Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update

Abstract
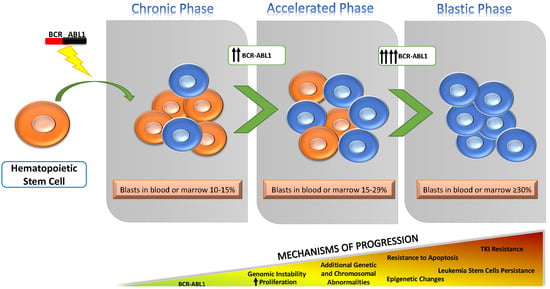
:
1. Introduction
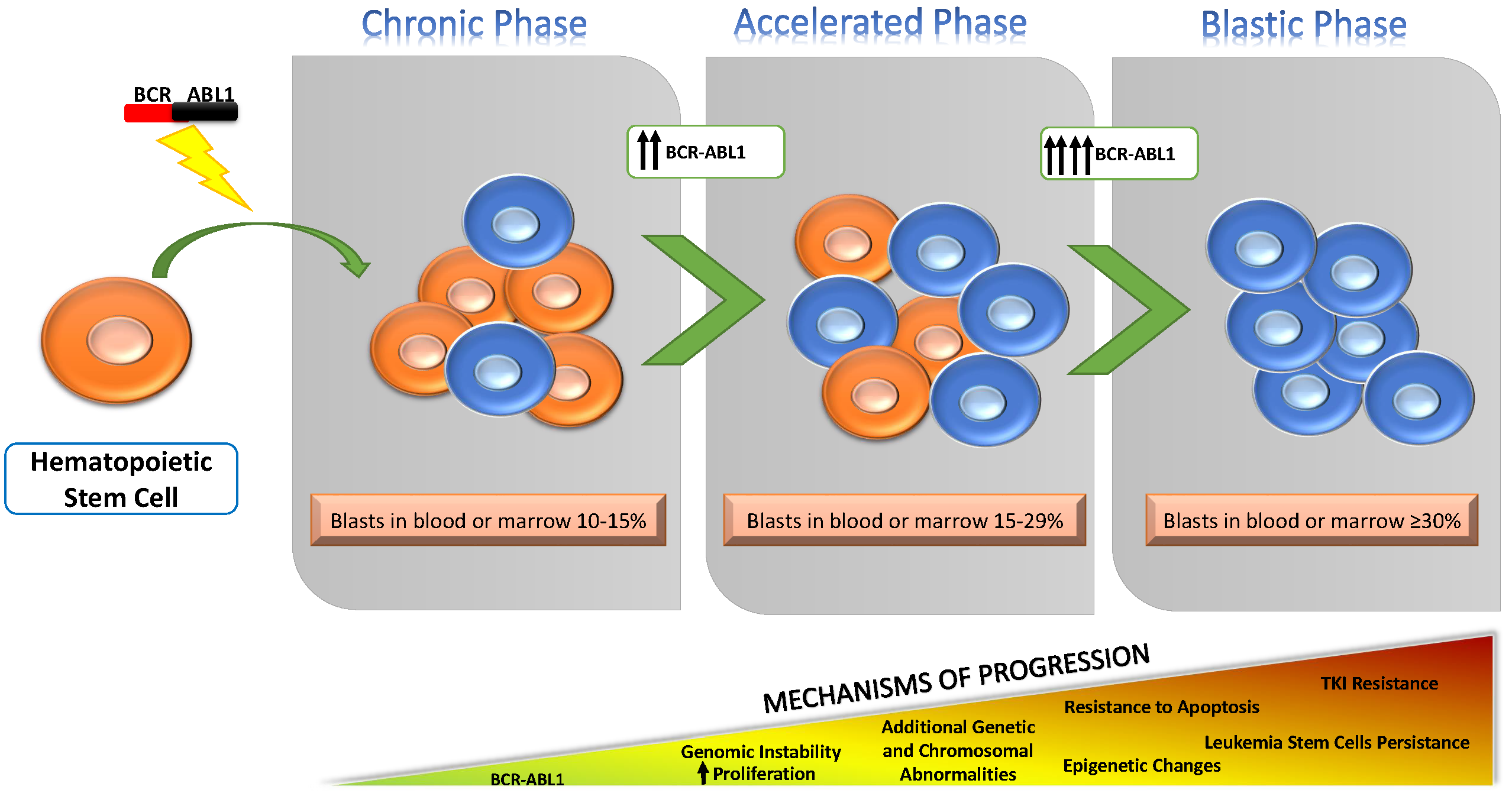
2. Overview of Known Mechanisms Underlying Progression to BP
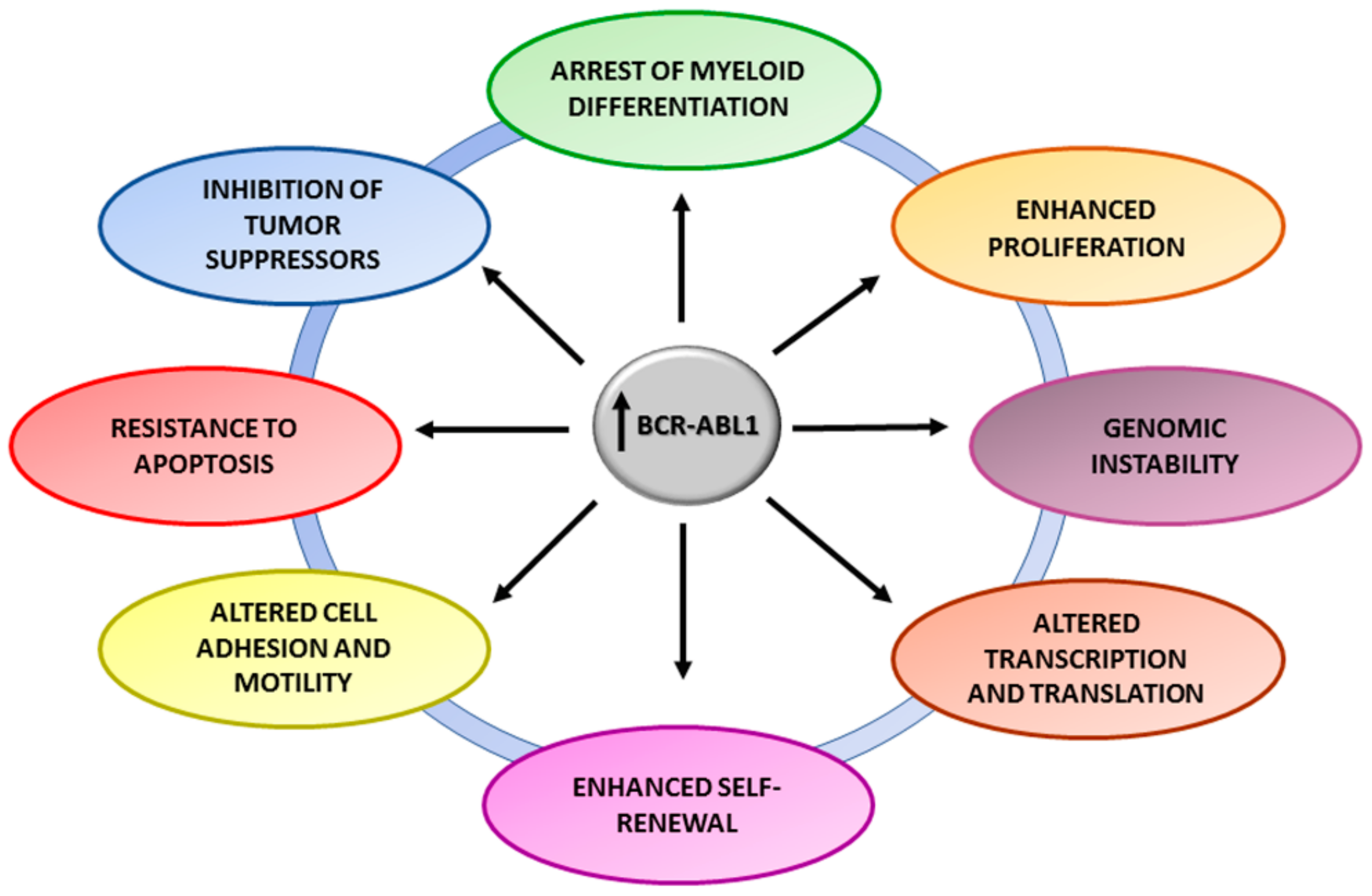
3. BCR-ABL1 and Genomic Instability
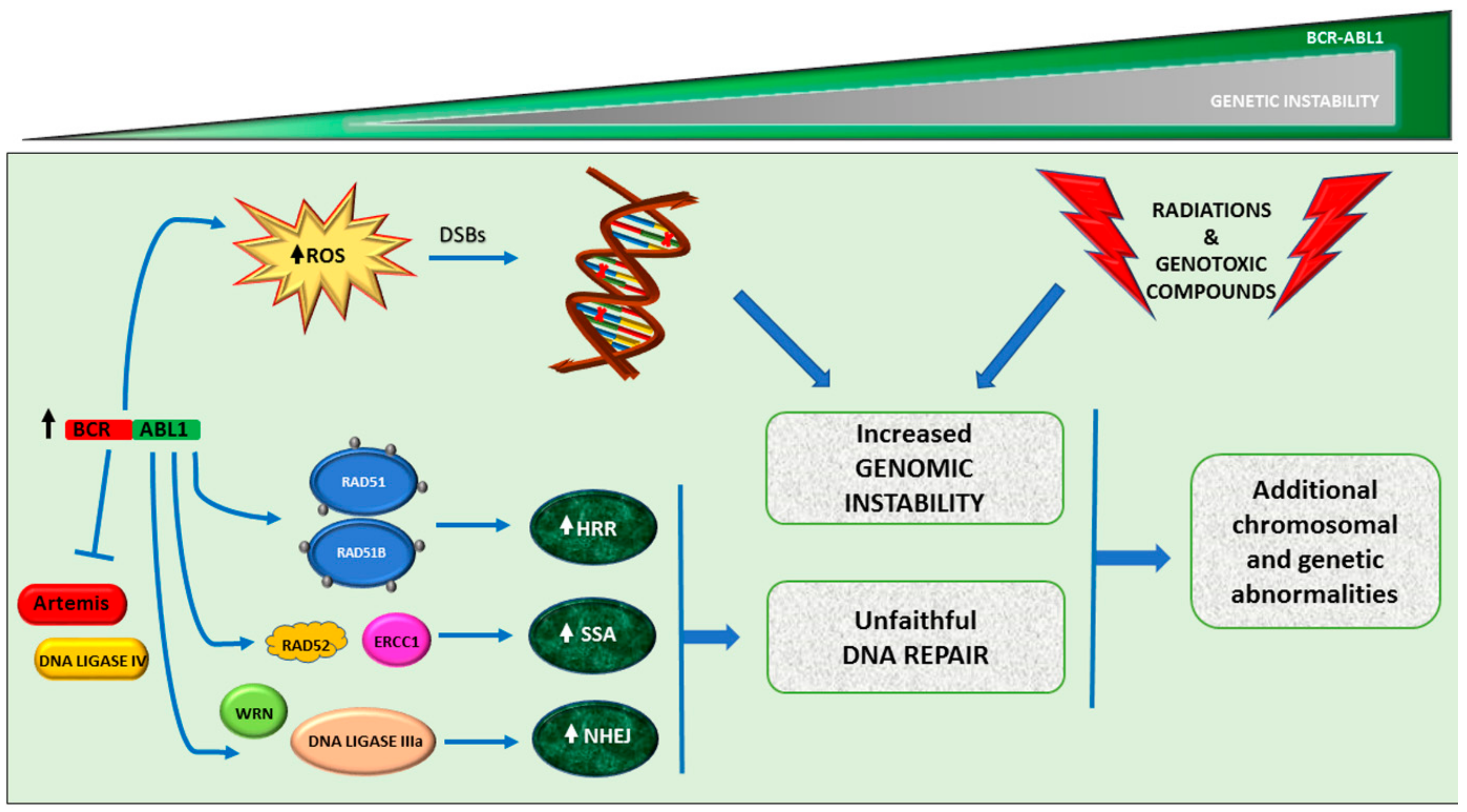
4. Additional Chromosomal Abnormalities
5. Gene Mutations and Submicroscopic Genetic Abnormalities
6. Non Genomic Loss of Tumor-Suppressor Function
7. Differentiation Arrest
8. Methylation Changes
9. Perturbations in RNA Transcription, Editing and Translation
10. The Role of MicroRNAs
11. Metabolic Changes
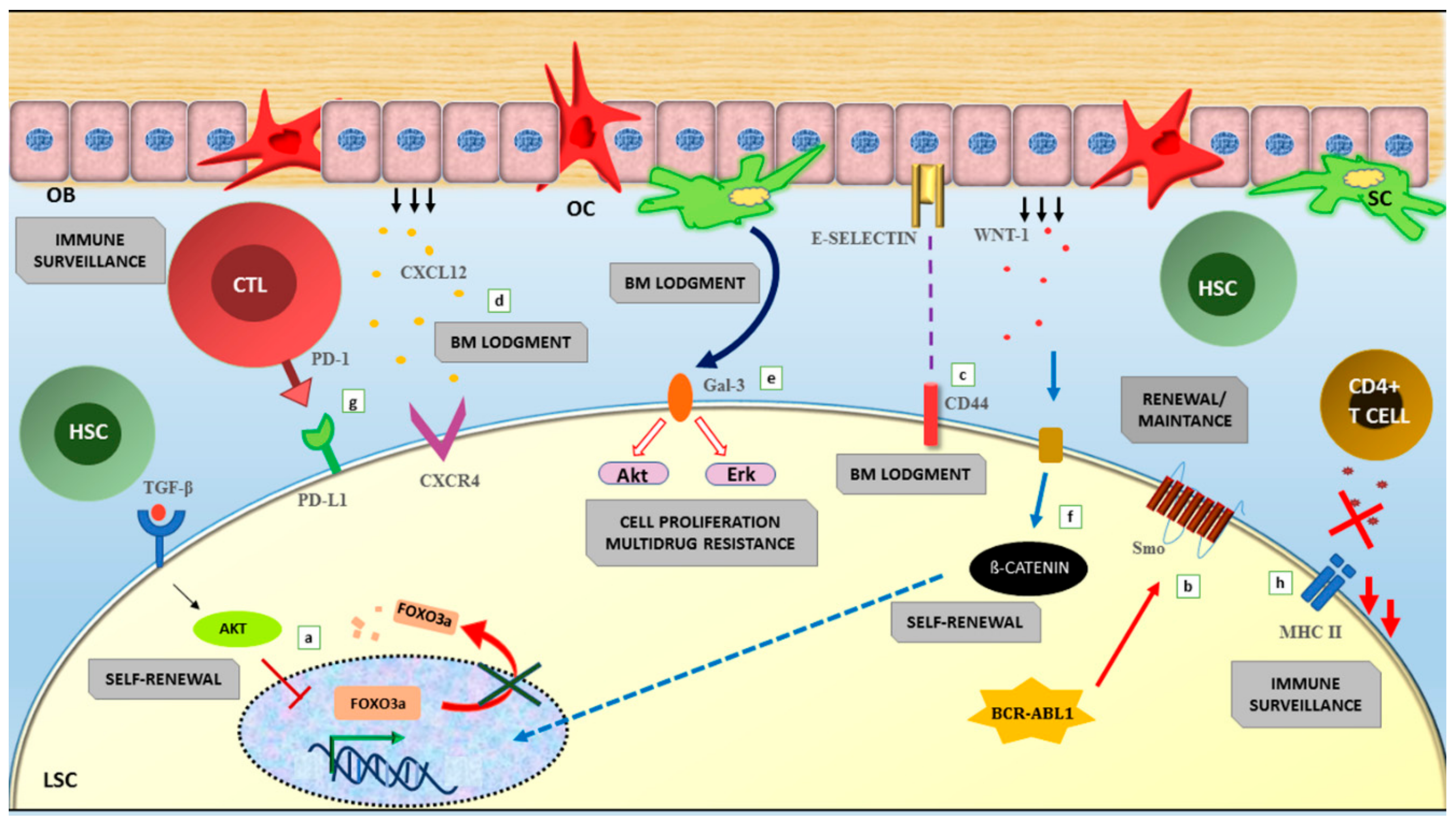
12. TKI Resistance and LSC Persistence: Two Sides of the Same Coin?
13. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Bruns, I.; Czibere, A.; Fischer, J.C.; Roels, F.; Cadeddu, R.P.; Buest, S.; Bruennert, D.; Huenerlituerkoglu, A.N.; Stoecklein, N.H.; Singh, R. The hematopoietic stem cell in chronic phase CML is characterized by a transcriptional profile resembling normal myeloid progenitor cells and reflecting loss of quiescence. Leukemia 2009, 23, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Ch’ang, H.-J.; Tien, H.-F.; Wang, C.-H.; Chuang, S.-M.; Chen, Y.C.; Shen, M.-C.; Lin, D.-T.; Lin, K.-H. Comparison of clinical and biologic features between myeloid and lymphoid transformation of Philadelphia chromosome positive chronic myeloid leukemia. Cancer Genet. Cytogenet. 1993, 71, 87–93. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris., N.L.; Stein, H.; Vardiman, J.W. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, WHO Classification of Tumours, 3rd ed.; IARC Press: Lyon, France, 2001; Volume 3. [Google Scholar]
- Reid, A.G.; De Melo, V.A.; Elderfield, K.; Clark, I.; Marin, D.; Apperley, J.; Naresh, K.N. Phenotype of blasts in chronic myeloid leukemia in blastic phase—Analysis of bone marrow trephine biopsies and correlation with cytogenetics. Leuk. Res. 2009, 33, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, D.M.; Longtine, J.A.; Fox, E.A.; Weinberg, D.S.; Pinkus, G.S. T-cell blast crisis in chronic myelogenous leukemia: Immunophenotypic and molecular biologic findings. Am. J. Clin. Pathol. 1997, 107, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, C.; Haferlach, T.; Kern, W.; Schnittger, S.; Berger, U.; Hehlmann, R.; Hiddemann, W.; Hochhaus, A. Occurrence of additional chromosome aberrations in chronic myeloid leukemia patients treated with imatinib mesylate. Leukemia 2003, 17, 461. [Google Scholar] [CrossRef] [Green Version]
- Gozzetti, A.; Bocchia, M.; Calabrese, S.; Pirrotta, M.T.; Crupi, R.; Raspadori, D.; Lauria, F. Promyelocytic blast crisis of chronic myelogenous leukemia during imatinib treatment. Acta Haematol. 2007, 117, 236–237. [Google Scholar] [CrossRef]
- Angriman, F.; Gutierrez Acevedo, M.N.; Rossi, M.S.; Gimenez Conca, A.D.; Otero, V.; Arbelbide, J.A.; Michelangelo, H. Promyelocytic Blastic Crisis in Chronic Myeloid Leukemia during Imatinib Treatment. Turk. J. Haematol. 2015, 32, 193–194. [Google Scholar] [CrossRef]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B. Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-naïve chronic myeloid leukemia patients trial. J. Clin. Oncol. 2016, 34, 2333. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; Le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; le Coutre, P. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: Results from the randomized BFORE trial. J. Clin. Oncol. 2018, 36, 231. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.M.; Ghorab, A.; Sasaki, K.; Jabbour, E.J.; Nogueras Gonzalez, G.; Kanagal-Shamanna, R.; Issa, G.C.; Garcia-Manero, G.; Dellasala, S.; et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer 2017, 123, 4391–4402. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.M.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A. Granulocyte–macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, Y.; Stuart, S.A.; Ikawa, T.; Jiang, Y.; Banno, A.; Hunton, I.C.; Young, D.J.; Naoe, T.; Murre, C.; Jamieson, C.H.M. BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17967–17972. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, F.; Sakata-Yanagimoto, M.; Komeno, Y.; Kato, N.; Uchida, T.; Haraguchi, K.; Kumano, K.; Harada, Y.; Harada, H.; Kitaura, J. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood 2010, 115, 2872–2881. [Google Scholar] [CrossRef] [Green Version]
- Wetzler, M.; Talpaz, M.; Van Etten, R.A.; Hirsh-Ginsberg, C.; Beran, M.; Kurzrock, R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J. Clin. Invest. 1993, 92, 1925–1939. [Google Scholar] [CrossRef] [Green Version]
- Gaiger, A.; Henn, T.; Horth, E.; Geissler, K.; Mitterbauer, G.; Maier-Dobersberger, T.; Greinix, H.; Mannhalter, C.; Haas, O.A.; Lechner, K. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood 1995, 86, 2371–2378. [Google Scholar] [CrossRef] [Green Version]
