Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate


Department of Neurology, National Cerebral and Cardiovascular Center, Suita 564-8565, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(24), 6176; https://doi.org/10.3390/ijms20246176
Submission received: 2 October 2019
/
Revised: 17 November 2019
/
Accepted: 3 December 2019
/
Published: 7 December 2019
(This article belongs to the Special Issue Vascular Cognitive Impairment: From Molecular Mechanisms to Preventive and Therapeutic Strategies)
Abstract
:Vascular cognitive impairment (VCI) or vascular dementia occurs as a result of brain ischemia and represents the second most common type of dementia after Alzheimer’s disease. To explore the underlying mechanisms of VCI, several animal models of chronic cerebral hypoperfusion have been developed in rats, mice, and primates. We established a mouse model of chronic cerebral hypoperfusion by narrowing the bilateral common carotid arteries with microcoils, eventually resulting in hippocampal atrophy. In addition, a mouse model of white matter infarct-related damage with cognitive and motor dysfunction has also been established by asymmetric common carotid artery surgery. Although most experiments studying chronic cerebral hypoperfusion have been performed in rodents because of the ease of handling and greater ethical acceptability, non-human primates appear to represent the best model for the study of VCI, due to their similarities in much larger white matter volume and amyloid β depositions like humans. Therefore, we also recently developed a baboon model of VCI through three-vessel occlusion (both the internal carotid arteries and the left vertebral artery). In this review, several animal models of chronic cerebral hypoperfusion, from mouse to primate, are extensively discussed to aid in better understanding of pathophysiology of VCI.
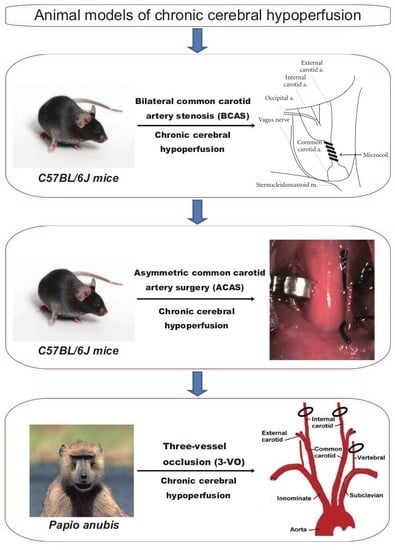
1. Introduction
Vascular cognitive impairment (VCI) or vascular dementia occurs as a result of persistently compromised blood flow to the brain and represents the second leading cause of dementia after Alzheimer’s disease [1,2]. However, the precise pathophysiological mechanisms underlying VCI remain unclear despite extensive research [3,4].
VCI results from systemic, cardiac, or local large or small vessel disease [5,6]. There are several causes of VCI: cerebral small vessel disease, multi-infarct dementia; strategic infarct (i.e., located in a functionally-critical brain area), hemorrhage/microbleed; angiopathy (including cerebral amyloid angiopathy), severe hypoperfusion (e.g., heart failure, cardiac arrhythmia), and hereditary vasculopathy (e.g., cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, CADASIL) [7,8]. No single animal model can replicate them all [9]. For instance, animal models of chronic cerebral hypoperfusion show demyelination and axonal damage, but do not show cardiovascular or other neurobiological changes of VCI, such as heart failure, hypertension, or arterial pathologies such as arteriosclerosis, fibrinoid necrosis, and lipohyalinosis [10]. However, in this review, we exclusively focused on chronic cerebral hypoperfusion animal models since subcortical vascular dementia, primarily characterized by ischemic white matter lesions due to chronic cerebral hypoperfusion, accounts for approximately half of all VCI cases in humans [10].
The cerebral blood flow (CBF) is diffusely or at least focally reduced in elderly patients with VCI [11]. Accordingly, for investigation of the mechanisms underlying VCI, several models of chronic cerebral hypoperfusion have been established in rats [12], mice [13,14], and non-human primates [15]. Models of chronic cerebral hypoperfusion can be generated by bilateral common carotid artery (CCA) occlusion in rats, bilateral CCA stenosis in mice, asymmetric CCA surgery in mice, and bilateral internal carotid artery and left vertebral artery occlusion (three-vessel occlusion; 3VO) in non-human primates. These animal models also provide unique platforms for the investigation of potential drugs or cell therapies, considering that no disease-modifying drugs for VCI have been developed till date [16].
In this review, several animal models of chronic cerebral hypoperfusion, ranging from mice to primates, are extensively discussed to further the understanding of pathophysiological mechanisms underlying VCI. Specifically, we have discussed the characteristic details accumulated since the establishment of rat, mice, and primate models of chronic cerebral hypoperfusion, with a focus on the methods, CBF, histopathological changes, cognition, motor functions, and practical applications.
2. Pathogenetic and Neurobiological Mechanism of Subcortical Vascular Cognitive Impairment
Subcortical VCI accounts for approximately half of all VCI cases in humans [10]. The pathogenetic and neurobiological mechanism of subcortical VCI is illustrated for a deeper understanding (Figure 1). Subcortical VCI is mainly caused by chronic cerebral hypoperfusion and small vessel disease. Vascular risk factors, such as hypertension, diabetes mellitus, and aging, cause small vessel changes, and atherosclerosis. Small vessel change may cause its occlusion and lacunar infarction, whereas loss of vasomotor reactivity due to angionecrosis/lipohyalinosis/fibrohyalinosis in the small vessels may cause chronic cerebral hypoperfusion in the white matter. Atherosclerosis also causes chronic cerebral hypoperfusion. Subsequent microglial/astroglial activation, matrix metalloproteinase activation, blood-brain barrier damage, and endothelial dysfunction are likely to induce neuroinflammation, oxidative stress, and oligoendroglial apoptosis, leading to white matter lesions [17,18,19,20].
Animal models of chronic cerebral hypoperfusion are used as subcortical VCI models and do not fully explain all aspects of VCI in humans. VCI also results from cardiovascular mechanisms such as heart failure, hypertension, cardiac arrythmia, arterial sclerosis, cerebral amyloid angiopathy, transient ischemia, focal ischemia, embolism, and hereditary vasculopathy (e.g., CADASIL) [8,17,18]. For instance, animal models of chronic cerebral hypoperfusion show white matter lesions and cognitive impairment, but do not correspond well to the cardiovascular mechanisms of VCI. To make up for this drawback, animal models of cardiovascular diseases such as heart failure (transverse aortic constriction model [21,22,23,24], myocardial infarction model [25]), hypertension (stroke prone spontaneously hypertensive rat model [26,27], chronically hypertensive rhesus monkey model [28]), atherosclerosis (Apolipoprotein E knockout mouse model [29,30], low-density lipoprotein receptor-knockout mouse model [31]), and embolic models [32,33] are essential for a deeper understanding of vascular dementia in humans [17,18].
3. Rat Model of Chronic Cerebral Hypoperfusion
Four animal models of chronic cerebral hypoperfusion are outlined in this review (Table 1). Difficulty, time, and cost of surgery of each animal model are also outlined (Table 2). First, we will discuss the rat model, which can be generated by bilateral CCA occlusion (BCAO). The rat model of chronic cerebral hypoperfusion has been used for several years and is one of the most widely used and established experimental model [7,34], exhibiting cognitive impairment and cholinergic deficits in addition to white matter rarefaction resembling those associated with cerebral white matter lesions induced by chronic cerebral hypoperfusion in humans. In fact, cerebral hypoperfusion, induced by carotid artery stenosis [35,36], atrial fibrillation [37,38], and heart failure [39], is associated with cognitive decline in the general population [40]. Interestingly, BCAO in the rat could also be used as a model of retinal ischemia [41]. Although the optic nerve damage is strong, it has been used as a hypoperfusive subcortical VCI model. However, this model also has some inherent limitations. For example, the visual pathway is invariably damaged by occlusion of the ophthalmic arteries [41], and this potentially compromises behavioral assessments. Furthermore, genetic studies may be hampered because of limited accessibility to molecular technologies for rats.
3.1. Method
Rat models of chronic cerebral hypoperfusion are prepared using male Wistar rats aged 9–12 weeks (weight, 300–350 g). Under anesthesia with 1–3% halothane inhalation, both CCAs are exposed through a midline cervical incision and permanently ligated [42]. Only CCAs should be ligated after careful incision of the carotid artery sheath followed by separation of the vagus nerve from CCA. Although the survival rate is generally ≥90%, stimulation of the vagus nerve during surgery should be avoided as far as possible. Moreover, the body temperature should be maintained using a heat retention pad.
This surgery is relatively easy since we just need to ligate both CCAs with sutures and it only takes 10 min to perform (Table 2). The cost is only incurred from the sutures.
3.2. Cerebral Blood Flow
CBF is strongly reduced in the acute ischemic phase after surgery. This is one of the limitations of rat models. CBF in the cortex and corpus callosum was shown to reduce to 30–50% of the original level at 1 to 3 days after surgery, with gradual recovery after 3–8 days following surgery [43]. In the chronic ischemic phase, CBF values slowly normalize by 1 week, although they remain significantly lower than the control values at 4 weeks after surgery. By 8 weeks to 3 months after surgery, CBF is almost restored to the original level. The blood flow reduction in the hippocampus and thalamus is slightly lesser than that in the cerebral cortex, reducing to 50–70% and recovering to the original level by 3 months [34].
