis of endometrial cells by regulating cytosolic calcium levels and mitochondrial membrane potential depolarization. J. Cell. Biochem. 2019, 120, 5072–5084. [Google Scholar] [CrossRef]
- Yu, F.S.; Yang, J.S.; Yu, C.S.; Lu, C.C.; Chiang, J.H.; Lin, C.W.; Chung, J.G. Safrole Induces Apoptosis in Human Oral Cancer HSC-3 Cells. J. Dent. Res. 2010, 90, 168–174. [Google Scholar] [CrossRef]
- Wang, M.; Ruan, Y.; Chen, Q.; Li, S.; Wang, Q.; Cai, J. Curcumin induced HepG2 cell apoptosis-associated mitochondrial membrane potential and intracellular free Ca2+ concentration. Eur. J. Pharmacol. 2011, 650, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Busselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tang, C.; Zhu, Y.; Xie, M.; He, D.; Pan, Q.; Zhang, P.; Hua, D.; Wang, T.; Jin, L.; et al. TrpC5 regulates differentiation through the Ca2+/Wnt5a signalling pathway in colorectal cancer. Clin. Sci. 2017, 131, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ning, K.; Sun, X.; Zhang, C.; Jin, L.-F.; Hua, D. Glycolysis is essential for chemoresistance induced by transient receptor potential channel C5 in colorectal cancer. BMC Cancer 2018, 18, 207. [Google Scholar] [CrossRef]
- Xie, J.; Wang, B.S.; Yu, D.H.; Lu, Q.; Ma, J.; Qi, H.; Fang, C.; Chen, H.Z. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int. J. Oncol. 2011, 38, 409–417. [Google Scholar] [CrossRef]
- Aoki, S.; Morita, M.; Hirao, T.; Yamaguchi, M.; Shiratori, R.; Kikuya, M.; Chibana, H.; Ito, K. Shift in energy metabolism caused by glucocorticoids enhances the effect of cytotoxic anti-cancer drugs against acute lymphoblastic leukemia cells. Oncotarget 2017, 8, 94271–94285. [Google Scholar] [CrossRef] [Green Version]
- Freedman, R.A.; Tolaney, S.M. Efficacy and safety in older patient subsets in studies of endocrine monotherapy versus combination therapy in patients with HR+/HER2−advanced breast cancer: A review. Breast Cancer Res. Treat. 2018, 167, 607–614. [Google Scholar] [CrossRef]
- Mason, R.P. Calcium channel blockers, apoptosis and cancer: Is there a biologic relationship? J. Am. Coll. Cardiol. 1999, 34, 1857–1866. [Google Scholar] [CrossRef]
- Onoda, J.M.; Nelson, K.K.; Taylor, J.D.; Honn, K.V. Invivo Characterization of Combination Antitumor Chemotherapy with Calcium Channel Blockers and cis-Diamminedichloroplatinum(II). Cancer Res. 1989, 49, 2844–2850. [Google Scholar] [PubMed]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M.; et al. The combined activation of KCa3.1 and inhibition of Kv11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2017, 118, 200. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-K.; Chou, C.-T.; Chang, H.-T.; Shu, S.-S.; Kuo, C.-C.; Tsai, J.-Y.; Liao, W.-C.; Wang, J.-L.; Lin, K.-L.; Lu, Y.-C.; et al. Effect of thapsigargin on Ca2+ fluxes and viability in human prostate cancer cells. J. Recept. Signal Transduct. 2011, 31, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Fan, C.; Yang, Y.; Di, S.; Hu, W.; Li, T.; Zhu, Y.; Han, J.; Xin, Z.; Wu, G.; et al. Thapsigargin sensitizes human esophageal cancer to TRAIL-induced apoptosis via AMPK activation. Sci. Rep. 2016, 6, 35196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackisch, C.; Hahm, H.A.; Tombal, B.; McCloskey, D.; Butash, K.; Davidson, N.E.; Denmeade, S.R. Delayed Micromolar Elevation in Intracellular Calcium Precedes Induction of Apoptosis in Thapsigargin-treated Breast Cancer Cells. Clin. Cancer Res. 2000, 6, 2844–2850. [Google Scholar]