- Elmaagacli, A.H.; Beelen, D.W.; Opalka, B.; Seeber, S.; Schaefer, U.W. The amount of BCR-ABL fusion transcripts detected by the real-time quantitative polymerase chain reaction method in patients with Philadelphia chromosome positive chronic myeloid leukemia correlates with the disease stage. Ann. Hematol. 2000, 79, 424–431. [Google Scholar] [CrossRef]
- Hosoya, N.; Sanada, M.; Nannya, Y.; Nakazaki, K.; Wang, L.; Hangaishi, A.; Kurokawa, M.; Chiba, S.; Ogawa, S. Genomewide screening of DNA copy number changes in chronic myelogenous leukemia with the use of high-resolution array-based comparative genomic hybridization. Genes Chromosom. Cancer 2006, 45, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Brazma, D.; Grace, C.; Howard, J.; Melo, J.V.; Holyoke, T.; Apperley, J.F.; Nacheva, E.P. Genomic profile of chronic myelogenous leukemia: Imbalances associated with disease progression. Genes Chromosom. Cancer 2007, 46, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Ogawa, S.; Müschen, M.; Kato, M.; Kawamata, N.; Meixel, A.; Nowak, V.; Kim, H.S.; Kang, S.; Paquette, R. SNP array analysis of tyrosine kinase inhibitor-resistant chronic myeloid leukemia identifies heterogeneous secondary genomic alterations. Blood 2010, 115, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Nadarajan, V.S.; Phan, C.-L.; Ang, C.-H.; Liang, K.-L.; Gan, G.-G.; Bee, P.-C.; Zakaria, Z. Identification of copy number alterations by array comparative genomic hybridization in patients with late chronic or accelerated phase chronic myeloid leukemia treated with imatinib mesylate. Int. J. Hematol. 2011, 93, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-T.; Ji, Y.; Kim, H.-J.; Ki, C.-S.; Jung, C.W.; Kim, J.-W.; Kim, S.-H. Sequential array comparative genomic hybridization analysis identifies copy number changes during blastic transformation of chronic myeloid leukemia. Leuk. Res. 2012, 36, 418–421. [Google Scholar] [CrossRef]
- van der Sligte, N.E.; Krumbholz, M.; Pastorczak, A.; Scheijen, B.; Tauer, J.T.; Nowasz, C.; Sonneveld, E.; de Bock, G.H.; Meeuwsen-de Boer, T.G.J.; van Reijmersdal, S. DNA copy number alterations mark disease progression in paediatric chronic myeloid leukaemia. Br. J. Haematol. 2014, 166, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Antoszewska-Smith, J.; Pawlowska, E.; Błasiak, J. Reactive oxygen species in BCR-ABL1-expressing cells–relevance to chronic myeloid leukemia. Acta Biochim. Pol. 2017, 64, 1–10. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef] [Green Version]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species–dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef]
- Pastink, A.; Eeken, J.C.J.; Lohman, P.H.M. Genomic integrity and the repair of double-strand DNA breaks. Mutat. Res. Mol. Mech. Mutagen. 2001, 480, 37–50. [Google Scholar] [CrossRef]
- Deutsch, E.; Jarrousse, S.; Buet, D.; Dugray, A.; Bonnet, M.-L.; Vozenin-Brotons, M.-C.; Guilhot, F.; Turhan, A.G.; Feunteun, J.; Bourhis, J. Down-regulation of BRCA1 in BCR-ABL–expressing hematopoietic cells. Blood 2003, 101, 4583–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podszywalow-Bartnicka, P.; Wolczyk, M.; Kusio-Kobialka, M.; Wolanin, K.; Skowronek, K.; Nieborowska-Skorska, M.; Dasgupta, Y.; Skorski, T.; Piwocka, K. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014, 13, 3727–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dkhissi, F.; Aggoune, D.; Pontis, J.; Sorel, N.; Piccirilli, N.; LeCorf, A.; Guilhot, F.; Chomel, J.-C.; Ait-Si-Ali, S.; Turhan, A.G. The downregulation of BAP1 expression by BCR-ABL reduces the stability of BRCA1 in chronic myeloid leukemia. Exp. Hematol. 2015, 43, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallmyr, A.; Tomkinson, A.E.; Rassool, F.V. Up-regulation of WRN and DNA ligase IIIα in chronic myeloid leukemia: Consequences for the repair of DNA double-strand breaks. Blood 2008, 112, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Poplawski, T.; Jozwiakowski, S.K.; Cramer, K.; Pytel, D.; Stoczynska, E.; Nowicki, M.O.; Blasiak, J.; Skorski, T. BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res. 2011, 71, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Wada, C.; Shionoya, S.; Fujino, Y.; Tokuhiro, H.; Akahoshi, T.; Uchida, T.; Ohtani, H. Genomic instability of microsatellite repeats and its association with the evolution of chronic myelogenous leukemia [see comments]. Blood 1994, 83, 3449–3456. [Google Scholar] [CrossRef] [Green Version]
- Slupianek, A.; Falinski, R.; Znojek, P.; Stoklosa, T.; Flis, S.; Doneddu, V.; Pytel, D.; Synowiec, E.; Blasiak, J.; Bellacosa, A. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia 2013, 27, 629. [Google Scholar] [CrossRef] [Green Version]
- Alhuraiji, A.; Kantarjian, H.; Boddu, P.; Ravandi, F.; Borthakur, G.; DiNardo, C.; Daver, N.; Kadia, T.; Pemmaraju, N.; Pierce, S. Prognostic significance of additional chromosomal abnormalities at the time of diagnosis in patients with chronic myeloid leukemia treated with frontline tyrosine kinase inhibitors. Am. J. Hematol. 2018, 93, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Smith, T.L.; McCredie, K.B.; Keating, M.J.; Walters, R.S.; Talpaz, M.; Hester, J.P.; Bligham, G.; Gehan, E.; Freireich, E.J. Chronic myelogenous leukemia: A multivariate analysis of the associations of patient characteristics and therapy with survival. Blood 1985, 66, 1326–1335. [Google Scholar] [CrossRef] [Green Version]