3.3. White Matter Lesions
The oligodendrocyte exhibits apoptosis and white matter rarefaction after 14 days. In the white matter, microglia are increased and activated from the first postoperative day. Reactivity changes in the astroglia are also observed after the third postoperative day. These activated glial cells produce inflammatory cytokines and free radicals and may play a role in white matter disorders [43]. White matter lesions primarily based on demyelination develop in the optic tract after 3 days and corpus callosum after 7 days.
3.4. Cortical Lesions
Cortical gray matter disorders vary with age, lineage, and surgical technique, and there is a discrepancy among reports [34]. Although no morphological abnormalities have been found in the cerebral cortex and hippocampus, one study reported mild cell shedding mainly in the hippocampal CA1 region [44].
3.5. Behavioral Analysis
In the BCAO rat model, because the optic nerve damage is strong, attention should be paid to interpretation of the results of behavioral assessments dependent on spatial working memory. Spatial learning ability assessed by the Morris water maze test was found to be impaired at 4 weeks after surgery, with more severe impairment at 20 weeks [45], while spatial working memory impairment was evident in the eight-arm radial maze test at 16 months. With regard to the object recognition test, abnormalities that were not observed at 30 days became evident at 60 and 90 days after surgery. Therefore, it is speculated that cognitive dysfunction cannot be attributed to acute-phase ischemic changes, whereas the influence of the chronic phase seems to be great.
3.6. Sensory/Motor Function
No major sensory/motor deficit has been reported in rat models [7].
4. Mouse Model of Chronic Cerebral Hypoperfusion (Bilateral Common Carotid Artery Stenosis)
As described above, the rat model of chronic cerebral hypoperfusion possesses some inherent drawbacks such as optic nerve damage and difficulty in genetic assessments. To overcome these limitations, more appropriate animal models are desirable. Accordingly, a mouse model of chronic cerebral hypoperfusion induced by bilateral CCA stenosis (BCAS) was established [13,46,47]. This model does not involve optic nerve damage and is therefore suitable for behavioral experiments; moreover, genetically modified animals can be used. In recent years, it has been increasingly used as a stable subcortical VCI model [18]. The mortality rate of the BCAS model, when performed by trained researchers, is less than 2%. Now that the BCAS model is used worldwide, it can be regarded as one of the most promising models of chronic cerebral hypoperfusion [18].
The severity of cerebral hypoperfusion can be easily controlled by regulation of the internal diameter of microcoils. This model mimics a neurological clinical condition of white matter rarefaction associated with white matter lesions induced by chronic cerebral hypoperfusion in humans. In fact, cerebral hypoperfusion, induced by carotid artery stenosis [35,36], atrial fibrillation [37,38], and heart failure [39], is associated with cognitive decline in the general population [40]. The model develops white matter lesions without cerebral infarctions and optic nerve damage, while hippocampal atrophy is induced for a longer time period. Interestingly, this may accurately reflect the hippocampal atrophy that is often observed in human patients with pure hypoperfusive subcortical VCI [48].
The apparent advantage of using this surgical technique is that it provides a unique platform to investigate the precise pathophysiological mechanism underlying VCI and explore potential drugs or cell therapies designed to develop novel treatments for dementia of vascular origin.
However, the mouse model of BCAS-induced chronic cerebral hypoperfusion has several limitations, including the lack of motor dysfunction and abrupt CBF reduction after the surgery [46,49].
To overcome these limitations, a better mouse model with motor impairment and a gradual reduction in CBF was recently established [14]; this will be discussed later. Nevertheless, a non-human primate model is always desirable because the rodent brain is different from the human brain in that it exhibits an extremely small volume of white matter [50] and no amyloid β accumulation.
4.1. Method
The mouse model of chronic cerebral hypoperfusion is developed in male C57BL/6J mice aged 10–12 weeks (weight, 24–29 g). We did not apply BCAS surgery to Wistar rat for several reasons. First, we don’t have appropriately sized microcoils for rat surgery. Second, the rat model of chronic cerebral hypoperfusion has some inherent drawbacks such as optic nerve damage and difficulty in genetic assessments. Third, the Wistar rat is more resistant to cerebral ischemia than the C57Bl/6J mouse [51]. Among seven mouse strains, we selected the C57Bl/6J mouse strain as it is most vulnerable to cerebral ischemia [13]. It has been hypothesized that the gradual recovery of cerebral blood flow is aided by blood supply through the posterior communicating artery. Therefore, a poorly-developed posterior communicating artery is suitable for maintaining chronic cerebral hypoperfusion over an extended period [14]. All procedures in this surgery were carried out in strict accordance with the guidelines for animal experimentation from the Animal Research Committee of Kyoto University. The protocol was approved by the Animal Research Committee, Kyoto University (Permit Number: MedKyo13270). All surgery was performed under anesthesia, and all efforts were made to minimize suffering.
Under anesthesia with 2% halothane inhalation, both CCAs are exposed through a midline cervical incision, followed by the application of a microcoil with an inner diameter of 0.18 mm (Figure 2A) [13]. Similar to the procedure used for the rat model of BCAO, the carotid artery sheath is dissected and the vagus nerve is separated from CCA so that the microcoil is applied only to CCA. The microcoil consists of piano wires with an internal diameter of 0.18 mm and is manufactured by mounting a microcoil (pitch, 0.5 mm; total length, 2.5 mm). Several microcoils with inner diameters varying from 0.16 mm to 0.22 mm are available for use. The survival rate at 1 week after surgery using a microcoil with a 0.18-mm inner diameter is ≥85%. The rectal temperature should be maintained between 36.5 °C and 37.5 °C during the procedure.
The difficulty of this surgery is moderate: microcoils have to be placed on both CCAs and it takes 10 min to perform. Microcoils can be purchased from Wuxi Samini Co., Ltd. (Wuxi, China). The cost of one is $25. Therefore, it costs $50 (two microcoils) per surgery (Table 2).
4.2. Cerebral Blood Flow
The mouse model also exhibits a strong reduction in CBF in the acute phase of ischemia after surgery. However, CBF in this model can be manipulated by changing the inner diameter of the microcoil. A microcoil with a 0.18-mm diameter, which is generally used from the perspective of reproducibility and confidence, results in a 30% drop in CBF at 2 h after surgery, following which there is gradual recovery (Figure 2B) [13]. A study showed that the blood flow in the cerebral cortex temporarily decreased to as much as 60% to 70% of the control value at 2 h after surgery, with gradual recovery to >80% at 1–3 months [52].
A major limitation of the mouse model is a relatively strong decrease in CBF in the acute phase after surgery. In other words, it remains unclear whether actual chronic cerebral hypoperfusion, characterized by both acute- and chronic-phase tissue changes, develops at 1–3 days after surgery. Accordingly, a model that does not exhibit such a strong reduction in CBF would be preferable.
4.3. White Matter Lesions
There is strong white matter rarefaction in the following order: corpus callosum > striatum > internal capsule > optic tract. This change appears in the corpus callosum, striatum, and internal capsule at 14 days after BCAS. Moreover, activation of the microglia and astrocyte proliferation are observed after 7 and 14 days following BCAS, respectively (Figure 2C) [13]. At the same time, the microglia exhibit a significant increase from 7 to 30 days after BCAS, whereas the astroglia increase from 14 to 30 days. Edema also appears to play a role in the development of white matter lesions. Activation of matrix metalloproteinase (MMP), which induces decomposition of the constituent proteins of the vascular basement membrane, impairment of the blood–brain barrier (BBB), and extravasation of serum proteins is also observed [53]. Among MMPs, MMP-2 has a strong ability to degrade myelin basic protein, and it is also believed to cause direct damage to the white matter.
4.4. Cortical Lesions
Gray matter lesions do not appear after BCAS using microcoils with an inner diameter of ≥0.18-mm in mice. However, microinfarctions in the cerebral cortex, cell shedding centered on the hippocampal CA1 region, and necrotic lesions scattered in the basal ganglia appear after most BCAS procedures using microcoils with an inner diameter of 0.16 mm, with a significant increase in the mortality rate [13].
The mouse model demonstrates good reproducibility in terms of glial activation, BBB disruption, and the development of white matter lesions and cognitive dysfunction, which appear within a month after surgery. Furthermore, in the longer term, significant hippocampal atrophy with pyknotic and apoptotic cells are observed at 8 months after surgery; this is evidence of a link between chronic cerebral hypoperfusion and neurodegeneration (Figure 2D,E) [52]. Hippocampal atrophy is induced by damage to the white matter that intimately connects with the hippocampus, and this damage can indirectly induce hippocampal atrophy through remote effects [52]. Actually, CBF in the hippocampus is significantly reduced after BCAS [54]. Interestingly, this may accurately reflect the hippocampal atrophy that is often observed in human patients with subcortical vascular dementia [48]. These results suggest that, in the long term, the mouse model of BCAS-induced chronic cerebral hypoperfusion replicates advanced stages of subcortical ischemic vascular dementia characterized by significant hippocampal neuronal loss.
4.5. Behavioral Analysis
Unlike the BCAO rat model, the BCAS mouse model shows constant blood flow in the common cervical and internal carotid arteries; therefore, the blood flow in the branched ocular artery is also preserved, resulting in preservation of the optic nerve. The white matter, which is the main component of the frontal-subcortical brain circuits, is strongly associated with the processing speed. Spatial working memory impairment is a characteristic feature of this model, because working memory depends on white matter integrity [52]. A decrease in spatial working memory is observed in the eight-arm radial maze test performed at 1 month after surgery, although reference memory impairment is not observed. This reproduces the cognitive profile of VCI in humans and is considered cognitive dysfunction due to impaired fiber communication between the frontal lobe and basal ganglia. However, in the longer term, i.e., at 5–6 months after BCAS, both reference and working memory are impaired in the Barnes and radial arm maze tests, respectively [52]; this is induced by both white matter lesions and hippocampal changes.