- Thews, O.; Hummel, M.; Kelleher, D.K.; Lecher, B.; Vaupel, P. Nifedipine improves blood flow and oxygen supply, but not steady-state oxygenation of tumours in perfusion pressure-controlled isolated limb perfusion. Br. J. Cancer 2002, 87, 1462–1469. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.J.; Hirst, D.G. Modification of tumour response by calcium antagonists in the SCVII/St tumour implanted at two different sites. Int. J. Radiat. Biol. 1989, 56, 355–367. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug Class | Drugs | Axis/Mechanism of Induction of Cell Death | Concentration Range | Apoptosis | Proliferation | Cell Line | References |
|---|---|---|---|---|---|---|---|
| Platinum agents (cytotoxic alkylating agent) | Cisplatin | [Ca2+]i ↑↑ by influx of extra cellular calcium. [Ca2+]i ↑↑ / ER stress / mitochondrial Ca2+ over load / caspase 3 activation. | 1 µM 5 µg/ml | ↑↑ | ↓↓ | MCF-7 SH-SY5Y HeLa-S3 | [31,71,72,73] |
| Anti-metabolites | 5-Fluorouracil | Ca2+-CaM-p53 activation, Ca2+ influx partially through L-type Ca2+ channel. Ca2+ entry through TRPV1 / mitochondrial ROS production / caspase 8. | 768 μM, 25 μM | ↑↑ | ↓↓ | HCT116 MCF-7 | [74,75] |
| Inorganic arsenic compounds | As2O3 | IP3R, RyR / [Ca2+]i ↑↑ / DNA damage / caspase 3. ↑↑ER–mitochondrial Ca2+ transfer. ↓↓ ERK1 and ERK2 | 1 µM | ↑↑ | ↓↓ | SH-SY5Y Pml-/- mice NB4 cells U937 | [31,76,77,78] |
| Anthracyclines | Doxorubicin | [Ca2+]i modulation - ERK1/2 inactivation, activation of pro apoptotic BIM pathway and mitochondrial Ca2+ overload. | 500 nM–1 µM | ↑↑ | ↓↓ | MDA-MB-231 | [79] |
| Taxanes | Paclitaxel Docetaxel | In activation of PMCA2/calcineurin A and activation of calcineurin A /NFAT pathway/ ↑↑ pro-apoptotic protein Fas ligand. External calcium influx, inhibition of bcl2/ IP3R-ER-[Ca2+]i. | 1 nM 10-6 M | ↑↑ | MDA-MB-231 MCF-7 MDA-MB-468 | [80,81] | |
| Natural compounds | Chalcones Quercetin EGCG Piceatannol Etoposide; (semi-synthetic) Resveratrol Curcumin Saikosaponin-d | Ca 2+ / ER stress / caspase 12. G-protein / IP3R-ER-[Ca2+]i, / modulation of p53 / transcription of pro-apoptotic genes. SERCA↓↓ activity / [Ca2+]i ↑↑ / increased mitochondrial Ca2+ uptake. SERCA ↓↓ / [Ca2+]i ↑↑ / ER stress / Autophagy mediated cell death. | 30–40 µM 50–100 µM 10 µM | ↑↑ | L1210 MDA-MB-231 MYCN2 HeLa SW480 (colon) MCF-7 | [55,82,83,84,85,86] | |
| Camptothecin analog | Topotecan | Increased [Ca2+]i, altered expression of calcium regulating proteins. | 0.01 µM | ↑↑ | ↓↓ | SH-SY5Y | [87] |
| Hormonal receptor modulator | Tamoxifen | [Ca2+]i ↑↑ by influx of extra cellular calcium and release of Ca2+ from multiple stores. VGCC | 5–10 µM | ↑↑ | MCF-7 MG63 ZR-75-1 SCC BFTC | [88,89,90,91,92] | |
| DNA methylation and HDAC modulators | TSA Azacitidine Digitoxin Pyrithion zinc Disulfiram | ↑↑SERCA3 / apoptosis. SOC / [Ca2+]i ↑↑ / CamK / via MeCP2 / reactivation of tumor suppressor genes. | 50 nM–5 µM | ↑↑ | KATO-III (gastric carcinoma) YB5 (colon) | [93,94] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int. J. Mol. Sci. 2019, 20, 3017. https://doi.org/10.3390/ijms20123017
Varghese E, Samuel SM, Sadiq Z, Kubatka P, Liskova A, Benacka J, Pazinka P, Kruzliak P, Büsselberg D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. International Journal of Molecular Sciences. 2019; 20(12):3017. https://doi.org/10.3390/ijms20123017
Chicago/Turabian StyleVarghese, Elizabeth, Samson Mathews Samuel, Zuhair Sadiq, Peter Kubatka, Alena Liskova, Jozef Benacka, Peter Pazinka, Peter Kruzliak, and Dietrich Büsselberg. 2019. "Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection" International Journal of Molecular Sciences 20, no. 12: 3017. https://doi.org/10.3390/ijms20123017