- Sokal, J.E.; Gomez, G.A.; Baccarani, M.; Tura, S.; Clarkson, B.D.; Cervantes, F.; Rozman, C.; Carbonell, F.; Anger, B.; Heimpel, H. Prognostic significance of additional cytogenetic abnormalities at diagnosis of Philadelphia chromosome-positive chronic granulocytic leukemia. Blood 1988, 72, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Marktel, S.; Marin, D.; Foot, N.; Szydlo, R.; Bua, M.; Karadimitris, A.; De Melo, V.A.; Kotzampaltiris, P.; Dazzi, F.; Rahemtulla, A. Chronic myeloid leukemia in chronic phase responding to imatinib: The occurrence of additional cytogenetic abnormalities predicts disease progression. Haematologica 2003, 88, 260–267. [Google Scholar]
- Ishihara, T.; Sasaki, M.; Oshimura, M.; Kamada, N.; Yamada, K.; Okada, M.; Sakurai, M.; Sugiyama, T.; Shiraishi, Y.; Kohno, S.; et al. A summary of cytogenetic studies on 534 cases of chronic myelocytic leukemia in Japan. Cancer Genet. Cytogenet. 1983, 9, 81–91. [Google Scholar] [CrossRef]
- Swolin, B.; Weinfeld, A.; Westin, J.; Waldenström, J.; Magnusson, B. Karyotypic evolution in Ph-positive chronic myeloid leukemia in relation to management and disease progression. Cancer Genet. Cytogenet. 1985, 18, 65–79. [Google Scholar] [CrossRef]
- Krulik, M.; Smadja, N.; De Gramont, A.; Gonzalez-Canali, G.; Audebert, A.A.; Dray, C.; Brissaud, P.; Debray, J. Sequential karyotype study on Ph-positive chronic myelocytic leukemia. Significance of additional chromosomal abnormalities during disease evolution. Cancer Genet. Cytogenet. 1987, 60, 974–979. [Google Scholar] [CrossRef]
- Mitelman, F.; Levan, G.; Nilsson, P.G.; Brandt, L. Non-random karyotypic evolution in chronic myeloid leukemia. Int. J. Cancer 1976, 18, 24–30. [Google Scholar] [CrossRef]
- Mitelman, F. The cytogenetic scenario of chronic myeloid leukemia. Leuk. Lymphoma 1993, 11, 11–15. [Google Scholar] [CrossRef]
- Cortes, J.E.; Talpaz, M.; Giles, F.; O’Brien, S.; Rios, M.B.; Shan, J.; Garcia-Manero, G.; Faderl, S.; Thomas, D.A.; Wierda, W. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood 2003, 101, 3794–3800. [Google Scholar] [CrossRef]
- Lippert, E.; Etienne, G.; Mozziconacci, M.-J.; Laibe, S.; Gervais, C.; Girault, S.; Gachard, N.; Tigaud, I.; Dastugue, N.; Huguet, F. Loss of the Y chromosome in Philadelphia-positive cells predicts a poor response of chronic myeloid leukemia patients to imatinib mesylate therapy. Haematologica 2010, 95, 1604–1607. [Google Scholar] [CrossRef]
- Luatti, S.; Castagnetti, F.; Marzocchi, G.; Baldazzi, C.; Gugliotta, G.; Iacobucci, I.; Specchia, G.; Zanatta, L.; Rege-Cambrin, G.; Mancini, M. Additional chromosomal abnormalities in Philadelphia-positive clone: Adverse prognostic influence on frontline imatinib therapy: A GIMEMA Working Party on CML analysis. Blood 2012, 120, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Leitner, A.; Hochhaus, A.; Müller, M.C.; Hanfstein, B.; Haferlach, C.; Göhring, G.; Schlegelberger, B.; Jotterand, M.; Reiter, A. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: Long-term observation of 1151 patients from the randomized CML Study IV. Blood 2011, 118, 6760–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cortes, J.E.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, R.K.; Geetha, N.; Sakthivel, K.M.; Aswathy, C.G.; Gopinath, P.; Raj, T.V.A.; Priya, G.; Nair, J.K.K.M.; Sreedharan, H. Genomic amplification of BCR-ABL1 fusion gene and its impact on the disease progression mechanism in patients with chronic myelogenous leukemia. Gene 2019, 686, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Fioretos, T.; Billström, R.; Mitelman, F. Aberrant cytogenetic evolution pattern of Philadelphia-positive chronic myeloid leukemia treated with interferon-α. Leukemia 1996, 10, 1134–1138. [Google Scholar]
- Serra, A.; Gottardi, E.; Della Ragione, F.; Saglio, G.; Iolascon, A. Involvement of the cyclin-dependent kinase-4 inhibitor (CDKN2) gene in the pathogenesis of lymphoid blast crisis of chronic myelogenous leukaemia. Br. J. Haematol. 1995, 91, 625–629. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.; Jones, D.; Talpaz, M.; Bekele, N.; O’brien, S.; Zhou, X.; Luthra, R.; Garcia-Manero, G.; Giles, F. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia 2006, 20, 1767. [Google Scholar] [CrossRef]
- Soverini, S.; Colarossi, S.; Gnani, A.; Rosti, G.; Castagnetti, F.; Poerio, A.; Iacobucci, I.; Amabile, M.; Abruzzese, E.; Orlandi, E. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: By the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 7374–7379. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.P.; Skaggs, B.J.; Branford, S.; Hughes, T.P.; Nicoll, J.M.; Paquette, R.L.; Sawyers, C.L. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J. Clin. Invest. 2007, 117, 2562–2569. [Google Scholar] [CrossRef] [Green Version]