4.6. Sensory/Motor Function
No major sensory/motor deficit can be observed before three months after BCAS [46]. However, despite the absence of lacunar infarcts, mice were found to exhibit impaired motor function at three months after BCAS, thus reproducing one of the key features of subcortical vascular dementia. It was speculated that frontal-subcortical circuit disruption may have resulted in not only cognitive impairment but also motor dysfunction [52].
4.7. Pharmacological Experiments and Applications to Genetically Modified Animals
Till date, the development of various medicines and therapies has involved the use of the BCAS mouse model. These include angiotensin II type 1 receptor [55], phosphodiesterase III inhibitor [56], bone marrow mononuclear cells [57], and environmental enrichment therapy [58,59], which reportedly protect the white matter. Their clinical application is yet awaited.
Furthermore, the BCAS procedure is easy to apply to genetically modified mice, and there have been some applications thus far. Cerebral hypoperfusion is also a common underlying mechanism and a major contributor to cognitive decline and degenerative processes in Alzheimer’s disease [60,61,62]. When chronic cerebral hypoperfusion was induced in mouse models of Alzheimer’s disease, represented by mice overexpressing a mutant form of the human amyloid precursor protein bearing both the Swedish and Indiana mutations (APP (Sw/Ind)-Tg mice), it was found that the coagulability of the amyloid protein was enhanced. These results suggest an interaction between chronic cerebral hypoperfusion and APP (Sw/Ind) overexpression in the cognitive decline caused by enhanced neuronal loss and altered amyloid β metabolism in mice [63]. Thus, the ease of application of BCAS in genetically modified animals is expected to clarify the pathology of ischemic white matter lesions and aid in the development of therapies for neurodegenerative diseases such as Alzheimer’s disease. To summarize, the mouse model of BCAS-induced chronic cerebral hypoperfusion is currently considered the best animal model of VCI because of its reproducibility and genetic applicability [18,64,65].
In the prospect of initiating translational research, we will begin an investigator-initiated clinical trial based on the evidence obtained from the BCAS mouse model. Adrenomedulin (AM), a vasoactive peptide hormone, is secreted from endothelial cells in various organs such as the brain, adrenal medulla, heart, and kidney [66]. AM has a variety of effects on vasculature including vasodilation, permeability regulation, inhibition of endothelial cell apoptosis and oxidative stress, smooth muscle cell proliferation regulation, and angiogenesis promotion [66]. By subjecting mice to overexpress circulating AM in BCAS, we previously assessed its effects of AM on cerebral perfusion, cerebral angioarchitecture, oxidative stress, white matter change, and cognitive function [67]. In addition, an experimental study using a mouse model of middle cerebral artery occlusion demonstrated the protective effects of AM against ischemic stroke by promoting angiogenesis and suppressing cerebral edema [68,69]. Based on these results, we will start an investigator-led clinical trial for patients with ischemic stroke to confirm the effects of AM on acute cerebral ischemia.
5. Mouse Model of Chronic Cerebral Hypoperfusion (Asymmetric Common Carotid Artery Surgery)
In the mouse model of chronic cerebral hypoperfusion induced by BCAS, the subcortical white matter is a frequent target of ischemic injury. Extensive white matter lesions are important substrates of VCI in humans. However, other procedures for generating rodent models of ischemic stroke mainly induce cerebral infarcts in the gray matter, while BCAS in mice only results in white matter rarefaction without infarcts [52]. The lack of animal models that consistently replicate white matter infarct-related damage may partially explain why many neuroprotective drugs for VCI have shown clinical failure despite success in preclinical experiments [16].
The other limitation of the BCAS mouse model is that the decline in CBF is relatively abrupt, and this does not replicate the normal pattern of VCI in humans. Furthermore, this model only exhibits cognitive impairment without motor dysfunction in the first 3 months after surgery. Thus, it does not describe all the features of subcortical vascular dementia [17].
Accordingly, a mouse model of white matter infarct-related damage with cognitive and motor dysfunction induced by asymmetric CCA surgery (ACAS) was established [14]. This model can be generated by surgical implantation of different devices in the right and left CCAs in C57BL/6J mice. Implantation of an ameroid constrictor in the right CCA results in gradual occlusion of the vessel over 28 days, whereas placement of a microcoil in the left CCA induces approximately 50% arterial stenosis. Moreover, gradual CBF reduction is observed in this model, with the induction of white matter infarcts, rarefaction, and gliosis. Furthermore, motor impairments, including gait disturbance and muscle weakness, develop before executive dysfunction mimicking a neurological clinical condition of chronic cerebral hypoperfusion in humans (e.g., carotid artery stenosis [35,36], atrial fibrillation [37,38], and heart failure [39]). The existing rodent models of chronic cerebral hypoperfusion partially recapitulate subcortical VCI by developing white matter rarefaction [17]. However, the BCAO rat model and BCAS mouse model do not show other characteristics of subcortical VCI, such as infarctions as well as gradual CBF reduction. Another advantage of the ACAS model would be right-to-left comparison of efficacy of certain drugs on ischemia because of the difference in ischemic depth between the hemispheres. The right hemisphere mimics the characteristics of multiple subcortical infarcts and the left hemisphere mimics leukoaraiosis, sequelae to chronic cerebral hypoperfusion. Prompt and reproducible generation of small subcortical infarcts and leukoaraiosis, in a timely manner would be an advantage in the analysis of subcortical VCI pathophysiology and search for drugs that target subcortical VCI. Thus, the ACAS model represents a novel animal model, which accurately replicates predominant white matter infarcts accompanied by motor deficits and dementia, similar to hypoperfusive subcortical VCI with infarctions and motor deficits, like Binswanger’s disease [14].
5.1. Method
Male C57BL/6J mice aged 10–12 weeks (weight, 24–29 g) are used. We did not apply ACAS surgery to Wistar rat for several reasons. First, we don’t have appropriately sized microcoils and ameroid constrictors for rat surgery. Second, the rat model of chronic cerebral hypoperfusion has some inherent drawbacks such as optic nerve damage and difficulty in genetic assessments. Third, a poorly-developed posterior communicating artery of C57Bl/6J mouse is more suitable for maintaining chronic cerebral hypoperfusion over an extended period than Wistar rat [13,51].
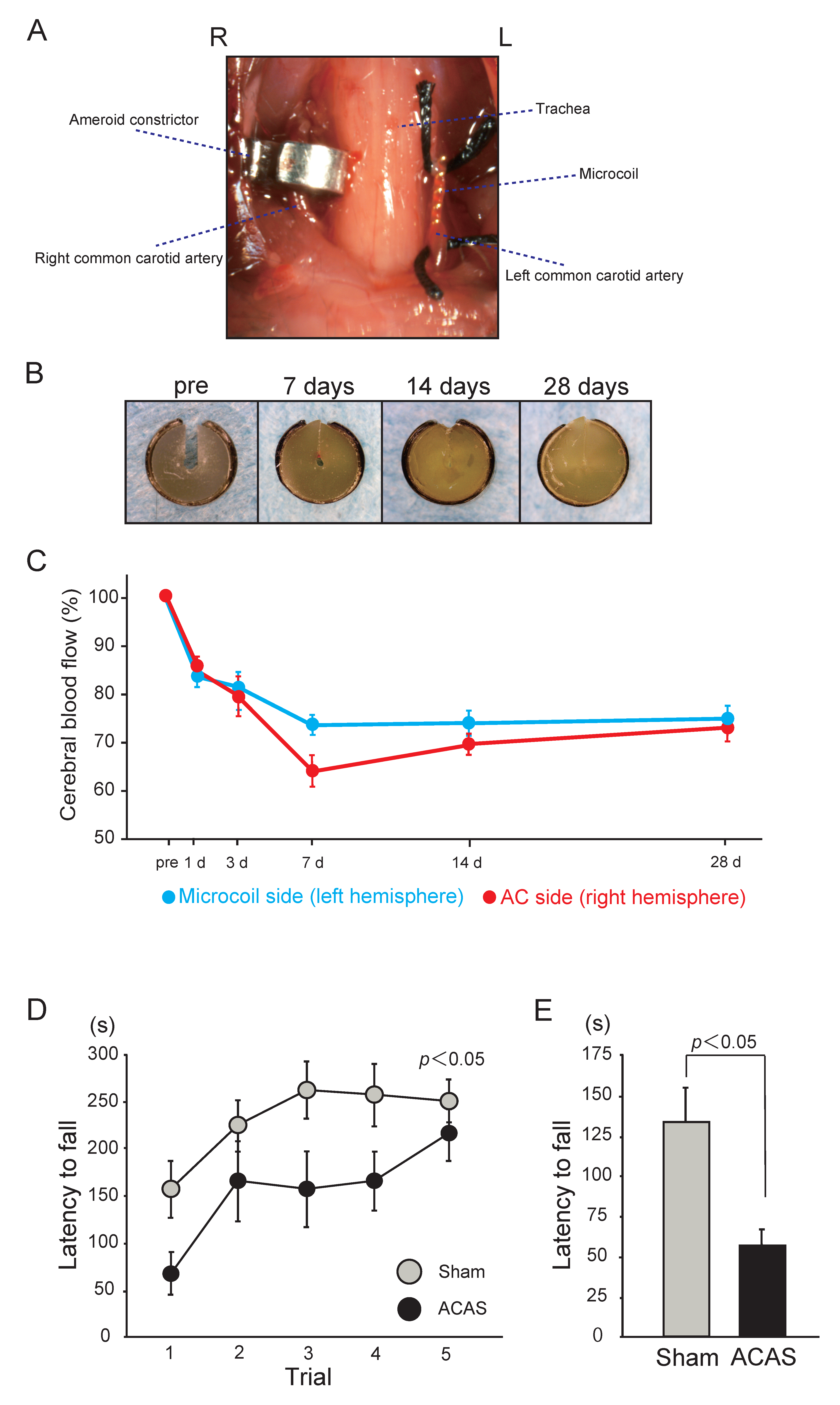
Under anesthesia with 1.5–2% isoflurane, both CCAs are exposed and freed from their sheaths through a midline cervical incision. Two 4-0 silk sutures are placed around the distal and proximal parts of the left CCA, which is gently lifted by the sutures and placed between the loops of the microcoil (inner diameter, 0.18 mm) just below the carotid bifurcation. The microcoil is implanted by rotation around the left CCA. Then, an ameroid constrictor is surgically implanted in the right CCA (Figure 3A,B) [14]. The ameroid constrictor consists of a titanium casing surrounding a hygroscopic casein material with an internal lumen. The casein component gradually absorbs water and consequently swells, leading to predictable narrowing and occlusion of the encased arterial lumen. The inner diameter, outer diameter, and length are 0.5, 3.25, and 1.28 mm, respectively. Placement of the ameroid constrictor in the right CCA produces the expected narrowing of the inner lumen. The ameroid constrictor gradually narrows the artery for up to 28 days after the procedure, following which it results in complete occlusion (Figure 3B). The rectal temperature should be maintained between 36.5 °C and 37.5 °C during the procedure. The reported mortality rate after ACAS is approximately 20.0% [14].