- Stagno, F.; Stella, S.; Berretta, S.; Massimino, M.; Antolino, A.; Giustolisi, R.; Messina, A.; Di Raimondo, F.; Vigneri, P. Sequential mutations causing resistance to both Imatinib Mesylate and Dasatinib in a chronic myeloid leukaemia patient progressing to lymphoid blast crisis. Leuk. Res. 2008, 32, 673–674. [Google Scholar] [CrossRef]
- Khorashad, J.S.; Kelley, T.W.; Szankasi, P.; Mason, C.C.; Soverini, S.; Adrian, L.T.; Eide, C.A.; Zabriskie, M.S.; Lange, T.; Estrada, J.C. BCR-ABL1 compound mutations in tyrosine kinase inhibitor–resistant CML: Frequency and clonal relationships. Blood 2013, 121, 489–498. [Google Scholar] [CrossRef]
- Soverini, S.; Bavaro, L.; Martelli, M.; De Benedittis, C.; Iurlo, A.; Orofino, N.; Pagano, L.; Criscuolo, M.; Bonifacio, M.; Scaffidi, L. Compound BCR-ABL1 Kinase Domain Mutants: Prevalence, Spectrum and Correlation with Tyrosine Kinase Inhibitor Resistance in a Prospective Series of Philadelphia Chromosome-Positive Leukemia Patients Analyzed By Next Generation Sequencing. Blood 2018, 132, 789. [Google Scholar] [CrossRef]
- Zabriskie, M.S.; Eide, C.A.; Tantravahi, S.K.; Vellore, N.A.; Estrada, J.; Nicolini, F.E.; Khoury, H.J.; Larson, R.A.; Konopleva, M.; Cortes, J.E. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014, 26, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, H.; Bar-Eli, M.; Advani, S.H.; Benchimol, S.; Cline, M.J. Alterations in the p53 gene and the clonal evolution of the blast crisis of chronic myelocytic leukemia. Proc. Natl. Acad. Sci. USA 1989, 86, 6783–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinstein, E.; Cimino, G.; Gale, R.P.; Alimena, G.; Berthier, R.; Kishi, K.; Goldman, J.; Zaccaria, A.; Berrebi, A.; Canaani, E. p53 in chronic myelogenous leukemia in acute phase. Proc. Natl. Acad. Sci. USA 1991, 88, 6293–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorski, T.; Nieborowska-Skorska, M.; Wlodarski, P.; Perrotti, D.; Martinez, R.; Wasik, M.A.; Calabretta, B. Blastic transformation of p53-deficient bone marrow cells by p210bcr/abl tyrosine kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13137–13142. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.; Ushijima, T.; Wakazono, K.; Oda, H.; Tanaka, Y.; Aizawa, S.; Ishikawa, T.; Yazaki, Y.; Hirai, H. Acquired loss of p53 induces blastic transformation in p210 bcr/abl-expressing hematopoietic cells: A transgenic study for blast crisis of human CML. Blood 2000, 95, 1144–1150. [Google Scholar] [CrossRef]
- Wendel, H.-G.; de Stanchina, E.; Cepero, E.; Ray, S.; Emig, M.; Fridman, J.S.; Veach, D.R.; Bornmann, W.G.; Clarkson, B.; McCombie, W.R. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc. Natl. Acad. Sci. USA 2006, 103, 7444–7449. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, H.G.; Jat, P.S.; Foti, A.; Bar-Eli, M.; Cline, M.J. Abnormalities of the retinoblastoma gene in the pathogenesis of acute leukemia. Blood 1991, 78, 3259–3268. [Google Scholar] [CrossRef]
- Beck, Z.; Kiss, A.; Tóth, F.D.; Szabó, J.; Bácsi, A.; Balogh, E.; Borbély, Á.; Telek, B.; Kovács, E.; Oláh, É. Alterations of P53 and RB genes and the evolution of the accelerated phase of chronic myeloid leukemia. Leuk. Lymphoma 2000, 38, 587–597. [Google Scholar] [CrossRef]
- Roche-Lestienne, C.; Deluche, L.; Corm, S.; Tigaud, I.; Joha, S.; Philippe, N.; Geffroy, S.; Laï, J.-L.; Nicolini, F.-E.; Preudhomme, C. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood 2008, 111, 3735–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.-J.; Wang, Y.-Y.; Li, G.; Ma, L.-Y.; Xiong, S.-M.; Weng, X.-Q.; Zhang, W.-N.; Wu, B.; Chen, Z.; Chen, S.-J. Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood 2012, 119, 2873–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, C.M.; Larson, R.A.; Anastasi, J.; Winter, J.N.; Thangavelu, M.; Vardiman, J.W.; Rowley, J.D.; Le Beau, M.M. t (3; 21)(q26; q22): A recurring chromosomal abnormality in therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990, 76, 2594–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucifora, G.; Begy, C.R.; Kobayashi, H.; Roulston, D.; Claxton, D.; Pedersen-Bjergaard, J.; Parganas, E.; Ihle, J.N.; Rowley, J.D. Consistent intergenic splicing and production of multiple transcripts between AML1 at 21q22 and unrelated genes at 3q26 in (3; 21)(q26; q22) translocations. Proc. Natl. Acad. Sci. USA 1994, 91, 4004–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucifora, G.; Rowley, J.D. AML1 and the 8; 21 and 3; 21 translocations in acute and chronic myeloid leukemia. Blood 1995, 86, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lugthart, S.; Gröschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.M.; van Zelderen-Bhola, S.L.; Jan Ossenkoppele, G.; Vellenga, E.; van den Berg-de Ruiter, E.; Schanz, U. Clinical, molecular, and prognostic significance of WHO type inv (3)(q21q26. 2)/t (3; 3)(q21; q26. 2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef]
- Nukina, A.; Kagoya, Y.; Watanabe-Okochi, N.; Arai, S.; Ueda, K.; Yoshimi, A.; Nannya, Y.; Kurokawa, M. Single-cell gene expression analysis reveals clonal architecture of blast-phase chronic myeloid leukaemia. Br. J. Haematol. 2014, 165, 414–416. [Google Scholar] [CrossRef]
- Goyama, S.; Yamamoto, G.; Shimabe, M.; Sato, T.; Ichikawa, M.; Ogawa, S.; Chiba, S.; Kurokawa, M. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 2008, 3, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Zhu, J.; Chen, F.; Lin, W.; Cai, J.; Zhong, J.; Zhong, H. RUNX1-Evi-1 fusion gene inhibited differentiation and apoptosis in myelopoiesis: An in vivo study. BMC Cancer 2015, 15, 970. [Google Scholar] [CrossRef] [Green Version]
- Sill, H.; Goldman, J.M.; Cross, N.C. Homozygous deletions of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood 1995, 85, 2013–2016. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Boluda, J.-C.; Cervantes, F.; Colomer, D.; Vela, M.-C.; Costa, D.; Paz, M.-F.; Esteller, M.; Montserrat, E. Genomic p16 abnormalities in the progression of chronic myeloid leukemia into blast crisis: A sequential study in 42 patients. Exp. Hematol. 2003, 31, 204–210. [Google Scholar] [CrossRef]
- Williams, R.T.; Sherr, C.J. The INK4-ARF (CDKN2A/B) Locus in Hematopoiesis and BCR-ABL–induced Leukemias. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2008; Volume 73, pp. 461–467. [Google Scholar]
- Nacheva, E.P.; Brazma, D.; Virgili, A.; Howard-Reeves, J.; Chanalaris, A.; Gancheva, K.; Apostolova, M.; Valgañon, M.; Mazzullo, H.; Grace, C. Deletions of immunoglobulin heavy chain and T cell receptor gene regions are uniquely associated with lymphoid blast transformation of chronic myeloid leukemia. BMC Genomics 2010, 11, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.; Jung, C.W.; Kim, J.W.; Kim, H.-J.; Kim, S.-H.; Shin, M.G.; Kim, Y.K.; Kim, H.J.; Suh, J.S.; Moon, J.H. Genome-wide high density single-nucleotide polymorphism array-based karyotyping improves detection of clonal aberrations including der (9) deletion, but does not predict treatment outcomes after imatinib therapy in chronic myeloid leukemia. Ann. Hematol. 2011, 90, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.-H. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110. [Google Scholar] [CrossRef]
- Georgopoulos, K.; Moore, D.D.; Derfler, B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science 1992, 258, 808–812. [Google Scholar] [CrossRef]
- Molnár, Á.; Wu, P.; Largespada, D.A.; Vortkamp, A.; Scherer, S.; Copeland, N.G.; Jenkins, N.A.; Bruns, G.; Georgopoulos, K. The Ikaros gene encodes a family of lymphocyte-restricted zinc finger DNA binding proteins, highly conserved in human and mouse. J. Immunol. 1996, 156, 585–592. [Google Scholar]
- Sun, L.; Goodman, P.A.; Wood, C.M.; Crotty, M.-L.; Sensel, M.; Sather, H.; Navara, C.; Nachman, J.; Steinherz, P.G.; Gaynon, P.S. Expression of aberrantly spliced oncogenic ikaros isoforms in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 1999, 17, 3753–3766. [Google Scholar] [CrossRef]
- Iacobucci, I.; Lonetti, A.; Messa, F.; Cilloni, D.; Arruga, F.; Ottaviani, E.; Paolini, S.; Papayannidis, C.; Piccaluga, P.P.; Giannoulia, P. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: Implications for a new mechanism of resistance. Blood 2008, 112, 3847–3855. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Howarth, A.; Clark, R.E. Ikaros transcripts Ik6/10 and levels of full-length transcript are critical for chronic myeloid leukaemia blast crisis transformation. Leukemia 2014, 28, 1745. [Google Scholar] [CrossRef]
- Soverini, S.; Score, J.; Iacobucci, I.; Poerio, A.; Lonetti, A.; Gnani, A.; Colarossi, S.; Ferrari, A.; Castagnetti, F.; Rosti, G. IDH2 somatic mutations in chronic myeloid leukemia patients in blast crisis. Leukemia 2011, 25, 178. [Google Scholar] [CrossRef]
- Grossmann, V.; Kohlmann, A.; Zenger, M.; Schindela, S.; Eder, C.; Weissmann, S.; Schnittger, S.; Kern, W.; Müller, M.C.; Hochhaus, A. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 2011, 25, 557. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Jankowska, A.M.; McDevitt, M.A.; O’Keefe, C.; Dujardin, S.; Cazzolli, H.; Przychodzen, B.; Prince, C.; Nicoll, J.; Siddaiah, H. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 2011, 117, e198–e206. [Google Scholar] [CrossRef] [PubMed]
- Magistroni, V.; Mauri, M.; D’Aliberti, D.; Mezzatesta, C.; Crespiatico, I.; Nava, M.; Fontana, D.; Sharma, N.; Parker, W.; Schreiber, A. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica 2019, 2017, 179937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Perrotti, D.; Cesi, V.; Trotta, R.; Guerzoni, C.; Santilli, G.; Campbell, K.; Iervolino, A.; Condorelli, F.; Gambacorti-Passerini, C.; Caligiuri, M.A. BCR-ABL suppresses C/EBPα expression through inhibitory action of hnRNP E2. Nat. Genet. 2002, 30, 48. [Google Scholar] [CrossRef]
- Radomska, H.S.; Huettner, C.S.; Zhang, P.U.; Cheng, T.A.O.; Scadden, D.T.; Tenen, D.G. CCAAT/enhancer binding protein α is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell. Biol. 1998, 18, 4301–4314. [Google Scholar] [CrossRef] [Green Version]