The difficulty of this surgery is relatively high because it requires two techniques: ameroid constrictor placement on the right CCA and microcoil placement on the left CCA. It takes 10 min to perform this surgery. The microcoil can be purchased from Wuxi Samini Co Ltd. (Wuxi, China) and the ameroid constrictor can be purchased from Tokyo Instruments Inc. (Tokyo, Japan). The cost of one microcoil is $25 and that of an ameroid constrictor is $100. Therefore, a cost of $125 (one microcoil and one ameroid constrictor) is incurred per surgery (Table 2).
5.2. Cerebral Blood Flow
The reduction in CBF is generally mild, without a sharp drop in the acute phase. The acute phase observed in the BCAS model was not seen in the ACAS model, and CBF on day 1 was significantly higher than that in the BCAS model (microcoil side, 83.7%; ameroid constrictor side, 87.2%; Figure 3C) [14]. However, CBF continued to decrease after ACAS and became significantly lower than that after BCAS at 7 days (microcoil side, 74.2%; ameroid constrictor side, 64.3%). Thereafter, CBF on the microcoil side almost exhibited a plateau until day 28, whereas that on the ameroid constrictor side showed slight recovery and approached the level on the microcoil side by day 28 (microcoil side, 75.3%; ameroid constrictor side, 73.4%). However, CBF on days 14 and 28 after ACAS remained significantly lower than that after BCAS. A significant right-to-left difference was observed only on day 7 after ACAS.
5.3. White Matter Lesions
With regard to white matter lesions, histopathological analysis of the corpus callosum and the anterior commissure using Klüver-Barrera staining reportedly showed grade 0 or grade 1 damage at 14 days after the procedure. However, at 32 days, moderate to severe rarefaction (grade 2–3) was observed in these regions [14]. In contrast, the white matter changes in the optic tract were less severe than those in the corpus callosum and anterior commissure.
Glial activation is also observed. Relative to that after sham surgery, the demyelination in the corpus callosum after ACAS was accompanied by significant proliferation of astrocytes and microglia. Similarly, the number of astrocytes on days 14 and 32 was higher with ACAS than with sham surgery, while the microglia continued to increase in number from days 14 to 32. The number of oligodendrocytes positive for glutathione-S-transferase-pi also tended to be lower with ACAS than with sham surgery on days 14 and 32. In addition, damaged SMI32-positive axons significantly increased on day 32 relative to the number on day 14 after both ACAS and sham surgery.
With regard to white matter infarction, 88% and 75% mice in the ACAS group showed ischemic stroke at 14 and 32 days after the procedure, respectively. At 14 and 32 days, multiple infarcts developed in subcortical areas such as the corpus callosum, caudoputamen, and internal capsule on the ameroid constrictor side in 81% mice [14]. Histopathological analysis showed multiple foci of infarct-related damage in the right subcortical regions, including the corpus callosum, internal capsule, hippocampal fimbria, and caudoputamen, in 81% mice. Mice displaying such damage showed significantly poorer results in locomotor and cognitive tests.
This animal model has a limitation: ACAS mice do not develop microvasculature pathology, while in most humans, VCI results from microvasculature pathology due to hypertension, diabetes, and genetic factors such as cerebral arteriopathy with subcortical infarcts and leukoencephalopathy [14].
5.4. Cortical Lesions
Cortical infarcts appeared in 31% mice and accounted for 15.6% of all infarcts developed in the ACAS group at 14 and 32 days. In contrast, no infarcts were seen on the microcoil side. Hematoxylin and eosin staining showed numerous accumulations of Iba1-positive microglia and glial fibrillary acidic protein-positive astrocytes in all infarctions. Hippocampal neuronal loss was observed only on the ameroid constrictor side in 69% mice. Activated microglia surrounded the area of neuronal loss in the hippocampus [14].
5.5. Behavioral Analysis
Executive dysfunction was induced, and motor impairments, including gait disturbance and muscle weakness, were also observed before the development of executive dysfunction [14]. The Y-maze test showed that spatial working memory was impaired at 14 days after ACAS. Relative to that in the sham group, the rate of alternation behavior, an indicator of spatial working memory, in the ACAS group was not decreased on day 14 but significantly decreased on day 28. This indicates that ACAS specifically results in spatial working memory impairment. The number of arm entries tended to be higher in the ACAS group than in the sham group on postoperative days 14 and 28. This indicates that spontaneous activity may be increased, rather than decreased, as a result of hyperactivity in mice subjected to ACAS. Furthermore, the Morris water maze test indicated that ACAS mice showed hippocampus-dependent reference memory impairment, reflecting existing hippocampal neuronal loss.
5.6. Sensory/Motor Function
Importantly, the mouse ACAS model also exhibits motor impairments, including muscle weakness and gait disturbance, earlier than the BCAS model [14]. The rotarod test showed that the latency to fall was significantly shorter in the ACAS group than in the sham group on day 14; this indicates that motor coordination and balance are significantly impaired after ACAS (Figure 3D). Similarly, the latency to fall in the wire hang test was significantly shorter in the ACAS group than in the sham group on day 14, indicating significantly impaired muscle strength after ACAS (Figure 3E).
6. Non-Human Primate Model of Chronic Cerebral Hypoperfusion
Finally, we introduce a non-human primate model of chronic cerebral hypoperfusion. Thus far, we described rat and mouse models; however, rodent brains are different from the human brain. First, they exhibit a very small volume of white matter, which is one the reasons why most clinical trials of neuroprotective drugs have failed [50]. Although most experiments on chronic cerebral hypoperfusion are performed in rodents because of the ease of handling and greater acceptability in terms of ethics, non-human primates appear to represent the best model for the evaluation of VCI because their cerebral architecture is similar to that in humans, with a considerably larger white matter volume [70,71]. Furthermore, aging baboon brains exhibit tau pathology and amyloid β deposition [72], both of which are characteristic features of Alzheimer’s disease and cannot be found in rodent brains. Accordingly, a baboon model of chronic cerebral hypoperfusion induced by three-vessel occlusion was developed [15]. In this model, glial activation and white matter changes, which precisely mimic the pathological changes in human hypoperfusive subcortical VCI induced by chronic cerebral hypoperfusion (e.g., carotid artery stenosis [35,36], atrial fibrillation [37,38], and heart failure [39]), could be seen. Thus, this model could be useful for investigation of the pathology underlying VCI.
6.1. Method
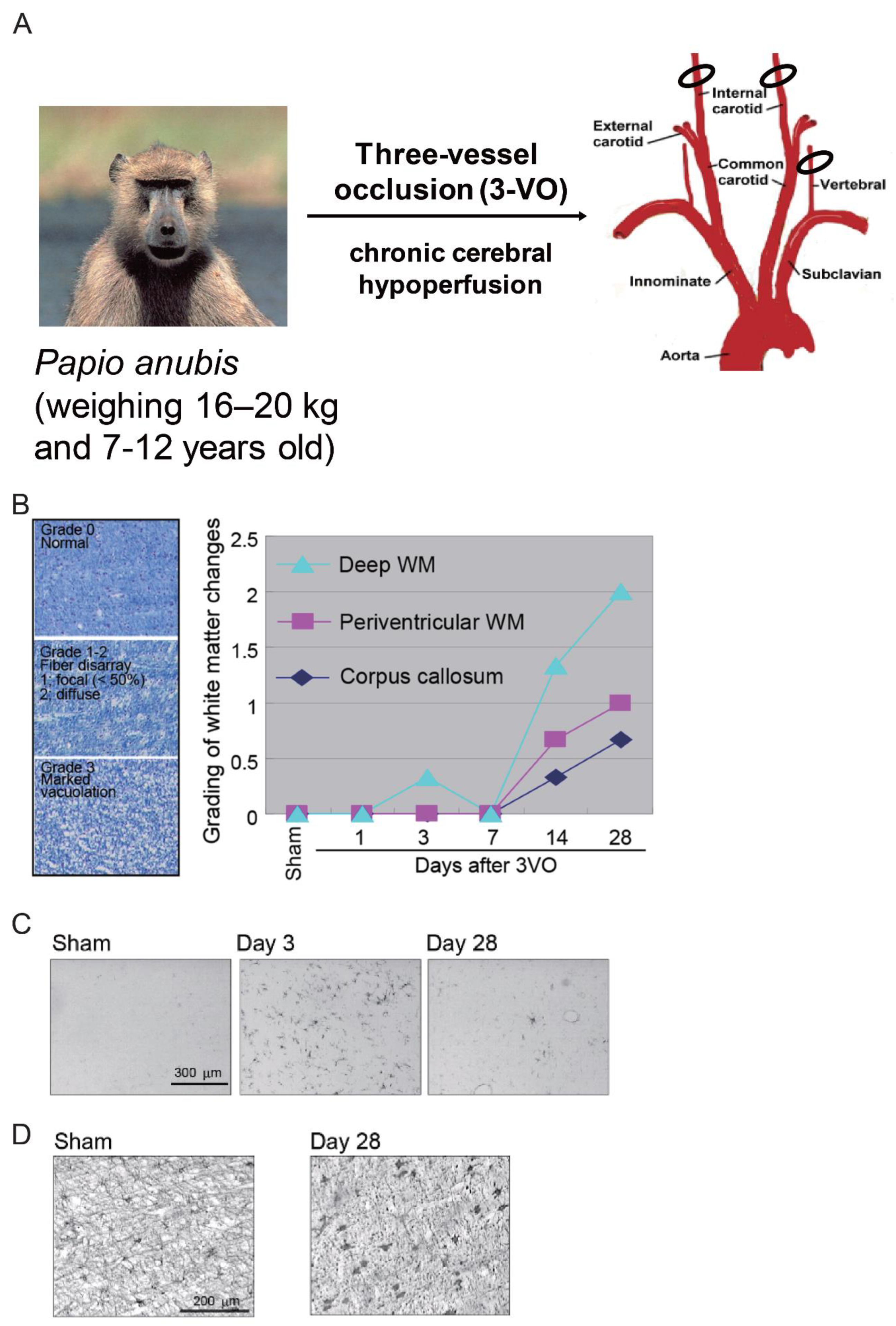
Adult baboons (Papio anubis) aged 7–12 years (weight, 16–20 kg) are used to generate this model. The baboons are subjected to permanent occlusion of both the internal carotid arteries and the left vertebral artery (three-vessel occlusion; 3VO) for 28 days (Figure 4A) [15].
This surgery is difficult: only a skilled veterinarian can perform it. The time and cost have not been well established (Table 2).
6.2. Cerebral Blood Flow
CBF changes in this model have not been reported and are still under investigation.
6.3. White Matter Lesions
White matter changes developed at 14 days after 3VO, predominantly in the deep white matter (Figure 4B). Marked vacuolation and moderate myelin loss also developed after 14 days, with changes continuing to occur up to 28 days.
Like rodent models of chronic cerebral hypoperfusion, the baboon model also showed glial activation and white matter changes after 3VO [15]. Microglial and astroglial cells were activated at three days after the procedure, with glial cell activation being more prominent in the deep white matter than in the corpus callosum and periventricular white matter (Figure 4C,D).
6.4. Cortical Lesions
Scattered infarcts in the cortices can be seen after 3VO. Degenerating neurons detected in the hippocampal CA1 and CA2 areas correlate with decreased neuron counts, with the greatest loss at 14 days after the procedure.
6.5. Behavioral Analysis
Behavioral changes are still under investigation.
6.6. Sensory/Motor Function
Immediately after 3VO, mild hemiparesis is temporally evident.
7. Conclusions
In conclusion, we described the benefits and drawbacks of four different animal models of chronic cerebral hypoperfusion. An appropriate animal model is essential for investigation of the precise mechanisms underlying VCI. The mouse model of chronic cerebral hypoperfusion induced by narrowing of the bilateral common carotid arteries with microcoils (BCAS) eventually results in hippocampal atrophy and is well validated and effective. The mouse model of chronic cerebral hypoperfusion induced by ACAS further replicates the phenotypes of human subcortical VCI, including multiple infarctions with motor and cognitive impairment. The baboon model of VCI induced by 3VO could be useful for assessing the pathophysiology of VCI because of the close resemblance to the human cerebral architecture. Finally, we expect that translational research based on the evidence generated using the mouse ACAS model or that the non-human primate model will help development of novel drugs for stroke. We also hope that these animal models will herald important breakthroughs in the development of novel treatments for VCI.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. K.W.; writing—original draft preparation, Y.H.; writing—review and editing, M.I.; writing—review and editing.
Funding
This work was funded by a Grant-in-Aid for Scientific Research on Priority Areas from the Japanese Ministry of Education, Science and Culture (K.W.).
Acknowledgments
We would like to thank Ohashi and Wada for their technical assistances. We would like to thank Editage (www.editage.com) for English language editing. This work was partially supported by Novartis Life Science Foundation and was also supported by the Grant-in-Aid for Scientific Research of Japan on Priority Areas from the Japanese Ministry of Education, Science and Culture (K.W.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| VCI | Vascular Cognitive Impairment |
| CBF | Cerebral Blood Flow |
| CCA | Common Carotid Artery |
| 3VO | Three-Vessel Occlusion |
| BCAO | Bilateral Common Carotid Artery Occlusion |
| BCAS | Bilateral Common Carotid Artery Stenosis |
| MMP | Matrix Metalloproteinase |
| ACAS | Asymmetric Common Carotid Artery Surgery |
References
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Tanizaki, Y.; Arima, H.; Yonemoto, K.; Doi, Y.; Ninomiya, T.; Sasaki, K.; Iida, M.; Iwaki, T.; Kanba, S.; et al. Incidence and survival of dementia in a general population of Japanese elderly: The Hisayama study. J. Neurol. Neurosurg. Psychiatry 2009, 80, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D. The underdiagnosis of the vascular contribution to dementia. J. Neurol. Sci. 2005, 229, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Pathology and pathophysiology of vascular cognitive impairment. A critical update. Panminerva Med. 2004, 46, 217–226. [Google Scholar] [PubMed]
- Korczyn, A.D.; Vakhapova, V.; Grinberg, L.T. Vascular dementia. J. Neurol. Sci. 2012, 322, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Jiwa, N.S.; Garrard, P.; Hainsworth, A.H. Experimental models of vascular dementia and vascular cognitive impairment: A systematic review. J. Neurochem. 2010, 115, 814–828. [Google Scholar] [CrossRef]
- Gooch, J.; Wilcock, D.M. Animal Models of Vascular Cognitive Impairment and Dementia (VCID). Cell. Mol. Neurobiol. 2016, 36, 233–239. [Google Scholar] [CrossRef]
- Helman, A.M.; Murphy, M.P. Vascular cognitive impairment: Modeling a critical neurologic disease in vitro and in vivo. Biochim. Et Biophys. Acta 2016, 1862, 975–982. [Google Scholar] [CrossRef]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef] [Green Version]
- Ihara, M.; Tomimoto, H.; Ishizu, K.; Mukai, T.; Yoshida, H.; Sawamoto, N.; Inoue, M.; Doi, T.; Hashikawa, K.; Konishi, J.; et al. Decrease in cortical benzodiazepine receptors in symptomatic patients with leukoaraiosis: A positron emission tomography study. Stroke 2004, 35, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edrissi, H.; Schock, S.C.; Cadonic, R.; Hakim, A.M.; Thompson, C.S. Cilostazol reduces blood brain barrier dysfunction, white matter lesion formation and motor deficits following chronic cerebral hypoperfusion. Brain Res. 2016, 1646, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Ohtani, R.; Ihara, M.; Tomimoto, H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004, 35, 2598–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Enmi, J.; Kitamura, A.; Yamamoto, Y.; Saito, S.; Takahashi, Y.; Iguchi, S.; Tsuji, M.; Yamahara, K.; Nagatsuka, K.; et al. A novel mouse model of subcortical infarcts with dementia. J. Neurosci 2015, 35, 3915–3928. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Akinyemi, R.O.; Hase, Y.; Firbank, M.J.; Ndung’u, M.N.; Foster, V.; Craggs, L.J.; Washida, K.; Okamoto, Y.; Thomas, A.J.; et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-Stroke dementia. Brain 2016, 139, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Markus, H.S. Do in vivo experimental models reflect human cerebral small vessel disease? A systematic review. J. Cereb. Blood Flow Metab. 2008, 28, 1877–1891. [Google Scholar] [CrossRef] [Green Version]