- Notari, M.; Neviani, P.; Santhanam, R.; Blaser, B.W.; Chang, J.-S.; Galietta, A.; Willis, A.E.; Roy, D.C.; Caligiuri, M.A.; Marcucci, G. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006, 107, 2507–2516. [Google Scholar] [CrossRef]
- Chang, J.S.; Santhanam, R.; Trotta, R.; Neviani, P.; Eiring, A.M.; Briercheck, E.; Ronchetti, M.; Roy, D.C.; Calabretta, B.; Caligiuri, M.A. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2–dependent suppression of C/EBPα-driven myeloid differentiation. Blood 2007, 110, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010, 140, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelkin, B.D.; Przepiorka, D.; Burke, P.J.; Thomas, E.D.; Baylin, S.B. Abnormal methylation of the calcitonin gene marks progression of chronic myelogenous leukemia. Blood 1991, 77, 2431–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.-P.J.; Zehnbauer, B.A.; Civin, C.I.; Collector, M.I.; Sharkis, S.J.; Davidson, N.E.; Kaufmann, S.H.; Baylin, S.B. The estrogen receptor CpG island is methylated in most hematopoietic neoplasms. Cancer Res. 1996, 56, 973–977. [Google Scholar] [PubMed]
- Issa, J.-P.J.; Zehnbauer, B.A.; Kaufmann, S.H.; Biel, M.A.; Baylin, S.B. HIC1 hypermethylation is a late event in hematopoietic neoplasms. Cancer Res. 1997, 57, 1678–1681. [Google Scholar]
- Nguyen, T.T.; Mohrbacher, A.F.; Tsai, Y.C.; Groffen, J.; Heisterkamp, N.; Nichols, P.W.; Mimi, C.Y.; Lübbert, M.; Jones, P.A. Quantitative measure of c-abl andp15 methylation in chronic myelogenous leukemia: Biological implications. Blood 2000, 95, 2990–2992. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Castillejo, J.A.; Jimenez, A.; Cervantes, F.; Boque, C.; Hermosin, L.; Leon, A.; Grañena, A.; Colomer, D.; Heiniger, A. Cadherin-13, a mediator of calcium-dependent cell-cell adhesion, is silenced by methylation in chronic myeloid leukemia and correlates with pretreatment risk profile and cytogenetic response to interferon alfa. J. Clin. Oncol. 2003, 21, 1472–1479. [Google Scholar] [CrossRef]
- Strathdee, G.; Holyoake, T.L.; Sim, A.; Parker, A.; Oscier, D.G.; Melo, J.V.; Meyer, S.; Eden, T.; Dickinson, A.M.; Mountford, J.C. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin. Cancer Res. 2007, 13, 5048–5055. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, J.; Gharibyan, V.; Estecio, M.R.H.; Kondo, K.; He, R.; Chung, W.; Lu, Y.; Zhang, N.; Liang, S.; Kantarjian, H.M. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS ONE 2011, 6, e22110. [Google Scholar] [CrossRef]
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861. [Google Scholar] [CrossRef] [Green Version]
- Trotta, R.; Vignudelli, T.; Candini, O.; Intine, R.V.; Pecorari, L.; Guerzoni, C.; Santilli, G.; Byrom, M.W.; Goldoni, S.; Ford, L.P. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 2003, 3, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Perrotti, D.; Bonatti, S.; Trotta, R.; Martinez, R.; Skorski, T.; Salomoni, P.; Grassilli, E.; Iozzo, R.V.; Cooper, D.R.; Calabretta, B. TLS/FUS, a pro-oncogene involved in multiple chromosomal translocations, is a novel regulator of BCR/ABL-mediated leukemogenesis. EMBO J. 1998, 17, 4442–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerzoni, C.; Bardini, M.; Mariani, S.A.; Ferrari-Amorotti, G.; Neviani, P.; Panno, M.L.; Zhang, Y.; Martinez, R.; Perrotti, D.; Calabretta, B. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood 2006, 107, 4080–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiring, A.M.; Neviani, P.; Santhanam, R.; Oaks, J.J.; Chang, J.S.; Notari, M.; Willis, W.; Gambacorti-Passerini, C.; Volinia, S.; Marcucci, G. Identification of novel posttranscriptional targets of the BCR/ABL oncoprotein by ribonomics: Requirement of E2F3 for BCR/ABL leukemogenesis. Blood 2008, 111, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Ly, C.; Arechiga, A.F.; Melo, J.V.; Walsh, C.M.; Ong, S.T. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. Cancer Res. 2003, 63, 5716–5722. [Google Scholar]
- Perrotti, D.; Turturro, F.; Neviani, P. BCR/ABL, mRNA translation and apoptosis. Cell Death Differ. 2005, 12, 534–540. [Google Scholar] [CrossRef]
- Jiang, Q.; Crews, L.A.; Barrett, C.L.; Chun, H.-J.; Isquith, J.M.; Zipeto, M.A.; Goff, D.J.; Minden, M.; Sadarangani, A.; Rusert, J.M. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 1041–1046. [Google Scholar] [CrossRef] [Green Version]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Santos, N.P.D.; Balaian, L.; Chun, H.-J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Crews, L.A.; Jiang, Q.; Zipeto, M.A.; Lazzari, E.; Ali, S.; Barrett, C.L.; Frazer, K.A.; Jamieson, C.H.M. An RNA editing fingerprint of cancer stem cell reprogramming. J. Transl. Med. 2015, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Fabbri, M.; Cimmino, A.; Calin, G.A.; Croce, C.M. MicroRNA expression and function in cancer. Trends Mol. Med. 2006, 12, 580–587. [Google Scholar] [CrossRef]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25. [Google Scholar] [CrossRef]