- Bink, D.I.; Ritz, K.; Aronica, E.; Van Der Weerd, L.; Daemen, M.J. Mouse models to study the effect of cardiovascular risk factors on brain structure and cognition. J. Cereb. Blood Flow Metab. 2013, 33, 1666–1684. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, P.B.; Counts, S.E.; Nyenhuis, D. Vascular cognitive impairment and dementia. Biochim. Et Biophys. Acta 2016, 1862, 860–868. [Google Scholar] [CrossRef]
- Venkat, P.; Chopp, M.; Chen, J. Models and mechanisms of vascular dementia. Exp. Neurol. 2015, 272, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Poulet, R.; Gentile, M.T.; Vecchione, C.; Distaso, M.; Aretini, A.; Fratta, L.; Russo, G.; Echart, C.; Maffei, A.; De Simoni, M.G.; et al. Acute hypertension induces oxidative stress in brain tissues. J. Cereb. Blood Flow Metab. 2006, 26, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnevale, D.; Mascio, G.; Ajmone-Cat, M.A.; D’Andrea, I.; Cifelli, G.; Madonna, M.; Cocozza, G.; Frati, A.; Carullo, P.; Carnevale, L.; et al. Role of neuroinflammation in hypertension-Induced brain amyloid pathology. Neurobiol. Aging 2012, 33, e219–e229. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Mascio, G.; D’Andrea, I.; Fardella, V.; Bell, R.D.; Branchi, I.; Pallante, F.; Zlokovic, B.; Yan, S.S.; Lembo, G. Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 2012, 60, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Montgolfier, O.; Pincon, A.; Pouliot, P.; Gillis, M.A.; Bishop, J.; Sled, J.G.; Villeneuve, L.; Ferland, G.; Levy, B.I.; Lesage, F.; et al. High Systolic Blood Pressure Induces Cerebral Microvascular Endothelial Dysfunction, Neurovascular Unit Damage, and Cognitive Decline in Mice. Hypertension 2019, 73, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Noyan-Ashraf, M.H.; Meissner, A.; Voigtlaender-Bolz, J.; Kroetsch, J.T.; Foltz, W.; Jaffray, D.; Kapoor, A.; Momen, A.; Heximer, S.P.; et al. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-Phosphate signaling. Circulation 2012, 126, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, K.; Hazama, F.; Yamori, Y.; Haebara, H.; Nagaoka, A. Pathogenesis and prevention of stroke in spontaneously hypertensive rats. Clin. Sci. Mol. Med. 1975, 2, 161s–163s. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.F.; McCabe, C.; Khatun, H.; Kaushal, N.; Bridges, L.R.; Holmes, W.M.; Barrick, T.R.; Graham, D.; Dominiczak, A.F.; Mhairi Macrae, I.; et al. An MRI-Histological study of white matter in stroke-Free SHRSP. J. Cereb. Blood Flow Metab. 2013, 33, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, T.L.; Blatt, G.J.; Killiany, R.J.; Moss, M.B. Neuropathology of progressive cognitive decline in chronically hypertensive rhesus monkeys. Acta Neuropathol. 2001, 101, 145–153. [Google Scholar]
- Grootendorst, J.; De Kloet, E.R.; Dalm, S.; Oitzl, M.S. Reversal of cognitive deficit of apolipoprotein E knockout mice after repeated exposure to a common environmental experience. Neuroscience 2001, 108, 237–247. [Google Scholar] [CrossRef]
- Lee, E.S.; Yoon, J.H.; Choi, J.; Andika, F.R.; Lee, T.; Jeong, Y. A mouse model of subcortical vascular dementia reflecting degeneration of cerebral white matter and microcirculation. J. Cereb. Blood Flow Metab. 2019, 39, 44–57. [Google Scholar] [CrossRef]
- Mulder, M.; Jansen, P.J.; Janssen, B.J.; Van De Berg, W.D.; Van Der Boom, H.; Havekes, L.M.; De Kloet, R.E.; Ramaekers, F.C.; Blokland, A. Low-Density lipoprotein receptor-Knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol. Dis. 2004, 16, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Alexis, N.E.; Dietrich, W.D.; Green, E.J.; Prado, R.; Watson, B.D. Nonocclusive common carotid artery thrombosis in the rat results in reversible sensorimotor and cognitive behavioral deficits. Stroke 1995, 26, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ding, F.; Deng, S.; Guo, X.; Wang, W.; Iliff, J.J.; Nedergaard, M. Focal Solute Trapping and Global Glymphatic Pathway Impairment in a Murine Model of Multiple Microinfarcts. J. Neurosci. 2017, 37, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, V.L.; Goldberg, A.P.; Rogers, M.W.; Anthony, L.; Terrin, M.L.; Guralnik, J.M.; Blackwelder, W.C.; Lam, D.F.H.; Sikdar, S.; Lal, B.K. Asymptomatic carotid stenosis is associated with mobility and cognitive dysfunction and heightens falls in older adults. J. Vasc. Surg. 2019. [Google Scholar] [CrossRef]
- Demarin, V.; Zavoreo, I.; Kes, V.B. Carotid artery disease and cognitive impairment. J. Neurol. Sci. 2012, 322, 107–111. [Google Scholar] [CrossRef]
- Chopard, R.; Piazza, G.; Gale, S.A.; Campia, U.; Albertsen, I.E.; Kim, J.; Goldhaber, S.Z. Dementia and Atrial Fibrillation: Pathophysiological Mechanisms and Therapeutic Implications. Am. J. Med. 2018, 131, 1408–1417. [Google Scholar] [CrossRef]
- De Bruijn, R.F.; Heeringa, J.; Wolters, F.J.; Franco, O.H.; Stricker, B.H.; Hofman, A.; Koudstaal, P.J.; Ikram, M.A. Association Between Atrial Fibrillation and Dementia in the General Population. JAMA Neurol. 2015, 72, 1288–1294. [Google Scholar] [CrossRef]
- Adelborg, K.; Horvath-Puho, E.; Ording, A.; Pedersen, L.; Sorensen, H.T.; Henderson, V.W. Heart failure and risk of dementia: A Danish nationwide population-based cohort study. Eur. J. Heart Fail. 2017, 19, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Wolters, F.J.; Zonneveld, H.I.; Hofman, A.; Van Der Lugt, A.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation 2017, 136, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fan, S.; Li, J.; Wang, Y.L. Bilateral Common Carotid Artery Occlusion in the Rat as a Model of Retinal Ischaemia. Neuro Ophthalmol. 2014, 38, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, M.; Ganesan, V.; Thomas, D.L.; Thornton, J.S.; Proctor, E.; King, M.D.; Van Der Weerd, L.; Gadian, D.G.; Lythgoe, M.F. The chronic vascular and haemodynamic response after permanent bilateral common carotid occlusion in newborn and adult rats. J. Cereb. Blood Flow Metab. 2006, 26, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Tomimoto, H.; Ihara, M.; Wakita, H.; Ohtani, R.; Lin, J.X.; Akiguchi, I.; Kinoshita, M.; Shibasaki, H. Chronic cerebral hypoperfusion induces white matter lesions and loss of oligodendroglia with DNA fragmentation in the rat. Acta Neuropathol. 2003, 106, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Li, N.; Li, S.; Han, R.; Huang, Q.; Hu, J.; Jin, K.; Ji, X. Limb Ischemic Conditioning Improved Cognitive Deficits via eNOS-Dependent Augmentation of Angiogenesis after Chronic Cerebral Hypoperfusion in Rats. Aging Dis. 2018, 9, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.X.; Zhang, J.J.; Zheng, P.; Zhang, Y. Altered expression of MAP-2, GAP-43, and synaptophysin in the hippocampus of rats with chronic cerebral hypoperfusion correlates with cognitive impairment. Brain Res. 2005, 139, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamasaki, N.; Miyakawa, T.; Kalaria, R.N.; Fujita, Y.; Ohtani, R.; Ihara, M.; Takahashi, R.; Tomimoto, H. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 2007, 38, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Ihara, M.; Taguchi, A.; Maki, T.; Washida, K.; Tomimoto, H. A mouse model of chronic cerebral hypoperfusion characterizing features of vascular cognitive impairment. Methods Mol. Biol. 2014, 1135, 95–102. [Google Scholar]
- Kim, G.H.; Lee, J.H.; Seo, S.W.; Kim, J.H.; Seong, J.K.; Ye, B.S.; Cho, H.; Noh, Y.; Kim, H.J.; Yoon, C.W.; et al. Hippocampal volume and shape in pure subcortical vascular dementia. Neurobiol. Aging 2015, 36, 485–491. [Google Scholar] [CrossRef]
- Fleischman, D.A.; Yang, J.; Arfanakis, K.; Arvanitakis, Z.; Leurgans, S.E.; Turner, A.D.; Barnes, L.L.; Bennett, D.A.; Buchman, A.S. Physical activity, motor function, and white matter hyperintensity burden in healthy older adults. Neurology 2015, 84, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Dewar, D.; Yam, P.; McCulloch, J. Drug development for stroke: Importance of protecting cerebral white matter. Eur. J. Pharmacol. 1999, 375, 41–50. [Google Scholar] [CrossRef]
- Kitagawa, K.; Matsumoto, M.; Yang, G.; Mabuchi, T.; Yagita, Y.; Hori, M.; Yanagihara, T. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: Evaluation of the patency of the posterior communicating artery. J. Cereb. Blood Flow Metab. 1998, 18, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Nishio, K.; Ihara, M.; Yamasaki, N.; Kalaria, R.N.; Maki, T.; Fujita, Y.; Ito, H.; Oishi, N.; Fukuyama, H.; Miyakawa, T.; et al. A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke 2010, 41, 1278–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.; Rosenberg, G.A. Rodent Models of Vascular Cognitive Impairment. Transl. Stroke Res. 2016, 7, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Enmi, J.; Iguchi, S.; Saito, S.; Yamamoto, Y.; Nagatsuka, K.; Iida, H.; Ihara, M. Substantial Reduction of Parenchymal Cerebral Blood Flow in Mice with Bilateral Common Carotid Artery Stenosis. Sci. Rep. 2016, 6, 32179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washida, K.; Ihara, M.; Nishio, K.; Fujita, Y.; Maki, T.; Yamada, M.; Takahashi, J.; Wu, X.; Kihara, T.; Ito, H.; et al. Nonhypotensive dose of telmisartan attenuates cognitive impairment partially due to peroxisome proliferator-Activated receptor-Gamma activation in mice with chronic cerebral hypoperfusion. Stroke 2010, 41, 1798–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, A.; Manso, Y.; Duncombe, J.; Searcy, J.; Koudelka, J.; Binnie, M.; Webster, S.; Lennen, R.; Jansen, M.; Marshall, I.; et al. Long-Term cilostazol treatment reduces gliovascular damage and memory impairment in a mouse model of chronic cerebral hypoperfusion. Sci. Rep. 2017, 7, 4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Ihara, M.; Ushiki, T.; Hirai, H.; Kizaka-Kondoh, S.; Hiraoka, M.; Ito, H.; Takahashi, R. Early protective effect of bone marrow mononuclear cells against ischemic white matter damage through augmentation of cerebral blood flow. Stroke 2010, 41, 2938–2943. [Google Scholar] [CrossRef] [Green Version]
- Hase, Y.; Craggs, L.; Hase, M.; Stevenson, W.; Slade, J.; Chen, A.; Liang, D.; Ennaceur, A.; Oakley, A.; Ihara, M.; et al. The effects of environmental enrichment on white matter pathology in a mouse model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Hase, Y.; Craggs, L.; Hase, M.; Stevenson, W.; Slade, J.; Lopez, D.; Mehta, R.; Chen, A.; Liang, D.; Oakley, A.; et al. Effects of environmental enrichment on white matter glial responses in a mouse model of chronic cerebral hypoperfusion. J. Neuroinflammation 2017, 14, 81. [Google Scholar] [CrossRef] [Green Version]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Ng, G.; Tan, E.K.; Liao, P.; Kandiah, N.; Zeng, L. Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci. Rep. 2016, 6, 23964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Yamashita, T.; Zhai, Y.; Nakano, Y.; Morihara, R.; Fukui, Y.; Hishikawa, N.; Ohta, Y.; Abe, K. Strong Impact of Chronic Cerebral Hypoperfusion on Neurovascular Unit, Cerebrovascular Remodeling, and Neurovascular Trophic Coupling in Alzheimer’s Disease Model Mouse. J. Alzheimers Dis. 2016, 52, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Ihara, M.; Okamoto, Y.; Maki, T.; Washida, K.; Kitamura, A.; Hase, Y.; Ito, H.; Takao, K.; Miyakawa, T.; et al. The influence of chronic cerebral hypoperfusion on cognitive function and amyloid beta metabolism in APP overexpressing mice. PLoS ONE 2011, 6, e16567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Okamoto, Y.; Maki, T.; Yamamoto, Y.; Oishi, N.; Yamahara, K.; Nagatsuka, K.; Takahashi, R.; Kalaria, R.N.; Fukuyama, H.; et al. Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase. Stroke 2014, 45, 3403–3411. [Google Scholar] [CrossRef] [Green Version]
- Bannai, T.; Mano, T.; Chen, X.; Ohtomo, G.; Ohtomo, R.; Tsuchida, T.; Koshi-Mano, K.; Hashimoto, T.; Okazawa, H.; Iwatsubo, T.; et al. Chronic cerebral hypoperfusion shifts the equilibrium of amyloid beta oligomers to aggregation-Prone species with higher molecular weight. Sci. Rep. 2019, 9, 2827. [Google Scholar] [CrossRef]
- Maki, T.; Takahashi, Y.; Miyamoto, N.; Liang, A.C.; Ihara, M.; Lo, E.H.; Arai, K. Adrenomedullin promotes differentiation of oligodendrocyte precursor cells into myelin-Basic-Protein expressing oligodendrocytes under pathological conditions in vitro. Stem Cell Res. 2015, 15, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Maki, T.; Ihara, M.; Fujita, Y.; Nambu, T.; Miyashita, K.; Yamada, M.; Washida, K.; Nishio, K.; Ito, H.; Harada, H.; et al. Angiogenic and vasoprotective effects of adrenomedullin on prevention of cognitive decline after chronic cerebral hypoperfusion in mice. Stroke 2011, 42, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, K.; Itoh, H.; Arai, H.; Suganami, T.; Sawada, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Yurugi-Kobayashi, T.; Park, K.; et al. The neuroprotective and vasculo-Neuro-Regenerative roles of adrenomedullin in ischemic brain and its therapeutic potential. Endocrinology 2006, 147, 1642–1653. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, T.; Ueta, Y.; Torii, K. Pre-Treatment of adrenomedullin suppresses cerebral edema caused by transient focal cerebral ischemia in rats detected by magnetic resonance imaging. Brain Res. Bull. 2011, 84, 69–74. [Google Scholar] [CrossRef]
- Stroke Therapy Academic Industry Roundtable (STAIR). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999, 30, 2752–2758. [Google Scholar] [CrossRef]
- Madigan, J.B.; Wilcock, D.M.; Hainsworth, A.H. Vascular Contributions to Cognitive Impairment and Dementia: Topical Review of Animal Models. Stroke 2016, 47, 1953–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndung’u, M.; Hartig, W.; Wegner, F.; Mwenda, J.M.; Low, R.W.; Akinyemi, R.O.; Kalaria, R.N. Cerebral amyloid beta(42) deposits and microvascular pathology in ageing baboons. Neuropathol. Appl. Neurobiol. 2012, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
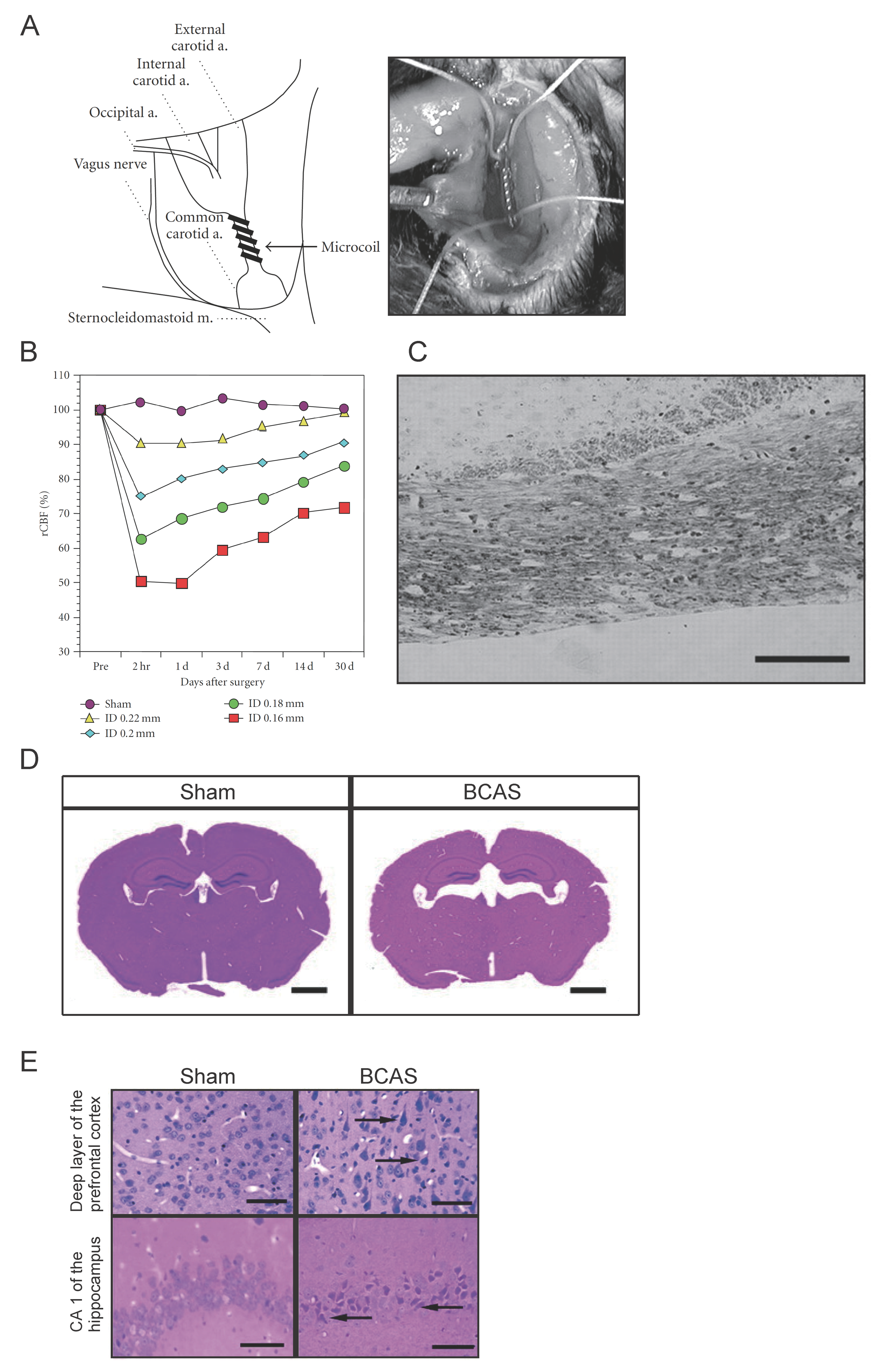
Figure 1.