- Poláková, K.M.; Lopotová, T.; Klamová, H.; Burda, P.; Trněný, M.; Stopka, T.; Moravcová, J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol. Cancer 2011, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Srutova, K.; Curik, N.; Burda, P.; Savvulidi, F.; Silvestri, G.; Trotta, R.; Klamova, H.; Pecherkova, P.; Sovova, Z.; Koblihova, J. BCR-ABL1 mediated miR-150 downregulation through MYC contributed to myeloid differentiation block and drug resistance in chronic myeloid leukemia. Haematologica 2018, 103, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kwon, H.Y.; Zimdahl, B.; Congdon, K.L.; Blum, J.; Lento, W.E.; Zhao, C.; Lagoo, A.; Gerrard, G.; Foroni, L. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature 2010, 466, 765. [Google Scholar] [CrossRef] [Green Version]
- Kharas, M.G.; Lengner, C.J.; Al-Shahrour, F.; Bullinger, L.; Ball, B.; Zaidi, S.; Morgan, K.; Tam, W.; Paktinat, M.; Okabe, R. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med. 2010, 16, 903. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.; Deininger, M.; Hochhaus, A.; Branford, S.; Radich, J.; Kaeda, J.; Baccarani, M.; Cortes, J.; Cross, N.C.P.; Druker, B.J. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: Review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006, 108, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.P.; Kaeda, J.; Branford, S.; Rudzki, Z.; Hochhaus, A.; Hensley, M.L.; Gathmann, I.; Bolton, A.E.; Van Hoomissen, I.C.; Goldman, J.M. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2003, 349, 1423–1432. [Google Scholar] [CrossRef]
- Patel, A.B.; O’Hare, T.; Deininger, M.W. Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol. Clin. 2017, 31, 589–612. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, D.N.M.; Jiang, X.; Brandwein, J.M.; Valencia-Serna, J.; Remant, K.C.; Uludağ, H. Current outlook on drug resistance in chronic myeloid leukemia (CML) and potential therapeutic options. Drug Discov. Today 2019. [Google Scholar]
- Burchert, A.; Wang, Y.; Cai, D.; Von Bubnoff, N.; Paschka, P.; Müller-Brüsselbach, S.; Ottmann, O.G.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 2005, 19, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Meng, F.; Lu, H.; Kong, L.; Bornmann, W.; Peng, Z.; Talpaz, M.; Donato, N.J. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood 2008, 111, 3821–3829. [Google Scholar] [CrossRef] [Green Version]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. J. Clin. Invest. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Esposito, N.; Colavita, I.; Quintarelli, C.; Sica, A.R.; Peluso, A.L.; Luciano, L.; Picardi, M.; Del Vecchio, L.; Buonomo, T.; Hughes, T.P. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome–positive cells derived from patients with chronic myeloid leukemia. Blood 2011, 118, 3634–3644. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J. A therapeutically targetable mechanism of BCR-ABL–independent imatinib resistance in chronic myeloid leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef] [Green Version]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586. [Google Scholar] [CrossRef] [Green Version]
- Khorashad, J.S.; Eiring, A.M.; Mason, C.C.; Gantz, K.C.; Bowler, A.D.; Redwine, H.M.; Yu, F.; Kraft, I.L.; Pomicter, A.D.; Reynolds, K.R. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood 2015, 125, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.J.; Joshua, J.O.; Ramasamy, S.; Paolo, N.; J, G.H.; Gregory, F.; Justin, J.E.; Yosef, L.; Ann-Kathrin, E.; Nash, Y.G. Preclinical and Clinical Efficacy of XPO1/CRM1 Inhibition by the Karyopherin Inhibitor KPT-330 in Ph+ Leukemias. Blood 2013, 122, 3034–3044. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.N.; Leng, X.; Perazzona, B.; Sun, X.; Lin, Y.-H.; Arlinghaus, R.B. Combination of jak2 and hsp90 inhibitors: An effective therapeutic option in drug-resistant chronic myelogenous leukemia. Genes Cancer 2016, 7, 201. [Google Scholar] [PubMed]
- Wagle, M.; Eiring, A.M.; Wongchenko, M.; Lu, S.; Guan, Y.; Wang, Y.; Lackner, M.; Amler, L.; Hampton, G.; Deininger, M.W. A role for FOXO1 in BCR–ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia 2016, 30, 1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.M.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of β-catenin and Bcr–Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065. [Google Scholar] [CrossRef]
- Eiring, A.M.; Jonathan, A.; Clinton, C.M.; Ethan, D.; Thomas, H.; Michael, W.D. Loss of G0S2 in Kinase-Independent TKI Resistance and Blastic Transformation of CML. Am Soc. Hematol. 2017, 4173. [Google Scholar]
- Zhu, Y.; Lu, L.; Qiao, C.; Shan, Y.; Li, H.; Qian, S.; Hong, M.; Zhao, H.; Li, J.; Yang, Z. Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor. Oncogene 2018, 37, 2837. [Google Scholar] [CrossRef]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef] [Green Version]