Schema of the pathogenetic and neurobiological mechanism of subcortical vascular cognitive impairment. Subcortical vascular cognitive impairment is mainly caused by chronic cerebral hypoperfusion and small vessel disease. Vascular risk factors, such as hypertension, diabetes mellitus, and aging, cause small vessel changes and atherosclerosis. Small vessel change may cause its occlusion and lacunar infarction, whereas loss of vasomotor reactivity due to angionecrosis/lipohyalinosis/fibrohyalinosis in the small vessels may cause chronic cerebral hypoperfusion in the white matter. Atherosclerosis also causes chronic cerebral hypoperfusion. Subsequent microglial/astroglial activation, matrix metalloproteinase activation, blood-brain barrier damage, and endothelial dysfunction are likely to induce neuroinflammation, oxidative stress, oligodendroglial apoptosis, and thence white matter lesions.
Figure 1.
Schema of the pathogenetic and neurobiological mechanism of subcortical vascular cognitive impairment. Subcortical vascular cognitive impairment is mainly caused by chronic cerebral hypoperfusion and small vessel disease. Vascular risk factors, such as hypertension, diabetes mellitus, and aging, cause small vessel changes and atherosclerosis. Small vessel change may cause its occlusion and lacunar infarction, whereas loss of vasomotor reactivity due to angionecrosis/lipohyalinosis/fibrohyalinosis in the small vessels may cause chronic cerebral hypoperfusion in the white matter. Atherosclerosis also causes chronic cerebral hypoperfusion. Subsequent microglial/astroglial activation, matrix metalloproteinase activation, blood-brain barrier damage, and endothelial dysfunction are likely to induce neuroinflammation, oxidative stress, oligodendroglial apoptosis, and thence white matter lesions.

Figure 2.
Mouse model of chronic cerebral hypoperfusion induced by bilateral common carotid artery (CCA) stenosis (BCAS). (A) The procedure for BCAS and microcoil placement in a C57BL/6J mouse. The microcoil is twined by rotation around CCA just proximal to the carotid bifurcation. (B) Cerebral blood flow (CBF) values after BCAS. The data are expressed as means ± standard deviations (SDs) or percentages of the preoperative values. * p < 0.05, ** p < 0.01 compared with control values. (C) There is myelin rarefaction and vacuole formation in the corpus callosum after placement of a microcoil with an inner diameter of 0.18 mm. Scale bar, 100 μm. (D) Representative hematoxylin and eosin-stained sections obtained from the sham (left) and BCAS (right) groups. Note the resultant dilation of the ventricle in the BCAS group. Scale bars, 1 mm. (E) Representative images of the deep layer of the frontal cerebral cortex (upper panels) and the hippocampal CA1 region (lower panels) for the sham (left panels) and BCAS (right panels) groups. The arrows indicate pyknotic neurons. Scale bar, 50 μm [13,52].
Figure 2.
Mouse model of chronic cerebral hypoperfusion induced by bilateral common carotid artery (CCA) stenosis (BCAS). (A) The procedure for BCAS and microcoil placement in a C57BL/6J mouse. The microcoil is twined by rotation around CCA just proximal to the carotid bifurcation. (B) Cerebral blood flow (CBF) values after BCAS. The data are expressed as means ± standard deviations (SDs) or percentages of the preoperative values. * p < 0.05, ** p < 0.01 compared with control values. (C) There is myelin rarefaction and vacuole formation in the corpus callosum after placement of a microcoil with an inner diameter of 0.18 mm. Scale bar, 100 μm. (D) Representative hematoxylin and eosin-stained sections obtained from the sham (left) and BCAS (right) groups. Note the resultant dilation of the ventricle in the BCAS group. Scale bars, 1 mm. (E) Representative images of the deep layer of the frontal cerebral cortex (upper panels) and the hippocampal CA1 region (lower panels) for the sham (left panels) and BCAS (right panels) groups. The arrows indicate pyknotic neurons. Scale bar, 50 μm [13,52].

Figure 3.
Mouse model of chronic cerebral hypoperfusion induced by asymmetric common carotid artery surgery (ACAS). (A) An image showing surgical implantation of the ameroid constrictor (AC) in the right common carotid artery (CCA) and the microcoil in the left CCA. (B) Representative images of AC at the indicated time points, showing gradual narrowing of the central lobe (lumen) with complete occlusion at 28 days. (C) Temporal profiles of the cerebral blood flow (CBF) in the cortex and subcortical area of surviving mice subjected to ACAS. (D) Behavioral performance of mice in the rotarod test after ACAS. Motor coordination and balance were tested with five consecutive trials on day 14. Data were analyzed by two-way repeated-measures analysis of variance. (E) Neuromuscular strength tested with the wire hang test on day 14. The error bar indicates the mean ± standard error of the mean [14].
Figure 3.
Mouse model of chronic cerebral hypoperfusion induced by asymmetric common carotid artery surgery (ACAS). (A) An image showing surgical implantation of the ameroid constrictor (AC) in the right common carotid artery (CCA) and the microcoil in the left CCA. (B) Representative images of AC at the indicated time points, showing gradual narrowing of the central lobe (lumen) with complete occlusion at 28 days. (C) Temporal profiles of the cerebral blood flow (CBF) in the cortex and subcortical area of surviving mice subjected to ACAS. (D) Behavioral performance of mice in the rotarod test after ACAS. Motor coordination and balance were tested with five consecutive trials on day 14. Data were analyzed by two-way repeated-measures analysis of variance. (E) Neuromuscular strength tested with the wire hang test on day 14. The error bar indicates the mean ± standard error of the mean [14].

Figure 4.
Baboon (non-human primate) model of chronic cerebral hypoperfusion induced by three-vessel occlusion (3VO). (A) The baboon is subjected to permanent occlusion of both the internal carotid arteries and the left vertebral artery (3VO) for 28 days. (B) White matter changes can be observed at 14 days after 3VO, predominantly in the deep white matter. Luxol fast blue staining shows marked vacuolation and moderate myelin loss after 14 days, with further changes continuing up to 28 days. Scale bar, 100 μm. (C) Immunohistochemical staining (CD68) shows microglial cell activation in the white matter in the 3VO group. Scale bar, 300 μm. (D) Immunohistochemical staining (glial fibrillary acidic protein) shows astroglial cell activation in the white matter in the 3VO group. Scale bar, 200 μm [15].
Figure 4.
Baboon (non-human primate) model of chronic cerebral hypoperfusion induced by three-vessel occlusion (3VO). (A) The baboon is subjected to permanent occlusion of both the internal carotid arteries and the left vertebral artery (3VO) for 28 days. (B) White matter changes can be observed at 14 days after 3VO, predominantly in the deep white matter. Luxol fast blue staining shows marked vacuolation and moderate myelin loss after 14 days, with further changes continuing up to 28 days. Scale bar, 100 μm. (C) Immunohistochemical staining (CD68) shows microglial cell activation in the white matter in the 3VO group. Scale bar, 300 μm. (D) Immunohistochemical staining (glial fibrillary acidic protein) shows astroglial cell activation in the white matter in the 3VO group. Scale bar, 200 μm [15].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of animal models of chronic cerebral hypoperfusion.
| Animal Models | Rat BCAO | Mouse BCAS | Mouse ACAS | Baboon 3-VO |
|---|---|---|---|---|
| CBF reduction (% preoperative level) | Rapid reduction to 30–50% of the original level with gradual recovery | Rapid reduction to 70% of the original level with gradual recovery by 0.18 mm microcoils | Gradual reduction to 60% (AC side) and 70% (microcoil side) of the original level without recovery | Under investigation |
| White matter lesion | Demyelination appears 14 days after surgery | Demyelination appears 14 days after surgery | Multiple infarcts and demyelination appears 14 days after surgery | Demyelination appears 14 days after surgery |
| Gray matter lesion | Rare | No gray matter infarction by microcoils with an inner diameter of 0.18 mm or more | Multiple infarcts only on the AC side | Rare |
| Cognitive dysfunction | Spatial working memory impairment | Spatial working memory impairment | Spatial working/reference memory impairment | Under investigation |
| Motor dysfunction | No motor deficits | No motor deficits before 3 months | Muscle weakness and gait disturbance | Temporal hemiparesis after surgery |
BCAO, bilateral common carotid artery occlusion; BCAS, bilateral common carotid artery stenosis; ACAS, asymmetric common carotid artery surgery; 3-VO, three-vessel occlusion; AC, ameroid constrictor.
Table 2.
Difficulty, time, and cost of surgery of each animal model.
| Animal Models | Rat BCAO | Mouse BCAS | Mouse ACAS | Baboon 3-VO |
|---|---|---|---|---|
| Difficulty of surgery | Relatively easy, ligation of both CCAs | Moderate, placement of microcoils on both CCAs | Relarively high, placement of AC on the right CCA and microcoil on the left CCA | Difficult, ligation of both ICAs and left VA |
| Surgery time | 10 min | 10 min | 10 min | Not established |
| Cost | No cost | $50 (microcoil $25 × 2) | $125 (AC $100 + microcoil $25) | Not established |
BCAO, bilateral common carotid artery occlusion; BCAS, bilateral common carotid artery stenosis; ACAS, asymmetric common carotid artery surgery; 3-VO, three-vessel occlusion; CCA, common carotid artery; ICA, internal carotid artery; VA, vertebral artery; AC, ameroid constrictor.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20, 6176. https://doi.org/10.3390/ijms20246176
AMA Style
Washida K, Hattori Y, Ihara M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. International Journal of Molecular Sciences. 2019; 20(24):6176. https://doi.org/10.3390/ijms20246176
Chicago/Turabian StyleWashida, Kazuo, Yorito Hattori, and Masafumi Ihara. 2019. "Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate" International Journal of Molecular Sciences 20, no. 24: 6176. https://doi.org/10.3390/ijms20246176
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.