- Chomel, J.-C.; Bonnet, M.-L.; Sorel, N.; Bertrand, A.; Meunier, M.-C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Sloma, I.; Bennaceur-Griscelli, A.; Rea, D.; Legros, L.; Marfaing-Koka, A.; Bourhis, J.-H.; Ame, S. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget 2016, 7, 35293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocchia, M.; Sicuranza, A.; Abruzzese, E.; Iurlo, A.; Sirianni, S.; Gozzini, A.; Galimberti, S.; Aprile, L.; Martino, B.; Pregno, P. Residual peripheral blood CD26+ leukemic stem cells in chronic myeloid leukemia patients during TKI therapy and during treatment-free remission. Front. Oncol. 2018, 30, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [Green Version]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest. 2011, 121, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Müller-Tidow, C.; Bhatia, R.; Brunton, V.G. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Invest. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Modi, H.; McDonald, T.; Chu, S.; Yee, J.-K.; Forman, S.J.; Bhatia, R. Role of BCR/ABL gene-expression levels in determining the phenotype and imatinib sensitivity of transformed human hematopoietic cells. Blood 2007, 109, 5411–5421. [Google Scholar] [CrossRef]
- Kumari, A.; Brendel, C.; Hochhaus, A.; Neubauer, A.; Burchert, A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood 2012, 119, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β–FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.M.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.P.; Whetton, A.D. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Krause, D.S.; Lazarides, K.; von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL–expressing leukemic stem cells. Nat. Med. 2006, 12, 1175. [Google Scholar] [CrossRef]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto-Sugitani, M.; Kuroda, J.; Ashihara, E.; Nagoshi, H.; Kobayashi, T.; Matsumoto, Y.; Sasaki, N.; Shimura, Y.; Kiyota, M.; Nakayama, R. Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 2011, 108, 17468–17473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Mumprecht, S.; Schürch, C.; Schwaller, J.; Solenthaler, M.; Ochsenbein, A.F. Programmed death 1 signaling on chronic myeloid leukemia–specific T cells results in T-cell exhaustion and disease progression. Blood 2009, 114, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riether, C.; Schürch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015, 22, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarafdar, A.; Hopcroft, L.E.M.; Gallipoli, P.; Pellicano, F.; Cassels, J.; Hair, A.; Korfi, K.; Jørgensen, H.G.; Vetrie, D.; Holyoake, T.L. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 2017, 129, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Marum, J.E.; Wang, P.P.S.; Stangl, D.; Yeung, D.T.; Mueller, M.C.; Dietz, C.T.; Walker, I.; Nataren, N.; Donaldson, Z.; Parker, W.T. Novel fusion genes at CML diagnosis reveal a complex pattern of genomic rearrangements and sequence inversions associated with the Philadelphia chromosome in patients with early blast crisis. Blood 2016, 128, 1219. [Google Scholar] [CrossRef]
- Marum, J.E.; Yeung, D.T.; Purins, L.; Reynolds, J.; Parker, W.T.; Stangl, D.; Wang, P.P.S.; Price, D.J.; Tuke, J.; Schreiber, A.W. ASXL1 and BIM germ line variants predict response and identify CML patients with the greatest risk of imatinib failure. Blood Adv. 2017, 1, 1369–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Copland, M.; Slupsky, J.R.; Clark, R.E. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood 2011, 117, 6660–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2017, 123, 1791–1799. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Pathway | Ref | Druggable? |
|---|---|---|
| PI3K/AKT/mTOR | Burchert, Leukemia 2005 [135] | PI3K/mTOR inhibitors |
| Lyn | Wu et al., Blood 2008 [136] | Src inhibitors |
| Autophagy | Bellodi et al., J Clin Invest 2009 [137] | hydroxychloroquine |
| SHP-1 | Esposito et al., Blood 2011 [138] | X |
| SIRT1 | Wang et al., Oncogene 2013 [139] | selisistat |
| PRKCH | Ma et al., Sci Transl Med 2014 [140] | MEK inh (trametinib) + Imatinib |
| STAT3 | Eiring et al., Leukemia 2015 [141] | BP-5-087 |
| CRM1/XPO1/RAN | Khorashad et al., Blood 2015 [142] Walker et al., Blood 2016 [143] | selinexor |
| JAK2 | Chakraborty et al., Genes Cancer 2016 [144] | ruxolitinib |
| FOXO1 | Wagle et al., Leukemia 2016 [145] | PI3K inhibitors |
| EZH2 | Scott et al., Cancer Discov 2016 [146] | EZH2 inhibitors |
| Wnt/b catenin | Eiring et al., Leukemia 2015 [141], Zhou et al., Leukemia 2017 [147] | C82 |
| MS4A3 | Eiring et al., ASH 2017 [148] | X |
| PFKFB3 | Zhu et al., Oncogene 2018 [149] | PFK-158 |
| Various miRNAs | X |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bavaro, L.; Martelli, M.; Cavo, M.; Soverini, S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. Int. J. Mol. Sci. 2019, 20, 6141. https://doi.org/10.3390/ijms20246141
Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. International Journal of Molecular Sciences. 2019; 20(24):6141. https://doi.org/10.3390/ijms20246141
Chicago/Turabian StyleBavaro, Luana, Margherita Martelli, Michele Cavo, and Simona Soverini. 2019. "Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update" International Journal of Molecular Sciences 20, no. 24: 6141. https://doi.org/10.3390/ijms20246141